

Abstract
BACKGROUND:
Screening programs for the most prevalent conditions occurring in a country is an evidence-based prevention strategy. The burden of autosomal recessive disease variations in Saudi Arabia is high because of the highly consanguineous population. The optimal solution for estimating the carrier frequency of the most prevalent diseases is carrier screening.
OBJECTIVES:
Identify the most influential recessive alleles associated with disease in the Saudi population.
DESIGN:
We used clinical whole-exome sequencing data from an in-house familial database to evaluate the most prevalent genetic variations associated with disease in a Saudi population.
SETTINGS:
King Abdullah International Medical Research Center (KAIMRC) and King Abdulaziz Medical City.
METHODS:
Whole exome sequencing data obtained from clinical studies of family members, a cohort of 1314 affected and unaffected individuals, were filtered using the in-house pipeline to extract the most prevalent variant in the dataset.
MAIN OUTCOME MEASURES:
Most prevalent genetic variations associated with disease in the Saudi population.
SAMPLE SIZE:
1314 affected and unaffected individuals.
RESULTS:
We identified 37 autosomal recessive variants and two heterozygous X-linked variants in 35 genes associated with the most prevalent disorders, which included hematologic (32%), endocrine (21%), metabolic (11%) and immunological (10%) diseases.
CONCLUSION:
This study provides an update of the most frequently occurring alleles, which support future carrier screening programs.
LIMITATIONS:
Single center that might represent the different regions but may be biased. In addition, most of the families included in the database are part of the proband's genetic identification for specific phenotypes.
CONFLICT OF INTEREST:
None.
INTRODUCTION
Carrier screening (CS) is widely implemented to identify reproductive carriers and reduce the consequences of single gene disorders.1 More than half of the Saudi population is in a consanguineous marriage, which is reflected in the increased number of autosomal recessive (AR) conditions.2 Saudi Arabia has the highest AR birth rate globally, with founder mutations accounting for 40% of the total mutation pool.2,3 The genetic pool or the founder effect is restricted to the family. Consequently, mating choices are limited in the clan or tribe, increasing the probability of mating with a carrier.4 The prevalence rate varies in regions; for example hemoglobinopathies, cystic fibrosis and Tay Sachs disease are prevalent in some regions or sub-populations, but not in others.5 Genetic screening programs usually target identified cases, however, carrier detection is important for disease prevention to secure a healthy progeny.6
Several countries with a high frequency of certain AR conditions have implemented CS in their healthcare system, including the United States, Mexico, Australia, Netherlands, Israel, United Kingdom, Cyprus, Italy, Malaysia and Saudi Arabia.1,5,6 A study by Delatycki et al identified the carrier frequency using data from 7100 clinical panels and 350 exome cases to estimate the most prevalent disease/variation associated with the Saudi population.4 Although the data is valuable, the landscape of CS is frequently unstable due to genetic drift, population admixture, multiracial and missing heritability, which are influenced by immigration and in- or outbreeding.7 Pan-ethnic screening, applied to targeted high prevalence diseases in subpopulations, resolves this problem.8,9 This approach has been proposed by the American College of Medical Genetics and Genomics (ACMG) and the American College of Obstetricians and Gynecologists (ACOG) to expand CS to couples willing to conceive, regardless of the ethnic background.8,9
Family-based analysis of whole-exome sequencing (WES) provides a high detection rate for prevalent and rare variations,10 covering 1% of the whole human genome and nearly 95% of the coding regions.11 In this study, we used clinical WES data from an in-house familial database and evaluated the most prevalent genetic variations associated with disease in the Saudi population. The data will provide a newly updated map for the most prevalent diseases and support the development and implementation of preventive measures.
