

Abstract
In the present work, a concise library of 1,3,5-triaryl-2-pyrazolines (2a–2q) was designed and synthesized by employing a multistep strategy, and the newly synthesized compounds were screened for their urease and α-glucosidase inhibitory activities. The compounds (2a–2q) were characterized using a combination of several spectroscopic techniques including FT-IR, 1H NMR, 13C NMR, and EI-MS. All the synthesized compounds, except compound 2i, were potent against urease as compared to the standard inhibitor thiourea (IC50 = 21.37 ± 0.26 μM). These analogs disclosed varying degrees of urease inhibitory activities ranging from 9.13 ± 0.25 to 18.42 ± 0.42 μM. Compounds 2b, 2g, 2m, and 2q having IC50 values of 9.36 ± 0.27, 9.13 ± 0.25, 9.18 ± 0.35, and 9.35 ± 0.35 μM, respectively, showed excellent inhibitory activity as compared to standard thiourea (IC50 = 21.37 ± 0.26 μM). A kinetic study of compound 2g revealed that compound 2g inhibited urease in a competitive mode. Among the synthesized pyrazolines, the compounds 2c, 2k, 2m, and 2o exhibited excellent α-glucosidase inhibitory activity with the lowest IC50 values of 212.52 ± 1.31, 237.26 ± 1.28, 138.35 ± 1.32, and 114.57 ± 1.35 μM, respectively, as compared to the standard acarbose (IC50 = 375.82 ± 1.76 μM). The compounds (2a–2q) showed α-glucosidase IC50 values in the range of 114.57 ± 1.35 to 462.94 ± 1.23 μM. Structure–activity relationship revealed that the size and electron-donating or -withdrawing effects of substituents influenced the activities, which led to the urease and α-glucosidase inhibiting properties. Compound 2m was a dual potent inhibitor against urease and α-glucosidase due to the presence of 2-CF3 electron-withdrawing functionality on the phenyl ring. To the best of our knowledge, these synthetic compounds were found to be the most potent dual inhibitors of urease and α-glucosidase with minimum IC50 values. The cytotoxicity of the compounds (2a–2q) was also investigated against human cell lines MCF-7 and HeLa. Compound 2l showed moderate cytotoxic activity against MCF-7 and HeLa cell lines. Moreover, in silico studies on most active compounds were also performed to understand the binding interaction of most active compounds with active sites of urease and α-glucosidase enzymes. Some compounds exhibited drug-like characteristics due to their lower cytotoxic and good ADME profiles.
1. Introduction
According to the contemporary research of drug discovery, enzyme inhibition plays an important role in this field. Urease and α-glucosidase are two imperative enzymes that are meticulously concomitant with various clinical fields.1,2 Urease (urea amidohydrolase, EC 3.5.1.5, nickel-based) comes from the superfamily of phosphotriesterase and amidohydrolase that initiates the hydrolysis of urea, eventually leading to their conversion into ammonia and carbon dioxide. The urease enzyme is widely spread from prokaryotes to eukaryotes. The urease enzyme’s hyperactivity releases an excessive amount of ammonia causing alkalinity in the stomach, consequently, enhancing the permeability of the gastric mucosa.3−10 Although humans lack the urease enzymes, urea is synthesized as the end product of protein metabolism in them that is usually eliminated from the body in the form of urine.11−13 Action of urease enzymes in cattle and many other animals also facilitates monitoring and regulating their nitrogen metabolism.14,15 Different pathogenic conditions can arise due to the elevated levels of these enzymes, especially, they assist in the flourishing of various bacterial infections, leading to some grave adverse effects.16 The growth of Helicobacter pylori (HP) is eased via the assistance of low pH of the stomach in humans, eventually causing the peptic and gastric ulcers, finally leading to cancer.17−21 Moreover, various metabolic dysregulations can arise due to the elevated level of ammonia abolishing the gastrointestinal tract (GIT) epithelium.22,23 Furthermore, kidney stone formation is also instigated via a high urease level.24
Excessive urease activity is generated in urea fertilization leading to intense environmental pollution ultimately causing major economic losses.25−30 This also leads to plant mutilation by controlling the intake of necessary nutrients, enhancing the soil pH and secondary ammonia toxicity.31,32 Thus, it is pivotal to distinguish new urease inhibitors with enhanced stability, prominent bioavailability, increased selectivity, extraordinary potency, and least toxicity.33−37
Diabetes mellitus (DM) is a prolonged metabolic disorder caused by hyperglycemia.38 Diabetes mellitus metabolic disease gives rise to solemn health disorders, for instance cardiovascular disorders, nephropathy, retinopathy, amputations, neuropathy, cancer, and reduced ability of wound healing. The control of glycemia is an illustrious method to cure diabetes mellitus.39−43 α-Glucosidase is a significant enzyme present on the epithelial wall of the small intestine that is involved in carbohydrate metabolism, and the inhibition of this enzyme lowers the glucose level in the blood facilitating the treatment of DM.44,45 Many detrimental effects such as gastrointestinal diseases, flatulence, and diarrhea are associated with typical available clinical drugs such as voglibose, miglitol, and acarbose.46,47 The inhibition of α-glucosidase via these inhibitors lowers the absorption of glucose that is released from the pancreas and salivary glands for the conversion of oligosaccharides into simple sugar via hydrolysis, consequently, lowering the postprandial blood glucose level, which is a beneficial approach to cure diabetes.48,49
Subsequently, because of the use of pyrazoline in the organic field due to its considerable bioavailability, it has always been interesting to design facile, effective, and eco-friendly methodologies for the synthesis of pyrazoline.50,51
Generally, a combination therapy is recommended for patients suffering from diabetes mellitus and peptic ulcer simultaneously. As diabetic patients show slow healing ability, peptic ulcer can cause various health problems tallying the damage of diabetes mellitus itself. Therefore, the combined urease and α-glucosidase inhibition could be an ideal therapeutic approach for patients who are suffering from both DM and peptic ulcer simultaneously (Figure 1).52−54
Figure 1.

Some identified representative motifs already reported in the literature.
Despite the important efforts made in the research, the management of malignancies in humans is still a major challenge in contemporary medicinal chemistry. Unfortunately, the significant toxicity exerted by most of the anticancer agents and the development of cellular drug resistance remain the primary hurdle for effective chemotherapy.55,56 Cancer is the second most fatal disease liable for ∼21% annual deaths globally.57 The malignant neoplastic disease is mainly caused by uncontrolled cell growth, metastasis, and invasion. Cancer cells proliferate quickly and their inhibition is most vital within the treatment of cancer exploiting metastatic tumor compounds.58 The difficulties in the diagnosis of the disease at the earliest stages, slender therapeutic indices of therapeutic agents, and, therefore, the development of multidrug resistance lead to a number of key obstacles, making cancer treatment complex and causing a high death rate worldwide.59,60 On the other hand, pyrazole-containing compounds have received considerable attention owing to their diverse chemotherapeutic potential including versatile antineoplastic activities. In addition, it was found that the introduction of a pyrazole nucleus between two aryl rings of chalcones played an integral role in increasing the cytotoxic potential.61
Recently, pyrazoline has been conferred as one of the most valuable heterocyclic scaffolds in medicinal chemistry on account of its wide spectrum in clinical drugs.54 It has been emerged as the most privileged source to design various synthons for drug synthesis (Figure 2). This class contains structure-targeting compounds in different fields such as synthetic and medicinal chemistry exhibiting a good affinity for various biotargets of heterocyclic compounds of this class.62−64 These scaffolds were utilized in the advancement of drug research and agricultural products.65 The pyrazolinic scaffolds also show a variety of applications in pharmacology due to their exceptional antioxidant, anti-inflammatory, antifungal, analgesic, antipyretic, antibacterial, antiangiogenic, antiviral, and antitumoral activities.66−68
Figure 2.
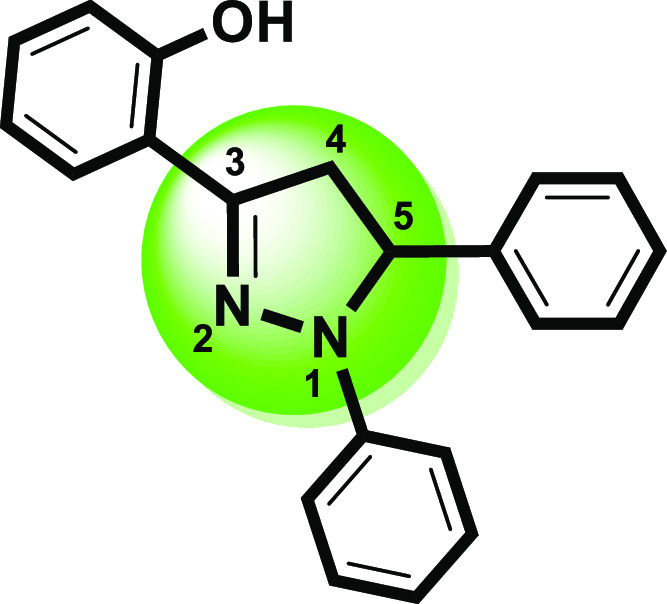
Structural framework of a versatile heterocycle (1,3,5-triaryl-2-pyrazoline).
Presently, our research group focusing on the designing of facile synthetic approaches of small compounds and exploring their potential biological activity.69−76 In recent years, pyrazolinic compounds have been drawing massive attention of our research group due to their remarkable inhibitory effects against urease and α-glucosidase enzymes.
Encouraged by the biological significance of pyrazoline structural motifs in the medicinal chemistry, herein, we report the synthesis, characterization, and biological evaluation of new pyrazoline derivatives as dual inhibitors of urease and α-glucosidase enzymes. The synthesized pyrazoline compounds with different substitution patterns were evaluated for their urease and α-glucosidase inhibitory activities, and these analogs were assessed, remarkably, to be the potent heterocyclic class among the already reported urease inhibitors. Furthermore, the synthesized compounds were investigated using cytotoxic and molecular modeling studies.
2. Results and Discussion
2.1. Chemistry
The title analogs (2a–2q) were synthesized from easily available and simple precursors as shown in Scheme 1. The first step involves a base-catalyzed condensation of 2′-hydroxyacetophenone with substituted aryl aldehydes in methanol to produce substituted 2′-hydroxychalcones (1a–1q). These intermediate compounds (1a–1q) were characterized by FTIR and NMR spectroscopy and subjected to the next step, which involves the cycloaddition of these intermediate 2′-hydroxychalcones (1a–1q) with phenylhydrazine hydrochloride in the presence of glacial acetic acid to furnish the target compounds (2a–2q) in good to excellent yields. The desired compounds (2a–2q) were purified through recrystallization by ethanol. The structures of novel pyrazoline analogs were elucidated using common spectral techniques such as FTIR, UV–Vis, NMR, and mass spectrometry. For example, the IR spectra of the pyrazoline derivatives depicted the absorption bands around 3357 for the OH group, 3090 for aromatic (C–H), 1592 for (C=N), and 1500 for (C=C). Similarly, the 1H NMR spectra of these compounds revealed a characteristic singlet signal around δ 12.00 ppm for the OH group. The aromatic protons were observed as multiplets in the region of δ 6.64–7.92 ppm. The presence of other protons in the pyrazoline moiety was observed as a doublet of doublets in the region of δ 2.98–3.04, 3.79–3.86, and 5.37–5.42 ppm corresponding to three protons in HA, HB, and HX patterns, respectively. Moreover, the molecular masses of all the newly synthesized compounds were corroborated by EI-MS, thus all the spectral data unequivocally confirm the proposed structures of the target 1,3,5-triaryl-2-pyrazolines.
Scheme 1. Synthesis of Chalcones (1a–1q) and Pyrazolines (2a–2q).


The spectral data of previously reported 2′-hydroxychalcones (1a–1q) are given in the following reference articles 1a, 1e, 1g, 1j,821b,831c,841d,851f,861h,871i,881k,891l,901m, 1n, 1o(91), 1p(92) and 1q.93 The spectral data of all the synthesized pyrazolines (2a–2q) are given here:
2.1.1. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-flouro-phenyl)-2-pyrazoline (2a)
Colorless solid; mp 145–147 °C; yield: 80%; UV λmax (MeOH) = 260 nm; FTIR (cm–1): 3284 (OH), 3012 (C–H), 1548 (C=N), 1493 (C=C), 1245 (C–N), 840 (C–F); 1H NMR (500 MHz, CDCl3): δ 10.23 (br s, 1H, OH), 7.98 (d, J = 10.0 Hz, 2H, Ar–H), 7.72 (d, J = 10.0 Hz, 2H, Ar–H), 7.10–7.01 (m, 3H, Ar–H), 6.91–6.80 (m, 6H, Ar–H), 5.10 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.91 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.50 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 168.4, 164.1, 163.6, 162.8, 146.7, 144.0, 138.0, 129.8, 129.0, 128.5, 127.7, 127.5, 127.1, 126.8, 122.3, 120.7, 118.0, 117.0, 116.7, 116.1, 115.8, 115.3, 66.0, 44.6; accurate mass (EI-MS) of [M]+: calcd for C21H17FN2O, 332.1325; found, 332.1316.
2.1.2. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-tertbutyl-phenyl)-2-pyrazoline (2b)
Dirty-white crystalline solid; mp 134–136 °C; yield: 75%; UV λmax (MeOH) = 345 nm; FTIR (cm–1): 3287 (O–H), 3021 (C–H), 1554 (C=N), 1475 (C=C), 1375 (C–H tertbutyl), 1285 (C–N); 1H NMR (500 MHz, CDCl3): δ 13.08 (br s, 1H, OH), 7.42 (d, J = 10.0 Hz, 2H, Ar–H), 7.22 (d, J = 10.0 Hz, 2H, Ar–H), 7.10–7.01 (m, 4H, Ar–H), 6.85–6.76 (m, 5H, Ar–H), 5.22 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.93 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.55 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 2.50 (d, J = 10.0 Hz, 2H, −CH2CH(Me)2), 1.87 (m, 1H, −CH2CH(Me)2), 0.93 (d, J = 10.0 Hz, 6H, −CH2CH(Me)2); 13C NMR (126 MHz, CDCl3): δ 169.0, 166.8, 148.4, 144.4, 137.6, 133.8, 132.4, 129.6, 128.7, 128.2, 127.6, 126.3, 119.7, 115.9, 114.6, 112.2, 65.3, 45.2, 43.8, 30.1, 22.7; accurate mass (EI-MS) of [M]+: calcd for C25H26N2O, 370.2045; found, 370.2035.
