

Abstract
The discovery of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and its development as a genome editing tool has revolutionized the field of molecular biology. In the DNA damage field, CRISPR has brought an alternative to induce endogenous double‐strand breaks (DSBs) at desired genomic locations and study the DNA damage response and its consequences. Many systems for sgRNA delivery have been reported in order to efficiently generate this DSB, including lentiviral vectors. However, some of the consequences of these systems are not yet well understood. Here, we report that lentiviral‐based sgRNA vectors can integrate into the endogenous genomic target location, leading to undesired activation of the target gene. By generating a DSB in the regulatory region of the ABCB1 gene using a lentiviral sgRNA vector, we can induce the formation of Taxol‐resistant colonies. We show that these colonies upregulate ABCB1 via integration of the EEF1A1 and the U6 promoters from the sgRNA vector. We believe that this is an unreported CRISPR/Cas9 on‐target effect that researchers need to be aware of when using lentiviral vectors for genome editing.
Keywords: CRISPR‐Cas9, drug resistance, gene activation, lentiviral integration, on‐target effects
Subject Categories: Chromatin, Transcription & Genomics; Methods & Resources
Lentivirus‐based sgRNA vectors can integrate into the endogenous genomic target location and activate the expression of the target gene.
Introduction
The discovery of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), their role in the prokaryotic immune system, and subsequent development as a genome editing tool has revolutionized the field of molecular biology (Mojica et al, 1993, 2005; van der Oost et al, 2009; Jinek et al, 2012; Cong et al, 2013; Mali et al, 2013). In recent years, many laboratories have developed CRISPR/Cas9 as a tool that can be applied to study many different biological questions (Lino et al, 2018). In the DNA damage field, CRISPR has brought an alternative to induce endogenous double‐strand breaks (DSBs) at desired genomic locations. This system allowed for the study of the DNA damage response and its consequences in different genome compartments or structures (Vítor et al, 2020). Combining imaging and high‐throughput technologies with DSB‐induced Cas9 systems allows one to examine processes such as transcription, chromatin dynamics, and DNA replication (Aymard et al, 2017; D’Alessandro & d’Adda di Fagagna, 2017; Clouaire & Legube, 2019; Miné‐Hattab & Chiolo, 2020).
The CRISPR/Cas9 system needs to be delivered in an accurate manner for efficient gene editing.
On the one hand, the Cas9 protein needs to be expressed in the host system or delivered in the form of a ribonucleoprotein (RNP) complex (Jinek et al, 2014). On the other hand, a target‐specific single‐guide RNA (sgRNA)—formed by CRISPR RNA (crRNA) and transactivating CRISPR RNA—needs to direct Cas9 to the target site (Doudna & Charpentier, 2014). It is important to choose the right delivery strategy for the sgRNA to survive the degradation processes in the cell and translocate into the nucleus to allow for gene editing. To date, we can classify sgRNA delivery methods into viral and nonviral, based on whether viral constructs are used for transfection (Lino et al, 2018).
Viral vectors include gamma‐retroviruses, adenovirus, adeno‐associated viruses (AAVs), and lentiviruses (LVs) (Warnock et al, 2011). Especially in LVs, Cas9 and sgRNA are relatively easy to clone and produce to efficiently transduced into the host cell. HIV‐1‐based lentiviral vectors convert single‐strand RNA into double‐strand DNA by reverse transcription and subsequent insertion into the genome of postmitotic cells (Lino et al, 2018). Lentiviral vectors have become important tools to deliver components of the CRISPR/Cas9 system for genome editing. Yet, one of the bigger challenges of these systems is the random integration of the construct into the genome (Kotterman et al, 2015). In fact, in gene therapy, stable viral integrations come with concerns regarding safety (Rothe et al, 2014). Among them, the deregulation of genes caused by the insertions and mutagenesis was found in gene therapy for immunodeficiencies in patients (Hacein‐Bey‐Abina et al, 2003). The nonviral methods are divided into physical and chemical. Physical methods include microinjections—where the sgRNAs are directly injected by a needle—and electroporation—where electric currents open the cell membrane for the delivery of molecules into the cell (Horii et al, 2014; de Melo & Blackshaw, 2018). Chemical delivery methods comprise a DNA or RNA form of the sgRNA that can be used to transfect the host by liposome‐based and non‐leptosomic reagents (Felgner et al, 1987; Liang et al, 2015). With RNA delivery methods, the transfection efficiency can be lower, but they are a safer alternative, as random viral integrations do not occur.
Even though targeting genomic regions with the CRISPR/Cas9 system are tightly controlled and specific, it is known that off‐target cutting activity can still occur (Cong et al, 2013; Hsu et al, 2013; Pattanayak et al, 2013). Other limitations of CRISPR include the requirement for a protospacer adjacent motif (PAM) to the target DNA sequence and the DNA‐damage toxicity triggered though the CRISPR‐induced DSB (Uddin et al, 2020). Nonetheless, valuable efforts have been made to understand and minimize these drawbacks. Many researchers are currently using lentiviral vectors for delivery of CRISPR/Cas9 components, as sgRNAs are relatively easy to clone into them (Lino et al, 2018). Lentiviral sgRNA‐delivery systems are used in functional genetic screens to find lethal interactions of specific biological processes (Mulero‐Sánchez et al, 2019). Even though many limitations are known regarding off‐targets or difference in efficiency between sgRNAs (Zhang et al, 2015; Kosicki et al, 2018; Cao et al, 2020), much less is known about how viral CRISPR/Cas9 delivery methods may affect genome integrity and gene expression when randomly integrated into the host genome.
Here, we show that an LV‐based sgRNA vector can integrate into the endogenous genomic target location, thereby affecting the expression of the target gene. By generating a DSB in the regulatory region of the ABCB1 gene with this system, we can produce Taxol‐resistant clones that upregulated ABCB1 through transcriptional activation via the EEF1A1 from the sgRNA vector. We speculate that targeted viral integration could result in deregulation of genes that may affect biological functions and therefore lead to false positive candidates, for instance, when performing functional genetic screens. Therefore, we believe that this unreported gene activation mechanism following CRISPR‐Cas9‐mediated genome editing needs to be taken into consideration when inducing DSBs with sgRNA lentiviral method.
