

Abstract
The activation of the nucleotide oligomerization domain (NOD)‐like receptor (NLR) family, pyrin domain‐containing protein 3 (NLRP3) inflammasome is related to the pathogenesis of a wide range of inflammatory diseases, but drugs targeting the NLRP3 inflammasome are still scarce. In the present study, we demonstrated that Licochalcone B (LicoB), a main component of the traditional medicinal herb licorice, is a specific inhibitor of the NLRP3 inflammasome. LicoB inhibits the activation of the NLRP3 inflammasome in macrophages but has no effect on the activation of AIM2 or NLRC4 inflammasome. Mechanistically, LicoB directly binds to NEK7 and inhibits the interaction between NLRP3 and NEK7, thus suppressing NLRP3 inflammasome activation. Furthermore, LicoB exhibits protective effects in mouse models of NLRP3 inflammasome‐mediated diseases, including lipopolysaccharide (LPS)‐induced septic shock, MSU‐induced peritonitis and non‐alcoholic steatohepatitis (NASH). Our findings indicate that LicoB is a specific NLRP3 inhibitor and a promising candidate for treating NLRP3 inflammasome‐related diseases.
Keywords: Licochalcone B, LPS‐induced septic shock, MSU‐induced peritonitis, NASH, NEK7, NLRP3 inflammasome
Subject Categories: Immunology, Pharmacology & Drug Discovery, Signal Transduction
Licochalcone B, a main component of the medicinal herb licorice, specifically inhibits the NLRP3 inflammasome by disrupting NEK7‐NLRP3 interaction and exhibits protective effects in mouse models of NLRP3 inflammasome‐mediated diseases.
Introduction
The nucleotide oligomerization domain (NOD)‐like receptor (NLR) family, pyrin domain‐containing protein 3 (NLRP3) inflammasome, is a protein complex formed by the NLR family member NLRP3, adaptor protein apoptosis‐associated speck‐like protein containing a CARD (ASC), and pro‐caspase‐1 (Mariathasan et al, 2004; Jo et al, 2016; Song et al, 2017). Upon activation, the NLRP3 inflammasome mediates the activation of caspase‐1 and the subsequent cleavage of pro‐interleukin (IL)‐1β and pro‐IL‐18, leading to the release of the pro‐inflammatory cytokines IL‐1β and IL‐18, respectively (Lim et al, 2020). Recent studies have reported that never in mitosis A (NIMA)‐related kinase‐7 (NEK7) can directly bind to NLRP3, and NEK7 is an essential for the activation of the NLRP3 inflammasome (He et al, 2016; Shi et al, 2016; Nozaki & Miao, 2019; Sharif et al, 2019). The NLRP3 inflammasome is the best‐characterised inflammasome that can be activated by factors derived not only from pathogens but also from the environment or host; thus, its dysregulation is related to the pathogenesis of a variety of human diseases. Several mutations in the NLRP3 gene can result in spontaneous activation of the NLRP3 inflammasome, which is central to the development of cryopyrin‐associated auto‐inflammatory syndromes (CAPS, a rare, hereditary, auto‐inflammatory disease; Broderick et al, 2015; Coll et al, 2015). In addition, the NLRP3 inflammasome also responds to some host‐derived danger signals, including monosodium urate crystals (MSU), amyloid starch, cholesterol crystals, high glucose, unsaturated fatty acids, and ceramide. These risk factors may result in the development and deterioration of a variety of chronic inflammatory diseases such as gout (Dalbeth et al, 2016, 2019), neurodegenerative diseases (Wu et al, 2021), atherosclerosis (Zhuang et al, 2019), and NASH (Mridha et al, 2017; Thomas, 2017; Gaul et al, 2021). Thus, NLPR3 inflammasome has been regarded as a potential drug target for the treatment of inflammatory diseases.
In recent years, some small‐molecule compounds, such as Tranilast (Huang et al, 2018), oridonin (He et al, 2018), MCC950 (Coll et al, 2015, 2019; Tapia‐Abellan et al, 2019), OLT1177 (Marchetti et al, 2018; Lonnemann et al, 2020), CY‐09 (Jiang et al, 2017), sulforaphane (Greaney et al, 2016), cardamonin (Wang et al, 2019b), carnosol (Shi et al, 2020), dehydrocostus lactone (Chen et al, 2020), echinatin (Xu et al, 2021), and cryptotanshinone (Liu et al, 2021), have been shown to have a potential inhibitory effect on the activation of the NLRP3 inflammasome in vitro. The above compounds have been tested in animal models of human diseases and showed potential therapeutic effects. Among these compounds, MCC950 is the best‐characterised NLRP3 inhibitor and was tested in a phase II clinical trial for rheumatoid arthritis, but was not developed further because its application resulted in hepatotoxicity (Mangan et al, 2018). Sulforaphane is an isothiocyanate found in broccoli sprout extracts (Fahey et al, 1997) and has been tested in humans with autism and exhibited negligible toxicity (Singh et al, 2014). Sulforaphane has been demonstrated to inhibit the NLRP1b, NLRP3, NAIP/NLRC4, and AIM2 inflammasomes independent of Nrf2 (Greaney et al, 2016), so it may not be a specific inhibitor of the NLRP3 inflammasome. OLT1177 (also known as dapansutrile) has been reported to specifically inhibit the NLRP3 inflammasome (Marchetti et al, 2018), and its safety and efficacy in the treatment of gout flares in an open‐label, proof‐of‐concept, phase 2a trial has been demonstrated (Klück et al, 2020). Moreover, OLT1177 has been tested in a phase 1B trial and the result showed that treatment with OLT1177 for 14 days was safe and well tolerated in patients with heart failure and reduced ejection fraction (Wohlford et al, 2020). Further studies are needed to confirm the clinical potential of OLT1177. Overall, inhibitors of NLRP3 inflammasome show great potential in the treatment of NLRP3‐mediated diseases.
Licochalcone B (LicoB) is a flavonoid bioactive ingredient found in licorice, the age‐old and widely used traditional herbal medicinal plant (Wang et al, 2015). Modern pharmacological research reports that LicoB has various biological activities (Wang et al, 2020), including anti‐inflammatory (Fu et al, 2013), anti‐oxidant (Fu et al, 2013) and anti‐tumour (Wang et al, 2019a) effects. LicoB has been shown to protect liver cells from alcohol‐induced cell damage by inhibiting cell apoptosis (Yuan et al, 2014), upregulating extracellular signal‐regulated kinase (Erk)‐nuclear factor erythroid related factor 2 (Nrf2) (Gao et al, 2017) and significantly inhibiting lipopolysaccharide (LPS)‐induced inducible nitric oxide synthase (iNOS) expression, nitric oxide (NO) production and expression of tumour necrosis factor‐α (TNF‐α) and monocyte chemotactic protein 1 (MCP‐1) (Furusawa et al, 2009). It has been shown that LicoB can inhibit the production of IL‐6, prostaglandin E2 (PGE2) and superoxide anions in the xanthine oxidase system and has a significant inhibitory effect on lipid peroxidation, and a strong scavenging effect on 2,2'‐azinobis‐(3‐ethylbenzthiazoline‐sulphonate) (ABTS) (+) radicals and 1,1‐Diphenyl‐2‐picrylhydrazyl radical and 2,2‐Diphenyl‐1‐(2,4,6‐trinitrophenyl)hydrazyl (DPPH) free radical activity (Haraguchi et al, 1998; Furusawa et al, 2009; Thiyagarajan et al, 2011). Although LicoB exhibits obviously beneficial effects, its underlying mechanism and direct targets remain to be elucidated.
In this study, we found that LicoB is a potential and effective NLRP3 inflammasome inhibitor. Our results demonstrated that LicoB directly binds to NEK7, interfering with the interaction between NLRP3 and NEK7, resulting in the inhibition of NLRP3 inflammasome activation. Moreover, LicoB displays significant therapeutic effects in several mouse models of NLRP3‐mediated diseases and may be developed as a promising candidate for the treatment of NLRP3 inflammasome‐related inflammatory diseases.
Results
LicoB inhibits both canonical and non‐canonical NLRP3 activation
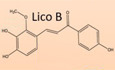
To identify potential candidates for the treatment of NLRP3‐mediated diseases, we screened inhibitors of NLRP3 and found that LicoB could block NLRP3 inflammasome activation (Fig EV1A). To further study the effect of LicoB on the activation of the NLRP3 inflammasome (Fig 1A), we first tested the cytotoxicity of LicoB in mouse bone marrow‐derived macrophages (BMDMs). Cell viability assays showed that LicoB did not exhibit any cytotoxicity at doses below 80 μM in BMDMs (Fig 1B). The BMDMs were first primed with LPS and then pre‐treated with a range of LicoB concentrations before being stimulated with nigericin at a dose of 10 μM or ATP at a dose of 5 mM, to induce NLRP3 inflammasome activation. The results showed that LicoB dose‐dependently inhibited caspase‐1 activation or IL‐1β secretion triggered by nigericin (Fig 1C–E) or ATP (Fig 1G–I) in LPS‐primed BMDMs, and the half‐maximal inhibitory concentration (IC50) of LicoB was approximately 18.1 μM (Fig 1E). Correspondingly, the protein levels of NLRP3 and pro‐IL‐1β in the whole cell lysates were not affected (Fig 1C and G).
Figure EV1. Licochalcone B inhibits NLRP3 inflammasome activation.
-
ABMDMs were primed with LPS for 4 h and then treated with LicoB 1 h prior to stimulation of nigericin for 45 min. Activity of caspase‐1 in culture supernatants of BMDMs were shown.
-
B, CBMDMs were primed with LPS for 4 h and then treated with LicoB for 1 h, prior to stimulation with nigericin for 45 min or ATP for 1 h. Western blot analyses of GSDMD, pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC in the whole cell lysate (WCL); activated caspase‐1 (p20) and cleaved IL‐1β (p17) in the culture SN of BMDMs. Coomassie Blue staining was used as the SN loading control, while lamin B was used as the lysate loading control.
-
D–FPMA‐primed THP‐1 cells were treated with LicoB 1 h prior to stimulation with nigericin for 45 min. Western blot analyses of pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC in the WCL; and activated caspase‐1 (p20) and cleaved IL‐1β (p17) (D) in the culture supernatants (SN) of THP‐1 cells. Caspase‐1 activity (E) and IL‐1β secretion (F) in the SN were measured.
-
G, HHuman PBMCs were primed with LPS for 4 h and then treated with LicoB 1 h prior to stimulation with nigericin for 45 min (G) or with ATP for 1 h (H). Caspase‐1 activity in supernatant was measured.
Data information: Error bars, mean ± SEM from three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001 and n.s.: not significant (one‐way ANOVA with Dunnett's post hoc test).
Source data are available online for this figure.
