

Abstract
Background
Bone is the most frequent site of metastases from breast cancer (BC), but no biomarkers are yet available to predict skeletal dissemination.
Methods
We attempted to identify a gene signature correlated with bone metastasis (BM) onset in circulating tumour cells (CTCs), isolated by a DEPArray-based protocol from 40 metastatic BC patients and grouped according to metastasis sites, namely “BM” (bone-only), “ES” (extra-skeletal) or BM + ES (bone + extra-skeletal).
Results
A 134-gene panel was first validated through targeted RNA sequencing (RNAseq) on sub-clones of the MDA-MB-231 BC cell line with variable organotropism, which successfully shaped their clustering. The panel was then applied to CTC groups and, in particular, the “BM” vs “ES” CTC comparison revealed 31 differentially expressed genes, including MAF, CAPG, GIPC1 and IL1B, playing key prognostic roles in BC.
Conclusion
Such evidence confirms that CTCs are suitable biological sources for organotropism investigation through targeted RNAseq and might deserve future applications in wide-scale prospective studies.
Subject terms: Breast cancer, Molecular medicine
Background
Breast cancer (BC) is a heterogeneous disease, whose histopathological and molecular features significantly influence clinical evolution [1–5]. Besides the most common prognostic factors, such as the expression of hormone receptors (HR) and human epidermal growth factor receptor 2 (Her-2) status [6], multigene assays have been shown to support management and decision-making processes in selected early-stage patients [7, 8].
Over the past few years, clinical and molecular monitoring of BC has been facilitated by developments within liquid biopsy technologies, since both the number of circulating tumour cells (CTCs) and their features have been shown to give additional prognostic information in both early and advanced BC [9, 10], paving the way to novel experimental approaches which provide a real-time and dynamic picture of these malignancies and their evolution during treatment [11]. Several techniques for BC CTC isolation have been proposed, relying on their phenotypical, physical or metabolic properties [12], although CellSearch® is still the only FDA-approved methodology for CTC enumeration [9, 10]. Longitudinal monitoring of CTC count has also been shown to reveal the onset of acquired resistance to treatment as well as disease progression, in parallel with diagnostic standards [13]. Phenotypical and molecular characterisation of BC CTCs, based on the application of “omics” technologies, have also been attempted [11, 14–16] with the purpose to identify prognostic/predictive signatures and novel therapeutic targets [15, 17].
A number of studies investigating BC metastatic behaviour in search of specific “organotropism” signatures have been performed on both BC cell lines and primary tumour samples [18, 19], whereas the potential application of CTCs to this field of research has been only partially explored [20–22]. Indeed, with respect to the “bone homing” process, molecular panels including Kang’s [7], Savci-Heijink’s [23] and Cosphiadi’s [24] have been defined in primary BC, although none of them has entered routine clinical practice.
Based on the assumption that CTCs reflect not only the disease extent but also its molecular heterogeneity and evolution [2, 10], we attempted to define and evaluate a specific “osteotropism” gene signature in BC CTCs which could enable the identification of a subset of breast malignancies capable of leading to “bone-only” metastases.
We thus developed a targeted RNAseq assay to screen a panel of genes critically involved in the metastatic cascade. The panel adequacy was first confirmed on cell models, including sub-clones of the MDA-MB-231 BC cell line characterised by variable organotropism (towards “bone” or “lung”) [25]. Then, the targeted RNAseq of CTCs from stage-IV BC patients explored the correlation between distant metastasis sites and CTC gene expression profile (GEP).
Methods
Patients
Forty stage-IV BC patients, attending the Medical Oncology Unit of the University Hospital “Policlinico of Bari”, were enrolled. Eligible patients were adult (≥18 years) subjects with metastatic BC, either systemic-treatment-naive or experiencing radiological disease progression during systemic anticancer treatment. In the latter case, patients were enrolled at least 21 days after the last cycle of therapy. Personal history of other synchronous or metachronous malignancies represented an exclusion criterion. Before enrollment, patients underwent a full-body computed tomography (CT) and a bone scan to define sites of distant metastases. When deemed necessary by the clinician, a 18F-fluorodeoxyglucose (FDG)-PET-CT was also performed, in agreement with current guidelines [26]. Clinical and pathological data from all patients were collected and recorded in anonymised form.
CTC identification and isolation
Viable CTCs were purified from 15 ml of peripheral blood, as previously described [27]. Briefly, following pre-enrichment through immunomagnetic sorting (AutoMACS Pro, Miltenyi Biotech, Bergisch Gladbach, Germany), cell samples were incubated with a mixture of monoclonal antibodies (Abs), against epithelial or mesenchymal markers, conjugated with different fluorochromes.
CTCs were loaded into a DEPArray V2 dielectrophoretic system (Menarini, Silicon Biosystem, Castel Maggiore, Italy), entrapped in single cages under the effect of a dielectric field and identified by a fluorescence microscope. Thus, selected CTCs were moved to the parking area, recovered as pools of ten cells and stored at −80 °C to preserve nucleic acid integrity until subsequent molecular analyses.
Cell lines
In order to set up the RNAseq method, human cell lines were used, including the healthy mammary gland epithelium-derived MCF-10A (ATCC CRL-10317) and the triple-negative MDA-MB-231 (ATCC HTB-26) BC cell line (P0), which shows no selective organotropism in vivo [28]. P7 and lung metastatic (LM) sub-clones of P0 cells, exhibiting bone and visceral tropism, respectively [25], and established at the “University of Sheffield” by serial passages in murine models of BC, were also employed.
BC cells were cultured at 37 °C in 5% CO2, in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4.5 g/L glucose, ultra-glutamine I (Lonza, Verviers, Belgium) and 10% fetal bovine serum (FBS). MCF-10A cells were cultured at 37 °C in 5% CO2 in DMEM Nutrient Mixture F-12 medium containing 10% FBS, 1% penicillin/streptomycin, 4 mM glutamine, 10 ng/ml epidermal growth factor (EGF) (Peprotech, London, UK), 100 IU/ml insulin and 0.5 µg/ml hydrocortisone (Sigma-Aldrich, Milan, Italy). Cell cultures were screened for Mycoplasma contamination (MP0040; Sigma) prior to molecular characterisation.
Definition of a putative “osteotropism gene panel”
In order to identify genes to be included in the putative BC osteotropism signature, a review of research articles published until May 2019 was performed by using PubMed, Scopus, ISI-Web of Science and Google Scholar databases. The applied keywords were “breast cancer”, “osteotropism”, “skeleton invasiveness”, “bone metastases” and “metastases”. The identified genes were subsequently screened for their function, correlation with metabolic pathways and alterations in invasive malignancies using UNIPROT (http://www.uniprot.org/), GENECARD (http://www.genecards.org/), OMIM (http://omim.org/) and iPATH2 (pathway.embl.de/iPath2.cgi#) web tools. Finally, the selected genes (Supplementary Table 1) were submitted via web interface for primer pool design and synthesis using the proprietary Ion Ampliseq Designer algorithm (https://www.ampliseq.com/browse.action).
RNAseq
Preliminary experiments with cell lines were performed to standardise the RNAseq method to be used for CTCs. In order to obtain RNA amounts comparable to those deriving from CTC pools, suspensions of 1 × 106 cells, from each line, were serially diluted until obtaining ten cells that subsequently underwent RNAseq analysis.