Results and Discussion
A LentiGuide‐induced DSB in the ABCB1 promoter leads to upregulation of ABCB1
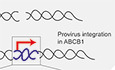
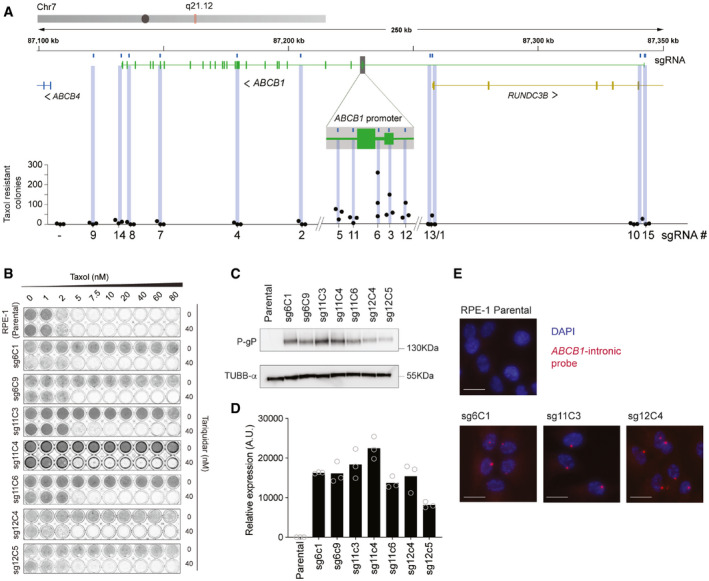
We have previously shown that in human retinal pigment ephitilial‐1 (RPE‐1), a major mechanism of Taxol resistance is transcriptional activation of the ABCB1 gene, that encodes for the multidrug resistance protein MDR1 or P‐Glycoprotein (PgP) (Tame et al, 2017; preprint: Manjón et al, 2021). Using the lentiviral system LentiGuide‐Puro from the Zhang Lab (Sanjana et al, 2014a), we cloned different sgRNAs targeting different noncoding regions across the ABCB1 locus to induce a DSB (Fig 1A). We chose noncoding regions to avoid the possibility that a break‐induced change in coding sequence could result in acquired Taxol resistance. Seven days after lentiviral infection and puromycin selection, we treated the RPE‐1 cells with 8 nM of Taxol in order to select cells that overexpressed PgP. Surprisingly, we observed that only cells treated with sgRNAs targeting the promoter of ABCB1 became resistant to Taxol (Figs 1A and EV1A), as we observed a considerable number of RPE‐1 colonies growing under Taxol pressure. Importantly, we also performed lentiviral infection with the same sgRNAs targeting ABCB1 in human mammary epithelial cells (HMEC) expressing Cas9. Here, we also observed that the sgRNAs targeting the promoter of ABCB1 lead to Taxol‐resistant colonies (Fig EV1B).
Figure 1. A DSB in the promoter of ABCB1 causes gene upregulation and Taxol‐resistance.
- Graphical representation of the ABCB1 genomic region and the location of the gRNA targeting the gene. RPE‐1 cells were infected with a lentivirus carrying one of the gRNAs and after 7 days of puromycin selection, one million cells were plated with 8 nM of Taxol for Colony Outgrowth Assay. Taxol‐resistant cells were counted and plotted in the graph, n = 3.
- Crystal violet staining of viability assay on RPE‐1 Parental cells and Taxol‐resistant clones obtained from A. For the clones’ nomenclature, sg# represents the sgRNA from where they are derived and C#, the clone number.
- Western blot showing the levels of the PgP and control (α‐TUBB) in RPE‐1 parental and Taxol‐resistant clones.
- ABCB1 mRNA levels determined by qRT‐PCR and normalized to GAPDH expression levels. Each dot represents a technical replicate (n = 3).
- Representative smRNA‐FISH images of RPE‐1 Parental and clones for the ABCB1 gene and DAPI. The images are projections of 0.5 µm sections and a total 5 µm in thickness. Scale bar, 15 µm.
Figure EV1. A DSB in the promoter of ABCB1 causes gene upregulation and Taxol‐resistance.
-
A, BExample replicate of crystal violet staining of Colony Formation Assays with 8 nM of Taxol for RPE‐1 WT cells quantified in Fig 1A (A) and HMEC‐1 (parental) and cells infected with Lenti‐guides targeting the ABCB1 gene (sgRNAs #1 to #15) (B).
-
CFlow cytometry plot of RPE‐1 cells infected with BFP‐dCas9 construct. BFP positive cells (in red) were sorted and grown to perform sgRNA infections and CFA under Taxol selection. X‐axis shows GFP expression (autofluorescence).
-
DExample replicate of crystal violet staining of Colony Formation Assays with 8 nM of Taxol for RPE‐1 Cas9 and RPE‐1 dCas9 cells infected with the indicated sgRNAs targeting ABCB1.
-
EQuantification of D, error bars show the SD of three biological replicates (n = 3).
In order to better understand the mechanisms responsible for the acquisition of the Taxol‐resistant phenotype, we decided to individually characterize these Taxol‐resistant clones that arose from sgRNA targeting of the ABCB1 promoter. Therefore, we expanded the RPE‐1‐resistant colonies observed in the colony outgrowth assays in the presence of Taxol. When performing a viability assay with increasing doses of Taxol, we observed that all clones were resistant to high concentrations of Taxol, and could be resensitized with Tariquidar, a PgP inhibitor (Fig 1B). As expected, with Western Blot and qRT‐PCR assays, we could confirm that the Taxol‐resistant clones expressed high levels of PgP protein as well as mRNA, respectively (Fig 1C and D). Thus, confirming that the mechanism of Taxol resistance was through ABCB1 upregulation. By performing intronic smRNA‐FISH, which allows for visualization of active transcription sites, we demonstrated that only one allele was actively transcribing ABCB1 (Fig 1E), confirming that ABCB1 copy number amplifications were not observed in these clones. In order to assess whether a DSB in ABCB1 was necessary to generate the Taxol‐resistant colonies, we generated RPE‐1 cells with a dCas9 construct. We then transduced the sgRNAs targeting ABCB1 both in RPE‐1 Cas9 and dCas9 cells followed by Puromycin and Taxol selection. Only cells with catalytically active Cas9 were able to generate Taxol‐resistant colonies (Fig EV1C–E). All together, these results indicate that a subset of cells, which undergo Cas9‐dependent DNA break formation in the ABCB1 regulatory region, acquire Taxol resistance through ABCB1 transcriptional activation.
The LentiGuide vector integrates and drives gene expression upon a DSB in the ABCB1 promoter
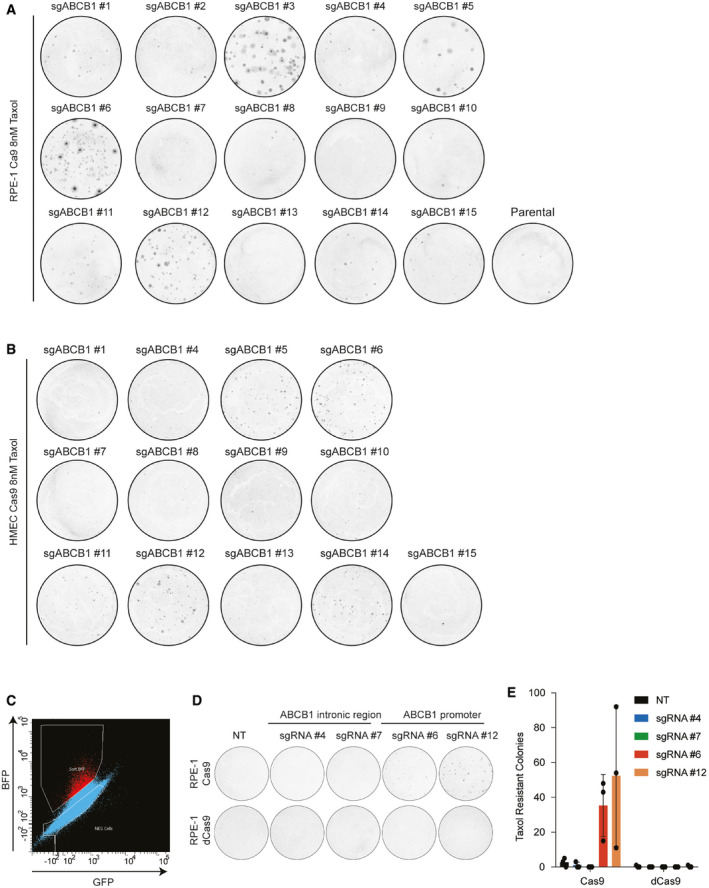
To exclude that DNA translocations or insertions might be induced by the DSB and could modify the activity of the ABCB1 promoter, we performed Targeted Locus Amplification (TLA), a chromosome conformation capture‐based technique, enabling the identification of single nucleotide variation and genomic rearrangements in a specific locus using a single PCR reaction (de Vree et al, 2014). We selectively amplified and sequenced the DNA flanking the ABCB1 promoter. We compared RPE‐1 parental cells with a Taxol‐resistant clone derived from the sgRNA #6 targeting the promoter of ABCB1 (sg6C9). Surprisingly, we found that our TLA experiments for the ABCB1 promoter amplified a 1.3‐kb region from chromosome 6 in the Taxol‐resistant clone (Fig 2A, green arrow). When zooming in on that region, we discovered that the promoter of the EEF1A1 gene was amplified in the sg6C9 Taxol‐resistant clone (Fig 2B). The read distribution over the EEF1A1 promoter is reminiscent of genomic insertions previously mapped with TLA (de Vree et al, 