

Abstract
The tumor microenvironment (TME) is a complex neighborhood that consists of immune cells, fibroblasts, pericytes, adipocytes, endothelial and neuronal cells, and the extracellular matrix proteins. TME also consists of physical factors, such as oxygen availability, changing pH, interstitial fluid pressure, and tissue stiffness. As cancer progresses, the physical properties and the cells in the TME change significantly, impacting the efficacy of the therapies and modulating drug resistance. This has led to the development of several new treatments targeting the TME. This review focuses on recent advances on the role of TME in drug resistance, with a particular focus on the ongoing clinical trials aiming at disrupting the TME-and the extracellular matrix-mediated protection against therapies.
Keywords: Tumor microenvironment, cancer-associated fibroblasts, extracellular matrix, drug resistance, stroma-cancer crosstalk, clinical trials
Cell types of the tumor microenvironment
In addition to the tumor cells themselves, the cell types that contribute to the changing tumor microenvironment (TME) (see Glossary) are the cancer-associated fibroblasts (CAFs), adipocytes, pericytes, neurons, endothelial and immune cells. Under non-pathological conditions these non-oncogenic cells are required for normal homeostasis. In cancer, although still genetically normal, their functions and behavior change significantly due to the effect of the tumor cells and the changing matrix and cytokine environment (Figure 1). Below we will describe some recent findings on how these cell types impact drug resistance.
Figure 1: Components of the tumor microenvironment (TME).
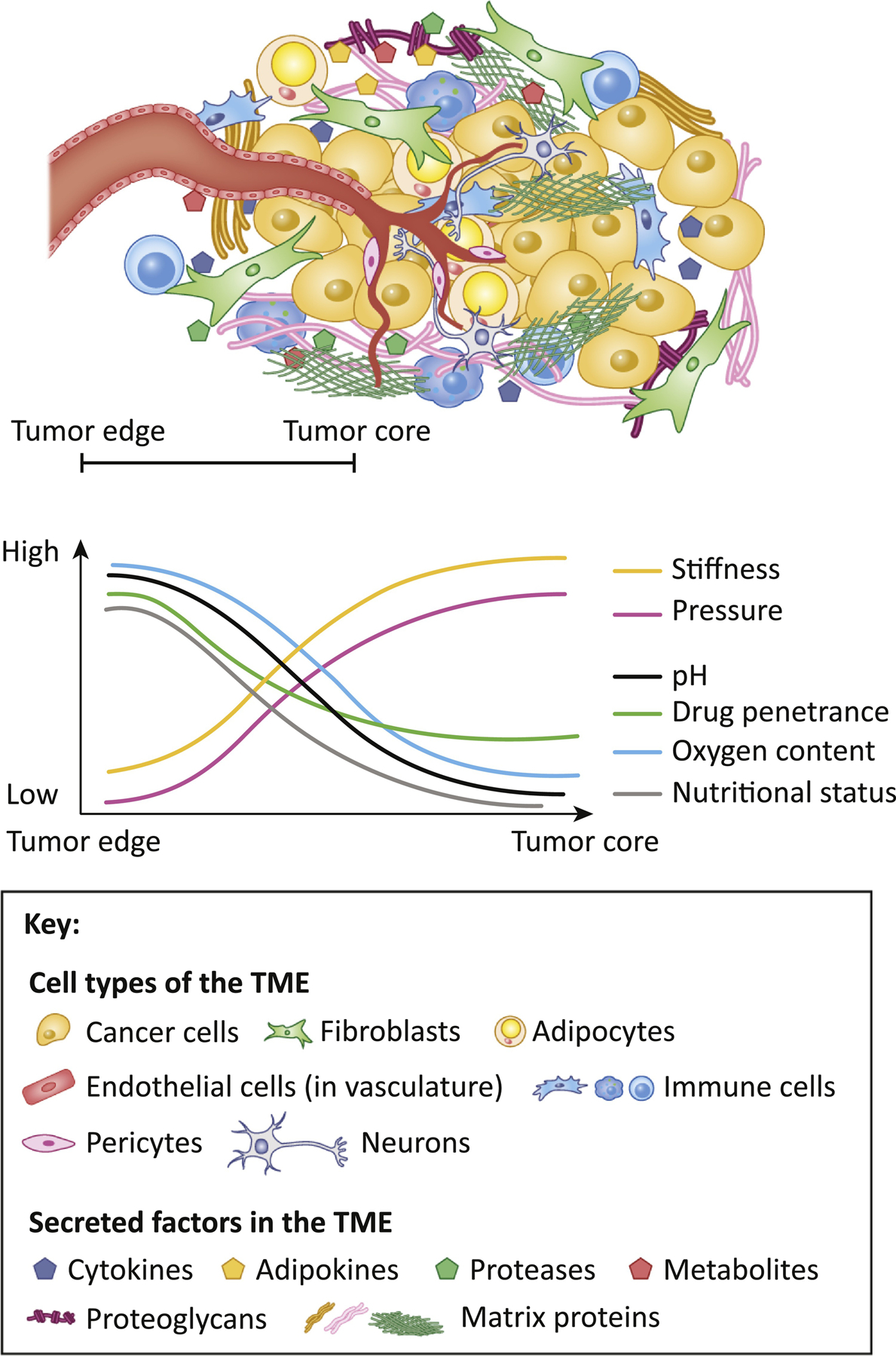
Cancer cells are surrounded by various cell types: fibroblasts, adipocytes, endothelial cells, pericytes, neurons and immune cells. The dense ECM that the tumors adhere to serves as a storage for growth factors, cytokines, lipids, proteases and metabolites, secreted by the cells of the TME. As tumor grows, the physical properties in the tumor core like oxygen and nutrient levels, pH, stiffness, and pressure become different from those on the edge of the tumor. These changes, so as the differences in matrix composition and alignment, influence the properties and behavior of other cell types in the TME, thus influencing drug penetrance, drug resistance and disease outcome.
CAFs:
CAFs, in addition to secreting extracellular matrix (ECM) proteins and cytokines, have recently been shown to secrete small metabolites that increase tumor fitness and promote drug resistance. For instance, in pancreatic cancer, the pancreatic stellate cells (PSCs) that produce the majority of the ECM and cytokines, recently were also shown to secrete alanine [1] and lipids that are taken up by the tumor cells to support the synthesis of phosphatidylcholines and lysophosphatic acid (LPA), thus favoring the proliferation and migration of the tumor cells [2]. Stellate cells were also shown to induce drug resistance by secretion of nucleosides that competed with Gemcitabine chemotherapy [3] [4]. It is also becoming evident that not all CAFs are tumor-promoting. Different populations of CAFs can have tumor-restricting effects, and targeting different CAF populations might have very different outcomes [5]. Tumor cells themselves can induce the CAFs to differentiate into these different populations with very distinct functions (e.g. inflammatory vs. myofibroblast-like CAFs), that in turn influence tumor and immune cell behavior [6, 7]. Furthermore, these different CAF subtypes and the ECM they secrete shape tumor cell heterogeneity, and are linked to differential drug responses and clinical outcomes in pancreatic [8] and breast cancer [9, 10]. CAFs have also been shown to secrete high amounts of cytokines such as Interleukine-6 and -8 (IL-6, IL-8), Transforming Growth Factor beta (TGF-β), and Leukemia Inhibitory Factor (LIF) [11] [12], most known for increasing stress tolerance and drug resistance [13–15], leading to efforts to target CAFs and CAF-secreted factors in cancer.
Adipocytes in the TME:
Obesity is a known risk factor for several cancers, and has been shown to be associated with therapy resistance [16–19]. Obesity can alter the TME by inducing chronic inflammation that has profound effects on all the cell types in the TME. In addition, adipocytes secrete lipids uptaken by the tumor cells. For instance, in hypoxia, tumor cells switch from glucose to lipid utilization, and adipocytes going through lipolysis ‘donate’ their lipids to the cancer cells, fueling their growth either at the primary site or during metastasis [20–22]. Obesity is also linked with increased IL-6 secretion, known to induce drug resistance [23].
Endothelial cells:
The stiff, expanded tumor stroma compresses the endothelial cell vasculature, contributing to intratumoral hypoxia. While still functional, the tumor vasculature is often leaky, with plasma proteins leaking out, and poor waste removal leading to the build-up of waste products and acidic pH. This greatly influences the tumor cells, their metabolism and energy utilization. Low pH, hypoxia and the ECM and cytokine environment also impact endothelial cell functions. Cytokines and pro- and anti-angiogenic signals embedded in the ECM guide endothelial cells to proliferate and to create new vasculature. This abnormal vasculature and endothelial cell behavior is considered to aid tumor cells, and to suppress the immune system [24].
Pericytes:
These are present in the microcapillaries and wrap the endothelial cells. They have a role in blood vessel formation and maintenance, stabilizing small blood vessels [25]. Recently it was shown that targeting the glioma stem-cell derived pericytes could disrupt the blood-tumor barrier and improve delivery of chemotherapy to the tumors [26].
Neural cells and perineural invasion:
Neurons are part of the TME, and newly formed neurons can infiltrate and proliferate in solid tumors. For instance, in prostate cancer tumors expressing neurotrophic factors attract neurons, causing neural progenitor cells to infiltrate and initiate neurogenesis [27]. The depletion of these progenitor cells could inhibit the early stages of tumor growth and metastasis [28]. Another aspect of the tumor-neuron crosstalk is the perineural invasion, where the tumor cells are attracted and invade the perineural space further driving cancer progression [29].
