

Abstract
Hormaomycins and belactosins are peptide natural products that contain unusual cyclopropane moieties. Bioinformatics analysis of the corresponding biosynthetic gene clusters showed that two conserved genes, hrmI/belK and hrmJ/belL, were potential candidates for catalyzing cyclopropanation. Using in vivo and in vitro assays, the functions of HrmI/BelK and HrmJ/BelL were established. HrmI and BelK, which are heme oxygenase-like dinuclear iron enzymes, catalyze oxidation of the ε-amino group of l-lysine to afford l-6-nitronorleucine. Subsequently, HrmJ and BelL, which are iron- and α-ketoglutarate-dependent oxygenases, effectively convert l-6-nitronorleucine into 3-(trans-2-nitrocyclopropyl)alanine through C4–C6 bond installation. These observations disclose a novel pathway of cyclopropane ring construction and exemplify the new chemistry involving metalloenzymes in natural product biosynthesis.
Keywords: biosynthesis, cyclopropane, metalloenzymes, natural products, peptides
Graphical Abstract
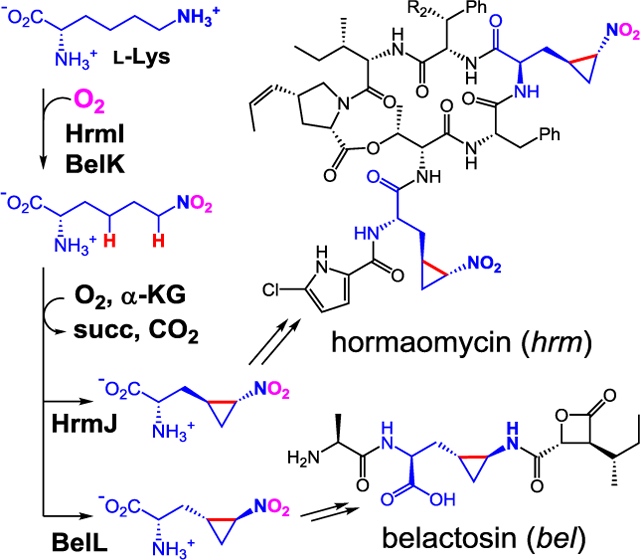
The biosynthetic pathway of amino/nitro-cyclopropylalanine moieties in hormaomycin and belactosin was fully elucidated. The pathway involves six-electron oxidation of the ε-amino group of l-lysine by heme oxygenase-like dinuclear iron enzymes (HrmI/BelK) and C–C bond formation by iron- and α-ketoglutarate-dependent oxygenases (HrmJ/BelL). This is the first example of Fe/α-KG oxygenase-catalyzed cyclopropane formation.
Cyclopropyl groups are found in various natural products, including terpenes, fatty acids, and peptides.[1] Owing to the rigidity and inherent ring strain of these three-membered ring structures, many cyclopropane-containing natural products have attractive biological activities. For example, duocarmycin exhibits potent antitumor activity via DNA alkylation.[2] Besides, coronatine produced by plant pathogenic Pseudomonas strains structurally mimics the plant hormone jasmonic acid–isoleucine, serving as a virulence factor to promote bacterial infection.[3] In addition, cyclopropyl scaffold is frequently seen in the FDA approved drugs.[4] In nature, several different strategies have been reported to enable cyclopropanation where different type enzymes with various cofactors including S-adenosylmethionine, pyridoxal 5′-phosphate, and flavin adenine dinucleotide are utilized (Figure S1).[1]
Hormaomycins and belactosins are natural products that contain cyclopropane amino acid residues 3-(trans-2-nitrocyclopropyl)-alanine and 3-(trans-2-aminocyclopropyl)-alanine, respectively (Figure 1). Hormaomycins, also known as takaokamycins, are depsipeptides originally isolated from Streptomyces griseoflavus W-384.[5] Hormaomycins have been reported to stimulate secondary metabolite production and show narrow-spectrum antibacterial specificity toward coryneform bacteria. Belactosins are proteasome inhibitors isolated from Streptomyces sp. KY11780 and Streptomyces UCK 14.[6] Belactosin C contains an ornithine residue within the dipeptide moiety, while belactosin A contains a 3-(trans-2-aminocyclopropyl)-alanine residue. Isotope tracer experiments have shown that the 3-(trans-2-nitrocyclopropyl)-alanine moiety in hormaomycins is derived from l-lysine (1) and 3-(trans-2-nitrocyclopropyl)-alanine (3), indicating that 3, in theory, is biosynthesized using 1 as the substrate.[7] In addition, analysis of the biosynthetic gene clusters of hormaomycin (hrm)[8] and belactosin (bel)[6a] showed that the peptide bonds in hormaomycin and belactosin are assembled by non-ribosomal peptide synthetases and ATP-dependent carboxylate-amine ligases, respectively. However, these cluster lacked candidate genes with the predicted functions suitable for nitro- and amino-cyclopropylalanine formation. Therefore, an unidentified pathway is likely involved in the biosynthesis of cyclopropane moieties in hormaomycins and belactosins. In contrast, bioinformatics analysis of hrm and bel clusters has revealed that two genes, hrmI/belK and hrmJ/belL, with uncharacterized functions are conserved, suggesting that these genes might be responsible for nitro- and amino-cyclopropylalanine formation.
