

Abstract
Signaling by neurotrophins such as the brain‐derived neurotrophic factor (BDNF) is known to modulate development of interneurons, but the circuit effects of this modulation remain unclear. Here, we examined the impact of deleting TrkB, a BDNF receptor, in parvalbumin‐expressing (PV) interneurons on the balance of excitation and inhibition (E‐I) in cortical circuits. In the mouse olfactory cortex, TrkB deletion impairs multiple aspects of PV neuronal function including synaptic excitation, intrinsic excitability, and the innervation pattern of principal neurons. Impaired PV cell function resulted in aberrant spiking patterns in principal neurons in response to stimulation of sensory inputs. Surprisingly, dampened PV neuronal function leads to a paradoxical decrease in overall excitability in cortical circuits. Our study demonstrates that, by modulating PV circuit plasticity and development, TrkB plays a critical role in shaping the evoked pattern of activity in a cortical network.
Keywords: GABAergic, neural circuits, neural plasticity
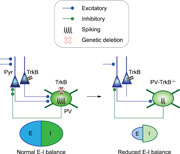
A model of how TrkB deletion in PV neurons weakens PV inhibition and ultimately reduces the excitation‐inhibition (E‐I) balance in the cortex. In this study, we found that genetic deletion of the TrkB receptor in PV neurons weakens the PV circuit by reducing the synaptic inputs, excitability, and the number of output synapses. Surprisingly, a weakened PV circuit ultimately leads to a reduction (not elevation) of E‐I balance in the anterior piriform cortex, altering cortical neural dynamics.
1. INTRODUCTION
Neurotrophins such as the brain‐derived neurotrophic factor (BDNF) affect multiple aspects of interneuron anatomy and physiology, including neurite growth and branching, synaptic strength, and long‐term synaptic plasticity, in major part by signaling through its receptor TrkB (Rutherford et al., 1997; X. Hong et al., 2008; X. Jin et al., 2003; Lu et al., 2005; Ohba et al., 2005). Neurotrophin signaling will, therefore, profoundly affect cortical function since inhibitory interneurons critically modulate diverse neural processes such as spike timing, tuning of receptive fields, and synaptic plasticity (Isaacson & Scanziani, 2011; Roux & Buzsáki, 2015). In addition to their role in normal physiology, BDNF and TrkB signaling is increasingly recognized as a potentially important therapeutic target in various neuropsychiatric disorders including Alzheimer's Disease and schizophrenia (Andero et al., 2014; Lu et al., 2013; Nagahara & Tuszynski, 2011).
Defects in the BDNF‐TrkB pathway and parvalbumin‐positive (PV) interneurons have been implicated in disorders such as schizophrenia (Hashimoto et al., 2005; Lewis et al., 2005) and autism spectrum disorders (Marín, 2012). PV interneurons exert particularly powerful inhibition on cortical principal neurons via perisomatic synapses (Freund & Katona, 2007; Hu et al., 2014). As such, activity‐dependent modulation of PV circuitry regulates diverse phenomena including excitation‐inhibition (E‐I) balance, gamma oscillations, and critical period initiation (Chevaleyre & Piskorowski, 2014; Takesian & Hensch, 2013). Genetic deletion of TrkB in PV neurons reduces the power of gamma oscillations in the hippocampus (Zheng et al., 2011) and visual cortex (Xenos et al., 2018), and induces motor hyperactivity and vestibular dysfunction (Lucas et al., 2014). Moreover, PV cell‐specific elimination of ErbB4, a receptor tyrosine kinase‐like TrkB, impacts both its synaptic input and output (Fazzari et al., 2010; Huang et al., 2021; Wen et al., 2010). How TrkB affects the function of PV neurons, however, is unclear. A key component in the BDNF‐TrkB signaling pathway could be glutamic acid decarboxylase 67 (GAD67), a GABA‐synthesizing enzyme that has been implicated in synaptic plasticity and dysfunction (Hartman et al., 2006; Hashimoto et al., 2005; Lau & Murthy, 2012; Lazarus et al., 2013; Sánchez‐Huertas & Rico, 2011). These studies raise the question of how TrkB signaling in PV neurons functions to control cortical circuit activity.
Here, we investigated the role of TrkB signaling in PV neurons in the anterior piriform cortex (APC), a simple three‐layer, non‐topographic cortex with similarities to the hippocampus and prefrontal cortex (Haberly, 2001; Shepherd, 2011; Wilson & Sullivan, 2011). APC is only one synapse downstream of the olfactory bulb (OB), making it a suitable region for studying plasticity. An advantage of the APC is its clear separation of the direct, sensory pathway (layer 1a) from the recurrent, associative pathway (layer 1b; Haberly, 2001). A lack of selective nearest‐neighbor interactions (Franks et al., 2011; Haberly, 2001) and the dispersed nature of information in this region suggests an important role for PV neurons in coordination of neural activity over large distances (Stokes & Isaacson, 2010; Suzuki & Bekkers, 2012). Using optogenetics, conditional genetics, electrophysiology, immunostaining, we investigated the cell‐autonomous effects of genetic deletion of TrkB within PV neurons, and the repercussion on downstream pyramidal cells. We show that genetic deletion of TrkB in PV neurons significantly impacts multiple aspects of PV neuron function, which markedly altered the network responses to activation of sensory inputs from OB.
2. MATERIALS AND METHODS
2.1. Animals and genetics
To generate the PV‐tomato mouse line, Pvalb Cre mouse (Jackson Laboratory 008069; referred to as PV‐Cre in this manuscript; Hippenmeyer et al., 2005) was crossed to a conditional Rosa26‐CAG::lox‐STOP‐lox‐tdTomato line (Ai9, Jackson Laboratory 007905; referred to as Tomato in this manuscript; Madisen et al., 2010). The Tomato expression is driven by the CMV‐IE enhancer/chicken beta‐actin/rabbit beta‐globin hybrid (CAG) promoter and should be activity‐independent. The effects of TrkB were examined by comparing littermates that contain one or both copies of the WT Ntrk2 gene (encoding TrkB protein) to those that completely lack them (TrkB‐null). We first generated two independent lines: (1) PV‐CreCre/+; TrkBF/+ (=PV‐TrkB+/−) and (2) TrkBF/F; Tomatotom/tom. The first line was generated by crossing homozygous PV‐CreCre/Cre to the homozygous TrkBF/F (from Dr. Louis Reichardt, UCSF (Grishanin et al., 2008). The second line was generated by crossing TrkBF/F to the Tomatotom/tom line and backcrossed to homozygosity for both alleles and identified using genotyping. The second round of breeding then involved crossing PV‐CreCre/+‐TrkBF/+ to TrkBF/F‐Tomatotom/tom (Figure 1a). PV‐CreCre/+; TrkBF/+; Tomatotom/+ is simplified as PV‐TrkB+/−, whereas PV‐CreCre/+; TrkBF/F; Tomatotom/+ is simplified as PV‐TrkB−/−. All offspring had one copy of the Tomato gene (Tomatotom/+). All procedures were performed in accordance with institutional (Harvard University Institutional Animal Care and Use Committee) and national guidelines. TrkB alleles were identified using the following primer pairs: TrkB‐n2: 5ʹ‐ATCTCGCCCTGGCTGAAGTG‐3ʹ and TrkB‐c8: 5ʹ‐ACTGACATCCGTAAGCCAGT‐3ʹ; WT product size, 369 bp; TrkBF product size, 450 bp. For Cre primers, forward (oIMR1084): GCG GTC TGG CAG TAA AAA CTA TC, reverse (oIMR1085): GTG AAA CAG CAT TGC TGT CAC TT; ~100 bp. PV‐Cre +/−‐TrkB f/f (referred to as PV‐TrkB−/−), compared to PV‐TrkB+/+ and PV‐TrkB+/− littermates, began displaying behavioral abnormalities like hindlimb clasping and hyperactive motor behavior at the end of postnatal Week 2, similar to recently reported observations (Lucas et al., 2014). We did not observe any overt seizure activity in the PV‐TrkB−/− mice. To measure motor activity, we tracked the animal's head position in an open‐box arena measuring 0.6 m by 0.6 m at a sampling rate of 10 Hz. The average speed of the animal was binned every second and reported.
Figure 1.
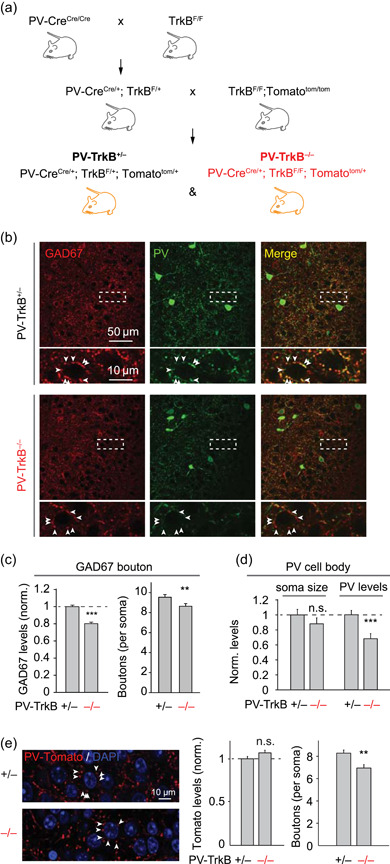
Loss of TrkB in PV cells affects the formation of PV synapses in APC. (a) Schematic showing how the triple genetic cross between PV‐Cre, TrkBF/F, and conditional Tomato (Ai9) mouse lines were made to generate the PV neuron‐specific knockout of TrkB together with Tomato reporter. (b–d) Representative confocal images (b) and a summary bar graph (mean ± SEM, c) showing that genetic deletion of TrkB in PV neurons (PV‐TrkB−/−) reduced the intensity and density of GAD67+ perisomatic punctae (arrowheads) surrounding pyramidal cells as compared to heterozygous control (PV‐TrkB+/−). GAD67 and PV colocalized in axons and boutons in APC. (d) Quantification of PV‐stained cell bodies revealed that TrkB removal decreased PV expression but not the size of the somata. (e) PV‐TrkB deletion reduced the density, but not intensity, of PV‐specific Tomato boutons around pyramidal cells. APC, anterior piriform cortex; PV, parvalbumin. *p < 0.05; **p < 0.01; ***p < 0.001
2.2. Stereotaxic injections
To express ChR2 specifically in PV cells, 100–300 nl of adeno‐associated virus carrying the ChR2‐YFP gene operating through the FLEX switch (AAV2/9.EF1a.DIO.hChR2(H134R)‐EYFP; Penn Vector Core) was injected stereotaxically (Stoelting) into APC of the PV‐Tomato mouse (Cre+) using these coordinates relative to bregma: lateral +2.7 mm, anterior +1.5 mm, ventral −3.5 mm. Two to three weeks after injection, acute APC slices were prepared for electrophysiology or brains were fixed for microscopy.