2.1.3. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-bromo-phenyl)-2-pyrazoline (2c)
Colorless crystalline solid; mp 149–151 °C; yield: 78%; UV λmax (MeOH) = 322 nm; FTIR (cm–1): 3295 (OH), 3015 (C–H), 1584 (C=N), 1480 (C=C), 1280 (C–N), 640 (C–F); 1H NMR (500 MHz, CDCl3): δ 13.02 (br s, 1H, OH), 7.98 (d, J = 10.0 Hz, 2H, Ar–H), 7.72 (d, J = 10.0 Hz, 2H, Ar–H), 7.10–7.01 (m, 3H, Ar–H), 6.91–6.80 (m, 6H, Ar–H), 5.10 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.91 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.52 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 167.6, 162.0, 146.8, 144.5, 133.1, 132.4, 129.7, 128.7, 128.5, 127.4, 125.0, 119.7, 118.5, 115.7, 114.2, 67.3, 45.5; accurate mass (EI-MS) of [M]+: calcd for C21H1779BrN2O, 392.0524; found, 392.0510.
2.1.4. 1-Phenyl-3(2-hydroxy-phenyl)-5-(3, 4, 5-trimethoxyphenyl)-2-pyrazoline (2d)
Light yellow crystalline solid; mp 142–144 °C; yield: 50%; UV λmax (MeOH) = 324 nm; FTIR (cm–1): 3295 (OH), 3015 (C–H), 1584 (C=N), 1480 (C=C), 1295 (C–N), 833 (OCH3); 1H NMR (500 MHz, CDCl3): δ 12.99 (br s, 1H, OH), 7.88 (d, J = 5.0 Hz, 2H, Ar–H), 7.70–7.64 (m, 2H, Ar–H), 7.27–7.28 (m, 3H, Ar–H), 7.00–6.90 (m, 4H, Ar–H), 5.09 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.90 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.50 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.83 (s, 3H, −OCH3), 3.81 (s, 6H, −OCH3); 13C NMR (126 MHz, CDCl3): δ 167.5, 164.8, 162.5, 147.7, 143.8, 136.6, 134.2, 129.1, 128.3, 127.8, 126.1, 123.8, 121.7, 118.4, 116.7, 114.3, 67.4, 56.7, 55.6, 44.1; accurate mass (EI-MS) of [M]+: calcd for C24H24N2O4, 404.1736; found, 404.1740.
2.1.5. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-methoxyphenyl)-2-pyrazoline (2e)
Light yellow solid; mp 157–159 °C; yield: 73%; UV λmax (MeOH) = 278 nm; FTIR (cm–1): 3485 (OH), 3040 (C–H), 1575 (C=N), 1487 (C=C), 1291 (C–N), 855 (OCH3); 1H NMR (500 MHz, CDCl3): δ 7.62 (d, J = 10.0 Hz, 2H, Ar–H), 7.32 (d, J = 10.0 Hz, 2H, Ar–H), 7.05–6.99 (m, 4H, Ar–H), 6.95–6.72 (m, 5H, Ar–H), 5.11 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.93 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.53 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.80 (s, 3H, OMe); 13C NMR (126 MHz, CDCl3): δ 166.7, 162.5, 144.7, 137.5, 132.6, 129.2, 128.8, 127.8, 127.5, 126.5, 121.4, 117.6, 114.3, 65.0, 55.6, 43.8; accurate mass (EI-MS) of [M]+: calcd for C22H20N2O2, 344.1525; found, 344.1517.
2.1.6. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-N,N-dimethylphenyl)-2-pyrazoline (2f)
Light yellow crystalline solid; mp 149–151 °C; yield: 55%; UV λmax (MeOH) = 352 nm; FTIR (cm–1): 3431 (OH); 1527 (C=N), 1387 (C–N), 750 (N(CH3)2); 1H NMR (500 MHz, CDCl3): δ 11.98 (br s, 1H, OH), 7.82 (d, J = 10.0 Hz, 2H, Ar–H), 7.10 (d, J = 10.0 Hz, 2H, Ar–H), 7.05–6.98 (m, 3H, Ar–H), 7.05–6.98 (m, 3H, Ar–H), 6.87–6.80 (m, 3H, Ar–H), 5.14 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.95 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.80 (s, 6H, −N(CH3)2), 3.56 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 166.7, 161.8, 147.7, 145.0, 139.0, 136.8, 132.8, 129.6, 128.5, 127.3, 126.1, 124.8, 121.7, 118.5, 116.8, 114.3, 64.1, 55.8, 45.1; accurate mass (EI-MS) of [M]+: calcd for C23H23N3O, 357.1841; found, 357.1830.
2.1.7. 1-Phenyl-3(2-hydroxy-phenyl)-5-(3,4-dimethoxyphenyl)-2-pyrazoline (2g)
White solid; mp 125–127 °C; yield: 76%; UV λmax (MeOH) = 259 nm; FTIR (cm–1): 3340 (OH), 2980 (C–H), 1595 (C=N), 1580 (C=C), 876 (OCH3); 1H NMR (500 MHz, CDCl3): δ 10.85 (br s, 1H, OH), 7.89 (dd, J = 10.0, 5.0 Hz, 1H, Ar–H), 7.78 (d, J = 5.0 Hz, 1H, Ar–H), 7.20 (dd, J = 10.0, 5.0 Hz, 1H, Ar–H), 6.95–6.90 (m, 4H, Ar–H), 6.80–6.68 (m, 5H, Ar–H), 5.10 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.94 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.86 (s, 3H, −OCH3), 3.82 (s, 3H, −OCH3), 3.55 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 167.3, 162.6, 146.4, 143.2, 137.3 130.0, 128.8, 127.7, 126.6, 124.8, 122.0, 117.3, 115.1, 114.5, 113.3, 68.0, 55.6, 55.0, 45.5; accurate mass (EI-MS) of [M]+: calcd for C23H22N2O3, 374.1630; found, 374.1622.
2.1.8. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-carboxyphenyl)-2-pyrazoline (2h)
Light yellow crystalline solid; mp 129–131 °C; yield: 76%; UV λmax (MeOH) = 374 nm; FTIR (cm–1): 3500 (COOH), 3284 (OH), 3010 (C–H), 1548 (C=N), 1493 (C=C), 1245 (C–N); 1H NMR (500 MHz, CDCl3): δ 11.6 (br s, 1H, OH), 8.89 (s, 1H, −COOH), 8.15 (d, J = 10.0 Hz, 2H, Ar–H), 7.80 (d, J = 10.0 Hz, 2H, Ar–H), 7.16–7.07 (m, 3H, Ar–H), 6.98–6.90 (m, 4H, Ar–H), 6.78–6.70 (m, 2H, Ar–H) 5.16 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.94 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.53 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 172.8, 168.1, 162.3, 159.5, 147.0, 142.5, 136.7, 129.2, 128.7, 127.4, 126.5, 124.0, 121.6, 117.3, 116.8, 114.3, 68.5, 47.6; accurate mass (EI-MS) of [M]+: calcd for C22H18N2O3, 358.1317; found, 358.1306.
2.1.9. 1-Phenyl-3(2-hydroxy-phenyl)-5-thiophene-2-pyrazoline (2i)
Light brown needles; mp 145–147 °C; yield: 81%; UV λmax (MeOH) = 300 nm; FTIR (cm–1): 3443.86 (OH); 3012 (CH), 1548 (C=N), 1493 (C=C), 1245 (C–N); 1H NMR (500 MHz, CDCl3): δ 12.88 (br s, 1H, OH), 7.98–7.90 (m, 2H, Ar–H), 7.62–7.55 (m, 2H, Ar–H), 7.21–7.13 (m, 2H, Ar–H), 7.11–6.99 (m, 6H, Ar–H), 5.11 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.95 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.50 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 168.4, 163.8, 159.8, 148.7, 142.0, 136.0, 129.4, 128.6, 127.8, 122.5, 117.5, 116.8, 114.7, 113.2, 67.8, 42.6; accurate mass (EI-MS) of [M]+: calcd for C19H16N2OS, 320.0983; found, 320.0970.
2.1.10. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-acetamidophenyl)-2-pyrazoline (2j)
Light brown crystals; mp 135 °C; yield: 45%; UV λmax (MeOH) = 380 nm; FTIR (cm–1): 3456 (OH), 3006 (CH), 1750 (C=O amide); 1535 (C=N), 1483 (C=C), 1230 (C–N); 1H NMR (500 MHz, CDCl3): δ 12.2 (br s, 1H, OH), 10.53 (br s, 1H, NHCOCH3), 8.80 (d, J = 10.0 Hz, 2H, Ar–H), 8.00 (d, J = 10.0 Hz, 2H, Ar–H), 7.81–7.70 (m, 2H, Ar–H), 7.52–7.47 (m, 2H, Ar–H), 7.08–6.97 (m, 5H, Ar–H), 5.12 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.94 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.53 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 2.18 (s, 3H, COCH3); 13C NMR (126 MHz, CDCl3): δ 171.3, 167.6, 162.6, 159.7, 147.5, 143.8, 133.4, 129.6, 128.7, 126.1, 121.7, 117.5, 116.3, 115.8, 114.3, 67.0, 47.6, 25.7; accurate mass (EI-MS) of [M]+: calcd for C22H19N3O2, 357.1477; found, 357.1480.
2.1.11. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-N,N diethyl phenyl)-2-pyrazoline (2k)
Pale yellow crystalline solid; mp 149–151 °C; yield: 65%; UV λmax = 345 nm; FTIR (cm–1): 3400 (OH), 3017 (C–H), 1530 (C=N), 1489 (C=C), 1235 (C–N); 1H NMR (500 MHz, CDCl3): δ 13.80 (s, 1H, OH), 8.10 (d, J = 10.0 Hz, 2H, Ar–H), 7.97 (d, J = 10.0 Hz, 2H, Ar–H), 7.74–7.63 (m, 3H, Ar–H), 7.46–7.40 (m, 2H, Ar–H), 7.01–6.95 (m, 4H, Ar–H), 5.15 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.95 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.52 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.40 (m, 4H, −CH2CH3), 1.16, (t, J = 10.0 Hz, 6H, −CH2CH3); 13C NMR (126 MHz, CDCl3): δ 166.8, 161.6, 147.8, 139.7, 138.6, 129.1, 128.5, 128.3, 127.2, 126.4, 123.8, 122.0, 118.6, 117.5, 116.8, 116.5, 113.5, 67.1, 48.1, 25.4, 20.6; accurate mass (EI-MS) of [M]+: calcd for C25H27N3O, 385.2154; found, 385.2141.
2.1.12. 1-Phenyl-3(2-hydroxy-phenyl)-5-phenyl-2-pyrazoline (2l)
Yellow solid; mp 107–109 °C; yield: 78%; UV λmax (MeOH) = 324 nm; FTIR (cm–1): 3330 (OH), 3006 (C–H), 1535 (C=N), 1483 (C=C), 1230 (C–N); 1H NMR (500 MHz, CDCl3): δ 11.10 (br s, 1H, OH), 8.10–7.80 (m, 6H, Ar–H), 7.42–7.26 (m, 5H, Ar–H), 7.05–6.96 (m, 3H, Ar–H), 5.08 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.90 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.48 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 166.8, 162.2, 146.8, 144.6, 137.9, 129.7, 128.6, 127.8, 127.2, 126.3, 117.4, 116.7, 115.4, 114.8, 113.3, 68.3, 49.0; accurate mass (EI-MS) of [M]+: calcd for C21H18N2O, 314.1419; found, 314.1423.
2.1.13. 1-Phenyl-3(2-hydroxy-phenyl)-5-(2-triflouromethyl-phenyl)-2-pyrazoline (2m)
Colorless crystalline solid; mp 141–143 °C; yield: 63%; UV λmax (MeOH) = 370 nm; FTIR (cm–1): 3456.56 (OH), 3019 (C–H), 1550 (C=N), 1454 (C=C), 1276 (C–N), 750 (C–F); 1H NMR (500 MHz, CDCl3): δ 12.51 (br s, 1H, OH), 8.72–8.55 (m, 2H, Ar–H), 7.85–7.74 (m, 2H, Ar–H), 7.64–7.50 (m, 4H, Ar–H), 7.32–7.22 (m, 2H, Ar–H), 7.10–6.98 (m, 3H, Ar–H), 5.16 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.96 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.56 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 168.0, 164.1, 163.7, 162.4, 146.5, 143.8, 139.6, 137.5, 129.3, 128.6, 127.4, 127.6, 127.3, 126.5, 122.6 (q, J = 270.8 Hz, CF3), 120.6, 118.6, 117.8, 116.3, 115.4, 113.3, 68.8; accurate mass (EI-MS) of [M]+: calcd for C22H17F3N2O, 382.1293; found, 382.1285.
2.1.14. 1-Phenyl-3(2-hydroxy-phenyl)-5-(3-triflouromethyl phenyl)-2-pyrazoline (2n)
Colorless crystalline solid; mp 145–147 °C; yield: 70%; UV λmax (MeOH) = 370 nm; FTIR (cm–1): 3456 (OH), 3019 (C–H), 1550 (C=N), 1454 (C=C), 1276 (C–N), 780 (C–F); 1H NMR (500 MHz, CDCl3): δ 12.61 (br s, 1H, OH), 8.55 (s, 1H, Ar–H), 8.48 (dd, J = 10.0, 5.0 Hz, 1H, Ar–H), 8.30 (m, 1H, Ar–H), 8.16 (dd, J = 10.0, 5.0 Hz, 1H, Ar–H), 7.99–7.90 (m, 3H, Ar–H), 7.70–7.52 (m, 3H, Ar–H), 7.01–6.91 (m, 3H, Ar–H), 5.17 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.96 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.55 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 169.1, 164.7, 163.5, 157.1, 145.2, 142.8, 138.6, 129.7, 128.4, 127.4, 127.7, 126.8, 127.2, 127.0, 122.8 (q, J = 270.8 Hz, CF3), 118.7, 117.3, 116.2, 115.8, 114.3, 68.0; accurate mass (EI-MS) of [M]+: calcd for C22H17F3N2O, 382.1293; found, 382.1280.