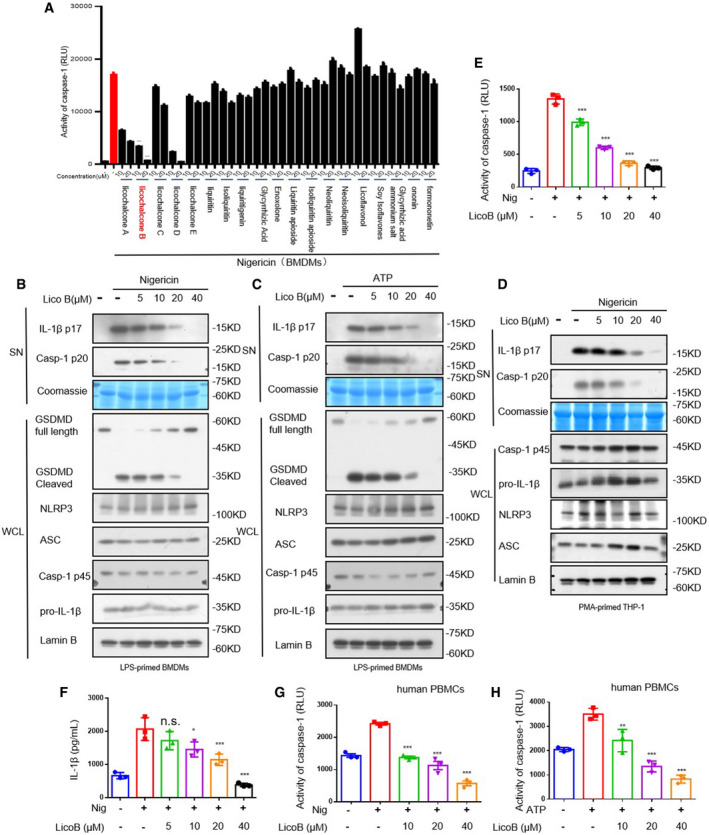
Figure 1. Licochalcone B (LicoB) inhibits NLRP3 inflammasome activation.
-
ALicochalcone B (LicoB) structure.
-
BCell Counting Kit 8 (CCK‐8) was used to assess the viability of BMDMs treated with different doses of LicoB for 24 h.
-
C–IBMDMs were primed with LPS for 4 h and then treated with LicoB for 1 h, prior to stimulation with nigericin for 45 min or ATP for 1 h. Western blot analyses of pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC (the arrow indicates ASC in the whole cell lysate (WCL); activated caspase‐1 (p20) and cleaved IL‐1β (p17) in the culture supernatants (SN) of BMDMs (C, G). Caspase‐1 activity (D, H), IL‐1β secretion (E, I), and LDH release (F, J) in the SN were measured. Coomassie Blue staining was used as the supernatant loading control, while lamin B was used as the lysate loading control.
Data information: Error bars, mean ± SEM from three biological replicates. **P < 0.01, ***P < 0.001, and n.s.: not significant (one‐way ANOVA with Dunnett's post hoc test).
Source data are available online for this figure.
Meanwhile, nigericin or ATP stimulation induced lactate dehydrogenase (LDH) release (marker of cell death) and gasdermin D (GSDMD) cleavage (indicator of pyro ptosis), and the release of LDH (Fig 1F and J) or GSDMD cleavage (Fig EV1, EV2, EV3B and C) was significantly blocked by LicoB. LicoB also impaired nigericin‐induced IL‐1β secretion and caspase‐1 cleavage in phorbol‐12‐myristate‐13‐acetate (PMA)‐differentiated THP‐1 cells (Fig EV1, EV2, EV3D–F) and human peripheral blood mononuclear cells (hPBMCs) (Fig EV1, EV2, EV3G and H). In summary, LicoB effectively blocks the nigericin‐ or ATP‐induced activation of the NLRP3 inflammasome in mouse and human immune cells.
Figure EV2. Licochalcone B inhibits K+ Efflux‐Independent NLRP3 Inflammasome Activation in BMDMs.
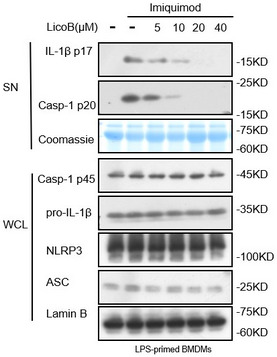
BMDMs were primed with LPS for 4 h and then treated with LicoB for 1 h prior to stimulation with imiquimod (70 μM) for 1 h. Western blot analyses of pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC in the whole cell lysate (WCL); activated caspase‐1 (p20) and cleaved IL‐1β (p17) in the culture SN of BMDMs were shown. Coomassie Blue staining was used as the SN loading control, while lamin B was used as the lysate loading control.
Source data are available online for this figure.
Figure EV3. Licochalcone B directly binds to NEK7 but does not affect the kinase activity of NEK7.
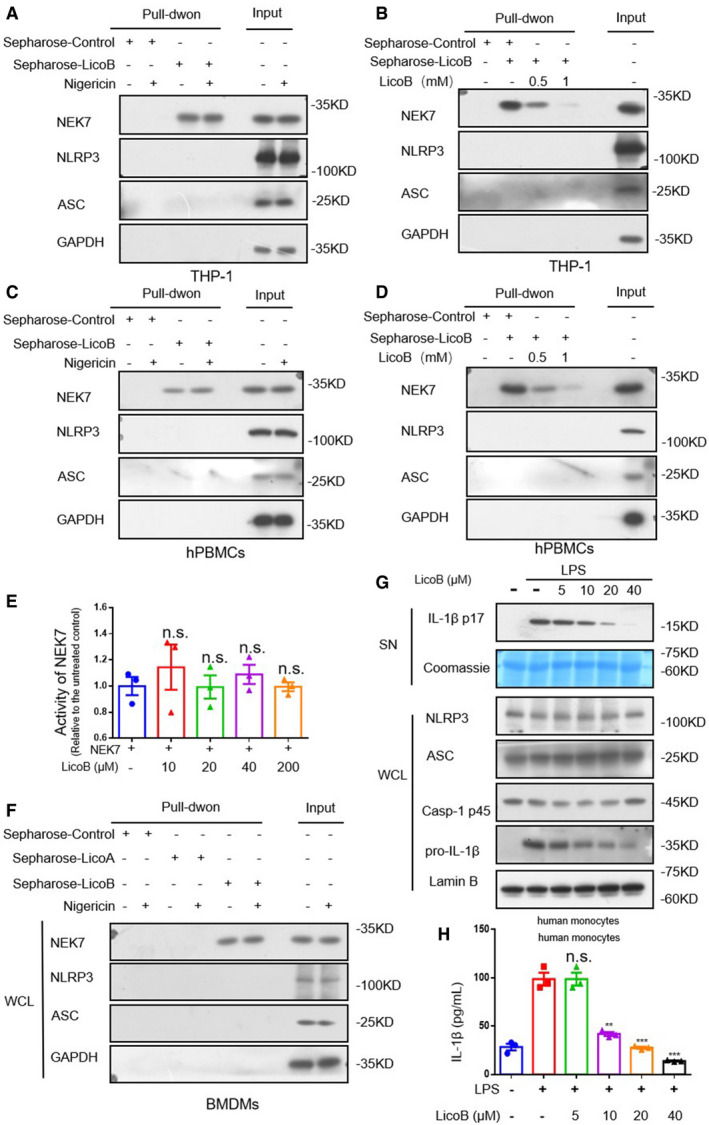
-
ACell lysates of PMA‐primed THP‐1 treated with nigericin or not were incubated with sepharose or LicoB‐sepharose. The pull‐down samples and input were analysed by Western blot.
-
BCell lysates of PMA‐primed THP‐1 were incubated with sepharose or LicoB‐sepharose in the presence of different concentrations of free LicoB (0.5 and 1 mM). The pull‐down samples and input were analysed by Western blot.
-
CCell lysates of LPS‐primed hPBMCs treated with nigericin or not were incubated with sepharose or LicoB‐sepharose. The pull‐down samples and input were analysed by Western blot.
-
DCell lysates of LPS‐primed hPBMCs were incubated with sepharose or LicoB‐sepharose in the presence of different concentrations of free LicoB (0.5 and 1 mM). The pull‐down samples and input were analysed by Western blot.
-
ENEK7 was incubated with β‐casein and ATP in the presence of different concentrations of LicoB. NEK7 kinase activity was measured using an ADP‐based phosphatase coupled kinase assay.
-
FCell lysates of LPS‐primed BMDMs treated with nigericin or not were incubated with Sepharose, Sepharose‐LicoA or Sepharose‐LicoB. The pull‐down samples and input were analysed by Western blot.
-
G, HHuman monocytes were treated with LicoB for 1 h, prior to stimulation with LPS (200 ng/ml) for 14 h. (G) Western blot analyses of pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC in the whole cell lysate (WCL); cleaved IL‐1β (p17) in the culture SN of BMDMs were shown. Coomassie Blue staining was used as the SN loading control, while lamin B was used as the lysate loading control. IL‐1β secretion (H) in the supernatant were measured by ELISA.
Data information: Error bars, mean ± SEM from three biological replicates. **P < 0.01, ***P < 0.001 and n.s.: not significant (one‐way ANOVA with Dunnett's post hoc test).
Source data are available online for this figure.
To test whether LicoB has a broad‐spectrum anti‐NLRP3 inflammasome effect, we investigated the effect of LicoB on NLRP3 inflammasome activation induced by multiple agonists. First, the effect of LicoB on canonical NLRP3 inflammasome activation was evaluated. It was found that LicoB effectively inhibited caspase‐1 activation and IL‐1β secretion induced by the canonical NLRP3 inflammasome agonists (Youm et al, 2013), such as nigericin, ATP, poly(I:C) or MSU (Fig 2A–C). We then evaluated the effect of LicoB on the activation of the non‐canonical NLRP3 inflammasome (Kayagaki et al, 2011, 2013, 2015). The results showed that LicoB dose‐dependently inhibited caspase‐1 cleavage and IL‐1β secretion induced by LPS transfection in Pam3CSK4‐primed BMDMs. Next, we evaluated the effect of LicoB on K+ efflux‐independent NLRP3 inflammasome activation induced by imiquimod (Groß et al, 2016). The results showed that LicoB dose‐dependently inhibited caspase‐1 cleavage and IL‐1β secretion induced by the K+ efflux‐independent NLRP3 inflammasome agonist imiquimod (Fig EV2). These results indicated that LicoB is a broad‐spectrum inhibitor of the NLRP3 inflammasome.
Figure 2. Licochalcone B is a specific inhibitor of the NLRP3 inflammasome.
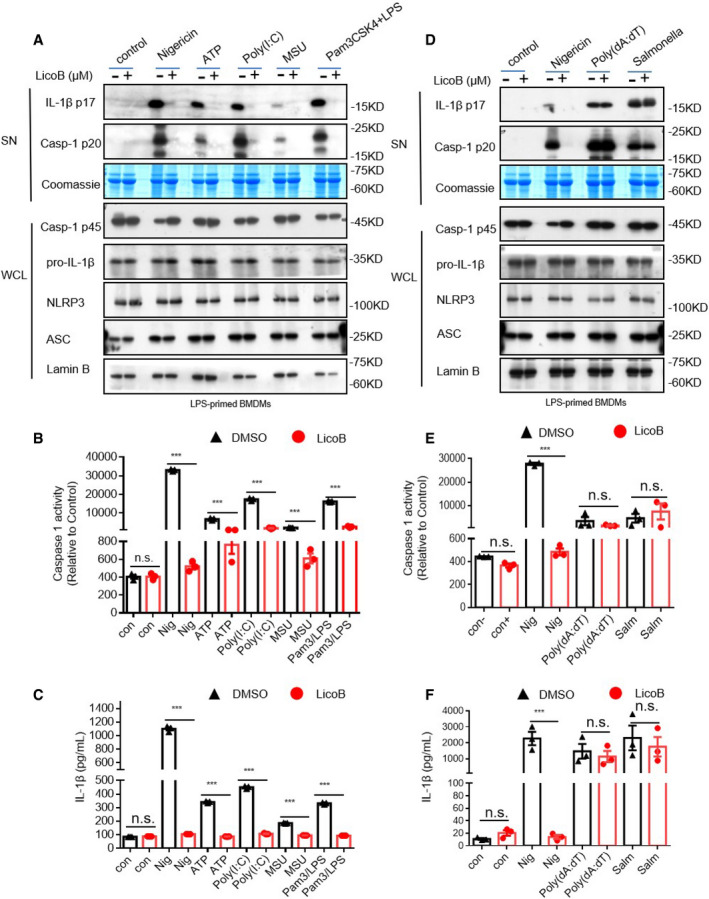
-
A–CBMDMs were primed with LPS and then treated with LicoB (20 μΜ) for 1 h, followed by stimulation with nigericin, ATP, poly (I:C) or MSU. Pam3CSK4‐primed BMDMs were treated with LicoB (20 μM) and then transfected with LPS. Western blot analyses of pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC in the whole cell lysate (WCL), and activated caspase‐1 (p20) and cleaved IL‐1β (p17) in the culture supernatants (SN) of BMDMs (A). Caspase‐1 activity (B) and IL‐1β secretion (C) in the SN were measured.