2.3. Electrophysiology and data analyses
Sagittal slices of the APC were prepared in ice‐cold slicing solution containing (in mM) 83 NaCl, 2.5 KCl, 0.5 CaCl2, 3.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 22 glucose, and 72 sucrose. Following a recovery period of 1–2 h, recordings were performed in artificial cerebrospinal fluid (ACSF) containing 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 22 glucose that is saturated with 95% O2 and 5% CO2 (carbogen) unless stated otherwise. Recording pipettes were pulled from borosilicate glass (WPI) and open‐tip resistance was typically 3–5 MΩ. Recordings were obtained from pyramidal or PV (Tomato+) cells using the whole‐cell patch‐clamp technique with an internal solution that is based either on Cs+ gluconate: (for voltage clamp, in mM) 130 d‐gluconic acid, 130 CsOH, 5 NaCl, 10 HEPES, 12 Di‐Tris‐P‐creatine, 1 EGTA, 3 Mg‐ATP and 0.2 Na‐GTP (pH 7.3; 290 mOsm) or K+ gluconate (components are the same except that 130 mM Cs+ gluconate was replaced by K+ gluconate, for current clamp). For recordings of EPSCs or IPSCs in PV neurons, a concentric bipolar electrode (FHC) was placed in the lateral olfactory tract (LOT) layer and tdTomato+ cells were patched and held at holding potentials (V h ) of −70 or 0 mV, respectively. To record synaptic potentials and spikes in pyramidal or PV cells, we used pipettes with K+ gluconate internal solution. To measure intrinsic excitability in PV cells, 1‐s long steady currents were injected to obtain the firing‐current (F‐I) curve. For spike waveform analysis, following detection of spikes, individual spikes were extracted using 5 ms before and 15 ms after the peak of upshoot. To measure spike activity in pyramidal cells, LOT was stimulated at an intensity that elicited a spike in the train in ~50% of the trials. Care was taken to avoid direct dendritic stimulation, which was indicated as spikes with near 0 ms latency. We set an upper limit for the stimulation strength used; if the cell did not fire any action potentials above 25 V, the cell was not included for spike analysis but was only used for the subthreshold analysis. For ChAQA experiments, ACSF contained 2.5 mM Sr2+ instead of Ca2+ and IPSCs were recorded using Cs+ gluconate internal solution with V h = 0 mV. ChR2+ PV cells were activated by brief (2 ms) pulses of blue LED light (473 nm) illuminating the back aperture of the objective. Amplitudes of quantal IPSCs with a latency of 100–1100 ms after the initial, single large IPSC were measured. Series resistance (<25 MΩ) was regularly monitored during recordings, and cells were rejected if resistance changed >20% during the experiment. Data were filtered at 2 kHz, digitized at 5–10 kHz, acquired through Axopatch 200B amplifier (Molecular Devices) and custom‐written scripts in Igor Pro 5 (Wavemetrics) or MATLAB (MathWorks). sIPSCs were analyzed using the Mini Analysis 6 program (Synaptosoft). Spike frequency, EPSC, and IPSC amplitudes were analyzed in Matlab (MathWorks). Significance was indicated if p < 0.05 using Student's t‐test or Mann–Whitney U‐test. Excitatory postsynaptic potentials (EPSPs) were quantified by measuring positive deflections of the amplitude within 20 ms following electric stimulus in the current clamp. To image the morphology of recorded cells, slices were fixed in 4% formaldehyde following recording. To visualize cells filled with biocytin, slices were incubated in streptavidin‐Alexa (488 or 568 nm, 1:500) at room temperature, mounted, and imaged with a confocal microscope (Zeiss 700).
2.4. Immunofluorescent imaging
Mice (~2 months old) were fixed by transcardial perfusion with 4% formaldehyde. Sagittal APC sections (100 µm) were blocked with PBS containing 5% normal goat serum and 0.1% Triton X‐100 and were incubated with an antibody against GAD67 (1:2000, Millipore), synaptotagmin‐2 (znp‐1 Syt2, 1:250, Developmental Studies Hybridoma Bank; Sommeijer & Levelt, 2012), PV (rabbit or mouse, 1:2000, Swant), or Homer1 (1:500, Synaptic Systems 160003) overnight at 4°C. Sections were decorated with secondary antibody conjugated with Alexa 488 or 568 dye and mounted in VectaShield (Vector Labs). Images were acquired with a confocal microscope (Zeiss 700 or 780) using a 40×, 1.3 NA oil‐immersion objective or Zeiss AxioImager Z2 with 10× objective at the Harvard Center for Biological Imaging. GAD67, PV‐Tomato, and Syt2 punctae were quantified using FIJI/ImageJ by putting 3 × 3 pixel boxes on perisomatic synapses. PV intensity and soma area were analyzed by hand‐drawn areas. For homer1 punctae analysis, PV neurons were defined as individual ROIs by using analyze particle tool in FIJI. The cells were checked by visual inspection. To pick up synapses, a threshold value for homer was determined finding a gray value that separates background fluorescence from Homer1+ punctae and set for all images. The minimal size of homer punctae was identified by measuring some typical punctae and used as a size filter. In each PV cell body, the number, area, intensity, and density of homer synapses were analyzed by using the analyze particle tool.
3. RESULTS
3.1. Generation of a triple transgenic cross between PV‐Cre, TrkB‐flox, and conditional tomato mouse lines
To examine the structure and function of PV cells in the mouse APC, we first examined a tomato reporter line for PV cells (generated by crossing the Pvalb‐ Cre line to a Cre‐dependent Tomato line, hereafter referred to as PV‐Tomato; see Section 2). The PV‐Tomato line appeared to faithfully report PV cell expression in APC since >90% of the Tomato+ cells colocalized with PV protein expression, ~50% of them colocalized with calbindin, and the Tomato+ cells did not overlap with markers for VIP‐ or CCK‐expressing interneurons (Figure S1; Suzuki & Bekkers, 2010). Whole‐cell patch‐clamp recording from Tomato+ PV cells showed that their maximal firing rate is >100 Hz, confirming their fast‐spiking phenotype (Figure 2). These data validate the PV‐tomato mouse line as a suitable reporter of PV cell identity in the APC.
Figure 2.
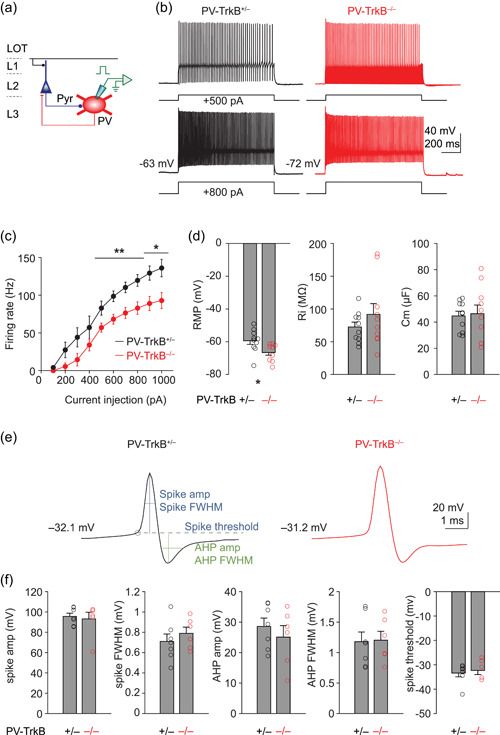
TrkB deletion dampens the excitability of PV neurons. (a) Schematic showing recording spike activity from PV cells (Tomato+) with K+ gluconate internal solution. (b, c) Representative spike activity (500 and 800 pA injection, b) and summary (c) showing injection of direct current into PV cells from PV‐TrkB+/− and revealed that the maximal firing rate of PV cells is reduced in PV‐TrkB−/− compared to PV‐TrkB+/− mice. (d) Effect of TrkB deletion on the resting membrane potential (RMP), input resistance (Ri), and capacitance (Cm) of PV cells. (e, f) Representative traces (e) and summary bar graphs (f) showing no detectable effect of TrkB deletion on extracted spike amplitude or full‐width half‐maximum (FWHM), afterhyperpolarization (AHP) amplitude or FWHM, or spike threshold. PV, parvalbumin. *p < 0.05; **p < 0.01; ***p < 0.001
To investigate the impact of TrkB on PV neuronal function, we compared littermates that contain one or two wild‐type copies of TrkB (PV‐TrkB+/+ or PV‐TrkB+/−; see Section 2) to those with homozygous deletion of TrkB in PV neurons (PV‐TrkB−/−; Figure 1a). Behaviorally, adult (2 months old) PV‐TrkB+/− mice appeared the same as wild‐type control (PV‐TrkB+/+), as qualitatively judged by normal observations in their home cages. In contrast, PV‐TrkB−/− mice began to exhibit hyperactive and circling behavior with head tilting by approximately P14 (Figure S3), corroborating with previous findings (Lucas et al., 2014; Xenos et al., 2018). These behavioral deficits were similar to those found in mice with conditional deletion of TrkB in the striatum (Baydyuk et al., 2011; Y. Li et al., 2012) or forebrain (Zörner et al., 2003). Moreover, PV‐TrkB−/− mice display hindlimb clasping behavior when held by their tail compared to PV‐TrkB+/− mice (Figure S3) in a similar manner to mice that are hypomorphic for Bdnf, which codes for the major ligand of TrkB, BDNF (Chang et al., 2006). There was no gross alteration in neuroanatomy as revealed by Tomato expression in PV cells and laminar structure in the cortex revealed by nuclear staining DAPI (Figure S2).