METHODS
We used the King Abdullah International Medical Research Center (KAIMRC) Genomic Database (KGD, KAIMRC Genomic Database) to extract the variants frequently occurring in the Saudi population. The data had been collected from 2014 to 2021, and populated with WES results from a mixed cohort of affected and unaffected individuals (n=1314) from 650 families with 2 173 863 filtered variants. The WES was performed for diagnostic purposes in the Genetic Department at King Abdulaziz Medical City, a College of American Pathologists accredited genetic laboratory. The results were obtained in variant calling files (VCF) and the pathogenic or likely pathogenic variants classified based on the ACMG scoring system 12 and the latest release of the ClinVar database.12 The database was extensively investigated, including the allele frequency from the local database, the Exome Sequencing Project (ESP, https://evs.gs.washington.edu/EVS/), the Genome Aggregation Database (gnomAD v2.1.1, https://gnomad.broadinstitute.org/), dbSNP/1000, the Saudi Human Genome Project database (SHGP dbm latest release, https://shgp.kacst.edu.sa/index.en.html) and other ethnically matched databases. The data include patient and variant information. The variant filtration pipeline includes all variants with a read depth more than 15×, with an allele frequency of more than 2%, based on the dbSNP/1000 genome, ESP, gnomAD (v2.1.1), and KGD. To avoid variant bias, only one family member was included, excluding affected individuals, the entire homozygous pathogenic variant and the autosomal dominant. This study was approved by the Institutional Review Board of King Abdullah International Medical Research Center (#RC19/315/R).
RESULTS
The data obtained from the KGD identified the variants with a high carrier frequency in the Saudi population, identifying 37 heterozygous AR variants and two variants carried by females as heterozygous X-linked variants. The pathogenic variants identified in the 35 genes and occurring in more than five individuals in the KGD, are available online on the Zenodo data repository at the following URL https://doi.org/10.5281/zenodo.5905071. The disease-related variants, reported to be associated with AR, had homozygous mutant patterns. The top 13 variants obtained from the KGD (Table 1) occurred in more than 10 of the 1314 individuals. The most prevalent diseases, in sequential order, were hematologic (32%), endocrine (21%), metabolic (11%) and immunologic (10%), occurring in more than 10% of the total number of variants (Figure 1).
Table 1.
The most prevalent pathogenic variations carried in the Saudi population, with the minor allele frequency obtained from Saudi Human Genome Project database (dbSNP) and the King Abdullah International Medical Research Center Genomic Database (KGD), and compared with the Genome Aggregation Database (gnomAD).
| Disorder | Gene | Disease associated | dbSNP | CDNA | KGD | SHGP db | GnomAD |
|---|---|---|---|---|---|---|---|
| Hematology | MPL | Thrombocytopenia | rs750046020 | c.317C>T | 2.46% | 2.96% | 0.01% |
| SPTA1 | Spherocytosis | rs377659326 | c.5263C>G | 1.31% | 0.72% | 0.00% | |
| HBB | Sickle cell anemia | rs334 | c.20A>T | 0.85% | 2.34% | 0.44% | |
| G6PD | Hemolytic anemia, G6PD deficient | rs5030868 | c.653C>T | 1.08% | 2.60% | 0.23% | |
| Endocrine | CYP21A2 | Congenital adrenal hyperplasia | rs7755898 | c.955C>T | 1.85% | 0.00% | 0% |
| rs776989258 | c.1447C>T | 0.85% | 1.11% | 0.05% | |||
| Metabolic | C6 | C6 deficiency | rs867425110 | c.2049C>G | 1.15% | 0.54% | 0% |
| ACY1 | Aminoacylase 1 deficiency | rs770702363 | c.575dupG | 0.85% | 0.88% | 0.01% | |
| ABCG5 | Sitosterolemia 2 | rs199689137 | c.1336C>T | 0.77% | 0.51% | 0.02% | |
| Immunology disorder | NCF1 | Chronic granulomatous disease | rs145360423 | c.579G>A | 1.00% | 0.42% | 0.06% |
| Otolaryngology | BDP1 | Deafness | rs199721728 | c.7873T>G | 1.00% | 2.44% | 0.08% |
| Ophthalmology | CYP1B1 | Glaucoma | rs28936700 | c.182G>A | 1.31% | 2.51% | 0.03% |
| Multisystem | LZTFL1 | Bardet-Biedl syndrome | rs1354476372 | c.3G>A | 1.00% | 0.33% | 0.00% |
Minor allele frequency calculated for the KGD sample size.
