

Significance
Mutations in a microglial protein, TREM2, represents a risk for Alzheimer’s disease (AD). We show that the soluble form of TREM2, sTREM2, can bind and inhibit fibrillization of Aβ peptides. sTREM2 increases uptake of Aβ fibrils into microglial and neuroglioma cell lines. Contrastingly, mutation R47H was found to have little effect on fibril nucleation and binding, but decreased uptake and functional responses. Our findings using integrative molecular modeling based on cross-linking mass spectrometry data for WT sTREM2–Aβ fibril complex demonstrate that TREM2 has at least two ligand-binding surfaces: one binding Aβ fibrils and the other anionic polyvalent ligands. R47H mutation lies on the latter surface. These findings inform mechanisms by which TREM2 modulates key processes in AD progression.
Keywords: soluble TREM2, amyloid-β, Alzheimer’s disease, integrative modeling, fibrillization kinetics
Abstract
Triggering receptor expressed on myeloid cells 2 (TREM2) is a single-pass transmembrane receptor of the immunoglobulin superfamily that is secreted in a soluble (sTREM2) form. Mutations in TREM2 have been linked to increased risk of Alzheimer’s disease (AD). A prominent neuropathological component of AD is deposition of the amyloid-β (Aβ) into plaques, particularly Aβ40 and Aβ42. While the membrane-bound form of TREM2 is known to facilitate uptake of Aβ fibrils and the polarization of microglial processes toward amyloid plaques, the role of its soluble ectodomain, particularly in interactions with monomeric or fibrillar Aβ, has been less clear. Our results demonstrate that sTREM2 does not bind to monomeric Aβ40 and Aβ42, even at a high micromolar concentration, while it does bind to fibrillar Aβ42 and Aβ40 with equal affinities (2.6 ± 0.3 µM and 2.3 ± 0.4 µM). Kinetic analysis shows that sTREM2 inhibits the secondary nucleation step in the fibrillization of Aβ, while having little effect on the primary nucleation pathway. Furthermore, binding of sTREM2 to fibrils markedly enhanced uptake of fibrils into human microglial and neuroglioma derived cell lines. The disease-associated sTREM2 mutant, R47H, displayed little to no effect on fibril nucleation and binding, but it decreased uptake and functional responses markedly. We also probed the structure of the WT sTREM2–Aβ fibril complex using integrative molecular modeling based primarily on the cross-linking mass spectrometry data. The model shows that sTREM2 binds fibrils along one face of the structure, leaving a second, mutation-sensitive site free to mediate cellular binding and uptake.
Alzheimer’s disease (AD) is the most common form of dementia and features the neuropathological hallmarks of extracellular Aβ plaques and intraneuronal tau neurofibrillary tangles (1, 2). Human genetic studies on heritable mutations in APP and PSEN causing early-onset familial AD (3) argue that pathogenic Aβ drives tau neurofibrillary tangle formation; in contrast, mutations in MAPT do not lead to Aβ pathology nor cause AD, but rather a rare genetic form of early-onset primary tauopathy (4). In support of the molecular genetics, a recent cross-sectional study in postmortem human AD brain samples demonstrated the presence and correlation of robust prion bioactivity for Aβ and tau proteins in nearly all cases (5), suggesting that even at death, Aβ in prion conformations are active in the late stages of disease. Together, these data establish the importance of pathogenic Aβ throughout AD progression and highlight the urgent need to better understand the cellular and molecular mechanisms that mitigate Aβ’s role in pathogenesis.