2.1.15. 1-Phenyl-3(2-hydroxy-phenyl)-5-(4-triflouromethyl phenyl)-2-pyrazoline (2o)
Colorless crystalline solid; mp 139–141 °C; yield: 45%; UV λmax (MeOH) = 370 nm; FTIR (cm–1): 3456 (OH), 3019 (C–H), 1550 (C=N), 1454 (C=C), 1276 (C–N), 823 (C–F); 1H NMR (500 MHz, CDCl3): δ 13.10 (br s, 1H, OH), 8.51 (d, J = 10.0 Hz, 2H, Ar–H), 8.21 (d, J = 10.0 Hz, 2H, Ar–H), 8.01–7.85 (m, 5H, Ar–H), 7.31–6.92 (m, 4H, Ar–H), 5.16 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.94 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.53 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 167.8, 164.6, 161.7, 155.6, 146.2, 143.5, 137.8, 129.6, 128.4, 127.7, 127.6, 127.3, 126.8, 122.7 (q, J = 270.8 Hz, CF3), 119.8, 118.7, 116.6, 116.1, 115.0, 114.3, 68.0; accurate mass (EI-MS) of [M]+: calcd for C22H17F3N2O, 382.1293; found, 382.1285.
2.1.16. 1-Phenyl-3(2-hydroxy-phenyl)-5(2,4-dimethyl-phenyl)-2-pyrazoline (2p)
Light yellow crystalline solid; mp 153–155 °C; yield: 77%; UV λmax (MeOH) = 335 nm; FTIR (cm–1): 3378 (OH), 3022 (C–H), 1521 (C=N), 1453 (C=C), 1267 (C–N); 1H NMR (500 MHz, CDCl3): δ 12.54 (br s, 1H, OH), 8.35 (d, J = 5.0 Hz, 1H, Ar–H), 8.10 (dd, J = 10.0, 5.0 Hz, 1H, Ar–H), 7.80–7.72 (m, 4H, Ar–H), 7.52 (d, J = 10.0 Hz, 1H, Ar–H), 7.12–7.01 (m, 3H, Ar–H), 6.98–6.90 (m, 2H, Ar–H), 5.11 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.89 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.47 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 2.40 (s, 3H, −CH3), 2.35 (s, 3H, −CH3); 13C NMR (126 MHz, CDCl3): δ 166.8, 163.8, 158.7, 146.2, 143.6, 137.6, 129.5, 128.6, 127.6, 127.4, 125.2, 121.8, 117.7, 115.1, 112.3, 67.3, 44.8, 22.2, 21.5; accurate mass (EI-MS) of [M]+: calcd for C23H22N2O, 342.1732; found, 342.1721.
2.1.17. 1-Phenyl-3(2-hydroxy-phenyl)-5(4-benzonitrile)-2-pyrazoline (2q)
Yellow crystalline solid; mp 154–156 °C; yield: 80%; UV λmax (MeOH) = 346 nm; FTIR (cm–1): 3406 (OH), 3087 (C–H), 2259 (C=N), 1550 (C=N), 1454 (C=C), 1276 (C–N); 1H NMR (500 MHz, CDCl3): δ 13.01 (br s, 1H, OH), 8.56 (d, J = 10.0 Hz, 2H, Ar–H), 8.28 (d, J = 10.0 Hz, 2H, Ar–H), 8.11–7.98 (m, 3H, Ar–H), 7.42–7.10 (m, 4H, Ar–H), 7.05–6.97 (m, 2H, Ar–H), 5.18 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.97 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic), 3.55 (dd, J = 15.0, 10.0 Hz, 1H, aliphatic); 13C NMR (126 MHz, CDCl3): δ 169.0, 163.4, 161.8, 146.8, 142.5, 129.7, 128.4, 127.6, 127.6, 126.3, 123.7, 119.8, 117.3, 116.7, 115.6, 114.2, 68.8, 45.6; accurate mass (EI-MS) of [M]+: calcd for C22H17N3O, 339.1372; found, 339.1375.
2.2. Biological Activities
2.2.1. Urease Inhibitory Activity
All synthesized pyrazoline analogs (2a–2q) were evaluated for their in vitro urease inhibitory activity. It is noteworthy that except compound 2i, the rest of the derivatives were found to be significant urease inhibitors with IC50 values in the range of 9.13 ± 0.25 to 18.42 ± 0.42 μM as compared to the standard thiourea (IC50 = 21.37 ± 0.26 μM) (Table 1). The varying activities of synthetic derivatives might be due to the varied substituents on aromatic rings. Compounds 2b, 2g, 2m, and 2q having IC50 values of 9.36 ± 0.27, 9.13 ± 0.25, 9.18 ± 0.35, and 9.35 ± 0.35 μM, respectively, showed excellent inhibitory activities due to the isobutyl, (−OCH3)2, −CF3, and −CN groups. Excitingly, analog 2m bearing a −CF3 group on the phenyl ring at the second position exhibited most potent activity having an IC50 value of 9.18 ± 0.35 μM compared to compounds 2n and 2o having −a CF3 group on the phenyl ring at the third and fourth positions having IC50 values of 18.42 ± 0.42 and 12.63 ± 0.56 μM, respectively. All these analogs showed excellent activity as compared to the standard thiourea (IC50 = 21.37 ± 0.26 μM). The substitution pattern of −F and −OCH3 functionalities at the aryl ring in compounds 2a and 2e is the crucial factor of enzyme inhibition of these compounds. The derivatives with electron-donating groups (EDGs) attached to the phenyl ring have a better urease inhibitory activity than the analogs with electron-withdrawing groups (EWGs) attached to phenyl rings (Table 1) and (Figure 3).
Table 1. Urease and α-Glucosidase Inhibitory Activities of Pyrazolines (2a–2q).
| compound no. | urease IC50 ± SEM (μM) | α-glucosidase IC50 ± SEM (μM) |
|---|---|---|
| 2a | 13.54 ± 0.34 | 445.83 ± 1.25 |
| 2b | 9.36 ± 0.27 | 462.94 ± 1.23 |
| 2c | 12.57 ± 0.43 | 212.52 ± 1.31 |
| 2d | 13.28 ± 0.25 | 421.34 ± 1.27 |
| 2e | 15.39 ± 0.38 | 384.73 ± 1.45 |
| 2f | 15.57 ± 0.42 | 407.28 ± 1.24 |
| 2g | 9.13 ± 0.25 | 419.45 ± 1.32 |
| 2h | 11.26 ± 0.51 | NA |
| 2i | 29.51 ± 0.35 | NA |
| 2j | 13.67 ± 0.47 | NA |
| 2k | 13.29 ± 0.25 | 237.26 ± 1.28 |
| 2l | NA*a | 457.82 ± 1.25 |
| 2m | 9.18 ± 0.35 | 138.35 ± 1.32 |
| 2n | 18.42 ± 0.42 | 434.26 ± 1.29 |
| 2o | 12.63 ± 0.56 | 114.57 ± 1.35 |
| 2p | 12.24 ± 0.38 | NA |
| 2q | 9.35 ± 0.35 | NA |
| thiourea (standard) | 21.37 ± 0.26 | |
| acarbose (standard) | 375.82 ± 1.76 |
NA* = no activity.
Figure 3.
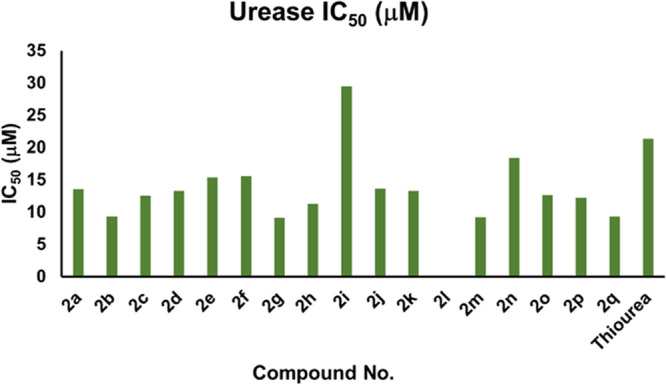
Graphical representation of IC50 values of analogs 2a–2q based on the urease enzyme.
2.2.2. α-Glucosidase Inhibitory Activity
In continuation to our previous studies on α-glucosidase,73c pyrazolines (2a–2q) were assessed for their antidiabetic activity as described in the Method section. All the tested compounds (2a–2q) showed moderate to good inhibition activity against the α-glucosidase enzyme. The table below illustrates the IC50 values of the tested compounds and reference acarbose (IC50 = 375.82 ± 1.76 μM). The compounds (2a–2q) showed IC50 values in the range of 114.57 ± 1.35 to 462.94 ± 1.23 μM (Table 1 and Figure 4). Among the synthesized pyrazolines, the compounds 2c, 2k, 2m, and 2o exhibited excellent α-glucosidase inhibitory activity with the lowest IC50 values of 212.52 ± 1.31, 237.26 ± 1.28, 138.35 ± 1.32, and 114.57 ± 1.35 μM, respectively. Therefore, compounds 2c, 2k, 2m, and 2o bearing −Br, −N(C2H5)2, 2-CF3, and 4-CF3, respectively, could be recommended as potential drug candidates for the treatment of diabetes mellitus in the future.
Figure 4.
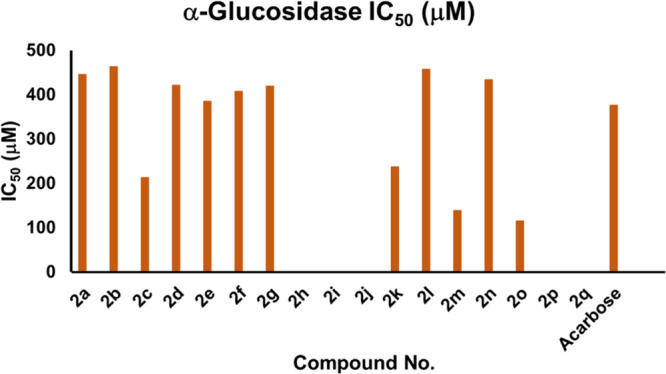
Graphical representation of IC50 values of analogs 2a–2q based on α-glucosidase enzyme.
Overall, it is concluded that among the synthesized pyrazoline series, compound 2m is a dual potent inhibitor against urease and α-glucosidase due to the presence of 2-CF3 electron-withdrawing functionality on the phenyl ring.
2.3. Kinetic Study
The mechanism of inhibition was further examined by a complete kinetic analysis of the most powerful derivative 2g (urease inhibitor) at various concentrations (0–20 μM) and substrates (0.5–4.0 mM). Enzymatic kinetics were used to establish the mode of inhibition and the inhibition constant (Ki). Lineweaver Burk plots used to determine the inhibition mode by measuring the kinetics of most active compound 2g are presented in Figure 5. To calculate Ki values, Lineweaver Burk secondary plots were drawn between the slope of each line and different inhibitor concentrations. To confirm the Ki value of the inhibitor, Dixon plots were constructed between the reciprocal of the rate of reaction and different concentrations of inhibitors. It was determined from kinetic study that 2g was a competitive inhibitor with a Ki value of 19.11 μM, a Km value of 0.93 mM, and a Vmax value of 5.76 μM/min at a 20 μM inhibitor concentration.
Figure 5.

Enzyme kinetics of analog 2g, (A) Lineweaver Burk plot, (B) secondary replot (Lineweaver Burk), and (C) Dixon plot for the urease inhibition activity. Note: Vmax is the maximum velocity of enzymatic activity at a 20 mM inhibitor concentration, Km is the Michaelis–Menten constant at a 20 mM inhibitor concentration, and Ki (μM) is the value calculated from the Dixon plot.
2.4. Structure–Activity Relationship Based on Urease, α-Glucosidase Inhibition Assay
All the synthetic analogs (2a–2q) were investigated for their urease and α-glucosidase inhibitory activities (in vitro). Structure–activity relationship (SAR) studies were carried out purely based on the central core and substitution patterns on the pyrazoline scaffold. Based on the SAR, it can be asserted that the variations observed in urease and α-glucosidase inhibitory activities of 1,3,5-triaryl-2-pyrazoline analogs (2a–2q) were owing to different substituents. Limited SAR was established based on the substitution pattern on the pyrazoline scaffold and are accountable for influencing bioactivities (Figure 6).
Figure 6.

Impact of the core nucleus and variable substituents on SAR of pyrazolines.
In this regard, it is noteworthy that compounds 2c (IC50 = 12.57 ± 0.43 μM for urease and 212.52 ± 1.31 μM for α-glucosidase), 2k (IC50 = 13.29 ± 0.25 μM for urease and 237.26 ± 1.28 μM for α-glucosidase), 2m (IC50 = 9.18 ± 0.35 μM for urease and 138.35 ± 1.32 μM for α-glucosidase), and 2o (IC50 = 12.63 ± 0.56 μM for urease and 114.57 ± 1.35 μM for α-glucosidase) were found to be the most potent dual inhibitors among the members of the series. All these synthetic analogs showed even higher activity than the standards thiourea (IC50 = 21.37 ± 0.26 μM) and acarbose (IC50 = 375.82 ± 1.76 μM). Compounds 2c, 2k, 2m, and 2o have various EDGs and EWGs at o- and p-positions of the aryl ring. The analogs 2m and 2o possess an inductively strong EWG, trifluoromethyl (−CF3) group, at the o-position and the p-position of the phenyl ring. The highest inhibition by 2m and 2o may be attributed to their position and strong interactions with the active pocket of the enzyme. The analog 2c bears an EWG (−Br) at the p-position on ring B. It seems that this atom is also suitably positioned on the ring to foster favorable interaction with the active pocket of the enzyme. The analog 2k has a −N(C2H5)2 group present on the pyrazoline framework and is accountable for enhanced inhibitory activity due to excellent interaction with the active pockets of the enzyme. Almost certainly, this functional group fits effectively into the large active pocket of the enzyme.
Interestingly, the next most potent inhibitor is compound 2g (IC50 = 9.13 ± 0.25 μM for urease) among the remaining analogs of the series. It is also more active than the standards, thiourea and acarbose. This compound 2g has methoxy at position-3 and -4 on ring B of the pyrazoline carbon skeleton and thus was found to be a highly potent urease and glucosidase inhibitor as well. The high inhibition potential might be due to the electronegativity of one methoxy group at 3rd and 4th positions of the phenyl ring, which suitably interacts with the enzyme active site.
Subsequently, the next synthetic analog with a comparable IC50 value is compound 2q (IC50 = 9.35 ± 0.35 μM). This compound also showed a strong inhibitory activity against the urease enzyme than the reference standard inhibitor thiourea. This might be a result of the strong chelation of benzonitrile moiety derivative 2q with the active electrophilic center of the urease enzyme.