2014). To confirm the fusion of the ABCB1 and EEF1A1, we performed PCRs on genomic DNA using either Forward and Reverse primers amplifying the ABCB1 break site or a Forward primer binding the promoter region of EEF1A1 together with a Reverse from the ABCB1 promoter. In this last event, only when EEF1A1 and ABCB1 are juxtaposed in the genome, this will result in a PCR product (Fig 2C). Remarkably, our data revealed that not only the Taxol‐resistant clone sg6C9 but also all the other clones derived from sgRNA #6 and some others from #3, #5, #11, and #12, all inducing a DSB in the promoter of ABCB1, gave a PCR product when using the ABCB1 and EEF1A1 primers (Fig 2D). We could also observe a higher band appearing when amplifying the sequence over the break site with Forward and Reverse ABCB1 primers (Fig 2E). These data confirm that the EEF1A1 promoter was integrated at the break site in the regulatory region of ABCB1. When we sequenced the PCR products from the different clones, we observed that there were other sequences belonging to the U6 promoter and the puromycin‐resistant cassette integrated (Table 1). We next decided to align the sequencing reads of the TLA experiment analyzing the sg6C9 Taxol‐resistant clone to the LentiGuide vector sequence that was used to clone the ABCB1‐targeting sgRNAs to induce the DSB. We found that in the sg6C9 Taxol‐resistant clone, there was a large region aligning with the LentiGuide vector belonging to the EF1α promoter, suggesting that the EEF1A1 integration found in the ABCB1 promoter belonged to the LentiGuide vector and not to the endogenous gene found on chromosome 6 (Fig 2F).
Figure 2. sgRNA integration and transcription activation of ABCB1 .
- TLA analysis for ABCB1 contacts in RPE‐1 parental and Taxol‐resistant clone sg6C9 covering the whole genome. Green arrow in sg6C9 shows a de novo interaction found between ABCB1 and a region in chromosome 6.
- TLA analysis for RPE‐1 parental and sg6C9. Zoom in in the region of chr6 with de novo interaction for sg6C9. Image modified from IGV viewer.
- Graphical representation of the PCR products to assess vector integration. Two different primer pairs were used to PCR the vector integration: (i) a common EEF1A1 Forward (F) primer with a specific Reverse for each break site (5/11/6/3/12R). Only when EEF1A1 is integrated in cis, we will obtain a PCR product. (ii) Forward and reverse primers are used to amplify each specific break site (5/11/6/3/12F and R). If EEF1A1 is integrated in the break site, the PCR product will be bigger.
- PCR products using the primers in C(1) over the ABCB1 and EEF1A1 regions in RPE‐1 parental and the different Taxol‐resistant clones.
- PCR products using the primers in C(2) over the specific break site in the ABCB1 promoter in RPE‐1 parental and the different Taxol‐resistant clones.
- TLA analysis for RPE‐1 parental and sg6C9. Reads are aligned to the lenti‐guide vector. The location of the EF1α promoter from the vector is highlighted in red. Image modified from IGV viewer.
- Percentage of RPE‐1 cells targeted with the ABCB1 promoter sgRNAs found with vector integration. Percentages were calculated from data in Fig 1A. Each dot represents a CFA replicate experiment. Horizontal bars represent the mean of lentiviral integration per sgRNA.
Table 1.
Blast search to find sequences producing significant alignments to PCR sequenced band of sg6C10 (EEF1A FWD + 6 REV). Top 10 sequences with best alignments found.
| Description | Scientific name | Max Score | Total Score | Query Cover | E value | Per. Identity | Acc. Len | Accession |
|---|---|---|---|---|---|---|---|---|
| Select seq MN996874.1 | Cloning vector sh‐AFP‐PPP2CA, complete sequence | 924 | 1318 | 56% | 0.0 | 99.41% | 8000 | MN996874.1 |
| Select seq MN996873.1 | Cloning vector sh‐PPP2CA.dna, complete sequence | 924 | 1318 | 56% | 0.0 | 99.41% | 7716 | MN996873.1 |
| Select seq MN996872.1 | Cloning vector sh‐ctrl, complete sequence | 924 | 1318 | 56% | 0.0 | 99.41% | 7717 | MN996872.1 |
| Select seq MN996871.1 | Cloning vector sh‐AFP, complete sequence | 924 | 1318 | 56% | 0.0 | 99.41% | 7716 | MN996871.1 |
| Select seq KJ175229.1 | Cloning vector pGL3‐U6‐sgRNA‐PG, complete sequence | 881 | 881 | 44% | 0.0 | 94.62% | 4952 | KJ175229.1 |
| Select seq MK801288.1 | Cloning vector RS474_ErbB‐RASER1C‐dCas9VP64, complete sequence | 680 | 1152 | 54% | 0.0 | 95.00% | 16350 | MK801288.1 |
| Select seq MH782475.1 | Cloning vector pMJA289, complete sequence | 680 | 1152 | 54% | 0.0 | 95.21% | 9466 | MH782475.1 |
| Select seq MH782474.1 | Cloning vector pMJA284, complete sequence | 680 | 1152 | 54% | 0.0 | 95.21% | 6739 | MH782474.1 |
| Select seq MH782473.1 | Cloning vector pMJA285, complete sequence | 680 | 1152 | 54% | 0.0 | 95.21% | 10035 | MH782473.1 |
| Select seq MG840314.1 | Cloning vector pLenti‐EF1a‐dCas9‐DNMT3B(E697A)‐2A‐bla, complete sequence | 680 | 1152 | 54% | 0.0 | 95.00% | 14877 | MG840314.1 |
We next decided to calculate the frequency of vector integration in the ABCB1 locus following sgRNA‐dependent DSB induction. Taxol‐resistant clones with vector integration are highly resistant to Taxol (Figs 1B and EV2A) and were obtained from big colonies in the colony formation assays (Fig EV1A). Indeed, PCRs performed in smaller colonies derived from sgRNA #12 did not show evidence of LentiGuide integration (Fig EV2B). This correlates with the fact that they are more sensitive to Taxol and have lower mRNA levels of ABCB1 (Fig EV2A and C). Therefore, in order to estimate the frequency of vector integration, we decided to only count the big colonies from the colony formation assays. We could observe some variability on frequency depending on the sgRNA used (Fig 2G). We estimated that from Puromycin‐selected cells, approximately 10 in a million cells will have the vector integrated in the DSB site (Fig 2G). This mechanism does not appear to be of high frequency, but the stringent selection system based on transcriptional activation of the ABCB1 gene allows us to detect these infrequent events.
Figure EV2. Small‐Taxol resistant colonies do not carry LentiGuide integration.

- Crystal violet staining of viability assay on RPE‐1 parental cells and the Taxol‐resistant clones obtained from A.