Figure 1.
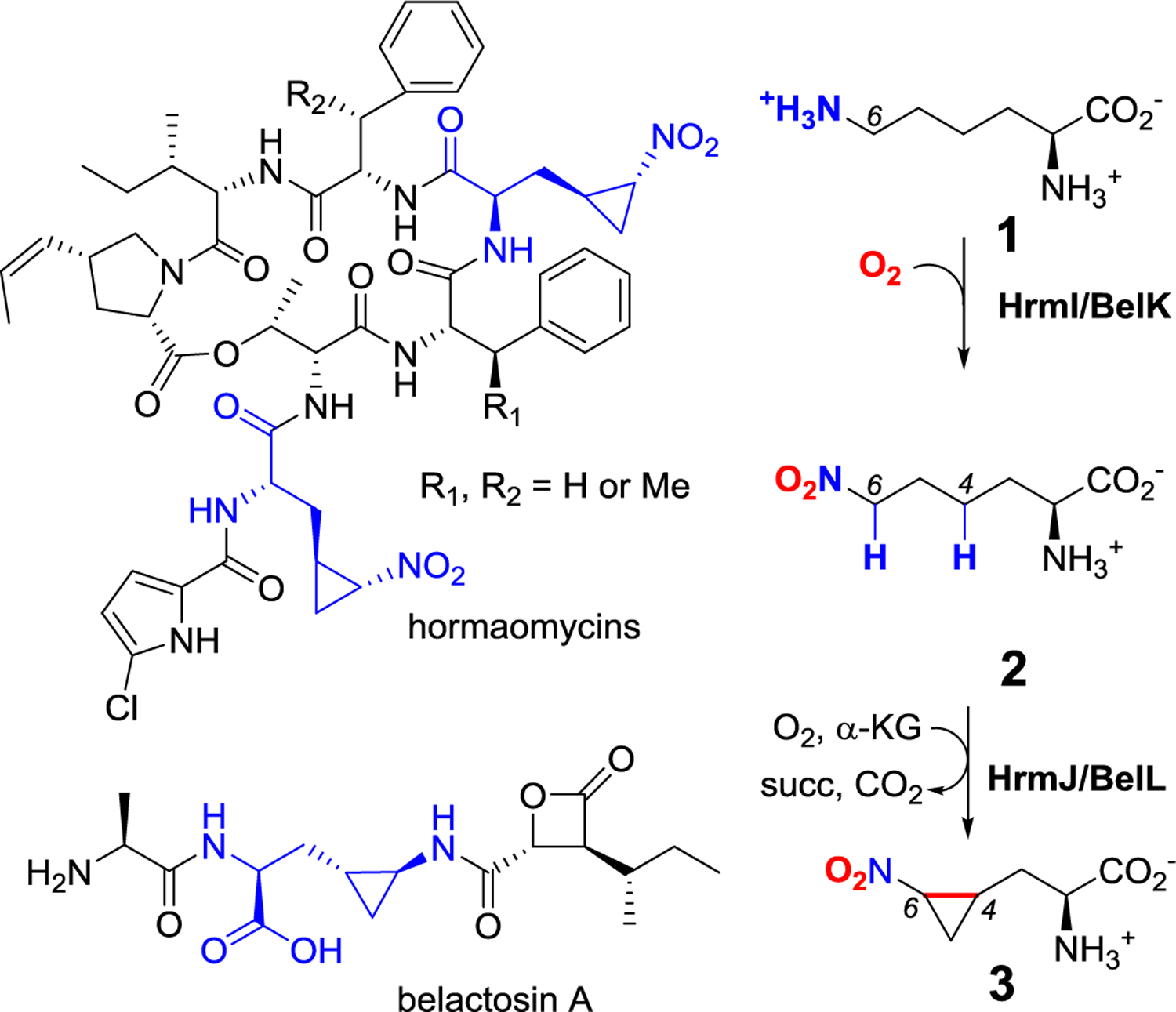
Structure of hormaomycins and belactosin A (left) and biosynthetic pathway of 3 identified in this study (right). Hormaomycins and belactosin A contain uncommon 3-(trans-2-nitrocyclopropyl)-alanine and 3-(trans-2-aminocyclopropyl)-alanine moieties, respectively (highlighted in blue).
