

Abstract
Developing non-natural reaction mechanisms is an attractive strategy for expanding the synthetic capabilities of substrate promiscuous enzymes. Here, we report an ‘ene’-reductase catalyzed asymmetric hydroalkylation of olefins using α-bromoketones as radical precursors. Radical initiation occurs via ground-state electron transfer from the flavin cofactor located within the enzyme active site, an underrepresented mechanism in flavin biocatalysis. Four rounds of site saturation mutagenesis were used to access a variant of the ‘ene’-reductase nicotinamide-dependent cyclohexanone reductase (NCR) from Zymomonas mobiles capable of catalyzing a cyclization to furnish β-chiral cyclopentanones with high levels of enantioselectivity. Additionally, wild-type NCR can catalyze intermolecular couplings with precise stereochemical control over the radical termination step. This report highlights the utility for ground-state electron transfers to enable non-natural biocatalytic C─C bond forming reactions.
Graphical Abstract
Introduction
Radicals are versatile synthetic intermediates that enable numerous fascinating processes and retrosynthetic disconnections.1-3 While the overall reactivity of radical intermediates is well understood,4 catalytic strategies for controlling the chemo- and stereoselectivity remain underdeveloped.5-7 Nature has evolved various biocatalysts, such as cobalamin and radical S-adenosylmethionine (SAM) enzymes, to catalyze the radical-mediated transformations with unparalleled chemo-, regio-, and enantioselectivity in the biosynthesis of complex natural products.8-11 However, these enzymes are not readily harnessed outside of their natural settings by organic synthetic chemists, owing to either operational difficulties or lack of catalytic versatility. We imagined that enzymes could be used more broadly to solve selectivity challenges in the radical literature by identifying and developing enzymes that can use non-natural mechanisms for radical formation.12,13
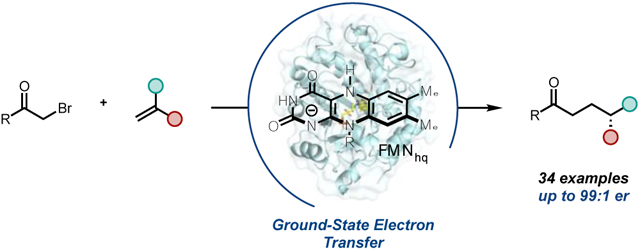
Flavin-dependent ‘ene’-reductases (EREDs) naturally catalyze the asymmetric reduction of electronically activated alkenes and are among the most widely used class of biocatalysts in chemical synthesis.14,15 Mechanistically, the reaction occurs via a stereoselective hydride transfer from the cofactor flavin hydroquinone (FMNhq) to the substrate’s electrophilic position followed by protonation by a conserved tyrosine residue in the enzyme active-site to give the hydrogenated product.14,15 Our group recently demonstrated that these enzymes can also catalyze asymmetric radical cyclizations when irradiated with visible light.16-19 This reaction occurs via photoexcitation of an enzyme templated charge transfer (CT) complexes formed between organohalides substrate and flavin hydroquinone (FMNhq) cofactor.16-18 This mechanism is effective for inducing electron transfer to alkyl iodides and chloroamides whose low reduction potentials make ground state electron transfer thermodynamically challenging (Figure 1A). However, as the energetics of CT complexes are governed by both electrostatic attraction and substrate electron affinity, it can be difficult to predict which substrates will form such complexes.20 Ground state electron transfer is an attractive alternative to photoinduced mechanisms because its viability is determined by the redox potentials of substrate and cofactor (E1/2 = −0.45 V versus saturated calomel electrode (SCE) for FMNhq21). We previously demonstrated the viability of this mechanism for asymmetric hydrodehalogenation of electronically activated α-bromophenylacetic esters.22 This mechanism has not, however, been utilized for forming less stabilized radicals or been deployed in C─C bond forming reactions.
Figure 1.

Strategies and challenges in using ‘ene’-reductases (EREDs) for non-natural radical cyclization.