Immune cells and the TME:
Given that cross-talk between the TME and immune cells has been covered in recent reviews [30], we will briefly highlight some of the recent studies that pertain to the ongoing clinical trials.
In order for the tumor to form, it needs to escape the immune system, which normally would eliminate abnormal cells. The immune TME is composed of dendritic cells, Cytotoxic T cells (CTLs), Regulatory T cells (Tregs), tumor-associated macrophages (TAMs), Natural killer (NK) cells, neutrophils and myeloid-derived suppressor cells (MDSCs) [31]. We have briefly described the functions of these cell types in Box 1, to help the reader follow the rationale for eliminating or attracting these various immune cells into the TME.
Box 1. Immune cell types in the TME.
Dendritic cells:
present antigens to T cells either in the tumor or at lymph nodes to activate T cells.
Cytotoxic T cells (CTLs):
Activated by dendritic antigen presenting cells. Recognize the (neo) antigens of tumor cells that are not present in normal cells. Also involved in auto-immune diseases, which is why Tregs evolved to dampen the T cells activity. CD8+ T cells give rise to CTLs that kill cancer cells, CTLs can be generated by priming naïve T cells or reprogramming memory T cells. CTLs must overcome intrinsic checkpoints (e.g. PD-L1, CTLA-4), extrinsic checkpoints (Tregs and myeloid cells), inflamed TME (immunosuppressive environment), immune-evasion by tumor cells, and physical blocks such as desmoplasia or fibrotic TME.
Memory T cells:
Provide long-term protection, can differentiate into CTLs.
Natural killer cells (subset of T cells):
Release cytotoxic cytokines to kill tumor cells.
Regulatory T cells (Tregs):
Critical for the development of adaptive immune system and for immune tolerance, these often maturate into TAMs, function by dampening CTL activity.
Macrophages:
Engulf and digest debris, microbes, cancer cells that are recognized as foreign. Recruit other cell types such as lymphocytes through antigen presentation to T and B cells. Can have inflammatory (M1) and anti-inflammatory (M2) roles through differential cytokine secretion, and this polarization is regulated by the microenvironment and cytokine milieu.
Tumor-associated macrophages (TAMs):
recruited to tumors through cytokine secretion (e.g. CSF1, IL-34 and CCL2), which have been shown to induce the tumor-promoting phenotype. Are often in the tumor-promoting M2 polarization state that is regulated by cytokines in the TME (e.g. TGFβ).
Myeloid-derived suppressor cells (MDSC):
Induced by chronic inflammation, they protect tumor cells and help form immunosuppressive TME. Their presence predicts higher stage and worse survival. They also predict resistance to immunotherapies, and give rise to TAMs, and suppress T cell functions. Tumor cells can recruit them and alter their activity and proliferation by secreting cytokines (e.g. IL-6 and GM-CSF).
Neutrophils:
Develop subtypes that can either promote or suppress tumor growth. G-CSF leads to neutrophil expansion and polarization towards T-cell suppressive phenotypes.
Chronic inflammation:
Induces infiltration of cytokine-activated myeloid cells and immune suppressive B, T and myeloid cells, suppresses cytotoxic T cells and modulate the TME to promote tumor growth.
Dense fibrotic stroma is considered immunosuppressive partially because of the high levels of TGFβ that can exclude CTLs [32] and attract immunosuppressive TAMs. Immunosuppressive TAM formation is also stimulated by stiffness, hypoxia and the common ECM component, Hyaluronic acid (HA) [33][34]. In addition to immunosuppression, TAMs can also support metastasis via secretion of the ECM remodeling enzymes, such as matrix metalloproteinases (MMPs) [35] in response to the cancer cells cytokine secretion. Another factor inducing the immunosuppressive state in the TME is the Colony stimulating factor 1 (CSF1), which is secreted by the tumor cells and the CAFs, leading to more immunosuppressive TME through the recruitment of TAMs [36]. TAMs are recruited and regulated by CSF1-ligand receptor pair (CSF1/R), and its expression in tumors is associated with worse outcome and worse response to chemotherapy [37]. Another chemokine that can attract TAMs is the cytokine C-X-C motif chemokine 12 (CXCL12) that is secreted by CAFs and endothelial cells. CXCL12 receptor CXCR4 is frequently expressed in tumors and immune cells, and its expression is up-regulated by hypoxia, inflammation and fibrosis. CXCL12 secretion attracts Tregs, MDSCs and immunosuppressive TAMs that express the CXCR4 receptor, thus increasing immune-suppression in hypoxic tumors [38].
Overall, intra-TME crosstalk, between the immune cells, ECM and the other cell types, poses a complex area of biology, and understanding this is of paramount importance for the development of successful immunotherapies in stroma-rich cancers.
The changing physical landscape of the TME in cancer progression
The physical and biochemical properties of the TME play a significant role in tumor progression and metastasis [40]. The TME is often referred to as a ‘wound that does not heal’, and is based on the tumors invoking programs closely resembling wound healing response in its recruitment and activation of the stroma to induce desmoplasia, with similarity to scar tissue [39]. This aberrant and fibrotic stroma influences the physical properties of the TME, and is very different from the normal stroma (Box 2).
Box 2. Basement membrane vs. stromal/interstitial matrix.
In normal tissues ECM comes in two varieties: 1) as stromal (or interstitial) ECM, or 2) as basement membrane. The stromal/interstitial ECM, that surrounds the mesenchymal cells and fills the space between organs, is secreted by mesenchymal fibroblasts and is composed mostly of collagens, fibronectin, elastin, glycosaminoglycans and proteoglycans. The basement membranes are a layer of deposited matrix proteins and consist mostly of laminins, collagen IV, nidogen, heparan sulphate proteoglycans perlecan and agrin. The basement membranes form the basal surface onto which polarized epithelial and endothelial cells adhere to. Basement membrane is required for normal epithelial cell polarity and function, but also restricts cell proliferation in the presence of oncogenic mutations, functioning as a barrier to carcinoma progression.
Matrix proteins themselves can increase tumor fitness and drug resistance through multiple mechanisms (Box 3). For example, the proteoglycan perlecan, secreted by the tumor-educated CAFs, can induce resistance to chemotherapy [41], and similarly, fibronectin can induce therapy resistance in breast cancer, contributing both to endocrine- [42] and chemotherapy resistance [43]. Recent data also showed that collagen remodeling contributes to melanoma metastasis, particularly during aging, where more aligned collagen increased metastases [44]. This is in line with the body of work from Patricia Keely’s group who showed that tumor-associated collagen signatures (TACS) are important predictors of tumor progression and therapy resistance (Box 4).
Box 3. ECM induced drug resistance.
ECM has been acknowledged for more than 20 years as a driver of drug resistance. Several groups showed that tumor drug responses differ between 2D and 3D cultures, and tumor cells cultured on different matrices (basement membrane vs. interstitial matrix) respond to drugs differently, some matrices inducing drug resistance while others sensitizing tumor cells to drug treatments [114, 115]. These insights have prompted efforts to generate better in vitro models, combining 3D cell culture models with different oxygen levels and different matrix components and adding different cell types from the TME to mimic more complex environments that could better predict tumor drug responses [116].
Box 4. Tumor-associated collagen signatures (TACS) in cancer and therapy resistance.
The composition and the organization (e.g. alignment) of matrix proteins (collagen most prominently) in the TME differs significantly from their normal tissue counterparts [117]. In particular in breast cancer it has been shown that patients with dense breast tissue have higher risk of developing breast cancer [118], and in animal models collagen density and organization can promote breast cancer initiation and progression. TACS have provided a useful marker to characterize survival and tumor invasiveness in breast cancer [119]. Furthermore, studies in melanoma have shown that treatment resistance correlates with the presence of bundled collagen [120] and stiffer ECM [121].
ECM proteins also induce survival signaling in the tumor cells through integrin engagement leading to the downstream activation of tyrosine kinases (c-Src and Focal adhesion kinase [FAK]) known to promote cell survival and drug resistance [14, 45–47]. These integrin-ECM adhesions and signaling also contribute to increased invasion, migration and metastasis in nutrient poor conditions [48]. Another less conventional role for ECM in increasing tumor cell fitness is to use it as a nutrient source [49]. Particularly Kirsten rat sarcoma viral-oncogene homolog (KRAS)-mutant cancers can use macropinocytosis to ingest any proteins in close proximity to increase their nutrient supplies under nutrient limiting and hypoxic conditions [50, 51]. Since ECM proteins are present in abundance in the TME, the cancer cells take advantage of this unconventional nutrient supply [52]. Therefore, the ECM environment regulates several aspects of the tumor cell behavior and is an attractive target for improving the efficacy of more traditional cancer therapies.
Interestingly, the source of the matrix also seems to play a role in tumor progression; the pancreatic cancer matrix proteome (matrisome) was recently characterized [53] and although the stromal fibroblasts secreted >90% of the tumor ECM mass, it was the tumor cell-secreted matrix proteins presence that correlated with the poorest patient survival. These data suggest that perhaps targeting the tumor-secreted ECM proteins might benefit patients more rather than targeting the stroma-secreted ones.
A less traditional cause for drug resistance are the extra-cellular vesicles secreted by multiple cell types in the TME; they carry a multitude of information (e.g. proteins, RNA, non-coding RNAs, and metabolites) that can provide protection against drugs as well as prepare pre-metastatic niches and modify the TME. These aspects of the TME have been recently reviewed so we will not cover this topic here further [54, 55].