On the basis of sequence similarity, HrmI/BelK belong to a newly recognized family of heme oxygenase-like dinuclear iron enzymes (HDOs),[9] while HrmJ/BelL are Fe(II) and α-ketoglutarate (Fe/α-KG) oxygenases.[10] Dinuclear iron enzymes have been shown to catalyze divergent reactivities, such as AurF/CmlI/SznF/RohS catalyzing N-oxygenation and UndA catalyzing fatty acid decarboxylation.[11] Furthermore, Fe/α-KG oxygenases also afford remarkable reactivities where the reaction outcomes are diverged from a substrate radical.[12] Some well-known reaction types involving these enzymes include hydroxylation, halogenation, desaturation, and epoxidation.[13]
The conversion of 1 to 3 involves multiple oxidation steps that includes oxidation of the ε-amino group and installation of a C4–C6 bond (Figure 1), making the above enzymes potential candidates. However, the use of either HDO or Fe/α-KG enzymes to install the cyclopropyl group has yet to be explored. In the present study, we established a biosynthetic pathway for 3-(trans-2-nitrocyclopropyl)-alanine formation by two types of metalloenzymes. In this pathway, the HDO enzyme (HrmI/BelK) catalyzes the conversion of 1 to l-6-nitronorleucine (2) in the presence of O2 and ascorbate, while the Fe/α-KG enzyme (HrmJ/BelL) enables the cyclopropanation of 2 to afford 3. Notably, the cyclopropyl moieties installed by HrmJ and Bel have the opposite configuration at C6 that is consistent with the stereochemistry of cyclopropyl group identified in belactosins and hormaomycins. To our knowledge, this study represents the first demonstration of using consecutive oxygenases to enable nitro-cyclopropyl group installation and rationalizes the necessity of a terminal nitro group to promote effective cyclopropanation.
To gain initial insight into the biosynthesis of amino- and nitro-cyclopropylalanine moiety, in vivo analysis was performed. Targeted genes, including hrmI, belK, hrmJ, and belL, were expressed using Escherichia coli BL21(DE3) as the host. The E. coli cells were harvested and then resuspended in water. After incubation with 1 on a rotary shaker at 37 °C for 3 h, the supernatant was analyzed by liquid chromatography–mass spectrometry (LC-MS). When hrmI was expressed, a new peak with a m/z value of 177.1 ([M+H]+, electrospray ionization (ESI), positive mode) was detected and coeluted with the synthetic standard of 2 (Figure S2). This peak was also observed using cells expressing belK gene. In contrast, incubation of 1 with cells expressing hrmJ or belL gene did not produce any specific product. These results suggested that the first step of both amino/nitro-cyclopropylalanine formation was oxidation of the ε-amino group of 1 to a nitro group by the dinuclear iron enzymes.
To support the in vivo results, in vitro assays were performed. HrmI and BelK were expressed in E. coli and purified as the N-His6-tagged proteins (Figure S3). Compound 1 was incubated with HrmI using ascorbate as the redox donor surrogate. As shown in Figure 2 (traces iii and iv), the formation of a new peak with the same m/z value and isotope ratio as that of synthetic 2 was detected. This peak was not observed in the control reactions omitting enzyme or substrate. Upon exposure to 18O2, the corresponding product showed m/z values of 177.1, 179.1, and 181.1 in a ratio of ~2:2:3 (Figure S4). The observed m/z change of +4 using 18O2 was consistent with the reported N-oxygenase reactivity.[11c] Detection of the +2 product might be associated with 16O2 contamination in the 18O2 experiment or another reaction pathway that has yet to be carefully examined. Analogously, BelK catalyzed the formation of 2 under similar reaction conditions. Formation of 2 by HrmI and BelK was further supported by 13C-NMR analysis of the reactions using 6-13C-1 as the substrate. In addition to the substrate peak (δ 36.8 ppm), a new peak (δ 72.6 ppm) corresponding to C6 of 2 was observed in the reactions containing HrmI or BelK (Figure 3). Altogether, these observations demonstrated that HrmI and BelK catalyzed nitro group formation using l-Lys (1) and suggested that a conserved cyclopropanation strategy is used in both hormaomycins and belactosins.
Figure 2.
