

Abstract
Mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS) is characterized by metabolic stroke, seizures, cognitive decline, lactic acidosis, ragged‐red fibers, headache, and vomiting, and in 80% of cases due to the mtDNA variant m.3243A>G. We report the case of a MELAS patient carrying a variant in subunit‐5 of the respiratory chain (MT‐ND5), rarely reported in MELAS. The patient is a 33‐year‐old male, who experienced a series of stroke‐like episodes (StLEs) since age 23 years, which manifested clinically as seizures transient sensory disturbances, weakness, and visual or cognitive impairment. Over 9 years, these StLEs were misinterpreted as ischemic strokes, respectively, as cerebral vasculitis. He presented with mild, recurrent elevations of the creatine kinase. Initially, anti‐seizure drugs and steroids appeared to be beneficial. Despite good recovery of each single StLE, the patient experienced a progressive decline of cognitive functions and activities of daily living. Cerebral imaging showed corresponding stroke‐like lesions in changing locations. At age 32y, genetic work‐up revealed the variant m.13513G>A in MT‐ND5. The patient profited significantly from a cocktail with anti‐oxidants/cofactors. This case shows that the variant m.13513G>A in MT‐ND5 can manifest as MELAS that StLEs recover spontaneously and that the course of MELAS is slowly progressive.
Keywords: MELAS, mtDNA, respiratory chain, seizures, stroke‐like episode
If MELAS does not coincide with the m.3243A>G variant, sequencing of the entire mtDNA is necessary to exclude other causes that are clinically or biochemically compatible with MELAS.
1. INTRODUCTION
Mitochondrial encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS) is a syndromic mitochondrial disorder most frequently due to mutations in mtDNA located genes. The most frequent of these mutations is the variant m.3243A>G in tRNA (Leu) accounting for about 80% of the MELAS cases. 1 MELAS is a multisystem disorder, predominantly affecting the brain and the muscle. 1 MELAS is diagnosed according to the Japanese 2 or Hirano criteria. 3 The variant m.13513G>A in MT‐ND5 only rarely causes MELAS and has not been reported in an Indian patient before.
2. CASE REPORT
A previously healthy, 33‐year‐old male presented at age 23 years with coma after right focal seizures with secondary generalization that had occurred in clusters one right after another for 2 days. Electroencephalography, cerebral computed tomography, and cerebral magnetic resonance imaging (cMRI) were non‐informative. He was put on levetiracetam (500 mg bid). The family history was positive for recurrent “strokes,” epilepsy, and death before 30 years in two maternal aunts. There was no parental consanguinity. Mother and grandmother from the mother's side were clinically unaffected (Figure 1).
FIGURE 1.
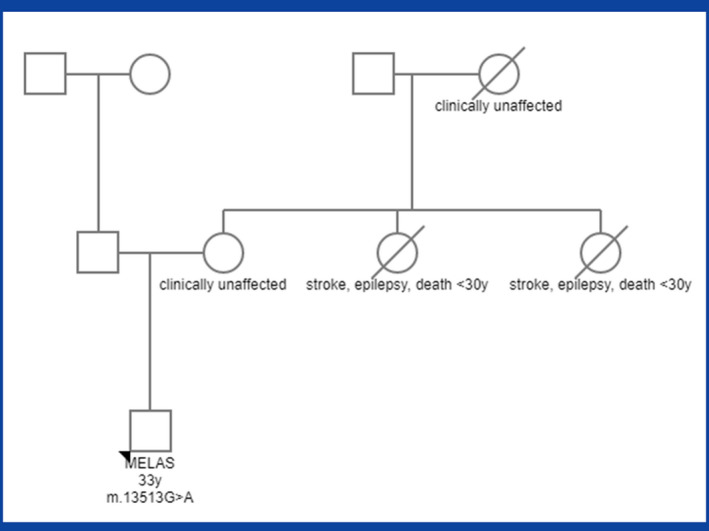
Family tree of the index patient
Eight months later, he developed sudden‐onset bilaterally impaired vision (complete loss left and partial loss right), which started with change in color vision and deteriorated to bare perception of light. Optic neuritis was suspected and methylprednisolone (1 g/day for 5 days) given. cMRI showed a diffusion weighted imaging (DWI) hyperintense and apparent diffusion coefficient (ADC) hypointense lesion in a right posterior distribution not confined to a vascular territory, resulting in cortical blindness (Figure 2). Cerebrospinal fluid (CSF) investigations were normal. Visual disturbance resolved within 1 week. Levetiracetam was increased to 1000 mg/day.