As a model, we targeted the cyclization of α-bromoketone 1 to afford β-stereogenic cyclopentanone 2 (Figure 1B). As α-bromoaliphatic ketones are more easily reduced by comparison to chloroamides (E1/2 = −0.74 V versus SCE for α-bromoacetophenone),23 we hypothesized that they would be amenable to reduction via ground state electron transfer. While this represents an endergonic electron transfer, we reasoned that subsequent fast and irreversible mesolytic cleavage of the C─Br bond would enable the reaction to proceed. The intermediacy of α-ketonyl radicals present distinct challenges by comparison to our previous biocatalytic cyclization involving α-amidyl radicals.16 Specifically, α-ketonyl radicals have lower reduction potentials and are more reactive, making them prone to undesired electron and hydrogen atom transfer pathways that ultimately form undesired hydrodebrominated products (Figure 1B).23 These side reactions also plague variants of this reaction mediated by small-molecule reagents.24-26 Additionally, the enhanced reactivity of this intermediate should also result in a transition state for the C─C bond-forming step that more closely resembles the starting material (per the Hammond Postulate), making it harder to achieve high levels of enantioselectivity, potentially accounting for the lack of general small molecules catalysts for this reaction (Figure 1B).27
Results and Discussion
Discovery and engineering of NCR for radical cyclization
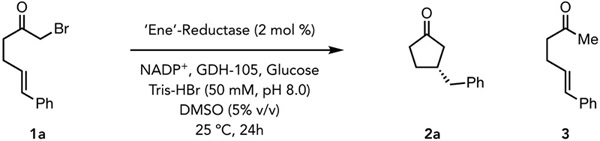
We began our studies by exploring the reductive cyclization of α-bromoketone 1a to β-stereogenic cyclopentanone 2a catalyzed by structurally diverse wild-type EREDs under ambient conditions with an NADPH regeneration system (GDH/NADP+/glucose) (Table 1). Several EREDs provided the desired cyclopentanone product 2a, although significant quantities of the hydrodehalogenation product 3 were observed in all cases. The ERED 12-oxophytodienoate reductase 1 (OPR1) from Solanum lycopersicum provided highest enantioselectivity (31% yield, 80:20 er) for product 2a, with 9% formation of the undesired hydrodebrominated product 3 (Table 1, entry 1). The nicotinamide-dependent cyclohexanone reductase (NCR) from Zymomonas mobilis afforded similar levels of enantioselectivity to OPR1 but with more formation of 3a (Table 1, entry 2). Interestingly, pentaerythritol tetranitrate reductase (PETNr) afforded the high yields for the desire product 2a, but with lower levels of enantioselectivity (Table 1, entry 4). Importantly, it is also found in commercial ERED collections. For instance, ERED-30 from the Prozomix library affords product 2a in 94% yield (65:35 er) and only 6% yield of the linear dehalogenated product 3 (Table 1, entry 4). Control experiments confirmed that both ERED and the NADPH regeneration system were required for reactivity (Table 1, entry 5 and 6).
Table 1.
Screening an initial panel of EREDs for cyclization.
| ||||
|---|---|---|---|---|
| Entry | ‘Ene’-Reductase | Yielda (%) of 2a | erb | Yield (%) of 3 |
| 1 | OPR1 | 31 | 80:20 | 9 |
| 2 | NCR | 20 | 79:21 | 19 |
| 3 | PETNr | 66 | 66:34 | 19 |
| 4 | ERED-30c | 94 | 65:35 | 6 |
| 5 | No enzyme | 0 | n.d.d | 0 |
| 6 | No NADPH regeneration | 0 | n.d. | 0 |
Reaction conditions: α-bromoketone (1 mg, 0.004 mmol), GDH-105 (0.5 mg), NADP+ (0.5 mg), glucose (5 mg) and purified ‘ene’-reductases (2 mol%) in 50 mM Tris-HBr buffer pH 8.0, with 5% DMSO (v/v) as cosolvent, final total volume is 500 μL. Reaction mixtures were shaken under anaerobic conditions at room temperature for 24 h.
Yield determined via LCMS relative to an internal standard (TBB).
Enantiomeric ratio (er) determined by HPLC on a chiral stationary phase.
ERED-30 (1 mol%) was screened from a genetically diverse EREDs library from Prozomix.
n.d., not determined.