-
D–FLPS‐primed BMDMs were treated with LicoB (20 μM) for 1 h and then stimulated with nigericin for 45 min, or poly(dA:dT)/Salmonella for 6 h. Western blot analyses of pro‐caspase‐1 (p45), pro‐IL‐1β, NLRP3 and ASC in the whole cell lysate (WCL); and activated caspase‐1 (p20) and cleaved IL‐1β (p17) in the culture supernatants (SN) of BMDMs (D). Caspase‐1 activity (E) and IL‐1β secretion (F) in the SN were measured.
Data information: Error bars, mean ± SEM from three biological replicates. ***P < 0.001 and n.s.: not significant (unpaired Student’s t‐test).
Source data are available online for this figure.
LicoB is a specific inhibitor of the NLRP3 inflammasome
We then investigated whether LicoB specifically inhibits the activation of the NLRP3 inflammasome. We conducted experiments on the influence of LicoB on the activation of AIM2 and NLRC4 inflammasomes (which can also mediate the cleavage of caspase‐1 and secretion of IL
‐1β; Fernandes‐Alnemri et al, 2009; Hornung et al, 2009; Miao et al, 2010; Zhao et al, 2011). LPS‐primed BMDMs were transfected with the dsDNA analog poly(dA:dT), to activate the AIM2 inflammasome, or stimulated with Salmonella typhimurium, to activate the NLRC4 inflammasome. The results showed that LicoB did not inhibit AIM2‐ or NLRC4‐mediated caspase‐1 activation and IL‐1β secretion (Fig 2D–F). The results showed that LicoB specifically inhibited the activation of the NLRP3 inflammasome.
LicoB blocks NLRP3‐dependent ASC oligomerization
After recognising that LicoB can specifically inhibit the activation of the NLRP3 inflammasome, we further explored the mechanism underlying this effect. First, an experiment was conducted to determine whether LicoB affects ASC oligomerization (a key step in NLRP3 inflammasome activation) (Lu et al, 2014). LPS‐primed BMDMs were stimulated with ATP, and the higher‐order complexes were detected using Western blot analysis. The results showed that LicoB attenuated the ATP‐induced oligomerisation of ASC in a dose‐dependent manner (Fig 3A), which is consistent with the blocking effect of LicoB on caspase‐1 cleavage and IL‐1β secretion (Fig 3B and C). Further investigation showed that LicoB effectively blocked ASC oligomerisation induced by other NLRP3 agonists, such as nigericin, poly(I:C), MSU and cytosolic LPS (Fig 3D). However, LicoB did not affect ASC oligomerisation induced by AIM2 or NLRC4 agonists (Fig 3E), which is consistent with previous results. These results suggested that LicoB might affect upstream events of ASC oligomerisation, to block NLRP3 inflammasome activation.
Figure 3. Licochalcone B inhibits ASC oligomerisation during NLRP3 inflammasome activation.
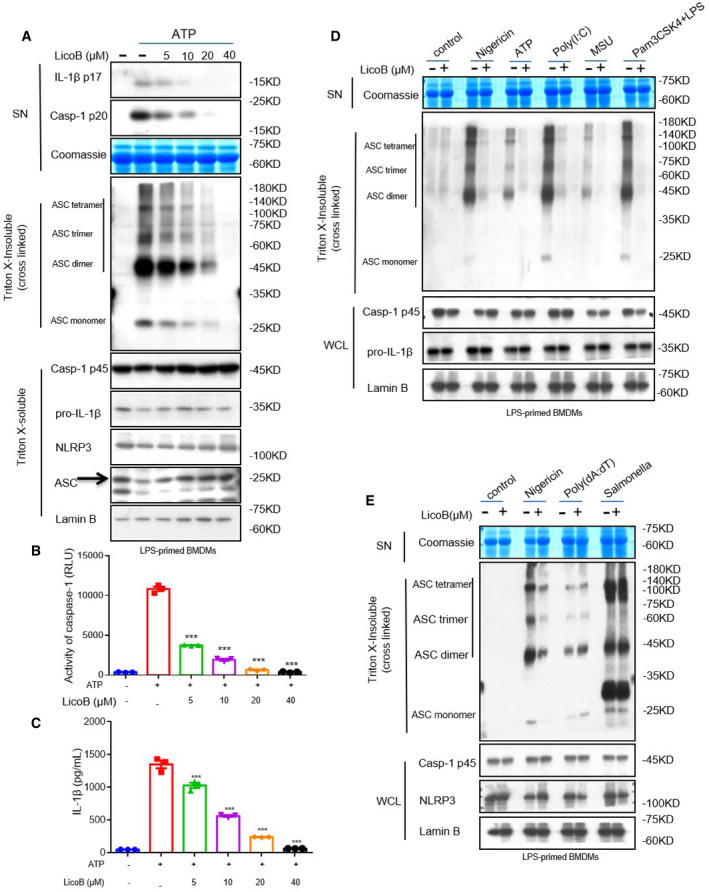
-
A–CLPS‐primed BMDMs were pre‐treated with the indicated dose of LicoB for 1 h and then stimulated with ATP for 1 h. Western blot analysis was used to detect cross‐linked ASC in the Triton X‐insoluble pellet, the arrow indicates ASC (A). Caspase‐1 activity (B) and IL‐1β secretion (C) in the SN were measured again to verify NLRP3 activation.
-
DWestern blot analysis of cross‐linked ASC in the Triton X‐insoluble pellet from LPS‐primed BMDMs pre‐treated with LicoB (20 μM) or vehicle and then stimulated with nigericin, ATP, poly(I:C) and MSU. Pam3CSK4‐primed BMDMs were treated with LicoB (20 μM) and then stimulated with LPS.
-
EWestern blot analysis of cross‐linked ASC in the Triton X‐insoluble pellet from LPS‐primed BMDMs pre‐treated with LicoB (20 μM) or vehicle and then stimulated with nigericin, poly(dA:dT) or Salmonella.
Data information: Error bars, mean ± SEM from three biological replicates. ***P < 0.001, and n.s.: not significant (one‐way ANOVA with Dunnett's post hoc test).
Source data are available online for this figure.
LicoB has no effect on K+ efflux, Ca2+ signalling or mitochondrial reactive oxygen species (mtROS) production during NLRP3 inflammasome activation
Since it has been reported that LicoB can inhibit nuclear factor kappa‐B (NF‐κB) signalling (Furusawa et al, 2009), we tested whether LicoB could affect LPS‐induced priming for NLRP3 inflammasome activation, according to a previously described method (Wang et al, 2019b). When BMDMs were treated with LicoB at a dose of 0–40 μM before or after LPS stimulation, we found that administration of LicoB before LPS treatment had a slight inhibitory effect on the production of pro‐IL‐1β and TNF‐α, while administration of LicoB after LPS had no effect on the production of pro‐IL‐1β, NLRP3 or TNF‐α (Fig 4A and B); in this case, LicoB inhibited IL‐1β secretion and caspase‐1 activation (Fig 1C and D). Collectively, these data suggested that LicoB has an inhibitory effect on the activation step (signal 2) of the NLRP3 inflammasome.
Figure 4. Licochalcone B does not affect K+ efflux, or Ca2+ signalling or mtROS production, but directly binds to NEK7 and inhibits the interaction between NEK7 and NLRP3.
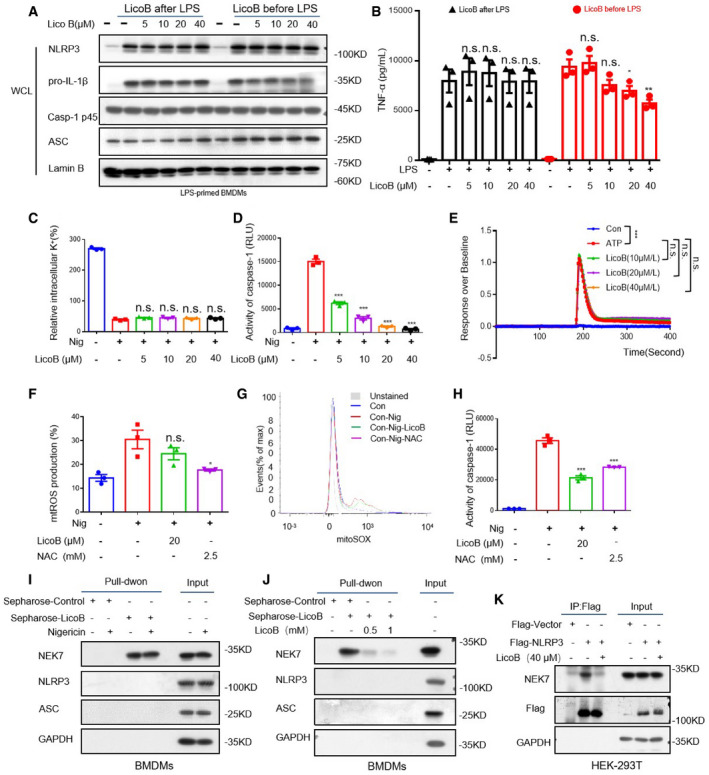
-
AWestern blot analysis of protein in whole cell lysates (WCL) from BMDMs stimulated with LPS for 3 h and then treated with LicoB for 1 h (LicoB after LPS), or BMDMs first treated with LicoB for 1 h and then stimulated with LPS for 3 h (LicoB before LPS).
-
BTNF‐α production in the supernatant (SN) was measured using ELISA in the indicated samples in (A).
-
C, DQuantification of intracellular K+ (C) and measurement of caspase‐1 activity in the SN (D) of LPS‐primed BMDMs pre‐treated with the indicated dose of LicoB for 1 h and then stimulated with nigericin for 45 min.
-
EThe FLIPR Tetra® system was used to determine the effect of LicoB on ATP‐induced Ca2+ flux in LPS‐primed BMDMs.
-
F, GPercentage of mtROS‐producing cells in LPS‐primed BMDMs pre‐treated with LicoB for 1 h and then stimulated with nigericin 45 min (F), followed by staining with MitoSOX™ (G).
-
HCaspase‐1 activity measurement in the supernatant (SN) of samples described in (F).
-
ICell lysates of LPS‐primed BMDMs treated with/without nigericin were incubated with sepharose or LicoB‐sepharose. The pull‐down samples and input were analysed using Western blot analysis.
-
JCell lysates of LPS‐primed BMDMs were incubated with sepharose or LicoB‐sepharose in the presence of different concentrations of free LicoB (0.5 and 1 mM). The pull‐down samples and input were analysed using Western blot.
-
KHEK‐293T cells were transfected with Flag‐NLRP3 or Flag‐vector and then treated with LicoB (40 μM). Immunoprecipitation was performed with anti‐DYKDDDDK (Flag) affinity gel agarose beads; Western blot analysis has been shown.
Data information: Error bars, mean ± SEM from three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001, and n.s.: not significant. One‐way ANOVA with Dunnett's post hoc test (B–D, F and H) and one‐way ANOVA with Sidak’s post hoc test (E).