Figure 1.
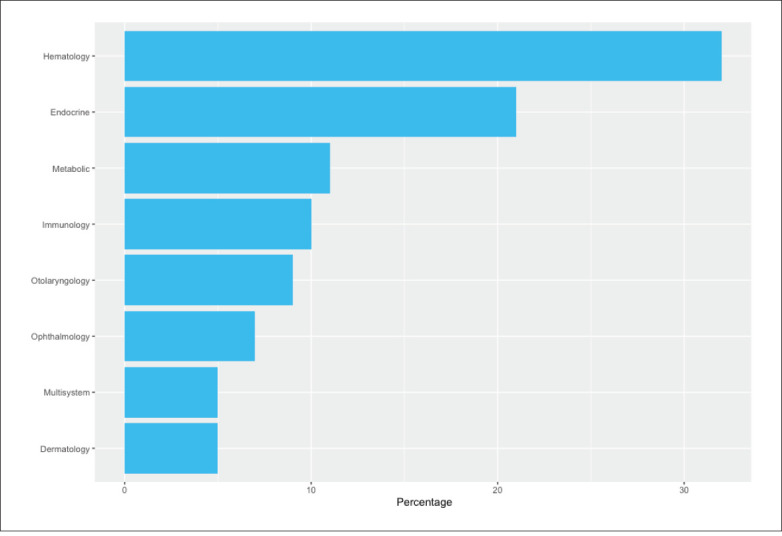
The prevalent disease types occurring in the Saudi population.
The data indicated that the most prevalent heterozygous mutation, caused by the missense mutation rs750046020 with a minor allele frequency (MAF) of 0.00007953 gnomAD, in the MPL proto-oncogene thrombopoietin receptor (MPL), is associated with congenital amegakaryocytic thrombocytopenia (OMIM:604498). Two variants, found in the cytochrome P450 family 21 subfamily A member 2 (CYP21A2) gene, are associated with congenital adrenal hyperplasia (CAH) (OMIM: 201910), and one variant occurring in 24 individuals was in the introducing stop codon rs7755898 (MAF=0.0003601). A missense variant rs776989258 (MAF=0.0005244) in 11 carriers was also observed in the CYP21A2 gene. This indicated that the pathogenic variants in the CYP21A2 gene constituted 20% of the total number of pathogenic variants obtained from the KGD (Figure 2). This variant was followed by the frequently carried missense variant rs377659326 (MAF=0.00001425) in spectrin alpha, erythrocytic 1 (SPTA1), which is associated with hereditary spherocytosis (HS) (OMIM: 270970). The third was the missense variant rs28936700 (MAF=0.0002919) in the cytochrome P450 Family 1 subfamily B member 1 (CYP1B1), which is associated with congenital glaucoma (OMIM: 231300).
Figure 2.
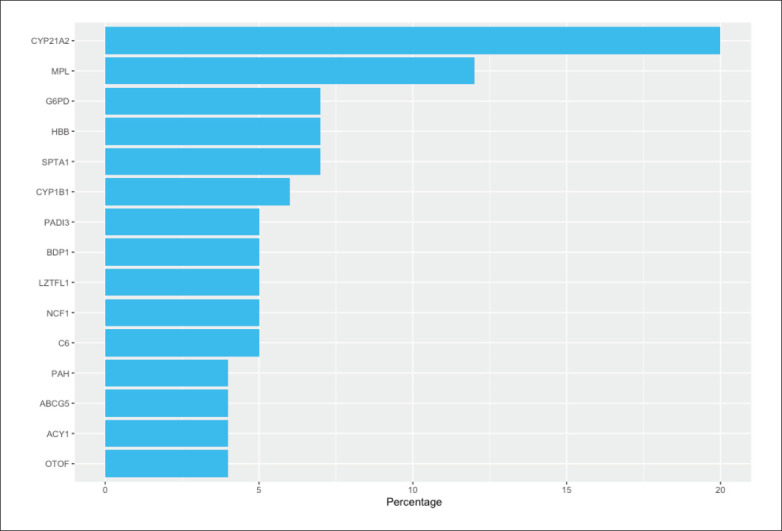
The most affected genes with most frequent pathogenic variants in the Saudi population.