Sequencing libraries were prepared by the Ion AmpliSeq™ Library Kit 2.0 as indicated in the Ion AmpliSeq™ RNA Library preparation user guide (Ion AmpliSeq™ Library Preparation, Quick Reference, Publication Number MAN0006735 Revision F.0). For all cell lines, ten-cell pools were lysed with 1 μl of the Reaction Buffer (19 μl of Lysis Buffer and 1 μl of RNase Inhibitor) (Takara Bio, Mountain View, USA) and incubated at 72 °C in a thermal cycler for 3 min. The lysed product was incubated at 50 °C for 10 min with 1 μl of SuperScript IV Reverse Transcriptase (SuperScript™ IV One-Step RT-PCR SystemKit, Thermo Fisher Scientific, Waltham, USA), 5 μl of Ion AmpliSeq™ HiFi Mix (Thermo Fisher Scientific) and 5 µl of the primer pool. The cDNA target amplification was performed at 98 °C for 15 s and at 60 °C for 4 min, increasing from 17 to 25 the number of cycles indicated in the “Amplify the Targets” section of the above-mentioned Ion AmpliSeq™ RNA Library preparation user guide, while the following steps were performed according to the manufacturer’s instructions.
Both the quality and quantity of libraries, purified by Agentcourt AMPure XP (Beckman Coulter, Indianapolis, USA), were evaluated by the Ion Library Taq Man Quantitation Kit (Life Technologies Carlsbad, California, USA) on the StepOne Plus system (Applied Biosystem, Foster City, California, USA). Finally, libraries were templated through the Ion OneTouch™ 2 System and Ion OneTouch™ ES, and sequenced on the NGS Ion Torrent PGM™ system by using Ion Torrent™ 318 chips. CTC RNAseq analyses were performed following the same protocol described for BC cell lines.
Analysis of RNAseq data
RNAseq data from both cell lines and CTCs were analysed by using the AmpliSeq RNA plugin available for Ion Torrent sequencing platforms and Partek Flow (Build version 9.0.20.0720; Partek Inc., St. Louis, MO).
Data analysis began with FASTQ files, with a single FASTQ file corresponding to each sequenced RNA sample and containing the information of sequenced reads as well as the quality score for each nucleotide. Once the quality of FASTQ files was checked, bases and reads with low quality were filtered out, and adaptors and barcodes were extracted from the data. Reads were aligned to the reference human genome (version hg19) and TMAP was used as an aligner.
The list of DEGs was obtained through the gene-specific analysis (GSA) method. Normalisation of read count was performed by the total number of counts (count per million) plus 0.0001, and all genes with less than ten normalised read counts were excluded from subsequent analyses. Each gene was associated with a relative (log2) fold change (FC) > 2, whose statistical significance was expressed in terms of P value. Only genes whose P value was ≤ 0.05 were considered differentially expressed.
Statistically significant DEGs were subsequently grouped in a hierarchical manner using the correlation distance and displayed in a heatmap. All correlation analyses were performed by calculating the Pearson coefficient and the adjustment for the multivariate analysis was conducted with the Benjamini and Hochberg method (false discovery rate, FDR < 0.25).
In both cell lines and CTCs, the analyses of DEGs were performed at the inter-group level, after classification of the samples according to the organotropism, for the former, and sites of distant metastasis, for the latter. The principal component analysis (PCA) method was adopted to visualise similarities and differences between the samples in the dataset while identifying potential outliers.
Once a list of ranked DEGs was obtained, Gene Ontology (GO) enrichment analysis was applied to annotate genes in classes or categories like “biological process”, “molecular function” and “cellular component” [29, 30], while the Kyoto Encyclopaedia of Genes and Genomes (KEGG) was used for pathway enrichment analysis [31]. In all cases, a P value < 0.05 was defined as a cut-off. Cytoscape (version 3.7.2) software [32] and its tool StringApp [33] were used for visualising networks and performing enrichment analysis by applying default parameters (confidence score cut-off 0.4).
Survival analysis
The potential prognostic meaning of the top-10 (4 ≤ FC ≤ −4) most deregulated genes, emerged from the comparison among CTC groups, was explored first in our patient series and then by applying the Molecular Taxonomy of BC International Consortium (METABRIC) dataset which includes 2509 primary breast tumours and matched clinical data [34–36]. In both cases, the association between putative biomarkers and overall survival (OS) was first explored by splitting the patients into “high” and “low” gene expression groups, and then visualised by Kaplan–Meier curves, plotted through “survminer” and “survival” packages of R software (v. 3.6.1), according to the relative log-rank P. In a similar fashion, we explored in our cohort the prognostic role of the above-mentioned genes with respect to the “time-to-BM diagnosis” and the “time-to-first skeletal-related event (SRE)” outcomes.
Results
Patients and CTC enumeration
Patients’ clinicopathological information is summarised in Table 1. As shown, 10 out of 40 subjects were systemic-treatment-naive, while the others were experiencing disease progression at the time of recruitment. The median number of viable CTCs from the whole cohort was 50 (range 10–110). Neither clinicopathological features nor the number of previous systemic treatments exhibited a significant correlation with the count of isolated CTCs (Spearman’s correlation coefficient between −0.30 and 0.14; data not shown). The majority of patients harboured also CTC clusters (Table 1), defined as circulating multicellular aggregates made up of ≥2 cells with distinct nuclei, and including at least one CTC [37, 38]. As expected, a significant correlation between the total number of viable CTCs and cluster count emerged (Spearman’s coefficient 0.34, P = 0.038).
Table 1.
Clinicopathological features of enrolled patients.
| PT ID | Age | Breast cancer subtype | Number of viable CTCs | Number of CTC clusters | Metastasis site | Number of previous therapy lines | Use of BTA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bone | Liver | Lung | Brain | OVS | Lymph nodes | ST | |||||||
| #1 | 36 | HR + HER2 + | 35 | 9 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | ≥3 | Yes |
| #2 | 69 | HR + HER2 + | 85 | 115 | Yes | Yes | Yes | No | No | Yes | No | ≥3 | Yes |
| #3 | 59 | LUMINAL A | 40 | 12 | Yes | No | No | No | No | Yes | No | <3 | No |
| #4 | 49 | LUMINAL B | 51 | 109 | Yes | No | No | No | No | No | No | <3 | No |
| #5 | 58 | HR + HER2 + | 53 | 82 | Yes | Yes | Yes | No | No | Yes | No | <3 | No |
| #6 | 44 | LUMINAL B | 45 | 42 | Yes | Yes | Yes | Yes | No | Yes | No | Naive | No |
| #7 | 69 | LUMINAL B | 61 | 23 | Yes | No | Yes | No | No | No | Yes | ≥3 | Yes |