As a wild-type enzyme could not be identified that afforded high yields and enantioselectivities; we chose to evolve an improved catalyst from NCR. This enzyme was selected as a starting point because it provided the appropriate balance of product yield, enantioselectivity, and high expression levels in 96-well plates. EREDs have not previously been evolved for radical biocatalytic reactions, so amino acid residues were chosen for site saturation mutagenesis (SSM) that line the active site pocket that are thought interact with either the substrate or flavin cofactor (the docking model of substrate 1a in wild-type NCR is shown in Supplemental Figure 1).28 For each round of SSM, enzyme libraries were co-expressed with the groES-groEL chaperone using Escherichia coli cells and screened in 96-well plates in the form of cell-free extracts (CFEs). After four rounds of SSM, we discovered four beneficial mutations Y343W, F269W, W342A, and I231S, which collectively furnishing a notable 7.7-fold improvement in yield and further enhancements in enantioselectivity; the undesired dehalogenated linear product 3 was also effectively diminished to a negligible level (Figure 2 and Supplemental Tables 2-10). Using the final quadruple mutant (Y343W/F269W/W342A/I231S), named NCR-C9, as the biocatalyst under optimized conditions, the (S)-configured cyclization product 2a was formed in 92% yield and 95:5 er, with a 99:1 ratio of cyclized/linear (2a/3) products (Figure 2). This reaction can be run on a preparative scale and afford product in 86% isolated yield with no changes in enantio- or chemoselectivity (Scheme 1).
Figure 2.

Directed evolution of NCR catalysts for non-natural asymmetric radical cyclization. A, Chemoselectivity (cyclization versus debrominative hydrogenation), stereoselectivity, and yields improvement of NCR variants over wild-type NCR. Standard reaction conditions: the reaction mixture (500 μL) consisted of model substrate (1a, 0.004 mmol), GDH-105 (0.5 mg), glucose (5.0 mg), NADP+ (0.5 mg), and purified NCR variants (1.0 mol%) in buffer (100 mM KPi buffer pH 8.0) with 5% DMSO as cosolvent at room temperature for 24 h. Yields were determined via HPLC relative to an internal standard (average of triplicate). Enantiomeric ratio (er) was determined via HPLC on a chiral stationary phase. B, Crystal structure of wild-type NCR (PDB: 4s3u). Residues for substrate binding (H172 and N175) are shown as blue sticks. Four beneficial sites Y343, F269, W342, and I231 are shown in orange sticks.
Scheme 1.

Asymmetric radical cyclization catalyzed by NCR-C9 mutant. Standard reaction conditions: the reaction mixture (500 μL) consisted of substrate (1, 0.004 mmol), GDH-105 (0.5 mg), glucose (5.0 mg), NADP+ (0.5 mg), and purified NCR-C9 mutant (1.0 mol%) in buffer (100 mM KPi buffer pH 8.0) with 5% DMSO as cosolvent at room temperature for 24 h. a Yields were determined via HPLC relative to an internal standard. b Enantiomeric ratio (er) was determined via HPLC on a chiral stationary phase. c Isolated yield was based on 0.08 mmol-scale reaction. d 2 mol% of wild-type NCR was used. e n.d., er not determined.
With the evolved non-natural radical enzyme NCR-C9 in hand, we explored the transformation scope and limitations (Scheme 1). The quadruple mutant NCR-C9 accommodated various para-substituents of the aromatic ring, with electron-donating (1e–f) and electron-withdrawing (1i) substituents affording β-stereogenic cyclopentanones in high yields (74−86%) and excellent enantioselectivities (> 94:6 er, Scheme 1). Notably, para-halogenated substrates (1g–h) were tolerated providing a handle for potential downstream manipulation (Scheme 1). Ortho-substituted (1b) and meta-substituted (1c–d) substrates were also well accepted by the evolved NCR-C9, giving the corresponding cyclized products 2c–d with good yields (57–81%) and high levels of stereoselectivity (> 90:10 er, Scheme 1). The aromatic substituent proved essential for the desired reactivity, with unsubstituted alkene or enone affording the hydrodehalogenated products exclusively (Scheme 1 and Supplemental Figure 2).