Changes in the composition of the ECM proteins modify the physical properties of the TME. It is well known that tumor elastic modulus (stiffness) is several times higher than normal tissue counterparts. This leads to more aggressive and migratory tumors, and, for example, stiffer breast tissue correlates with increased cancer risk, suggesting that mechano-sensing also drives cancer progression [56]. Interestingly, there appears to be a threshold after which the stiffness becomes tumor suppressive again, suggesting a bell-shaped effect on cancer growth and invasion [57]. Stiffer TME also impacts intracellular signaling triggered by integrins, FAK and c-Src kinases. Secretion of matrix crosslinking enzymes, like collagen prolyl hydroxylases, lysyl hydroxylases, lysyl oxidases (LOXs), and weaker ECM linkers, such as Hyaluronan and Proteoglycan Link Protein 1 (HAPLN1), by the tumor cells and the CAFs can significantly increase tissue stiffness [58]. These enzymes fold and align the collagen fibers and crosslink them with elastin molecules, making the ECM mechanically durable and stiff, and increase cancer progression and metastasis [58].
Another physical feature of the TME is the high interstitial fluid pressure (IFP). One of the molecules that increases this is HA, a predominant glycosaminoglycan in the TME, highly expressed in pancreatic, breast, lung and colorectal cancer [59]. HA increases the IFP by trapping water molecules. A by-product of this increased pressure is collapsing vasculature. The collapsed vasculature further increases IFP, creating a vicious cycle leading to intratumoral hypoxia, known to contribute to cancer progression and resistance to therapies by a variety of mechanisms, including direct regulation of ECM homeostasis through the activity of Hypoxia inducible factors (HIFs) [60]. Hypoxia can dramatically increase the synthesis rate of ECM proteins [61], as well as other non-structural matricellular proteins like thrombospondins, osteonectins, tenascins, osteopontin, periostin, and fibulins [62]. HIF-1α was shown to upregulate the expression of several hydroxylases [63] and LOX’s under hypoxic conditions [64], contributing to ECM remodeling and stiffening TME. Furthermore, hypoxia also influences the remodeling of ECM via enhanced expression of proteolytic enzymes like MMPs, and the components of the plasminogen activation system [65]. These dynamic changes in the ECM lead to a more effective uptake of cytokines, growth factors and adipokines secreted by different cell types residing in the TME.
Targeting TME in cancer
Over the years several approaches have been used to target the TME. These approaches include targeting cancer cell-ECM adhesions, matrix proteins, cytokines, and stromal cells directly (Figure 2). Given the sometimes tumor-promoting sometimes tumor-restricting role of the ECM and other TME components in cancer, the preclinical and clinical trials targeting the TME have had mixed results. Yet, the number of clinical trials targeting the TME has been growing (Table 1), and the biomarkers known to be indicative of clinical outcomes have steadily grown as well [66]. The mixed results from the stroma targeting are likely to be explained by its ability to form a physical barrier around the tumor, trapping the tumor cells and preventing metastatic spread, but at the same time providing tumor cells with nutrients and protecting them from drug treatments. In the next chapters we will discuss the current ongoing trials that are targeting different aspects of the TME.
Figure 2: Current strategies to target tumor stroma in clinical trials.
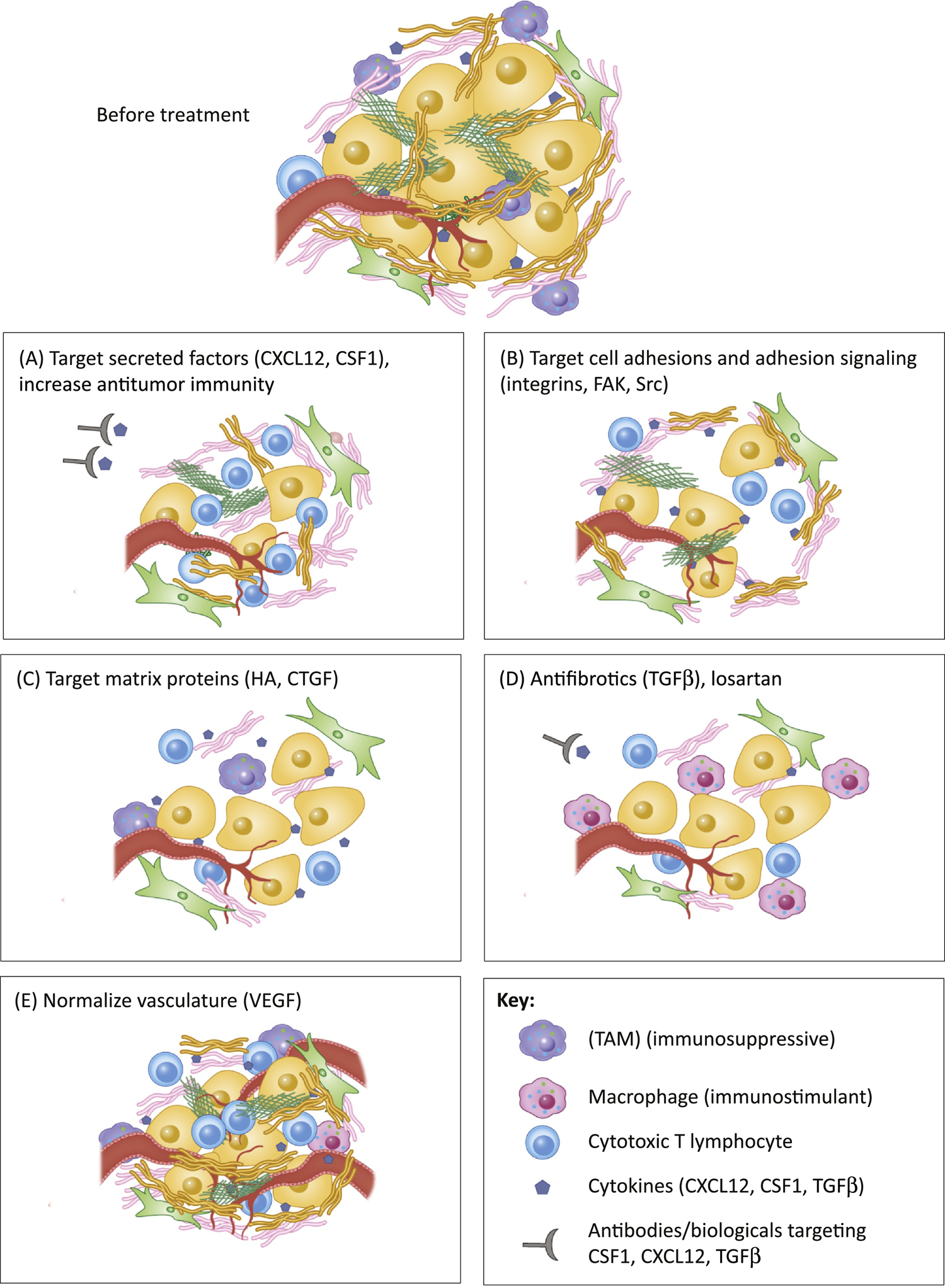
Different strategies currently used in the clinic or in clinical trials to target stroma in cancer include A) increasing anti-tumor immunity by targeting secreted factors (e.g. CXCL12, CSF1) that decrease the presence of TAMs and increase the infiltration of cytotoxic T lymphocytes; B) targeting ECM-cell adhesion molecules such as integrins, Src and FAK kinases (often in combination with other drugs) to reduce tumor cell fitness and tumor drug resistance to other therapies; C) targeting matrix proteins such as Hyaluronic acid and connective tissue growth factor (CTGF) to normalize tumor stroma and interstitial pressure and to normalize vasculature and reduce hypoxia, and to increase delivery of chemotherapies and increase the access of immune cells; D) use of anti-fibrotics and TGFβ inhibitors to reduce fibrosis, increase drug delivery, normalize vasculature, reverse hypoxia and increase immune cell infiltration; E) use of VEGF inhibitors, to normalize vasculature and increase cytotoxic T lymphocyte and immune cell infiltration.
Table 1.
Clinical trials targeting stroma in cancer; all trials vs. trials that are active, recruiting patients and not yet recruiting were included in this table.