Microglia are the innate immune effector cell in the brain with myriad functions in healthy aging and neurological diseases. Recent human genetic studies have discovered mutations in several genes encoding microglia-specific proteins that increase risk for AD, thus supporting the notion that microglia are central to AD pathogenesis. Genetic variants of triggering receptor expressed on myeloid cells 2 (TREM2), a cell-surface receptor expressed on myeloid cells and microglia, increase the risk of AD by threefold, implicating microglia and the innate immune system as important determinants in AD pathogenesis (6). TREM2 consists of an extracellular Ig-like domain, a transmembrane domain, and a cytoplasmic tail. Proteolytic cleavage of TREM2 at His157 releases soluble TREM2 (sTREM2) that can be detected in the cerebrospinal fluid (7). While the function of sTREM2 is uncertain, it is believed to promote microglia survival, proliferation, and phagocytosis, making it important for cell viability and innate immune functions in the brain (6, 8, 9). Full-length membrane-bound TREM2 binds to its adaptor protein, DAP12, on the surface of microglia to transmit downstream signaling in response to clustering induced by multivalent ligands (10). Most of the studied mutations are in the Ig-like domain of TREM2. Misfolding, retention, and aberrant shedding are postulated to be caused by some mutations, while other variants have altered ability to interact with their binding partners (8, 11, 12).
The R47H mutation in TREM2 constitutes one of the strongest single allele genetic risk factors for AD. The R62H, D87N, and T96K mutations in TREM2 were also linked to AD after extensive analyses of TREM2 polymorphisms (13–16). Several in vivo studies show that TREM2 regulates polarization of microglial processes toward Aβ deposits, leading to plaque compaction and pacification in human AD brain samples and mouse models (17–19). Genetic deletion of TREM2 expression in transgenic mice injected with exogenous Aβ fibrils leads to accelerated amyloid plaque seeding (20). The prominent phenotype in plaque-associated microglia suggests that the effects of AD-risk mutations or genetic deletions are driven by loss of full-length TREM2 signaling. However, a recent in vivo study using exogenously injected recombinant sTREM2 showed reduced amyloid burden and behavioral rescue in mice (21). New clues for the potential importance of sTREM2 in AD have been revealed in clinical studies on living AD patients. sTREM2 can be measured in the cerebrospinal fluid (CSF) and it increases during early stages of AD symptomology (22, 23), suggesting that sTREM2 may be a biomarker for microglia activation. Recent studies indicate that AD patients with relatively high levels of sTREM2 in the CSF have slower rates of amyloid accumulation and reduced cognitive decline (24, 25). These human data support the hypothesis that microglia and sTREM2 play a protective role in early stages of AD progression.
While most risk variants of TREM2 exist in the ligand-binding Ig-like domain, the AD-associated point mutation H157Y falls within the stalk region and is known to increase the shedding of full-length TREM2, which possibly results in higher titers of sTREM2 (6). Elevated ectodomain shedding reduces cell-surface full-length TREM2 available for TREM2-mediated phagocytosis and plaque compaction as well as down-stream signal transduction. Although more work is needed, such data begin to suggest there is a delicate balance between the functions of membrane-bound and secreted TREM2, and hence opposing cellular effects of TREM2 variants can emerge (i.e., reduced versus enhanced shedding, which result in similar phenotypic outcomes by reducing cell-surface TREM2) (6, 26).
sTREM2 binds to diverse ligands, including phospholipids, apolipoproteins, DNA, and Aβ. Although the full physiological and pathological roles of these interactions remain to be revealed (11, 12, 27, 28), there is general agreement that the extracellular domain of TREM2 (sTREM2) binds to oligomeric forms of Aβ42. However, the observed apparent affinities vary over many orders-of-magnitude (7, 29–31). Most studies were conducted with dimeric Fc fusion proteins, tetrameric constructs, or biotinylated protein bound to the tetrameric streptavidin, which might artificially increase the avidity of the protein for oligomeric forms of Aβ peptides (7, 29–31). Moreover, the studies that report the highest affinities relied on biolayer interferometry or surface plasmon resonance, in which oligomeric protein constructs were immobilized on a surface and Aβ peptides were allowed to diffuse over the surface. Aβ oligomers were found to bind, but they either did not dissociate at all, or they dissociated slowly, leading to affinity estimates in the picomolar to nanomolar range (7, 30, 31). However, the extent of binding of Aβ to the surface did not saturate at concentrations that were orders-of-magnitude greater than the reported dissociation constants, suggesting that the slow off-rate was instead due to precipitation of insoluble Aβ on the bilayer surface (7). In another study, Aβ was fused to the dimeric protein glutathione S-transferase (29). Furthermore, there is inconsistency in the studies involving monomeric Aβ42, with some studies finding nanomolar to low micromolar dissociation constants for the interaction of monomeric Aβ42 and TREM2 ectodomain (29, 30), in contrast to two other studies that reported weak or no interaction (7, 31).
