

Abstract
A prototype of a self-contained, automated, disposable device for chemically-amplified protein-based detection of influenza virus from nasal swab specimens was developed and evaluated in a clinical setting. The device required only simple specimen manipulation without any dedicated instrumentation or specialized training by the operator for interpretation. The device was based on a sandwich immunoassay for influenza virus nucleoprotein; it used an enzyme-labeled antibody and a chromogenic substrate to provide an amplified visible signal, in a two-dimensional paper network format. All reagents were stored within the device. Device performance was assessed at Seattle Children's Hospital; clinical staff collected nasal swab samples from 25 patients, and then operated test devices on site to detect influenza A and B in those specimens. The total test time from device initiation to result was approximately 35 minutes. Device performance for influenza A detection was ∼70% accurate using in-house qRT-PCR influenza A as a gold-standard comparison. The ratio of valid to total completed device runs yielded a success rate of 92%, and the negative predictive value for both the influenza A and B assay was 81%. The ability to diagnose respiratory infections rapidly and close to the patient was well received by hospital staff, inspiring further optimization of device function.
Influenza, or the flu, is a common illness caused by a set of viruses that have infected humans for at least the past two millennia1. Pandemic and seasonal influenza outbreaks, such as the 1918 Spanish Flu, the 2009 H1N1 strain, and the recent H5N1 strain, have been threats to public health and a burden on the global economy2–4. Future influenza pandemics, caused by the emergence of novel subtypes of influenza, are unfortunately an ongoing concern5. Rapid, sensitive, and accurate diagnosis of influenza, especially for patients at an early stage of infection, is valuable for guiding therapy, as well as critical for outbreak control. Early diagnosis can reduce disease severity, healthcare costs, erroneous use of antibiotics, and the spread of the disease.
Influenza A and B viruses are the primary causes of seasonal epidemics of moderate to severe respiratory illness. Influenza C virus usually causes only mild respiratory illness6, while influenza D virus primarily affects cattle. Antiviral treatment is only available for influenza A. Differential diagnosis of influenza A or influenza B is therefore important, and depends on the detection of differences in the viral nucleoprotein (NP). Laboratory-based influenza tests, including those based on the polymerase chain reaction (PCR) and viral culture, are now considered the gold standard for diagnosis of influenza. However, these techniques are expensive and labor intensive7–9, require laboratory facilities and trained personnel, and take several hours to generate results9–11. They are considered unsuitable for settings outside a well-equipped laboratory. In contrast, FDA-approved protein-based rapid diagnostic tests (RDTs) for influenza exist for use at the point-of-care (POC)9,10. These tests provide fast diagnosis by using antibodies to detect the presence of influenza NP, and an increasing number of reports have shown RDTs can help identify and control the spread of influenza in institutions, schools or semi-closed communities10,12. The usage of RDTs has reduced the cost of patient visits and hospitalization time, as well as helped limit the erroneous use of antibiotics and antivirals for patients in a pediatric emergency room13. However, RDTs are limited by poor clinical sensitivity, due to the low number of target proteins in patient specimens8–10,14,15; and by the need for multiple sample processing steps, including vortexing, pipetting, and mixing, that are still performed on the benchtop.
To address the gap between expensive laboratory-based tests and low-sensitivity RDTs, we developed a POC device for influenza that is potentially much more sensitive than current RDTs, and are self-contained, automated, and disposable. The device is easily operated by a minimally-trained user, as it accepts an unprocessed patient specimen (nasal swab) and returns easily-interpreted visual results. The sample-to-result simplicity of the device is a result of the integration and automation of sample processing steps. Viral lysis, target protein capture, secondary labeling, rinsing, and enzymatic turnover of a colorimetric substrate all occur without user intervention. The two-dimensional paper network (2DPN) format is a useful platform for integration and automation of biochemical assay steps16–18. The device we describe here integrates aqueous reagent storage, a sample introduction system, and a 2DPN that stores dry reagents and automates sequential reagent delivery for separate but simultaneous influenza A & B assays. A major advance of this work is the integrated system design, in which the action of closing the device is the single, simple activation step that initiates the test.
The device reported here is a novel, point-of-care system for detection of the nucleoprotein (NP) from influenza virus. The device was tested in the Emergency Department at Seattle Children's Hospital during the 2015-2016 influenza season. Clinical staff collected nasal swab samples from 25 patients and operated the devices on-site in the emergency department. The total test time was approximately 35 minutes, and results indicated ∼70% accuracy in the detection of Flu A using qRT-PCR as the gold standard.
Materials and Methods
Assay Chemistry
A schematic of the sandwich immunoassay implemented to detect NP in nitrocellulose (NC) and the process control are shown in Figure 1. The target nucleoprotein capture antibodies, which were type-specific for either influenza A or B (0.4 μL, 1 mg/mL, mouse monoclonal anti-influenza A/B NP IgG, Hytest Ltd), and a process control capture antibody (0.3 μL, 0.4 mg/mL, goat anti-mouse IgG, Jackson ImmunoResearch) were immobilized on 5-mm-wide NC membrane (Millipore, HF120) strips, and spaced 4 mm apart via printing with a reagent dispenser (BioDot, XYZ3060), followed by drying at 37°C for 2 hr in a box with desiccant. Then the strips were stored in a sealed pouch with desiccant overnight in a humidity-controlled room (20% RH); they were cut to final size with a Matrix™ 2360 programmable shear (Kinematic Automation, CA). Diaminobenzidine (DAB), in the presence of hydrogen peroxide (H2O2), is enzymatically converted from colorless substrate to a brown precipitate by horseradish peroxidase (HRP). The two detection antibodies, which were biotinylated and type-specific for either influenza A or B (biotinylated mouse monoclonal anti-influenza AB NP, Hytest Ltd) were conjugated with Pierce™ streptavidin poly-HRP (Thermo Fisher Scientific, Waltham, MA), which contains 2-8 HRP molecules per streptavidin tetramer.