Protein engineering campaigns result in catalysts that are better suited for the reaction on which they were evolved. To probe the specialization in this protein engineering campaign, we tested alternative substrates and cyclization modes. The (Z)-isomer of the model substrate (Z)-1a was accepted by the NCR-C9, although with diminished yield and enantioselectivity (62% yield, 69:31 er, S-isomer over access) by comparison to the (E)-configured substrate (E)-1a, indicating the importance of geometric configuration of the substrate during the NCR-catalyzed radical cyclization reaction (Scheme 1). In contrast, (E)-1a and (Z)-1a perform similarly with wild-type NCR, indicating that the protein engineering campaign has enhanced the preference for the (E)-olefin isomer. In the realm of alternative cyclization modes, the specialization was less pronounced. While NCR-C9 was ineffective for a 6-exo-trig cyclization, the wild-type enzyme could catalyze the reaction in low yield, indicating the parent enzyme is more promiscuous. In contrast, both variants afforded similar results for 7-exo-trig cyclization (Scheme 1). In addition, both the wild-type and evolved variants can catalyze the 6-endo-trig cyclization providing γ-substituted cyclohexanone 2m in moderate yield; however, 5-endo-trig cyclization was inaccessible to both enzymes, fitting to the Baldwin rules (Scheme 1 and Supplemental Figure 3). Collectively, these results indicate that specialization is apparent within individual cyclization modes but does not necessarily hold when looking at other manifolds.
Intermolecular radical C─C bond formation catalyzed by wild-type NCR
Having evolved a selective enzyme for a radical cyclization, we further questioned whether EREDs could catalyze an intermolecular radical hydroalkylation. Zhao and coworkers have previously reported that EREDs can catalyze the proposed reaction when irradiated with blue LEDs.29 However, as α-bromoacetophenone has a modest reduction potential (−0.74 V vs. SCE),23 we hypothesized that this reaction can occur without photoexcitation. In a dark manifold, the central challenge is identifying a gating mechanism for ensuring that both the bromoketone and alkene are bound within the protein active site prior to electron transfer. In the absence of such a mechanism, we would expect to form the hydrodehalogenated product primarily. In the photoenzymatic coupling of chloroamides and alkenes, we observed an enzyme templated ternary CT complex between FMNhq, chloroamide, and α-methylstyrene.30 As this complex is more strongly absorbing by comparison to the binary complex (FMNhq and chloroamide), productive chemistry primarily occurs through a CT complex that has both coupling partners bound within the protein active site.30 In the absence of this photoregulation mechanism, it was unclear whether radical formation would be substrate gated.
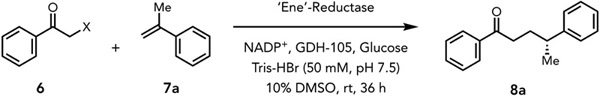
We tested a panel of EREDs on the intermolecular hydroalkylation of α-bromo acetophenone 6a with α-methyl styrene 7a to provide γ-stereogenic ketone 8a (Table 2). GluER-T36A, the best variant for the coupling of chloroamides with alkenes, afforded product in 62% yield with 97:3 er (Table 2, entry 1). Remarkably, wild-type NCR catalyzed the desired transformation in a quantitative yield (using alkene as the limiting reagent) with excellent enantioselectivity (97:3 er) for the (R)-enantiomer, outperforming other tested wild-type EREDs (Table 2, entry 2). Decreasing the catalyst loading to 1 mol % afforded decreased yields but with no loss in enantioselectivity (Table 2, entry 3). Interestingly, irradiation with a blue kessil lamp did not afford an increase in yield (Table 2, entry 4). This result indicates that a photoexcited electron transfer mechanism is not available to this substrate/enzyme combination. The (S)-enantiomer of 8a can also be accessed using Yersina bercovieri alkene reductase (YersER) as the biocatalyst, albeit in moderate yield and enantioselectivity (44% yield, 83:17 er) (Table 2, entry 5). In all cases, <15% of hydrodehalogenated product was observed, suggesting a mechanism for gating radical formation. After optimizing the experimental parameters, wild-type NCR (1.5 mol% loading) catalyzed the non-natural intermolecular hydroalkylation providing the (R)-8a in 99% yield (82% isolated yield) and excellent enantioselectivity (99:1 er) (Table 2, entry 7). Control experiments confirmed that both NCR and the NADPH regeneration system are necessary for this biotransformation (Supplemental Table 12). Interestingly, α-chloroacetophenone is an effective substrate under these reaction conditions (49% yield, 97:3 er), despite being less reducing by 600 mV (−1.44 V vs. SCE),23 indicating that photoinduced electron transfer is not required for radical initiation (Table 2, entry 6).29
Table 2.