The variant creating the stop codon rs867425110 (MAF=C=0.0003/1 KOREAN, C=0.0046/1 Qatari) db-SNP, found in complement C6 (C6), is associated with C6 deficiency (OMIM: 612446). Another stop codon initiated variation rs145360423 (MAF=0.0006412) was found in the neutrophil cytosolic factor 1 (NCF1), and is associated with chronic granulomatous disease (OMIM: 233700). A variant affecting the initiated codon rs1354476372 (MAF=0.000007435) was found in the 5 prime of the untranslated region (UTR) in the leucine zipper transcription factor like 1 (LZTFL1) gene, which is associated with Bardet-Biedl syndrome (BBS) (OMIM: 615994). A loss of function in the B double prime 1, subunit of RNA polymerase iii transcription initiation factor IIIB (BDP1) gene, caused by the stop codon rs199721728 (MAF=0.0007889), is associated with deafness (OMIM: 618257). The insertion of two base pairs in the aminoacylase1 (ACY1) gene rs770702363 (MAF=0.0001309) is associated with aminoacylase 1 deficiency (OMIM: 609924). A missense variant, observed in the most prevalent variant in KGD, rs334 (MAF=0.004374), found in the hemoglobin subunit beta (HBB), is associated with sickle cell anemia (OMIM: 603903). Ten female carriers had an X-linked mutation rs5030868 (MAF=0.002301), which is associated with G6PD deficiency (hemolytic anemia) (OMIM: 300908). Finally, a stop codon rs199689137 (MAF=0.0001702), causing a gain in the function in the ATP binding cassette subfamily G member 5 (ABCG5), is associated with sitosterolemia 2 (OMIM: 618666). The variations have been compared to a global population MAF using the GnomAD, in parallel with results of the SHGP database.
DISCUSSION
Globally, multi-ethnic databases such as the Exome Aggregation Consortium (ExAC)13 and the gnomAD14 still lack a Middle East genetic representation.15 The KAIMRC Genomic Database, the first WES published database in the region, although still underpowered and diverse, represents the Middle East region. The results obtained from the KGD provide additive value to the most prevalent diseases occurring in the Saudi population (Figure 1). This result is confirmed by the SHGP database, which indicates a higher trend in the Saudi population compared to the GnomAD. The data also identified new variants, observed in KGD but not in SHGP database, such as the rs7755898 in CYP21A2, and other variants in the KGD have a different presentation compared to the SHGP-db observations and in a higher frequency than the GnomAD. Multiple research projects have been conducted to identify the most prevalent disease or genetic variation in the Saudi population,4,16,17 resulting in screening programs that improved the healthcare service and reduced disease-related complications in Saudi Arabia.18
On the basis of the mutation frequency in the Saudi population, the KGD results provide an estimate of disease prevalence in the Saudi population. It is possible, however, that the actual prevalence of disease will be affected by several factors, including whether or not a screening program is implemented in the country. Most of the variations identified are associated with AR. However, the carrier frequency is still outstanding, due to the WES being requested for cases and not carriers. This family-based database facilitates the investigation of healthy carriers, estimating the carrier frequency for several diseases.