- PCR products using primers binding to the EEF1A1 and the ABCB1 sequence (upper gel) or spanning the DSB break site induced with sgRNA #12 (lower gel) and its graphical representation. A parental WT RPE‐1 cell line, two small Taxol‐resistant colony (sg12C1, sg12C3), and one big Taxol‐resistant colony (sg12C4) obtained from colony formation assays were used for PCR reaction.
- ABCB1 mRNA levels determined by qRT‐PCR and normalized to GAPDH expression levels and WT. Mean is determined by three technical replicates (n = 3).
In order to know whether this phenomenon could be reproduced in other cell lines, we also performed PCRs over the DSB site in Taxol‐resistant HMEC clones derived from the sgRNA #12 targeting the ABCB1 promoter (Fig EV1B). Strikingly, in all the Taxol‐resistant clones tested, we detected a higher PCR product than the expected one based on the endogenous ABCB1 sequence (Fig EV3A). We therefore conclude that in both RPE‐1 and HMEC cells, the LentiGuide‐Puro vector had been integrated into the ABCB1 promoter, most likely due to the presence of the CRISPR‐induced DSB in that region. As the U6 promoter is an RNA Pol III promoter, most likely this will not result in mRNA and protein translation. Therefore, most probably the EEF1A1 promoter from this vector induced the transcriptional activation of ABCB1.
Figure EV3. sgRNA integration and transcription activation of ABCB1 .

- Top: PCR products using primers spanning the sgRNA #12 break site on the ABCB1 promoter in HMEC parental and the different Taxol‐resistant clones. Bottom: Graphical representation of the expected PCR product size (535 bp) in the endogenous ABCB1 locus.
- Schematic depicting the procedure for generating RPE‐1 CRISPRa cell lines to upregulate ABCG2.
- ABCG2 mRNA levels determined by qRT‐PCR and normalized to GAPDH expression levels. Error bars show the SD of three independent experiments (n = 3).
- Histogram from Flow Cytometry analysis of CRISPRa‐ABCG2 cells treated with Hoechst (sgABCG2), CRISPRa‐NT treated with Hoechst (sgNT), and cells without Hoechst (no‐Hoechst).
- Flow Cytometry plots of RPE‐1 Cas9 cells treated with Hoechst and infected with sgRNA NT (upper panel) or sgRNA targeting the promoter of ABCG2 (lower panel). Low Hoechst cells from sgRNA ABCG2 (in pink) were sorted and expanded for PCR experiments.
- Graphical representation of the ABCG2 locus with zoom in the regulatory region. PCR primers to amplify the break site and the expected product (385 bp) are indicated.
- PCR products using primers spanning the break site on the ABCG2 promoter in RPE‐1 Cas9 cells with sgRNA NT or sgRNA ABCG2 Hoechst‐low sorted polyclonal population.
- SnapGene image of purified and sequenced 750‐bp band from sgRNA ABCG2 on G aligned to the LentiGuide‐Puro sequence.
In order to understand whether LentiGuide vector integration was specific to the ABCB1 gene, we set out to investigate whether this phenomenon could happen in other genomic regions. Therefore, we generated an sgRNA targeting the regulatory region of ABCG2. ABCG2 gene encodes for another drug efflux pump similar to ABCB1, which has been described to be responsible for Hoechst 33342 dye efflux (Scharenberg et al, 2002). Hoechst is a fluorescent compound, which can be incorporated by cells, which allows for fluorescence‐activated cell sorting (FACS). We generated an RPE‐1 cell line with overexpression of ABCG2 (CRISPRa‐ABCG2), which confirmed lower intracellular levels of Hoechst by FACS (Fig EV3B–D). We next induced a DSB in the promoter of ABCG2 in RPE‐1 Cas9 cells with a lentiviral system followed by HoechstLow cell sorting (Fig EV3E). Strikingly, only the population targeted with the ABCG2 sgRNA showed increased numbers of HoechstLow cells. When performing PCRs over the DSB site in this polyclonal HoechstLow population, we could observe higher DNA bands (Fig EV3F and G). Sanger sequence of the purified 750‐bp band demonstrated that the EEF1A1 promoter of the LentiGuide construct was integrated in the break site (Fig EV3H). Altogether, these data indicate that vector integration can also happen in other genic regions, which will lead to gene upregulation.
Chromatin changes in the ABCB1 promoter region upon LentiGuide vector integration
We next decided to compare the chromatin landscape of the ABCB1 promoter in RPE‐1 parental cells and the Taxol‐resistant clones with the LentiGuide‐Puro integration. It is known that in RPE‐1 parental cells, ABCB1 is found in a repressive chromatin environment, consequently leading to transcriptional repression (preprint: Manjón et al, 2021). We therefore performed chromatin immunoprecipitation followed by massive parallel sequencing (ChIP‐seq) for the repressive histone modification H3K9me3. ChIP‐sequencing tracks of this chromatin mark showed that in RPE‐1 parental cells, ABCB1 is located in a region that contains intermediate level of H3K9me3, flanked by regions with high levels of H3K9me3 (Fig 3A, black track). Importantly, the seven Taxol‐resistant clones derived from different sgRNAs also displayed a similar H3K9me3 pattern (Fig 3A). In order to better quantify the levels of H3K9me3, we performed ChIP‐qPCRs of the ABCB1 promoter spanning up to 10 kb surrounding the transcriptional start site (Fig 3B). Interestingly, we observed that while some Taxol‐resistant clones had lower levels of H3K9me3 (sg6C1, sg6C9, and sg12C5), others had similar levels compared to the parental cell line (sg11C3‐4‐6 and sg12C4) (Figs 3B and EV4A). It has been suggested that DNA methylation of the ABCB1 promoter can regulate its transcriptional status (Chen et al, 2005; Reed et al, 2010). Therefore, we set out to study whether the DNA methylation pattern was altered in the Taxol‐resistant clones. By performing a DNA methylation array, we could assess the relative methylation status of 11 CpG islands located in close proximity to the sgRNAs targeting regions within the ABCB1 promoter (Fig 3C and D). In parental RPE‐1 cells, only two CpG islands were nearly fully methylated (CpG 1 and 2) and maintained the same status in four out of seven Taxol‐resistant clones (Fig 3D). The remaining CpG islands were hemi‐methylated in the parental cell line and vary between clones (Fig 3D). Therefore, even though some Taxol‐resistant clones showed lower levels of repressive chromatin marks as compared to their parental Taxol‐sensitive counterpart, others that maintained these levels still resulted in ABCB1 gene activation. Possibly, the DNA damage‐repair process induced by the sgRNA‐Cas9 system could cause the erasure of repressive chromatin marks, but this does not fully explain the observed ABCB1 gene activation pattern. Therefore, we suggest that the endogenous ABCB1 promoter remains repressed in (at least a subset of) the Taxol‐resistant clones containing the LentiGuide‐Puro integration, and that transcriptional activation occurs via the EEF1A1 promoter present in that vector.
Figure 3. Chromatin landscape in the ABCB1 promoter region upon LentiGuide vector integration.