Furthermore, the replacement of the aromatic ring with other heterocyclic rings such as thiophene in compound 2i (IC50 = 29.51 ± 0.35 μM) showed a decreased inhibition activity as compared to the standard (Table 1). The urease inhibition by compound 2i relied on the ability of a sulfur atom to coordinate within the active site of the urease enzyme. Afterward, the next compounds manifesting inhibitory activities are 2h (IC50 = 11.26 ± 0.51 μM) and 2j (IC50 = 13.67 ± 0.47 μM), where the aromatic ring B is substituted at position-4 with −COOH and acetamido (NHCOCH3) groups, respectively. Interestingly, the highly hydrophilic p-substituted analogs 2h and 2j exhibited excellent activity against urease.
Additionally, compounds 2a (IC50 = 13.54 ± 0.34 μM for urease and 445.83 ± 1.25 μM for α-glucosidase), 2b (IC50 = 9.36 ± 0.27 μM for urease and 462.94 ± 1.23 μM for α-glucosidase), 2d (IC50 = 13.28 ± 0.25 μM for urease and 421.34 ± 1.27 μM for α-glucosidase), 2e (IC50 = 15.39 ± 0.38 μM for urease and 384.73 ± 1.45 μM for α-glucosidase), 2f (IC50 = 15.57 ± 0.42 μM for urease and 407.28 ± 1.24 μM for α-glucosidase), 2g (IC50 = 9.13 ± 0.25 μM for urease and 419.45 ± 1.32 μM for α-glucosidase), 2l (IC50 = 457.82 ± 1.25 μM for α-glucosidase), 2n (IC50 = 18.42 ± 0.42 μM for urease and 434.26 ± 1.29 μM for α-glucosidase), and 2p (IC50 = 12.24 ± 0.38 μM for urease) were found to be more active against the envisioned enzyme urease and less active against α-glucosidase. These findings reflect that various EWGs and EDGs (i.e., −F, −isobutyl, −(OCH3)3, p-OCH3, −N(CH3)2, −(OCH3)2, −(OCH3)2, phenyl, m-CF3, and −(CH3)2) on the aryl ring are accountable for their excellent inhibitory activities because of increased interaction with the urease enzyme. Moreover, these findings also suggest that highly hydrophilic and hydrophobic (i.e., −F, −isobutyl, −(OCH3)3, p-OCH3, −N(CH3)2, −(OCH3)2, −(OCH3)2, phenyl, m-CF3, and −(CH3)2) groups on the aromatic ring are accountable for low inhibitory activities because of reduced favorable interactions with the active site of the target enzyme (α-glucosidase) due to conformational, steric, and electronic factors. Comparatively, the synthesized derivatives (2a–2q) are more active against urease as compared to the α-glucosidase enzyme.
Overall, it should be noted that these studies reveal that the nature, position, and number of substituents on the aryl ring greatly influence the activities of these analogs. In addition, it is evident from the results that bulky and polar functional groups present at the p-position on the phenyl ring play an essential role in controlling the inhibitory potential of the pyrazoline scaffold against the tested enzymes.
2.5. Cytotoxicity in Treated Cell Lines
The cytotoxic effects of 2a–2q on human breast carcinoma, MCF-7 and cervical cancer, HeLa cell lines were investigated. There was a dose-dependent increase in cytotoxicity of 2l on both cancer cell lines as shown in Figure 7. Interestingly, MCF-7 was shown to be more sensitive as compared to HeLa cells after treatment with 2l having EC50 of 120 μM/mL and EC50 of 510 μM/mL, respectively. Similarly, 2k exhibited a lower EC50 (250 μM/mL) on MCF-7 as compared to HeLa cell (EC50 500 μM/mL). A similar cytotoxic effect was observed on MCF-7 and HeLa cells after treatment with 2a–2j and 2m–2q, where both cell lines possessed EC50 > 500 as shown in Table 2. Furthermore, 2l is more sensitive toward MCF-7 cells as compared to the capecitabine (CP) drug with an EC50 of 980 μM/mL. MCF-7 was more sensitive toward 2l as compared to HeLa cells in our studies. Therefore, 2l was selected for MCF cells to further investigate the mode of cell death (apoptosis).
Figure 7.
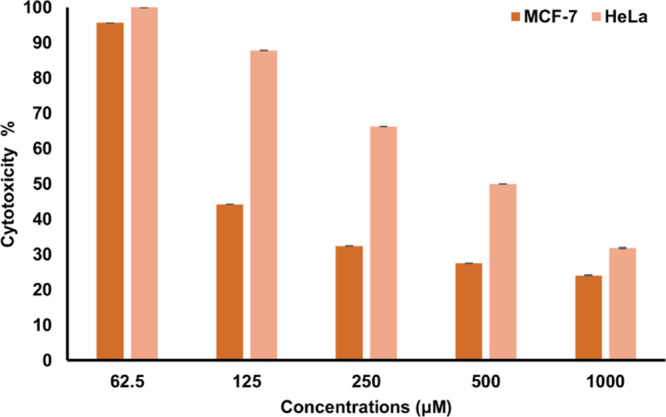
Cytotoxicity of 2l on MCF-7 and HeLa cells after 72 h.
Table 2. EC50 Values of 1–17 (μM/m).
| EC50 |
||
|---|---|---|
| compound no. | MCF-7 | HeLa |
| 2a | >500 | >500 |
| 2b | >500 | >500 |
| 2c | >500 | >500 |
| 2d | >500 | >500 |
| 2e | >500 | >500 |
| 2f | >500 | >500 |
| 2g | >500 | >500 |
| 2h | >500 | >500 |
| 2i | >500 | >500 |
| 2j | >500 | >500 |
| 2k | 250 ± 0.09 | >500 |
| 2l | 120 ± 0.10 | 510 |
| 2m | >500 | >500 |
| 2n | >500 | >500 |
| 2o | >500 | >500 |
| 2p | >500 | >500 |
| 2q | >500 | >500 |
2.5.1. Apoptosis Study (Early Apoptosis)
In order to determine the mode of MCF-7 cell death, Annexin V-FITC was used. MCF-7 cells were treated with 2l at a concentration of EC50 at 72 h for 6 and 9 h along with positive (CP) and negative control (without treatment). 2l was shown to induce early apoptosis by exposure of phosphoserine (PS) from inward to outward of the plasma membrane (PM) of cancer cell, which was detectable in cells by the green stain of Annexin-FITC after 6 h incubation treatment. Remarkably, both early (Annexin-FITC V+) and late apoptosis propidium iodide positive (PI+) cells were visible after 9 h. Besides, most of the cell death occurred due to apoptosis (AnnexinV+/PI+) with very few cells undergoing necrosis (Annexin V–/PI+) as shown in Figure 8. Apoptosis (programmed cell death) is a highly regulated mechanism noticeable by various morphological features of cell such as cytoplasmic shrinkage and reduction of cell size, permeabilization of the PM, nuclear condensation, and DNA fragmentation.94 Externalization of the PS protein to the outer leaflet of a cell membrane is among the early steps in the induction of apoptosis, which provides a signal to immune cells, probably phagocytes to further engulfment of apoptotic cells.95 Excitingly, the results revealed the cytotoxic effects of 2l by apoptosis in the MCF-7 cell line.
Figure 8.
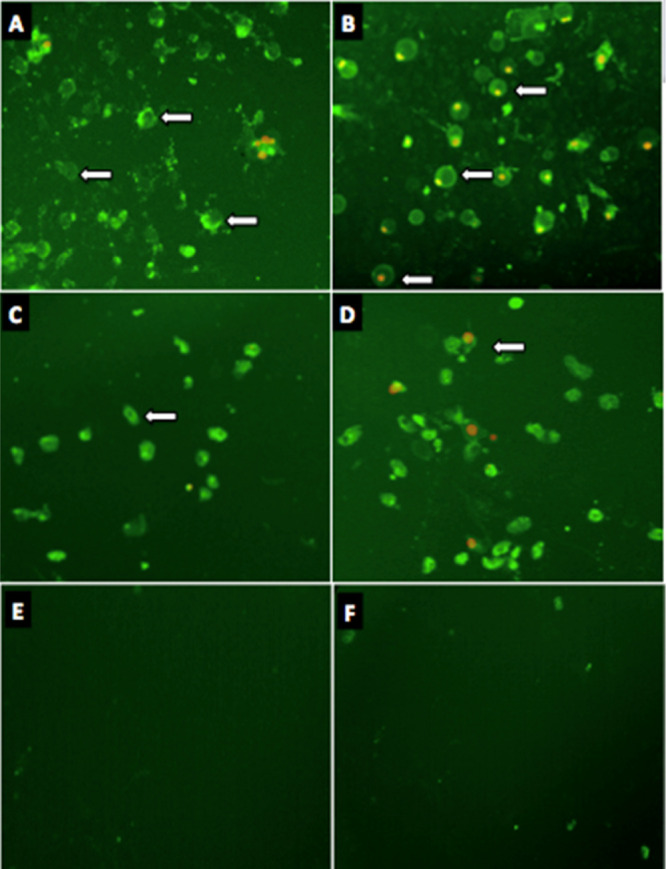
Green stain (annexin-FITC) signifies induction of early apoptosis and red (PI) late apoptosis in MCF-7 cells treated with; (A,B) 2l for 6 and 9 h; (C,D) CP for 6 and 9 h; and (E,F) negative control for 6 and 9 h incubation.
2.5.2. Apoptosis Study (Late Apoptosis)
In order to further validate the cytotoxicity by induction of apoptosis in the MCF-7 cell line, a fluorescence terminal nick end labeling of DNA (TUNEL) detection system was used. The in situ fluorometric determination of DNA fragmentation via TUNEL is a sensitive and accurate method of detection of apoptosis in cancer cells. Rows A and B in Figure 9 present the cells treated with 2l and CP alone, respectively, at a concentration of EC50 of 72 h. The nuclei of MCF-7 cells treated with 2l were morphologically detectable (green) due to the polymerization reaction of labeled nucleotides by a transferase enzyme (TdT) at the site of fragmented DNA after 36 h incubation treatment. Accordingly, DNA fragmentation is considered as a hallmark of apoptosis, which also plays a major role in the detection of apoptosis in cancer cells.96 Also, green-stained nuclei were observed in CP-treated cells. However, nuclei of negative control (no treatment) and the DNA of MCF-7 cells remain intact and the lack of green stain is noticeable under an Image Xpress Micro XLS Widefield High-Content Analysis System (HCS) (Sunnyvale, USA).
Figure 9.
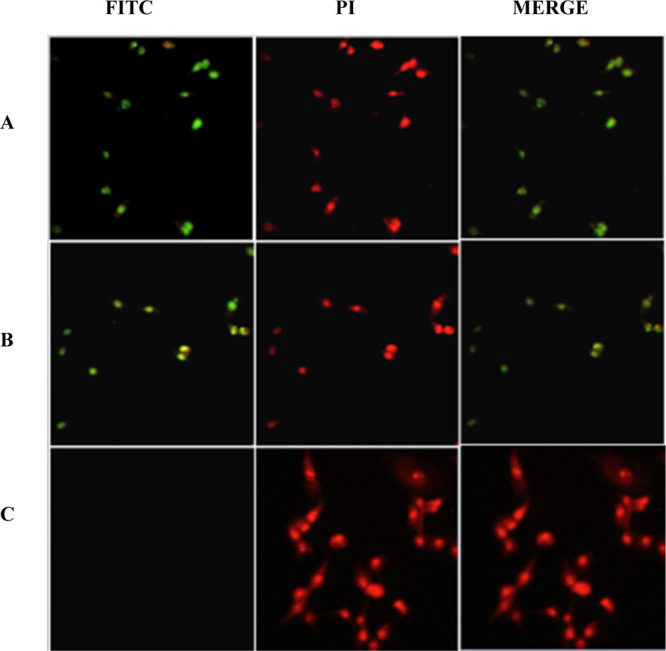
(A–D) Presence of green stain (FITC), which indicates DNA fragmentation in MCF-7 cells treated with: (A) 2l; (B) CP; and (C) negative control (untreated) for 36 h incubation.
2.6. In Silico Studies
2.6.1. Docking Analysis
ChemDraw was used to create 2D structures of compounds (2a-2q), which were then transformed into 3D using Chem Pro3D software. The ligands were docked to the protein using AutoDock 4.2.0 after being energetically minimized using Merck Molecular Force Field (MMFF). Table 3 and Figure 10 show the docking results, with just the best conformers and dock scores for each ligand. The ligand with the lowest docking score will produce the most stable drug–receptor combination. Hydrogen bonding, hydrophobic type interactions, and van der Waal’s interactions were investigated in the drug–receptor complexes of several powerful ligands.
Table 3. Binding Energies (Kcal mol–1) of Synthesized Analogs (2a–2q) against Urease and α-Glucosidase.
| docking study against urease |
docking study against α-glucosidase |
|||
|---|---|---|---|---|
| compound no. | lowest binding energy (Kcal mol–1) | mean binding energy (Kcal mol–1) | lowest binding energy (Kcal mol–1) | mean binding energy (Kcal mol–1) |
| 2a | –7.98 | –7.63 | –6.68 | –6.14 |
| 2b | –8.74 | –7.70 | –7.20 | –6.59 |
| 2c | –8.65 | –8.59 | –7.29 | –6.64 |
| 2d | –8.24 | –7.51 | –6.31 | –5.72 |
| 2e | –8.23 | –7.87 | –6.78 | –6.16 |
| 2f | –8.47 | –8.00 | –6.78 | –6.16 |
| 2g | –9.16 | –8.31 | –6.59 | –5.95 |
| 2h | –8.40 | –7.81 | –6.25 | –5.59 |
| 2i | –7.81 | –7.49 | –7.19 | –6.35 |
| 2j | –8.68 | –8.15 | –7.58 | –6.79 |
| 2k | –8.41 | –7.86 | –6.65 | –6.05 |
| 2l | –7.85 | –7.38 | –7.67 | –6.69 |
| 2m | –7.74 | –7.14 | –6.53 | –6.04 |
| 2n | –7.74 | –7.32 | –6.51 | –5.99 |
| 2o | –7.78 | –7.33 | –7.48 | –5.91 |
| 2p | –7.65 | –7.14 | –6.57 | –5.93 |
| 2q | –8.32 | –7.96 | –7.35 | –7.35 |
| standard | –8.31 (AHA) | –8.11 (AHA) | –7.98 (MIG) | –7.61 (MIG) |
Figure 10.