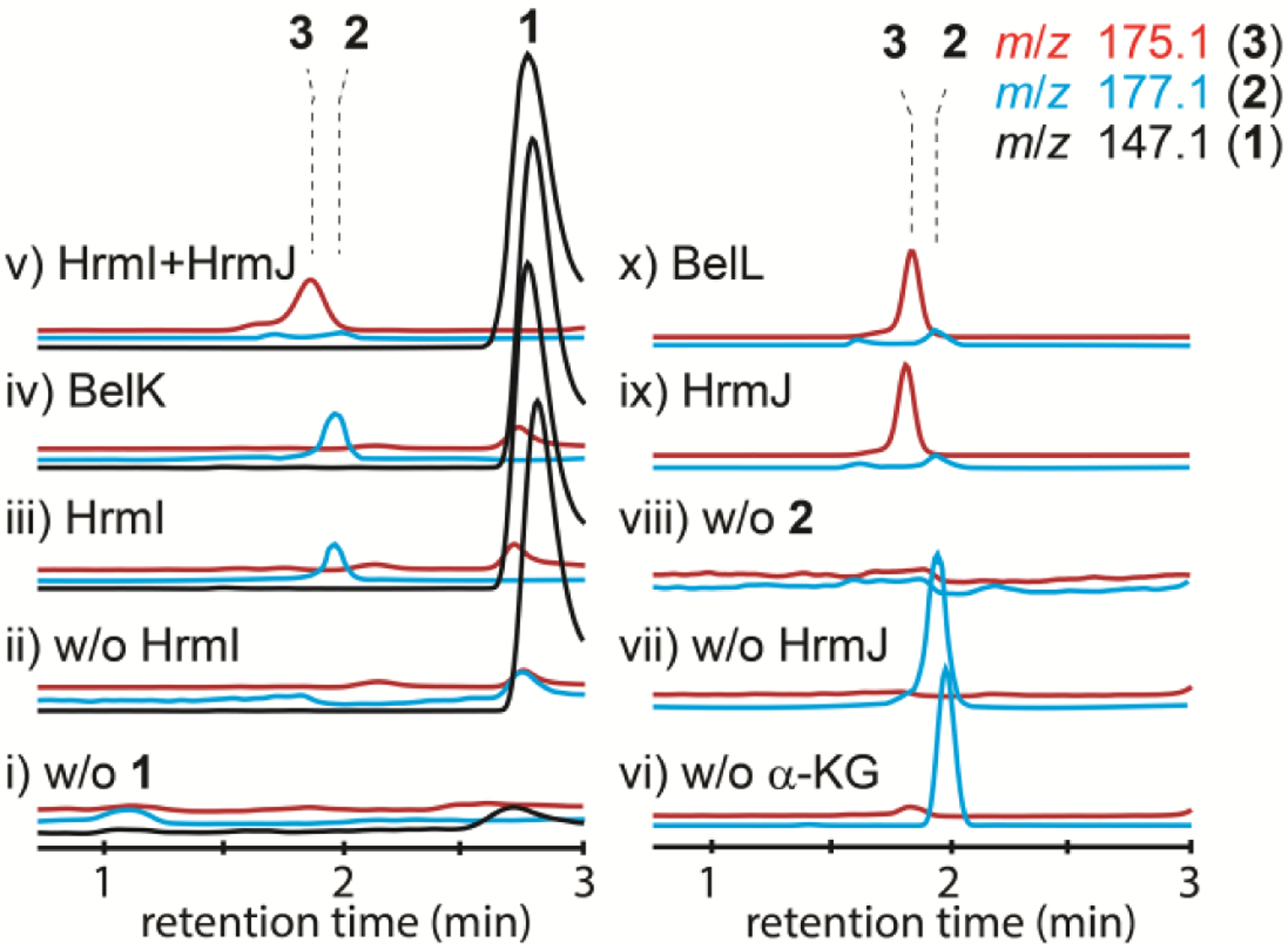
LC-MS chromatograms of i) HrmI reaction without 1, ii) incubation of 1 without enzyme, iii) HrmI reaction with 1, iv) BelK reaction with 1, v) HrmI+HrmJ reaction with 1, vi) HrmJ reaction without α-KG, vii) incubation of 2 without enzyme, viii) HrmJ reaction without 2, ix) HrmJ reaction with 2, and x) BelL reaction with 2. Chromatograms were monitored with m/z 147.1 (black traces for 1), 177.1 (blue traces for 2), and 175.1 (red traces for 3).
Figure 3.