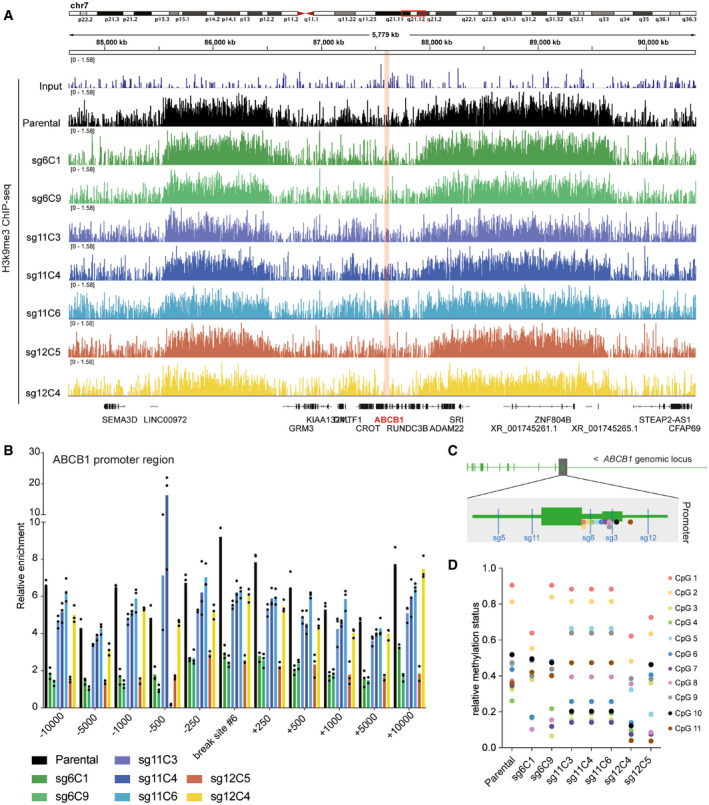
- H3K9me3 ChIP‐sequencing tracks from the q21.12 arm of chromosome 7 in RPE‐1 cells (parental and Taxol‐resistant clones). The ABCB1 gene region is shown in pink. Each colored track shows the H3k9me3 profile of a different Taxol‐resistant clone.
- H3K9me3 ChIP‐qPCR in the ABCB1 promoter region. Bar graph shows primer pairs amplifying the sgRNA#6 break and spanning this region (+/− base pairs) for each Taxol‐resistant clone compared to the parental. Bar plots show the mean of H3K9me3 relative enrichment. Each dot represents a technical replicate (n = 3).
- Graphical representation of the ABCB1 promoter region. The location of the sgRNAs targeting the promoter is shown in blue. The location of the eleven CpG islands analyzed in the methyl array are shown in colored dots.
- Relative methylation status (1: methylated, 0: nonmethylated) of the eleven CpG islands shown in C for RPE‐1 parental and the Taxol‐resistant clones.
Figure EV4. Chromatin landscape of the ABCB1 promoter in the Taxol‐resistant clones.
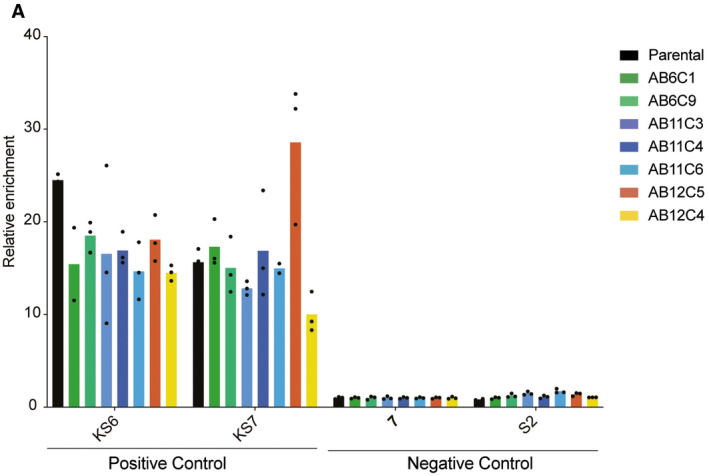
H3K9me3 ChIP‐qPCR showing two primer pairs amplifying positive regions for H3K9me3 (KS6 and KS7) and two primer pairs amplifying negative regions for H3K9me3 (7 and S2) in RPE‐1 parental and Taxol‐resistant clones. Each dot represents a technical replicate (n = 3).
Altogether, in this study, we show that a lentiviral sgRNA delivery system used to induce a DSB close to the transcriptional start site of a gene can result in the integration of the vector in the break site and subsequent activation of the gene. When generating a DBS in the regulatory region of ABCB1 with this system, we were able to find cells with genetic alterations that contained the U6 and EE1A1 promoters of the lentiviral vector. We believe that the DSB increased the probability of the vector to integrate into this location. In RPE‐1 cells, ABCB1 is repressed and the cells are sensitive to Taxol. The integration of these promoters allowed for gene activation and produced a Taxol‐resistant phenotype (Fig 4). Furthermore, as CRISPR enables the induction of DNA breaks at specific endogenous loci, more and more researchers are using several Cas9 systems to study of DSB repair and its biological consequences (Vítor et al, 2020; Van Den Berg et al, 2018; Schep et al, 2021). As we show here, inducing a DSB in a gene regulatory region can have consequences in gene expression, thus leading to incorrect interpretation of the results. Therefore, to study long‐term consequences of the DNA damage response, we suggest to employ nonintegrative systems such as synthetic gRNAs delivered in the form of RNA complexes (Yan & Schubert, 2017).
Figure 4. Model of sgRNA integration in endogenous ABCB1 locus.
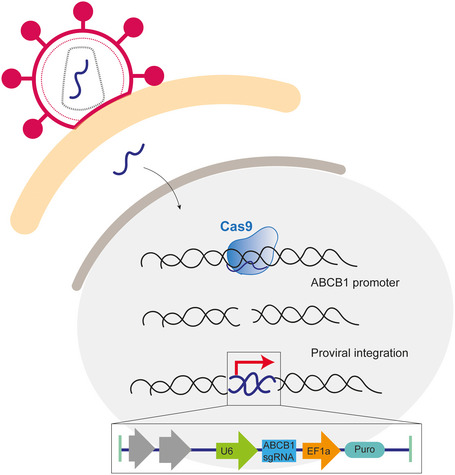
When a lenti‐guide Puro vector is used to deliver a gRNA to induce a DSB, the gRNA can be integrated into the break site. The DSB was induced in the promoter of ABCB1 and therefore the highly active promoters of the vector were driving the expression of the ABCB1 gene. In this case, we were selecting for cells that upregulated ABCB1 and therefore the frequency of this event was higher.
Materials and Methods
Cell lines and cell culture conditions
RPE‐1 cells are hTert‐immortalized human retinal pigment epithelium nontumoral cells. HMEC cells are hTert‐immortalized human mammary epithelial nontumoral cells. RPE‐1 and HMEC Cas9 cells were generated by lentiviral transduction with a lenti‐Cas9‐Blast (Addgene #73310) and Blasticidine S selection with 10 µg/ml. RPE‐1 dCas9 cells were generated by cotransduction with the viral particles containing SunTag‐dCas9‐BFP (Addgene #60910) and selected on BFP positivity. RPE‐1 and HMEC cell lines were maintained in DMEM/F‐12 + Glutamax (Gibco, Life Technology) supplemented with 1% penicillin/streptomycin and 6% fetal bovine serum (FBS, S‐FBS‐EU‐015, Serana).
Compound treatments
Taxol and Tariquidar were dissolved in DMSO and prepared at stock concentrations before usage at varying final concentrations as indicated in each figure. Hoechst 33342 was used at a final concentration of 0.1 µg/ml.
sgRNA designed and cloning
The sgRNAs targeting ABCB1 were cloned into a LentiGuide‐Puro (Addgene plasmid # 52963) using the BsmBI restriction site following the original protocol (Sanjana et al, 2014b; Shalem et al, 2014). sgRNA sequences are summarized in Table 2.