| Clinical Trials Cancer Only Trials | |||
|---|---|---|---|
| Target | Compound | # of all Trials: | # of Active, Recruiting and Not yet recruiting trials: |
| Vitamin D | Vitamin D, not specified | 396 | 114 |
| Paricalcitol | 43 | 12 | |
| Cholecalciferol | 141 | 40 | |
| Hyaluronan | PEGPH20 | 22 | 13 |
| Integrins | Integrin targeting trials | 146 | 53 |
| Src | Src targeting, other | 117 | 37 |
| Saracatinib | 26 | 1 | |
| FAK | FAK targeting, not specified | 41 | 12 |
| Defactinib/VS-6063 | 14 | 6 | |
| TGFβ | TGFβ targeting trials, not specified | 168 | 77 |
| Losartan | 12 | 6 | |
| Galunisertib | 21 | 12 | |
| AVID200 | 2 | 2 | |
| Fresolimumab | 7 | 1 | |
| LY3200882 | 3 | 3 | |
| Vactosertib | 8 | 7 | |
| Anti-PD-L1/TGFβRII Fusion Protein M7824 | 17 | 16 | |
| CXCR4 | CXCR4 | 61 | 27 |
| CXCL12/SDF1a | CXCL12/SDF1a | 45 | 14 |
| CSF1 | All CSF1 targeting trials | 256 | 95 |
| CSF1R | All CSF1R targeting trials | 42 | 30 |
| VEGF | Sevacizumab | 3 | 1 |
| Avastin/Bevacizumab | 1912 | 488 | |
| Vanucizumab (angiopoetin) | 4 | 2 | |
| Cabozantinib (c-MET, VEGFR2, AXL, RET) | 145 | 108 | |
| Axitinib | 131 | 51 | |
| Regorafenib | 171 | 89 | |
| Apatinib | 287 | 194 | |
| IBI305 | 38 | 35 | |
| Ramucirumab | 112 | 63 | |
| Cediranib | 103 | 30 | |
| CTGF | Pamrevlumab | 1 | 1 |
Targeting tumor cell adhesions
Targeting integrin-ECM interactions has been an attractive target for cancer therapy given that integrins are involved in cancer progression, invasion, metastasis and drug resistance [67] [68]. Integrins are heterodimeric adhesion receptors that bind ECM proteins, adhesion molecules and TGFβ. They regulate signals from the outside of the cells to the inside [69] and signal to c-Src and FAK. Therefore, small molecules and blocking antibodies have been developed to target integrins and their downstream pathways. Integrin heterodimers αV/β3 and αV/β5 are increased in melanoma and glioblastoma, and associated with invasion and poor survival, and inhibitors for these heterodimers have been developed and tested in pre-clinical and clinical trials [67]. However, despite showing promise in early preclinical trials, the larger trials have failed [68]. Targeting integrin αV/β6 has also shown mixed results, showing tumor shrinkage in some models and increasing tumor growth in others [67]. However, it was recently shown in preclinical models that targeting β1 integrin prevented the disseminated tumor cells from adhering to the perivascular niche and sensitized them to chemotherapy [70]. Thus, the rationale to target integrins in cancer remains. Unfortunately, so far, the clinical trials have not met their primary endpoints. This might be due to the large amount and overlapping functions of integrins, and also the overlapping downstream pathways that integrin heterodimers regulate. Therefore, many current clinical trials are targeting the kinases downstream from integrins, c-Src and FAK.
Targeting FAK
FAK is activated by the stiff and abundant ECM in several cell types in the TME [71]. It has a role in cell motility, adhesion, survival and drug resistance, and its differential activation status in the stromal cells vs. the tumor cells might play a role in its therapeutic efficacy [72]. Of note, FAK targeting in the endothelial cells can sensitize tumor cells to chemotherapy [73]. Several FAK inhibitors have shown encouraging clinical efficacy when used in combinations with other therapies. In a mouse model of pancreatic ductal adenocarcinoma (PDAC), FAK inhibitor, defactinib, decreased the number of tumor-infiltrating immunosuppressive cells, tumor fibrosis, and the formation of liver metastases [74]. Defactinib is currently in phase I/II clinical trials for advanced PDAC in combination with Pembrolizumab (PD-1) and Gemcitabine (Clinical Trial Numberi: NCT02546531), and in ovarian cancer with carboplatin and paclitaxel (NCT02546531). A Phase II study is ongoing in PDAC to combine Defactinib with Pemrolizumab in patients with resectable PDAC (Clinical Trial Number: NCT03727880), so as several other trials listed in Table 1.
Targeting c-Src
Src-family kinases (SFK) are non-receptor tyrosine kinases that transduce mitogenic, survival, angiogenic and migratory signals from receptor tyrosine kinases, G-protein coupled receptors, steroid hormone receptors, and ECM receptors [75]. There are 11 members of SFK among which Src, Fyn and Yes are ubiquitously expressed. Activation of SFK leads to the induction of several downstream signaling survival pathways, and increased activation of SFKs is associated with cancer progression. Therefore, several inhibitors have been developed to target this pathway. Dasatinib is an FDA-approved inhibitor of SFK for the treatment of chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphocytic leukemia (ALL), and there are several ongoing clinical trials of dasatinib in solid malignancies (Table 1). In addition, AZD0530 (saracatinib), bosutinib and imatinib have been used in solid tumor therapies. Dasatinib and imatinib target CSF-1 signaling in tumor-associated macrophages [76, 77], suggesting a possible role of c-Src inhibition in the modulation of TME. Further supporting this option are the studies demonstrating that pharmacological or genetic (shRNA) inhibition of c-Src decreases survival of breast cancer cells that have metastasized to the bone marrow but not to the brain or liver [78]. The bone marrow environment provided CXCL12 to support c-Src-dependent survival of the cancer cells. These data indicate a critical role for TME in shaping cancer cell responses to SFK inhibitors. Therefore, understanding the mechanisms of TME-mediated SFK activation in cancer cells and in the other cell types in the TME will provide critical information on how to normalize TME-mediated activation of oncogenic signaling in cancer cells.
Targeting Vascular Endothelial Growth Factor (VEGF)
Aberrant secretion of pro-angiogenic signals such as VEGF in the TME often leads to abnormal vasculature aiding tumor cells and suppressing the immune system [24]. VEGF blocking therapies have been used to try to normalize the tumor vasculature and to improve therapy responses [79]. However, as a monotherapy, VEGF inhibitors have not been able to result in tumor shrinkage, but they show great promise used as combination therapies, and FDA recently approved a combination of Programmed Death-Ligand 1 (PD-L1) and VEGF1 blockade for the treatment of metastatic nonsquamous non-small cell lung carcinoma (Clinical Trial Number: NCT02366143), and numerous other trials are ongoing (Table 1). Another approach for normalizing vasculature could be the use of lysyl oxidase (LOX) inhibitors that prevent collagen cross-linking. However, LOX function blocking antibodies, although showing promise in preclinical trials, failed in the clinical trials for pancreatic and colorectal cancer. It is likely that success of the approaches involving stroma targeting strongly relies on both the timing, and the combination of the drugs used.
Targeting stromal cytokines and chemokines
Several cell types in the TME secrete CSF1 and CXCL12. This secretion attracts immunosuppressive cells making immunotherapy approaches less successful. Data indicate that high presence of the CSF1R positive MDSCs in the TME is associated with poor clinical responses to chemotherapy and reduced overall survival, and that CSF1R blockade can promote an immuno-stimulatory TME [80]. More recently it was shown that targeting TAMs by blocking CSF1R in breast cancer models led to an increase in interferon signaling in the tumors, enhancing chemotherapy efficacy and targeting immunosuppressive neutrophils in this model [81]. CSF1R antagonists including cabiralizumab are in clinical trials in pancreatic cancer combined with immune checkpoint blockade ii (also see Clinical Trial Number: NCT03599362, NCT03697564, NCT03336216, NCT02526017). It is thought that depleting TAMs may increase cytotoxic T-cell responses and sensitize tumors to anti PD-1 treatments. However, recent data also indicate that inhibition of CSF1R can change CAF secretome and result in accumulation of tumor-promoting MDSCs and that combination treatments are needed for the CSF1R inhibitors to be most effective [82].
Blocking the CXCL12 receptor, CXCR4, has been shown to reduce desmoplasia, increase CTL infiltration and improve immunotherapy efficacy in metastatic breast cancer models that are normally resistant to immunotherapies [83]. Interestingly, the immunosuppressive effects in this model were dependent on the CXCR4 signaling in CAFs and pericytes. In immuno-competent ovarian cancer mouse model, CXCR4 blockade led to increased apoptosis and necrosis in tumor cells, reduction of intraperitoneal dissemination, and increased antitumor immune response [84]. These preclinical data led to the initiation of several clinical trials testing CXCR4 blockade in pancreatic cancer (Table 1), and also to the development of newer CXCR4 inhibitors, such as BL8040 which is currently in clinical trials for PDAC in combination with Pembrolizumab (Clinical Trial Number: NCT02826486) and 5FU/nalirinotecan (Clinical Trial Number: NCT02907099, NCT02826486), and as a basket study (Clinical Trial Number: NCT03193190). Similar approaches are used in renal cell carcinoma, squamous cell carcinoma, and melanoma, where several trials are combining CSF1R or CXCR4 inhibitors with Axitinib (VEGFR inhibitor) or with Pembrolizumab (Clinical Trial Number: NCT04058145) and have showed encouraging early results iii). Another approach to increase anti-tumor immunity by targeting stroma is by anchoring intratumorally administered cytokines (IL-2 and IL-12) to collagen binding lumican. This was shown to potentiate systemic immunotherapy and reduce toxicity [85].
Targeting ECM
High MMP expression has been linked to poor prognosis given that MMPs can modulate the ECM, enabling tumor cell migration and metastasis. Several small molecule inhibitors targeting MMPs have been developed that have been tested in clinical trials (Clinical Trial Number: NCT00004147, NCT00003721, NCT00001683, NCT00020683). Disappointingly, these trials failed for lack of efficacy, poor oral bioavailability and toxicity, prompting the development of safer and more specific biologics, such as function-blocking antibodies, currently in clinical trials (Clinical Trial Number: NCT02864381, NCT02545504, NCT03486730, NCT03631836) [86].
Other targets
Epidemiological studies suggest that vitamin D supplementation can reduce cancer risk, but this might be due to its effect on the TME rather than on cancer cells. Recent reports have described a role for the vitamin D receptor in the TME in pancreatic stellate cells, where treatment with the vitamin D analog calcipotriol resulted in a normalization of the stroma, reduced fibrosis and increased drug penetration, it also suppressed PDAC metastasis by inhibiting secretion of cytokines from the pancreatic stellate cells [87]. Vitamin D has a protective effect also in colorectal carcinoma through its high receptor expression in the CAFs [88] as well as by suppressing the secretion of tumorigenic microRNAs from the CAFs [89]. These pre-clinical studies prompted numerous clinical trials that are investigating whether addition of Vitamin D or its analogs would result in better disease outcomes in cancer (Table 1). However, even the preclinical data have been somewhat mixed in their outcomes, some studies showing positive outcomes, yet others finding no correlation with improved patient outcome [90]. Recently the largest-ever clinical trial that investigated vitamin D’s effect in cancer prevention, failed to find any link [91]. Perhaps one reason for this outcome is the different effects of the vitamin D in tumor cells vs. stromal cells. A synthetic lethal screen found that vitamin D receptor knock-down sensitized tumor cells to gemcitabine treatment [92]. This is likely because vitamin D supplementation has been shown to activate the anti-oxidant responses and DNA repair pathways [93], thus promoting resistance to chemotherapies. Therefore, successful Vitamin D receptor targeting in the CAFs vs the tumor cells might increase Vitamin D therapeutic efficacy.