FIGURE 2.
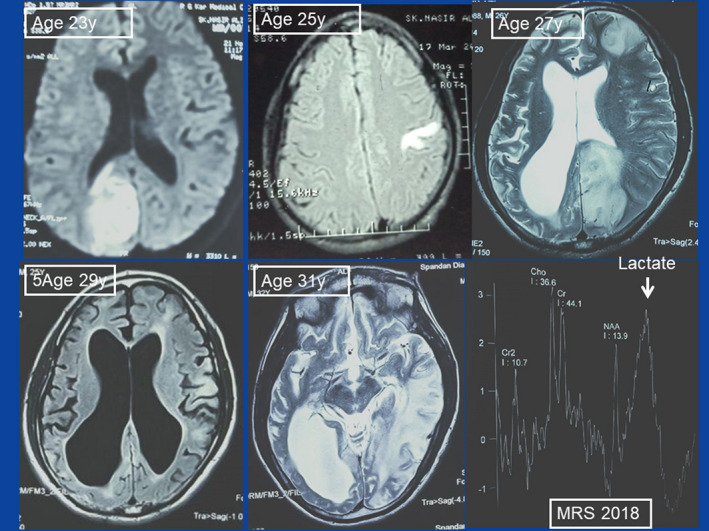
MRI findings at various stages of the disease. Upper middle: the third StLE manifested as cortical hyperintensity in the left post‐central gyrus (3/12). Upper left: the fourth StLE (11/12) showed up as right occipital lesion. Upper right: the fifth StLE manifested as lesion in the left frontal and the left occipital lobe. Lower left: MRI in 5/16 showed internal hydrocephalus, cortical atrophy, and a left parietal and left frontal lesion. Lower middle: MRI in 8/18 showed a hyperintense lesion in the left parieto‐occipital region. All lesions were not confined to a vascular territory. Lower right: MR spectroscopy in 8/18 revealed an elevated lactate peak
At age 25 years, he experienced sudden‐onset paresthesias over the right upper limb associated with difficulties in buttoning/unbuttoning his shirt. cMRI revealed a cortical lesion in the left post‐central gyrus suggestive of embolic stroke, demyelinating central nervous system (CNS) disorder, or CNS vasculitis (Figure 2). Creatine kinase (CK) was elevated (Table 1). Antinuclear antibodies (ANA) were positive. Renal ultrasound suggested bilateral renal parenchymal disease. Both serum and CSF lactate were normal (Table 1). Though cerebral digital subtraction angiography (DSA) was normal, CNS vasculitis was suspected and the patient was put on steroids, without benefit.
TABLE 1.
Variability of serum/CSF lactate and pyruvate and serum CK values over the course of illness
| Month/year | Lactate (serum) | Pyruvate (serum) | L/P (serum) | CK (serum) | Lactate (CSF) | Pyruvate (CSF) | L/P (serum) | L/P (CSF) |
|---|---|---|---|---|---|---|---|---|
| 11/2009 | nd | nd | nd | nd | nd | nd | nd | |
| 7/2010 | nd | nd | nd | nd | nd | nd | nd | |
| 3/2011 | 1.03 | nd | nd | 286 | 1.55 | nd | nd | |
| 11/2012 | 2.13 | nd | nd | 376 | 0.52 | nd | nd | |
| 3/2014 | 0.94 | nd | nd | 366 | 1.42 | nd | nd | |
| 4/2015 | 1.08 | nd | nd | 412 | 1.83 | nd | nd | |
| 5/2016 | 1.86 | nd | nd | 356 | 2.02 | nd | nd | |
| 8/2018 | 2.98 | 0.045 | 66.2 | 472 | 5.42 | 0.078 | 69.48 | 69.48 |
| 8/2019 | 2.75 | 0.060 | 45.83 | 219 | 4.07 | 0.086 | 47.32 | 47.32 |
| Reference limits | 0.5–2.2 mmol/L | 0.030–0.107 mmol/L | >25 | <170 U/L | <2.22 mmol/L | 0.06–0.19 mmol/L | >25 | <2.22 |
Abbreviations: L/P, lactate/pyruvate ratio; nd, not done.