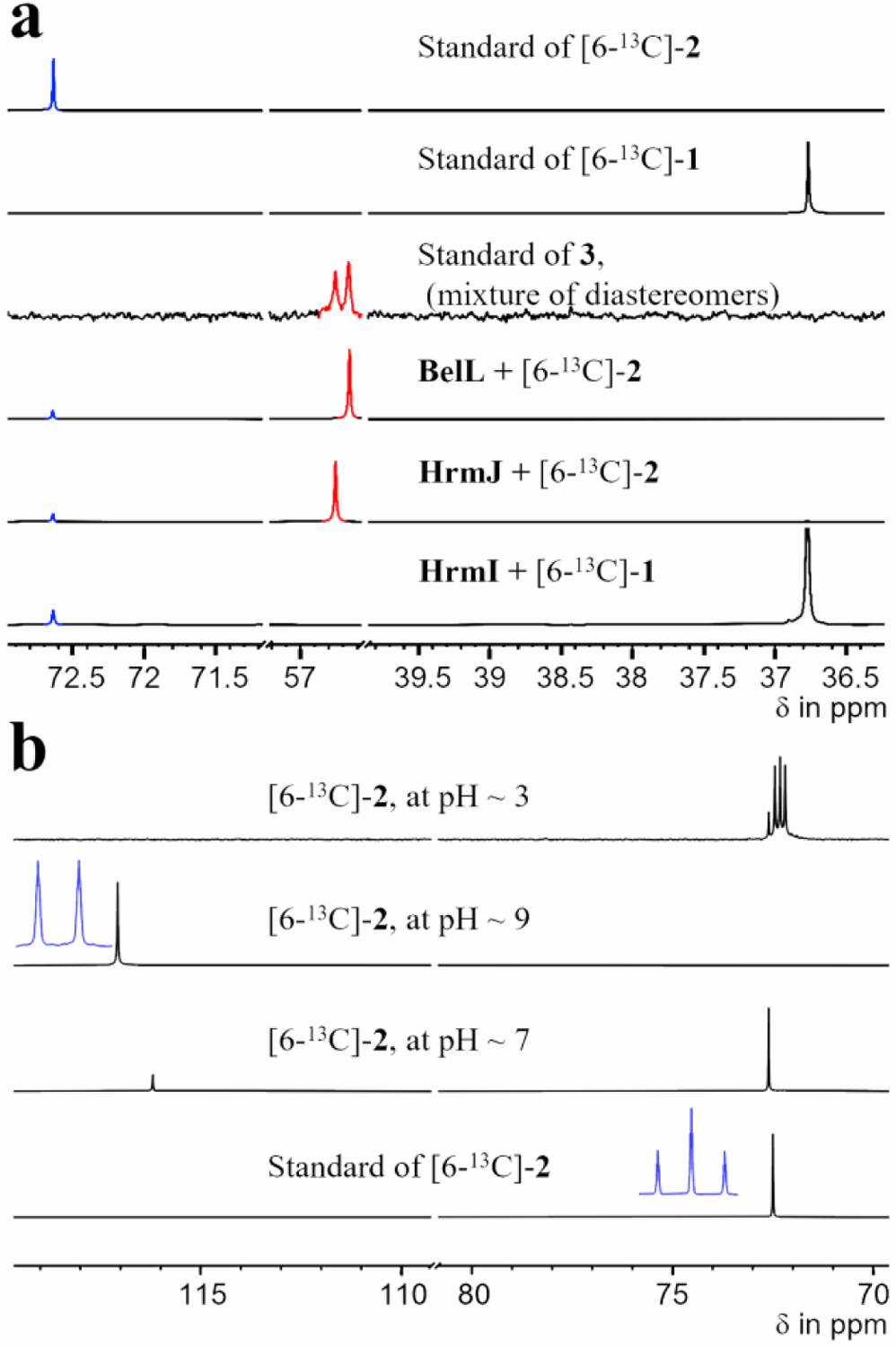
(a) 13C-NMR spectra of HrmI, HrmJ, BelK, and BelL-catalyzed reactions using 6-13C-1 or 6-13C-2 as substrate. (b) 13C-NMR spectra of 6-13C-2 under different pH conditions. Insets (highlighted in blue) are 13C-NMR spectra recorded in C–H coupling mode.
Having established the functions of HrmI and BelK as ε-N-oxygenases to afford 2, we speculated that Fe/α-KG oxygenases HrmJ and BelL catalyze cyclopropanation. First, the HrmI and HrmJ coupled reaction was conducted using 1 as the substrate. A new peak with an m/z value corresponding to that of 3 was produced (Figure 2, trace v). Furthermore, in the presence of α-KG and O2, both HrmJ and BelL catalyzed the efficient conversion of 2 to a new product with m/z 175.1 ([M+H]+, ESI, Figure 2, traces ix and x). To confirm that this m/z change of −2 (177.1 → 175.1) was associated with cyclopropane moiety formation, but not with desaturation or other reactivities commonly found in Fe/α-KG oxygenases,[13] we chemically synthesized 3-(trans-2-nitrocyclopropyl)-alanine (3) as an authentic standard and showed that the enzymatic reaction product had the same retention time as the synthetic standard. To our knowledge, this is the first demonstration of Fe/α-KG oxygenase-catalyzed cyclopropane formation. As belactosin contains 3-(trans-2-aminocyclopropyl)-alanine but not 3, we next examined whether BelL can catalyze the cyclopropanation of l-Lys (1) to synthesize 3-(trans-2-aminocyclopropyl)-alanine. Incubation of BelL with 1 in the presence of α-KG and O2 did not result in the product with an m/z value change of −2 to the substrate (Figure S5). Combined with the N-oxygenase activity of HrmI and BelK, these results showed that a conserved strategy is used to generate 3 in two biosynthetic pathways. Bioinformatics analysis revealed that bel cluster but not hrm cluster contained a gene for dimethylsulfoxide reductase family of molybdoenzymes (belN) near belK/L genes. We proposed that reduction of 3, putatively by belN, is involved in the later stage of belactosin biosynthesis.