Targeting Hyaluronic acid (HA) has delivered some successes in preclinical trials and led to phase 3 trial in pancreatic cancer. HA rich tumors are correlated with poorer prognosis, have extremely high interstitial fluid pressure (IFP), poor perfusion and poor drug accumulation [94]. Therefore, HA has posed an attractive target in cancer therapy. The ablation of stromal HA normalizes IFP and re-expands the vasculature. In combination with gemcitabine, the treatment resulted in a near doubling of overall survival in mouse models of PDAC [95–98]. These data led to a phase 2 trial of PEGPH20 hyalurodinase with gemcitabine/nab-paclitaxel in untreated metastatic PDAC. This trial showed improved progression free survival in patients with high HA content in their tumors [99]. A phase 1b/II trial was initiated combining PEGPH20 with FOLFIRINOX (FOLinic acid-Fluorouracil-IRINotecan-OXaliplatin) [100] but was closed early when it showed poorer survival in the PEGPH20 group. The unexpected results from this trial could be due to the fact that the patients in the combination group had experienced more treatment-related toxicities, which resulted in lower drug doses, dose delays and reduced exposure. The next phase III trial used gemcitabine nab-paclitaxel as the chemotherapy backbone, and included patients with HA high tumors. Unfortunately, this Phase 3 trial was also recently stopped early as it did not meet the primary end-point, increasing survival iv. One might speculate that perhaps some of these disappointing results can be explained by a recent study that showed that Hyaluronidase treatment was linked to increased tumor metabolism and led to a robust increase in tumor cell glycolysis through degradation of Thioredoxin-Interacting Protein (TXNIP) RNA required for glucose transporter internalization. This resulted in upregulation of the glucose transporter 1 (GLUT1) at the plasma membrane, increased glucose uptake, and increased migration and metastasis of the tumor cells [101]. These data might suggest that breaking down HA with hyaluronidase might have unwanted consequences, leading to increased metastasis.
Anti-fibrotics and TGFβ
Anti-fibrotics and TGF-β inhibitors work partially by reducing fibrosis and are being tested in several clinical trials (Table 1). TGFβ induces fibrosis through stimulating ECM secretion and its own secretion in a feed-forward fashion [102]. It is deposited to ECM in its latent form and needs to be processed to obtain its activated form. Integrins can help activate the latent TGFβ [103] and the efforts to target integrins were in part aimed at inhibiting TGFβ signaling [67]. Through reducing fibrosis the anti-fibrotics and TGF-β inhibitors are thought to allow other cancer drugs to penetrate the tumors more effectively [104].
Ajulemic acid is a synthetic cannabinoid derivative that was shown to have anti-fibrotic and anti-inflammatory effects, and is being tested in phase III clinical trials as an anti-fibrotic agent in several diseases under the trade name Lenabasum. In pancreatitis it can normalize the activated stellate cells [105]. Its mechanism of action has been shown to be through activation of the cannabinoid receptor 2 that leads to production of eicosanoids, decreasing inflammatory cytokines, and inhibiting TGFβ production. Thus, there is some interest to test this compound in combination with chemotherapy in stroma-rich cancers.
Recently halofuginone, an antifibrotic agent, was used to normalize the tumor stroma in a mouse model of PDAC. This treatment increased drug delivery by decreasing fibroblast activation and reducing ECM elements. Treatment also altered the immune landscape in PDAC, with increased number and distribution of activated inflammatory macrophages and cytotoxic T cells. This led to a widespread intra-tumoral necrosis and reduced tumor volume [106]. However, no clinical trials are currently using this compound.
Targeting TGFβ-expressing CAFs with pirfenidone, an anti-fibrotic agent and a TGF-β antagonist, in a triple-negative breast cancer mouse model inhibited tumor fibrosis and TGFβ-signaling and in combination with doxorubicin prevented metastasis [107] suggesting that the stroma-targeting agents are most effective when used in combination with other cancer therapies. Pirfenidone is currently in a phase 1 clinical trial in advanced stage non-small cell lung cancer in combination with chemotherapy (Clinical Trial Number: NCT03177291).
Angiotensin II receptor agonists, such as Losartan, are normally used to treat high blood pressure, but they also inhibit TGFβ signaling, resulting in reduced desmoplasia through reduced collagen I and HA deposition, and increasing vascular integrity and improved drug delivery to tumors [108–110]. Losartan has also been implicated in reduced mortality in certain cancers [109, 111], and is being evaluated in clinical trials in locally advanced PDAC in combination with FOLFIRINOX, nivolumab and radiotherapy (Clinical Trial Number: NCT03563248). The results from a previous similar trial (Clinical Trial Number: NCT01821729) were encouraging, showing a surgical resection rate of 61% in the treated patients (surgical removal of the tumor is critical to cure PDAC), with overall median progression-free survival of 17.5 months, and median overall survival of 31.4 months [112]. In ovarian cancer, tumor fibrosis and angiotensin-driven fibrogenic signaling are inversely correlated with survival, and Losartan treatment enhanced chemotherapy efficacy in ovarian cancer xenograft models by normalizing the tumor stroma. The authors also found in a retrospective analysis that patients receiving angiotensin system inhibitors concurrently with standard treatment for ovarian cancer exhibited longer overall survival compared with patients on other anti-hypertensives [113].
Concluding remarks and Future perspectives
In conclusion, the efforts to target stroma in cancer are still not as successful as hoped for, with very mixed results coming from the clinical trials. This is likely because the TME-cancer crosstalk is still inadequately understood and perhaps normalizing the stroma, targeting the stroma-induced pathways in tumor cells, or the tumor-secreted ECM, rather than completely ablating the stroma, might be a better strategy for improved outcomes. Furthermore, different cancers have very different stromal and matrix environment that likely impact therapeutic efficacies (see Outstanding Questions). On one hand, the stroma would need to be more penetrable for better drug delivery, but on the other hand, not ablate its ability to restrict metastasis. The results coming from the clinical trials using anti-fibrotics could do this by allowing other tumor targeting agents and immune cells to penetrate and reach the cancer cells that have been previously protected.
Outstanding questions.
How to effectively normalize the stroma?
Several trials are targeting stroma (e.g. VEGF, PEGPH20, Losartan) in combination with immune checkpoint inhibitors, particularly in cancers that have been traditionally considered immune cold. Will these approaches change the current treatment strategies for these extremely hard-to-treat, drug resistant cancers?
How to translate measurement of stromal density and composition (biomarkers) into clinical applications? What are the best ways to stratify patients for clinical trials targeting stroma?
How do chemotherapy and targeted therapy shape different components of the TME?
Stroma targeting has also the potential to increase immune cell infiltration and activation, particularly in traditionally immune cold cancers such as breast and pancreatic cancer, and this is an exciting avenue of research, with several clinical trials testing checkpoint inhibitors with stroma targeting drugs. There are still many hurdles to be crossed, particularly with patient stratification and biomarker identification, and understanding which parts of the tumor stroma should be targeted and which aspects are more helpful at restricting tumor cells in their original location. Also, a more thorough understanding of the underlying biology is necessary to optimize the use of stroma-targeting agents. Lastly, it is important to understand how the current therapies change the TME, to evaluate whether therapies evoke pro-, or anti-tumorigenic behavior in TME, or both, and which cell types are affected. This might allow us to identify cell type specific vulnerabilities that could be used to target tumor cells more efficiently.
Highlights.
Tumor microenvironment is a habitat for cancer cells, immune cells, fibroblasts, endothelial cells, pericytes, neural cells, adipocytes and the extracellular matrix (ECM) proteins, significantly influencing the efficacy of the modern cancer therapeutic agents, in some cases inducing resistance, in others sensitizing the tumors to therapy.
Stiff and abundant stroma is able to restrict and promote cancer cell dissemination both at the same time, due to the dynamic remodeling of the ECM. Anti-cancer drugs aiming at complete depletion of the stroma have led to enhanced rate of metastases, but too abundant stroma blocks drug penetrance and leads to tumor growth, therefore efforts to normalize the tumor stroma are tested increasingly in the clinic.
Current treatment approaches of the stroma-rich cancers include targeting proteins and cytokines secreted and expressed by the stromal cells, targeting cancer cell–stroma signaling interactions, and targeting stroma-immune cell crosstalk increasing anti-tumor immunity.
ACKNOWLEDGEMENTS
This work was supported by Susan G Komen grant #CCR18547665 and American Cancer Society grant #RSG-19-0201-CSM to T.M. and Kaleidoscope of Hope Ovarian Cancer Foundation and Olipass Inc grants for M.I.
GLOSSARY
- Cancer-associated fibroblasts (CAFs)
cancer-associated fibroblasts, a cell type within the tumor microenvironment that promotes tumorigenic features by remodeling the extracellular matrix or by secreting cytokines.
- CSF1
The colony-stimulating factor 1, also known as macrophage colony-stimulating factor, a secreted cytokine regulating the survival, proliferation and differentiation of macrophages and monocytes.