Hematologic disorders are the most prevalent variant in the Saudi population. The rs750046020 in the MPL gene has been reported as a pathogenic variant associated with thrombocytosis in the Saudi population.19 The second (rs7755898) observation, associated with CAH, has been included in the National New-Born Screening Program.18 It is known that mutations in the CYP21A2 gene are associated with more than 95% of the CAH cases, resulting in 21-hydroxylase deficiency (21-OHD).20 The disease is associated with compound heterozygous pattern of inheritance.21,22 A variant in the SPTA1 gene, c.5263C>G, usually found as compound heterozygous, causes a hematological disease affecting the red blood cell (RBC) cytoskeleton, namely hereditary hemolytic anemia (HHA).23 The mutation c.182G>A in the CYP1B1 has been reported as the most common variant associated with congenital glaucoma.24
The SHGP database identified most of the variants in Table 1, with minor differences in the MAF. A variant, such as rs867425110 in C6, is associated with a pathogenic phenotype.25 The rs145360423 in the NCF1 is in the SHGP database and the Omani population database.26 The variant found in the LZTFL1 is associated with a retinitis pigmentosa diagnosis, relatively unique to BBS.27,28 The rs199721728, identified in the BDP1 gene, is associated with hearing loss in a number of populations, including the Saudi population.29 Rs770702363, found in the ACY1, is associated with aminoacylase 1 deficiency.30 In addition, several genes, previously linked to hematological disorders, are also associated with HBB,31 G6BD32-34 and ABCG5,35 were identified in several populations, including Saudi, with a higher MAF compared to the other variants (Table 1).
The current study expanded our knowledge base regarding the most prevalent genetic variations associated with disease in the Saudi population. This update enhances the existing list of diseases included in premarital programs and supports new screening methodologies, including CS panels and biochemical testing. The significance of the database will increase with contributions from the whole nation, facilitating the identification of carriers in subpopulations to create a national pan-ethnic CS program. Such a program will provide insight for at-risk couples belonging to a geographical region or specific tribe, through prompt action and preventive measures before conception. The program can be established through fertility clinics or genetic counselors. Discussion options include aspects such as preimplantation genetic diagnosis (PGD), pregnancy termination or family planning. Additional value for implementing CS programs is reducing the risk of having babies with an inherited disease and reducing the burden on healthcare providers and families. An updated platform for the most prevalent cases and carrier states will support up-to-date screening programs and raise awareness regarding the most prevalent conditions and variations in Saudi Arabia.
The limitations of this study include the use of a single center, although the center represents different regions of the country. The sample size may not be representative of the nation and could be biased by the single center. In addition, most of the families included in the database are part of proband's genetic identification for certain phenotypes.
ACKNOWLEDGMENTS
The patients and their families who participated in this study.
APPENDIX 1

Funding Statement
None.
STATEMENT OF ETHICS
This study was approved by the Institutional Research Board of the King Abdullah International Medical Research Center #RC19/315/R. All patients have been consented to be enrolled in this study, a written consent form was obtained from all subjects and/or their parents or legal guardians in the case of minors aged 18 years old or younger.
AUTHORS CONTRIBUTIONS:
M.A, T.A and Ahmed.A designed the study, interpreted the clinical data, and wrote the article. L.A, M.A, and G.A collected the samples, genotyped the cases and assisted in the statistical analysis. Abdulrahman.A, W.E, F.A, Farouq.A, and M.A, contributed in sample collection, clinical correlation and manuscript revision. All authors have read and approved the final manuscripts.