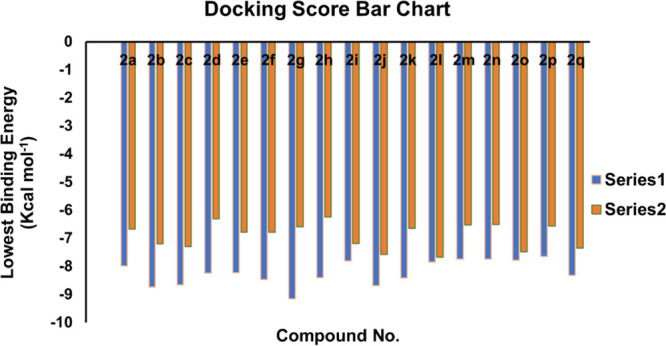
Graphical depiction of docking scores (Kcal/mol) of synthesized pyrazolines (2a–2q).
Root mean square deviation (RMSD) is used to assess the validity of docking research. To calculate the RMSD value, the cocrystalline ligand was removed from the active pockets of the selected substrate and redocked. The docked poses of AHA and MIG overlapped with the poses in the X-ray crystal structure (PDBs: 4H9M and 5NN6) at RMSD values of 0.6 and 0.8 Å, respectively, which validates the docking process.
2c, an important ligand (lowest binding energy, −8.65 Kcal mol-1), expressed promising inhibitory potential against the urease enzyme. This ligand 2c developed conventional hydrogen bonding with the His593 amino acid residue of biological-targeted moiety 4H9M. In addition to hydrogen bonding, this ligand 2c also built up several important interactions such as hydrophobic π-alkyl and electrostatic π-anion with Met637, Asp633, Ala636, Ala436, and Arg439 inside the active pockets of under study protein 4H9M as shown in Figure 11a,b.
Figure 11.

(a) Putative binding interactions of ligand 2c against urease. (b) Interactions of the ligand 2c in 3D space. Interactions with specific amino acid residues are shown in the box. The 3D ribbon represents the enzyme-stick model of the lowest energy conformers of the inhibitor 2c along with amino acids of urease interacting with it.
In the same fashion, this ligand 2c (lowest binding energy −7.29 Kcal mol–1) is also a potential inhibitor of glucosidase enzyme. This ligand 2c performed its action by building conventional hydrogen bonds with Arg600 and Asp616 amino acid residues of the targeted biological specimen 5NN6. Trp481, Trp376, Phe649, and Met519 amino acid residues also developed hydrophobic π–π T-shaped, hydrophobic π–π stacked, and hydrophobic alkyl type associations inside the active pockets of under study molecule 5NN6. The inhibitory effect of this ligand also increases due to the electrostatic π anion interaction against Asp282 and Asp518 inside 5NN6 as shown in Figure 12a,b.
Figure 12.

(a) Putative binding interactions of ligand 2c against α-Glucosidase. (b) Interactions of the ligand 2c in 3D space. Interactions with specific amino acid residues are shown in the box. The 3D ribbon represents the enzyme-stick model of the lowest energy conformers of the inhibitor 2c along with amino acids of α-glucosidase interacting with it.
Another potent ligand 2g (lowest binding energy −9.16 Kcal mol–1) reveals its inhibitory potential against urease enzyme via various intermolecular attractions. This ligand 2g exhibits hydrogen bonding with Arg609, Arg439, and Gln635 amino acid residues of under study protein 4H9M. This ligand also undergoes electrostatic π anion type interactions with Asp633 of 4H9M. Ala636, Arg439, Ala440, and His492 amino acid residues of 4H9M protein associate with ligand 2g via hydrophobic alkyl, hydrophobic π alkyl, hydrophobic π–sigma, and hydrophobic π–π T-shaped type interactions as shown in Figure 13a,b.
Figure 13.

(a) Putative binding interactions of ligand 2g against Urease. (b) Interactions of the ligand 2g in 3D space. Interactions with specific amino acid residues are shown in the box. The 3D ribbon represents the enzyme-stick model of the lowest energy conformers of the inhibitor 2g along with amino acids of urease interacting with it.
Similarly, ligand 2o (lowest binding energy −7.48 Kcal mol–1) is a potent inhibitor against the glucosidase enzyme. The flouro group of the ligand 2o involves in hydrogen bonding with Ser523 of 5NN6. The hydroxyl group of the ligand 2o also undergoes hydrogen bonding with Arg600 and Asp616 amino acid residues of the targeted molecule 5NN6. Aromatic delocalized π electronic cloud of the ligand 2o develops electrostatic π cation, electrostatic π anion, and hydrophobic π–π stacked type associations with Arg600, Asp282, Trp376, and Asp518 amino acid residues of targeted biological specimen 5NN6. Met519 amino acid of the 5NN6 protein builds up hydrophobic alkyl and hydrophobic π alkyl type interactions with ligand 2o as shown in Figures 14a,b.
Figure 14.

(a) Putative binding interactions of ligand 2o against α-glucosidase. (b) Interactions of the ligand 2o in 3D space. Interactions with specific amino acid residues are shown in the box. The 3D ribbon represents the enzyme-stick model of the lowest energy conformers of the inhibitor 2o along with amino acids of α-glucosidase interacting with it.
2.6.2. Drug-Likeness Study
The expected chemoinformatics properties, for instance, solubility, polarizability, and polar surface area were evaluated computationally. Already reported data were used to establish a standard value for the number of atoms (20–70) and molar molecular weight (160–480).73c,75 Inferences have shown that expected values of 2c, 2g, and 2o are better than those of all other synthesized compounds as well as standard values. Furthermore, the Lipinski’s rule of five (RO5) does not state anything about structural features or specific chemistry present in drugs or non-drugs. The computational results predicted that 2c, 2g, and 2o possess 2, 4, and 2 hydrogen bond acceptor (HBA) (≤10), 1, 1, and 1 hydrogen bond donor (HBD) (≤5), 0.42, 2.23, and 0.40 mg/L Log S, and PSA 30.47, 45.73, and 30.47 A2 (≤120 A2) values, respectively, which considerably indicated their drug-like behavior with drug-like model scores 0.01, 0.27, and −0.32. Besides, their molecular weights (392.05, 374.16, and 382.13 g/mol, respectively) were also better than the standard value (Table 4 and Figure 15). These findings illustrate that many derivatives show drug-like effects without violating any of the rules regarding their medicinal potential.
Table 4. Physicochemical Properties of Synthesized Pyrazoline Analogs (2a–2q).
| compound no. | mol. formula | mol. wt (g/mol) | no. HBAa | no. HBDb | mol. Log Pc | mol. Log Sd (mg/L) | mol. PSAe A2 | drug-likeness model score |
|---|---|---|---|---|---|---|---|---|
| 2a | C21H17FN2O | 332.13 | 2 | 1 | 5.79 | 0.65 | 30.47 | 0.19 |
| 2b | C25H26N2O | 370.20 | 2 | 1 | 7.49 | 0.45 | 30.47 | 0.96 |
| 2c | C21H17BrN2O | 392.05 | 2 | 1 | 6.56 | 0.42 | 30.47 | 0.01 |
| 2d | C24H24N2O4 | 404.17 | 5 | 1 | 5.72 | 1.63 | 53.53 | 0.42 |
| 2e | C22H20N2O2 | 344.15 | 3 | 1 | 5.68 | 1.27 | 38.01 | 0.24 |
| 2f | C23H23N3O | 357.18 | 2 | 1 | 5.79 | 1.02 | 33.27 | 0.08 |
| 2g | C23H22N2O3 | 374.16 | 4 | 1 | 5.40 | 2.23 | 45.73 | 0.27 |
| 2h | C22H18N2O3 | 358.13 | 4 | 2 | 5.53 | 2.13 | 58.88 | 0.41 |
| 2i | C19H16N2OS | 320.10 | 3 | 1 | 5.25 | 8.50 | 31.49 | –0.36 |
| 2j | C22H19N3O2 | 357.15 | 3 | 3 | 4.37 | 4.05 | 64.53 | 0.57 |
| 2k | C25H27N3O | 385.22 | 2 | 1 | 6.53 | 0.51 | 33.20 | 0.23 |
| 2l | C21H18N2O | 314.14 | 2 | 1 | 5.73 | 0.76 | 30.47 | –0.45 |
| 2m | C22H17F3N2O | 382.13 | 2 | 1 | 6.55 | 0.46 | 30.47 | –0.69 |
| 2n | C22H17F3N2O | 382.13 | 2 | 1 | 6.60 | 0.30 | 30.47 | –0.44 |
| 2o | C22H17F3N2O | 382.13 | 2 | 1 | 6.67 | 0.40 | 30.47 | –0.32 |
| 2p | C23H22N2O | 342.17 | 2 | 1 | 6.82 | 0.49 | 30.47 | –0.06 |
| 2q | C22H17N3O | 339.14 | 3 | 1 | 5.33 | 6.10 | 47.52 | –0.29 |
The table above depicts all of Lipinski’s RO5 components, that is, the number of HBAs.
Number of HBDs.
Octanol–water partition coefficient.
Measured solubility.
Total polar surface area.
Figure 15.
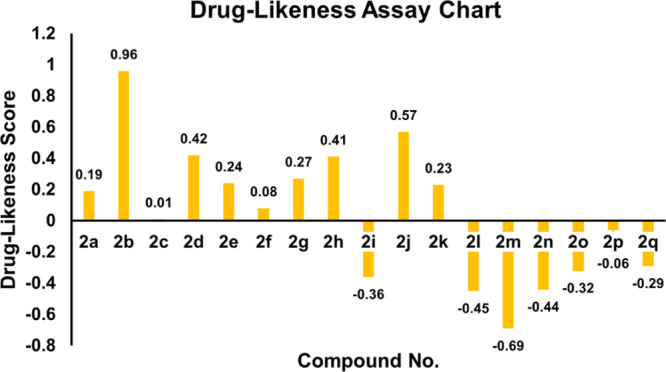
Drug-likeness score bar chart.
3. Conclusions
In conclusion, we have synthesized a series of 1,3,5-triaryl-2-pyrazoline derivatives using precedent methodologies and evaluated their inhibitory potential against urease and α-glucosidase. Their cytotoxic activities were also investigated. The synthesized compounds showed varying degrees of urease inhibitory activity ranging from 9.13 ± 0.25 to 18.42 ± 0.42 μM. Among the series of compounds, the compound 2g exhibited the lowest IC50 value (9.13 ± 0.25 μM) against urease, and other compounds in the series also showed excellent antiurease activity as compared to standard thiourea (IC50 = 21.37 ± 0.26 μM). Compounds 2a, 2b, 2c, 2d, 2e, 2f, 2h, 2j, 2k, 2m, 2n, 2o, 2p, and 2q having IC50 values of 13.54 ± 0.34, 9.36 ± 0.27, 12.57 ± 0.43, 13.28 ± 0.25, 15.39 ± 0.38, 15.57 ± 0.42, 11.26 ± 0.51, 13.67 ± 0.47, 13.29 ± 0.25, 9.18 ± 0.35, 18.42 ± 0.42, 12.63 ± 0.56, 12.24 ± 0.38, and 9.35 ± 0.35 μM, respectively, showed excellent inhibitory activity as compared to standard thiourea (IC50 = 21.37 ± 0.26 μM). The inhibition mechanism of biologically assayed compound 2g (competitive inhibition) was investigated by Lineweaver Burk and Dixon plots to determine the Vmax, Km, and Ki values. The compounds (2a–2q) also exhibited α-glucosidase IC50 values in the range of 114.57 ± 1.35 to 462.94 ± 1.23 μM. Furthermore, the compounds 2c, 2k, 2m, and 2o exhibited excellent α-glucosidase inhibitory activity with the lowest IC50 = 212.52 ± 1.31, 237.26 ± 1.28, 138.35 ± 1.32, and 114.57 ± 1.35 μM, respectively, as compared to the standard acarbose (IC50 = 375.82 ± 1.76 μM). Additionally, compound 2m acts as a potential dual inhibitor against urease and α-glucosidase. Exploring the urease and α-glucosidase inhibiting activities of the synthesized pyrazolines, we herein report that pyrazolines are potent dual urease and α-glucosidase inhibitors. Moreover, the cytotoxicity studies were performed and findings revealed that compound 2l displayed moderate and selective anticancer activity against MCF-7 and HeLa cell lines compared to CP. It is noteworthy to mention that the synthesized compounds are comparatively more active against urease than the α-glucosidase enzyme. In addition, compound 2l significantly induced apoptosis in both cell lines. The SAR study established that the nature, as well as the position of varying groups attached to the aryl group, played crucial roles in defining the urease and α-glucosidase inhibition activities. The molecular docking results agreed with the in vitro biological assay data. Pharmacological studies revealed that synthesized analogs 2a–2q obey Lipinski’s rule. Drug-likeness parameter assessment showed that these analogs show considerable lead-like characteristics with the least toxicity and can serve as templates in drug designing.
4. Experimental Section
4.1. Materials and Methods
All the chemicals and solvents were purchased from Merck and Sigma-Aldrich and were utilized as received. The melting points were determined on an electrothermal digital instrument, which are uncorrected. The IR spectra were recorded on a Bio-Rad spectrophotometer. The NMR spectra were obtained using a Bruker spectrometer (1H, 500 MHz, 13C, 126 MHz). NMR chemical shift values were defined in δ (ppm) units. TLC was used to check/monitor reaction progress and completion and spots were visualized under a UV lamp (254 nm). The QUARTZ cell was used to record the absorption spectra in ethanol on the Jasco UV-VIS V-660 instrument. Accurate mass measurements were carried out with the Fisons VG sector-field instrument (EI) and a FT-ICR mass spectrometer.
4.2. General Procedures for the Syntheses of Chalcones (1a–1q) and 1,3,5-Triaryl-2-pyrazolines (2a–2q)
In the first step, chalcones were synthesized by Claisen–Schmidt condensation according to the literature procedure.75,76 A mixture of 2′-hydroxyacetophenone (1.0 mmol) and aqueous NaOH solution (30%, 5.0 mL) was dissolved in distilled methanol (15 mL) and stirred for 30 min at ambient temperature, followed by the dropwise addition of aromatic aldehyde (1.0 mmol). Reactions were monitored by TLC using ethyl acetate/n-hexane (1:3) as a solvent system. After the reaction completion (checked by TLC), the reaction content was poured onto the ice-water and neutralized by HCl (10%). The product obtained was filtered, washed with cold water, and was recrystallized from ethanol to obtain the purified compounds (1a–1q) in good yields. In the second step, pyrazolines were synthesized according to the literature.77 Appropriate chalcones (1 mmol) were dissolved in glacial acetic acid. To this mixture, phenylhydrazine HCl (1 mmol) was added and the reaction mixture was refluxed for 16–18 h. The reaction was monitored by TLC using ethyl acetate/n-hexane (1:3) as a solvent system. After the reaction completion, the reaction mixture was then poured onto crushed ice accompanied by vigorous stirring. It was left at room temperature to obtain a crystalline compound. The obtained solid was filtered and recrystallized from ethanol to obtain a pure product (2a–2q) in good yields.