Figure 1.
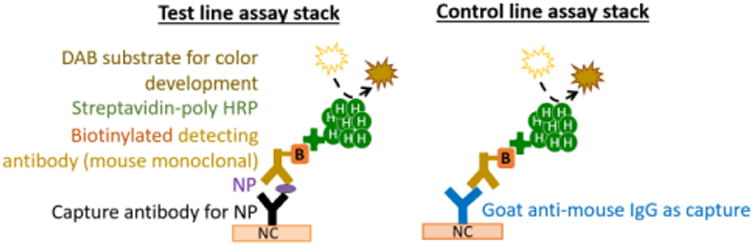
Molecular stacks for the detection of NP in a sandwich immunoassay format. At the test lines, capture antibodies bind NP (either A or B, depending on the specificity of the antibody) from lysed virus. The surface-bound NP is subsequently labeled by a mouse-derived antibody pre-conjugated to multiple HRP molecules, and visualized by DAB color development. At the control line, a goat anti-mouse IgG is immobilized; it will bind to the same indicator Ab/HRP conjugate in the absence of NP, and detection Ab complex is then visualized by DAB color development.
Dry Reagent Storage
Three separate dry storage pads were used. Two of the pads (the conjugate pads) contained a single, type of biotinylated antibody-streptavidin HRP conjugate specific for either influenza A or B; the third pad contained DAB. The pads were made from a glass fiber membrane, GF 8950 pads (Ahlstrom, Helsinki, Finland), with dimensions 5 mm × 10 mm (∼20 μL fluid capacity). The pads were blocked by soaking in an aqueous solution contained 10 mM PBS, 1% (w/v) bovine serum albumin (BSA), and 0.05% (v/v) Tween-20 in a Petri dish for 1 hour. Once blocked, the pads were dried in a desiccated oven at 37 °C for 2 hours, then transferred to a desiccator for storage until use.
To prepare antibody-HRP conjugates for dry storage, Pierce™ streptavidin poly-HRP (final concentration 2 μg/mL) and anti-influenza A NP IgG (final concentration 4 μg/mL) or anti-influenza B NP IgG (final concentration 1.5 μg/mL) described above were added to a 20 μL preservation mixture containing 0.01 M FeSO4-EDTA (Fe-EDTA), 4% trehalose (Sigma Aldrich, Saint Louis, MO), and 0.1% BSA (Sigma Aldrich, Saint Louis, MO) in PBS. Our previous study showed 0.01 M Fe-EDTA was optimal for long-term dry stability of the HRP-antibody in trehalose19. The FeSO4 was prepared in water by mixing equimolar concentrations of FeSO4 and EDTA solution, which prevented precipitation of the FeSO4 in phosphate-buffered saline (PBS)19. Both types of antibody-HRP conjugate pads were prepared in polystyrene microwell plates (Fisher Scientific, Pittsburgh, PA), with a chemical-resistant polypropylene mesh (McMaster-Carr, Elmhurst, IL) used as a spacer between the pad and the bottom of the well to prevent sticking after drying. Antibody-HRP in the preservation mix was added to the pads in 20 μL volumes. The pads were dried at 30°C under vacuum (Genevac Inc., Gardiner NY) for 1.5 hours, placed in a foil pouch (Ted Pella, Redding, CA) with a one-gram, blue-indicating silica gel packet (Delta Adsorbents, Roselle, IL), sealed, and then stored in a desiccator at room temperature.
To prepare DAB for dry storage, DAB tablets (Sigma Aldrich, Saint Louis, MO) were dissolved in water and mixed with trehalose to achieve a final concentrations of 2 mg/mL DAB and 4% trehalose in a 20 μL preservation solution. DAB pads were prepared under the same conditions as the antibody-HRP conjugate pads.
Aqueous Reagents Storage
Three aqueous reagents were used. The lysis buffer (80 μL) contained 10 mM PBS (P3563-10PAK, Sigma Aldrich, Saint Louis, MO), 0.05% (v/v) Tween-20, 1% (w/v) BSA (A-3294, Sigma Aldrich, Saint Louis, MO), and 12% (v/v) Triton X-100 (T-9284, Sigma Al-drich, Saint Louis, MO). The rinse buffer (80 μL) contained 10 mM PBS, 0.05% (v/v) Tween-20, and 1% (w/v) BSA. The signal amplification buffer (280 μL) contained 10 mM PBS, 0.05% Tween-20, and 0.02% sodium percarbonate, which becomes H2O2 in solution. The aqueous reagents were stored in the device in containers constructed from PMMA and aluminum foil tape (#7631A71, McMaster-Carr, Santa Fe Springs, CA), as described below.
Device Part Fabrication
Materials for the devices were cut into desired shapes and sizes using a CO2 laser (Universal Laser Systems, Scottsdale, AZ). AutoCAD (Autodesk Inc., CA) design files are available (Figure S-1). Porous membrane materials were laser-cut from sheets. Sheets of glass fiber GF8950 were cut into 2DPN distributor pads and rectangular 5 mm × 10 mm dry storage pads. Sheets of cellulose CFSP223 (EMD Millipore, Billerica, MA) were cut into 7 mm × 12 mm wicking pads. Sheets of nitrocellulose HF120 (HF12002XSS, EMD Millipore, Billerica, MA) were cut into 5 mm × 18 mm lateral flow (LF) test strips. Plastic materials for the device mechanical structure were laser-cut from 12″ × 12″ polymethyl methacrylate (PMMA) sheets (U.S. Plastic Corp, Lima, OH). Structural materials were cut from either 1/16″ or 1/8″-thick sheets, minimizing thictenkness where possible without compromising structural strength of the final device.