Table 2.
gRNA sequences targeting ABCB1.
| Name | Target gene | gRNA sequence |
|---|---|---|
| LentiGuide‐Puro‐ABCB1 #1 | ABCB1 | GCTGCTTTAAAAGGTCCGCG |
| LentiGuide‐Puro‐ABCB1 #2 | ABCB1 | AGAAAGCTCCATCAACCGCA |
| LentiGuide‐Puro‐ABCB1 #3 | ABCB1 | GCTGGGCAGGAACAGCGCCG |
| LentiGuide‐Puro‐ABCB1 #4 | ABCB1 | TGTGACTGCTGATCACCGCA |
| LentiGuide‐Puro‐ABCB1 #5 | ABCB1 | GCTTTCCTGCCCCAGACAGG |
| LentiGuide‐Puro‐ABCB1 #6 | ABCB1 | CCTCCCGGTTCCAGTCGCCG |
| LentiGuide‐Puro‐ABCB1 #7 | ABCB1 | CTGCTCCTCCAAATGAAAGG |
| LentiGuide‐Puro‐ABCB1 #8 | ABCB1 | GGTTTCCCCCTGTAAATAGA |
| LentiGuide‐Puro‐ABCB1 #9 | ABCB1 | CCTATTGTCCTGCTATGGCG |
| LentiGuide‐Puro‐ABCB1 #10 | ABCB1 | ATACAATCCAAGAAAAACAA |
| LentiGuide‐Puro‐ABCB1 #11 | ABCB1 | ACAAACTTCTGCTCTAAGCA |
| LentiGuide‐Puro‐ABCB1 #12 | ABCB1 | TCAATGCCCGTGTTTTTCCA |
| LentiGuide‐Puro‐ABCB1 #13 | ABCB1 | ATATTATCCCTGTTAATGCA |
| LentiGuide‐Puro‐ABCB1 #14 | ABCB1 | CCAAGAAGAATGAAGCCAGA |
| LentiGuide‐Puro‐ABCB1 #15 | ABCB1 | CTAAGCCATGTAACTCTTCG |
| LentiGuide‐Puro‐ABCG2 #5 | ABCG2 | GCGATAAGCGCCCTGCGACC |
HEK cells transfection and virus production
In order to generate the lentiviral sgRNAs, two million HEK 293T cells were plated followed by DNA transfection. In 200 µl of Opti‐MEM™ (Reduced Serum Medium), 2 µg of psPAX and 2 µg of pMD2G packaging plasmids were cotransfected with 2 µg of the corresponding sgRNA vector. Sixteen microliters of FuGENE® 6 were used as lipid‐based transfection reagent. Twenty‐four‐hour posttransfection, the medium was exchanged and for 24 h, the virus was collected for further experiments.
sgRNA lentiviral infection and colony formation assays
Of 400,000 RPE‐1 or HMEC cells were infected with a specific lenti‐sgRNA in 1:4 ratio. Next day, cells were trypsinized and 10 µg/ml of Puromycin was added. Cells were allowed to grow under Puromycin selection for 7 days. Following selection, 1 million cells were seeded and treated with 8 nM of Taxol and allowed to grow out for 15 days. Plates were fixed in 80% methanol and stained with 0.2% Crystal Violet solution. After fixation, the number of Taxol‐resistant cells was counted.
Viability assays
For viability assays, 1,000 cells were plated in a 96‐well plate and treated for 7 days with indicated drug concentrations. Subsequently, plates were fixed in 80% methanol and stained with 0.2% Crystal Violet solution.
RNA isolation and qRT‐PCR analysis
RNA isolation was performed by using Qiagen RNeasy kit and quantified using NanoDrop (Thermo Fisher Scientific). cDNA was synthesized using Bioscript reverse transcriptase (Bioline), Random Primers (Thermo Fisher), and 1,000 ng of total RNA according to the manufacturer's protocol. Primers were designed with a melting temperature close to 60 degrees to generate 90–120‐bp amplicons, mostly spanning introns. cDNA was amplified for 40 cycles on a cycler (model CFX96; Bio‐Rad Laboratories) using SYBR Green PCR Master Mix (Applied Biosystems). Target cDNA levels were analyzed by the comparative cycle (Ct) method and values were normalized against GAPDH expression levels. qRT‐PCR oligo sequences are summarized in Table 3.
Table 3.
RT‐qPCR primers.
| RT‐qPCR primers | FWD | REV |
|---|---|---|
| ABCB1 | ACAGCACGGAAGGCCTAATG | GTCTGGCCCTTCTTCACCTC |
| GAPDH | TGCACCACCAACTGCTTA | GGATGCAGGGATGATGTTC |
| ABCG2 | TTTCCAAGCGTTCATTCAAAAA | TACGACTGTGACAATGATCTGAGC |
Western Blots
For western blot experiments, equal amounts of cells were lysed with Laemmli buffer and separated by SDS–polyacrylamide gel electrophoresis followed by transfer to a nitrocellulose membrane. Membranes were blocked in 5% milk in PBST for 1 h at RT before overnight incubation with primary antibody in PBST with 5% milk at 4°C. Membranes were washed three times with PBST followed by incubation with secondary antibody in PBST with 5% milk for 2 h at RT. Antibodies were visualized using enhanced chemiluminescence (ECL) (GE Healthcare). The following antibodies were used for western blot experiments: α‐Tubulin (Sigma t5168), MDR(PgP) (sc‐8313). For secondary antibodies, peroxidase‐conjugated goat anti‐rabbit (P448 DAKO, 1:2,000), goat anti‐mouse (P447 DAKO, 1:2,000), and rabbit anti‐goat (P449) were used.
smRNA FISH
RPE‐1 cells were plated on glass coverslips and washed twice with BS before fixation in 4% PFA in PBS for 10 min at room temperature. After two additional washes in 1× PBS, coverslips were incubated in 70% ethanol at 4°C overnight. Coverslips were incubated for prehybridization in wash buffer (2× saline‐sodium citrate (SSC) with deionized formamide (Sigma) 10%) for 2–5 min at room temperature. RNA FISH probe mix wash was dissolved in hybridization buffer (wash buffer supplemented with 10% dextran sulfate). Thirty‐eight probes labeled with Cy5 were targeted to the intronic regions of ABCB1 (Biosearch Technologies). Coverslips were incubated in hybridization solution for at least 4 h at 37°C. Then coverslips were washed twice for 30 min with wash buffer followed by a quick rinse with 2× SSC. Finally, coverslips were washed once for 5 min in 1× PBS before mounting on slides using Prolong gold DAPI mounting medium (Life Technologies). Images were acquired with the use of a DeltaVision Elite (Applied Precision) equipped with a 60× 1.45 numerical aperture (NA) lens (Olympus) and cooled CoolSnap CCD camera. ABCB1 transcription start site quantification was performed manually double blind.
TLA analysis
TLA was performed as previously described with minor modifications. TLA libraries were sequenced on an MiSeq and were mapped to genome using bwa bwasw (Li & Durbin, 2010) to enable partial mapping of sequence reads. Reads were mapped to hg19 reference of the human genome.