| #8 | 49 | LUMINAL B | 83 | 26 | No | No | Yes | No | No | Yes | No | Naive | No |
| #9 | 59 | TRIPLE NEG | 60 | 56 | Yes | No | No | No | No | No | No | <3 | Yes |
| #10 | 58 | LUMINAL A | 58 | 178 | No | No | No | Yes | No | No | No | <3 | No |
| #11 | 52 | LUMINAL A | 11 | 250 | Yes | Yes | No | No | Yes | Yes | Yes | <3 | Yes |
| #12 | 60 | HR- HER2 + | 32 | 87 | No | No | Yes | No | No | Yes | Yes | <3 | No |
| #13 | 57 | LUMINAL A | 66 | 70 | No | Yes | No | No | No | No | No | ≥3 | No |
| #14 | 53 | LUMINAL A | 45 | 6 | Yes | No | No | No | No | No | No | Naive | No |
| #15 | 67 | unknown | 56 | 8 | Yes | Yes | Yes | No | No | Yes | Yes | ≥3 | Yes |
| #16 | 74 | HR + HER2 + | 20 | 14 | Yes | No | Yes | No | No | No | No | <3 | No |
| #17 | 73 | LUMINAL B | 44 | 8 | Yes | No | No | No | No | No | No | Naive | No |
| #18 | 77 | LUMINAL A | 62 | 21 | Yes | No | Yes | No | No | Yes | Yes | Naive | No |
| #19 | 61 | HR + HER2 + | 77 | 114 | No | No | No | Yes | No | No | No | <3 | No |
| #20 | 63 | TRIPLE NEG | 43 | 3 | No | No | Yes | Yes | No | No | No | Naive | No |
| #21 | 47 | HR + HER2 + | 78 | 40 | Yes | No | Yes | No | No | No | No | Naive | No |
| #22 | 76 | LUMINAL B | 44 | 35 | Yes | No | No | No | No | No | No | <3 | No |
| #23 | 77 | LUMINAL A | 48 | 105 | Yes | No | No | No | No | Yes | Yes | ≥3 | Yes |
| #24 | 52 | LUMINAL B | 110 | 66 | Yes | No | No | Yes | No | No | No | <3 | No |
| #25 | 65 | LUMINAL A | 30 | 66 | No | No | Yes | No | No | No | No | Naive | No |
| #26 | 66 | LUMINAL B | 18 | 0 | Yes | Yes | No | No | No | Yes | No | <3 | No |
| #27 | 51 | TRIPLE NEG | 10 | 0 | Yes | No | Yes | No | No | No | No | Naive | No |
| #28 | 67 | LUMINAL B | 52 | 69 | No | No | No | No | No | Yes | No | <3 | No |
| #29 | 72 | unknown | 34 | 2 | Yes | No | Yes | Yes | No | No | No | ≥3 | Yes |
| #30 | 73 | LUMINAL B | 30 | 61 | No | Yes | Yes | Yes | No | Yes | No | <3 | No |
| #31 | 64 | LUMINAL A | 67 | 78 | Yes | No | Yes | No | No | No | No | Naive | No |
| #32 | 78 | unknown | 68 | 5 | No | Yes | Yes | No | No | Yes | Yes | ≥3 | No |
| #33 | 78 | LUMINAL B | 35 | 136 | Yes | No | No | No | No | No | No | <3 | No |
| #34 | 67 | HR + HER2 + | 50 | 9 | Yes | No | Yes | Yes | No | Yes | No | ≥3 | No |
| #35 | 66 | LUMINAL B | 21 | 6 | Yes | No | No | No | No | Yes | No | <3 | No |
| #36 | 72 | LUMINAL B | 55 | 0 | Yes | No | No | No | No | No | No | <3 | No |
| #37 | 72 | HR- HER2 + | 60 | 0 | Yes | Yes | Yes | No | No | Yes | No | <3 | No |
| #38 | 55 | HR- HER2 + | 29 | 1 | Yes | No | Yes | No | No | Yes | No | <3 | No |
| #39 | 57 | TRIPLE NEG | 71 | 12 | Yes | Yes | Yes | No | Yes | Yes | No | <3 | No |
| #40 | 72 | LUMINAL A | 35 | 1 | No | No | No | No | No | Yes | Yes | ≥3 | No |
BTA bone targeting agents (bisphosphonates and/or denosumab), HR hormone receptor, OVS other visceral sites, ST soft tissues (including pleura, peritoneum, subcutaneous tissue and muscles).
With regard to metastatic disease, at the time of CTC collection 7 patients (17.5%) exhibited bone-only metastases (defined as “BM”), 22 subjects (55.0%) had both skeletal and extra-skeletal metastases (BM + ES) and 11 patients (27.5%) presented with metastatic disease in sites other than bone (ES) (Table 1).
Assay set-up on BC cell lines
The literature review process described in the “Methods” led to the identification of 134 genes involved in several biological processes and functions, such as the epithelial-to-mesenchymal transition (EMT), angiogenesis, cell adhesion and motility, cell–cell signalling, intracellular signal transduction, remodelling of the extracellular matrix, modulation of immune response and immune escape (Supplementary Table 1).
CTC GEP analysis was first set up on MCF-10A and BC cell lines. The transcriptome heatmap of unsupervised hierarchical clustering of cell lines, based on normalised read counts, showed that “healthy” cell samples successfully separated from BC ones, while P7 clearly diverged from LM cells (Supplementary Fig. 1), validating the gene panel adequacy. Such divergences were further confirmed by the differential gene expression analysis, performed to identify deregulated genes among P0 and both of its sub-clones (Supplementary Table 2). Moreover, GO enrichment analysis applied to DEGs identified a number of significantly deregulated processes in osteotropic P7 cells compared to LM including, among upregulated ones, connective tissue development, cartilage development and ossification (Supplementary Table 3).
Targeted RNAseq of CTCs reveals metastasis site-related GEP
CTCs isolated from stage-IV BC patients were analysed in their GEP by using the same experimental approach described for cell lines. Due to the low quality of bases and reads emerging from the FASTQ file check, patients #29 and #40 were excluded from subsequent analyses.
The PCA, performed to evaluate the contribution of the transcript levels to CTC clustering, demonstrated a separation of “BM” CTCs from the remaining groups, namely “BM + ES” and “ES” (Fig. 1a), with the exception of one sample (#33) derived from a patient who, at the time of enrollment, had suspicious sub-centimetre lung nodules deserving close follow-up.
Fig. 1. Targeted RNAseq of CTCs reveals metastasis site-related GEP.

a The PCA, performed to visualise similarities and differences among CTC samples, showed a clear-cut separation of “BM” CTCs (in blue, N = 7) from the others (in yellow: “ES” CTCs, N = 10; in red: “BM + ES” cells, N = 21). In the three-dimensional PCA plot, each sample is represented as a sphere; the closer the spheres in the spreadsheet, the higher the similarity among CTC GEP. b Volcano plot of differential gene expression analysis performed by comparing “BM” vs “ES” CTCs. c The represented heatmap shows the expression profile of the 31 deregulated genes identified by comparing “BM” vs “ES” CTCs (−2 < FC < 2, P ≤ 0.05, FDR < 0.25). BM bone metastases only, BM + ES bone and extra-skeletal metastases, CTCs circulating tumour cells, ES extra-skeletal metastases, FC fold change, FDR false discovery rate, GEP gene expression profile, PCA principal component analysis.
By taking into account a −2 ≤ fold change (FC) ≥ 2 and a false discovery rate (FDR) threshold of 0.25, 31 DEGs were identified in “BM” CTCs compared with those from “ES” subjects (Table 2a), as shown by the volcano plot and the heatmap in Fig. 1b, c). Moreover, 24 DEGs emerged from the comparison between CTC samples belonging to “BM” and “BM + ES” patients, among which 6 were found upregulated and 18 downregulated in the former (Table 2b and Supplementary Fig. 2A), while no significantly deregulated genes were identified in CTCs from “BM + ES” versus “ES” patients (Supplementary Fig. 2B).
Table 2.
Lists of DEGs obtained comparing the GEP of BC CTCs.