Integrated System Assembly
Non-moving plastic parts were bonded by pipetting methylene chloride (Fisher Scientific, Fair Lawn, NJ) between contacting faces (Figure S-1). Moving plastic materials were treated with a dry Teflon lubricant (DuPont/Finish Lin Tech, Bay Shore, NY) on contacting surfaces to provide durable lubrication. The same lubricant was applied to the base of each plastic material designed to puncture aqueous reagent storage containers.
The aqueous reagent storage containers were designed individually for the desired liquid storage volume. The swab head was inserted into a relatively small volume (80 μL) of lysis buffer, so the shape of the lysis buffer container was designed to closely conform to the shape of the swab to ensure its complete submersion. The rinse buffer and amplification buffer containers were designed to hold 80 μL and 280 μL of buffer, respectively. All three aqueous reagent containers were sealed with aluminum foil tape (7631A71, McMaster-Carr, Elmhurst, IL), on one side, loaded with reagent, then sealed on the other side with another piece of aluminum foil tape.
Usability Study to Improve Device Design
Prior to device testing at Seattle Children's Hospital, a usability study was conducted by PATH through observation and interviews using the System Usability Scale (SUS)20,21, in which 16 participants (eight laboratory technicians and eight naïve users) operated the mock device. The goals of the study were to evaluate usability, identify potential failure modes, and observe user interactions with the device. The participants ran a mock device that was built specifically for the usability assessment. It contained no sharps, or active reagents for NP detection, but it included lateral flow strips with visible test and control lines. Participants were interviewed regarding their experiences using a semi-structured interview guide and a SUS questionnaire based on the Likert scale response system. The interview data were analyzed and interpreted according to industry standard interpretations20–22.
Device Testbed
A testbed was built to facilitate device function within the hospital clinical workflow; it also enabled consistent image capture of test results, and monitored user activity (Figure 2). A PMMA base (McMaster-Carr) was laser cut with a slot to enable the consistent placement of the disposable influenza device relative to the cell phone camera. A Nexus 5X smartphone (LG) was positioned 5 inches above the base with a custom, laser-cut PMMA holder. A Hero 4 Session video camera (GoPro Inc.) was mounted so as to view user interactions with the device. A 22.4 Ah USB battery pack (Innori, City of Industry, CA) was also mounted to the base to ensure that the smartphone and GoPro remained adequately charged during the month-long clinical evaluation.
Figure 2.

Imaging system used to hold the disposable device, record signal development in the device, and to monitor user activities during device operation at Seattle Children's Hospital.
Swab Specimen Collection
Seattle Children's Hospital Institutional Review Board (IRB) approved the protocols for subject consent, sample collection, handling, and the analysis of specimens. The clinical samples were collected as part of NIH grant R01AI096184 (Yager, PI). Mid-nasal swab specimens were collected from patients using FLOQSwab contoured flocked swabs (adult: 56380CS01 or infant: 56780CS01, Co-pan Diagnostics, Inc., Murrietta, CA). The 25 specimens were collected between February and March of the 2016 flu season.
Swab Specimen Transport and Swab Storage Tube
Clinic workflow dictated constraints on specimen handling, transport, and storage prior to device tests. Devices were tested in a separate room from patients. Therefore, after collection from patients, loaded nasal swab were stored and transported in made swab storage tubes; these tubes were fabricated from off-the-shelf lab supplies and contained either 300 or 600 μL PBST with 0.05% sodium azide. Storage time varied, typically ranging from 5 min to 2 hr, except for two devices (tests Flu23 & Flu33), which were run ∼15 h after specimen collection.
Image Capture and Analysis
Automated image capture was performed on the Nexus 5X smartphone with a custom mobile app written using the Android Application Programming Interface (API) 23. The app recorded standard (3×8-bit sRGB) JPEG images of the test strips every 30 seconds for 20 minutes and every minute for 35 more minutes. The operator was asked to start the app just before the integrated device was activated to ensure recording of the entire test and control line development process. Test strips were also scanned using a flatbed scanner (ScanMaker i900, MicroTek International, Cerritos, CA) in 3×16-bit-depth sRGB mode (the 16-bit blue channel was used for image analysis) at 600 dpi within five days of use.
Test results were interpreted to be valid only when control lines on both NC detection strips were visible by eye or by digital interpretation of the cell phone images thereof; tests were invalid when one or both control lines were not visible. Device results were visually interpreted (an average of fifty minutes after the device was activated) and recorded by the clinician operator at the end of each device test at Seattle Children's Hospital. The test devices were retrieved within five days of usage, and test strips were extracted from the used devices for scanning in the Yager lab. Smartphone images, or scanner images in the nine cases where the cell phone image recording was incomplete, were algorithmically interpreted in an automated analysis procedure described below. The automated interpretation, the visual interpretations by the clinical team at Children's Hospital, and gold-standard qRT-PCR results were then compared.