At age 26 years, he presented with left hemiparesis and left temporal hemianopia. cMRI displayed a T2‐ and FLAIR‐hyperintense lesion in a right occipital distribution with normal DSA. Work‐up for embolic stroke and CNS vasculitis was negative. Serum/CSF lactate and the lactate:pyruvate ratio were normal (Table 1).
At age 27 years, he presented with a single generalized tonic‐clonic seizure associated with marked memory impairment, executive dysfunction and attention deficits. cMRI revealed old encephalomalacic changes in the right occipital lobe and new onset T2‐ and FLAIR‐hyperintensities in a left frontal and left occipital distribution with marked generalized cortical atrophy (Figure 2). Prednisolone (50 mg/day with slow tapering) was added for suspected, recurrent, vasculitic infarcts.
At age 29 years, he was admitted with extensive cognitive dysfunction (Addenbrooke's score: 64, mini‐mental state examination [MMSE]; 10), grossly impaired activities of daily living and poor quality of life. cMRI revealed marked cortical atrophy, with diffuse encephalomalacic changes (Figure 2).
Re‐evaluation at age 32 years revealed elevated CK, serum/CSF lactate, and lactate:pyruvate ratio (Table 1). Genetic work‐up revealed the variant m.13513G>A in MT‐ND5 (p. Asp393Asn). The heteroplasmy rate was 11% in lymphocytes. MELAS was diagnosed and coenzyme‐Q, L‐arginine, riboflavin, thiamine, L‐carnitine, and lipoic acid were prescribed. Additionally, he was discharged with clonazepam, clobazam, levetiracetam, phenytoin, lamotrigine, telmisartan, and memantine. Within 7 months, the Addenbrooke's score improved from 64 to 72 and the MMSE from 10 to 16.
3. DISCUSSION
The presented patient is interesting for several aspects. First, MELAS was diagnosed with a latency of 9 years although he presented with typical StLEs. Delayed diagnosis of mitochondrial disorders (MIDs) is common as many physicians are not familiar with the syndrome and appropriate diagnostic facilities are not available everywhere. Second, the patient experienced at least five StLEs, which resolved spontaneously or upon levetiracetam and steroids. Recurrence of stroke‐like lesions (SLLs) can be regarded as pathognomonic for MELAS. Third, MELAS was caused by the variant m.13513G>A in MT‐ND5, which not only causes MELAS 4 but also Leber's hereditary optic neuropathy, 5 Leigh syndrome, 6 neonatal death, 5 or MELAS/Leigh overlap syndrome. 7 Only ten other cases with MELAS and the m.13513G>A variant have been reported (Table 2). Three of them were described with fatal congenital acidosis, seizures, language delay, kidney dysplasia, and cerebellar atrophy (Table 2). Novel features in the index case are recurrent StLEs and cognitive decline.
TABLE 2.
Phenotypic spectrum of patients carrying the variant m.13513G>A
| Reference | Yahata (2017) 14 | Karnebeek (2011) 4 | Vasques‐Justes (2019) 5 | Jimenez‐Legido (2019) 15 | Wang (2010) 7 | Index patient |
|---|---|---|---|---|---|---|
| Age | 16 years | 1 day to 10 years | 16 years | 18 months, 20 months | 13–22 years | 33 years |
| Gender | m | m (n=1), f (n=2) | m | m (n=1), f (n=1) | m (n=1), f (n=2) | m |
| Heteroplasmy | 64% | 0–66% | np | 64%, 72% | 46–61% | 11% |
| Phenotype | MELAS | MELAS | LHON | Leigh syndrome | MELAS/Leigh | MELAS |
| No phenotypic details provided |
Fatal congenital acidosis cerebellar atrophy Seizures kidney dysplasia language delay clumsiness |
Hypertrophic cardiomyopathy, myopathy | np | np | Seizures cortical atrophy recurrent StLEs cognitive decline myopathy |
Abbreviations: f, female; LHON, Leber's hereditary optic neuropathy; M, male; MELAS, mitochondrial encephalopathy, lactic acidosis, and stroke‐like episode; np, not provided.