| (a) | (b) | ||||||
|---|---|---|---|---|---|---|---|
| Gene symbol | P value | FDR step up | Fold change | Gene symbol | P value | FDR step up | Fold change |
| CAPG | 1.73E-02 | 2.02E-01 | 30.79 | ADGRL2 | 7.40E-03 | 6.23E-02 | 3.52 |
| HRAS | 1.34E-02 | 2.02E-01 | 11.67 | SPTBN1 | 1.08E-02 | 8.03E-02 | 2.67 |
| IL1B | 2.46E-02 | 2.02E-01 | 5.37 | SOX9 | 1.77E-03 | 2.73E-02 | 2.45 |
| FGFR4 | 1.99E-02 | 2.02E-01 | 5.28 | TTYH1 | 2.04E-03 | 2.91E-02 | 2.38 |
| MAF | 2.38E-02 | 2.02E-01 | 4.39 | PRDX1 | 3.51E-03 | 4.06E-02 | 2.26 |
| SERPINB2 | 2.79E-02 | 2.02E-01 | 4.38 | VEGFA | 1.30E-02 | 9.24E-02 | 2.2 |
| CTSK | 1.91E-02 | 2.02E-01 | 4.06 | KCNQ3 | 1.02E-02 | 7.85E-02 | −2.23 |
| MAFA | 2.77E-02 | 2.02E-01 | 3.92 | MMP28 | 1.25E-03 | 2.31E-02 | −2.3 |
| COL3A1 | 8.53E-03 | 2.02E-01 | 3.75 | TNFSF11 | 6.19E-03 | 5.88E-02 | −2.44 |
| TTYH1 | 1.37E-03 | 2.02E-01 | 3.62 | MYL3 | 7.35E-04 | 2.26E-02 | −2.61 |
| AURKB | 3.38E-02 | 2.08E-01 | 3.48 | HMGA2 | 8.62E-03 | 6.93E-02 | −2.77 |
| HMMR | 1.66E-02 | 2.02E-01 | 2.97 | SOX6 | 2.96E-03 | 3.64E-02 | −2.86 |
| NAP1L3 | 2.59E-02 | 2.02E-01 | 2.88 | SMAD3 | 6.36E-03 | 5.88E-02 | −2.86 |
| EPHB3 | 3.04E-02 | 2.02E-01 | 2.82 | NCKAP5L | 1.51E-02 | 9.91E-02 | −2.89 |
| SYNM | 5.47E-02 | 2.64E-01 | 2.74 | MMP12 | 9.59E-04 | 2.26E-02 | −3.06 |
| GIPC1 | 3.06E-02 | 2.02E-01 | 2.43 | IBSP | 2.46E-05 | 4.55E-03 | −3.45 |
| RERGL | 3.03E-02 | 2.02E-01 | 2.38 | FOXF1 | 1.65E-04 | 1.02E-02 | −3.55 |
| ITGB4 | 3.38E-02 | 2.08E-01 | 2.34 | KLHL25 | 5.88E-05 | 5.44E-03 | −3.93 |
| PRDX1 | 2.23E-02 | 2.02E-01 | 2.25 | COL6A3 | 1.43E-02 | 9.77E-02 | −4.06 |
| ST3GAL1 | 8.12E-03 | 2.02E-01 | 2.2 | SMAD2 | 3.39E-04 | 1.26E-02 | −4.08 |
| MEF2C | 3.79E-02 | 2.24E-01 | 2.19 | MYL6B | 6.75E-03 | 5.95E-02 | −5.13 |
| DKK1 | 5.46E-02 | 2.64E-01 | 2.18 | ZNF703 | 1.09E-03 | 2.26E-02 | −6.23 |
| MAPK1 | 4.30E-02 | 2.32E-01 | 2.17 | TPSD1 | 2.75E-03 | 3.64E-02 | −6.99 |
| FGF5 | 3.01E-02 | 2.02E-01 | 2.15 | ZNF26 | 1.55E-02 | 9.91E-02 | −7.17 |
| SOX9 | 4.92E-02 | 2.57E-01 | 2.04 | ||||
| FGFR3 | 1.30E-02 | 2.02E-01 | −2.75 | ||||
| HPRT1 | 1.24E-02 | 2.02E-01 | −2.79 | ||||
| SMAD2 | 2.62E-02 | 2.02E-01 | −2.86 | ||||
| HMGA2 | 2.33E-03 | 2.02E-01 | −4.11 | ||||
| MCM2 | 3.91E-02 | 2.24E-01 | −4.56 | ||||
| ANLN | 9.02E-03 | 2.02E-01 | −12.02 | ||||
(a) Upregulated and downregulated genes emerged from the comparison between “BM” vs “ES” CTCs.
(b) Upregulated and downregulated genes emerged from the comparison between “BM” vs “BM + ES” CTCs.
According to our preliminary categorisation of gene functional classes (Supplementary Table 1), the majority of these DEGs belonged to EMT, Wnt/β-catenin signalling, extracellular matrix remodelling and cell motility categories.
Interestingly, among the 31 deregulated genes observed in “BM” samples (compared to “ES” ones), CAPG (FC: 30.79; P value 1.73E-02), IL1B (FC: 5.37; P value: 2.46E-02), MAF (FC: 4.39; P value: 2.38E-02) and GIPC1 (FC: 2.43; P value 3.06E-02) were found overexpressed in CTCs derived from patients with skeletal relapse, in agreement with previous data describing the upregulation of these markers in osteotropic primary breast tumours [25, 39, 40].
In order to gain a deeper knowledge about the played biological functions and the reciprocal interactions existing among the identified DEGs, a gene regulatory network was constructed by using the Search Tool for the Retrieval of Interacting Genes database (STRING). Given a list of the 31 DEGs as input, this database assembled the protein–protein interactions (PPI) network and their topological information was visualised by Cytoscape. Figure 2a shows the PPI network of all DEGs (PPI enrichment in the amount of 3.97E-13) consisting of all proteins hierarchically located and interactions among them.
Fig. 2. Functional enrichment analysis of deregulated genes resulting from the comparison of “BM” vs “ES” CTCs.

In (a), the PPI network obtained by StringApp Cytoscape application (confidence score cut-off = 0.4; maximum additional interactors = 0; query default set-up) is shown. b The bar plot in this figure represents the most significantly enriched pathways of DEGs that emerged from KEGG analysis. BM bone metastases only, CTCs circulating tumour cells, DEGs differentially expressed genes, ES extra-skeletal metastases, FDR false discovery rate, KEGG Kyoto Encyclopaedia of Genes and Genomes, PPI protein–protein interaction.
Afterwards, genes on PPI network underwent GO enrichment analysis which showed that several upregulated DEGs found in “BM” CTCs (e.g. MAPK1, SOX9, IL1B, FGFR4, COL3A1, FGF5 and MEF2C) were enriched with greater statistical significance in biological processes related to cell signal transduction and proliferation including “cell surface receptor signalling pathway”, “regulation of intracellular signal transduction” and “MAPK cascade” (Table 3). On the other hand, downregulated DEGs (SMAD2, FGFR3, HMGA2 and MCM2) were enriched in different biological processes correlated with bone rearrangement, such as “chondrocyte differentiation” and “chondrocyte proliferation”, “mesoderm formation”, “skeletal system development”, “skeletal system morphogenesis”, “mesenchyme development” and “cell differentiation” (Table 3). Notably, in accordance with GO analysis, KEGG pathway analysis suggested that significantly upregulated genes were mainly enriched in key proliferative and bone-related processes, namely “MAPK signalling”, “regulation of actin cytoskeleton”, “PI3K-Akt signalling”, “Ras signalling”, “osteoclast differentiation” and “breast cancer” (Fig. 2b).
Table 3.
GO enrichment analysis of DEGs emerged from the comparison between “BM” vs “ES” CTCs.