ImageJ (NIH, Bethesda, MD) was used to prepare images of each test for line determination by algorithmic analysis. Each test image (smartphone or scanner) was straightened and cropped to the edges of the LF strips, and inverted. The algorithm, written in Mathematica 10.4 (Wolfram, Champaign, IL), was then run on the entire set of prepared images. First, the pixel intensity in the blue channel was extracted from within two rectangular regions of interest, which were fixed in location in each image relative to the size of the image (as a percentage of image height and width) due to the different resolutions of the cell phone and scanner images. For both image types, the algorithm located a rectangle associated with each of the test strips that contained 100% of the image height, and 35% of the image width (all the pixels between 7.5% and 42.5%, and 57.5% and 92.5%, of the image width for Flu A, and Flu B). The intensity values in these rectangles were row-averaged to generate a line intensity profile running the length of each strip in each image. The entire set of line intensity profiles was then filtered to remove noisy signals. The effect of uneven lighting was corrected through background subtraction. A sixth-order polynomial line, which was fit to the line intensity profile in background regions only, was subtracted from the entire line intensity profile. Again, due to the differences in image resolutions, the background regions were defined in percentages of image height (all the pixels between 0% and 25%, 40% and 65%, and 85% and 100% of the image height). After background subtraction, the average and standard deviation of the remaining background in these regions were calculated, and a threshold was set at three standard deviations from the mean. Test lines were distinguished from flow artifacts and nitrocellulose damage as excursions above the threshold with an integrated intensity value greater than 56.
qRT-PCR
To confirm the protein-based results, influenza A and B RNA was quantified in the remaining sample fluid using the UltraSense™ One-Step Quantitative RT-PCR assay mix (Life Technologies, Carlsbad, CA) and primer and probe sequences (published previously23,24) in 20 μL reactions run on a CFX96 Touch (Bio-Rad). The thermal protocol used was: 50°C hold for 15 minutes, 95°C hold for 2 minutes, 40 cycles of 95°C for 15 seconds and 60°C for 55 seconds. Genomic RNA copy numbers were determined relative to standard curve analysis using influenza A or B control RNA of known copy number.
Results and Discussion
Device Operation
Figure 3 summarizes the complete user experience of the prototype device, which included swab collections, storage, and transfer; sample introduction to the device (swab was returned to the buffer storage tube); device activation; and interpretation of visible results. Figure 4 depicts the assay chemistry and internal device components, which automate the assay after device activation.
Figure 3.

Device operation at Seattle Children's Hospital. Clinical staff collected patient samples and stored the swab in a swab 4 storage tube. The actual device test occurred in a room nearby within the hospital. The cumulative time required for test completion was ∼35 min. The image system continuously recorded the images of the lateral flow test strips for ∼55 min.
Figure 4.
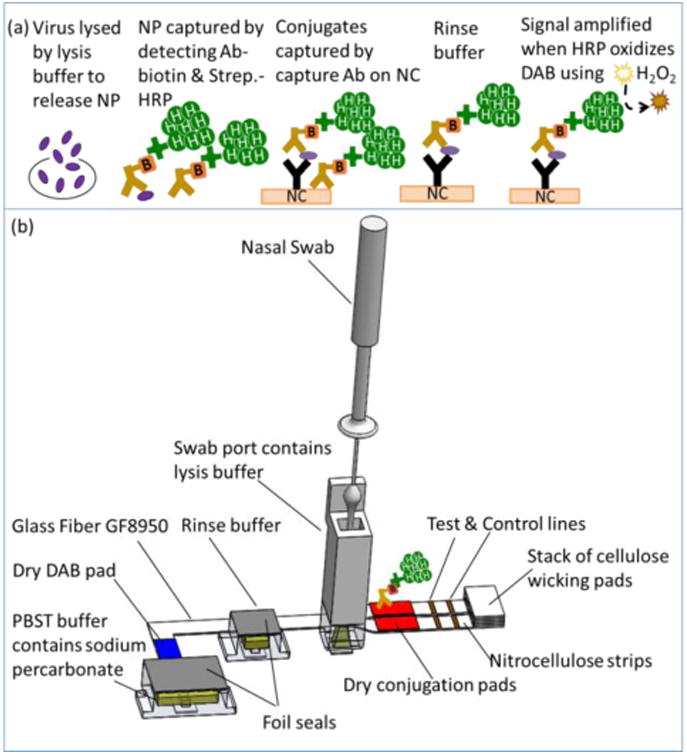
Internal device operation. a) Cartoon of the NP detection assay chemistry at the analyte capture lines, which is based on NP binding to HRP-conjugated antibodies. Next, the NP conjugate was captured by the membrane bound capture antibody, followed by HRP turnover of the chromogenic substrate, DAB in the presence of H2O2. b) 3D model of the internal components of device. Sample introduction by swab into a swab port containing lysis buffer is followed by activation of the device, which causes puncture of aqueous reagent container and release of aqueous reagents into legs of the 2DPN. The 2DPN automates rehydration of dry reagents, splitting the sample into two channels (one for influenza A detection, the other for influenza B detection), and sequential delivery of subsequent assay reagents.
Swab introduction to the device was performed by inserting a swab into the swab port of the device, followed by a “one Hz twirl” in the on-board lysis buffer for ten seconds25 to release virus from swab. As there were different swabs in use for adult and infant patients, we chose a lysis buffer with a total volume and a detergent concentration that accommodated both swab types. A volume of 80 μL was sufficient to submerge the heads of both swab types. Previous assay development work led to the use Triton X-100 at a final concentration of 5% for both swab types (Figure S-2). The lysis buffer caused viral lysis and release of NP upon swab introduction. After sample introduction, device activation was accomplished by simply closing the device; all subsequent device assay operations were automated.