Source data are available online for this figure.
To further clarify the mechanism underlying the effect of LicoB on the activation of the NLRP3 inflammasome, we further investigated the effect of LicoB on K+ efflux, which is an important upstream event in NLRP3 activation (Muñoz‐Planillo et al, 2013; He et al, 2016). Our data showed that there was a significant decrease in the intracellular K+ concentration upon nigericin stimulation in LPS‐primed BMDMs, while LicoB treatment had no effect on this nigericin‐induced intracellular K+ reduction (Fig 4C and D). We further studied the effect of LicoB on Ca2+ flow during the activation of NLRP3 inflammasomes (Murakami et al, 2012). When we used ATP to stimulate LPS‐induced BMDM, there was a significant increase in the Ca2+ flow, but LicoB treatment did not inhibit it (Fig 4E). In addition, we tested whether LicoB influences mtROS production, which is another important upstream signalling pathway for NLRP3 inflammasome assembly (Zhou et al, 2011; Zhao et al, 2020). In this experiment, N‐acetyl‐L‐cysteine (NAC), a reported ROS inhibitor, was used as the positive control (Sayin et al, 2014). The results showed that NAC inhibited nigericin‐induced mtROS production, but LicoB had no such effect (Fig 4F–H). Therefore, these results indicated that the inhibitory effect of LicoB on NLRP3 inflammasome activation may be unrelated to K+ efflux, Ca2+ flux or mtROS production.
LicoB directly binds to NEK7 and inhibits NEK7‐NLRP3 interaction
We then examined whether LicoB directly binds to NLRP3 inflammasome components to block inflammasome assembly. LicoB was incubated with cyanogen bromide‐activated Sepharose® 4B overnight to form a complex (sepharose‐LicoB), which was incubated with LPS‐primed BMDMs cell lysate, PMA‐primed THP‐1 cell lysate or LPS‐primed hPBMCs cell lysate with or without nigericin stimulation. The proteins that interacted with LicoB were analysed and detected by means of immunoblotting. The results showed that NEK7, which is considered to be an essential component of the NLRP3 inflammasome (He et al, 2016; Schmid‐Burgk et al, 2016; Shi et al, 2016), was pulled down, but NLRP3 and ASC were not (Figs 4I and EV3A and C), and the binding of LicoB to NEK7 was not affected by nigericin (Figs 4I and EV3EA and C). Moreover, free LicoB interfered with the interaction between NEK7 and sepharose‐LicoB (Figs 4J and EV3EB and D), suggesting that LicoB directly binds to NEK7.
The interaction between NEK7 and NLRP3 is an important step in the assembly of the NLRP3 inflammasome, which is crucial for NLRP3 oligomerisation and recruitment of ASC to NLRP3 (He et al, 2016; Schmid‐Burgk et al, 2016; Shi et al, 2016; Sharif et al, 2019; Yang et al, 2020). Next, to further study how LicoB inhibits the activation of the human classic NLRP3 inflammasome, we conducted a semi‐endogenous co‐immunoprecipitation experiment to investigate whether LicoB affects the interaction between NEK7 and NLRP3. The results showed that the interaction between NLRP3 and NEK7 in HEK‐293T cells was inhibited upon LicoB treatment (Fig 4K). This suggests that LicoB can directly bind to NEK7 and inhibit the interaction between NEK7‐NLRP3, thus blocking the activation of the NLRP3 inflammasome.
Moreover, we investigated whether LicoB affected the kinase activity of NEK7. The in vitro kinase assay was performed with NEK7 and β‐casein as a substrate, with a Universal Kinase Activity Kit. Our experimental results showed that LicoB did not affect the kinase activity of NEK7 (Fig EV3E). It has been reported that Licochalcone A inhibits the NLRP3 inflammasome by blocking the production of mtROS (Yang et al, 2018). Licochalcone A and LicoB show very high structural similarity. So we tested whether Licochalcone A bound to NEK7, and the result showed that LicoA did not interact with NEK7 (Fig EV3F).
LicoB inhibits alternative inflammasome activation
Gaidt et al (2016) demonstrated the alternative inflammasome activation in human monocytes, lipopolysaccharide alone induced an alternative inflammasome, and the activation was propagated by TLR4‐TRIF‐RIPK1‐FADD‐CASP8 signalling upstream of NLRP3. We tested the effect of LicoB on alternative NLRP3 inflammasome activation in human monocytes. The result showed that pre‐treatment of LicoB inhibited the IL‐1β secretion induced by 14 h of LPS treatment in human monocytes, but the expression of pro‐IL‐1β was also inhibited (Fig EV3G and H), suggesting that LicoB may affect the alternative NLRP3 inflammasome in human monocytes at least partially via inhibition of production of pro‐IL‐1β (LPS‐induced transcriptional priming).
LicoB inhibits the activation of the NLRP3 inflammasome in vivo and protects against LPS‐induced septic shock
Next, we tested whether LicoB has a protective role in LPS‐induced septic shock mediated by NLRP3 inflammasome activation (Sutterwala et al, 2006; Hou et al, 2020). To assess the effect of LicoB on LPS‐induced lethality, mice were pre‐treated with LicoB or MCC950 for 1 h, then intraperitoneally (i.p.) injected with LPS, and the survival was observed. We found that pre‐treatment with LicoB improved mouse survival in a dose‐dependent manner, as compared to that in the control group (Fig 5A). Interestingly, mice pre‐treated with the combination of LicoB and MCC950 (the two compounds were dissolved and mixed together for i.p.) exhibited the same level of resistance to LPS‐mediated lethality, as compared to that upon treatment with the well‐known NLRP3 inhibitor MCC950 alone (Fig 5A), suggesting that LicoB effectively attenuated LPS‐induced lethality by targeting the NLRP3 inflammasome. Furthermore, we tested the effect of LicoB on pro‐inflammatory cytokine secretion in vivo. Mice were pre‐treated with LicoB or MCC950 for 1 h followed by i.p. injection of LPS. The levels of pro‐inflammatory cytokines in the peritoneal lavage fluid and serum were then measured. The results showed that LicoB effectively inhibited the production of IL‐1β (Fig 5B and C) induced by LPS, but had no significant effect on the release of TNF‐α (Fig 5D and E). At the same time, LicoB reduced the number of peritoneal exudative cells (Fig 5F) and peritoneal macrophages (Fig 5G). Taken together, these data demonstrated that LicoB inhibits the activation of the NLRP3 inflammasome in vivo and protects against LPS‐induced septic shock. In addition, we evaluated the safety of LicoB in vivo. Compared to the control group, the mice that received 40 mg/kg of LicoB for 34 days did not show any changes in biochemical parameters such as serum alanine aminotransferase (ALT) (Fig 5H), aspartate aminotransferase (AST) (Fig 5I) and creatinine (CRE) (Fig 5J) levels and body weight (Fig 5K), indicating that LicoB is well tolerated in mice and may be safe for use in the treatment of related diseases.
Figure 5. Licochalcone B inhibits NLRP3 inflammasome activation in vivo and ameliorates LPS‐induced septic shock.
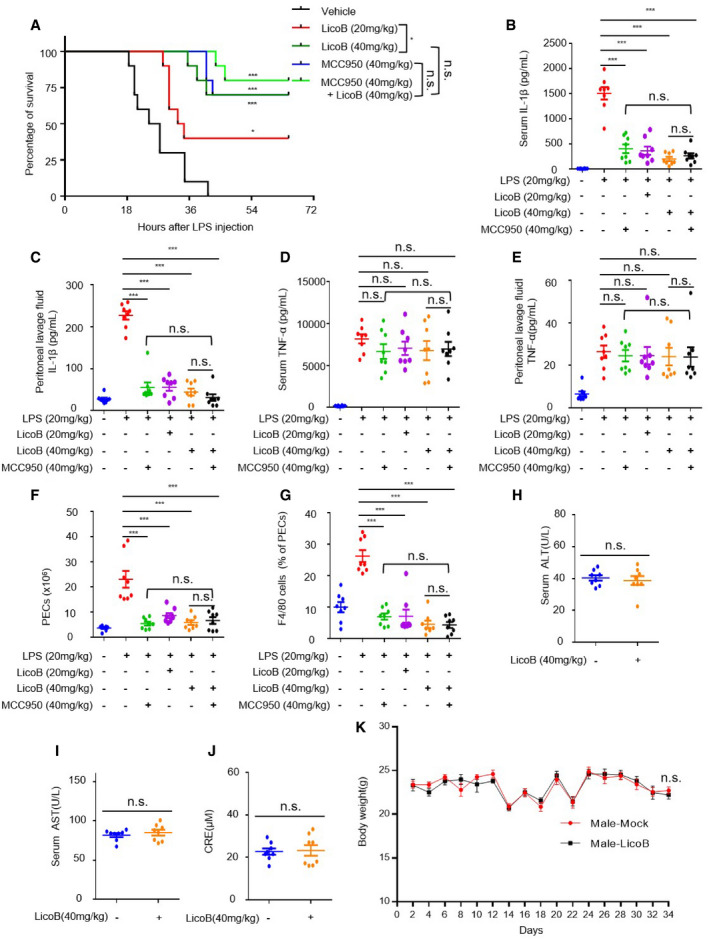
-
ASurvival of WT mice pre‐treated with vehicle, LicoB (20 mg/kg), LicoB (40 mg/kg), MCC950 (40 mg/kg) or LicoB (40 mg/kg) + MCC950 (40 mg/kg), followed by i.p. injection with LPS (20 mg/kg); mice survival was monitored for 72 h (n = 10 mice per group).
-
B–GMice were pre‐treated with LicoB or MCC950 for 1 h, then i.p. injected with LPS (20 mg/kg) and treated for 3 h. The levels of IL‐1β (B, C) and TNF‐α (D, E) in the serum and peritoneal lavage fluid were measured using ELISA. Quantification of peritoneal exudate cells (PECs) (F) and macrophages (F4/80+ cells) (G) using flow cytometry (n = 8 mice per group).
-
H–KC57BL/6 mice received vehicle or LicoB (40 mg/kg) for 17 consecutive doses, once every two days for 34 days. Following that treatment, the serum levels of ALT (H), AST (I) and CRE (J) were measured, along with the body weight (K) (n = 8 mice per group).
Data information: Error bars, mean ± SEM from biological replicates. The significance of the differences was analysed using log‐rank test (A), one‐way ANOVA with Sidak’s post hoc test (B–G), unpaired Student’s t‐test (H–K). *P < 0.05, **P < 0.01, ***P < 0.001, and n.s.: not significant vs. control group.
LicoB suppresses MSU‐induced peritonitis
Intraperitoneal injection of MSU‐induced peritonitis, which is NLRP3‐dependent, characterized by IL‐1β production and neutrophil influx (Martinon et al, 2006). Mice were pre‐treated with LicoB or MCC950 for 1 h followed by i.p. injection of MSU. The levels of pro‐inflammatory cytokines in the peritoneal lavage fluid and serum were then measured. As expected, LicoB treatment significantly reduced MSU‐induced IL‐1β production (Fig 6A and B), as well as the number of peritoneal exudates (Fig 6C) and neutrophils (Fig 6D) in mice. These results indicate that LicoB prevents MSU‐induced peritonitis via inhibition of the NLRP3 inflammasome in vivo.
Figure 6. LicoB suppresses NLRP3‐dependent peritonitis.