Screening an initial panel of EREDs for intermolecular hydroalkylation.
| |||||
|---|---|---|---|---|---|
| Entry | X | ‘Ene’-Reductases | Loading | Yield (%)a | erb |
| 1 | Br | GluER-T36A | 2 mol% | 62 | 97:3 |
| 2 | Br | NCR | 2 mol% | 99 | 97:3 |
| 3 | Br | NCR | 1 mol% | 61 | 97:3 |
| 4c | Br | NCR | 1 mol% | 64 | 97:3 |
| 5 | Br | YersER | 1 mol% | 44 | 17:83 |
| 6 | Cl | NCR | 2 mol% | 49 | 97:3 |
| 7d | Br | NCR | 1.5 mol% | 99 (82e) | 99:1 |
Reaction conditions: α-haloacetophenone (6, 0.015 mmol, 3 equvi), α-methylstyrene (7a, 0.005 mmol, 1 equiv), NADP+ (0.2 mg), GDH-105 (0.3 mg), glucose (5 mg) and purified ‘ene’-reductases in 50 mM Tris-HBr buffer pH 7.5, with 10% DMSO as cosolvent, total volume is 500 μL. Reaction mixtures were shaken under anaerobic conditions at room temperature for 36 h.
Yield determined via LCMS relative to an internal standard (TBB).
Enantiomeric ratio (er) determined by HPLC on a chiral stationary phase.
Irradiation with a blue LED.
Total reaction volume is 1200 μL.
Yield of isolated product from a 0.1 mmol-scale reaction.
A variety of α-bromo aryl ketones are well tolerated by the reaction (Scheme 2). α-bromoketones possessing either electron-donating (6b–c) or electron-withdrawing (6d–i) substituents at the para-position were efficiently converted to the desired enantioenriched γ-chiral ketones (8b–i) in yields of 43–98% with excellent enantioselectivity (> 97:3 er). The meta-substituted substrates 6j–k were also well accepted by the wild-type NCR, providing the corresponding products 8j–k with good yields (72–96%) and high levels of enantioselectivity (> 97:3 er). Beyond acetophenone derivatives, simple α-bromo alkyl ketone 6n was also successful in the reaction, giving rise to the product 8n in 63% yield and 95:5 er, highlighting the generality of the ground-state electron transfer mechanism. Unfortunately, α-bromo acetic acid and α-bromo esters were unreactive (Supplemental Figure 4).
Scheme 2.

Asymmetric intermolecular radical hydroalkylation catalyzed by wide-type NCR. Standard reaction conditions: the reaction mixture (1.2 mL) consisted of substituted styrene (7, 0.005 mmol, 1 equiv), α-bromo carbonyl (6, 0.015 mmol, 3 equiv), GDH-105 (0.3 mg), glucose (5.0 mg), NADP+ (0.2 mg), and purified wild-type NCR protein (1.5 mol% to alkene) in buffer (50 mM Tris-HBr buffer pH 7.5) with 10% DMSO as cosolvent at room temperature for 36 h. a Yields were determined via HPLC relative to an internal standard. b Enantiomeric ratio (er) was determined via HPLC on a chiral stationary phase. c An excess of styrene (7, 0.015 mmol, 3 equiv) to α-bromo carbonyl (6, 0.005 mmol, 1 equiv) was used.
The alkene scope is also broad for the NCR-catalyzed intermolecular hydroalkylation (Scheme 2). Various ortho- and meta-substituted α-methyl styrenes were well accepted by the enzyme, providing the corresponding γ-chiral ketones 8o–r in yields of 58%–88% with excellent enantioselectivity (up to 99:1 er, Scheme 2). Para-substituted α-methyl styrenes were less tolerated in the reaction. The smaller para-F substituent (8s, 99% yield, 95:5 er) outperformed larger groups such as para-Me (8t) or para-Cl (8u) substituent, which afforded only moderate yields (59–69%) and poor enantioselectivity (Scheme 2). Beyond styrenyl alkenes, wild-type NCR could also accept heterocycles, including an electron-deficient α-methylvinylpyridine and electron-rich α-methylvinylthiophene, providing the respective products 8w–x in 61–74% yield and high enantioselectivity (up to 99:1 er). This enzyme, however, was limited to small substituents at the α-position of styrene (8y-z). Larger groups, such as i-propyl and n-propyl, were poorly reactive (Scheme 2). We hypothesize that this limitation could be overcome using protein engineering. Finally, aliphatic alkenes, such as α-methylvinylcyclohexane, were not reactive (Scheme 2 and Supplemental Figure 4).