The device housing was composed of five parts, which were a horizontal slider, a vertical slider, a base plate that provided a track for each slider, a middle plate that supported the aqueous reagent containers and 2DPN, and a cover plate that fully encloses the internal components, all assembled as shown in Figure S-3. Activation of the device required only one step once a sample was introduced; as shown in Figure S-3c, closing the device with gentle pressure from the user's thumbs pushed the vertical slider along the 45° tracks in the base plate. This vertical movement caused the sharps to puncture the foil seals on the aqueous reagent containers, thus releasing fluid to the 2DPN.
Vacuum-dried reagents, stored in the 2DPN materials, were rehydrated by the lysed sample and other aqueous reagents as they passed through the paper fluidic channel. System operations were automated in the 2DPN, which was specifically designed to rehydrate dry reagents, split the sample into parallel influenza A and B tests, and sequence fluid delivery (Figure 4). As illustrated in Figure 4, the lysate, which contained free NP, flowed into two channels through dry conjugation pads, which rehydrated antibody-HRP conjugates that captured the free NP. The lysate with these complexes flowed through test lines, where the complexes were captured by antibodies localized to the test line on the NC membrane. Rinse buffer sequentially flowed through the test and control lines, to minimize non-specific adsorption of antibody-HRP conjugates. A mixture of H2O2 and rehydrated DAB sequentially flowed through the test and control lines, thereby allowing visualization of the test and control lines through enzymatic conversion of the colorless DAB to a brown precipitate by HRP in the presence of H2O2. A time series of images illustrating the assay delivery sequence using colored dye is shown in Figure S-4. This assay chemistry was chosen because it showed 10-fold better sensitivity over conventional gold nanoparticles in preliminary working using sequential dipping of nitrocellulose strips into microwell26.
Usability Study
Users rated the device high on learnability, meaning that they thought people could learn to use the device quickly. On average, first time users took 8 minutes to run the device, which included time for the users to review the instructions, think through the device operation procedure, read and interpret device results. Repeated runs of the mock device were performed much more quickly and averaged 3 min. User feedback regarding the device design was largely positive and the device was considered to be simple to use overall. The average usability score was 74.38, which placed the device well within the acceptable range on the SUS. Naïve users scored the device higher than laboratory technicians, with SUS scores of 78.13 (SD 10.36) and 70.63 (SD 10.29) respectively.
The study results suggested that improved usability and prevention of failure modes could be achieved through better user instructions and minor device modifications (e.g. distinct color on the device to better differentiate parts, adjusting read window geometry to allow easier photo taking, etc.).
Suggestions from this study formed the basis of changes made to the device design, coloring, and user instructions for the fully integrated device prototype for use in a clinical setting. The performance study was conducted at Seattle Children's Hospital by six clinical staff members who had no involvement in the prototype development.
Performance Study in a Clinical Setting
Following an IRB-approved protocol, the device performance study was performed in a “test room” in the Emergency Department, but separate from the patient examination rooms. A custom, sealed swab storage tube was developed and used to transfer the swab from the examination rooms (where samples were collected) to the test room, and provided subsequent swab storage for validation testing using qRT-PCR. Reagents that would typically require cold storage were stored dry in the device; this was necessitated by the limitation of cold storage space in the test room, but also allow for ultimate use in “austere environments”. Automatic image capture and video monitoring (as shown in Figure 2) for recording of device operation by clinical staff and the test and control lines and development, were developed and used in response to the unpredictable nature of patient interactions with clinical staff, which translated to variable delays between sample acquisition, running the tests, and reading the results.
Prototype devices were operated by clinical staff as soon as possible (∼5 min to, in two cases, 15 h) after human nasal swab specimens were collected. In total, 25 device tests were completed. No device operational failures (e.g. user error, broken devices, or failed chemical functions of the device operation) occurred during the clinical test period. For the first four device tests (Flu01-04), the buffer volume in the original swab storage tube was 300 μL. Devices, Flu03 & Flu04, were invalid, with one or both control lines missing on the LF strips. In each of those cases, we observed that the patient sample was highly viscous (contained viscous nasal secretions), or appeared to contain high amounts of blood. We believe the high viscosity of those samples disrupted the capillary flow of the sample through the test strip. Therefore, for the remaining 21 tests, we increased the volume of buffer in the swab storage tube to 600 μL. There were no invalid device tests after this change, suggesting that further dilution of the patient samples improved flow through the paper network. The ratio of valid to total completed device runs in the entire set yielded a success rate of 92 % (23/25), while, a success rate was 100 % (21/21) for the subset of tests with swab storage volume of 600 μL.
Examples of image analysis for strong and weak test line intensity, as well as a negative sample result are shown in Figure 5. For each test, the fluid flow direction was downward in the images, with the test line (T) positioned above the control line (C). Variable room conditions, such as the occasional loss of room light, led to an incomplete image record for some runs. All image analysis results are shown in Figure S-5. Figure 6 summarizes the results of all 25 devices and compares them with qRT-PCR tests. There were no false positives observed. Out of the ten qRT-PCR influenza A-positive samples, seven were correctly identified by our device. For five qRT-PCR influenza B positive samples, our device correctly detected one to be positive. It is important to note that the influenza copy number (Figure S-6) reflects the remaining amount of virus in the swab storage tube after device test. The purpose is to determine whether the sample was influenza positive or negative.
Figure 5.