3.2. TrkB loss reduces the number of PV synapses made on pyramidal neurons
The behavioral abnormalities of PV‐TrkB−/− mice suggested dysfunctional neural circuits in the brain. Whole‐brain deletion of TrkB (Alcántara et al., 1997) or conditional removal of TrkB in CaMKII+ (excitatory) neurons (Xu et al., 2000) results in neuronal loss. Does PV‐TrkB knockout lead to overall neuronal death in APC? To address this question, we quantified the DAPI+ neuronal density in APC L2. L2 DAPI+ cells are largely excitatory neurons and comprise the majority of neurons in APC. Knockout of TrkB in PV neurons did not significantly alter the number of DAPI+ cells in L2 of APC (PV‐TrkB+/− vs. PV‐TrkB−/−: 7505 ± 551 vs. 8777 ± 446 cells per mm2, n = 12, p = 0.09, Student's t‐test). This suggests that TrkB deletion in PV neurons did not induce cell death of excitatory neurons in the APC.
Next, we assessed whether there were defects in PV circuits in APC. We first addressed whether TrkB regulates PV synapse formation and development by examining perisomatic innervation on pyramidal cells as evaluated by the expression of GAD67, PV, and synaptotagmin‐2 (Syt2; Figures 1 and S4). GAD67 colocalized with PV and appeared punctate in the perisomatic region of principal neurons (Figure 1a). Homozygous deletion of TrkB in PV cells reduced the intensity (to 82.4 ± 3% of PV‐TrkB+/− control, n = 50 cells, p < 0.0001; Figure 1a,b) and density (from 9.5 ± 0.3 to 8.7 ± 0.3 compared to PV‐TrkB+/− control, n = 50 cells, p < 0.02; Figure 1a,b) of GAD67+ punctae around pyramidal cell bodies. We also found that PV expression was drastically reduced in PV‐TrkB−/− neurons (from 100 ± 6% to 68 ± 7%, n = 17 cells, p < 0.001, Figure 1c). PV cell body size was unaffected by TrkB deletion (100 ± 7% vs. 88 ± 8%, n = 17 cells, p = 0.28, Figure 1c), consistent with the lack of change in electrical capacitance of PV cells (Figure 2d). We next measured the effects of TrkB deletion on Syt2, a presynaptic protein selectively expressed on PV cells (Sommeijer & Levelt, 2012). The levels (to 69.4 ± 3.4% of PV‐TrkB+/− control, n = 151 punctae, p < 0.001; Figure S4) and density (PV‐TrkB+/− vs. PV‐TrkB−/−, 7.6 ± 0.3 vs. 6.6 ± 0.3, n = 37 vs. 28 cells, p < 0.02; Figure S4) of Syt2 were also markedly reduced by TrkB deletion. To understand whether the reduction in synaptic density is caused by a reduction in GAD67 and/or Syt2 staining levels (leading to some synapses going below detection threshold), we also evaluated PV synapses as marked by Tomato, whose expression should not be activity‐dependent. As we performed a triple cross between PV‐Cre, TrkB‐flox, and conditional Tomato (see Section 2), the PV‐TrkB+/− (het) mouse was used as a control to compare against PV‐TrkB−/−. TrkB deletion reduced the density of Tomato+ boutons (PV‐TrkB+/− vs. PV‐TrkB−/−: 8.3 ± 0.3 vs. 6.9 ± 0.3, n = 50 vs. 56 cells, p < 0.002; Figure 1d) surrounding the pyramidal cell's soma without altering the intensity levels (PV‐TrkB−/−: 107 ± 3% of PV‐TrkB+/−, p = 0.09; Figure 1d). Moreover, we found that TrkB deletion in PV neurons significantly reduced the number of Tomato+ PV cells (PV‐TrkB+/− vs. PV‐TrkB−/−: 22.3 ± 1.4 vs. 9.4 ± 1.9 cells per mm2, n = 12 vs. 11 sections, p = 0.0004, Wilcoxon rank‐sum test). As the expression of Tomato should not be activity‐dependent, this result suggests that PV neurons died when TrkB was absent. To assess whether TrkB deletion also affected PV cell morphology, we quantified the number of dendrites emanating from each PV cell body. TrkB deletion did not significantly alter the number of dendrites on PV cells (PV‐TrkB+/− vs. PV‐TrkB−/−: 2.3 ± 0.1 vs. 2.1 ± 0.2 dendrites per soma, n = 95 vs. 29 cells, p = 0.29, Wilcoxon rank‐sum test), suggesting that TrkB does not play a major role in determining the dendritic arborization during postnatal development. Altogether, deletion of TrkB resulted in the death of PV neurons without alteration of their dendritic arborization or death of excitatory neurons in APC.
3.3. TrkB deletion weakens inhibition received by pyramidal neurons
Does the reduction in the number of PV cells and synapses translate into functional differences? To answer this question, we next examined how deletion of TrkB impacts inhibitory transmission seen by pyramidal neurons by recording spontaneous IPSCs (sIPSCs) in these cells (Figure 3a). The homozygous WT TrkB mouse (PV‐TrkB+/+) was used as a control because (1) we aimed to assay for the maximal effect of TrkB, (2) we did not need to label PV cells fluorescently, and (3) the effects on sIPSC could be minimal. TrkB deletion in PV cells significantly increased sIPSC interevent interval (PV‐TrkB+/+ vs. PV‐TrkB−/−, 573 ± 33 vs. 676 ± 37 ms, n = 8 vs. 11 cells, p < 0.05; Figure 3b,d) but did not alter their amplitude (PV‐TrkB+/+ vs. PV‐TrkB−/−, 39.6 ± , 1.6 pA vs. 44.4 ± 2.3 pA, n = 8 vs. 11 cells, p = 0.10; Figure 3b,c), similar to what is observed in the hippocampus (Zheng et al., 2011). These results suggested a change in the overall inhibitory drive received by pyramidal cells, but do not reveal whether this is due to changes specifically in PV synapses. As we previously found that GAD67 knockout diminished quantal amplitude at GABAergic synapses (Lau & Murthy, 2012) and TrkB deletion affected GAD67 expression (Figure 1), we hypothesized that TrkB deletion would impair the quantal size of PV synapses.
Figure 3.
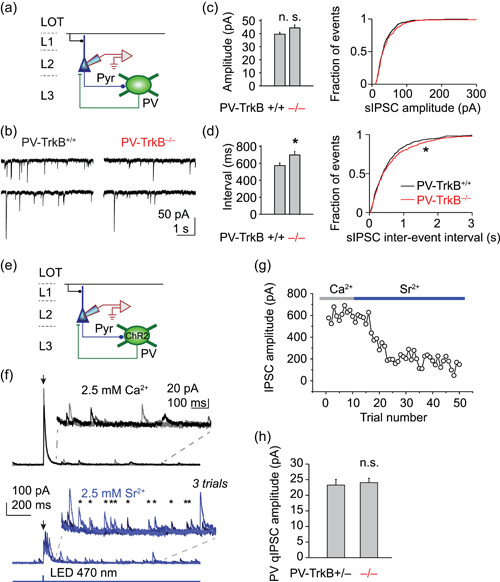
Reduced inhibitory synaptic activity in PV‐TrkB mutant mouse. (a–d) PV‐TrkB deletion reduced the number of inhibitory events in pyramidal cells. (a) Schematic showing whole‐cell patch‐clamp recordings of spontaneous IPSCs in layer 2 (L2) pyramidal cells in APC (CsCl internal solution with APV and CNQX to block NMDA and AMPA receptors, respectively; V h = −70 mV). Representative traces (b) and bar graphs (mean ± SEM) and summary cumulative probability distributions (c, d) showing that TrkB removal increased the interval (d) between spontaneous release events without altering the amplitude of inhibitory transmission (c). (e–h) PV‐TrkB mutant displayed normal quantal size. (e) Schematic showing IPSC recordings in a pyramidal cell by inducing synaptic release from PV cells with ChR2 (Cs gluconate internal solution; V h = 0 mV). (f) Three superimposed recording traces showing whole‐field illumination (2‐ms pulse) triggered synchronous release of GABA in 2.5 mM extracellular Ca2+ (top) but asynchronous release (individual release indicated by *) in presence of 2.5 mM Sr2+ (bottom). (g) Time course of reduction of the initial amplitude (within 20 ms) of EPSC by substitution of Sr2+ for Ca2+ (stimulation every 10 s). (h) Quantal amplitude of PV synapses was not altered by PV‐TrkB deletion. APC, anterior piriform cortex; LOT, lateral olfactory tract; n.s., nonsignificant; PV, parvalbumin. *p < 0.05; **p < .0.01; ***p < 0.001
To specifically monitor PV synaptic output, we stereotaxically injected an adeno‐associated virus (AAV) into the APC to express channelrhodopsin2 (flex‐ChR2‐YFP) in PV cells (Figure S5). In acute slices of APC taken 2–3 weeks after viral infection, whole‐field illumination (470 nm LED, 2 ms pulse width) elicited large IPSCs in principal neurons in normal ACSF, presumably from activation of many PV‐positive axons (Figures S4 and 3f). Strontium (Sr2+) has been previously shown to be able to support the synaptic release of neurotransmitters but induces asynchronous release of vesicles, allowing us to monitor the strength of single vesicles (quantal amplitude; Daw et al., 2009). Replacement of extracellular Ca2+ by Sr2+ induced asynchronous release of GABA from PV synapses (Nahmani & Turrigiano, 2014) as evidenced by the increase in the number of events from 200 to 1000 ms after the light pulse (Figure 3f) and the drop in amplitude of the initial peak (Figure 3g). Using this method, which we termed ChR2‐assisted quantal analysis (ChAQA), we determined the quantal size of PV synapses to be ~20 pA. Contrary to expectation, homozygous deletion of TrkB did not significantly alter the quantal amplitude of PV transmission compared to control (PV‐TrkB+/− vs. PV‐TrkB−/−, 26.3 ± 1.8 vs. 24.1 ± 1.4 pA, n = 8, p = 0.73; Figure 3h). It is possible that is that a 20% reduction in the levels of GAD67 was not sufficient to decrease the filling of vesicles with GABA. Taken together, these data suggest that deletion of TrkB in PV neurons reduces the number of synapses they make on principal neurons, without altering the elementary quantal amplitude.