Mechanistic Experiments
To elucidate the mechanism of termination for the benzylic radical in the NCR-catalyzed intra- and intermolecular C─C bond-forming reactions, we conducted a set of deuterium-labeling experiments (Figure 3 and Supplemental Figure 6-11). For the cyclization mediated by NCR-C9, when cofactor flavin was labeled in situ using d1-glucose and glucose dehydrogenase (GDH), the product β-benzyl cyclopentanone 2a was obtained in 83% yield and 95:5 er with 47% deuterium incorporation at the benzylic position (Figure 3A). Alternatively, when the model cyclization reaction was performed in deuterated buffer, the product 2a was observed in high yield and enantioselectivity (93% yield, 95:5 er) with only 11% of benzylic deuterium incorporation (Figure 3A). Collectively, these deuterium-labeling data suggest that the primary mechanism of quenching the benzylic radical would be HAT from flavin (FMNsq), as proposed in Supplemental Figure 5A. In the experiment where in situ deuterium-labeled flavin was used, the moderate deuterium incorporation (47 D%) of product 2a can be rationalized by deuterium atom transfer being competitive with the deuterium exchange at the flavin N5 position with H2O buffer, suggesting a possible kinetic isotope effect in the radical termination event (Supplemental Figure 12).31
Figure 3.

Deuterium labeling experiments. A. Deuterium labeling experiments for the cyclization mediated by NCR-C9. B. Deuterium labeling experiments for the intermolecular hydroalkylation catalyzed by wide-type NCR. C. α-methylstyrene (7a) affects the depletion of α-bromoacetophenone (6a) in NCR-catalyzed reaction. For experimental details, see Supplemental Figure 6-15.
For the intermolecular hydroalkylation catalyzed by wide-type NCR, when flavin was labeled in situ using d1-glucose and GDH, the γ-chiral ketone product 8a was formed in 42% yield with 99:1 er and 80% deuterium incorporation at the benzylic position (Figure 3B). Additionally, when this reaction was carried out in a deuterated buffer, the yield of product 8a improved to 94% (99:1 er), but hardly any deuterium incorporation at the benzylic position of 8a was observed (Figure 3B). Collectively, these results suggest the benzylic radical is primarily terminated via HAT from flavin (FMNsq), which also set the γ-stereocenter of the hydroalkylation products (Supplemental Figure 5B).
Finally, we were interested in whether the presence of alkene influenced radical formation. This was determined by measuring the consumption of α-bromo acetophenone 6a with and without α-methylstyrene 7a. In the presence of 3 equivalents of 7a, 6a was consumed at 7.6 mmol/hour. In the absence of alkene, that initial rate decreased to 3.0 mmol/hour, which was a 2.5-fold decrease to the initial rate (Figure 3C and Supplemental Figure 13-15). These experiments indicate that acetophenone consumption is enhanced by the presence of α-methylstyrene, empirically suggesting a gating mechanism. We hypothesize that an interaction between the π-bond of the alkene and the σ*C─Br could be responsible for the rate enhancement. Electron transfer in these reactions is understood to be endergonic and reversible. The first irreversible step is mesolytic cleavage of the ketyl radical to form the α-acyl radical. A hyperconjugative interaction between the alkene and either α-bromoacetophenone or corresponding radical anion would weaken the C─Br bond, making the mesolytic cleavage more facile. Alternatively, the proposed interaction could decrease the reduction potential of α-haloacetophenone by polarizing the C─X bond causing a buildup of positive charge on the α-carbon. This could potentially account for the ground state reactivity observed with α-chloroacetophenone despite its lower reduction potential.