4.3. Enzyme Inhibition Assay
4.3.1. Urease Inhibitory Assay
A reported method was used to perform the urease inhibitory potential of newly synthesized pyrazoline derivatives (2a–2q).1,78 Reaction mixtures containing 55 μL buffer solution comprising of 100 mM urea and 25 μL enzyme solution were incubated with 5 μL newly synthesized compounds (1 mM concentration) at 30 °C in 96-well plates for 15 min. The production of ammonia was determined via utilizing the indophenol procedure to analyze the activity of urease. Momentarily, both 70 μL alkali reagent (0.5% w/v NaOH and 0.1% active chloride NaOCl) and 45 μL phenol reagent (1% w/v phenol and 0.005% w/v sodium nitroprusside) were added to each well. The enhanced absorbance was measured at 630 nm via utilizing a microplate reader (Molecular Device, USA) after 50 min. All steps were performed three times in a final volume of 200 μL. The final measurements (change in absorbance per min) were obtained via utilizing the SoftMax Pro software (Molecular Device, USA) for the abstraction of the results. Assays were performed at pH 8.2 (0.01 M K2HPO4·3H2O, 1 mM EDTA, and 0.01 M LiCl). Acetohydroxamic acid was used as the standard inhibitor of urease.
Percentage of inhibition was calculated from the following formula
4.3.2. α-Glucosidase Inhibitory Assay
The α-glucosidase inhibitory activity of newly designed derivatives of pyrazolines (2a–2q) was determined by following the already reported procedure with minor amendments.73c 250 mL of acarbose at different concentrations (250–1000 mg/mL) was incubated with 500 mL of 1.0 U/mL of α-glucosidase solution in 100 mM phosphate buffer (pH 6.8) for 20 min at 37 °C. 250 mL of 4-nitrophenyl-b-d-glucopyranoside solution and 250 mL of 1% starch42 dissolved in 100 mM phosphate buffer (pH 6.8), respectively, were then added to the reaction mixture and incubated for 1 h at 37 °C.20 Then, 1.0 mL of the 3,5-dinitrosalicylic acid coloring reagent was added to the reaction mixture and it was boiled for 10 min. The absorbance of the final reaction mixture was determined for α-glucosidase at 405 nm. The preparation of blank was carried out for correcting absorbance. Acarbose solution was utilized as positive control. The inhibitory activity was stated as a percentage of a control sample without the inhibitors.47
4.4. Kinetic Study
The kinetics of the binding mechanism of most dominant compound 2g (inhibitor), which has an IC50 value comparable with thiourea (standard), was investigated. Five different inhibitor concentrations (0–20 μM) were reacted with different substrate concentrations (urea, 0.5–4.0 mM) to determine whether the inhibitor is competitive, noncompetitive (mixed), and uncompetitive after calculating/assessing the Km (app) Vmax (app) values from the Lineweaver Burk plot, and the Ki values (inhibition constant) were also determined by the Dixon plot using PRISM 7.0.79
4.5. Cytotoxicity Study
Culture ware and other plastic consumables were purchased from Nunc, Denmark. Cancer cell lines were purchased from American Type Cell Culture, ATCC, USA. The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent was obtained from thermofisher scientific, USA, ApoAlert Annexin V, Clontech, USA, DeadEnd Fluorometric TUNEL Promega, USA. All required chemicals and cell culture reagents were obtained from Sigma-Aldrich, USA.
4.5.1. Cell Culturing
The human cell lines MCF-7 (human breast adenocarcinoma) and HeLa (cervical cancer) were grown in RPMI 1640 medium (Gibco Life Technologies) containing 10% fetal calf serum (FCS; Gibco) and 1% antibiotics (100 IU/mL penicillin and 100 μg/mL streptomycin) in a humidified incubator at 37 °C in 5% CO2 and 95% air.
4.6. Cytotoxicity Studies
4.6.1. Cell Viability in Treated Cell Lines (MTT Assay)
Cytotoxicity study was carried out using the MTT assay.80 The cell viability was determined by reading the absorbance of the metabolized product (purple formazan) of the MTT reagent by living cells. The cells were plated in a 96-well plate at a density of 6000 cells/well and incubated at 37 °C in a CO2 incubator for 24 h. Next, the old medium was replaced with fresh media containing treatments at given concentrations (0, 15.62, 31.25, 62.5, 125, 250, 500, and 1000 μM/mL). After 72 h of incubation, 20 μL of MTT (5 mg/mL) was added to each well and incubated for 4 h. Subsequently, after removal of media containing treatments, 100 μL of DMSO was added to each well to dissolve crystals of formazan. Using an Elisa reader, the absorbance was measured at 570 nm (Multiskan, Thermo fisher Scientific USA). The treatment was repeated three times, with the results calculated as percentage of growth inhibition values based on the mean of two independent values (SEM). The EC50 (half effective inhibitory concentration) value was calculated by Graphpad Prism 7 using a non-linear regression model (curve fit) based on a sigmoidal dose–response curve (variable) (GraphPad, San Diego, California, USA).
4.6.2. Mode of Cell Death Study
4.6.2.1. Determination of Early Apoptosis in Treated Cells
The apoptotic effect of the cytotoxic crystal 2l (having lower EC50 value) on the MCF-7 was evaluated according to the previously reported method.81 Briefly, MCF-7 cells were plated at a cell density of 10,000 cells/well and incubated at 37 °C for 24 h. After 24 h, the medium was removed and replaced with fresh media containing 2l at a concentration of EC50 along with positive (cisplatin) and negative controls (without treatment). Next, the cells were incubated in 200 μL of binding buffer containing Annexin V-FITC (green) and PI (red) for 10 min at 37 °C. The cells were observed under an Image Xpress Micro XLS Widefield High-Content Analysis System (HCS) (Sunnyvale, USA).
4.6.2.2. Determination of Late Apoptosis in Treated Cells
A TUNEL assay was carried out using the DeadEnd Fluorometric TUNEL system (Promega, USA) for investigating in situ DNA fragmentation occurring in treated cells. The assay was performed according to the manufacturer’s protocol. Briefly, The MCF-7 cells were plated in Labtek Chamber Slides (Nunc, Denmark) with a density of 2 × 104 and incubated at 37 °C for 24 h. Next, both treated and control (without treatment) cells were fixed by 4% paraformaldehyde solution in PBS (pH 7.4) for 25 min at 4 °C. Subsequently, the cells were washed with phosphate buffer saline (PBS) for 5 min. After this, fixed cells were permeabilized for 5 min using 0.2% triton X-100 in PBS. Then, the permeabilized cells were washed with PBS; each step of washing was done thrice for 5 min. The nick ends of DNA or fragmented ends were labeled by adding a 50 μL reaction mixture, containing equilibration Buffer, Nucleotide Mix, and enzyme rTdT in a ratio and incubated in the dark at 37 °C. 20× SSC (1:10) was diluted with deionized water and added enough to fill a standard chamber. Additionally, propidium iodide (PI) solution was freshly prepared (1 μg/mL in PBS) and added to stain the nuclear DNA for 15 min at room temperature in the dark. The Image Xpress Micro XLS Widefield High-Content Analysis System (HCS) (Sunnyvale, USA) was used to detect the green fluorescence of FITC-labeled apoptotic cells.
4.7. Molecular Modeling Assay
AutoDock 4.2.0 was applied in order to study the protein–ligand associations. The X-ray crystallographic structure of Jack bean urease (JBU) (in complex with acetohydroxamic acid, AHA) (PDB ID: 4H9M) and α-glucosidase (in complex with MIG ((2R,3R,4R,5S)-1-(2-hydroxyethyl)-2-(hydroxymethyl)piperidine-3,4,5-triol) (PDB ID: 5NN6, species Homo sapiens) (www.rcsb.org) was obtained, from RCSB protein data bank (PDB), as a model. 3D grids of AHA and MIG in the binding pocket of urease and α-glucosidase were estimated by using discovery studio 4.0. The protein structure was improved, and side-chain hydrogens were added.73a,73b
4.8. Drug-Likeness Study
The Molsoft tool has been utilized to determine the drug-like characteristics of the predicted compounds. Lipinski’s RO5 was used to establish a framework for assessing the bioavailability and pharmacokinetics of a given molecule when administered orally, using a set of physiochemical parameters.73c,75
Acknowledgments
The authors would like to acknowledge the Deanship of Scientific Research at Umm Al-Qura University, for supporting this work by Grant code: 22UQU4320545DSR03. The authors would like to extend their sincere appreciation to Taif University Researchers Supporting Project number (TURSP-2020/312), Taif University, Taif, Saudi Arabia. The authors thank the Higher Education Commission of Pakistan for providing research grant under the project numbers NRPU-6484 and NRPU-15800.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c06694.
FTIR spectra of some of the synthesized compounds and 1H NMR and 13C NMR spectra of the some of the synthesized compounds (PDF)
Author Contributions
R.M. contributed in experiments and first-draft preparation; A.S. contributed in main idea, supervision, and final writing of the manuscript; R.I.A. contributed in experiments and first-draft preparation; E.U.M. contributed in main idea, cosupervision, and final writing of the manuscript; M.A.A. contributed in first-draft preparation and data analysis and collection; N.N. contributed in first-draft preparation, data analysis and collection, and drug-likeness study; A.J. contributed in docking studies; M.M.A.-R. contributed in first-draft preparation and data analysis and collection; G.-S.C. performed enzyme inhibition and cytotoxicity studies; and S.A.A. contributed in main idea, cosupervision, and final writing of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Arshad T.; Khan K. M.; Rasool N.; Salar U.; Hussain S.; Asghar H.; Ashraf M.; Wadood A.; Riaz M.; Perveen S.; Taha M.; Ismail N. H. 5-Bromo-2-aryl benzimidazole derivatives as non-cytotoxic potential dual inhibitors of α -glucosidase and urease enzymes. Bioorg. Chem. 2017, 72, 21–31. 10.1016/j.bioorg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Rahim F.; Taha M.; Ullah H.; Wadood A.; Selvaraj M.; Rab A.; Sajid M.; Shah S. A. A.; Uddin N.; Gollapalli M. Synthesis of new arylhydrazide bearing Schiff bases/thiazolidinone: α-Amylase, urease activities and their molecular docking studies. Bioorg. Chem. 2019, 91, 103112. 10.1016/j.bioorg.2019.103112. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Imran M.; Muddassar M.; Hussain R.; Khan M. U.; Ahmad S.; Mehboob M. Y.; Ashfaq S. Benzenesulfonohydrazides inhibiting urease: Design, synthesis, their in vitro and in silico studies. J. Mol. Struct. 2020, 1220, 128740. 10.1016/j.molstruc.2020.128740. [DOI] [Google Scholar]
- Shi W.-K.; Deng R.-C.; Wang P.-F.; Yue Q.-Q.; Liu Q.; Ding K.-L.; Yang M.-H.; Zhang H.-Y.; Gong S.-H.; Deng M.; Liu W.-R.; Feng Q.-J.; Xiao Z.-P.; Zhu H.-L. 3-Arylpropionylhydroxamic acid derivatives as Helicobacter pylori urease inhibitors: Synthesis, molecular docking and biological evaluation. Bioorg. Med. Chem. 2016, 24, 4519–4527. 10.1016/j.bmc.2016.07.052. [DOI] [PubMed] [Google Scholar]
- Hameed A.; Khan K. M.; Zehra S. T.; Ahmed R.; Shafiq Z.; Bakht S. M.; Yaqub M.; Hussain M.; de la Vega de León A.; Furtmann N.; Bajorath J.; Shad H. A.; Tahir M. N.; Iqbal J. Synthesis, biological evaluation and molecular docking of N-phenyl thiosemicarbazones as urease inhibitors. Bioorg. Chem. 2015, 61, 51–57. 10.1016/j.bioorg.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Follmer C. Ureases as a target for the treatment of gastric and urinary infections. J. Clin. Pathol. 2010, 63, 424–430. 10.1136/jcp.2009.072595. [DOI] [PubMed] [Google Scholar]
- Imran M.; Waqar S.; Ogata K.; Ahmed M.; Noreen Z.; Javed S.; Bibi N.; Bokhari H.; Amjad A.; Muddassar M. Identification of novel bacterial urease inhibitors through molecular shape and structure based virtual screening approaches. RSC Adv. 2020, 10, 16061–16070. 10.1039/d0ra02363a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton K. A.; Brooks C. L.; Morgan D. R.; Krakowka S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991, 59, 2470–2475. 10.1128/iai.59.7.2470-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayerdörffer E.; Ottenjann R. The role of antibiotics in Campylobacter pylori associated peptic ulcer disease. Scand. J. Gastroenterol. 1988, 23, 93–100. 10.3109/00365528809091721. [DOI] [PubMed] [Google Scholar]