Figure 4.
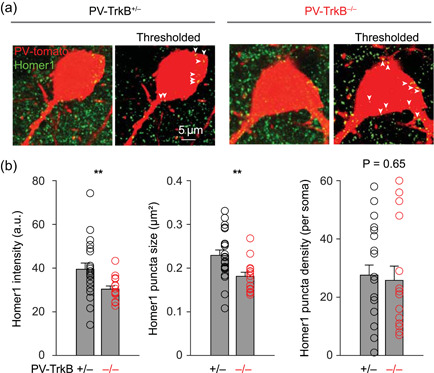
TrkB deletion alters excitatory synapses on PV cell bodies. (a) Representative images of raw and thresholded images of Homer1+ excitatory synapses on PV cell bodies (Tomato+). (b) Summary bar graphs showing that TrkB deletion reduced the intensity and size but not the density of Homer1 punctae. PV, parvalbumin. **p < 0.01
3.4. TrkB deletion reduces intrinsic excitability of PV neurons
We next examined whether deletion of TrkB affects the intrinsic excitability of PV neurons. We recorded from identified PV cells (Tomato+) and injected a series of direct currents from 100 to 1000 pA, in 100 pA steps, to construct a firing rate versus current curve (F‐I curve; Figure 2a). In PV‐TrkB+/− control neurons, firing frequency increased gradually and approached a near‐plateau with a current step of 1000 pA; maximal firing rate was upwards of 130 Hz (136 ± 12 Hz, n = 7, Figure 2b,c). In contrast, PV‐TrkB−/− shifted the F‐I curve downwards thereby reducing the maximal firing rate of PV cells (1000 pA injection, PV‐TrkB+/− vs. PV‐TrkB−/−, 136 ± 12 vs. 93 ± 11 Hz, n = 6, p < 0.05; Figure 2d). We found that the downshift of the PV FI curve was likely due to a hyperpolarized resting membrane potential (RMP) as PV‐TrkB−/− reduced PV cell RMP by ~7 mV without altering input resistance or capacitance (Figure 2d). As the maximal firing rate of PV cells can be determined by a variety of active conductance including sodium and potassium currents, we next analyzed the spike height, full width at half maximum (FWHM) of both spike upshoot and afterhyperpolarization (AHP). PV‐TrkB−/− did not detectably alter the amplitude or FWHM of either the spike or AHP nor the spike threshold (Figure 2e,f). These data suggest that TrkB critically regulates the neuronal excitability of PV neurons by regulating the DC offset for firing action potentials.
3.5. TrkB deletion reduces sensory input‐mediated recruitment of PV cells
BDNF signaling has been shown to regulate synaptic properties in the APC (Nanobashvili et al., 2005), but how it regulates the activity of PV neurons is unclear. Homer1 is a major component of the excitatory postsynaptic density and can be found in the soma of PV neurons (Favuzzi et al., 2017). To examine how TrkB affects excitatory synapses on PV neurons, we stained and quantified the Homer1‐positive synapses that were present on the PV cell bodies. We applied a threshold on both PV‐Tomato and the Homer1 signals and measured the intensity, size, and density of Homer1 punctae that were within the boundary of the PV cell body, similar to a previously published method (Exposito‐Alonso et al., 2020). On average, there were ~28 Homer1+ synapses on each PV cell body. TrkB deletion decreased the intensity and size, but not density, of individual Homer1 punctae (Figure 4a,b). This suggests that TrkB signaling is important for maintaining excitatory synapses on PV neurons.
Our results so far showed that the excitatory synapses were altered by the removal of TrkB in PV cells. To understand how TrkB affects excitatory inputs for PV neurons while excitatory and inhibitory circuits interact, we recorded from PV neurons while we electrically stimulated the main sensory input for APC, the LOT (five pulses at 10 Hz), mimicking mitral and tufted cell activity upstream in the OB (Figure 5a,b). LOT stimulation elicited polysynaptic EPSCs in PV cells that were transiently facilitating, but subsequently depressing in PV‐TrkB+/− control animal (fifth pulse amplitude: 55.9 ± 12% of first pulse, Figure 5c‐e). EPSC amplitude in the PV‐TrkB−/− mice depressed significantly faster than in control mice (fifth pulse, PV‐TrkB+/− vs. PV‐TrkB−/−, 56 ± 8% vs. 31 ± 6%, n = 7 vs. 10 cells, p = 0.02; Figure 5c‐e). The difference in circuit dynamics of EPSCs cannot be explained by a difference in stimulation strength as the first EPSC amplitude was similar (310 ± 65 vs. 286 ± 70 pA, n = 7 vs. 8 cells, p = 0.80) using similar electrical stimulation intensity (13.4 ± 2.2 vs. 17.5 ± 1.9 V, n = 7 vs. 8 cells, p = 0.09). Thus, TrkB deletion stifles excitatory recruitment of PV cells, suggesting that it is more difficult to induce PV cell spiking by a burst of sensory activity.
Figure 5.
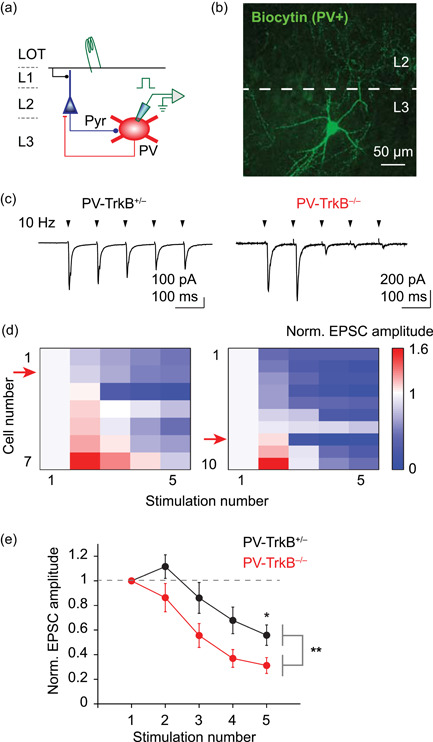
Genetic ablation of TrkB affects the excitatory drive of PV neurons. (a) Schematic of recording and stimulation configuration. Stimulation of main input layer LOT activates pyramidal cells, which in turn recruit PV cells, providing feedback inhibition. EPSCs were measured in PV cells in response to electrical stimulation of LOT (Cs gluconate internal solution, V h = −70 mV). (b) Representative confocal image of a recorded PV cell filled with biocytin in L3 (maximum‐intensity projection). Note that multiple dendrites emanate from the soma and extensive axonal arborization target the L2. (c) Five shocks delivered to the LOT at 10 Hz (arrowheads) elicited EPSCs that are, in general, depressing in amplitude in both PV‐TrkB+/− and PV‐TrkB−/− mice. Stimulus artifacts were removed for clarity. (d) Heat map showing the relative amplitude of EPSCs, normalized to the first pulse, for individual cells (rows) in response to five electrical stimulations (columns). Most cells exhibited depressing (blue), rather than facilitating (red), amplitudes of EPSCs. Red arrow indicates the representative cell illustrated in (c). (e) Quantification revealed that EPSCs in PV cells of PV‐TrkB−/− mice depressed more rapidly than compared to PV‐TrkB+/−. LOT, lateral olfactory tract; PV, parvalbumin. *p < 0.05; **p < 0.01
3.6. PV‐TrkB deletion results in altered sensory input‐induced activation of pyramidal neurons
Our data thus far suggest that TrkB deletion weakens the input, output, and excitability of PV cells. Based on these results, a prediction is that pyramidal cell firing would be aberrantly owing to altered APC circuit excitability. We tested this hypothesis by examining the integrative properties in pyramidal cells in response to LOT stimulation when TrkB is lacking in PV cells (Figure 6a). To assay for network excitability in the APC, we examined (1) spike pattern with perithreshold stimulation in response to electrical shocks of LOT and (2) EPSPs with subthreshold stimulation. With a weaker PV circuit whose recruitment depressed rapidly (Figures 4 and 5), we expected spike probability to increase in the latter half of the pulse train in the knockout animals. We varied the LOT stimulation intensity in current‐clamp mode and settled on an intensity that induced spikes in ~50% of the trials. In PV‐TrkB+/− control mice, spikes occurred in the latter half of the pulse train in control mice (Figure 6e,f), consistent with facilitation and a previous report (Stokes & Isaacson, 2010). Surprisingly, in PV‐TrkB−/− mice, spike probability in the fourth and fifth pulses were zero, and spikes could only be induced in the early part of the train (peak of spike probability for PV‐TrkB+/−: 0.27 at fifth pulse; the peak of spike probability for PV‐TrkB−/−: 0.35 at second pulse; Figure 6b‐d). Three out of seven (3/7) pyramidal cells could not be driven to spike in the PV‐TrkB−/− mice at a maximal stimulation intensity (Figure 6c). The appearance of spiking activity early in the train can be partly explained by a larger response on the first stimulation with comparable stimulation strength (first EPSP amplitude for PV‐TrkB+/− vs. PV‐TrkB−/−, 4.0 ± 0.6 vs. 8.3 ± 1.5 mV, n = 8 vs. 7 cells, p = 0.014; stimulation intensity, 23.2 ± 2.7 vs. 20.4 ± 2.7 V, n = 8 vs. 7 cells, p = 0.48). Can the alteration in spike pattern in PV‐TrkB−/− be caused by the underlying excitatory drive by EPSPs? Next, we recorded the subthreshold EPSPs in the same pyramidal cells by reducing the stimulation intensity by ~20% (PV‐TrkB+/− vs. PV‐TrkB−/− stimulation intensity, 18.6 ± 2.3 vs. 16.2 ± 2.2 V, n = 8 vs. 7 cells, p = 0.74). Subthreshold stimulation of LOT elicited EPSPs in pyramidal cells that generally facilitated in amplitude in control PV‐TrkB+/− mouse. In contrast, stimulation of sensory inputs evoked EPSPs in PV‐TrkB−/− mice that lacked facilitation, suggesting lower integrated excitatory activity in the network (fifth pulse normalized to first pulse, PV‐TrkB+/− vs. PV‐TrkB−/−: 135 ± 21% vs. 73 ± 15%, n = 7 vs. 7 cells, p = 0.04; Figure 6e‐g). These data indicate that blunted PV neuronal function, caused by TrkB deletion, greatly altered the integrative properties in pyramidal cells and piriform circuitry.