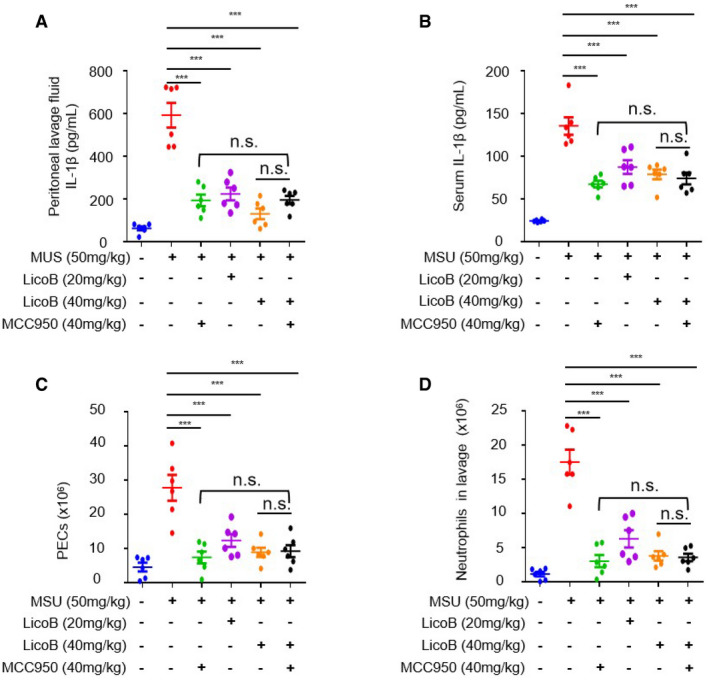
-
A–DMice were pre‐treated with LicoB or MCC950 for 1 h, then i.p. injected with MSU (50 mg/kg) and treated for 6 h (n = 6 mice per group). The levels of IL‐1β in the peritoneal lavage fluid (A) and serum (B) were measured using ELISA. Quantification of peritoneal exudate cells (PECs) (C) and neutrophils (Ly6G and CD11b) (D) using flow cytometry.
Data information: Error bars, mean ± SEM from biological replicates. ***P < 0.001, and n.s.: not significant vs. control group (one‐way ANOVA with Sidak’s post hoc test).
LicoB exhibits therapeutic effects in a NASH mouse model
It has been reported that the NLRP3 inflammasome is an important mediator of NASH induced by a methionine‐ and choline‐deficient (MCD) diet (Szabo & Petrasek, 2015), and blocking the activation of the NLRP3 inflammasome can effectively reduce liver inflammation and liver fibrosis, while improving the pathological characteristics of NASH (Mridha et al, 2017). We tested the protective effects of LicoB on MCD‐induced NASH, a novel mouse NASH model that shows several features of human NASH, such as hepatic steatosis, inflammatory cell infiltration and fibrosis (Varela‐Rey et al, 2009; Huang et al, 2018). The mice were continuously fed with MCD for 6 weeks, and LicoB and/or MCC950 at a dose of 40 mg/kg were administered to them by gavage every two days. MCC950 treatment alone was used as a positive control (Mridha et al, 2017). As compared to the mice fed with methionine‐ and choline‐sufficient (MCS) feed, mice fed with MCD showed obvious liver morphological changes, liver steatosis, balloon dilatation and fibrosis (Fig 7A) and significantly increased serum ALT (Fig 7B) and AST (Fig 7C) levels. As expected, the aforementioned disease phenotypes were robustly alleviated or ameliorated upon LicoB or MCC950 treatment (Fig 7A–C). In addition, we found that the therapeutic effect of the combined administration on MCD diet‐induced NASH pathology and fibrosis was similar to that of LicoB or the well‐characterised NLRP3 inhibitor MCC950 (Fig 7A–C). To test whether LicoB blocks NLRP3 inflammasome activation in livers with NASH, the protein level of active caspase‐1 in liver tissue treated with or without LicoB was analysed. The results showed that as compared to those in the model group, treatment of MCD diet‐fed mice with LicoB or MCC950 significantly inhibited the activation of caspase‐1 and expression of the pro‐fibrotic marker alpha smooth muscle actin (α‐SMA) in the liver (Fig 7D). In addition, we evaluated the transcription levels of genes involved in liver fibrogenesis. The results showed that the MCD‐induced mRNA expression of col1a1 (Fig 7E), and pro‐inflammatory genes TNF‐α (Fig 7F), IL‐1β (Fig 7G) and IL‐18 (Fig 7H) were inhibited upon LicoB treatment. The activation of NLRP3 inflammasomes is accompanied by the maturation and secretion of IL‐18, which is an important indicator protein for specific inhibitors of NLRP3 inflammasomes. For this, we also detected the amount of IL‐18 in serum of mice by ELISA and the data showed that LicoB reduced the serum level of IL‐18 (Fig 7I). Overall, these results suggested that LicoB alleviates liver inflammation and improves NASH pathology in an experimental NASH mouse model via inhibition of the NLRP3 inflammasome.
Figure 7. Licochalcone B exhibits therapeutic effect in the non‐alcoholic steatohepatitis (NASH) model.
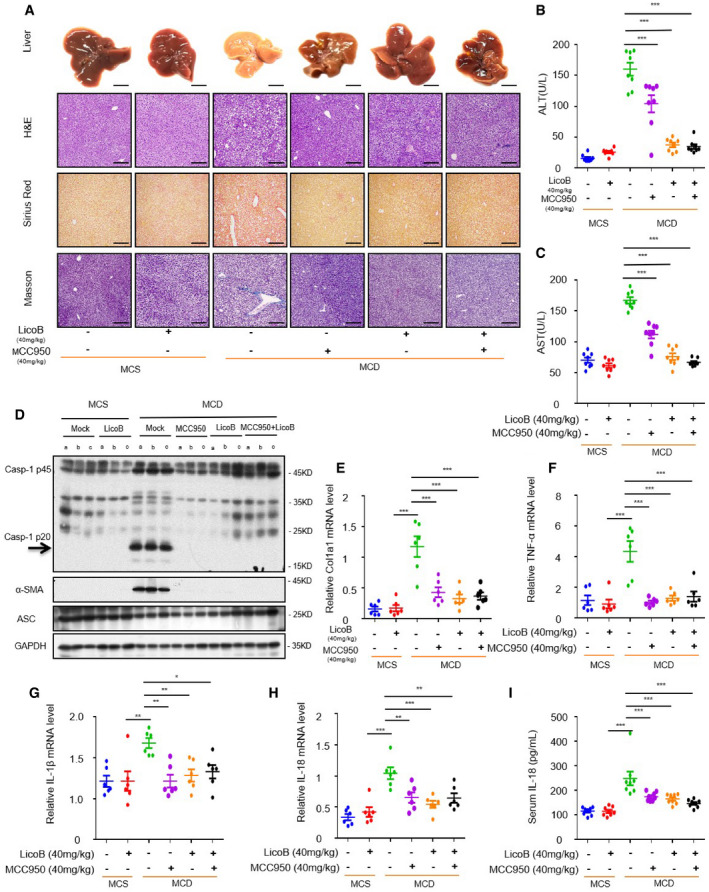
-
A–HUnder the same growth conditions (permitted water, free food and a 12 h/12 h dark/light cycle at 20 ± 2°C), eight‐week‐old male C57BL/6 mice were continuously fed with methionine‐ and choline‐supplemented (MCS) or methionine‐ and choline‐deficient (MCD) diets for 6 weeks, and at the same time, gavaged with LicoB, MCC950 or a combination of LicoB and MCC950 (n = 8 mice per group (B, C and I) or n = 6 mice per group (E–H)). (A) Representative liver section images stained with H&E, Sirius Red and Masson’s trichrome stains. Scale bar: 0.5 cm (top row) and 200 μm (3 bottom rows). (B, C) The serum levels of ALT and AST (n = 8 mice per group) were measured using ELISA. (D) Western blot analyses of pro‐caspase‐1, cleaved caspase‐1, α‐SMA, and ASC in the mice livers, as described in (A). (E‐H) Real‐time quantitative PCR was used to detect the mRNA levels of Col1a1 (E), TNF‐α (F), IL‐1β (G) and IL‐18 (H) in the mice livers, as described in (A) (n = 6 mice per group).
-
IThe levels of IL‐18 (I) in the serum were measured using ELISA (n = 8 mice per group).
Data information: Error bars, mean ± SEM from biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001, and n.s.: not significant vs. control group (one‐way ANOVA with Dunnett's post hoc test).
Source data are available online for this figure.
Discussion
The NLRP3 inflammasome participates in a variety of infections and sterile triggers, which seem to be closely related to common human inflammatory and degenerative diseases (Diehl & Day, 2017; Kazankov et al, 2019; Younossi et al, 2019; Sheka et al, 2020). In view of its role in pathogenesis, blocking the activation of the NLRP3 inflammasome is considered a promising intervention strategy for the treatment of related complex human diseases (Shi et al, 2016; Huang et al, 2018). The findings of the present study demonstrated that LicoB specifically inhibits NLRP3 inflammasome activation both in vitro and in vivo, suggesting that LicoB can be a potent candidate for the treatment of NLRP3‐mediated inflammatory diseases.
A previous study reported that LicoB has an inhibitory effect on the NF‐κB pathway in the RAW 264.7 cell model (Furusawa et al, 2009). In our study, we found that administration of LicoB before LPS priming slightly inhibited the production of pro‐IL‐1β and TNF‐α in BMDMs, while administration of LicoB after LPS priming had no effect on pro‐IL‐1β or NLRP3 expression (Fig 4A and B); in this case, LicoB inhibited the nigericin‐ or ATP‐induced IL‐1β secretion and caspase‐1 activation (Fig 1C and G), suggesting that LicoB indeed plays a role in the activation step (Signal 2) of the NLRP3 inflammasome.
In addition to the NLRP3 inflammasome, activation of NLRC4 and AIM2 inflammasomes also leads to caspase‐1 activation and IL‐1β secretion (Fernandes‐Alnemri et al, 2009; Hornung et al, 2009; Miao et al, 2010; Zhao et al, 2011). By testing the effect of LicoB on the activation of different types of inflammasomes, we found that LicoB did not affect the activation of AIM2 or NLRC4, but only specifically blocked NLRP3 inflammasome activation (Fig 2D), suggesting that LicoB is a specific inhibitor of the NLRP3 inflammasome.
Recent studies have reported that NEK7 is an essential component of the NLRP3 inflammasome; it interacts with NLRP3 and mediates NLRP3 inflammasome assembly and activation (Fry et al, 2012; He et al, 2016; Schmid‐Burgk et al, 2016; Van Hauwermeiren & Lamkanfi, 2016). By identifying the target protein of LicoB, we found that LicoB can bind to NEK7 and inhibit the interaction between NLRP3 and NEK7, which is essential for the activation of the NLRP3 inflammasome (Cover art). This may explain why LicoB specifically inhibits the activation of the NLRP3 inflammasome. NEK7 specifically mediates only NLRP3 inflammasome activation, but does not participate in the activation of NLRC4 and AIM2 inflammasomes. It has been demonstrated that oridonin covalently binds to Cys279 of NLRP3 and block NEK7‐NLRP3 interaction (He et al, 2018). A recent study demonstrates that berberine directly targets NEK7 to inhibit the interaction between NLRP3 and NEK7 (Zeng et al, 2021). These studies demonstrate that inhibition of NEK7‐NLRP3 interaction may be a good strategy (Shi et al, 2016) to inhibit NLRP3 inflammasome activation.