- CXCL12
Stromal cell-derived factor 1 (also called SDF1a), active on T-lymphocytes and monocytes but not neutrophils via activation of the C-X-C chemokine receptor CXCR4 to induce a rapid and transient rise in the level of intracellular calcium ions and chemotaxis.
- Desmoplasia
Excessive deposition of ECM proteins causing dense fibrosis around tumors or other affected organs.
- Extracellular matrix (ECM)
consists of proteins that form the 3D like-meshwork of proteoglycans and fibrous proteins, such as laminins, collagens and fibronectin. It provides structural and biochemical support to the tissues it is in contact with and also acts a storage and sequester of growth factors that the cells in the TME secrete.
- Fibrosis
the formation of an excessive ECM deposition occurring as a consequence of inflammation, activation of the stromal cells or other injury.
- Hyaluronic acid (HA)
a predominant glycosaminoglycan in the TME, highly expressed in pancreas, breast, lung and colorectal cancers.
- Neurogenesis
A process by which cells of the nervous system are generated from stem cells.
- PEGPH20
pegvorhyaluronidase alfa, the name of a drug degrading hyaluronan, potentially increasing drug delivery in stroma-rich cancers.
- Pericytes
Fibroblast-like cells present in intervals in the capillaries and venules and wrap around the endothelial cells of the microvessels. They are important for blood vessel formation and maintenance, and they also maintain the blood-brain barrier and regulate immune cell entry into the central nervous system.
- Perineural invasion
Invasion of cancer cells to the area surrounding the nerve. Can make the resection of the lesions more difficult, frequently seen in head and neck, prostate and colorectal cancer.
- Stiffness
mechanical property of tumors defining the rigidity of the TME and the extent of tumors’ resistance to deformation, depending on the composition and organization of the ECM strands and other structural components of the TME.
- TME
tumor microenvironment consisting of the cancer, immune cells, fibroblasts, endothelial cells, adipocytes and the extracellular matrix (ECM) proteins.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
RESOURCES:
REFERENCES
- 1.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, Asara JM, Evans RM, Cantley LC, Lyssiotis CA, and Kimmelman AC, Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature, 2016. 536(7617): p. 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, Vringer E, Schug M, Novo D, Hwang RF, Evans RM, Nixon C, Dorrell C, Morton JP, Norman JC, Sears RC, Kamphorst JJ, and Sherman MH, A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov, 2019. 9(5): p. 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, Hong HS, Kremer DM, Nelson BS, Kemp S, Zhang L, Chang D, Biankin A, Shi J, Frankel TL, Crawford HC, Morton JP, Pasca di Magliano M, and Lyssiotis CA, Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab, 2019. 29(6): p. 1390–1399 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, Fenoglio S, Luengo A, Lees JA, Vander Heiden MG, Lauffenburger DA, and Hemann MT, Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, Esaki N, Ushida K, Mii S, Shiraki Y, Ando K, Weng L, Ishihara S, Ponik SM, Conklin MW, Haga H, Nagasaka A, Miyata T, Matsuyama M, Kobayashi T, Fujii T, Yamada S, Yamaguchi J, Wang T, Woods SL, Worthley D, Shimamura T, Fujishiro M, Hirooka Y, Takahashi M, and Enomoto A, Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res, 2019. [DOI] [PubMed] [Google Scholar]
- 6.Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, Chio II, Hwang CI, Tiriac H, Baker LA, Engle DD, Feig C, Kultti A, Egeblad M, Fearon DT, Crawford JM, Clevers H, Park Y, and Tuveson DA, Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med, 2017. 214(3): p. 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson DA, IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov, 2019. 9(2): p. 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, Ebright RY, Karabacak MN, Kulkarni AS, Liu A, Vincent Jordan N, Franses JW, Philipp J, Kreuzer J, Desai N, Arora KS, Rajurkar M, Horwitz E, Neyaz A, Tai E, Magnus NKC, Vo KD, Yashaswini CN, Marangoni F, Boukhali M, Fatherree JP, Damon LJ, Xega K, Desai R, Choz M, Bersani F, Langenbucher A, Thapar V, Morris R, Wellner UF, Schilling O, Lawrence MS, Liss AS, Rivera MN, Deshpande V, Benes CH, Maheswaran S, Haber DA, Fernandez-Del-Castillo C, Ferrone CR, Haas W, Aryee MJ, and Ting DT, Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell, 2019. 178(1): p. 160–175 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, Huang D, Zhao J, Yang L, Liao D, Su F, Li M, Liu Q, and Song E, CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell, 2018. 172(4): p. 841–856 e16. [DOI] [PubMed] [Google Scholar]
- 10.Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, Sams SB, Pillai MM, Elias AD, Robinson WA, Sartorius CA, and Kabos P, Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin Cancer Res, 2017. 23(7): p. 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, Ridinger-Saison M, DelGiorno KE, Antal CE, Liang G, Atkins AR, Erikson G, Sun H, Meisenhelder J, Terenziani E, Woo G, Fang L, Santisakultarm TP, Manor U, Xu R, Becerra CR, Borazanci E, Von Hoff DD, Grandgenett PM, Hollingsworth MA, Leblanc M, Umetsu SE, Collisson EA, Scadeng M, Lowy AM, Donahue TR, Reya T, Downes M, Evans RM, Wahl GM, Pawson T, Tian R, and Hunter T, Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature, 2019. 569(7754): p. 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plava J, Cihova M, Burikova M, Matuskova M, Kucerova L, and Miklikova S, Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol Cancer, 2019. 18(1): p. 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohrer LR, Chuntova P, Bade LK, Beadnell TC, Leon RP, Brady NJ, Ryu Y, Goldberg JE, Schmechel SC, Koopmeiners JS, McCarthy JB, and Schwertfeger KL, Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res, 2014. 74(1): p. 374–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marusyk A, Tabassum DP, Janiszewska M, Place AE, Trinh A, Rozhok AI, Pyne S, Guerriero JL, Shu S, Ekram M, Ishkin A, Cahill DP, Nikolsky Y, Chan TA, Rimawi MF, Hilsenbeck S, Schiff R, Osborne KC, Letai A, and Polyak K, Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res, 2016. 76(22): p. 6495–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, and Settleman J, Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell, 2014. 26(2): p. 207–21. [DOI] [PubMed] [Google Scholar]
- 16.Lengyel E, Makowski L, DiGiovanni J, and Kolonin MG, Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer, 2018. 4(5): p. 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, Babykutty S, Huang Y, Jung K, Rahbari NN, Han X, Chauhan VP, Martin JD, Kahn J, Huang P, Desphande V, Michaelson J, Michelakos TP, Ferrone CR, Soares R, Boucher Y, Fukumura D, and Jain RK, Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov, 2016. 6(8): p. 852–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM, Klemm DJ, Woolthuis CM, Stranahan AW, Park CY, and Jordan CT, Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell, 2016. 19(1): p. 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duong MN, Cleret A, Matera EL, Chettab K, Mathe D, Valsesia-Wittmann S, Clemenceau B, and Dumontet C, Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res, 2015. 17: p. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, and Lengyel E, Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med, 2011. 17(11): p. 1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T, Holst J, Saunders DN, and Hoy AJ, Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab, 2017. 5: p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal C, Yong-Gonzales V, Abu-Akeel M, Merghoub T, Jones DR, Zhu XG, Arora A, Ariyan CE, Birsoy K, Wolchok JD, Panageas KS, Hollmann T, Bravo-Cordero JJ, and White RM, Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov, 2018. 8(8): p. 1006–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, Pinter M, Babykutty S, Chin SM, Vardam TD, Huang Y, Rahbari NN, Roberge S, Wang D, Gomes-Santos IL, Puchner SB, Schlett CL, Hoffmman U, Ancukiewicz M, Tolaney SM, Krop IE, Duda DG, Boucher Y, Fukumura D, and Jain RK, Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med, 2018. 10(432). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munn LL and Jain RK, Vascular regulation of antitumor immunity. Science, 2019. 365(6453): p. 544–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Dijk CG, Nieuweboer FE, Pei JY, Xu YJ, Burgisser P, van Mulligen E, el Azzouzi H, Duncker DJ, Verhaar MC, and Cheng C, The complex mural cell: pericyte function in health and disease. Int J Cardiol, 2015. 190: p. 75–89. [DOI] [PubMed] [Google Scholar]
- 26.Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X, Huang Z, Zhai K, Ke SQ, Ping YF, Feng H, Rich JN, Yu JS, Bao S, and Bian XW, Targeting Glioma Stem Cell-Derived Pericytes Disrupts the Blood-Tumor Barrier and Improves Chemotherapeutic Efficacy. Cell Stem Cell, 2017. 21(5): p. 591–603 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, and Frenette PS, Autonomic nerve development contributes to prostate cancer progression. Science, 2013. 341(6142): p. 1236361. [DOI] [PubMed] [Google Scholar]
- 28.Mauffrey P, Tchitchek N, Barroca V, Bemelmans A, Firlej V, Allory Y, Romeo PH, and Magnon C, Progenitors from the central nervous system drive neurogenesis in cancer. Nature, 2019. 569(7758): p. 672–678. [DOI] [PubMed] [Google Scholar]
- 29.Chen SH, Zhang BY, Zhou B, Zhu CZ, Sun LQ, and Feng YJ, Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am J Cancer Res, 2019. 9(1): p. 1–21. [PMC free article] [PubMed] [Google Scholar]