Figure 6.
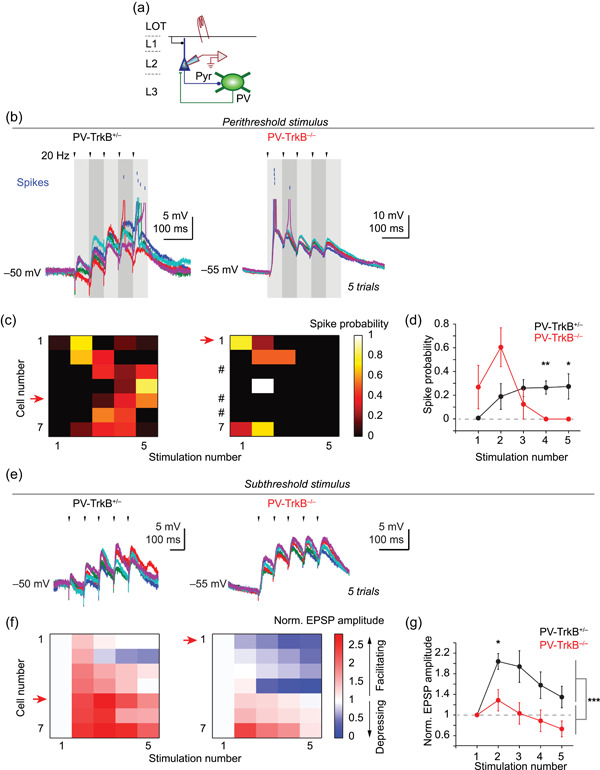
TrkB deletion in PV neurons disrupts the integrative properties of pyramidal cells in APC. (a) Schematic showing the recording of EPSPs and spikes in pyramidal cells in response to LOT stimulation in current‐clamp mode (K+ gluconate internal solution). (b–d) Perithreshold stimulation of LOT revealed different spike patterns in the PV‐TrkB null. (b) Five superimposed traces showing perithreshold stimulation‐induced spiking in pyramidal cells. Voltage traces were truncated at approximately −20 to −30 mV, and spikes were represented as blue lines above. (c,d) Heat map of individual cells (sorted by the normalized size of the second EPSP; c) and average line graph (d) showing that spike probability is higher in the latter part of the pulse train in PV‐TrkB+/− mice whereas it is higher in the earlier part for PV‐TrkB−/− mice. # denotes cells that did not fire action potentials even at the upper limit of stimulus intensity (25 V). (e–g) Subthreshold stimulation of LOT revealed altered circuit dynamics in the same pyramidal cells as in (b–d). (e) Five consecutive traces of LOT‐induced postsynaptic response in a representative cell for each genotype (arrowheads, electrical stimulation). (f, g) Heat map (sorted in the same order as in c; f) of individual cells and average (±SEM) of all cells (g) showed that EPSPs summated over the course of the 5‐train stimuli in PV‐TrkB+/− but not PV‐TrkB−/− mice. APC, anterior piriform cortex; LOT, lateral olfactory tract; PV, parvalbumin. *p < 0.05; **p < 0.01; ***p < 0.001
3.7. PV‐TrkB deletion causes widespread alteration in cortical E‐I balance
Our data thus far suggest that TrkB deletion in PV cells weakened their activation and output, which in turn, surprisingly, dampened network activation of pyramidal cells. Is this caused by a change in the network E‐I balance? To assess the E‐I ratio in APC, we recorded EPSC and IPSC in the same cell and varied the frequency of electrical stimulation of LOT, mimicking varying input from the OB (Figure 7a). In PV‐TrkB+/− control mice, LOT stimulation elicited EPSCs that were facilitating and IPSCs that were depressing (Figure 7b). Hence, the E‐I ratio increased over the course of a pulse train for both 10 and 20 Hz stimulation (Figure 7c). By contrast, although the E‐I ratio increased over the pulse train for PV‐TrkB−/− mice, the values are markedly lower than that of the PV‐TrkB+/− control for both 10 and 20 Hz stimulation. The difference in E‐I dynamics cannot be explained by experimental conditions since the amplitudes for the first EPSC or IPSC did not differ (EPSC1: 51 ± 18 vs. 37 ± 15 pA, p = 0.23; IPSC1: 101 ± 30 vs. 116 ± 23 pA, p = 0.57; stimulation intensity: 11.3 ± 1 vs. 11.3 ± 1.4 V, p = 0.99; all n = 8). These data are consistent with results from spike and EPSP recordings (Figure 6) where PV‐TrkB deletion resulted in reduced network excitability of pyramidal neurons. Taken together, these findings strongly indicate that PV‐TrkB deletion caused a widespread reduction in E‐I ratio in APC that results in difficulty in driving pyramidal neurons to spike.
Figure 7.
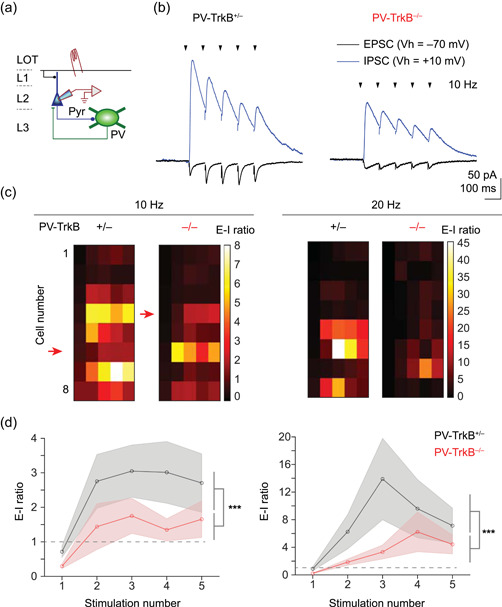
TrkB deletion in PV neurons reduces the excitation‐inhibition (E‐I) ratio of synaptic inputs in pyramidal cells. (a) Schematic showing recording from pyramidal cells. (b) Representative traces showing EPSC (V h = −70 mV) and IPSC (V h = +10 mV) resulting from 10 Hz stimulation of LOT. (c) Heat map showing the E‐I ratio measured for each individual pyramidal cell for 10 and 20 Hz stimulation, each row sorted by the cell. (d) Summary line graphs showing that although the E‐I ratio increased over the pulse train for both genotypes, the E‐I ratio was largely lower in the PV‐TrkB−/− mice. LOT, lateral olfactory tract; PV, parvalbumin
4. DISCUSSION
Genetic and expression studies have implicated activity‐dependent regulation of interneurons via modulation of PV, TrkB, and GAD67 proteins. However, little is known about how these molecular changes affect physiology at the network level where excitatory and inhibitory circuits are interacting. In this study, we used a combination of optogenetic, genetic, molecular, and electrophysiological methods to establish a causal role for TrkB on PV circuit function. We found that the loss of TrkB in PV neurons reduces their activation leading to altered network activity in response to sensory input (Figure 8).
Figure 8.
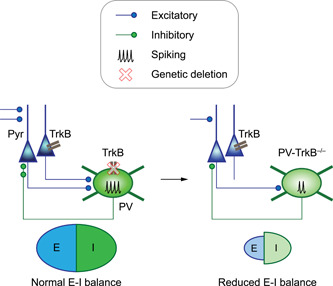
Model of how TrkB deletion in PV neurons weakens PV inhibition and ultimately reduces the excitation‐inhibition (E‐I) balance in the cortex. In this study, we found that genetic deletion of the TrkB receptor in PV neurons weakens the PV circuit by reducing the synaptic inputs, excitability, and the number of output synapses. Surprisingly, a weakened PV circuit ultimately leads to a reduction (not elevation) of E‐I balance in the anterior piriform cortex, altering cortical neural dynamics. PV, parvalbumin
4.1. Cell‐autonomous role of TrkB in on the function of PV neuronal circuits
Specific deletion of TrkB in PV cells significantly weakened their excitatory input (Figures 4 and 5), their intrinsic excitability (Figure 2), and their synaptic output (Figures 1 and 3), and the combined effect of these three changes will reduce the influence of PV neurons in the circuit they are embedded in. We show that TrkB deletion reduces the number of synapses formed by PV neurons onto pyramidal cells using measurements from three different markers: GAD67, Tomato, and Syt2. The reduction in GAD67‐positive PV boutons is consistent with the report that even loss of one copy of the GAD67 allele impairs the maturation and development of perisomatic innervation of pyramidal cells by PV neurons (Chattopadhyaya et al., 2007). Because BDNF is released in an activity‐dependent fashion (Jiao et al., 2011; Kuczewski et al., 2008; W. Li et al., 2012; Yang et al., 2009), our results support the hypothesis that TrkB signaling regulates GAD67 levels during PV circuit maturation. Corroborating this histological result is our finding that the frequency of spontaneous IPSCs in pyramidal neurons is reduced by the loss of TrkB (Figure 1).