| Term | Description | Genes | P value |
|---|---|---|---|
| Upregulated | |||
| GO.0065008 | Regulation of biological quality | MAPK1, SOX9, PRDX1, IL1B, CAPG, CTSK, FGFR4, COL3A1, AURKB, MAFA, EPHB3, MEF2C, DKK1, GIPC1, SERPINB2, HRAS | 1.90E-04 |
| GO.0000165 | MAPK cascade | MAPK1, SOX9, IL1B, FGFR4, FGF5, MEF2C, HRAS | 1.90E-04 |
| GO.0006468 | Protein phosphorylation | MAPK1, SOX9, IL1B, FGFR4, FGF5, AURKB, EPHB3, MEF2C, HRAS | 4.00E-04 |
| GO.1902533 | Positive regulation of intracellular signal transduction | MAPK1, SOX9, IL1B, FGFR4, COL3A1, FGF5, MEF2C, DKK1, HRAS | 4.40E-04 |
| GO.0050804 | Modulation of chemical synaptic transmission | MAPK1, IL1B, MEF2C, DKK1, GIPC1, HRAS | 6.40E-04 |
| GO.0007166 | Cell surface receptor signalling pathway | ITGB4, MAPK1, SOX9, IL1B, FGFR4, COL3A1, FGF5, EPHB3, MEF2C, DKK1, SERPINB2, HRAS | 6.60E-04 |
| GO.1902531 | Regulation of intracellular signal transduction | MAPK1, SOX9, PRDX1, IL1B, FGFR4, COL3A1, FGF5, AURKB, MEF2C, DKK1, HRAS | 6.60E-04 |
| GO.0009967 | Positive regulation of signal transduction | MAPK1, SOX9, IL1B, FGFR4, COL3A1, FGF5, MEF2C, DKK1, GIPC1, HRAS | 9.70E-04 |
| GO.0032872 | Regulation of stress-activated MAPK cascade | MAPK1, PRDX1, IL1B, DKK1, HRAS | 9.70E-04 |
| GO.0008283 | Cell population proliferation | MAPK1, SOX9, PRDX1, FGF5, AURKB, MEF2C, HRAS | 0.0014 |
| GO.0051781 | Positive regulation of cell division | IL1B, FGF5, AURKB, GIPC1 | 0.0014 |
| GO.0007167 | Enzyme-linked receptor protein signalling pathway | MAPK1, SOX9, FGFR4, COL3A1, FGF5, EPHB3, HRAS | 0.0017 |
| GO.0043408 | Regulation of MAPK cascade | MAPK1, PRDX1, IL1B, FGFR4, MEF2C, DKK1, HRAS | 0.0017 |
| GO.0043406 | Positive regulation of MAP kinase activity | MAPK1, IL1B, MEF2C, DKK1, HRAS | 0.0017 |
| GO.0071363 | Cellular response to growth factor stimulus | MAPK1, SOX9, FGFR4, COL3A1, FGF5, MEF2C | 0.0019 |
| Downregulated | |||
| GO.0035988 | Chondrocyte proliferation | FGFR3, HMGA2 | 0.0034 |
| GO.0048513 | Animal organ development | SMAD2, MCM2, ANLN, HPRT1, FGFR3, HMGA2 | 0.0045 |
| GO.0001706 | Endoderm formation | SMAD2, HMGA2 | 0.0241 |
| GO.0001707 | Mesoderm formation | SMAD2, HMGA2 | 0.0241 |
| GO.0002062 | Chondrocyte differentiation | FGFR3, HMGA2 | 0.0241 |
| GO.0035265 | Organ growth | SMAD2, FGFR3 | 0.0241 |
| GO.0001501 | Skeletal system development | SMAD2, FGFR3, HMGA2 | 0.0241 |
| GO.1903047 | Mitotic cell cycle process | MCM2, ANLN, HMGA2 | 0.0241 |
| GO.0065003 | Protein-containing complex assembly | SMAD2, MCM2, ANLN, HPRT1 | 0.0241 |
| GO.0022607 | Cellular component assembly | SMAD2, MCM2, ANLN, HPRT1, HMGA2 | 0.0241 |
| GO.0008283 | Cell population proliferation | HPRT1, FGFR3, HMGA2 | 0.0292 |
| GO.0031497 | Chromatin assembly | MCM2, HMGA2 | 0.0307 |
| GO.0030154 | Cell differentiation | SMAD2, ANLN, HPRT1, FGFR3, HMGA2 | 0.0307 |
| GO.0048705 | Skeletal system morphogenesis | SMAD2, FGFR3 | 0.0374 |
| GO.0060485 | Mesenchyme development | SMAD2, HMGA2 | 0.0374 |
Survival analysis
Based on RNASeq data emerging from “BM vs ES” CTC comparison, we arbitrarily defined a top-ten group of deregulated genes by considering their FC values (4 ≤ FC ≤ −4). Thus, the potential prognostic meaning of these genes (i.e. CAPG, HRAS, IL1B, FGFR4, MAF, SERPINB2, CTSK, ANLN, MCM2 and HMGA2) was explored in both metastatic and early BC.
First, the enrolled patients were included in two different groups namely “altered” (N = 24) and “not altered” (N = 14), according to the presence (or not) of at least one top-ten gene deregulation in CTCs. Kaplan–Meier curves in Supplementary Fig. 3 show the lack of a significant correlation between such patient classification and median OS, calculated from either the time of BC diagnosis or distant metastasis onset, until death from any cause or last follow-up visit. A non-significant difference between the two groups was also found in terms of median time-to-BM onset (altered = 56 months, not altered = 108 months, P = 0.28) (Supplementary Fig. 3) and median time-to-first SRE (altered = not reached, not altered = 5 months, P = 0.16) (Supplementary Fig. 4). Once we tested patient dichotomisation according to the presence of at least two top-ten gene deregulations (≥2 group = 14 patients; 0–1 group = 24 patients), we did not find significant correlations between CTC GEP and the above-mentioned outcomes (data not shown).
Then, METABRIC dataset [27–29] was employed to investigate the potential prognostic meaning of the top-ten gene expression alterations. We focused on BC patients whose primary tumours had been screened for the expression of the ten genes of interest (N = 481). Since only nine stage-IV BC patients were included in this cohort, we decided to focus on the early-stage population (stage I–III, N = 472) for subsequent analyses. By applying the above-mentioned criterion to assign patients to either “altered” or “not altered” group, a statistically significant longer median survival emerged in the “altered” group of early-stage patients, compared to “not altered” one (199 vs 112 months, P = 0.014) (Fig. 3).
Fig. 3. Survival probability analysis based on METABRIC BC dataset.
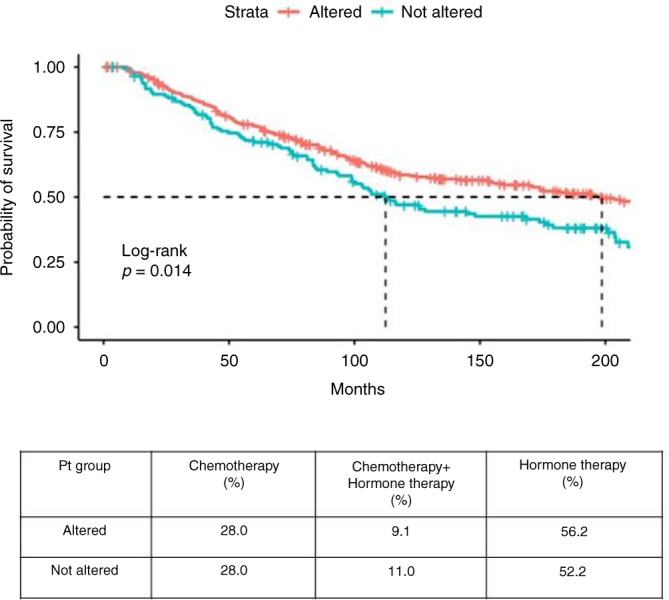
The expression of the top-ten most deregulated genes (CAPG, HRAS, IL1B, FGFR4, MAF, SERPINB2, CTSK, HMGA2, ANLN, MCM2) obtained from the comparison of “BM” vs “ES” CTCs was used to stratify stage I to III METABRIC BC patients (N = 472) in “altered” (red) and “not altered” (blue) groups. Data in cBioPortal were analysed to generate OS tables and create Kaplan–Meier curves, plotted through “survminer” and “survival” packages (v. 3.6.1) of R software. A statistically significant longer median survival was observed in “altered” group, compared to “not altered” one (199 vs 112 months, P = 0.014). BM bone metastases only, CTCs circulating tumour cells, BC breast cancer, ES extra-skeletal metastases.
No information about the sites of distant metastases was available in METABRIC dataset, for which we could not speculate about the BM-predictive capability of our gene panel. Results from this analysis suggest that the identified top-ten gene deregulations might successfully select a subset of early breast malignancies with better prognosis, whose organotropism might deserve further, prospective evaluation.