Another difference between belactosins and hormaomycins is the stereochemistry of the 3-(trans-cyclopropyl)-alanine moiety at C4 and C6 (Figure 1). As HrmI and BelK only catalyze N-oxygenation to afford 2, the downstream Fe/α-KG oxygenases (HrmJ and BelL) likely catalyze stereoselective cyclopropanation. Alternatively, stereoinversion by an unidentified enzymatic transformation in the later stage of the biosynthesis is involved. Importantly, the synthetic standard of 3 contains two pairs of diastereomers in a ratio of ~5:4, which were unable to be separated under the current LC-MS conditions. To distinguish the above hypotheses and establish the stereopreferences of the two enzymes, HrmJ and BelL-catalyzed reactions were conducted using 6-13C-2 as the substrate. An obvious difference between these reactions was observed by 13C-NMR. The C6 position of the HrmJ-catalyzed product had a chemical shift (δ 56.8 ppm) that is different from the BelL reaction product (δ 56.7 ppm) (Figure 3). The chemical shifts of these two peaks were in accordance with those of synthetic standard 3, which contained two pairs of diastereomers. Under current conditions, while HrmI and BelK only resulted in ~10% conversion (1 → 2), >95% of substrate was converted to 3 by the HrmJ and BelL catalyzed reactions (Figures 2 and 3). These results strongly indicated that HrmJ and BelL are responsible for stereoselective cyclopropanation with different stereochemistry.
In vitro characterization showed that HrmJ and BelL were involved in cyclopropanation. Oxidative C–C bond formation has been reported in Fe/α-KG oxygenases, including KabC and DPS.[14] In these reactions, an olefin or a benzene appended on the substrate serves as a radical or carbocation receptor to enable C–C bond formation. We speculated that a similar reaction might occur in the HrmJ and BelL-catalyzed cyclopropanations. In this case, deprotonation at C6 forms a C=N moiety in situ, which is used to interact with the resulting radical, cation, or lactone generated via C4–H cleavage (Scheme 1). To investigate this hypothesis, the acidity of the C6-proton was investigated using 13C-NMR by treating 6-13C-2 with LiOH in D2O (Figure 3). The appearance of a new signal with a chemical shift of ~120 ppm and a concomitant decrease in the substrate signal (δ ~73 ppm) were detected. In C–H coupling mode, the newly formed peak was observed as a doublet, which indicates only one proton is retained at C6. These observations are consistent with the formation of a C=N moiety. Subsequent addition of HCl to the reaction resulted in reappearance of the substrate peak. Based on the chemical shift change and number of protons observed in the C–H coupling 13C-NMR, these results were in agreement with the elevated acidity of the proton adjacent to the nitro group where a C=N moiety is formed under slightly basic conditions.[15] Deuterium incorporation from solvent (D2O) into C6 of 2 after reacidification by HCl, which resulted in splitting of the 13C signal (δ ~73 ppm), also supported proton abstraction at C6. This suggested that C6 proton can be abstracted by an amino acid residue in HrmJ/BelL.