MELAS was diagnosed according to the Hirano 3 and Japanese criteria. 2 According to the Japanese criteria, 2 the patient was diagnosed as definite MELAS (the presence of >1 item of category‐A [headache with vomiting, seizures, hemiplegia, cortical blindness/hemianopia, and acute MRI lesion] and >1 item of category‐B [high serum/brain lactate or reduced respiratory chain enzyme activity, abnormalities on muscle biopsy, and pathogenic MELAS mutation]). According to the Hirano criteria, probable MELAS was diagnosed (StLE <40 years, seizures, dementia, lactic acidosis, or ragged‐red fibers [RRFs] and >1 of normal early development, recurrent headache, or recurrent vomiting). 3 Only one item was missing for diagnosing definite MELAS.
Arguments against MELAS are the absence of myopathy, the initially normal serum/CSF lactate (Table 1), and the unusual presentation of the SLLs on cMRI. Usually, SLLs are hyperintense on DWI/ADC. Stroke in MELAS is most commonly seen in a cortical distribution, but subcortical involvement is also frequently encountered. 8 The absence of short stature, headache, and vomiting is no argument against MELAS as not all patients develop these features. 9
Whether the detected variant was truly causative remains speculative as no specific investigations to confirm pathogenicity (biochemical investigations, single‐fiber studies, cybrid studies, and assay on buccal or urinary epithelial cells) had been carried out. 10 There is also no proof for segregation of the disease with the variant. An argument for causality, however, is that the variant has been previously reported in association with MIDs (Table 2). In a study of two MELAS patients carrying the variant m.15315A>G, it was clearly demonstrated that this variant was responsible for reduced activity of complex‐I of the respiratory chain as the amino acid change D393N is crucial for the function of complex‐I. 11 The low heteroplasmy rate does not exclude pathogenicity as only a mildly affected tissue was investigated. A further argument in favor of the pathogenicity is that the amino acid change Asp>Asn at position D393 may lead to loss of the quinine reactive site and subsequently to reduced activity of the oxidative phosphorylation. 12
Reasons for the delayed diagnosis are unawareness of the condition, initially normal serum/CSF lactate, positive ANA, and misleading cMRI. A further reason for the delay in diagnosis could be the fact that the entire sequence of mtDNA is not performed routinely and that once the m.3243A>G mutation is excluded, the suspicion of MELAS in the mind of neurologists might fade away. However, even if there is absence of the m.3243A>G variant, work‐up for defects of the mtDNA should be reinforced. The reason for spontaneous recovery of visual impairment at the second StLE remains elusive but it can be speculated that either steroids were effective, as has been previously reported, 13 or that the anti‐oxidative/regenerative capacity was still preserved. Spontaneous resolution of a SLL is common in MELAS. Positive ANA were most likely a secondary phenomenon as immune mechanisms can be triggered by the metabolic breakdown. Whether improvement on the Addenbrooke's score and the MMSE was attributable to the cocktail or due to spontaneous recovery after recovery from the StLE remains speculative. The specific efficacy of the cocktail is not validated, which is particularly the case for lipoic acid, of which the application as a pre‐synthetized compound is ineffective given the fact that lipoic acid is synthetized in situ during the formation of pyruvate dehydrogenase, respectively, the alpha‐ketoglutarate dehydrogenase.
This case shows that the variant m.13513G>A in MT‐ND5 may manifest phenotypically as MELAS. MELAS should be suspected upon the typical presentation of SLLs. If MELAS does not coincide with the m.3243A>G variant, sequencing of the entire mtDNA is necessary to exclude other causes that are clinically or biochemically compatible with MELAS. This concept should be fully acquired by neurologists and neuropediatricians.
CONFLICT OF INTEREST
None of the authors declares any conflict of interest.
AUTHOR CONTRIBUTIONS
Ritwik Ghosh contributed to literature search, discussion, critical comments, drafting, and final approval. Souvik Dubey, Subhas Bhuin, Durjoy Lahiri, Biman Kanti Ray, and Josef Finsterer contributed to literature search, discussion, critical comments, and final approval.
ETHICAL APPROVAL
Publication of the case was approved by the institutional review board. The patient's caregivers consented with the publication.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENT
None.