Discussion
BC is one of the most osteotropic malignancies, with ~14% of women with early-stage disease experiencing subsequent skeletal dissemination [41] which occurs in up to 70% of patients with advanced tumours.
Several research groups have looked for putative prognostic factors, including clinicopathological features and molecular signatures, able to stratify BC patients according to the risk of future skeletal involvement [19], with the purpose to personalise treatment and follow-up strategies in consideration of the favourable results of adjuvant bisphosphonate studies [42–44]. Despite the extensive research on this topic, none of the identified signatures has entered routine clinical practice to date, but most studies were based on the examination of primary or secondary tumour-derived samples, with consequent limitations related to the intrinsic spatio-temporal heterogeneity of cancer [2, 45].
Recently, the identification of BC “organotropism” signatures, as well as novel prognostic and predictive biomarkers, has been pursued through the phenotypical and molecular characterisation of CTCs, which dynamically reproduce cancer features and their variations over time [2, 10].
By applying a previously described protocol to isolate viable CTCs from metastatic BC patients [27], in this study we performed a targeted RNAseq of these cells, aiming at the identification of a GEP specifically correlated with BM onset.
In a study by Aceto et al. [22], CTCs derived from patients with metastatic HR+ BC underwent RNAseq, to unravel mechanisms involved in acquired endocrine resistance, showing activation of the androgen receptor pathway in CTCs from “bone-predominant” BC. In the present study, we focused on patients with “bone-only” metastases, regardless of the tumour HR status, assuming that molecular osteotropism signatures might be at least partially shared across BC sub-groups [18].
Moreover, to develop a cost-effective method, potentially applicable to large-scale analyses, we developed a targeted RNAseq protocol, based on a literature-derived gene panel, whose adequacy was preliminarily verified on BC cell lines with different organotropism, observing a clear clustering of the samples according to their biological behaviour. Interestingly, we found MMP1, FST and GIPC1 significantly upregulated in the bone-homing cell population (P7), as compared to parental P0 cells, in agreement with previous findings and proteomics data [7, 39, 46].
We then focused on CTCs, grouped according to the sites of distant metastases detected at the time of patient enrollment, and analysed as 10-cell pools, to overcome the drawbacks of single-cell gene expression analyses, such as insufficient RNA quantity and quality [47–49], as well as issues related to intra-individual heterogeneity [50].
As expected [51], we did not find a perfect overlap between BC cell lines and CTC GEP but, notably, the PCA plot showed a sharp separation of “BM” CTCs from the others. Moreover, 31 DEGs emerged from the comparison of “BM” vs “ES” CTCs, such as CAPG, GIPC1, IL1B and MAF, whose overexpression in osteotropic BC emerged from previous studies on wide patient series [25, 39, 40]. Major functional classes including deregulated genes were the “EMT” one, which encompassed 8 out of 31 genes (i.e. MAF, MAFA, GIPC1, PRDX1, MEF2C, SOX9, SMAD2, HMGA2) and the Wnt/β-catenin signalling (including 5 out 31 genes), whose direct and indirect participation in the BM cascade has been widely reported [18, 52–58].
With respect to KEGG enrichment analysis, different pathways were found upregulated in “BM” CTCs, including those involved in osteoclast differentiation, which is a key step of the BM “vicious circle” [18, 52]. Notably, one of the overexpressed genes found in “BM” versus “ES” CTCs was CTSK, whose role in osteoclast differentiation is well documented [59].
In the last part of the present work, we attempted to find out any potential prognostic meaning of the identified panel, focusing on the top-ten most deregulated genes emerged from “BM vs ES CTCs” comparison. Patient classification according to the presence (“altered”), or not (“not altered”), of at least one gene deregulation in matched CTC samples, did not exhibit a significant correlation with median survival, although this analysis might have been limited by the small sample size and the heterogeneity of our patient cohort, especially in terms of metastasis-free-interval and number of previous treatment lines [60].
Hence, we moved to the METABRIC dataset [34–36], in which integrated transcriptomic/genomic data relative to 2509 primary breast tumours are publicly available, together with matched long-term clinical follow-up data. Interestingly, in stage I–III BC subjects whose primary tumours had been screened for the expression of the top-ten genes, we observed a significantly longer median survival in the “altered” group, as compared to the “not altered” one. This suggests that the combination of the above-mentioned genes in a single panel might provide prognostic information in early BC, although we could not speculate about its capability to predict the organotropism of such malignancies, for which prospective, long-term investigation in early BC patients is required. Moreover, functional analyses exploring the role of the top-ten genes in the metastatic cascade are planned in our laboratory, and will certainly be useful to clarify the apparently discrepant results obtained in metastatic versus early-stage patients. In addition, we are aware that the biological source of METABRIC data is different from the one we employed and, hence, future comparative analyses between primary tumour and CTC samples are mandatory.
With respect to transcriptomic data, we recognise that RNAseq performed on small amounts of nucleic acids, as in the case of CTCs, raises several issues due to both technical limitations and biological variables (e.g., biases of transcript coverage, low capture efficiency and sequencing coverage, stochastic transcription, high drop-outs and bursting events) [61, 62]. For these reasons, when analysing RNAseq data from CTCs it is crucial to use appropriate computational methods to overcome the difficulties in normalisation and DEG identification. However, to date, none of the currently used pipelines for RNAseq data analysis is able to simultaneously satisfy sensitivity and specificity constraints [63]. Some recent studies have attempted to compare different DEG software tools, concluding that those methods exhibiting high sensitivity are often less accurate, due to the presence of false positives; on the other hand, the highly specific tools generally identify less DEGs and are limited by higher false-negative rates [61, 63–66].
In our work, in order to reduce the chances of false positives, we set the parameters to a FDR (used in the hypothesis testing to correct type I error) <0.25. Furthermore, for DEG analysis we used Partek® Flow® GSA, capable of considering the following response distributions: Normal, Lognormal, Lognormal with shrinkage, Negative Binomial, Poisson, and ANOVA (https://documentation.partek.com/display/FLOWDOC/Gene-specific+Analysis). In addition, an a posteriori analysis performed using the DESeq2 tool, also contained in the Partek® Flow® package, has independently validated our previous results confirming the top-10 (4 ≤ FC ≤ −4) most deregulated genes among DEGs, with the exception of CTSK.
However, it has to be underlined that DESeq2 is specifically designed and commonly used for bulk RNAseq data but, once applied to CTCs, it tends to have greater specificity at the expense of sensitivity [65]. Moreover, it is well recognised that RNAseq data derived from small nucleic acid amounts might miss a fraction of transcripts, resulting in zero read counts, due to stochastic gene expression and low capture efficiency, affecting the subsequent differential gene expression analysis [61, 62]. Previous studies on this topic even claim that the missing rate can reach nearly 30%, resulting in a loss of valuable information [62].
For instance, we were surprised to find no significant differences in the expression of CXCR4, MMP1, CTGF and IL11 genes between “BM” and “ES” CTCs, in consideration of the previous literature supporting their role in BC osteotropism [6, 45]. Hence, our data analysis could have incorporated a certain rate of errors due to false signals, but gold-standard computational tools have yet to be developed.
In conclusion, despite the above-mentioned limitations of RNAseq, this method is still one of the most widely used technologies for gene expression profiling [67] and, for this reason, we deemed it appropriate for investigating BC osteotropism, in line with previous reports [15, 16, 21, 47].
However, rather than performing a more expensive whole trascriptome RNAseq, we adopted a targeted analysis that encompasses a selected number of genes. This strategy might render CTC GEP investigation more feasible, economically sustainable, and even applicable to a clinical setting, although a prospective validation, over a wider patient cohort involving early BC patients to be longitudinally monitored, is mandatory before moving from the bench to bedside.