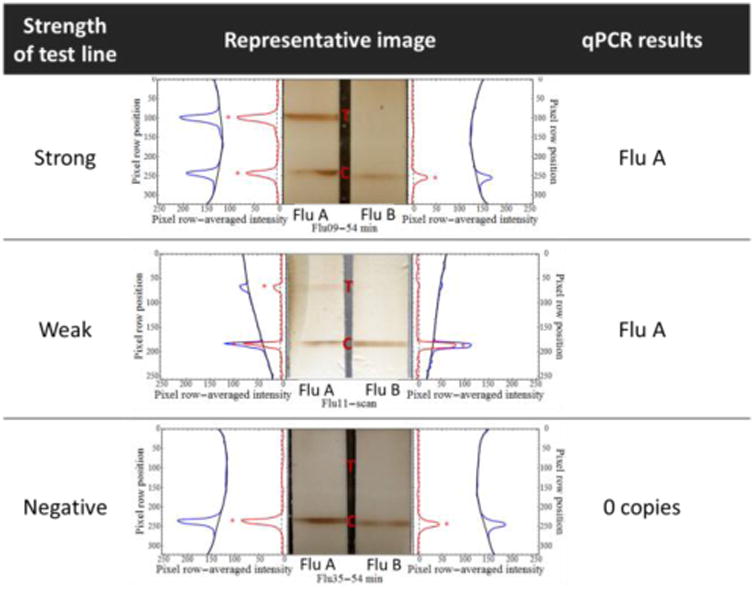
Examples of the extracted signal profiles for strong, weak, and negative results. The row-averaged pixel intensities in the blue channel of test strip images were used to generate plot profiles for each test strip (blue lines). Sixth-order polynomials (black lines) were fit to the background regions of each plot profile. Test lines were detected as thresholded (dotted black lines) excursions (red asterisks) of the background-subtracted plot profiles (red lines) beyond three standard deviations of the mean background-subtracted intensity (dotted black lines).
Figure 6.
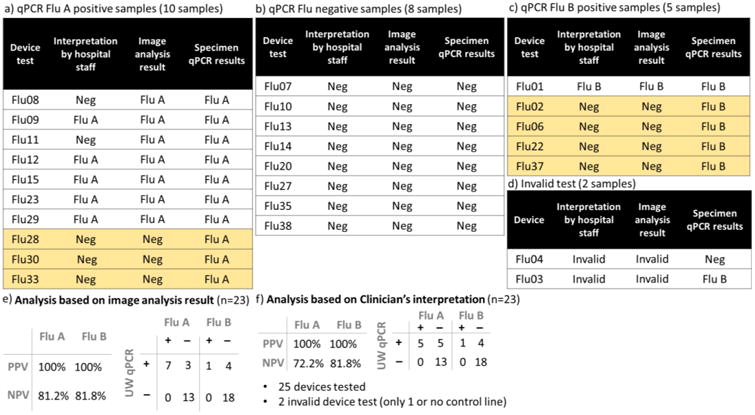
Summary of 25 device results vs. gold-standard qRT-PCR test results. The qRT-PCR was used to determine whether the sample was influenza positive or negative. Yellow boxes show disagreement between qRT-PCR test results and the device results.
Figure 6 lists the diagnostic interpretations given by hospital staff immediately after each device test. Figure 6 also lists the diagnostic interpretations provided by an algorithmic image analysis run on a phone camera image (or a scanned image if the phone camera image was not available) for each device test. The algorithmic interpretations returned negative predictive value for both the influenza A and B assays of 81% (Figure 6e), while the clinical staff interpretations returned negative predictive values for both the influenza A and B assays of 72% (Figure 6f). The clinical sensitivity of our device for Flu A detection is 70%, and for Flu B is 20%. The specificity for both tests is 100%. Though the clinical sample size was small, these first clinical results are encouraging.
In this study, the algorithm interpretations yielded a better negative predictive value than did the clinical staff interpretations. Specifically, we noted that positive test Flu08 and Flu11 (Figure S-5) had faint test lines, which were interpreted as negative by clinical staff, but as positive by our algorithmic analysis. Nonetheless, the automated image analysis was limited by the use of heuristic thresholds, and by the quality of the images of the LF strips. The algorithm was designed to remove noisy signals from across the set of images given a priori knowledge of test line locations and of typical sources of noise, such as uneven room lighting, non-specific enzyme amplification, and damage to the nitrocellulose. For phone images, changes in room lighting led to the occasional, artificial decrease in intensity (appearing as horizontal white lines) adjacent to the position of the test line (e.g. Figure S-5, Flu22, 30 etc.), which increased the likelihood of false negative interpretations. For scanned images, we observed a strong brown residue on the left edge of scanned Flu A strips (e.g. Figure S-5, Flu12), and occasional scratches made during removal of the strips from the device for scanning (e.g. Figure S-5, Flu12), which increased the likelihood of false positive interpretation. Algorithmically, the heuristic approaches to these problems were to 1) model and subtract the background from each row-averaged line intensity profile using a sixth-order polynomial, 2) filter out the majority of noisy signals with a threshold set to three standard deviations of subtracted background intensities, and 3) filter out noisy signals due to damaged nitrocellulose with a second threshold on excursions above the first threshold. However, device Flu28 (Figure S-5), which appeared to generate a positive influenza A test line, but only on the very edge of the strip, was interpreted as negative for influenza A by the algorithm. Therefore, the heuristic thresholds may impact the general applicability of the algorithm, and will need to be tested further in a variety of conditions.
We have also demonstrated, subsequent to the performance study reported here, that changes in the geometry of the 2DPN can substantially improve the sensitivity of the test (data not shown). The geometry of material overlaps (e.g., between the reagent storage pad and the rest of the paper network) impacts the concentration gradient of reagents after rehydration from dry storage, which in turn has a significant impact on assay performance and signal development.