Our results also demonstrated that LicoB displays remarkable therapeutic efficacy in several experimental mouse models, such as LPS‐mediated septic shock, MSU‐induced peritonitis and MCD diet‐induced NASH. As shown in Figs 5, 6, 7, when compared to treatment with LicoB or MCC950 alone, treatment with their combination exhibited similar protective effects in lethal LPS‐induced sepsis, MSU‐induced peritonitis and MCD diet‐induced NASH. Taken together, these results suggested that LicoB administration ameliorated the aforementioned inflammatory diseases by suppressing NLRP3 inflammasome activation. Of note, LicoB has been shown to be safe because the mice that received the same dose as has been used for all examined mouse models for 34 days did not exhibit any biochemical, morphological side effects or weight changes, suggesting that LicoB is well tolerated in mice. Collectively, our research demonstrated that LicoB is a specific NLRP3 inflammasome inhibitor with great potential to be developed as a clinical drug for the treatment of NLRP3 inflammasome‐mediated diseases. However, it may not be soon to enter clinical trials and reach the level of development of other NLRP3 inhibitors (such as OLT1177) that are now in clinical trials, and further studies are needed to evaluate its therapeutic potential.
Materials and Methods
Study design
This study mainly evaluated the therapeutic effect of LicoB on diseases mediated by the NLRP3 inflammasome and also explored its mechanism of inhibiting the NLRP3 inflammasome. The effect of LicoB on activation of the NLRP3 inflammasome in mouse BMDMs, THP‐1 and human PBMCs, and the underlying mechanism of action were studied using immunoblotting, enzyme‐linked immunosorbent assay (ELISA), co‐immunoprecipitation, pull‐down assay and histological staining. The therapeutic effect was also evaluated in mouse models of NLRP3 inflammasome‐mediated diseases.
Mice
Eight‐week‐old C57BL/6 mice were purchased from SPF Biotechnology Company Limited (Beijing, China). The investigator is blinded to animal experiments, and the mice were randomly selected and grouped that were housed in a specific sterile facility under controlled conditions (12 h/12 h light/dark cycle; 20 ± 2°C). All animal experiments were approved by the Experimental Animal Welfare and Ethics of the Fifth Medical Centre of the Chinese People’s Liberation Army General Hospital.
Reagents
Licochalcone B (HY‐N0373), MCC950 (HY‐12815A), Licochalcone A (HY‐N0372), Imiquimod (HY‐B0180) and NAC (HY‐B0215) were purchased from MedChemExpress (State of New Jersey, US). Dimethyl sulfoxide (DMSO), ATP, nigericin, poly (I:C), poly (dA:dT), β‐casein, and PMA and Cyanogen bromide‐activated Sepharose® 4B (C9142‐5G) were purchased from Sigma‐Aldrich (Jefferson City, USA). Ultrapure LPS, tlrl‐msu Monosodium Urate Crystals, Pam3CSK4 and MitoSOX™ were purchased from InvivoGen (State of New Jersey, US). Penicillin‐streptomycin 100× sterile (CC004) was purchased from MacGene (Beijing, China). Salmonella was provided by Prof. Tao Li from the National Centre of Biomedical Analysis (Beijing, China). Caspase‐Glo® 1 Inflammasome Assay (G9951) and CytoTox 96® Non‐Radioactive Cytotoxicity Assay (G1780) were purchased from Promega (Wisconsin, USA). Anti‐mouse caspase‐1 antibody (1:1,500, p20, AG‐20B‐0042) was purchased from Adipogen (USA). Anti‐NEK7 antibody (1:1,500, ab133514) and recombinant human NEK7 protein (ab60703) were purchased from Abcam (UK). Anti‐human cleaved IL‐1β (1:1,000, p17, 12703S), anti‐mouse α‐SMA (1:1,000, 19245s), anti‐mouse IL‐1β (1:1,000, p17, 12242S), anti‐human caspase‐1 (1:1,500, 4199S) and anti‐NLRP3 (1:1,000, 15101S) antibodies were purchased from Cell Signaling Technology (Massachusetts, USA). Anti‐mouse IL‐1β (1:1,000, p17, AF‐401‐NA) antibody and Universal Kinase Activity Kit (EA004) were purchased from R&D Systems (Minnesota, USA). Anti‐ASC (1:1,000, sc‐22514‐R) antibody and Protein A/G PLUS‐Agarose (sc‐2003) were purchased from Santa Cruz Biotechnology (Delaware, USA). Blocking Buffer and Coupling Buffer were purchased from Changzhou Tiandi Renhe Biotechnology Co., Ltd. ImunoSep Human CD14+ cell positive selection kit, Falcon Round Bottom Polystyrene Tubes and ImunoSep Buffer were purchased from Beijing Nuowei Biotechnology Co., Ltd. Anti‐GAPDH (1:2,000, 60004‐1‐1g), anti‐DYKDDDDK tag (1:1,500, 20543‐1‐AP) and anti‐lamin B1 (1:1,500, 66095‐1‐Ig) antibodies were purchased from Proteintech (Chicago, USA).
Plasmids
pCMV‐Flag‐vector and pCMV‐NLRP3‐Flag were provided by Prof. Tao Li from the National Centre of Biomedical Analysis.
Cell preparation and stimulation
BMDMs were isolated from the bone marrow of 8‐week‐old mice and cultured with 50 ng/ml recombinant M‐CSF (416‐ML‐050, R&D Systems) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (04‐001‐1ACS, Biological Industries, USA) and 1% double antibiotics (penicillin‐streptomycin) for 5 days. According to the principles of the Declaration of Helsinki and approved by the responsible ethics committee (the Experimental Welfare and Ethics committee of the Fifth Medical Centre of the Chinese People’s Liberation Army General Hospital), written informed consent was obtained, and hPBMCs were isolated from the peripheral blood of healthy volunteers and then seeded into cell culture plates and cultured overnight in RPMI‐1640 medium (CM10040, MacGene) supplemented with 10% foetal bovine serum and 1% double antibiotics (penicillin‐streptomycin) before stimulation. Human primary monocytes (Gaidt et al, 2016) isolated from fresh hPBMCs using CD14 MACS microbeads or Monocyte Isolation Kit (711410, Precision Biomedicals) according to the provider’s protocol and were cultivated in RPMI Medium 1640 supplemented with l‐glutamine (MCE), sodium pyruvate (ATCC), 10% foetal bovine serum (04‐001‐1ACS, Biological Industries) and double antibiotics (penicillin‐streptomycin). THP‐1 cells were cultured overnight in RPMI‐1640 medium with 100 nM PMA before stimulation. The HEK‐293T cells in our laboratory were tested to be free of mycoplasma, frozen in liquid nitrogen and cultured in DMEM medium after resuscitation. The cells were incubated at 37°C in a humidified incubator (Heracell™ 150i, Thermo Scientific (Massachusetts, USA)) filled with 5% CO2 and 95% air.
In the experiment involving activation of the inflammasome pathway, BMDMs were seeded into 12‐well cell culture plates at a density of 1 × 106 cells/ml and allowed to grow overnight (12–18 h), following which the cells were treated with 50 ng/ml LPS or 400 ng/ml Pam3CSK4 (for non‐canonical inflammatory activation) for 4 h. The cells were then treated with LicoB for 1 h, followed by treatment with different stimulants, as follows: 10 μM nigericin for 45 min; 5 mM ATP or 70 μM Imiquimod for 1 h; 200 μg/ml MSU or 200 ng/ml salmonella for 6 h; 2 μg/ml poly (I:C), 1 μg/ml ultra‐LPS or 2 μg/ml poly(dA:dT) transfected into the BMDMs using Lipofectamine® 2000 (11668‐019, Invitrogen (California, USA)) for 6 h. hPBMCs were seeded into 12‐well cell culture plates at a density of 6 × 106 cells/ml and allowed to grow overnight. After that, the cells were treated LPS 4 h before with LicoB for 1 h and then stimulated with 10 μM nigericin for 45 min or 5 mM ATP for 1 h. Human primary monocytes were seeded into cell culture plate and treated LPS (200 ng/ml) 14 h after with LicoB for 1 h (for Alternative inflammatory activation). THP‐1 cells primed with PMA (100 nM) were seeded into 12‐well cell culture plates at a density of 5 × 106 cells/ml. After 8–12 h, the cells were treated with LicoB for 1 h and then stimulated with 10 μM nigericin for 45 min.
Immunoprecipitation and pull‐down assays
Cultured HEK‐293T cells were seeded into 6‐well cell culture plates and allowed to grow overnight. Following that, the cells were first transfected with Flag‐tagged plasmids (Flag‐vector, Flag‐NLRP3) for 24 h and then treated with LicoB for 6 h. NP‐40 lysis buffer (Huang et al, 2018) (pH 7.4, 150 mM NaCl, 50 mM Tris, 2 mM EDTA, 0.5% Nonidet P‐40) with complete protease inhibitors (1× EDTA‐free Roche protease inhibitor cocktail, TargetMol, C0001) was used to lyse the cells, following which the lysate was collected and centrifuged at 15,000 × g for 15 min at 4°C. The anti‐DYKDDDDK tag antibody was incubated with Protein A/G PLUS‐Agarose at 4°C overnight. According to the manufacturer’s instructions, for immunoprecipitation, the supernatant was incubated with affinity beads combined with anti‐Flag‐M2 (DYKDDDDK tag Antibody‐Protein A/G PLUS‐Agarose) antibody for 3 h. The incubated beads were then centrifuged at 750 × g for 5 min, spun and washed with NP‐40 lysis buffer for 5 min. The above operation was repeated five times, after which 1× SDS–PAGE loading radio immunoprecipitation assay (RIPA) buffer was added to the samples and they were boiled at 105°C for 15 min. The target protein that was pulled down was then analysed using Western blot.
For pull‐down assays, LicoB was conjugated with cyanogen bromide‐activated Sepharose® 4B at 37°C overnight. BMDMs (1.0 × 106 cells), hPBMCs (6 × 106 cells/ml) or PMA‐primed THP‐1 (5 × 106 cells/ml) were seeded into a medium culture dish and allowed to grow overnight, after that BMDMs and hPBMCs were treated with LPS for 4 h, and with or without nigericin for 45 min. Following the treatment, the supernatant was discarded, and the cells were lysed in RIPA lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.5, 1% Triton X‐100, 1% sodium deoxycholate) with complete protease inhibitors (1× EDTA‐free Roche protease inhibitor cocktail), and the cell lysate was collected and subjected to centrifugation at 15,000 × g for 15 min at 4°C. The supernatants were incubated with LicoB‐conjugated Sepharose® 4B or LicoA‐conjugated Sepharose® at 4°C overnight. The incubated beads were then subjected to centrifugation at 750 × g for 5 min, spun and washed with lysis buffer for 5 min. The above operation was repeated approximately 12 times until the supernatant was clear and transparent. The sample was then mixed with 1× SDS–PAGE loading RIPA buffer and boiled at 105°C for 15 min. The target protein that was pulled down was analysed using Western blot.
Caspase‐1 activity assay
The Caspase‐Glo® 1 (Caspase‐Glo® 1 Inflammasome Assay) reagent was prepared according to the manufacturer’s instructions, and the assay was performed according to a previously described protocol for the determination of caspase‐1 activity (Xu et al, 2021).
In vitro kinase assay
NEK7 (ab60703, Abcam) was incubated with β‐casein (9000‐71‐9, Sigma‐Aldrich) as a substrate in the presence of different concentrations of LicoB. NEK7 kinase activity was measured with a Universal Kinase Activity Kit according to the manufacturer’s instructions (R&D Systems; Shi et al, 2016).
LDH assay
According to the manufacturer’s instructions, we used the CytoTox 96® 1 Non‐Radioactive Cytotoxicity Assay to assess the amount of LDH secreted by cells into the supernatant.