Scheme 1.
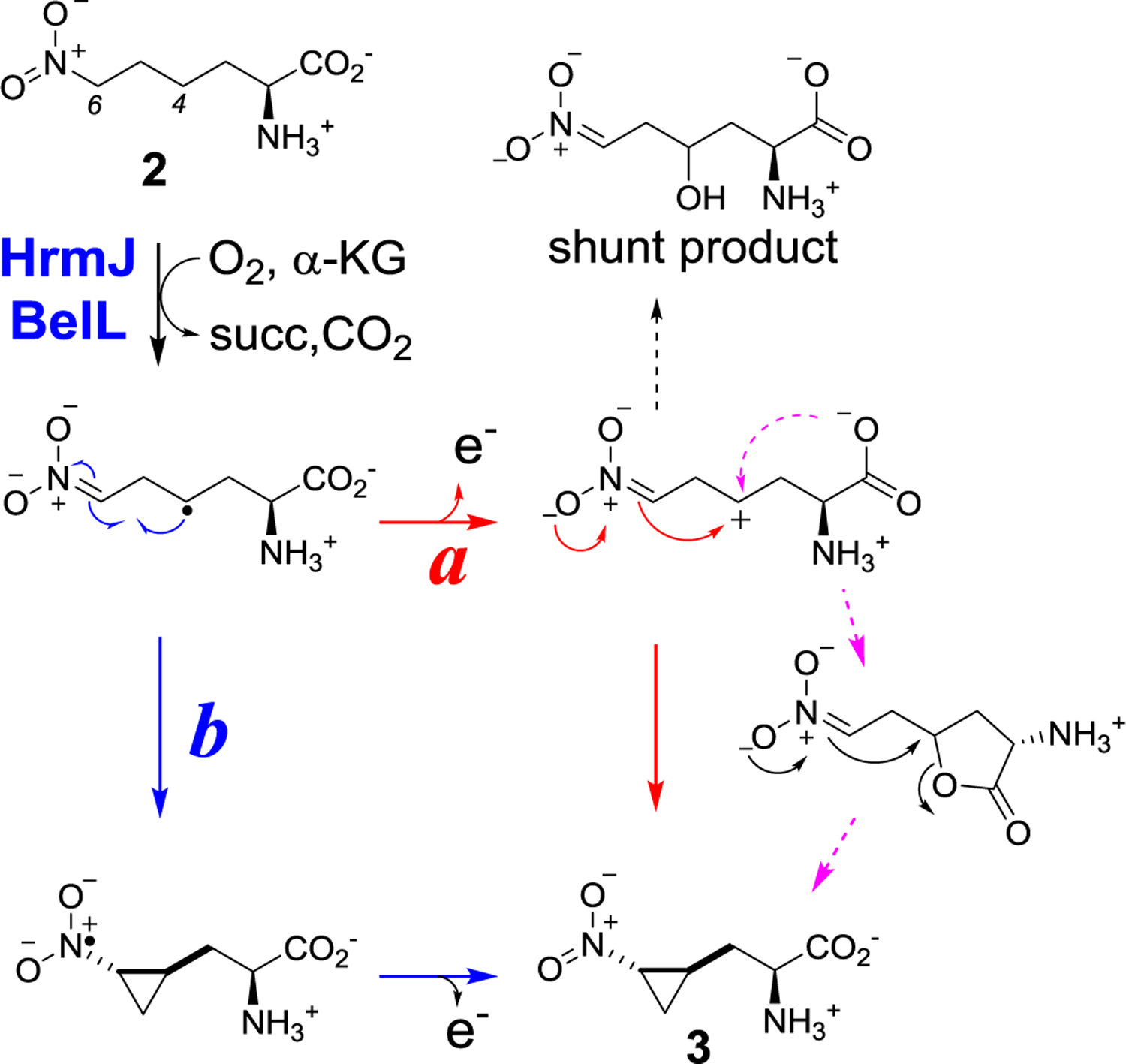
Proposed reaction mechanism for HrmJ and BelL-catalyzed reactions.
Furthermore, examination of the HrmJ-catalyzed reaction using 1 showed that a peak with an m/z change of +16 (147.1 → 163.1) was detected by LC-MS (Figure S6). This m/z change was likely associated with C4-hydroxylation, but not C6. The hemiaminal generated via C6-hydroxylation is prone to hydrolysis to the corresponding aldehyde. The sluggish reactivity toward 1 suggested that 2 was a preferred substrate. Further evidence for C4-hydroxylation was provided by observation of a minor shunt product with an m/z change of +16 (177.1 → 193.1) in the reaction using 2. Under H218O conditions (~50% 18O), ~18.5% 18O incorporation was detected (Figure S7). Notably, although observation of 18O incorporation from water favors the polar pathway (pathway a) where the cation or lactone is quenched by water, the possibility of Fe(IV)-oxo or Fe(III)-OH tautomerization with water cannot be ruled out.
To assess whether a similar approach is widely distributed for cyclopropanation, we searched for the gene clusters containing both HrmI and HrmJ homologs in the public genome database. Many putative biosynthetic gene clusters were identified in different bacterial strains, mainly in Actinobacteria and Pseudomonas (Figure S8). Of these recombinant enzymes, WP_052441272 (HrmI ortholog) and WP_042425400 (HrmJ ortholog) found in Streptacidiphilus anmyonensis were prepared and confirmed to have the same activities as BelK and BelL, respectively (Figure S9). While belactosins and hormaomycins are the only reported natural products with nitro- or amino-cyclopropylalanine moieties, above analysis suggested that a similar strategy of utilizing consecutive oxygenases to install cyclopropane moieties might be involved in other potential biosynthetic pathways.
In conclusion, this study demonstrated that HrmI and BelK catalyzed l-6-nitronorleucine formation, authenticating their functions as N-oxygenases. Subsequently, HrmJ and BelL triggered asymmetric cyclopropanation via C4–C6 bond formation. Although diverse strategies for cyclopropyl ring construction have long been desirable,[1] using two consecutive enzymes to activate a substrate for effective cyclopropanation represents a new approach to install cyclopropane moieties. Furthermore, preliminary evidence of a plausible reaction pathway was supported by observation of in situ C=N moiety formation and a hydroxylation shunt product. During the revision of this paper, Shimo et al. also reported the characterization of HrmI/J and BelK/L.[16] While mechanistic details of the HrmI/J and BelK/L-catalyzed reactions have yet to be carefully investigated, our results provide a new pathway for cyclopropane construction and further demonstrate the novel strategies involving metalloenzymes in natural product biosynthesis.