- Holter H.; Linderstrøm-Lang K. Micromethods and their application in the study of enzyme distribution in tissues and cells. Physiol. Rev. 1951, 31, 432–448. 10.1152/physrev.1951.31.4.432. [DOI] [PubMed] [Google Scholar]
- Taha M.; Ismail N. H.; Khan A.; Shah S. A. A.; Anwar A.; Halim S. A.; Fatmi M. Q.; Imran S.; Rahim F.; Khan K. M. Synthesis of novel derivatives of oxindole, their urease inhibition and molecular docking studies. Bioorg. Med. Chem. Lett. 2015, 25, 3285–3289. 10.1016/j.bmcl.2015.05.069. [DOI] [PubMed] [Google Scholar]
- Shehzad M. T.; Khan A.; Islam M.; Halim S. A.; Khiat M.; Anwar M. U.; Hussain J.; Hameed A.; Pasha A. R.; Khan F. A.; Al-Harrasi A.; Shafiq Z. Synthesis, characterization and molecular docking of some novel hydrazonothiazolines as urease inhibitors. Bioorg. Chem. 2020, 94, 103404. 10.1016/j.bioorg.2019.103404. [DOI] [PubMed] [Google Scholar]
- Shehzad M. T.; Khan A.; Islam M.; Hameed A.; Khiat M.; Halim S. A.; Anwar M. U.; Shah S. R.; Hussain J.; Csuk R.; Khan S.; Al-Harrasi A.; Shafiq Z. Synthesis and urease inhibitory activity of 1,4-benzodioxane-based thiosemicarbazones: Biochemical and computational approach. J. Mol. Struct. 2020, 1209, 127922. 10.1016/j.molstruc.2020.127922. [DOI] [Google Scholar]
- Mobley H. L.; Island M. D.; Hausinger R. P. Molecular biology of microbial ureases. Microbiol. Rev. 1995, 59, 451–480. 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith D. P.; Gleeson M. J.; Lee H.; Longuet R.; Deman E.; Earle N. Randomized, double-blind trial of Lithostat (Acetohydroxamic Acid) in the palliative treatment of infection-induced urinary calculi. Eur. Urol. 1991, 20, 243–247. 10.1159/000471707. [DOI] [PubMed] [Google Scholar]
- Pan L.; Wang C.; Yan K.; Zhao K.; Sheng G.; Zhu H.; Zhao X.; Qu D.; Niu F.; You Z. Synthesis, structures and Helicobacter pylori urease inhibitory activity of copper(II) complexes with tridentate aroylhydrazone ligands. J. Inorg. Biochem. 2016, 159, 22–28. 10.1016/j.jinorgbio.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Zonia L. E.; Stebbins N. E.; Polacco J. C. Essential role of urease in germination of nitrogen-limited Arabidopsis thaliana seeds. Plant Physiol. 1995, 107, 1097–1103. 10.1104/pp.107.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhengping W.; Van Cleemput O.; Demeyer P.; Baert L. Effect of urease inhibitors on urea hydrolysis and ammonia volatilization. Biol. Fertil. Soils 1991, 11, 43–47. 10.1007/bf00335833. [DOI] [Google Scholar]
- Li W.-Y.; Ni W.-W.; Ye Y.-X.; Fang H.-L.; Pan X.-M.; He J.-L.; Zhou T.-L.; Yi J.; Liu S.-S.; Zhou M.; Xiao Z.-P.; Zhu H.-L. N-monoarylacetothioureas as potent urease inhibitors: Synthesis, SAR, and biological evaluation. J. Enzyme Inhib. Med. Chem. 2020, 35, 404–413. 10.1080/14756366.2019.1706503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aispuro-Pérez A.; López-Ávalos J.; García-Páez F.; Montes-Avila J.; Picos-Corrales L. A.; Ochoa-Terán A.; Bastidas P.; Montaño S.; Calderón-Zamora L.; Osuna-Martínez U. Synthesis and molecular docking studies of imines as α-glucosidase and α-amylase inhibitors. Bioorg. Chem. 2020, 94, 103491. 10.1016/j.bioorg.2019.103491. [DOI] [PubMed] [Google Scholar]
- Hamad A.; Khan M. A.; Rahman K. M.; Ahmad I.; Ul-Haq Z.; Khan S.; Shafiq Z. Development of sulfonamide-based Schiff bases targeting urease inhibition: Synthesis, characterization, inhibitory activity assessment, molecular docking and ADME studies. Bioorg. Chem. 2020, 102, 104057. 10.1016/j.bioorg.2020.104057. [DOI] [PubMed] [Google Scholar]
- Kanwal; Khan M.; Arshia; Khan K. M.; Parveen S.; Shaikh M.; Fatima N.; Choudhary M. I. Syntheses, in vitro urease inhibitory activities of urea and thiourea derivatives of tryptamine, their molecular docking and cytotoxic studies. Bioorg. Chem. 2019, 83, 595–610. 10.1016/j.bioorg.2018.10.070. [DOI] [PubMed] [Google Scholar]
- Rauf A.; Shahzad S.; Bajda M.; Yar M.; Ahmed F.; Hussain N.; Akhtar M. N.; Khan A.; Jończyk J. Design and synthesis of new barbituric- and thiobarbituric acid derivatives as potent urease inhibitors: Structure activity relationship and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 6049–6058. 10.1016/j.bmc.2015.05.038. [DOI] [PubMed] [Google Scholar]
- Scavenging D. R. Synthesis of benzophenone hydrazone analogs and their DPPH radical scavenging and urease inhibitory activities. J. Chem. Soc. Pak. 2015, 37, 479. [Google Scholar]
- Bremner J. M.Recent research on problems in the use of urea as a nitrogen fertilizer. Nitrogen Economy in Tropical Soils, 2nd ed.; Springer: Dordrecht, 1995; pp 321–329. [Google Scholar]
- Witte C.-P.; Tiller S. A.; Taylor M. A.; Davies H. V. Leaf urea metabolism in potato. Urease activity profile and patterns of recovery and distribution of 15N after foliar urea application in wild-type and urease-antisense transgenics. Plant Physiol. 2002, 128, 1129–1136. 10.1104/pp.010506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciurli S.; Benini S.; Rypniewski W.; Wilson K.; Miletti S.; Mangani S. The complex of Bacillus pasteurii urease with acetohydroxamate anion from X-ray data at 1.55 Å resolution. J. Coord. Chem. Rev. 1999, 190, 331–355. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy P.; Parlow M.; Zitzer J. B.; Vakil N. B.; Mobley H. L. T.; Levy M.; Phadnis S. H.; Dunn B. E. Helicobacter pylori containing only cytoplasmic urease is susceptible to acid. Infect. Immun. 1998, 66, 5060–5066. 10.1128/iai.66.11.5060-5066.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioacchini P.; Nastri A.; Marzadori C.; Giovannini C.; Vittori Antisari L.; Gessa C. Influence of urease and nitrification inhibitors on N losses from soils fertilized with urea. Biol. Fertil. Soils 2002, 36, 129–135. 10.1007/s00374-002-0521-1. [DOI] [Google Scholar]
- Ahmed D.; Younas S.; Anwer-Mughal Q. M. Study of alpha-amylase and urease inhibitory activities of Melilotus indicus (Linn.) All. Pak. J. Pharm. Sci. 2014, 27, 57–61. [PubMed] [Google Scholar]
- Collins C. M.; D’Orazio S. E. F. Bacterial ureases: structure, regulation of expression and role in pathogenesis. Mol. Microbiol. 1993, 9, 907–913. 10.1111/j.1365-2958.1993.tb01220.x. [DOI] [PubMed] [Google Scholar]
- Lee A. The microbiology and epidemiology of Helicobacter pylori infection. Scand. J. Gastroenterol. 1994, 29, 2–6. 10.3109/00365529409105352. [DOI] [PubMed] [Google Scholar]
- Allende-Montalbán R.; Martín-Lammerding D.; Delgado M. d. M.; Porcel M. A.; Gabriel J. L. Urease inhibitors effects on the nitrogen use efficiency in a maize–wheat rotation with or without water deficit. Agriculture 2021, 11, 684. 10.3390/agriculture11070684. [DOI] [Google Scholar]
- Islam M.; Khan A.; Shehzad M. T.; Khiat M.; Halim S. A.; Hameed A.; Shah S. R.; Basri R.; Anwar M. U.; Hussain J.; Csuk R.; Al-Harrasi A.; Shafiq Z. Therapeutic potential of N-substituted thiosemicarbazones as new urease inhibitors: Biochemical and in silico approach. Bioorg. Chem. 2021, 109, 104691. 10.1016/j.bioorg.2021.104691. [DOI] [PubMed] [Google Scholar]
- Mamidala R.; Bhimathati S. R. S.; Vema A. Discovery of Novel Dihydropyrimidine and hydroxamic acid hybrids as potent Helicobacter pylori Urease inhibitors. Bioorg. Chem. 2021, 114, 105010. 10.1016/j.bioorg.2021.105010. [DOI] [PubMed] [Google Scholar]
- Khan M. U.; Aslam M.; Shahzad S. A.; Khan Z. A.; Khan N. A.; Ali M.; Naz S.; Rahman J.; Farooq U. Design and synthesis of thiobarbituric acid analogues as potent urease inhibitors. J. Mol. Struct. 2021, 1231, 129959. 10.1016/j.molstruc.2021.129959. [DOI] [Google Scholar]
- Özil M.; Tuzcuoğlu Ö.; Baltaş N.; Emirik M. Synthesis and molecular docking studies of potent urease inhibitors based on benzoxazole scaffold. ChemistrySelect 2021, 6, 5307–5312. 10.1002/slct.202100928. [DOI] [Google Scholar]
- Moghaddam F. M.; Daneshfar M.; Daneshfar Z.; Iraji A.; Samandari-Najafabad A.; Faramarzi M. A.; Mahdavi M. Synthesis and characterization of 1-amidino-O-alkylureas metal complexes as α-glucosidase Inhibitors: Structure-activity relationship, molecular docking, and kinetic studies. J. Mol. Struct. 2022, 1250, 131726. 10.1016/j.molstruc.2021.131726. [DOI] [Google Scholar]
- Azimi F.; Azizian H.; Najafi M.; Hassanzadeh F.; Sadeghi-Aliabadi H.; Ghasemi J. B.; Ali Faramarzi M.; Mojtabavi S.; Larijani B.; Saghaei L.; Mahdavi M. Design and synthesis of novel quinazolinone-pyrazole derivatives as potential α-glucosidase inhibitors: Structure-activity relationship, molecular modeling and kinetic study. Bioorg. Chem. 2021, 114, 105127. 10.1016/j.bioorg.2021.105127. [DOI] [PubMed] [Google Scholar]
- Xie H.-X.; Zhang J.; Li Y.; Zhang J.-H.; Liu S.-K.; Zhang J.; Zheng H.; Hao G.-Z.; Zhu K.-K.; Jiang C.-S. Novel tetrahydrobenzo[b]thiophen-2-yl)urea derivatives as novel α-glucosidase inhibitors: Synthesis, kinetics study, molecular docking, and in vivo anti-hyperglycemic evaluation. Bioorg. Chem. 2021, 115, 105236. 10.1016/j.bioorg.2021.105236. [DOI] [PubMed] [Google Scholar]
- Moghimi S.; Salarinejad S.; Toolabi M.; Firoozpour L.; Esmaeil Sadat Ebrahimi S.; Safari F.; Madani-Qamsari F.; Mojtabavi S.; Faramarzi M. A.; Karima S.; Pakrad R.; Foroumadi A. Synthesis, in-vitro evaluation, molecular docking, and kinetic studies of pyridazine-triazole hybrid system as novel α-glucosidase inhibitors. Bioorg. Chem. 2021, 109, 104670. 10.1016/j.bioorg.2021.104670. [DOI] [PubMed] [Google Scholar]
- Taha M.; Alrashedy A. S.; Almandil N. B.; Iqbal N.; Anouar E. H.; Nawaz M.; Uddin N.; Chigurupati S.; Wadood A.; Rahim F.; Das S.; Venugopal V.; Nawaz F.; Khan K. M. Synthesis of indole derivatives as diabetics II inhibitors and enzymatic kinetics study of α-glucosidase and α-amylase along with their in-silico study. Int. J. Biol. Macromol. 2021, 190, 301–318. 10.1016/j.ijbiomac.2021.08.207. [DOI] [PubMed] [Google Scholar]
- Radhi A. J.; Zimam E. H.; Jafer E. A. New barbiturate derivatives as potent in vitro α-glucosidase inhibitors. Egypt. J. Chem. 2021, 64, 117–123. 10.21608/ejchem.2019.16210.1991. [DOI] [Google Scholar]
- Bashir B.; Shahid W.; Ashraf M.; Saleem M.; Aziz-ur-Rehman S.; Muzaffar S.; Imran M.; Amjad H.; Bhattarai K.; Riaz N. Identification of phenylcarbamoylazinane-1,3,4-oxadiazole amides as lipoxygenase inhibitors with expression analysis and in silico studies. Bioorg. Chem. 2021, 115, 105243. 10.1016/j.bioorg.2021.105243. [DOI] [PubMed] [Google Scholar]
- Gani R. S.; Kudva A. K.; Timanagouda K.; Raghuveer S. B. H.; Mujawar S. B. H.; Joshi S. D.; Raghu S. V. Synthesis of novel 5-(2,5-bis(2,2,2-trifluoroethoxy)phenyl)-1,3,4-oxadiazole-2-thiol derivatives as potential glucosidase inhibitors. Bioorg. Chem. 2021, 114, 105046. 10.1016/j.bioorg.2021.105046. [DOI] [PubMed] [Google Scholar]
- Lakshmana Senthil S.; Chandrasekaran R.; Arjun H. A.; Anantharaman P. In vitro and in silico inhibition properties of fucoidan against α-amylase and α-D-glucosidase with relevance to type 2 diabetes mellitus. Carbohydr. Polym. 2019, 209, 350–355. 10.1016/j.carbpol.2019.01.039. [DOI] [PubMed] [Google Scholar]
- Alqahtani A. S.; Hidayathulla S.; Rehman M. T.; ElGamal A. A.; Al-Massarani S.; Razmovski-Naumovski V.; Alqahtani M. S.; El Dib R. A.; AlAjmi M. F. alpha-amylase and alpha-glucosidase enzyme inhibition and antioxidant potential of 3-oxolupenal and katononic acid isolated from Nuxia oppositifolia. Biomolecules 2020, 10, 61. 10.3390/biom10010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff H. D.; Repin N.; Fabek H.; El Khoury D.; Gidley M. J. Dietary fibre for glycaemia control: Towards a mechanistic understanding. Bioact. Carbohydr. Diet. Fibre 2018, 14, 39–53. 10.1016/j.bcdf.2017.07.005. [DOI] [Google Scholar]
- Xiao J. B.; Högger P. Dietary polyphenols and type 2 diabetes: current insights and future perspectives. Curr. Med. Chem. 2015, 22, 23–38. 10.2174/0929867321666140706130807. [DOI] [PubMed] [Google Scholar]
- Kumar L.; Lal K.; Yadav P.; Kumar A.; Paul A. K. Synthesis, characterization, α-glucosidase inhibition and molecular modeling studies of some pyrazoline-1H-1,2,3-triazole hybrids. J. Mol. Struct. 2020, 1216, 128253. 10.1016/j.molstruc.2020.128253. [DOI] [Google Scholar]
- Kumar L.; Lal K.; Kumar A.; Paul A. K.; Kumar A. Pyrazoline tethered 1,2,3-triazoles: Synthesis, antimicrobial evaluation and in silico studies. J. Mol. Struct. 2021, 1246, 131154. 10.1016/j.molstruc.2021.131154. [DOI] [Google Scholar]