Conclusions
In conclusion, we have discovered that the flavin-dependent enzyme NCR is capable of catalyzing C─C bond-forming radical cyclization and that the activity and selectivity of the reaction can be significantly improved using directed evolution. The engineered quadruple mutant NCR-C9 shows broad substrate scope and excellent chemo- and stereoselectivity in the 5-exo-trig cyclization reaction, allowing the enantioselective synthesis of a variety of β-chiral cyclopentanones (Scheme 1). Moreover, we also developed the challenging asymmetric intermolecular radical C─C bond-forming reactions using wild-type NCR as the biocatalyst, offering a valuable option for efficient preparation of γ-chiral ketones with high optical purity (Scheme 2). While the β-chiral center of cyclopentanone product was constructed by the C─C bond-forming step in the cyclization catalyzed by NCR-C9, the γ-stereocenter of intermolecular hydroalkylation products produced by wild-type NCR was set via a stereoselective HAT process, thus demonstrating the versatile roles of enzymes with unparalleled capability in controlling stereoselectivity (Supplemental Figure 5). Overall, our study showcases how non-natural reactivities of native EREDs can be exploited, which also can be further enhanced by protein engineering, if needed, to expand the biocatalyst toolbox and address the long-standing selectivity challenge in radical chemistry. We foresee EREDs and other flavin-dependent enzymes can be further leveraged to generate new synthetically applicable reactivities; this is particularly true as other fields, such as photocatalysis,16-18,29,32 are increasingly merging with biocatalysis.33,34
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (NIH) National Institute of General Medical Sciences (NIGMS) (R01 GM127703) and the Princeton Catalysis Initiative. HL acknowledges the BioLEC Distinguished Postdoctoral Fellowship Program for support. The authors thank Dr. Yucong Zheng for his insightful discussions and assistance with docking. The authors also thank Josh Turek-Herman for generously providing the NCR single-site library, Jacob DeHovitz for sharing reference compounds, Jingzhe Cao for sharing intermediates, and Simon Charnock (Prozomix) for generously providing their ERED library. Dr. Ivan Keresztes, Anthony M. Condo, and Dr. John Eng are acknowledged for their help for measuring HRMS.
Footnotes
Supporting Information.
Detailed experimental procedures, characterization data (NMR spectra and HPLC traces). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Plesniak MP; Huang H-M; Procter DJ Radical Cascade Reactions Triggered by Single Electron Transfer. Nat. Rev. Chem 2017, 1, 1–16. [Google Scholar]
- (2).Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chemie Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (4).Ishibashi H Controlling the Regiochemistry of Radical Cyclizations. Chem. Rec 2006, 6, 23–31. [DOI] [PubMed] [Google Scholar]
- (5).Liu X; Ye X; Bureš F; Liu H; Jiang Z Controllable Chemoselectivity in Visible-Light Photoredox Catalysis: Four Diverse Aerobic Radical Cascade Reactions. Angew. Chemie Int. Ed 2015, 54, 11443–11447. [DOI] [PubMed] [Google Scholar]
- (6).Proctor RSJ; Colgan AC; Phipps RJ Exploiting Attractive Non-Covalent Interactions for the Enantioselective Catalysis of Reactions Involving Radical Intermediates. Nat. Chem 2020, 12, 990–1004. [DOI] [PubMed] [Google Scholar]
- (7).Sibi MP; Manyem S; Zimmerman J Enantioselective Radical Processes. Chem. Rev 2003, 103, 3263–3295. [DOI] [PubMed] [Google Scholar]
- (8).Jäger CM; Croft AK Anaerobic Radical Enzymes for Biotechnology. ChemBioEng Rev. 2018, 5, 143–162. [Google Scholar]
- (9).Tang MC; Zou Y; Watanabe K; Walsh CT; Tang Y Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Walsh CT; Moore BS Enzymatic Cascade Reactions in Biosynthesis. Angew. Chemie Int. Ed 2019, 58, 6846–6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yokoyama K; Lilla EA C─C Bond Forming Radical SAM Enzymes Involved in the Construction of Carbon Skeletons of Cofactors and Natural Products. Nat. Prod. Rep 2018, 35, 660–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hyster TK Radical Biocatalysis: Using Non-Natural Single Electron Transfer Mechanisms to Access New Enzymatic Functions. Synlett. 2020, 31, 248–254. [Google Scholar]