REFERENCES
- 1.Hernande-Nieto C, Alkon-Meadows T, Lee J, Cacchione T, Iyune-Cojab E, Garza-Galvan M, et al.. Expanded carrier screening for preconception reproductive risk assessment: Prevalence of carrier status in a Mexican population. Prenat Diagn. 2020;40(5):635–43. [DOI] [PubMed] [Google Scholar]
- 2.Alkuraya FS.. Genetics and genomic medicine in Saudi Arabia. Mol Genet Genomic Med. 2014;2(5):369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al.. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am J Hum pGenet [Internet]. 2019;104(6):1182–201. Available from: 10.1016/j.ajhg.2019.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abouelhoda M, Sobahy T, El-Kalioby M, Patel N, Shamseldin H, Monies D, et al.. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet Med. 2016;18(12):1244–9. [DOI] [PubMed] [Google Scholar]
- 5.Delatycki MB, Alkuraya F, Archibald A, Castellani C, Cornel M, Grody WW, et al.. International perspectives on the implementation of reproductive carrier screening. Prenat Diagn. 2020;40(3):301–10. [DOI] [PubMed] [Google Scholar]
- 6.Westemeyer M, Saucier J, Wallace J, Prins SA, Shetty A, Malhotra M, et al.. Clinical experience with carrier screening in a general population : support for a comprehensive pan-ethnic approach. Genet Med [Internet]. 2020;22(8). Available from: 10.1038/s41436-020-0807-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez AD, Hirschman C.. The Changing Racial and Ethnic Composition of the US Population: Emerging American Identities. Popul Dev Rev. 2009;35(1):1–51. doi: 10.1111/j.1728-4457.2009.00260.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross SJ, Pletcher BA, Monaghan KG.. Carrier screening in individuals of Ashkenazi Jewish descent. Genet Med. 2008;10(1):54–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ACOG.. preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obs Gynecol [Internet]. 2009;114:950–953. Available from: http://library1.nida.ac.th/termpaper6/sd/2554/19755.pdfhttps://pubmed.ncbi.nlm.nih.gov/19888064/ [DOI] [PubMed] [Google Scholar]
- 10.Alfares A, Alsubaie L, Aloraini T, Alaskar A, Althagafi A, Alahmad A, et al.. What is the right sequencing approach? Solo VS extended family analysis in consanguineous populations. BMC Med Genomics. 2020;13(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alfares A, Aloraini T, subaie L Al, Alissa A, Qudsi A Al, Alahmad A, et al.. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med [Internet]. 2018;20(11):1328–33. Available from: 10.1038/gim.2018.41 [DOI] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015. May;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lek M, Karczewski KJ, Minikel E V, Samocha KE, Banks E, Fennell T, et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abou Tayoun AN, Rehm HL.. Genetic variation in the Middle East—an opportunity to advance the human genetics field. Genome Med. 2020;12(1):12–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alfadhel M, Al Othaim A, Al Saif S, Al Mutairi F, Alsayed M, Rahbeeni Z, et al.. Expanded Newborn Screening Program in Saudi Arabia: Incidence of screened disorders. J Paediatr Child Health. 2017. Jun 1;53(6):585–91. [DOI] [PubMed] [Google Scholar]
- 17.Alfares A, Alkuraya F.. An Overview of Mendelian Disorders In Saudi Arabia. Riyadh, Saudi Arabia. 2016:26–37
- 18.Gosadi IM.. National screening programs in Saudi Arabia: Overview, outcomes, and effectiveness. J Infect Public Health [Internet]. 2019;12(5):608–14. Available from: 10.1016/j.jiph.2019.06.001 [DOI] [PubMed] [Google Scholar]
- 19.Alsultan A, Al-Suliman AM, Aleem A, AlGahtani FH, Alfadhel M.. Utilizing whole-exome sequencing to characterize the phenotypic variability of sickle cell disease. Genet Test Mol Biomarkers. 2018;22(9):561–7. [DOI] [PubMed] [Google Scholar]