Supplementary information
Acknowledgements
The authors are grateful to Luigia Stefania Stucci for her precious help in recruiting participants and to Claudia Felici for her valuable technical support. The authors would like to thank also “DIVELLA SpA” and particularly Enzo Divella, for the generous donation of the Ion Torrent used for NGS analyses.
Author contributions
Conception and design: SD, DL, RP and FS; development of methodology: PC, DL and RP; acquisition of the data: JB, PC, SD, EL, CP, FS and SW; analysis and interpretation of the data: SD and DL; writing, review, and/or revision of the manuscript: all authors; study supervision: FS.
Funding information
This work was supported by the Apulia Region (“Oncogenomic”, “Jonico-Salentino”, “Precision Medicine” and “Tecnopolo per la Medicina di Precisione-GR Puglia 2117/2018” projects).
Data availability
The datasets generated during this study are available at Gene Expression Omnibus (GEO) data repository, with GEO accession number GSE160314.
Ethics approval and consent to participate
The patients were enrolled after written informed consent. The study was approved by the Ethics Committee of the University Hospital “Policlinico of Bari” (Project identification code: 44100) and performed in accordance with the principles of the Declaration of Helsinki.
Consent to publish
Not applicable.
Competing interests
JB: speaker fees from Amgen and advisory board Novartis; DL, SD, RP, PC, SW, CP, EL, RC and FS declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Domenica Lovero, Stella D’Oronzo.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01481-z.
References
- 1.Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12:6243s–6249s. doi: 10.1158/1078-0432.CCR-06-0931. [DOI] [PubMed] [Google Scholar]
- 2.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 3.Leone BA, Vallejo CT, Romero AO, Machiavelli MR, Pérez JE, Leone J, et al. Prognostic impact of metastatic pattern in stage IV breast cancer at initial diagnosis. Breast Cancer Res Treat. 2017;161:537–48. doi: 10.1007/s10549-016-4066-7. [DOI] [PubMed] [Google Scholar]
- 4.Wang R, Zhu Y, Liu X, Liao X, He J, Niu L. The clinicopathological features and survival outcomes of patients with different metastatic sites in stage IV breast cancer. BMC Cancer. 2019;19:1091. doi: 10.1186/s12885-019-6311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Oronzo S, Gregory W, Nicholson S, Khong Chong Y, Brown J, Coleman R. Natural history of stage II/III breast cancer, bone metastasis and the impact of adjuvant zoledronate on distribution of recurrence. J Bone Oncol. 2021;28:100367. doi: 10.1016/j.jbo.2021.100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waks AG, Winer EP. Breast cancer treatment: a review. J Am Med Assoc. 2019;321:288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 7.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 8.Markopoulos C, Hyams DM, Gomez HL, Harries M, Nakamura S, Traina T, et al. Multigene assays in early breast cancer: insights from recent phase 3 studies. Eur J Surg Oncol. 2020;46:656–66. doi: 10.1016/j.ejso.2019.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Thery L, Meddis A, Cabel L, Proudhon C, Latouche A, Pierga JY, et al. Circulating tumor cells in early breast cancer. JNCI Cancer Spectr. 2019;3:pkz026. doi: 10.1093/jncics/pkz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cristofanilli M, Pierga JY, Reuben J, Rademaker A, Davis AA, Peeters DJ, et al. The clinical use of circulating tumor cells (CTCs) enumeration for staging of metastatic breast cancer (MBC): international expert consensus paper. Crit Rev Oncol Hematol. 2019;134:39–45. doi: 10.1016/j.critrevonc.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Alimirzaie S, Bagherzadeh M, Akbari MR. Liquid biopsy in breast cancer: a comprehensive review. Clin Genet. 2019;95:643–60. doi: 10.1111/cge.13514. [DOI] [PubMed] [Google Scholar]
- 12.Palmirotta R, Lovero D, Cafforio P, Felici C, Mannavola F, Pellè E, et al. Liquid biopsy of cancer: a multimodal diagnostic tool in clinical oncology. Ther Adv Med Oncol. 2018;10:1758835918794630. doi: 10.1177/1758835918794630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schochter F, Friedl TWP, deGregorio A, Krause S, Huober J, Rack B, et al. Are circulating tumor cells (CTCs) ready for clinical use in breast cancer? An overview of completed and ongoing trials using CTCs for clinical treatment decisions. Cells. 2019;8:1412. doi: 10.3390/cells8111412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klotz R, Thomas A, Teng T, Han SM, Iriondo O, Li L, et al. Circulating tumor cells exhibit metastatic tropism and reveal brain metastasis drivers. Cancer Discov. 2020;10:86–103. doi: 10.1158/2159-8290.CD-19-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braun M, Markiewicz A, Kordek R, Sadej R, Romanska H. Profiling of invasive breast carcinoma circulating tumour cells-are we ready for the ‘liquid’ revolution? Cancers. 2019;11:143. doi: 10.3390/cancers11020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lang JE, Ring A, Porras T, Kaur P, Forte VA, Mineyev N, et al. RNA-seq of circulating tumor cells in stage II-III breast cancer. Ann Surg Oncol. 2018;25:2261–70. doi: 10.1245/s10434-018-6540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Xu R, Yuan H, Zhang Y, Cheng S. Single-cell RNA sequencing reveals novel gene expression signatures of trastuzumab treatment in HER2+ breast cancer: a pilot study. Medicine. 2019;98:e15872. doi: 10.1097/MD.0000000000015872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Hoffmann AD, Liu H, Liu X. Organotropism: new insights into molecular mechanisms of breast cancer metastasis. npj Precis Oncol. 2018;2:4. doi: 10.1038/s41698-018-0047-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Oronzo S, Brown J, Coleman R. The role of biomarkers in the management of bone-homing malignancies. J Bone Oncol. 2017;9:1–9. doi: 10.1016/j.jbo.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Molina J, Jiang J, Ferber M, Pruthi S, Jatkoe T, et al. Gene expression markers in circulating tumor cells may predict bone metastasis and response to hormonal treatment in breast cancer. Mol Clin Oncol. 2013;1:1031–8. doi: 10.3892/mco.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boral D, Vishnoi M, Liu HN, Yin W, Sprouse ML, Scamardo A, et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat Commun. 2017;8:196. doi: 10.1038/s41467-017-00196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aceto N, Bardia A, Wittner BS, Donaldson MC, O’Keefe R, Engstrom A, et al. AR expression in breast cancer CTCs associates with bone metastases. Mol Cancer Res. 2018;16:720–7. doi: 10.1158/1541-7786.MCR-17-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savci-Heijink CD, Halfwerk H, Koster J, van de Vijver MJ. A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res Treat. 2016;156:249–59. doi: 10.1007/s10549-016-3741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cosphiadi I, Atmakusumah TD, Siregar NC, Muthalib A, Harahap A, Mansyur M. Bone metastasis in advanced breast cancer: analysis of gene expression microarray. Clin Breast Cancer. 2018;18:e1117–22. doi: 10.1016/j.clbc.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Nutter F, Holen I, Brown HK, Cross SS, Evans CA, Walker M, et al. Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: evidence using a novel bone-seeking cell line. Endocr Relat Cancer. 2014;21:327–41. doi: 10.1530/ERC-13-0158. [DOI] [PubMed] [Google Scholar]
- 26.Coleman R, Hadji P, Body JJ, Santini D, Chow E, Terpos E, et al. Bone health in cancer: ESMO clinical practice guidelines. Ann Oncol. 2020;31:1650–63. doi: 10.1016/j.annonc.2020.07.019. [DOI] [PubMed] [Google Scholar]