- 30.Datta M, Coussens LM, Nishikawa H, Hodi FS, and Jain RK, Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am Soc Clin Oncol Educ Book, 2019. 39: p. 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palucka AK and Coussens LM, The Basis of Oncoimmunology. Cell, 2016. 164(6): p. 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y, Chalouni C, Ziai J, Senbabaoglu Y, Santoro S, Sheinson D, Hung J, Giltnane JM, Pierce AA, Mesh K, Lianoglou S, Riegler J, Carano RAD, Eriksson P, Hoglund M, Somarriba L, Halligan DL, van der Heijden MS, Loriot Y, Rosenberg JE, Fong L, Mellman I, Chen DS, Green M, Derleth C, Fine GD, Hegde PS, Bourgon R, and Powles T, TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature, 2018. 554(7693): p. 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, and Zheng L, Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood, 2007. 110(2): p. 587–95. [DOI] [PubMed] [Google Scholar]
- 34.McWhorter FY, Davis CT, and Liu WF, Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci, 2015. 72(7): p. 1303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liguori M, Solinas G, Germano G, Mantovani A, and Allavena P, Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (Basel), 2011. 3(4): p. 3740–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, and Ruttinger D, Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer, 2017. 5(1): p. 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruffell B and Coussens LM, Macrophages and therapeutic resistance in cancer. Cancer Cell, 2015. 27(4): p. 462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, Fan C, Huang P, Bardeesy N, Zhu AX, Jain RK, and Duda DG, CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology, 2015. 61(5): p. 1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dvorak HF, Tumors: wounds that do not heal-redux. Cancer Immunol Res, 2015. 3(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roma-Rodrigues C, Mendes R, Baptista PV, and Fernandes AR, Targeting Tumor Microenvironment for Cancer Therapy. Int J Mol Sci, 2019. 20(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vennin C, Melenec P, Rouet R, Nobis M, Cazet AS, Murphy KJ, Herrmann D, Reed DA, Lucas MC, Warren SC, Elgundi Z, Pinese M, Kalna G, Roden D, Samuel M, Zaratzian A, Grey ST, Da Silva A, Leung W, Australian Pancreatic Genome I, Mathivanan S, Wang Y, Braithwaite AW, Christ D, Benda A, Parkin A, Phillips PA, Whitelock JM, Gill AJ, Sansom OJ, Croucher DR, Parker BL, Pajic M, Morton JP, Cox TR, and Timpson P, CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun, 2019. 10(1): p. 3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sampayo RG, Toscani AM, Rubashkin MG, Thi K, Masullo LA, Violi IL, Lakins JN, Caceres A, Hines WC, Coluccio Leskow F, Stefani FD, Chialvo DR, Bissell MJ, Weaver VM, and Simian M, Fibronectin rescues estrogen receptor alpha from lysosomal degradation in breast cancer cells. J Cell Biol, 2018. 217(8): p. 2777–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amrutkar M, Aasrum M, Verbeke CS, and Gladhaug IP, Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer, 2019. 19(1): p. 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaur A, Ecker BL, Douglass SM, Kugel CH 3rd, Webster MR, Almeida FV, Somasundaram R, Hayden J, Ban E, Ahmadzadeh H, Franco-Barraza J, Shah N, Mellis IA, Keeney F, Kossenkov A, Tang HY, Yin X, Liu Q, Xu X, Fane M, Brafford P, Herlyn M, Speicher DW, Wargo JA, Tetzlaff MT, Haydu LE, Raj A, Shenoy V, Cukierman E, and Weeraratna AT, Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov, 2019. 9(1): p. 64–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, and Brugge JS, Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell, 2012. 21(2): p. 227–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanker AB, Estrada MV, Bianchini G, Moore PD, Zhao J, Cheng F, Koch JP, Gianni L, Tyson DR, Sanchez V, Rexer BN, Sanders ME, Zhao Z, Stricker TP, and Arteaga CL, Extracellular Matrix/Integrin Signaling Promotes Resistance to Combined Inhibition of HER2 and PI3K in HER2(+) Breast Cancer. Cancer Res, 2017. 77(12): p. 3280–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dangi-Garimella S, Krantz SB, Barron MR, Shields MA, Heiferman MJ, Grippo PJ, Bentrem DJ, and Munshi HG, Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res, 2011. 71(3): p. 1019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rainero E, Howe JD, Caswell PT, Jamieson NB, Anderson K, Critchley DR, Machesky L, and Norman JC, Ligand-Occupied Integrin Internalization Links Nutrient Signaling to Invasive Migration. Cell Rep, 2015. 10(3): p. 398–413. [DOI] [PubMed] [Google Scholar]
- 49.Muranen T, Iwanicki MP, Curry NL, Hwang J, DuBois CD, Coloff JL, Hitchcock DS, Clish CB, Brugge JS, and Kalaany NY, Starved epithelial cells uptake extracellular matrix for survival. Nat Commun, 2017. 8: p. 13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, and Bar-Sagi D, Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, and Thompson CB, The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell, 2015. 162(2): p. 259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN, Berthezene P, Rubis M, Secq V, Garcia S, Moutardier V, Lombardo D, Iovanna JL, Tomasini R, Guillaumond F, Vander Heiden MG, and Vasseur S, Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun, 2017. 8: p. 16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian C, Clauser KR, Ohlund D, Rickelt S, Huang Y, Gupta M, Mani DR, Carr SA, Tuveson DA, and Hynes RO, Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci U S A, 2019. 116(39): p. 19609–19618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han L, Lam EW, and Sun Y, Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol Cancer, 2019. 18(1): p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Namee NM and O’Driscoll L, Extracellular vesicles and anti-cancer drug resistance. Biochim Biophys Acta Rev Cancer, 2018. 1870(2): p. 123–136. [DOI] [PubMed] [Google Scholar]
- 56.Trichet L, Le Digabel J, Hawkins RJ, Vedula SR, Gupta M, Ribrault C, Hersen P, Voituriez R, and Ladoux B, Evidence of a large-scale mechanosensing mechanism for cellular adaptation to substrate stiffness. Proc Natl Acad Sci U S A, 2012. 109(18): p. 6933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Fanous MJ, Kilian KA, and Popescu G, Quantitative phase imaging reveals matrix stiffness-dependent growth and migration of cancer cells. Sci Rep, 2019. 9(1): p. 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oudin MJ and Weaver VM, Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb Symp Quant Biol, 2016. 81: p. 189–205. [DOI] [PubMed] [Google Scholar]
- 59.Li X and Thompson CB, Targeting hyaluronan accumulation in the tumor microenvironment. Oncotarget, 2018. 9(100): p. 37349–37351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrova V, Annicchiarico-Petruzzelli M, Melino G, and Amelio I, The hypoxic tumour microenvironment. Oncogenesis, 2018. 7(1): p. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Myllyharju J and Schipani E, Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res, 2010. 339(1): p. 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Labrousse-Arias D, Martinez-Ruiz A, and Calzada MJ, Hypoxia and Redox Signaling on Extracellular Matrix Remodeling: From Mechanisms to Pathological Implications. Antioxid Redox Signal, 2017. 27(12): p. 802–822. [DOI] [PubMed] [Google Scholar]
- 63.Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, and Semenza GL, Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem, 2013. 288(15): p. 10819–29. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, Fraley SI, Wong CM, Khoo US, Ng IO, Wirtz D, and Semenza GL, Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci U S A, 2011. 108(39): p. 16369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kietzmann T, Samoylenko A, Roth U, and Jungermann K, Hypoxia-inducible factor-1 and hypoxia response elements mediate the induction of plasminogen activator inhibitor-1 gene expression by insulin in primary rat hepatocytes. Blood, 2003. 101(3): p. 907–14. [DOI] [PubMed] [Google Scholar]
- 66.Eble JA and Niland S, The extracellular matrix in tumor progression and metastasis. Clin Exp Metastasis, 2019. 36(3): p. 171–198. [DOI] [PubMed] [Google Scholar]
- 67.Raab-Westphal S, Marshall JF, and Goodman SL, Integrins as Therapeutic Targets: Successes and Cancers. Cancers (Basel), 2017. 9(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cooper J and Giancotti FG, Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell, 2019. 35(3): p. 347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamidi H and Ivaska J, Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer, 2018. 18(9): p. 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, Shor RE, Goddard ET, Dai J, Schweitzer EM, Lim AR, Crist SB, Cheresh DA, Nelson PS, Hansen KC, and Ghajar CM, Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol, 2019. 21(2): p. 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kleinschmidt EG and Schlaepfer DD, Focal adhesion kinase signaling in unexpected places. Curr Opin Cell Biol, 2017. 45: p. 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sulzmaier FJ, Jean C, and Schlaepfer DD, FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer, 2014. 14(9): p. 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong PP, Alexopoulou A, Elia G, Clear A, Ledoux A, Hunter J, Perkins N, Gribben JG, and Hodivala-Dilke KM, Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature, 2014. 514(7520): p. 112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, and DeNardo DG, Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med, 2016. 22(8): p. 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patel A, Sabbineni H, Clarke A, and Somanath PR, Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci, 2016. 157: p. 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brownlow N, Mol C, Hayford C, Ghaem-Maghami S, and Dibb NJ, Dasatinib is a potent inhibitor of tumour-associated macrophages, osteoclasts and the FMS receptor. Leukemia, 2009. 23(3): p. 590–4. [DOI] [PubMed] [Google Scholar]
- 77.Dewar AL, Cambareri AC, Zannettino AC, Miller BL, Doherty KV, Hughes TP, and Lyons AB, Macrophage colony-stimulating factor receptor c-fms is a novel target of imatinib. Blood, 2005. 105(8): p. 3127–32. [DOI] [PubMed] [Google Scholar]