Cell counting Kit 8 assay
We used the Cell Counting Kit 8 (CCK‐8) to determine cell viability. BMDMs were seeded into a 96‐well plate at a density of 1 × 106 cells/ml and incubated overnight at 37°C. The cells were then treated with different concentrations of LicoB (0–80 μM) for 24 h. CCK‐8 reagent was then added to the cell culture medium and incubated with the cells for 15 min, following which the optical density was measured at the wavelength of 450 nm (Liu et al, 2021).
ASC oligomerisation assays
The ASC oligomerisation assay was performed as described previously. BMDM cells were treated with 50 ng/ml LPS for 4 h, followed by LicoB treatment for 1 h, and then stimulated with different stimuli, as described in the “Cell preparation and stimulation” section. The cell supernatant was then discarded, and the cells were lysed in Triton lysis buffer (0.5% Triton X‐100, 50 mM Tris–HCl, 150 mM NaCl) with protease inhibitors (1× EDTA‐free Roche protease inhibitor cocktail). The cell lysate was collected and centrifuged at 6,000 g for 15 min at 4°C. The supernatant was then discarded, and the cells were washed twice with ice‐cold phosphate‐buffered saline (1× PBS) and then re‐suspended in 200 μl of 1× PBS. Four microliters of 200 mM (100×) disuccinimidyl sulphate (DSS) was then added to the re‐suspended sample and incubated for a further 30 min at 37°C, with gentle vortexing once every 10 min. Finally, the cross‐linked sample was centrifuged, re‐suspended in 1× SDS–PAGE loading Triton buffer, boiled at 105°C for 15 min and analysed using Western blot.
ELISA
The cytokines IL‐1β, TNF‐α, ALT, AST, CRE and human IL‐1β in supernatants from cell culture or serum were assayed using ELISA kits (R&D Systems; Nanjing Jiancheng Bioengineering Institute, China), according to the manufacturer’s instructions.
Western blot
The protein in the cell culture supernatants or tissue lysate was extracted for Western blot analysis, as described previously (Liu et al, 2021).
Intracellular K+ measurements
The protocol for measurement of intracellular K+ levels has been described previously (Xu et al, 2021). To determine the intracellular K+ concentration, BMDMs were first seeded into 12‐well cell culture plates overnight and then treated with 50 ng/ml LPS for 4 h. The cells were then treated with LicoB for 1 h and stimulated with nigericin for 45 min. Following that, the cell supernatant was discarded, and the cells were washed three times with potassium‐free buffer (pH 7.2, 10 mM Na2HPO4, 1.7 mM NaH2PO4 and 139 mM NaCl). This was followed by addition of ultrapure HNO3 to the cells for lysis and collection of the cell lysate. The samples were boiled at 105°C for 30 min. The samples were then cooled to room temperature by addition of ddH2O to a total volume of 5 ml, following which the intracellular K+ concentration of the samples was measured using inductively coupled plasma mass spectrometry (ICP‐MS).
Intracellular Ca2+ measurement
The protocol for the measurement of intracellular Ca2+ has been described previously (Coll et al, 2015; Liu et al, 2021). BMDM cells were seeded into a 384‐well plate at a density of 2.5 × 104 cells/ml, allowed to grow overnight and then treated with LPS for 4 h, followed by stimulation with ATP for 45 min, with or without LicoB. A trace showing ATP‐induced Ca2+ flux was measured using the FLIPR Tetra® system (Molecular Devices, USA).
Measurement of mtROS
We followed an experimental procedure from a previous report for the measurement of mtROS (Liu et al, 2021). BMDM cells were seeded in a medium culture dish at a density of 1 × 106 cells/ml and allowed to grow overnight (12–18 h). The cells were then treated with LPS for 4 h, collected, transferred to a test tube, treated with or without LicoB for 1 h and stimulated with nigericin for 45 min. The samples were then subjected to centrifugation at 400 × g for 5 min, and the cell supernatant was discarded. The cells were then washed twice with Hank’s balanced salt solution (HBSS) and stained with 4 μM MitoSOX™ Red mitochondrial superoxide indicator (M36008, Invitrogen) at 37°C for 10 min. This was followed by rinsing of the cells twice with HBSS again and re‐suspending them in 200 μl HBSS. The obtained samples were then subjected to flow cytometric analysis using the FACSCanto™ II cell analyser (BD Biosciences, USA).
LPS‐induced septic shock
For the septic shock model, 8‐week‐old C57BL/6 female mice were given i.p. injections of vehicle, LicoB (20 or 40 mg/kg), MCC950 (40 mg/kg) or LicoB (40 mg/kg) in combination with MCC950 (40 mg/kg) (n = 10) for 1 h, followed by an i.p. injection of LPS (20 mg/kg) (O55:B5; Sigma‐Aldrich). The number of deaths and survival of the mice were monitored continuously for 72 h. To analyse cytokine secretion, mice (n = 8 per group) were treated in the same way as mentioned above. Three hours later, the mice were sacrificed by means of cervical dislocation, blood was collected from them, serum samples were separated, the peritoneal cavity was washed with 10 ml ice‐cold 1× PBS, and the peritoneal lavage fluid was collected. The levels of IL‐1β and TNF‐α in the serum and peritoneal lavage fluid were measured using ELISA. The cell counter was used to measure the number of peritoneal exudative cells in the peritoneal lavage fluid. Flow cytometry was used to determine the number of peritoneal macrophages (F4/80 cells) in the peritoneal lavage fluid.
Toxicology
Eight‐week‐old C57BL/6 male mice were given i.p. injections of LicoB (40 mg/kg) or vehicle (5% DMSO, 5% Tween 80, and normal saline) every two days for a total of 34 days. Body weight was measured before each injection. Blood was collected from the animals and serum samples were separated on the 34th day. ALT, AST and CRE levels were measured according to the manufacturer’s instructions.
MSU‐induced peritonitis model in vivo
Eight‐week‐old C57BL/6 female mice were given i.p. injections of vehicle, LicoB (20 or 40 mg/kg), MCC950 (40 mg/kg) or LicoB (40 mg/kg) in combination with MCC950 (40 mg/kg) (dissolved in vehicle containing 5% DMSO, 5% Tween‐80 and 90% normal saline) (n = 6) for 1 h, followed by an i.p. injection of MSU crystals (50 mg/kg, dissolved in 1×PBS). 6 h later, the mice were sacrificed by means of cervical dislocation, blood was collected from them, serum samples were separated, the peritoneal cavity was washed with 10 ml ice‐cold 1× PBS, and the peritoneal lavage fluid was collected. The levels of IL‐1β in the serum and peritoneal lavage fluid were measured using ELISA. The cell counter was used to measure the number of peritoneal exudative cells in the peritoneal lavage fluid. Flow cytometry was used to determine the number of polymorphonuclear neutrophils (Ly6G and CD11b) by staining in the peritoneal lavage fluid (Martinon et al, 2006; Tang et al, 2017).
MCD diet‐induced NASH model
Male C57BL/6 mice aged 8 weeks were fed with MCD (MCDAA, Dytz Biotechnology Co., Ltd. (Beijing, China)) and MCS (MCSAA, Dytz Biotechnology Co. Ltd.) feed for six weeks. The model group was fed an MCD diet, while the control group was fed an MCS diet. At the same time, the different treatment groups were given vehicle (5% DMSO, 5% Tween‐80, and normal saline), LicoB (40 mg/kg, LicoB was dissolved in DMSO and then added with 5% DMSO and then 95% (5.26% Tween‐80 in saline) to prepare a 2 mg/ml solution, no precipitation was observed during the preparation. For a 20‐gram mouse, it was gavaged with 0.4 ml, MCC950 (40 mg/kg) or LicoB (40 mg/kg) combined with MCC950 (40 mg/kg) for six weeks by gavage every two days (n = 8 per group). At the end of the experiment, the mice were anaesthetised, liver tissues were collected from them for analysis and detection of mRNA and protein, and the serum was separated for detection of ALT, AST and IL‐18.
Statistical analyses
For comparison of two groups, unpaired Student's t‐test was used, and for comparison of multiple groups, one‐way ANOVA with Dunnett's post hoc test or Sidak’s post hoc test (GraphPad Prism, USA) was used. Data are presented as mean ± SEM. Log‐rank test was used for survival analysis.
Author contributions
ZB, XX, XZhan, PZ and GX supervised the project and acquired funding for the study. ZB, XZhan and QL designed the experiments. QL performed most of the experiments and analysed the data. HF, HW and YHWang provided assistance with the investigation. GX, QL, ZL and WM collected the human plasma samples. RL provided assistance with animal resources. ZB, GX, XZhan, WS and ZWang designed the NLRP3 mechanistic studies and supervised the experiment. ZF, LR and ZWei performed the LPS‐induced septic shock or MSU‐induced peritonitis animal experiments and analysed the data. YWang, LL, XH, WD, TL and PL helped perform the MCD‐induced mouse model. JW, YG and XZhao assisted with the histological experiments and analysed the data. QL, ZB and XZhan wrote and revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 7
Acknowledgments
We thank Prof. Tao Li (National Centre of Biomedical Analysis) for providing Salmonella typhimurium. This work was supported by the National Key New Drug Creation and Manufacturing Program, Ministry of Science and Technology (2017ZX09301022), Beijing Nova Program (Z181100006218001), National Natural Science Foundation of China (81874368, 81630100, 81930110, 81721002, 82003984, 81903891) and National Key Research and Development Program of China (2018YFC1706502).
EMBO Reports (2022) 23: e53499.
Contributor Information
Xiaoyan Zhan, Email: xyzhan123@163.com.
Xiaohe Xiao, Email: pharmacy_302@126.com.
Zhaofang Bai, Email: baizf2008@hotmail.com.
Data availability
No data were deposited in a public database.