- (13).Sandoval BA; Hyster TK Emerging Strategies for Expanding the Toolbox of Enzymes in Biocatalysis. Curr. Opin. Chem. Biol 2020, 55, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Toogood HS; Scrutton NS Discovery, Characterization, Engineering, and Applications of Ene-Reductases for Industrial Biocatalysis. ACS Catal. 2018, 8, 3532–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Winkler CK; Faber K; Hall M Biocatalytic Reduction of Activated C═C-Bonds and Beyond: Emerging Trends. Curr. Opin. Chem. Biol 2018, 43, 97–105. [DOI] [PubMed] [Google Scholar]
- (16).Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science. 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Black MJ; Biegasiewicz KF; Meichan AJ; Oblinsky DG; Kudisch B; Scholes GD; Hyster TK Asymmetric Redox-Neutral Radical Cyclization Catalysed by Flavin-Dependent ‘Ene’-Reductases. Nat. Chem 2020, 12, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Clayman PD; Hyster TK Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization. J. Am. Chem. Soc 2020, 142, 15673–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ye Y; Fu H; Hyster TK Activation Modes in Biocatalytic Radical Cyclization Reactions. J. Ind. Microbiol. Biotechnol 2021. 10.1093/jimb/kuab021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Crisenza GEM; Mazzarella D; Melchiorre P Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Stewart RC; Massey V Potentiometric Studies of Native and Flavin-Substituted Old Yellow Enzyme. J. Biol. Chem 1985, 260, 13639–13647. [PubMed] [Google Scholar]
- (22).Sandoval BA; Meichan AJ; Hyster TK Enantioselective Hydrogen Atom Transfer: Discovery of Catalytic Promiscuity in Flavin-Dependent ‘Ene’-Reductases. J. Am. Chem. Soc 2017, 139, 11313–11316. [DOI] [PubMed] [Google Scholar]
- (23).Tanner DD; Singh HK Reduction of α-Halo Ketones by Organotin Hydrides. An Electron-Transfer-Hydrogen Atom Abstraction Mechanism. J. Org. Chem 1986, 51, 5182–5186. [Google Scholar]
- (24).Jung J; Kim J; Park G; You Y; Cho EJ Selective Debromination and α-Hydroxylation of α-Bromo Ketones Using Hantzsch Esters as Photoreductants. Adv. Synth. Catal 2016, 358, 74–80. [Google Scholar]
- (25).Tucker JW; Narayanam JMR; Krabbe SW; Stephenson CRJ Electron Transfer Photoredox Catalysis: Intramolecular Radical Addition to Indoles and Pyrroles. Org. Lett 2010, 12, 368–371. [DOI] [PubMed] [Google Scholar]
- (26).Maji T; Karmakar A; Reiser O Visible-Light Photoredox Catalysis: Dehalogenation of Vicinal Dibromo-, α-Halo-, and α,α-Dibromocarbonyl Compounds. J. Org. Chem 2011, 76, 736–739. [DOI] [PubMed] [Google Scholar]
- (27).Curran DP; Chang C. tai. Atom Transfer Cyclization Reactions of α-Iodo Esters, Ketones, and Malonates: Examples of Selective 5-Exo, 6-Endo, 6-Exo, and 7-Endo Ring Closures. J. Org. Chem 1989, 54 (13), 3140–3157. [Google Scholar]
- (28).Reich S; Hoeffken HW; Rosche B; Nestl BM; Hauer B Crystal Structure Determination and Mutagenesis Analysis of the Ene Reductase NCR. ChemBioChem 2012, 13, 2400–2407. [DOI] [PubMed] [Google Scholar]
- (29).Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H Photoenzymatic Enantioselective Intermolecular Radical Hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]
- (30).Page CG; Cooper SJ; DeHovitz JS; Oblinsky DG; Biegasiewicz KF; Antropow AH; Armbrust KW; Ellis JM; Hamann LG; Horn EJ; Oberg KM; Scholes GD; Hyster TK Quaternary Charge-Transfer Complex Enables Photoenzymatic Intermolecular Hydroalkylation of Olefins. J. Am. Chem. Soc 2021, 143, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sandoval BA; Kurtoic SI; Chung MM; Biegasiewicz KF; Hyster TK Photoenzymatic Catalysis Enables Radical-Mediated Ketone Reduction in Ene-Reductases. Angew. Chemie Int. Ed 2019, 58, 8714–8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sandoval BA; Clayman PD; Oblinsky DG; Oh S; Nakano Y; Bird M; Scholes GD; Hyster TK Photoenzymatic Reductions Enabled by Direct Excitation of Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc 2021, 143, 1735–1739. [DOI] [PubMed] [Google Scholar]
- (33).Rudroff F; Mihovilovic MD; Gröger H; Snajdrova R; Iding H; Bornscheuer UT Opportunities and Challenges for Combining Chemo- and Biocatalysis. Nat. Catal 2018, 1, 12–22. [Google Scholar]
- (34).Schmermund L; Jurkaš V; Özgen FF; Barone GD; Büchsenschütz HC; Winkler CK; Schmidt S; Kourist R; Kroutil W Photo-Biocatalysis: Biotransformations in the Presence of Light. ACS Catal. 2019, 9, 4115–4144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.