PCR to assess vector integration
500,000 RPE‐1 or HMEC cells were lysed using DirectPCR Cell (Viagen). Two hundred milliliters µl of reagent and ~20 µl of proteinase K (20 mg/ml) were added to each sample. Samples were incubated at 55°C for 4–6 h, followed by 85°C for 45 min to inactivate the proteinase K. Phusion polymerase was used for DNA amplification in the following protocol: 4 µl 5× HF, 0.4 µl dNTPs, 1 µl of FWD primer, 1 µl of REV primer, 1 µl of DNA, 0.2 µl of Phusion polymerase and 12.4 µl of H2O. PCR cycles (×30): 98°C 30″ ‐ 98°C 10″ ‐ 58°C 30″ ‐ 72°C 2′ ‐ 72°C 10′. DNA samples were loaded in a 1.5% agarose gel and visualized using UV light (BioRad). Primers used for PCR can be found in Table 4.
Table 4.
PCR primers to assess vector integration.
| Break site primers | FWD | REV |
|---|---|---|
| sgRNA ABCB1 #3 | CTTCTCCCGTGAAGACCAAG | TAAATGCGAATCCCGAGAAA |
| sgRNA ABCB1 #5 | GCAGAGCACCATGATCAAAA | caagtccgggtatttgaagg |
| sgRNA ABCB1 #6 | CTTCTTTGCTCCTCCATTGC | GCTTCTTGAGGCGTGGATAG |
| sgRNA ABCB1 #12 | GGGAAATTTTCTCGGGATTC | AAGCTCTGATGTGAGTTAGCATTG |
| sgRNA ABCG2 #5 | taacttgctctgggtgcgag | gtttccccaggtcggggttc |
| EEF1A1 FWD | CACGGCGACTACTGCACTTA |
ChIP‐seq and ChIP‐qRT‐PCR of RPE‐1 hTERT cells
Chromatin immunoprecipitations (ChIP) were performed as described previously (Prekovic et al, 2021) with minor adjustments. Approximately 7 × 106 cells per condition were fixed, 50 μl of Protein A magnetic beads (Invitrogen) and 5 μg of antibody H3K9me3 (abcam ab8898). For ChIP‐seq, samples were processed for library preparation (Part# 0801‐0303, KAPA Biosystems kit), sequenced using an Illumina Hiseq2500 genome analyzer (65 bp reads, single end), and aligned to the Human Reference Genome (hg19) using Burrows‐Wheeler Aligner (bwa) version 0.5.9. Mapped reads were filtered based on mapping quality of 20 using samtools version 0.1.19. For ChIP‐qPCR analysis, DNA was amplified for 40 cycles on a cycler (model CFX96; Bio‐Rad Laboratories) using SYBR Green PCR Master Mix (Applied Biosystems). Target DNA levels were analyzed by the comparative cycle (Ct) method and values were normalized against input DNA and positive control region (specific for each chromatin mark). ChIP‐qPCR oligo locations are summarized in Table 5.
Table 5.
ChIP‐qPCR primers.
| FWD | REV | |
|---|---|---|
| ChIP‐qPCR controls | ||
| 7 (negative control) | TGCCACACACCAGTGACTTT | ACAGCCAGAAGCTCCAAAAA |
| S2 (negative control) | CTAGGAGGGTGGAGGTAGGG | GCCCCAAACAGGAGTAATGA |
| KS6 (positive control) | TGAAGACACATCTGCGAACC | TCGCGCACTCATACAGTTTC |
| KS7 (positive control) | CAATTGGCCCATATCTTTACG | CATGTTCTCGAAAGCAAGCA |
| ChIP‐qPCR ABCB1 | ||
| 250− | CCATTCCGACCTGAAGAGAAA | CTATTACTGCTCTCTGGCTTC |
| 500− | TTCTGCTCTAAGCAGGGATATTG | CTAGCCTCCAGCTCTGAAATAAA |
| 1,000− | GGCGACCAACACCACTT | GTCTTGGTGTGCCTCTTTCT |
| 5,000− | AGAGGTGCTTGTGATGGTAATG | GCCACCATTCCTGACTTAGAT |
| 10,000− | CAGGGTCACCTGGTTTAGATTG | GGATGAACTGTATCCTTCCTGTC |
| 250+ | GAAGAGCCGCTACTCGAATG | ATCTGTGGTGAGGCTGATTG |
| 500+ | CTACAGGACGTAGTTAAGGGAAAT | AGGAGGCAGAAAGGTGATACAG |
| 1,000+ | TTCCTGTCCACTATTTACTTCAAAC | GCTCTGATGTGAGTTAGCATTG |
| 5,000+ | GACTTACTAGCTGTGTGGCTTT | GAGCAGTGGAGTATTTCTTCAAATG |
| 10,000+ | GGCAAAGGCAAACCTTCTTAC | CACCCAAGTGGCAATTTCTG |
Generation of dCas9 and CRISPRa cell lines
For dCas9 experiments, RPE cells were transduced with viral particles containing SunTag‐dCas9‐BFP (Addgene# 60910). After 1 week of culturing, fluorescence‐activated cell sorting (FACS) was used to select for cells that were BFP positive. Polyclonal CRISPRa cell lines were obtained, which were subsequently transduced with viral particles containing sgRNAs targeting ABCB1 (#4, #7, #6 and #12). For CRISPRa experiments, a previously generated RPE‐1 cell line containing SunTag‐dCas9‐BFP (Addgene# 60910) and scFV‐VP69‐GFP (Addgene# 60904) was used (Tame et al, 2017). The indicated cell line was transduced with viral particles containing pools of sgRNAs targeted at the promoter of ABCG2. Cells were selected for 2 weeks with puromycin to obtain stable polyclonal cell lines for the sgRNA expression. The sgRNAs used can be found in Table 6.
Table 6.
sgRNAs for CRISPRa ABCG2 upregulation with overhangs for BsmBI cloning.
| RT‐qPCR controls | FWD | REV |
|---|---|---|
| ABCG2 CRISPRa #1 | CACCGGTACCACCGCCCTCCCTCG | AAACCGAGGGAGGGCGGTGGTACC |
| ABCG2 CRISPRa #2 | CACCGGCCGGAGCGCCGAAGCACC | AAACGGTGCTTCGGCGCTCCGGCC |
| ABCG2 CRISPRa #3 | CACCGGGGAGACCCGGACATCCAG | AAACCTGGATGTCCGGGTCTCCCC |
| ABCG2 CRISPRa #4 | CACCGACATCCAGGGGACGAGCTC | AAACGAGCTCGTCCCCTGGATGTC |
| ABCG2 CRISPRa #5 | CACCGAAACCCGGGGCGCTGGGGA | AAACTCCCCAGCGCCCCGGGTTTC |
DNA methylation array
DNA methylation was measured with the Infinium MethylationEPIC BeadChip (Illumina Inc., San Diego, CA) according to the manufacturer’s protocol. In short, 500 ng of genomic DNA was bisulfite converted using the EZ‐96 DNA Methylation Deep‐Well Kit (Zymo Research, Irvine, CA, USA). The samples were plated in a randomized order. The bisulfite conversion was performed according to the manufacturers’ protocol with the following modifications. For binding of the DNA, 15 µl of MagBinding Beads was used. The conversion reagent incubation was done according to the following cycle protocol: 16 cycles of 95°C for 30 s followed by 50°C for 1 h. After the cycle protocol, the DNA is incubated for 10 min at 4°C. Next, DNA samples were hybridized on the Infinium MethylationEPIC BeadChip (Illumina Inc., San Diego, CA) according to the manufacturer’s protocol.
Author contributions
Conceived and designed study: AGM, RHM. Experiments: AGM, SL, HT, AF, WZ. Data processing, bioinformatics: EdW. Discussion and interpretation of data: AGM, RHM. Manuscript writing: AGM, RHM, with input from all authors.