Related optimization of the flow rate of reagents can improve assay performance and signal development, especially with respect to background signal. Several studies have shown that the flow rate in LF strips significantly impacts the assay sensitivity18,27–29. Flow rate can be tuned by incorporating fluidic sinks28, barriers29 or other architecture modifications27,29 to achieve an improved assay sensitivity.
As the patient samples are biologically complex, it is uncertain how much virus from the sample into the device was released to either the storage buffer in the storage tube or the lysis buffer in the device, or how consistent the release was into either buffer over all tests. Also uncertain is the degree to which nasal swab sample components could impact fluid flow or inhibit the assays. Therefore, difficulties remain in correlating test line intensity with virus copy number; more patient samples would be required to further explore any possible correlation. As previously mentioned, to address the issue that some individual patient samples were more viscous and seemed to adversely affect sample flow in the device, we reduced input sample viscosity by increasing the buffer volume in which the swab was immersed from 300 to 600 μL. This substantial increase in dilution of the sample (i.e., “normalization” of the input sample viscosity to be closer to that of buffer for all samples) resolved the flow issue, such that after this change, 100% of the devices ran successfully (i.e., resulted in two visible control lines). The trade-off for diluting the sample further is that the virus copy number/volume of fluid will be lower (and thus the NP protein concentration). In our case, this trade off was deemed to be acceptable, since we perform signal amplification downstream. We noticed that some of our tests showed only a faint Flu B control line (Figure S-5, Flu22 & 27). As the assay chemistry and 2DPN design in our current devices were pre-optimized using purified recombinant proteins/influenza viruses, a lesson is that if available, clinical samples are preferred to guide device development (and prevent issues such as faint control lines).
A direct comparison of our assay chemistry with the commercially available RDTs was not part of our performance study in the clinical setting, nor was it included in our IRB. Thus we directly compared our assay to RDTs using influenza A re-combinant NP. Specifically, our NP assay, implemented in a related dipstick format compares well (3-fold improvement) with a high-performing RDT (BD Directigen™ EZ Flu A+B), as shown in Figure S-7. We also tested the Alere BinaxNOW® Influenza A&B, which is CLIA-waived and requires minimal user steps by rotating the swab in the liquid, pipetting the liquid on the card, and then closing the card. Our dipstick format shows better sensitivity (20-fold improvement); the caveat is that the Alere BinaxNOW® Influenza A&B is optimized for complex patient samples and not purified proteins (Figure S-7). In addition, we reference additional results26 in which the assay was benchmarked with whole virus and showed 10-fold better sensitivity compared to BD Directigen™ EZ Flu A+B (manuscript in preparation). These results highlight the potential improvement in sensitivity that we can achieve in our integrated device using our enzymatically-amplified flu reagent system. We expect that future sensitivity improvements in the integrated device can be accomplished by optimizing our current 2DPN geometry and flow rate as we discussed above (manuscript in preparation).
In summary, the main strengths of our integrated device are threefold. First, usability studies were conducted and guided the current iteration (and will continue to guide future iterations) of our device. Second, sample preparation was substantially simplified from current RDTs by (a) elimination of the requirement for users to pipet sample and (b) improvement of ease of use through a simple activation step. There is high value in removing extraneous user steps and in particular any step that could produce substantial variations in the sample volume processed by the device and in turn the resulting signal magnitude. Further, the clinical staff found our device easy and robust to use. From their perspective, the elimination of pipetting and other sample preparation steps makes it possible for busy caregivers to conduct effective testing at the POC.
Finally, the imaging capture system (Figure 2) used for the device performance study in the clinical setting was based on clinician feedback on setting requirements. Our long-term goal is to add an adapter to our integrated device that connects the device to the smartphone for image acquisition after the test is complete. This, plus further work on the image analysis algorithm, could be used for automated interpretation of the test result.
Conclusions
While the current device performed well overall in the clinical setting, there is more work to be done to optimize its performance. Improvements to the sensitivity of the device for both Flu A and Flu B targets would be of especially high value. Because our Flu B assay did not perform as well as our Flu A assay in the performance study, we are already exploring influenza B antibodies with higher affinities to improve the Flu B assay sensitivity. Improvements to the ability of the device to provide subtype information are important as well. Thus, a demonstration of the detection of influenza HA using novel, recombinant affinity proteins developed by our collaborators is underway in our lab30.
The aims of the work described here were 1) to demonstrate a multiplexed sandwich immunoassay, with signal amplification, for enhanced-sensitivity detection of proteins from pathogens in a clinical sample, and 2) embody that test in a fully integrated, sample-to-result influenza diagnostic device for simple, robust, easy-to use operation by clinical staff working during the 2016 influenza season. We achieved reasonable sensitivity levels in a prototyped device that, after sample introduction, was activated with a single user step and had a short sample-to-result time. The prototype device used a combination of dry storage of sensitive biological reagents and wet storage of stable buffers, which eliminated the need for cold shipment and storage. The prototype device had a materials cost of less than $5 (which could be greatly reduced in mass fabrication), and a reagent cost of less than $1 per device. These features could enable broad use of this device, including in low-resource settings.
The work was motivated by the need for higher-sensitivity POC influenza diagnostic; the level of integration and automation achieved with this prototype device represents a significant step towards the development of rapid sensitive protein-based diagnostic devices for POC settings.
Supplementary Material
Acknowledgments
This work was supported by NIH (R01AI096184) to Dr. Paul Yager at the University of Washington with subcontracts to Dr. Janet Englund at Seattle Children's, Dr. Elain Fu at Oregon State University, Dr. David Moore at GE Global Research Center, and Dr. Gonzalo Domingo at PATH. All teams contributed materially to the concepts behind the design presented. Particular thanks are owed to Seattle Children's Emergency Medicine for their support on patient sample collection and device test.