Knockout of TrkB did not affect quantal size at PV synapses (Figure 3). Although we anticipated a drop in the quantal size because genetic deletion of GAD67 reduces the inhibitory quantal size by ~50% (Lau & Murthy, 2012), the lack of change can be explained by the much smaller reduction in expression for GAD67. TrkB deletion results in only a ~20% drop in GAD67 expression (Figure 1), and this modest change may not be sufficient to reduce the vesicular concentration of GABA in PV neurons. Such a nonlinear relation between cytosolic GABA and vesicular GABA may arise due to the complex interactions between GAD65/67, the vesicular GABA transporter, and even vesicle recycling machinery (Jin et al., 2003; Wang et al., 2013). The marked reduction in Syt2 staining at PV boutons suggests that other presynaptic properties may be affected, for example, the number of total vesicles in a bouton. We also show that TrkB deletion reduces the intrinsic excitability of PV cells, which reduces the maximal firing rates. These changes correlate with a reduction in PV protein expression as well as GAD67 expression. Interestingly, the levels of PV and GAD67 have been shown to co‐fluctuate in the hippocampus (Donato et al., 2013), and all of these changes could be related to reduced PV cell firing. Our observed changes in PV expression and IPSC frequency are consistent with findings in the hippocampus (Zheng et al., 2011), and extend them by revealing multiple synergistic alterations at the input, output, and excitability levels. TrkB deletion in PV neurons results in an ~50% decrease in the number of PV neurons, with an ~20% decrease in the number of synapses (Figure 1c,e). This suggests that TrkB deletion markedly affected PV neuronal survival while having a more modest effect on the number of PV synapses. Moreover, the strength of individual quantum of PV vesicles was not changed (Figure 3). Altogether, these results suggest that despite the strong reduction of PV neuronal number, other compensatory mechanisms are in place to maintain a certain number and strength of PV synapses. The mechanisms by which this is implemented remain to be discovered.
4.2. Effects of weakened PV circuit on cortical excitability
The constellation of changes to PV cells resulting from deletion of TrkB has significant consequences for network activity in APC. A previous study in the hippocampus indicated that gamma rhythms induced by carbachol in slices are altered by loss of TrkB in PV neurons (Zheng et al., 2011), but it remains unclear how the input‐output transform within a network is altered. The APC receives direct sensory input from the OB, as well as indirect inputs from other sensory regions such as the anterior olfactory nucleus (Haberly, 2001). Input from the OB arrives as bursts of spikes locked to sniffing (Wachowiak, 2011) and we mimicked this with a burst stimulus to the LOT. We found that pyramidal cells in wild‐type mice could accumulate synaptic excitation over multiple stimuli and spike at later times during trains of stimuli. In mice lacking TrkB in PV cells, synaptic inputs early in the train could already reach the threshold for spikes. Interestingly, this led to reduced spiking later in the stimulus train. Therefore, reduced PV cell function can lead to aberrant excitation of pyramidal cells during burst inputs that mimic in vivo patterns of mitral cell activity. Almost half (3/7) of the pyramidal cells could not be driven to spike in the PV‐TrkB null mice, while the remaining pyramidal cells elicit spikes earlier in the train (Figure 6). The difficulty to excite pyramidal cells to spike correlates well with the reduced ability to accumulate EPSPs (Figure 6), and reduced ability to increase E‐I over the course of a pulse train (Figures 7 and 8). Our results corroborate a recent study showing that expression of a dominant‐negative version of TrkB in PV cells in the prefrontal cortex reduces the firing rate of PV cells and muted gamma oscillations (Guyon et al., 2021), and extends it by showing that altered excitatory circuit dynamics in the sensory cortex. At the same time, Guyon et al. found that dominant‐negative TrkB reduces spike threshold spike half‐width while we did not observe these changes in APC. Moreover, PV‐TrkB deletion results in fewer inhibitory contacts with pyramidal neurons in cortical layer 5 but not the cerebellum (Xenos et al., 2018). Taken together, our findings and others' studies highlight region‐specific roles of TrkB.
4.3. BDNF and TrkB signaling in regulation of synapses and behavior
In a previous report, the knockout efficiency of TrkB in cortical PV neurons was estimated to be ~75% using immunofluorescent staining (Xenos et al., 2018). Our PV‐TrkB−/− began displaying hyperactive behavior around P14, when the expression of PV, and therefore, Cre, starts. Moreover, PV‐TrkB−/− mice display hindlimb clasping behavior when held by their tail compared to PV‐TrkB+/− mice (Figure S2) in a similar manner to mice that are hypomorphic for Bdnf, which codes for the major ligand of TrkB, BDNF (Chang et al., 2006). We, therefore, believe that the knockout efficiency of TrkB in PV neurons is high. It is possible, however, that the hyperactive motor behavior could be due to the removal of TrkB in the other brain regions and may not be attributable to the APC. This is because of evidence that selective removal of TrkB in either the cerebellum (Rico et al., 2002), striatum (Baydyuk et al., 2011; Y. Li et al., 2012), or forebrain (Zörner et al., 2003) can result in hyperactive behavior.
Is BDNF is the endogenous ligand for the TrkB receptor that induces these circuit effects? Classically, BDNF is known to be the major ligand for the TrkB receptor that binds with high affinity to promote neuronal survival and differentiation; BDNF binds to the low‐affinity receptor p75NTR to induce different intracellular signals and effects (Lu et al., 2005). NT4 is another ligand for TrkB but its expression in the brain normally is low (Fan et al., 2000). Acute application of BDNF depresses EPSCs in cortical slices, which in turn is entirely blocked by an anti‐TrkB antibody (Jiang et al., 2004). In the striatum, knockout of TrkB recapitulates the effects of BDNF knockout on spine density and intracellular signaling such as DARPP32 (Li et al., 2012). Together, these studies indicate that BDNF is the major ligand for TrkB. However, a very recent paper demonstrated a new phenomenon where cholesterol and even anti‐depressant drugs can directly bind to TrkB (Casarotto et al., 2021), suggesting new ways to influence TrkB signal activity.
What are the signaling pathways that could mediate BDNF/TrkB regulation of inhibitory synapses? BDNF‐induced suppression of inhibitory currents requires signaling via PLCγ and PKC in the cortex (Zhao & Levine, 2014), and PKC in the superior colliculus (Henneberger et al., 2002). In contrast, in the cerebellum, BDNF‐induced enhancement of inhibitory currents requires PLCγ and CaMKII but not PKC (Cheng & Yeh, 2005) and requires cdk5 (Huang et al., 2012). An ultimate effect of TrkB activation is that it promotes the assembly and maintenance of GABAergic synapses by increasing the amount of scaffold proteins, gephyrin, and contactin‐1 (Chen et al., 2011), but its signaling mechanism is unknown. Altogether, it is possible that in our system TrkB acts via kinases like CaMKII or PKC to exert its effects on synapses, but these remain to be tested.
4.4. Implication of BDNF/TrkB signaling and PV circuits in neuropsychiatric disorders
In schizophrenia, one of the most consistently found correlate between protein expression and post‐mortem brains and animal models is the altered expression of BDNF, TrkB, PV, and GAD67 (Behrens et al., 2007; Belforte et al., 2010; Hashimoto et al., 2005; Keilhoff et al., 2004; Lewis et al., 2005). Following TrkB removal in the PV cells, the multiple effects of reduced GAD67 and PV expression, PV neuronal firing, and input excitation are consistent with these findings. It is possible that dysfunction of the BDNF/TrkB pathway leads to aberrant homeostasis of PV circuits, an endophenotype found in various neuropsychiatric disorders (Lisman et al., 2008; Ramocki & Zoghbi, 2008). Supporting this is the implication of BDNF/TrkB signaling in a wide variety of neuropsychiatric disorders including schizophrenia, depression, and autism spectrum disorders (Ebert & Greenberg, 2013; Li & Pozzo‐Miller, 2014). By positively modulating inhibitory circuits, BDNF/TrkB signaling can strongly influence the E‐I state of a network. The BDNF/TrkB transduction pathway can, therefore, be harnessed as a new treatment target for neuropsychiatric disorders with known E‐I imbalance (Kavalali & Monteggia, 2020).
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The TrkBF/F mouse was a kind gift from Dr. Louis Reichardt, UCSF. This study was supported by the National Institutes of Health grant DC011291 (to V.N.M.) and the NARSAD Young Investigator Award 271644 (to C.G.L.) and the Hong Kong Research Grants Council (RGC/ECS 21103818 to C.G.L., and RGC/GRF 11104320 to C.G.L.). The authors thank David Gire for assistance with measurements of mouse behavior.
Lau, C. G. , Zhang, H. , & Murthy, V. N. (2022). Deletion of TrkB in parvalbumin interneurons alters cortical neural dynamics. J Cell Physiol, 237, 949–964. 10.1002/jcp.30571
Contributor Information
Chunyue Geoffrey Lau, Email: geoff.lau@cityu.edu.hk.
Venkatesh N. Murthy, Email: vnmurthy@fas.harvard.edu.