- 20.Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al.. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2018;103(11):4043–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coeli FB, Soardi FC, Bernardi RD, de Araújo M, Paulino LC, Lau IF, et al.. Novel deletion alleles carrying CYP21A1P/A2chimeric genes in Brazilian patients with 21-hydroxylase deficiency. BMC Med Genet. 2010;11(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elmougy F, Sharaf S, Hafez M, Khattab A, Abou-Yousef H, Elsharkawy M, et al.. CYP21A2 genetic profile in 14 Egyptian children with suspected congenital adrenal hyperplasia: a diagnostic challenge. Ann N Y Acad Sci. 2018;1415(1):11–20. [DOI] [PubMed] [Google Scholar]
- 23.Kalfa TA, Chonat S, Risinger M, Sakthivel H, Niss O, Rothman JA, et al.. The spectrum of SPTA1-associated hereditary spherocytosis. Front Physiol. 2019;10:815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsaif HS, Khan AO, Patel N, Alkuraya H, Hashem M, Abdulwahab F, et al.. Congenital glaucoma and CYP1B1: an old story revisited. Hum Genet [Internet]. 2019;138(8-9):1043–9. Available from: 10.1007/s00439-018-1878-z [DOI] [PubMed] [Google Scholar]
- 25.Bertoli-Avella AM, Beetz C, Ameziane N, Rocha ME, Guatibonza P, Pereira C, Calvo M, Herrera-Ordonez N, Segura-Castel M, Diego-Alvarez D, Zawada M.. Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. European Journal of Human Genetics. 2021. Jan;29(1):141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajab A, Hamza N, Al Harasi S, Al Lawati F, Gibbons U, Al Alawi I, et al.. Repository of mutations from Oman: The entry point to a national mutation database [version 1; referees: 2 approved]. F1000Research. 2015;4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaefer E, Delvallée C, Mary L, Stoetzel C, Geoffroy V, Marks-Delesalle C, et al.. Identification and characterization of known biallelic mutations in the IFT27 (BBS19) gene in a novel family with Bardet-Biedl syndrome. Front Genet. 2019;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weihbrecht K.. Bardet-Biedl syndrome. In: Genetics and Genomics of Eye Disease. Elsevier. 2020;117–36. [Google Scholar]
- 29.Girotto G, Abdulhadi K, Buniello A, Vozzi D, Licastro D, d’Eustacchio A, et al.. Linkage study and exome sequencing identify a BDP1 mutation associated with hereditary hearing loss. PLoS One. 2013;8(12):e80323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sass JO, Mohr V, Olbrich H, Engelke U, Horvath J, Fliegauf M, et al.. Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am J Hum Genet. 2006;78(3):401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okumura J V, Silva DGH, Torres LS, Belini-Junior E, Barberino WM, Oliveira RG, et al.. Inheritance of the Bantu/Benin haplotype causes less severe hemolytic and oxidative stress in sickle cell anemia patients treated with hydroxycarbamide. J Hum Genet. 2016;61(7):605–11. [DOI] [PubMed] [Google Scholar]
- 32.Alagoz M, Kherad N, Gunger E, Kaymaz S, Yuksel A.. The New CIC Mutation Associates with Mental Retardation and Severity of Seizure in Turkish Child with a Rare Class I Glucose-6-Phosphate Dehydrogenase Deficiency. J Mol Neurosci. 2020;1–8. [DOI] [PubMed]
- 33.Farrell John J., Al-Nafaie Awatif N., Al-Ali Amein K., Al-Rubaish Abdullah M., Naserullah Zaki, Alsuliman Ahmed, Steinberg Martin H., Clinton T.. Baldwin; The Evolutionary Impact Of Malaria On The Saudi Arabian Genome. Blood 2013; 122 (21): 1001. [Google Scholar]
- 34.Mahfouz NA, Kizhakkedath P, Ibrahim A, El Naofal M, Ramaswamy S, Harilal D, Qutub Y, Uddin M, Taylor A, Alloub Z, AlBanna A.. Utility of clinical exome sequencing in a complex Emirati pediatric cohort. Computational and structural biotechnology journal. 2020. Jan 1;18:1020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andolfo I, Russo R, Gambale A, Iolascon A.. Hereditary stomatocytosis: an underdiagnosed condition. Am J Hematol. 2018;93(1):107–21. [DOI] [PubMed] [Google Scholar]