- 78.Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, and Massague J, Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell, 2009. 16(1): p. 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin JD, Seano G, and Jain RK, Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol, 2019. 81: p. 505–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, and Ruttinger D, Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell, 2014. 25(6): p. 846–59. [DOI] [PubMed] [Google Scholar]
- 81.Salvagno C, Ciampricotti M, Tuit S, Hau CS, van Weverwijk A, Coffelt SB, Kersten K, Vrijland K, Kos K, Ulas T, Song JY, Ooi CH, Ruttinger D, Cassier PA, Jonkers J, Schultze JL, Ries CH, and de Visser KE, Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol, 2019. 21(4): p. 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, Hashimoto A, Vonteddu P, Behera R, Goins MA, Mulligan C, Nam B, Hockstein N, Denstman F, Shakamuri S, Speicher DW, Weeraratna AT, Chao T, Vonderheide RH, Languino LR, Ordentlich P, Liu Q, Xu X, Lo A, Pure E, Zhang C, Loboda A, Sepulveda MA, Snyder LA, and Gabrilovich DI, Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell, 2017. 32(5): p. 654–668 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, Huang P, Lindeman N, Langer R, and Jain RK, Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci U S A, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, Forbes B, Edelblute B, Collette B, Xing D, Kowalski M, Mingari MC, Vianello F, Birrer M, Orsulic S, Dranoff G, and Poznansky MC, CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res, 2011. 71(16): p. 5522–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, Lutz EA, Kang B, Irvine DJ, Spranger S, and Wittrup KD, Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci Transl Med, 2019. 11(498). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alaseem A, Alhazzani K, Dondapati P, Alobid S, Bishayee A, and Rathinavelu A, Matrix Metalloproteinases: A challenging paradigm of cancer management. Semin Cancer Biol, 2019. 56: p. 100–115. [DOI] [PubMed] [Google Scholar]
- 87.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, and Evans RM, Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell, 2014. 159(1): p. 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferrer-Mayorga G, Gomez-Lopez G, Barbachano A, Fernandez-Barral A, Pena C, Pisano DG, Cantero R, Rojo F, Munoz A, and Larriba MJ, Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut, 2017. 66(8): p. 1449–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kong F, Li L, Wang G, Deng X, Li Z, and Kong X, VDR signaling inhibits cancer-associated-fibroblasts’ release of exosomal miR-10a-5p and limits their supportive effects on pancreatic cancer cells. Gut, 2019. 68(5): p. 950–951. [DOI] [PubMed] [Google Scholar]
- 90.Wu X, Hu W, Lu L, Zhao Y, Zhou Y, Xiao Z, Zhang L, Zhang H, Li X, Li W, Wang S, Cho CH, Shen J, and Li M, Repurposing vitamin D for treatment of human malignancies via targeting tumor microenvironment. Acta Pharm Sin B, 2019. 9(2): p. 203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Gordon D, Copeland T, D’Agostino D, Friedenberg G, Ridge C, Bubes V, Giovannucci EL, Willett WC, Buring JE, and Group VR, Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N Engl J Med, 2019. 380(1): p. 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhattacharjee V, Zhou Y, and Yen TJ, A synthetic lethal screen identifies the Vitamin D receptor as a novel gemcitabine sensitizer in pancreatic cancer cells. Cell Cycle, 2014. 13(24): p. 3839–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jeon SM and Shin EA, Exploring vitamin D metabolism and function in cancer. Exp Mol Med, 2018. 50(4): p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stylianopoulos T, Munn LL, and Jain RK, Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer, 2018. 4(4): p. 292–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, Nadjsombati S, and Frost GI, Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther, 2010. 9(11): p. 3052–64. [DOI] [PubMed] [Google Scholar]
- 96.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR, Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell, 2012. 21(3): p. 418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, Lolkema MP, Jiang P, Kultti A, Thompson CB, Maneval DC, Jodrell DI, Frost GI, Shepard HM, Skepper JN, and Tuveson DA, Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut, 2013. 62(1): p. 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li X, Shepard HM, Cowell JA, Zhao C, Osgood RJ, Rosengren S, Blouw B, Garrovillo SA, Pagel MD, Whatcott CJ, Han H, Von Hoff DD, Taverna DM, LaBarre MJ, Maneval DC, and Thompson CB, Parallel Accumulation of Tumor Hyaluronan, Collagen, and Other Drivers of Tumor Progression. Clin Cancer Res, 2018. 24(19): p. 4798–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, Braiteh F, Ritch PS, Zalupski MM, Bahary N, Oberstein PE, Wang-Gillam A, Wu W, Chondros D, Jiang P, Khelifa S, Pu J, Aldrich C, and Hendifar AE, HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol, 2018. 36(4): p. 359–366. [DOI] [PubMed] [Google Scholar]
- 100.Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, Thumar J, Chiorean EG, Shields AF, Behl D, Mehan PT, Gaur R, Seery T, Guthrie KA, and Hochster HS, Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J Clin Oncol, 2019. 37(13): p. 1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, Jelinek D, Whiteley AE, Maxwell MB, Wilde BR, Banerjee U, Coller HA, Shackelford DB, Braas D, Ayer DE, de Aguiar Vallim TQ, Lowry WE, and Christofk HR, Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell, 2018. 175(1): p. 117–132 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Curran CS and Keely PJ, Breast tumor and stromal cell responses to TGF-beta and hypoxia in matrix deposition. Matrix Biol, 2013. 32(2): p. 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, and Sheppard D, The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell, 1999. 96(3): p. 319–28. [DOI] [PubMed] [Google Scholar]
- 104.de Gramont A, Faivre S, and Raymond E, Novel TGF-beta inhibitors ready for prime time in onco-immunology. Oncoimmunology, 2017. 6(1): p. e1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Burstein SH, Ajulemic acid: potential treatment for chronic inflammation. Pharmacol Res Perspect, 2018. 6(2): p. e00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elahi-Gedwillo KY, Carlson M, Zettervall J, and Provenzano PP, Antifibrotic Therapy Disrupts Stromal Barriers and Modulates the Immune Landscape in Pancreatic Ductal Adenocarcinoma. Cancer Res, 2019. 79(2): p. 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takai K, Le A, Weaver VM, and Werb Z, Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget, 2016. 7(50): p. 82889–82901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coulson R, Liew SH, Connelly AA, Yee NS, Deb S, Kumar B, Vargas AC, O’Toole SA, Parslow AC, Poh A, Putoczki T, Morrow RJ, Alorro M, Lazarus KA, Yeap EFW, Walton KL, Harrison CA, Hannan NJ, George AJ, Clyne CD, Ernst M, Allen AM, and Chand AL, The angiotensin receptor blocker, Losartan, inhibits mammary tumor development and progression to invasive carcinoma. Oncotarget, 2017. 8(12): p. 18640–18656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, and Jain RK, Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A, 2011. 108(7): p. 2909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popovic Z, Huang P, Bawendi MG, Boucher Y, and Jain RK, Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun, 2013. 4: p. 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Busby J, McMenamin U, Spence A, Johnston BT, Hughes C, and Cardwell CR, Angiotensin receptor blocker use and gastro-oesophageal cancer survival: a population-based cohort study. Aliment Pharmacol Ther, 2018. 47(2): p. 279–288. [DOI] [PubMed] [Google Scholar]
- 112.Murphy JE, Wo JY, Ryan DP, Clark JW, Jiang W, Yeap BY, Drapek LC, Ly L, Baglini CV, Blaszkowsky LS, Ferrone CR, Parikh AR, Weekes CD, Nipp RD, Kwak EL, Allen JN, Corcoran RB, Ting DT, Faris JE, Zhu AX, Goyal L, Berger DL, Qadan M, Lillemoe KD, Talele N, Jain RK, DeLaney TF, Duda DG, Boucher Y, Fernandez-Del Castillo C, and Hong TS, Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol, 2019. 5(7): p. 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, Nia HT, Zhang Y, Stylianopoulos T, Kumar AS, Mpekris F, Datta M, Sun Y, Wu L, Gao X, Yeku O, Del Carmen MG, Spriggs DR, Jain RK, and Xu L, Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci U S A, 2019. 116(6): p. 2210–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weigelt B, Ghajar CM, and Bissell MJ, The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev, 2014. 69–70: p. 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Serebriiskii I, Castello-Cros R, Lamb A, Golemis EA, and Cukierman E, Fibroblast-derived 3D matrix differentially regulates the growth and drug-responsiveness of human cancer cells. Matrix Biol, 2008. 27(6): p. 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Langhans SA, Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol, 2018. 9: p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schedin P and Keely PJ, Mammary gland ECM remodeling, stiffness, and mechanosignaling in normal development and tumor progression. Cold Spring Harb Perspect Biol, 2011. 3(1): p. a003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boyd NF, Lockwood GA, Byng JW, Tritchler DL, and Yaffe MJ, Mammographic densities and breast cancer risk. Cancer Epidemiol Biomarkers Prev, 1998. 7(12): p. 1133–44. [PubMed] [Google Scholar]
- 119.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, and Keely PJ, Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol, 2011. 178(3): p. 1221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brighton HE, Angus SP, Bo T, Roques J, Tagliatela AC, Darr DB, Karagoz K, Sciaky N, Gatza ML, Sharpless NE, Johnson GL, and Bear JE, New Mechanisms of Resistance to MEK Inhibitors in Melanoma Revealed by Intravital Imaging. Cancer Res, 2018. 78(2): p. 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, and Sahai E, Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell, 2015. 27(4): p. 574–88. [DOI] [PMC free article] [PubMed] [Google Scholar]