- Sato T.; Kitahara F.; Nakamura T.; Kojima Y.; Fujino M. A. [Peptic ulcer in patients with diabetes mellitus]. Nihon Rinsho. Jpn. J. Clin. Med. 2002, 60, 1580–1584. [PubMed] [Google Scholar]
- Tachecí I.; Bures J. [Peptic ulcer disease in patients with diabetes mellitus]. Vnitr. Lek. 2011, 57, 347–350. [PubMed] [Google Scholar]
- Gasbarrini A.; Ojetti V.; Pitocco D.; Luca A. D.; Franceschi F.; Candelli M.; Torre E. S.; Pola P.; Ghirlanda G.; Gasbarrini G. Helicobacter pylori infection in patients affected by insulin-dependent diabetes mellitus. Eur. J. Gastroenterol. Hepatol. 1998, 10, 469–472. 10.1097/00042737-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Xu J.; Yang J.; Ran Q.; Wang L.; Liu J.; Wang Z.; Wu X.; Hua W.; Yuan S.; Zhang L.; Shen M.; Ding Y. Synthesis and biological evaluation of novel 1-O- and 14-O-derivatives of oridonin as potential anticancer drug candidates. Bioorg. Med. Chem. Lett. 2008, 18, 4741–4744. 10.1016/j.bmcl.2008.06.097. [DOI] [PubMed] [Google Scholar]
- Cozzi P. The discovery of a new potential anticancer drug: a case history. Il Farmaco 2003, 58, 213–220. 10.1016/s0014-827x(03)00014-4. [DOI] [PubMed] [Google Scholar]
- Evan G. I.; Vousden K. H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Krishna R.; Mayer L. D. Multidrug resistance (MDR) in cancer. Eur. J. Pharm. Sci. 2000, 11, 265–283. 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- Calafiore A. M.; Di Mauro M.; Teodori G.; Di Giammarco G.; Cirmeni S.; Contini M.; Iacò A. L.; Pano M. Impact of aortic manipulation on incidence of cerebrovascular accidents after surgical myocardial revascularization. Ann. Thorac. Surg. 2002, 73, 1387–1393. 10.1016/s0003-4975(02)03470-7. [DOI] [PubMed] [Google Scholar]
- Dinesha; Viveka S.; Naik P.; Nagaraja G. K. Synthesis, characterization of new imidazoquinonyl chalcones and pyrazolines as potential anticancer and antioxidant agents. Med. Chem. Res. 2014, 23, 4189–4197. 10.1007/s00044-014-0998-9. [DOI] [Google Scholar]
- Rostom S. A. F.; Badr M. H.; Abd El Razik H. A.; Ashour H. M. A.; Abdel Wahab A. E. Synthesis of some pyrazolines and pyrimidines derived from polymethoxy chalcones as anticancer and antimicrobial agents. Arc. Pharm. 2011, 344, 572–587. 10.1002/ardp.201100077. [DOI] [PubMed] [Google Scholar]
- Varghese B.; Al-Busafi S. N.; Suliman F. O.; Al-Kindy S. M. Z. Unveiling a versatile heterocycle: pyrazoline - a review. RSC Adv. 2017, 7, 46999–47016. 10.1039/c7ra08939b. [DOI] [Google Scholar]
- Chandra T.; Garg N.; Lata S.; Saxena K. K.; Kumar A. Synthesis of substituted acridinyl pyrazoline derivatives and their evaluation for anti-inflammatory activity. Eur. J. Med. Chem. 2010, 45, 1772–1776. 10.1016/j.ejmech.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Karabacak M.; Altıntop M.; İbrahim Çiftçi H.; Koga R.; Otsuka M.; Fujita M.; Özdemir A. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules 2015, 20, 19066–19084. 10.3390/molecules201019066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K. A.; Govindaraju M. Pyrazolines: versatile molecules of synthetic and pharmaceutical applications-a review. Int. J. ChemTech Res. 2015, 8, 313–322. [Google Scholar]
- Ardiansah B. Pharmaceutical importance of pyrazoline derivatives: a mini review. J. Pharm. Sci. Res. 2017, 9, 1958–1960. [Google Scholar]
- Nehra B.; Rulhania S.; Jaswal S.; Kumar B.; Singh G.; Monga V. Recent advancements in the development of bioactive pyrazoline derivatives. Eur. J. Med. Chem. 2020, 205, 112666. 10.1016/j.ejmech.2020.112666. [DOI] [PubMed] [Google Scholar]
- Matiadis D.; Sagnou M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020, 21, 5507. 10.3390/ijms21155507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal E. U.; Sadiq A.; Murtaza S.; Rafique H.; Zafar M. N.; Riaz T.; Khan B. A.; Hameed A.; Khan K. M. Synthesis, structure-activity relationship and molecular docking of 3-oxoaurones and 3-thioaurones as acetylcholinesterase and butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2017, 25, 100–106. 10.1016/j.bmc.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Mughal E. U.; Javid A.; Sadiq A.; Murtaza S.; Zafar M. N.; Khan B. A.; Sumrra S. H.; Tahir M. N.; Kanwal K. M.; Khan K. M. Synthesis, structure-activity relationship and molecular docking studies of 3-O-flavonol glycosides as cholinesterase inhibitors. Bioorg. Med. Chem. 2018, 26, 3696–3706. 10.1016/j.bmc.2018.05.050. [DOI] [PubMed] [Google Scholar]
- Mughal E. U.; Sadiq A.; Ashraf J.; Zafar M. N.; Sumrra S. H.; Tariq R.; Mumtaz A.; Javid A.; Khan B. A.; Ali A.; Javed C. O. Flavonols and 4-thioflavonols as potential acetylcholinesterase and butyrylcholinesterase inhibitors: Synthesis, structure-activity relationship and molecular docking studies. Bioorg. Chem. 2019, 91, 103124. 10.1016/j.bioorg.2019.103124. [DOI] [PubMed] [Google Scholar]
- Mughal E. U.; Sadiq A.; Ayub M.; Naeem N.; Javid A.; Sumrra S. H.; Zafar M. N.; Khan B. A.; Malik F. P.; Ahmed I. Exploring 3-Benzyloxyflavones as new lead cholinesterase inhibitors: synthesis, structure–activity relationship and molecular modelling simulations. J. Biomol. Struct. Dyn. 2021, 39, 6154–6167. 10.1080/07391102.2020.1803136. [DOI] [PubMed] [Google Scholar]
- a Asgari M. S.; Azizian H.; Nazari Montazer M.; Mohammadi-Khanaposhtani M.; Asadi M.; Sepehri S.; Ranjbar P. R.; Rahimi R.; Biglar M.; Larijani B.; Amanlou M.; Mahdavi M. New 1,2,3-triazole-(thio)barbituric acid hybrids as urease inhibitors: Design, synthesis, in vitro urease inhibition, docking study, and molecular dynamic simulation. Arch. Pharm. 2020, 353, 2000023. 10.1002/ardp.202000023. [DOI] [PubMed] [Google Scholar]; b Türkeş C.; Akocak S.; Işık M.; Lolak N.; Taslimi P.; Durgun M.; Gülçin İ.; Budak Y.; Beydemir Ş. Novel inhibitors with sulfamethazine backbone: synthesis and biological study of multi-target cholinesterases and α-glucosidase inhibitors. J. Biomol. Struct. Dyn. 2021, 18, 1–13. 10.1080/07391102.2021.1916599. [DOI] [PubMed] [Google Scholar]; c Ashraf J.; Mughal E. U.; Sadiq A.; Naeem N.; Muhammad S. A.; Qousain T.; Zafar M. N.; Khan B. A.; Anees M. Design and synthesis of new flavonols as dual α-amylase and α-glucosidase inhibitors: Structure-activity relationship, drug-likeness, in vitro and in silico studies. J. Mol. Struct. 2020, 1218, 128458. 10.1016/j.molstruc.2020.128458. [DOI] [Google Scholar]
- Ashraf J.; Mughal E. U.; Sadiq A.; Bibi M.; Naeem N.; Ali A.; Massadaq A.; Fatima N.; Javid A.; Zafar M. N.; Khan B. A.; Nazar M. F.; Mumtaz A.; Tahir M. N.; Mirzaei M. Exploring 3-hydroxyflavone scaffolds as mushroom tyrosinase inhibitors: synthesis, X-ray crystallography, antimicrobial, fluorescence behaviour, structure-activity relationship and molecular modelling studies. J. Biomol. Struct. Dyn. 2021, 39 (18), 7107–7122. 10.1080/07391102.2020.1805364. [DOI] [PubMed] [Google Scholar]
- Ashraf J.; Mughal E. U.; Alsantali R. I.; Obaid R. J.; Sadiq A.; Naeem N.; Ali A.; Massadaq A.; Javed Q.; Javid A.; Sumrra S. H.; Zafar M. N.; Ahmed S. A. Structure-based designing and synthesis of 2-phenylchromone derivatives as potent tyrosinase inhibitors: In vitro and in silico studies. Bioorg. Med. Chem. 2021, 35, 116057. 10.1016/j.bmc.2021.116057. [DOI] [PubMed] [Google Scholar]
- Alsharif M. A.; Naeem N.; Mughal E. U.; Sadiq A.; Jassas R. S.; Kausar S.; Altaf A. A.; Zafar M. N.; Mumtaz A.; Obaid R. J.; Alsantali R. I.; Ahmed S.; Ahmed I.; Altass H. M.; Ahmed S. A. Experimental and theoretical insights into the photophysical and electrochemical properties of flavone-based hydrazones. J. Mol. Struct. 2021, 1244, 130965. 10.1016/j.molstruc.2021.130965. [DOI] [Google Scholar]
- Hassan S. Synthesis, antibacterial and antifungal activity of some new pyrazoline and pyrazole derivatives. Molecules 2013, 18, 2683–2711. 10.3390/molecules18032683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohiuddin G.; Khan K. M.; Salar U.; Kanwal M. A.; Lodhi M. A.; Wadood A.; Riaz M.; Perveen S. Biology-oriented drug synthesis (BIODS), in vitro urease inhibitory activity, and in silico study of S-naproxen derivatives. Bioorg. Chem. 2019, 83, 29–46. 10.1016/j.bioorg.2018.10.021. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Qadir M. A.; Hameed A.; Arshad M. N.; Asiri A. M.; Muddassar M. Azomethines, isoxazole, N-substituted pyrazoles and pyrimidine containing curcumin derivatives: Urease inhibition and molecular modeling studies. Biochem. Biophys. Res. Commun. 2017, 490, 434–440. 10.1016/j.bbrc.2017.06.059. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Gul-e-Saba I. M.; Ismail N.; Mohamad H.; Sung Y. Y.; Muhammad T. Induction of apoptosis by Aaptos sp., fractions in human breast cancer cell line, MCF-7. Int. J. Pharm. Sci. Res. 2018, 9, 328–337. [Google Scholar]
- Patel S.; Shah U. Synthesis of flavones from 2-hydroxy acetophenone and aromatic aldehyde derivatives by conventional methods and green chemistry approach. Asian J. Pharm. Clin. Res. 2017, 10, 403–406. 10.22159/ajpcr.2017.v10i2.15928. [DOI] [Google Scholar]
- Balamurugan C.; Kamalakkannan D.; Suresh R.; Sakthinathan S.; Vanangamudi G.; Thirunarayanan G. Synthesis, structure-parameter correlation and antimicrobial evoluation of 1-(4-isobutylphenyl)-3-phenyl-2-propenone compounds. Int. J. Life Sci. Res. 2016, 4, 7–22. [Google Scholar]
- Chen F. C.; Chang C. T. 30. Synthesis of 7-halogenoflavone and related compounds. J. Chem. Soc. 1958, 146–150. 10.1039/jr9580000146. [DOI] [Google Scholar]
- Gaur R.; Yadav D.; Kumar S.; Darokar M.; Khan F.; Bhakuni R. Molecular modeling based synthesis and evaluation of in vitro anticancer activity of indolyl chalcones. Curr. Top. Med. Chem. 2015, 15, 1003–1012. 10.2174/1568026615666150317222059. [DOI] [PubMed] [Google Scholar]
- Coutinho D. L. M.; Fernandes P. S. Synthesis and biological activity of some 2-Aryl-4H-l-benzopyran-4-ones. J. Indian Chem. Soc. 1992, 69, 265–267. [Google Scholar]
- Yang H.-M.; Shin H.-R.; Cho S.-H.; Bang S.-C.; Song G.-Y.; Ju J.-H.; Kim M.-K.; Lee S.-H.; Ryu J.-C.; Kim Y. Structural requirement of chalcones for the inhibitory activity of interleukin-5. Bioorg. Med. Chem. 2007, 15, 104–111. 10.1016/j.bmc.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Cabrera M.; Lavaggi M. L.; Croce F.; Celano L.; Thomson L.; Fernández M.; Pintos C.; Raymondo S.; Bollati M.; Monge A.; López de Ceráin A.; Piro O. E.; Cerecetto H.; González M. Identification of chalcones as in vivo liver monofunctional phase II enzymes inducers. Bioorg. Med. Chem. 2010, 18, 5391–5399. 10.1016/j.bmc.2010.05.033. [DOI] [PubMed] [Google Scholar]
- Amole K. L.; Bello I. A.; Oyewale A. O. Synthesis, Characterization and Antifungal Study of Five New Derivatives of E-1-(2-Hydroxyphenyl)chalcone. Chem. Afr. 2019, 2, 1–14. 10.1007/s42250-018-0027-3. [DOI] [Google Scholar]
- Kukharenko A. V.; Avramenko G. V. Synthesis of boron-containing complexes of 1-(2-hydroxyphenyl)-3-phenyl-2-propen-1-one. Russ. J. Gen. Chem. 2001, 71, 1562–1564. 10.1023/a:1013994718545. [DOI] [Google Scholar]
- Chen J.; Liu C.-F.; Rao G.-W. Progress in the synthesis, angiogenesis activity and mechanism of chalcone derivatives. Mini-Rev. Org. Chem. 2020, 17, 814–827. 10.2174/1570193x17666191223161941. [DOI] [Google Scholar]
- da Silva Lima D. C.; do Vale C. R.; Véras J. H.; Bernardes A.; Pérez C. N.; Chen-Chen L. Absence of genotoxic effects of the chalcone (E)-1-(2-hydroxyphenyl)-3-(4-methylphenyl)-prop-2-en-1-one) and its potential chemoprevention against DNA damage using in vitro and in vivo assays. PLoS One 2017, 12, e0171224 10.1371/journal.pone.0171224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshima T.; Takeishi M.; Arai T. Red fluorescence from tautomers of 2′-hydroxychalcones induced by intramolecular hydrogen atom transfer. New J. Chem. 2009, 33, 1393–1401. 10.1039/b823431k. [DOI] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S.; Suzuki J.; Segawa K.; Fujii T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. 10.1038/cdd.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z.; Galkowski D.; Zhao H. Analysis of apoptosis by cytometry using TUNEL assay. Methods 2008, 44, 250–254. 10.1016/j.ymeth.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.