To help elucidate the role of sTREM2 and its interaction with Aβ, we evaluated the binding of sTREM2, without any nonnative oligomerization domains added to the studied construct, to specific forms of Aβ40 and Aβ42. We used NMR to show that sTREM2 does not bind to monomeric Aβ, even at high micromolar concentrations. Next, we examined the binding of sTREM2 to fibrils, formed under well-defined conditions to provide a relatively homogenous structure, as assessed by solid-state NMR (32). Additionally, because oligomeric forms of Aβ are heterogeneous and kinetically labile, we opted to determine how sTREM2 affects the formation of intermediates in the fibrillization of Aβ and show that it has a profound effect on the secondary nucleation step of the process. We find that the R47H variant binds to Aβ40 and Aβ42 fibrils with a similar affinity and inhibits their fibrilization just as the WT sTREM2 does. Finally, we show that WT sTREM2, but not the mutant R47H, strongly enhances the uptake of Aβ fibrils in human neural and microglial cells.
A second goal of this report was to define the structural underpinnings of the interaction between sTREM2 and Aβ fibrils. Although individual structures of sTREM2 and Aβ40 fibrils have been reported (8, 33), the structures of the complex are not available. The molecular surface of sTREM2 is particularly interesting with regards to its function (8, 29). The crystal structure of the ectodomain of TREM2 (TREM2ECD) revealed an immunoglobulin fold motif with a highly asymmetric distribution of charged and hydrophobic residues. The surface of the hydrophobic and aromatic protrusion at the top of the structure (Fig. 1, red dotted area) has a highly positive electrostatic potential adjacent to it is a relatively flat surface of positively charged residues (Fig. 1, black dotted area, surface 1). Surface 1 appears suited for binding to acidic moieties (like in Protein Data Bank [PDB] ID code 6B8O) (8). R47 lies near the basic patch, consistent with the R47H mutation disrupting the conformation of the CD loop (8), which comprises a large portion of surface 1. Molecular dynamics simulations suggest that disease-promoting mutations disrupt the apolar character and electrostatic surface of this region of the protein (34). The R47H mutation is also known to disrupt sTREM2’s ability to bind to and signal in response to acidic phospholipids (29). Thus, the data indicate that this surface is important for binding or signaling in response to anionic lipids. In contrast, the determinants of binding to Aβ peptides are uncertain, with different studies coming to differing conclusions concerning the effect of AD mutants on binding or uptake of Aβ fibrils (7, 29–31). Recently, it was suggested that different surfaces might be involved in binding different TREM2 ligands (29). Indeed, sTREM2 has a second unusual, variegated electrostatic surface (surface 2 in Fig. 1), with an extended band of positively charged residues flanked by acidic patches near the top and bottom of the structure, which might interact with different binding partners. Here, we use integrative structural modeling guided by chemical cross-linking mass spectrometry (XL-MS) to map the structure of the fibrillar Aβ–sTREM2 complex, and how it is affected by the R47H substitution. The resulting model suggests that the patch of hydrophobic and basic residues on sTREM2 that contains R47 does not directly interact with Aβ40 fibrils. Instead, sTREM2 is predicted to interact with Aβ primarily via surface 2, while projecting surface 1 away from the amyloid fibrils, with implications for both cellular uptake and signaling.