Supplementary Material
Acknowledgements
This study was supported in part by Grants-in-Aid for Research on Innovative Areas from MEXT, Japan (JSPS KAKENHI Grant Number JP16H06452 to T.D.), Grants-in-Aid for Scientific Research from JSPS (JP18H03937 to T.D. and JP18K05449 to Y.O.), and National Institutes of Health (GM127588 to W-c.C). We are grateful to Kyowa Hakko Bio Co. Ltd., Tokyo, Japan, for providing Streptomyces sp. KY11780. We thank Simon Partridge, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
The authors declare no competing financial interests.
References
- [1].a) Thibodeaux CJ, Chang W.-c., Liu H.-w., Chem. Rev 2012, 112, 1681–1709; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wessjohann LA, Brandt W, Thiemann T, Chem. Rev 2003, 103, 1625–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Boger DL, Garbaccio RM, Bioorg. Med. Chem 1997, 5, 263–276. [DOI] [PubMed] [Google Scholar]
- [3].Katsir L, Schilmiller AL, Staswick PE, He SY, Howe GA, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Talele TT, J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- [5].a) Rössner E, Zeeck A, König WA, Angew. Chem. Int. Ed 1990, 29, 64–65; [Google Scholar]; b) Zlatopolskiy BD, Loscha K, Alvermann P, Kozhushkov SI, Nikolaev SV, Zeeck A, de Meijere A, Chem. Eur. J 2004, 10, 4708–4717.15372686 [Google Scholar]
- [6].a) Wolf F, Bauer JS, Bendel TM, Kulik A, Kalinowski J, Gross H, Kaysser L, Angew. Chem. Int. Ed 2017, 56, 6665–6668; [DOI] [PubMed] [Google Scholar]; b) Asai A, Hasegawa A, Ochiai K, Yamashita Y, Mizukami T, J. Antibiot 2000, 53, 81–83. [DOI] [PubMed] [Google Scholar]
- [7].a) Brandl M, Kozhushkov Sergei I., Zlatopolskiy Boris D., Alvermann P, Geers B, Zeeck A, de Meijere A, Eur. J. Org. Chem 2005, 123–135; [Google Scholar]; b) Kozhushkov SI, Zlatopolskiy BD, Brandl M, Alvermann P, Radzom M, Geers B, de Meijere A, Zeeck A, Eur. J. Org. Chem 2005, 854–863. [Google Scholar]
- [8].Hofer I, Crusemann M, Radzom M, Geers B, Flachshaar D, Cai X, Zeeck A, Piel J, Chem. Biol 2011, 18, 381–391. [DOI] [PubMed] [Google Scholar]
- [9].Rajakovich LJ, Zhang B, McBride MJ, Boal AK, Krebs C, Jr JMB., Comprehensive Natural Products III Vol. 5, Elsevier, Amsterdam, 2020, pp 215–250. [Google Scholar]
- [10].Hausinger RP, Crit. Rev. Biochem. Mol. Biol 2004, 39, 21–68. [DOI] [PubMed] [Google Scholar]
- [11].a) Ng TL, Rohac R, Mitchell AJ, Boal AK, Balskus EP, Nature 2019, 566, 94–99; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) He HY, Henderson AC, Du YL, Ryan KS, J. Am. Chem. Soc 2019, 141, 4026–4033; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Komor AJ, Jasniewski AJ, Que L, Lipscomb JD, Nat. Prod. Rep 2018, 35, 646–659; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Manley OM, Fan R, Guo Y, Makris TM, J. Am. Chem. Soc 2019, 141, 8684–8688; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang B, Rajakovich LJ, Van Cura D, Blaesi EJ, Mitchell AJ, Tysoe CR, Zhu X, Streit BR, Rui Z, Zhang W, Boal AK, Krebs C, Bollinger JM Jr., J. Am. Chem. Soc 2019, 141, 14510–14514; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Hedges JB, Ryan KS, Angew. Chem. Int. Ed 2019, 58, 11647–11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Martinez S, Hausinger RP, J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Gao SS, Naowarojna N, Cheng R, Liu X, Liu P, Nat. Prod. Rep 2018, 35, 792–837; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Herr CQ, Hausinger RP, Trends Biochem. Sci 2018, 43, 517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Chekan JR, McKinnie SMK, Moore ML, Poplawski SG, Michael TP, Moore BS, Angew. Chem. Int. Ed 2019, 58, 8454–8457; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen TY, Xue S, Tsai WC, Chien TC, Guo YS, Chang W.-c., ACS Catal. 2021, 11, 278–282; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lau W, Sattely ES, Science 2015, 349, 1224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wheland GW, Farr J, J. Am. Chem. Soc 1943, 65, 1433. [Google Scholar]
- [16].Shimo S, Ushimaru R, Engelbrecht A, Harada M, Miyamoto K, Kulik A, Uchiyama M, Kaysser L, Abe I, J. Am. Chem. Soc 2021, 143, 18413–18418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.