Ghosh R, Dubey S, Bhuin S, Lahiri D, Ray BK, Finsterer J. MELAS with multiple stroke‐like episodes due to the variant m.13513G>A in MT‐ND5 . Clin Case Rep. 2022;10:e05361. doi: 10.1002/ccr3.5361
DATA AVAILABILITY STATEMENT
All data used for the manuscript are available from the corresponding author.
REFERENCES
- 1. El‐Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015;116(1–2):4‐12. [DOI] [PubMed] [Google Scholar]
- 2. Yatsuga S, Povalko N, Nishioka J, et al; Taro Matsuoka for MELAS Study Group in Japan . MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 1820;2012:619‐624. [DOI] [PubMed] [Google Scholar]
- 3. Hirano M, Ricci E, Richard Koenigsberger M, et al. MELAS: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125‐135. [DOI] [PubMed] [Google Scholar]
- 4. Van Karnebeek CD, Waters PJ, Sargent MA, et al. Expanding the clinical phenotype of the mitochondrial m.13513G>A mutation with the first report of a fatal neonatal presentation. Dev Med Child Neurol. 2011;53:565‐568. [DOI] [PubMed] [Google Scholar]
- 5. Vázquez‐Justes D, Carreño‐Gago L, García‐Arumi E, et al. Mitochondrial m.13513G>A point mutation in ND5 in a 16‐year‐old man with leber hereditary optic neuropathy detected by next‐generation sequencing. J Pediatr Genet. 2019;8:231‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jimenez‐Legido M, Bernardino‐Cuesta B, Lopez‐Marin L, et al. Two new cases of Leigh syndrome caused by mutation m.13513G> A in the MTND5 gene. Rev Neurol. 2019;68:312‐314. [DOI] [PubMed] [Google Scholar]
- 7. Wang Z, Qi XK, Yao S, et al. Phenotypic patterns of MELAS/LS overlap syndrome associated with m.13513G>A mutation, and neuropathological findings in one autopsy case. Neuropathology. 2010;30:606‐614. [DOI] [PubMed] [Google Scholar]
- 8. Finsterer J. Mitochondrial metabolic stroke: phenotype and genetics of stroke‐like episodes. J Neurol Sci. 2019;400:135‐141. [DOI] [PubMed] [Google Scholar]
- 9. Liu XL, Bao XH, Ma YN, Chang XZ, Qin J, Wu XR. Clinical, pathological and molecular biological characteristics of mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episode in children. Zhonghua Er Ke Za Zhi. 2013;51:130‐135. [PubMed] [Google Scholar]
- 10. Finsterer J, Zarrouk‐Mahjoub S, Shoffner JM. MERRF classification: implications for diagnosis and clinical trials. Pediatr Neurol. 2018;80:8‐23. [DOI] [PubMed] [Google Scholar]
- 11. Corona P, Antozzi C, Carrara F, et al. A novel mtDNA mutation in the ND5 subunit of complex I in two MELAS patients. Ann Neurol. 2001;49(1):106‐110. doi: [DOI] [PubMed] [Google Scholar]
- 12. Sun CB, Bai HX, Xu DN, Xiao Q, Liu Z. Mitochondrial 13513G>A mutation with low mutant load presenting as isolated Leber's hereditary optic neuropathy assessed by next generation sequencing. Front Neurol. 2021;4(12):601307. doi: 10.3389/fneur.2021.601307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finsterer J, Frank M. Glucocorticoids for mitochondrial disorders. Singapore Med J. 2015;56:122‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yahata N, Matsumoto Y, Omi M, Yamamoto N, Hata R. TALEN‐mediated shift of mitochondrial DNA heteroplasmy in MELAS‐iPSCs with m.13513G>A mutation. Sci Rep. 2017;7(1):15557. doi: 10.1038/s41598-017-15871-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jimenez‐Legido M, Bernardino‐Cuesta B, Lopez‐Marin L, et al. Dos nuevos casos de sindrome de Leigh por mutacion m.13513G>A en el gen MTND5 Two new cases of Leigh syndrome caused by mutation m.13513G> A in the MTND5 gene. Rev Neurol. 2019;68(7):312‐314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used for the manuscript are available from the corresponding author.