References
- Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E (2015) The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol 10: 395–424 [DOI] [PubMed] [Google Scholar]
- Chen Y, Li R, Wang Z, Hou X, Wang C, Ai Y, Shi W, Zhan X, Wang J‐B, Xiao X et al (2020) Dehydrocostus lactone inhibits NLRP3 inflammasome activation by blocking ASC oligomerization and prevents LPS‐mediated inflammation in vivo . Cell Immunol 349: 104046 [DOI] [PubMed] [Google Scholar]
- Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson AAB et al (2019) MCC950 directly targets the NLRP3 ATP‐hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15: 556–559 [DOI] [PubMed] [Google Scholar]
- Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A et al (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez‐Ruiz F, Stamp LK (2019) Gout. Nat Rev Dis Primers 5: 69 [DOI] [PubMed] [Google Scholar]
- Dalbeth N, Merriman TR, Stamp LK (2016) Gout. Lancet 388: 2039–2052. [DOI] [PubMed] [Google Scholar]
- Diehl AM, Day C (2017) Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 377: 2063–2072 [DOI] [PubMed] [Google Scholar]
- Fahey JW, Zhang Y, Talalay P (1997) Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci USA 94: 10367–10372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes‐Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458: 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry AM, O'Regan L, Sabir SR, Bayliss R (2012) Cell cycle regulation by the NEK family of protein kinases. J Cell Sci 125: 4423–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Chen J, Li YJ, Zheng YF, Li P (2013) Antioxidant and anti‐inflammatory activities of six flavonoids separated from licorice. Food Chem 141: 1063–1071 [DOI] [PubMed] [Google Scholar]
- Furusawa J, Funakoshi‐Tago M, Mashino T, Tago K, Inoue H, Sonoda Y, Kasahara T (2009) Glycyrrhiza inflata‐derived chalcones, Licochalcone A, Licochalcone B and Licochalcone D, inhibit phosphorylation of NF‐kappaB p65 in LPS signaling pathway. Int Immunopharmacol 9: 499–507 [DOI] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid‐Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, Hornung V (2016) Human monocytes engage an alternative inflammasome pathway. Immunity 44: 833–846 [DOI] [PubMed] [Google Scholar]
- Gao XP, Qian DW, Xie Z, Hui H (2017) Protective role of licochalcone B against ethanol‐induced hepatotoxicity through regulation of Erk signaling. Iran J Basic Med Sci 20: 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaul S, Leszczynska A, Alegre F, Kaufmann B, Johnson CD, Adams LA, Wree A, Damm G, Seehofer D, Calvente CJ et al (2021) Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol 74: 156–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney AJ, Maier NK, Leppla SH, Moayeri M (2016) Sulforaphane inhibits multiple inflammasomes through an Nrf2‐independent mechanism. J Leukoc Biol 99: 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groß C, Mishra R, Schneider K, Médard G, Wettmarshausen J, Dittlein D, Shi H, Gorka O, Koenig P‐A, Fromm S et al (2016) K(+) Efflux‐independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 45: 761–773 [DOI] [PubMed] [Google Scholar]
- Haraguchi H, Ishikawa H, Mizutani K, Tamura Y, Kinoshita T (1998) Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg Med Chem 6: 339–347 [DOI] [PubMed] [Google Scholar]
- He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, Liu Q, Liang G, Deng X, Jiang W et al (2018) Oridonin is a covalent NLRP3 inhibitor with strong anti‐inflammasome activity. Nat Commun 9: 2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 458: 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Xu G, Wang Z, Zhan X, Li H, Li R, Shi W, Wang C, Chen Y, Ai Y et al (2020) Glaucocalyxin A alleviates LPS‐mediated septic shock and inflammation via inhibiting NLRP3 inflammasome activation. Int Immunopharmacol 81: 106271 [DOI] [PubMed] [Google Scholar]
- Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, Deng X, Liang G, Zhang H, Jiang W et al (2018) Tranilast directly targets NLRP3 to treat inflammasome‐driven diseases. EMBO Mol Med 10: e8689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, Wang A, Tao J, Wang C, Liu Q et al (2017) Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214: 3219–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo EK, Kim JK, Shin DM, Sasakawa C (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13: 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A et al (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: 1246–1249 [DOI] [PubMed] [Google Scholar]
- Kazankov K, Jørgensen SMD, Thomsen KL, Møller HJ, Vilstrup H, George J, Schuppan D, Grønbæk H (2019) The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 16: 145–159 [DOI] [PubMed] [Google Scholar]
- Klück V, Jansen TLTA, Janssen M, Comarniceanu A, Efdé M, Tengesdal IW, Schraa K, Cleophas MCP, Scribner CL, Skouras DB et al (2020) Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open‐label, dose‐adaptive, proof‐of‐concept, phase 2a trial. The Lancet Rheumatology 2: e270–e280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RR, Wieser ME, Ganga RR, Barathi VA, Lakshminarayanan R, Mohan RR, Hainsworth DP, Chaurasia SS (2020) NOD‐like receptors in the eye: uncovering its role in diabetic retinopathy. Int J Mol Sci 21: 899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhan X, Xu G, Wang Z, Li R, Wang Y, Qin Q, Shi W, Hou X, Yang R et al (2021) Cryptotanshinone specifically suppresses NLRP3 inflammasome activation and protects against inflammasome‐mediated diseases. Pharmacol Res 164: 105384 [DOI] [PubMed] [Google Scholar]
- Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D'Alessandro A, Dinarello CA, Korte M (2020) The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 117: 32145–32154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 156: 1193–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E (2018) Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discovery 17: 588–606 [DOI] [PubMed] [Google Scholar]
- Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, Carta S, Tengesdal I, Nemkov T, D’Alessandro A et al (2018) OLT1177, a β‐sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA 115: E1530–e1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose‐Girma M, Erickson S, Dixit VM (2004) Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 430: 213–218 [DOI] [PubMed] [Google Scholar]
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241 [DOI] [PubMed] [Google Scholar]
- Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107: 3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, Haczeyni F, Teoh N‐H, Savard C, Ioannou GN et al (2017) NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66: 1037–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz‐Planillo R, Kuffa P, Martínez‐Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38: 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA 109: 11282–11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Miao EA (2019) A licence to kill during inflammation. Nature 570: 316–317 [DOI] [PubMed] [Google Scholar]
- Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO (2014) Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6: 221ra215 [DOI] [PubMed] [Google Scholar]
- Schmid‐Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, Hornung V (2016) A genome‐wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 291: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Wang LI, Wang WL, Magupalli VG, Andreeva L, Qiao QI, Hauenstein AV, Wu Z, Núñez G, Mao Y et al (2019) Structural mechanism for NEK7‐licensed activation of NLRP3 inflammasome. Nature 570: 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S (2020) Nonalcoholic steatohepatitis: a review. JAMA 323: 1175–1183 [DOI] [PubMed] [Google Scholar]
- Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S et al (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17: 250–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Xu G, Zhan X, Gao Y, Wang Z, Fu S, Qin N, Hou X, Ai Y, Wang C et al (2020) Carnosol inhibits inflammasome activation by directly targeting HSP90 to treat inflammasome‐mediated diseases. Cell Death Dis 11: 252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Connors SL, Macklin EA, Smith KD, Fahey JW, Talalay P, Zimmerman AW (2014) Sulforaphane treatment of autism spectrum disorder (ASD). Proc Natl Acad Sci USA 111: 15550–15555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Liu Z‐S, Xue W, Bai Z‐F, Wang Q‐Y, Dai J, Liu X, Huang Y‐J, Cai H, Zhan X‐Y et al (2017) NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell 68: 185–197 [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara‐Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW et al (2006) Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase‐1. Immunity 24: 317–327 [DOI] [PubMed] [Google Scholar]
- Szabo G, Petrasek J (2015) Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 12: 387–400 [DOI] [PubMed] [Google Scholar]
- Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, Cui J, Bai LI, Wang J, Jiang W et al (2017) CLICs‐dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun 8: 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia‐Abellan A, Angosto‐Bazarra D, Martinez‐Banaclocha H, de Torre‐Minguela C, Ceron‐Carrasco JP, Perez‐Sanchez H, Arostegui JI, Pelegrin P (2019) MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol 15: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiyagarajan P, Chandrasekaran CV, Deepak HB, Agarwal A (2011) Modulation of lipopolysaccharide‐induced pro‐inflammatory mediators by an extract of Glycyrrhiza glabra and its phytoconstituents. Inflammopharmacology 19: 235–241 [DOI] [PubMed] [Google Scholar]
- Thomas H (2017) NAFLD: A critical role for the NLRP3 inflammasome in NASH. Nat Rev Gastroenterol Hepatol 14: 197 [DOI] [PubMed] [Google Scholar]
- Van Hauwermeiren F, Lamkanfi M (2016) The NEK‐sus of the NLRP3 inflammasome. Nat Immunol 17: 223–224 [DOI] [PubMed] [Google Scholar]
- Varela‐Rey M, Embade N, Ariz U, Lu SC, Mato JM, Martínez‐Chantar ML (2009) Non‐alcoholic steatohepatitis and animal models: understanding the human disease. Int J Biochem Cell Biol 41: 969–976 [DOI] [PubMed] [Google Scholar]
- Wang C, Chen L, Xu C, Shi J, Chen S, Tan M, Chen J, Zou L, Chen C, Liu Z et al (2020) A comprehensive review for phytochemical, pharmacological, and biosynthesis studies on Glycyrrhiza spp. Am J Chin Med 48: 17–45 [DOI] [PubMed] [Google Scholar]
- Wang J, Liao AM, Thakur K, Zhang JG, Huang JH, Wei ZJ (2019a) Licochalcone B extracted from Glycyrrhiza uralensis fisch induces apoptotic effects in human hepatoma cell HepG2. J Agric Food Chem 67: 3341–3353 [DOI] [PubMed] [Google Scholar]
- Wang L, Yang R, Yuan B, Liu Y, Liu C (2015) The antiviral and antimicrobial activities of licorice, a widely‐used Chinese herb. Acta Pharmaceutica Sinica B 5: 310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu G, Gao Y, Zhan X, Qin N, Fu S, Li R, Niu M, Wang J, Liu Y et al (2019b) Cardamonin from a medicinal herb protects against LPS‐induced septic shock by suppressing NLRP3 inflammasome. Acta Pharmaceutica Sinica B 9: 734–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlford GF, Van Tassell BW, Billingsley HE, Kadariya D, Canada JM, Carbone S, Mihalick VL, Bonaventura A, Vecchié A, Chiabrando JG et al (2020) Phase 1B, Randomized, double‐blinded, dose escalation, single‐center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II‐III systolic heart failure. J Cardiovasc Pharmacol 77: 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A‐G, Zhou X‐G, Qiao G, Yu LU, Tang Y, Yan LU, Qiu W‐Q, Pan R, Yu C‐L, Law B‐K et al (2021) Targeting microglial autophagic degradation in NLRP3 inflammasome‐mediated neurodegenerative diseases. Ageing Res Rev 65: 101202 [DOI] [PubMed] [Google Scholar]
- Xu G, Fu S, Zhan X, Wang Z, Zhang P, Shi W, Qin N, Chen Y, Wang C, Niu M et al (2021) Echinatin effectively protects against NLRP3 inflammasome–driven diseases by targeting HSP90. JCI Insight 6: e134601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Lee HE, Yeon SH, Kang HC, Cho YY, Lee HS, Zouboulis CC, Han SH, Lee JH, Lee JY (2018) Licochalcone A attenuates acne symptoms mediated by suppression of NLRP3 inflammasome. Phytother Res 32: 2551–2559 [DOI] [PubMed] [Google Scholar]
- Yang XD, Li W, Zhang S, Wu D, Jiang X, Tan R, Niu X, Wang Q, Wu X, Liu Z et al (2020) PLK4 deubiquitination by Spata2‐CYLD suppresses NEK7‐mediated NLRP3 inflammasome activation at the centrosome. EMBO J 39: e102201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm Y‐H, Grant R, McCabe L, Albarado D, Nguyen K, Ravussin A, Pistell P, Newman S, Carter R, Laque A et al (2013) Canonical Nlrp3 inflammasome links systemic low‐grade inflammation to functional decline in aging. Cell Metab 18: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, Wai‐Sun Wong V, Yilmaz Y, George J, Fan J et al (2019) Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 69: 2672–2682 [DOI] [PubMed] [Google Scholar]
- Yuan X, Li T, Xiao E, Zhao H, Li Y, Fu S, Gan L, Wang Z, Zheng Q, Wang Z (2014) Licochalcone B inhibits growth of bladder cancer cells by arresting cell cycle progression and inducing apoptosis. Food Chem Toxicol 65: 242–251 [DOI] [PubMed] [Google Scholar]
- Zeng Q, Deng H, Li Y, Fan T, Liu Y, Tang S, Wei W, Liu X, Guo X, Jiang J et al (2021) Berberine directly targets the NEK7 protein to block the NEK7‐NLRP3 interaction and exert anti‐inflammatory activity. J Med Chem 64: 768–781 [DOI] [PubMed] [Google Scholar]
- Zhao N, Li CC, Di B, Xu LL (2020) Recent advances in the NEK7‐licensed NLRP3 inflammasome activation: Mechanisms, role in diseases and related inhibitors. J Autoimmun 113: 102515 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600 [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
- Zhuang T, Liu J, Chen X, Zhang L, Pi J, Sun H, Li LI, Bauer R, Wang H, Yu Z et al (2019) Endothelial Foxp1 suppresses atherosclerosis via modulation of Nlrp3 inflammasome activation. Circ Res 125: 590–605 [DOI] [PubMed] [Google Scholar]