- 27.D’Oronzo S, Lovero D, Palmirotta R, Stucci LS, Tucci M, Felici C, et al. Dissection of major cancer gene variants in subsets of circulating tumor cells in advanced breast cancer. Sci Rep. 2019;9:17276. doi: 10.1038/s41598-019-53660-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoneda T, Williams PJ, Hiraga T, Niewolna M, Nishimura R. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res. 2001;16:1486–95. doi: 10.1359/jbmr.2001.16.8.1486. [DOI] [PubMed] [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–61. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: network analysis and visualization of proteomics data. J. Proteome Res. 2019;18:623–32. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costa C, Muinelo-Romay L, Cebey-López V, Pereira-Veiga T, Martínez-Pena I, Abreu M, et al. Analysis of a real-world cohort of metastatic breast cancer patients shows circulating tumor cell clusters (CTC-clusters) as predictors of patient outcomes. Cancers. 2020;12:1111. doi: 10.3390/cancers12051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis AA, Zhang Q, Gerratana L, Shah AN, Zhan Y, Qiang W, et al. Association of a novel circulating tumor DNA next-generating sequencing platform with circulating tumor cells (CTCs) and CTC clusters in metastatic breast cancer. Breast Cancer Res. 2019;21:137. doi: 10.1186/s13058-019-1229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westbrook JA, Cairns DA, Peng J, Speirs V, Hanby AM, Holen I, et al. CAPG and GIPC1: breast cancer biomarkers for bone metastasis development and treatment. J Natl Cancer Inst. 2016;108:djv360. [DOI] [PMC free article] [PubMed]
- 40.Pavlovic M, Arnal-Estapé A, Rojo F, Bellmunt A, Tarragona M, Guiu M, et al. Enhanced MAF oncogene expression and breast cancer bone metastasis. J. Natl Cancer Inst. 2015;107:djv256. doi: 10.1093/jnci/djv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pulido C, Vendrell I, Ferreira AR, Casimiro S, Mansinho A, Alho I, et al. Bone metastasis risk factors in breast cancer. Ecancermedicalscience. 2017;11:715. doi: 10.3332/ecancer.2017.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Early Breast Cancer Trialists’ Collaborative, G. Adjuvant bisphosphonate treatment in early breast cancer: meta-analyses of individual patient data from randomised trials. Lancet. 2015;386:1353–61. doi: 10.1016/S0140-6736(15)60908-4. [DOI] [PubMed] [Google Scholar]
- 43.D’Oronzo S, Silvestris E, Paradiso A, Cives M, Tucci M. Role of bone targeting agents in the prevention of bone metastases from breast cancer. Int J Mol Sci. 2020;21:3022. [DOI] [PMC free article] [PubMed]
- 44.Coleman R. Clinical benefits of bone targeted agents in early breast cancer. Breast. 2019;48:S92–6. doi: 10.1016/S0960-9776(19)31133-6. [DOI] [PubMed] [Google Scholar]
- 45.Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501:355–64. doi: 10.1038/nature12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bellahcène A, Bachelier R, Detry C, Lidereau R, Clézardin P, Castronovo V. Transcriptome analysis reveals an osteoblast-like phenotype for human osteotropic breast cancer cells. Breast Cancer Res Treat. 2007;101:135–48. doi: 10.1007/s10549-006-9279-8. [DOI] [PubMed] [Google Scholar]
- 47.Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24:496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014;8:1905–18. doi: 10.1016/j.celrep.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossi E, Zamarchi R. Single-cell analysis of circulating tumor cells: how far have we come in the -Omics Era? Front Genet. 2019;10:958. doi: 10.3389/fgene.2019.00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magbanua MJM, Rugo HS, Wolf DM, Hauranieh L, Roy R, Pendyala P, et al. Expanded genomic profiling of circulating tumor cells in metastatic breast cancer patients to assess biomarker status and biology over time (CALGB 40502 and CALGB 40503, alliance) Clin Cancer Res. 2018;24:1486–99. doi: 10.1158/1078-0432.CCR-17-2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE. 2012;7:e33788. doi: 10.1371/journal.pone.0033788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu MY, Li CJ, Yiang GT, Cheng YL, Tsai AP, Hou YT, et al. Molecular regulation of bone metastasis pathogenesis. Cell. Physiol. Biochem. 2018;46:1423–38. doi: 10.1159/000489184. [DOI] [PubMed] [Google Scholar]
- 53.Zhuang X, Zhang H, Li X, Li X, Cong M, Peng F, et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol. 2017;19:1274–85. doi: 10.1038/ncb3613. [DOI] [PubMed] [Google Scholar]
- 54.Kasoha M, Bohle RM, Seibold A, Gerlinger C, Juhasz-Böss I, Solomayer EF. Dickkopf-1 (Dkk1) protein expression in breast cancer with special reference to bone metastases. Clin Exp Metastasis. 2018;35:763–75. doi: 10.1007/s10585-018-9937-3. [DOI] [PubMed] [Google Scholar]
- 55.Johnson RW, Merkel AR, Page JM, Ruppender NS, Guelcher SA, Sterling JA. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin Exp Metastasis. 2014;31:945–59. doi: 10.1007/s10585-014-9682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eyre R, Alférez DG, Santiago-Gómez A, Spence K, McConnell JC, Hart C, et al. Microenvironmental IL1beta promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat Commun. 2019;10:5016. doi: 10.1038/s41467-019-12807-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holen I, Lefley DV, Francis SE, Rennicks S, Bradbury S, Coleman RE, et al. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget. 2016;7:75571–84. doi: 10.18632/oncotarget.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tulotta C, Lefley DV, Freeman K, Gregory WM, Hanby AM, Heath PR, et al. Endogenous production of IL1B by breast cancer cells drives metastasis and colonization of the bone microenvironment. Clin Cancer Res. 2019;25:2769–82. doi: 10.1158/1078-0432.CCR-18-2202. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, He W, Zhang S. Seeking for correlative genes and signaling pathways with bone metastasis from breast cancer by integrated analysis. Front Oncol. 2019;9:138. doi: 10.3389/fonc.2019.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lobbezoo DJ, van Kampen RJ, Voogd AC, Dercksen MW, van den Berkmortel F, Smilde TJ, et al. Prognosis of metastatic breast cancer: are there differences between patients with de novo and recurrent metastatic breast cancer? Br J Cancer. 2015;112:1445–51. doi: 10.1038/bjc.2015.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang T, Li B, Nelson CE, Nabavi S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinforma. 2019;20:40. doi: 10.1186/s12859-019-2599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang MQ, Weissman SM, Yang W, Zhang J, Canaann A, Guan R. MISC: missing imputation for single-cell RNA sequencing data. BMC Syst. Biol. 2018;12:114. doi: 10.1186/s12918-018-0638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Rouchka EC, Brock GN, Yan J, O’Toole TE, Tieri DA, et al. A combined approach with gene-wise normalization improves the analysis of RNA-seq data in human breast cancer subtypes. PLoS ONE. 2018;13:e0201813. doi: 10.1371/journal.pone.0201813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soneson C, Robinson MD. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods. 2018;15:255–61. doi: 10.1038/nmeth.4612. [DOI] [PubMed] [Google Scholar]
- 65.Mou T, Deng W, Gu F, Pawitan Y, Vu TN. Reproducibility of methods to detect differentially expressed genes from single-cell RNA sequencing. Front Genet. 2019;10:1331. doi: 10.3389/fgene.2019.01331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holland CH, Tanevski J, Perales-Patón J, Gleixner J, Kumar MP, Mereu E, et al. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 2020;21:36. doi: 10.1186/s13059-020-1949-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu Z, Qiu S, Shao K, Hou Y. Progress and challenges of sequencing and analyzing circulating tumor cells. Cell Biol Toxicol. 2018;34:405–15. doi: 10.1007/s10565-017-9418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus (GEO) data repository, with GEO accession number GSE160314.