REFERENCES
- Alcántara, S. , Frisén, J. , del Río, J. A. , Soriano, E. , Barbacid, M. , & Silos‐Santiago, I. (1997). TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy‐induced cell death. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 17(10), 3623–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andero, R. , Choi, D. C. , & Ressler, K. J. (2014). BDNF‐TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Progress in Molecular Biology and Translational Science, 122, 169–192. 10.1016/B978-0-12-420170-5.00006-4 [DOI] [PubMed] [Google Scholar]
- Baydyuk, M. , Russell, T. , Liao, G.‐Y. , Zang, K. , An, J. J. , Reichardt, L. F. , & Xu, B. (2011). TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proceedings of the National Academy of Sciences of the United States of America, 108(4), 1669–1674. 10.1073/pnas.1004744108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens, M. M. , Ali, S. S. , Dao, D. N. , Lucero, J. , Shekhtman, G. , Quick, K. L. , & Dugan, L. L. (2007). Ketamine‐induced loss of phenotype of fast‐spiking interneurons is mediated by NADPH‐oxidase. Science, 318(5856), 1645–1647. 10.1126/science.1148045 [DOI] [PubMed] [Google Scholar]
- Belforte, J. E. , Zsiros, V. , Sklar, E. R. , Jiang, Z. , Yu, G. , Li, Y. , Quinlan, E. M. , & Nakazawa, K. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia‐like phenotypes. Nature Neuroscience, 13(1), 76–83. 10.1038/nn.2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarotto, P. C. , Girych, M. , Fred, S. M. , Kovaleva, V. , Moliner, R. , Enkavi, G. , Biojone, C. , Cannarozzo, C. , Sahu, M. P. , Kaurinkoski, K. , Brunello, C. A. , Steinzeig, A. , Winkel, F. , Patil, S. , Vestring, S. , Serchov, T. , Diniz, C. , Laukkanen, L. , Cardon, I. , … Castrén, E. (2021). Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell, 184(5), 1299–1313. 10.1016/j.cell.2021.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Q. , Khare, G. , Dani, V. , Nelson, S. , & Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron, 49(3), 341–348. 10.1016/j.neuron.2005.12.027 [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya, B. , Di Cristo, G. , Wu, C. Z. , Knott, G. , Kuhlman, S. , Fu, Y. , Palmiter, R. D. , & Huang, Z. J. (2007). GAD67‐mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron, 54(6), 889–903. 10.1016/j.neuron.2007.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, A. I. , Nguyen, C. N. , Copenhagen, D. R. , Badurek, S. , Minichiello, L. , Ranscht, B. , & Reichardt, L. F. (2011). TrkB (tropomyosin‐related kinase B) controls the assembly and maintenance of GABAergic synapses in the cerebellar cortex. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 31(8), 2769–2780. 10.1523/JNEUROSCI.4991-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, Q. , & Yeh, H. H. (2005). PLCgamma signaling underlies BDNF potentiation of Purkinje cell responses to GABA. Journal of Neuroscience Research, 79(5), 616–627. 10.1002/jnr.20397 [DOI] [PubMed] [Google Scholar]
- Chevaleyre, V. , & Piskorowski, R. (2014). Modulating excitation through plasticity at inhibitory synapses. Frontiers in Cellular Neuroscience, 8, 93. 10.3389/fncel.2014.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw, M. I. , Tricoire, L. , Erdelyi, F. , Szabo, G. , & McBain, C. J. (2009). Asynchronous transmitter release from cholecystokinin‐containing inhibitory interneurons is widespread and target‐cell independent. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 29(36), 11112–11122. 10.1523/JNEUROSCI.5760-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato, F. , Rompani, S. B. , & Caroni, P. (2013). Parvalbumin‐expressing basket‐cell network plasticity induced by experience regulates adult learning. Nature, 504(7479), 272–276. 10.1038/nature12866 [DOI] [PubMed] [Google Scholar]
- Ebert, D. H. , & Greenberg, M. E. (2013). Activity‐dependent neuronal signalling and autism spectrum disorder. Nature, 493(7432), 327–337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exposito‐Alonso, D. , Osório, C. , Bernard, C. , Pascual‐García, S. , Del Pino, I. , Marín, O. , & Rico, B. (2020). Subcellular sorting of neuregulins controls the assembly of excitatory‐inhibitory cortical circuits. eLife, 9, 9. 10.7554/eLife.57000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, G. , Egles, C. , Sun, Y. , Minichiello, L. , Renger, J. J. , Klein, R. , Liu, G. , & Jaenisch, R. (2000). Knocking the NT4 gene into the BDNF locus rescues BDNF deficient mice and reveals distinct NT4 and BDNF activities. Nature Neuroscience, 3(4), 350–357. 10.1038/73921 [DOI] [PubMed] [Google Scholar]
- Favuzzi, E. , Marques‐Smith, A. , Deogracias, R. , Winterflood, C. M. , Sánchez‐Aguilera, A. , Mantoan, L. , Maeso, P. , Fernandes, C. , Ewers, H. , & Rico, B. (2017). Activity‐dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron, 95(3), 639–655. 10.1016/j.neuron.2017.06.028 [DOI] [PubMed] [Google Scholar]
- Fazzari, P. , Paternain, A. V. , Valiente, M. , Pla, R. , Luján, R. , Lloyd, K. , Lerma, J. , Marín, O. , & Rico, B. (2010). Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature, 464(7293), 1376–1380. 10.1038/nature08928 [DOI] [PubMed] [Google Scholar]
- Franks, K. M. , Russo, M. J. , Sosulski, D. L. , Mulligan, A. A. , Siegelbaum, S. A. , & Axel, R. (2011). Recurrent circuitry dynamically shapes the activation of piriform cortex. Neuron, 72(1), 49–56. 10.1016/j.neuron.2011.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund, T. F. , & Katona, I. (2007). Perisomatic inhibition. Neuron, 56(1), 33–42. 10.1016/j.neuron.2007.09.012 [DOI] [PubMed] [Google Scholar]
- Grishanin, R. N. , Yang, H. , Liu, X. , Donohue‐Rolfe, K. , Nune, G. C. , Zang, K. , Xu, B. , Duncan, J. L. , Lavail, M. M. , Copenhagen, D. R. , & Reichardt, L. F. (2008). Retinal TrkB receptors regulate neural development in the inner, but not outer, retina. Molecular and Cellular Neurosciences, 38(3), 431–443. 10.1016/j.mcn.2008.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon, N. , Zacharias, L. R. , van Lunteren, J. A. , Immenschuh, J. , Fuzik, J. , Märtin, A. , Xuan, Y. , Zilberter, M. , Kim, H. , Meletis, K. , Lopes‐Aguiar, C. , & Carlén, M. (2021). Adult trkB signaling in parvalbumin interneurons is essential to prefrontal network dynamics. The Journal of Neuroscience, 41, 3120–3141. 10.1523/JNEUROSCI.1848-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberly, L. B. (2001). Parallel‐distributed processing in olfactory cortex: New insights from morphological and physiological analysis of neuronal circuitry. Chemical Senses, 26(5), 551–576. [DOI] [PubMed] [Google Scholar]
- Hartman, K. N. , Pal, S. K. , Burrone, J. , & Murthy, V. N. (2006). Activity‐dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nature Neuroscience, 9(5), 642–649. 10.1038/nn1677 [DOI] [PubMed] [Google Scholar]
- Hashimoto, T. , Bergen, S. E. , Nguyen, Q. L. , Xu, B. , Monteggia, L. M. , Pierri, J. N. , Sun, Z. , Sampson, A. R. , & Lewis, D. A. (2005). Relationship of brain‐derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. The Journal of Neuroscience, 25(2), 372–383. 10.1523/JNEUROSCI.4035-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger, C. , Jüttner, R. , Rothe, T. , & Grantyn, R. (2002). Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. Journal of Neurophysiology, 88(2), 595–603. 10.1152/jn.2002.88.2.595 [DOI] [PubMed] [Google Scholar]
- Hippenmeyer, S. , Vrieseling, E. , Sigrist, M. , Portmann, T. , Laengle, C. , Ladle, D. R. , & Arber, S. (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLOS Biology, 3(5), e159. 10.1371/journal.pbio.0030159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, E. J. , McCord, A. E. , & Greenberg, M. E. (2008). A biological function for the neuronal activity‐dependent component of Bdnf transcription in the development of cortical inhibition. Neuron, 60(4), 610–624. 10.1016/j.neuron.2008.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. , Gan, J. , & Jonas, P. (2014). Interneurons. Fast‐spiking, parvalbumin+ GABAergic interneurons: From cellular design to microcircuit function. Science, 345(6196), 1255263. 10.1126/science.1255263 [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Jiang, H. , Zheng, Q. , Fok, A. H. K. , Li, X. , Lau, C. G. , & Lai, C. S. W. (2021). Environmental enrichment or selective activation of parvalbumin‐expressing interneurons ameliorates synaptic and behavioral deficits in animal models with schizophrenia‐like behaviors during adolescence. Molecular Psychiatry. 10.1038/s41380-020-01005-w [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Ko, H. , Cheung, Z. H. , Yung, K. K. L. , Yao, T. , Wang, J.‐J. , Morozov, A. , Ke, Y. , Ip, N. Y. , & Yung, W.‐H. (2012). Dual actions of brain‐derived neurotrophic factor on GABAergic transmission in cerebellar Purkinje neurons. Experimental Neurology, 233(2), 791–798. 10.1016/j.expneurol.2011.11.043 [DOI] [PubMed] [Google Scholar]
- Isaacson, J. S. , & Scanziani, M. (2011). How inhibition shapes cortical activity. Neuron, 72(2), 231–243. 10.1016/j.neuron.2011.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, B. , Kitamura, A. , Yasuda, H. , Sohya, K. , Maruyama, A. , Yanagawa, Y. , Obata, K. , & Tsumoto, T. (2004). Brain‐derived neurotrophic factor acutely depresses excitatory synaptic transmission to GABAergic neurons in visual cortical slices. The European Journal of Neuroscience, 20(3), 709–718. 10.1111/j.1460-9568.2004.03523.x [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Zhang, Z. , Zhang, C. , Wang, X. , Sakata, K. , Lu, B. , & Sun, Q.‐Q. (2011). A key mechanism underlying sensory experience‐dependent maturation of neocortical GABAergic circuits in vivo. Proceedings of the National Academy of Sciences of the United States of America, 108(29), 12131–12136. 10.1073/pnas.1105296108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, H. , Wu, H. , Osterhaus, G. , Wei, J. , Davis, K. , Sha, D. , Floor, E. , Hsu, C.‐C. , Kopke, R. D. , & Wu, J.‐Y. (2003). Demonstration of functional coupling between gamma‐aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proceedings of the National Academy of Sciences of the United States of America, 100(7), 4293–4298. 10.1073/pnas.0730698100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X. , Hu, H. , Mathers, P. H. , & Agmon, A. (2003). Brain‐derived neurotrophic factor mediates activity‐dependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures. The Journal of Neuroscience, 23(13), 5662–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali, E. T. , & Monteggia, L. M. (2020). Targeting homeostatic synaptic plasticity for treatment of mood disorders. Neuron, 106(5), 715–726. 10.1016/j.neuron.2020.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhoff, G. , Becker, A. , Grecksch, G. , Wolf, G. , & Bernstein, H.‐G. (2004). Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience, 126(3), 591–598. 10.1016/j.neuroscience.2004.03.039 [DOI] [PubMed] [Google Scholar]