Footnotes
Supporting Information: The Supporting Information PDF contains the following: details of the device fabrication, an experimental protocol for the determination of swab volume and Triton X-100 concentration in the lysis buffer, device activation by a single step, a color dye experiment to show the automated assay steps using a 2DPN, details of image analysis for the results for 25 patient specimens, influenza qRT-PCR assay results for 25 patient specimens, and additional limit of detection comparisons of our assay (dipstick format) to commercially available RDTs using recombinant NP as a target.
References
- 1.Wright PF, Neumann G, Kawaoka Y. Fields Virology 5th Edition. 2007:1693–1740. [Google Scholar]
- 2.Molinari NAM, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. Vaccine. 2007;25(27):5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 3.Morens DM, Taubenberger JK, Harvey Ha, Memoli MJ. Crit Care Med. 2010;38(4):e10–e20. doi: 10.1097/CCM.0b013e3181ceb25b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Organization, W. H. WHO | Influenza (Seasonal) World Health Organization; 2014. [Google Scholar]
- 5.Taubenberger JK, Morens DM. Public Health Rep. 2010;125 Suppl:16–26. [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. CDC -Seasonal Influenza (Flu) http://www.cdc.gov/flu/
- 7.Mahony JB. Clin Microbiol Rev. 2008;21(4):716–747. doi: 10.1128/CMR.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bose ME, Sasman A, Mei H, Mccaul KC, Kramp WJ, Chen LM, Shively R, Williams TL, Beck ET, Henrickson KJ. Influenza Other Respi Viruses. 2014;8(4):474–481. doi: 10.1111/irv.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurt AC, Alexander R, Hibbert J, Deed N, Barr IG. J Clin Virol. 2007;39(2):132–135. doi: 10.1016/j.jcv.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Centers for Disease Control and Prevention. Guidance for Clinicians on the Use of RT-PCR and Other Molecular Assays for Diagnosis of Influenza Virus Infection. 2014 [Google Scholar]
- 11.Mahony JB. Clin Microbiol Rev. 2008;21(4):716–747. doi: 10.1128/CMR.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases. World Health Organization; 2010. p. 27. [Google Scholar]
- 13.Bonner AB, Monroe KW, Talley LI, Klasner AE, Kimberlin DW. Pediatrics. 2003;112(2):363–367. doi: 10.1542/peds.112.2.363. [DOI] [PubMed] [Google Scholar]
- 14.Chartrand C, Leeflang MMG, Minion J, Brewer T, Pai M. Annals of Internal Medicine. 2012:500–511. doi: 10.7326/0003-4819-156-7-201204030-00403. [DOI] [PubMed] [Google Scholar]
- 15.Peterson S, Dugas AF, Rothman RE. Annals of Emergency Medicine. 2013:573–576. [Google Scholar]
- 16.Fridley GE, Le HQ, Fu E, Yager P. Lab Chip. 2012;12(21):4321–4327. doi: 10.1039/c2lc40785j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu E, Liang T, Spicar-Mihalic P, Houghtaling J, Ramachandran S, Yager P. Anal Chem. 2012;84(10):4574–4579. doi: 10.1021/ac300689s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grant BD, Smith CA, Karvonen K, Richards-Kortum R. Analytical Chemistry. 2016:2553–2557. doi: 10.1021/acs.analchem.5b03999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramachandran S, Fu E, Lutz B, Yager P. Analyst. 2014;139(6):1456–1462. doi: 10.1039/c3an02296j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bangor A, Kortum PT, Miller JT. Int J Hum Comput Interact. 2008;24(6):574–594. [Google Scholar]
- 21.Bangor A, Kortum P, Miller J. J usability Stud. 2009;4(3):114–123. [Google Scholar]
- 22.Sauro J, Lewis JR. Chapter 2 - Quantifying User Research. 2012;11 [Google Scholar]
- 23.Kuypers J, Wright N, Ferrenberg J, Huang ML, Cent A, Corey L, Morrow R. J Clin Microbiol. 2006;44(7):2382–2388. doi: 10.1128/JCM.00216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casado B, Pannell LK, Iadarola P, Baraniuk JN. Proteomics. 2005;5(11):2949–2959. doi: 10.1002/pmic.200401172. [DOI] [PubMed] [Google Scholar]
- 25.Panpradist N, Toley BJ, Zhang X, Byrnes S, Buser JR, Englund JA, Lutz BR. PLoS One. 2014;9(9) doi: 10.1371/journal.pone.0105786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abe K, Huang S, Yager P. Meeting of Pacifichem. Hawaii: 2015. [Google Scholar]
- 27.Parolo C, Medina-Sánchez M, de la Escosura-Muñiz A, Merkoçi A. Lab Chip. 2013;13(3):386–390. doi: 10.1039/c2lc41144j. [DOI] [PubMed] [Google Scholar]
- 28.Choi JR, Liu Z, Hu J, Tang R, Gong Y, Feng S, Ren H, Wen T, Yang H, Qu Z, Pingguan-Murphy B, Xu F. Anal Chem. 2016 doi: 10.1021/acs.analchem.6b00195. acs.analchem.6b00195. [DOI] [PubMed] [Google Scholar]
- 29.Fu E, Lutz B, Kauffman P, Yager P. Lab Chip. 2010;10(7):918. doi: 10.1039/b919614e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson Ia, Baker D. Science. 2011;332(6031):816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.