Supporting information
Expanded View Figures PDF
Acknowledgements
This work was supported by ZonMW‐TOP grant 91215067 (RHM, AGM, and AF). EdW and HT are supported by an ERC StG (HAP‐PHEN 637587). EdW is also supported by an ERC CoG FuncDis3D 865459. The Oncode Institute is partly supported by KWF Dutch Cancer Society.
Conflict of interest
Elzo de Wit is cofounder of Cergentis B.V. which commercializes the TLA technology.
EMBO Reports (2022) 23: e53902.
Data availability
All sequencing raw and processed data files generated in this study are available in GEO with the GEO accession code GSE185725 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185725).
References
- Aymard F, Aguirrebengoa M, Guillou E, Javierre BM, Bugler B, Arnould C, Rocher V, Iacovoni JS, Biernacka A, Skrzypczak M et al (2017) Genome‐wide mapping of long‐range contacts unveils clustering of DNA double‐strand breaks at damaged active genes. Nat Struct Mol Biol 24: 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JX, Wang YL, Wang ZX (2020) Advances in precise regulation of CRISPR/Cas9 gene editing technology. Yi Chuan = Hered 42: 1168–1177 [DOI] [PubMed] [Google Scholar]
- Chen KG, Wang YC, Schaner ME, Francisco B, Durán GE, Juric D, Huff LM, Padilla‐Nash H, Ried T, Fojo T et al (2005) Genetic and epigenetic modeling of the origins of multidrug‐resistant cells in a human sarcoma cell line. Cancer Res 65: 9388–9397 [DOI] [PubMed] [Google Scholar]
- Clouaire T, Legube G (2019) A snapshot on the cis chromatin response to DNA double‐strand breaks. Trends Genet 35: 330–345 [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro G, d’Adda di Fagagna F (2017) Transcription and DNA damage: holding hands or crossing swords? J Mol Biol 429: 3215–3229 [DOI] [PubMed] [Google Scholar]
- de Vree PJP, de Wit E, Yilmaz M, van de Heijning M, Klous P, Verstegen MJAM, Wan YI, Teunissen H, Krijger PHL, Geeven G et al (2014) Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol 32: 1019–1025 [DOI] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E (2014) The new frontier of genome engineering with CRISPR‐Cas9. Science 346: 1258096 [DOI] [PubMed] [Google Scholar]
- Felgner P, Gadek T, Holm M, Roman R, Chan H, Wenz M, Northrop J, Ringold G, Danielsen M (1987) Lipofection: a highly efficient, lipid‐mediated DNA‐transfection procedure. Proc Natl Acad Sci USA 84: 7413–7417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein‐Bey‐Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval J‐L, Fraser CC, Cavazzana‐Calvo M et al (2003) A serious adverse event after successful gene therapy for X‐linked severe combined immunodeficiency. N Engl J Med 348: 255–256 [DOI] [PubMed] [Google Scholar]
- Horii T, Arai Y, Yamazaki M, Morita S, Kimura M, Itoh M, Abe Y, Hatada I (2014) Validation of microinjection methods for generating knockout mice by CRISPR/Cas‐mediated genome engineering. Sci Rep 4: 4513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O et al (2013) DNA targeting specificity of RNA‐guided Cas9 nucleases. Nat Biotechnol 31: 827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S et al (2014) Structures of Cas9 endonucleases reveal RNA‐mediated conformational activation. Science 343: 1247997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, Bradley A (2018) Repair of double‐strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36: 765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman MA, Chalberg TW, Schaffer DV (2015) Viral vectors for gene therapy: translational and clinical outlook. Annu Rev Biomed Eng 17: 63–89 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R (2010) Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S et al (2015) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208: 44–53 [DOI] [PubMed] [Google Scholar]
- Lino CA, Harper JC, Carney JP, Timlin JA (2018) Delivering crispr: a review of the challenges and approaches. Drug Deliv 25: 1234–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA‐guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjón AG, Hupkes DP, Liu NQ, Friskes A, Joosten S, Teunissen H, Aarts M, Prekovic S, Zwart W, De Wit E et al (2021) Perturbations in 3D genome organization can promote acquired drug resistance. bioRxiv 10.1101/2021.02.02.429315 [PREPRINT] [DOI] [PubMed] [Google Scholar]
- de Melo J, Blackshaw S (2018) In vivo electroporation of developing mouse retina. Methods Mol Biol 1715: 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miné‐Hattab J, Chiolo I (2020) Complex chromatin motions for DNA repair. Front Genet 11: 800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez‐Villaseñor C, García‐Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60: 174–182 [DOI] [PubMed] [Google Scholar]
- Mojica FJM, Juez G, Rodriguez‐Valera F (1993) Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol Microbiol 9: 613–621 [DOI] [PubMed] [Google Scholar]
- Mulero‐Sánchez A, Pogacar Z, Vecchione L (2019) Importance of genetic screens in precision oncology. ESMO Open 4: e000505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJJ (2009) CRISPR‐based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci 34: 401–407 [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR (2013) High‐throughput profiling of off‐target DNA cleavage reveals RNA‐programmed Cas9 nuclease specificity. Nat Biotechnol 31: 839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekovic S, Schuurman K, Mayayo‐Peralta I, Manjón AG, Buijs M, Yavuz S, Wellenstein MD, Barrera A, Monkhorst K, Huber A et al (2021) (2021) Glucocorticoid receptor triggers a reversible drug‐tolerant dormancy state with acquired therapeutic vulnerabilities in lung cancer. Nat Commun 121: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed K, Hembruff SL, Sprowl JA, Parissenti AM (2010) The temporal relationship between ABCB1 promoter hypomethylation, ABCB1 expression and acquisition of drug resistance. Pharmacogenomics J 10: 489–504 [DOI] [PubMed] [Google Scholar]
- Rothe M, Modlich U, Schambach A (2014) Biosafety challenges for use of lentiviral vectors in gene therapy. Curr Gene Ther 13: 453–468 [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014a) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014b) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharenberg CW, Harkey MA, Torok‐Storb B (2002) The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 99: 507–512 [DOI] [PubMed] [Google Scholar]
- Schep R, Brinkman EK, Leemans C, Beijersbergen RL, Medema RH, Van B, Correspondence S (2021) Impact of chromatin context on Cas9‐induced DNA double‐strand break repair pathway balance [DOI] [PMC free article] [PubMed]
- Shalem O, Sanjana NE, Hartenian E, Shi XI, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tame MA, Manjón AG, Belokhvostova D, Raaijmakers JA, Medema RH (2017) TUBB3 overexpression has a negligible effect on the sensitivity to taxol in cultured cell lines. Oncotarget 8: 71536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin F, Rudin CM, Sen T (2020) CRISPR gene therapy: applications, limitations, and implications for the future. Front Oncol 10: 1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Berg J, Manjón AG, Manjón M, Kielbassa K, Feringa FM, Freire R, Medema RH (2018) A limited number of double‐strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res 46: 10132–10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vítor AC, Huertas P, Legube G, de Almeida SF (2020) Studying DNA double‐strand break repair: an ever‐growing toolbox. Front Mol Biosci 7: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock JN, Daigre C, Al‐Rubeai M (2011) Introduction to viral vectors. Methods Mol Biol 737: 1–25 [DOI] [PubMed] [Google Scholar]
- Yan S, Schubert M, Young M, Wang B (2017) Applications of Cas9 nickases for genome engineering
- Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH (2015) Off‐target effects in CRISPR/Cas9‐mediated genome engineering. Mol Ther ‐ Nucleic Acids 4: e264 [DOI] [PMC free article] [PubMed] [Google Scholar]