- Kuczewski, N. , Porcher, C. , Ferrand, N. , Fiorentino, H. , Pellegrino, C. , Kolarow, R. , Lessmann, V. , Medina, I. , & Gaiarsa, J.‐L. (2008). Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. The Journal of Neuroscience, 28(27), 7013–7023. 10.1523/JNEUROSCI.1673-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, C. G. , & Murthy, V. N. (2012). Activity‐dependent regulation of inhibition via GAD67. The Journal of Neuroscience, 32(25), 8521–8531. 10.1523/JNEUROSCI.1245-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus, M. S. , Krishnan, K. , & Huang, Z. J. (2013). GAD67 deficiency in parvalbumin interneurons produces deficits in inhibitory transmission and network disinhibition in mouse prefrontal cortex. Cerebral Cortex, 25, 1290–1296. 10.1093/cercor/bht322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, D. A. , Hashimoto, T. , & Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nature Reviews Neuroscience, 6(4), 312–324. 10.1038/nrn1648 [DOI] [PubMed] [Google Scholar]
- Li, W. , Calfa, G. , Larimore, J. , & Pozzo‐Miller, L. (2012). Activity‐dependent BDNF release and TRPC signaling is impaired in hippocampal neurons of Mecp2 mutant mice. Proceedings of the National Academy of Sciences of the United States of America, 109(42), 17087–17092. 10.1073/pnas.1205271109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , & Pozzo‐Miller, L. (2014). BDNF deregulation in Rett syndrome. Neuropharmacology, 76(Pt C), 737–746. 10.1016/j.neuropharm.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Yui, D. , Luikart, B. W. , McKay, R. M. , Li, Y. , Rubenstein, J. L. , & Parada, L. F. (2012). Conditional ablation of brain‐derived neurotrophic factor‐TrkB signaling impairs striatal neuron development. Proceedings of the National Academy of Sciences of the United States of America, 109(38), 15491–15496. 10.1073/pnas.1212899109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman, J. E. , Coyle, J. T. , Green, R. W. , Javitt, D. C. , Benes, F. M. , Heckers, S. , & Grace, A. A. (2008). Circuit‐based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends in Neurosciences, 31(5), 234–242. 10.1016/j.tins.2008.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, B. , Nagappan, G. , Guan, X. , Nathan, P. J. , & Wren, P. (2013). BDNF‐based synaptic repair as a disease‐modifying strategy for neurodegenerative diseases. Nature Reviews Neuroscience, 14(6), 401–416. 10.1038/nrn3505 [DOI] [PubMed] [Google Scholar]
- Lu, B. , Pang, P. T. , & Woo, N. H. (2005). The yin and yang of neurotrophin action. Nature Reviews Neuroscience, 6(8), 603–614. 10.1038/nrn1726 [DOI] [PubMed] [Google Scholar]
- Lucas, E. K. , Jegarl, A. , & Clem, R. L. (2014). Mice lacking TrkB in parvalbumin‐positive cells exhibit sexually dimorphic behavioral phenotypes. Behavioural Brain Research, 274, 219–225. 10.1016/j.bbr.2014.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen, L. , Zwingman, T. A. , Sunkin, S. M. , Oh, S. W. , Zariwala, H. A. , Gu, H. , Ng, L. L. , Palmiter, R. D. , Hawrylycz, M. J. , Jones, A. R. , Lein, E. S. , & Zeng, H. (2010). A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience, 13(1), 133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín, O. (2012). Interneuron dysfunction in psychiatric disorders. Nature Reviews Neuroscience, 13(2), 107–120. 10.1038/nrn3155 [DOI] [PubMed] [Google Scholar]
- Nagahara, A. H. , & Tuszynski, M. H. (2011). Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nature Reviews Drug Discovery, 10(3), 209–219. 10.1038/nrd3366 [DOI] [PubMed] [Google Scholar]
- Nahmani, M. , & Turrigiano, G. G. (2014). Deprivation‐induced strengthening of presynaptic and postsynaptic inhibitory transmission in layer 4 of visual cortex during the critical period. The Journal of Neuroscience, 34(7), 2571–2582. 10.1523/JNEUROSCI.4600-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanobashvili, A. , Jakubs, K. , & Kokaia, M. (2005). Chronic BDNF deficiency permanently modifies excitatory synapses in the piriform cortex. Journal of Neuroscience Research, 81(5), 696–705. 10.1002/jnr.20578 [DOI] [PubMed] [Google Scholar]
- Ohba, S. , Ikeda, T. , Ikegaya, Y. , Nishiyama, N. , Matsuki, N. , & Yamada, M. K. (2005). BDNF locally potentiates GABAergic presynaptic machineries: Target‐selective circuit inhibition. Cerebral Cortex, 15(3), 291–298. 10.1093/cercor/bhh130 [DOI] [PubMed] [Google Scholar]
- Ramocki, M. B. , & Zoghbi, H. Y. (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature, 455(7215), 912–918. 10.1038/nature07457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico, B. , Xu, B. , & Reichardt, L. F. (2002). TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nature Neuroscience, 5(3), 225–233. 10.1038/nn808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, L. , & Buzsáki, G. (2015). Tasks for inhibitory interneurons in intact brain circuits. Neuropharmacology, 88, 10–23. 10.1016/j.neuropharm.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, L. C. , DeWan, A. , Lauer, H. M. , & Turrigiano, G. G. (1997). Brain‐derived neurotrophic factor mediates the activity‐dependent regulation of inhibition in neocortical cultures. The Journal of Neuroscience, 17(12), 4527–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Huertas, C. , & Rico, B. (2011). CREB‐dependent regulation of GAD65 transcription by BDNF/TrkB in cortical interneurons. Cerebral Cortex, 21(4), 777–788. 10.1093/cercor/bhq150 [DOI] [PubMed] [Google Scholar]
- Shepherd, G. M. (2011). The microcircuit concept applied to cortical evolution: From three‐layer to six‐layer cortex. Frontiers in Neuroanatomy, 5, 30. 10.3389/fnana.2011.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommeijer, J.‐P. , & Levelt, C. N. (2012). Synaptotagmin‐2 is a reliable marker for parvalbumin positive inhibitory boutons in the mouse visual cortex. PLOS One, 7(4), e35323. 10.1371/journal.pone.0035323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, C. C. A. , & Isaacson, J. S. (2010). From dendrite to soma: Dynamic routing of inhibition by complementary interneuron microcircuits in olfactory cortex. Neuron, 67(3), 452–465. 10.1016/j.neuron.2010.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, N. , & Bekkers, J. M. (2010). Distinctive classes of GABAergic interneurons provide layer‐specific phasic inhibition in the anterior piriform cortex. Cerebral Cortex, 20(12), 2971–2984. 10.1093/cercor/bhq046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, N. , & Bekkers, J. M. (2012). Microcircuits mediating feedforward and feedback synaptic inhibition in the piriform cortex. The Journal of Neuroscience, 32(3), 919–931. 10.1523/JNEUROSCI.4112-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takesian, A. E. , & Hensch, T. K. (2013). Balancing plasticity/stability across brain development. Progress in Brain Research, 207, 3–34. 10.1016/B978-0-444-63327-9.00001-1 [DOI] [PubMed] [Google Scholar]
- Wachowiak, M. (2011). All in a sniff: Olfaction as a model for active sensing. Neuron, 71(6), 962–973. 10.1016/j.neuron.2011.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Tu, P. , Bonet, L. , Aubrey, K. R. , & Supplisson, S. (2013). Cytosolic transmitter concentration regulates vesicle cycling at hippocampal GABAergic terminals. Neuron, 80(1), 143–158. 10.1016/j.neuron.2013.07.021 [DOI] [PubMed] [Google Scholar]
- Wen, L. , Lu, Y.‐S. , Zhu, X.‐H. , Li, X.‐M. , Woo, R.‐S. , Chen, Y.‐J. , Yin, D.‐M. , Lai, C. , Terry, A. V. , Vazdarjanova, A. , Xiong, W.‐C. , & Mei, L. (2010). Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin‐positive interneurons. Proceedings of the National Academy of Sciences of the United States of America, 107(3), 1211–1216. 10.1073/pnas.0910302107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, D. A. , & Sullivan, R. M. (2011). Cortical processing of odor objects. Neuron, 72(4), 506–519. 10.1016/j.neuron.2011.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xenos, D. , Kamceva, M. , Tomasi, S. , Cardin, J. A. , Schwartz, M. L. , & Vaccarino, F. M. (2018). Loss of TrkB signaling in parvalbumin‐expressing basket cells results in network activity disruption and abnormal behavior. Cerebral Cortex, 28(10), 3399–3413. 10.1093/cercor/bhx173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, B. , Zang, K. , Ruff, N. L. , Zhang, Y. A. , McConnell, S. K. , Stryker, M. P. , & Reichardt, L. F. (2000). Cortical degeneration in the absence of neurotrophin signaling: Dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron, 26(1), 233–245. 10.1016/s0896-6273(00)81153-8 [DOI] [PubMed] [Google Scholar]
- Yang, J. , Siao, C.‐J. , Nagappan, G. , Marinic, T. , Jing, D. , McGrath, K. , Chen, Z.‐Y. , Mark, W. , Tessarollo, L. , Lee, F. S. , Lu, B. , & Hempstead, B. L. (2009). Neuronal release of proBDNF. Nature Neuroscience, 12(2), 113–115. 10.1038/nn.2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , & Levine, E. S. (2014). BDNF‐endocannabinoid interactions at neocortical inhibitory synapses require phospholipase C signaling. Journal of Neurophysiology, 111(5), 1008–1015. 10.1152/jn.00554.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, K. , An, J. J. , Yang, F. , Xu, W. , Xu, Z.‐Q. D. , Wu, J. , Hökfelt, T. G. M. , Fisahn, A. , Xu, B. , & Lu, B. (2011). TrkB signaling in parvalbumin‐positive interneurons is critical for gamma‐band network synchronization in hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 108(41), 17201–17206. 10.1073/pnas.1114241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörner, B. , Wolfer, D. P. , Brandis, D. , Kretz, O. , Zacher, C. , Madani, R. , Grunwald, I. , Lipp, H.‐P. , Klein, R. , Henn, F. A. , & Gass, P. (2003). Forebrain‐specific trkB‐receptor knockout mice: Behaviorally more hyperactive than “depressive. Biological Psychiatry, 54(10), 972–982. 10.1016/s0006-3223(03)00418-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.