Fig. 1.
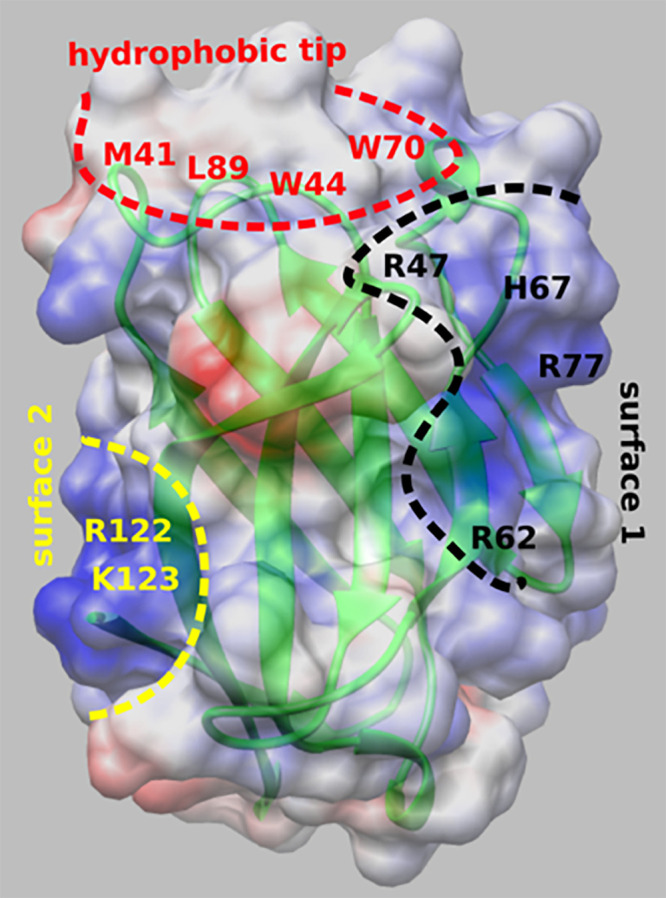
Crystal structure of sTREM2 (PDB ID code 5UD7) (8), showing electrostatic potential map of the ectodomain. The white, red, and blue colors in the map correspond to the neutral, acidic, and basic residues, respectively. The map was generated using CHIMERA v1.14 (69). The hydrophobic and aromatic protrusion in sTREM2 is highlighted with a red dashed curve (hydrophobic tip). The flat surface of basic residues adjacent to the hydrophobic tip is shown with black dashed curve (surface 1). Another patch of basic residues, opposite to surface 1, is highlighted with a yellow dashed curve (surface 2). Key residues in these three regions are indicated.
Results
Interactions of sTREM2 with Different Aβ Species.
We first investigated the interactions of sTREM2 with different Aβ species. Previous studies have come to inconsistent conclusions concerning the binding affinity of sTREM2 constructs for monomeric Aβ (7, 29–31). We therefore decided to study the interaction between monomeric Aβ and sTREM2 by NMR spectroscopy, a highly sensitive and reliable method to map molecular interactions. The 15N HSQC spectrum of 15N Aβ42 displayed well-dispersed peaks (Fig. 2A), comparable to the previously reported spectrum of monomeric Aβ42 (35). The addition of one equivalent of sTREM2 did not cause any chemical shift perturbation or changes of peak volumes. Thus, our data indicate that sTREM2 does not bind monomeric Aβ42, even at a concentration of 100 μM. We observed similar results for monomeric Aβ40, as sTREM2 did not bind to monomeric Aβ40 at a concentration of 100 μM (Fig. 2B).
Fig. 2.

(A) 15N HSQC spectra of 100 μM 15N mAβ42 alone (blue) and with 100 μM sTREM2 (red). (B) 15N HSQC spectra of 100 μM 15N mAβ40 alone (blue) and with 100 μM sTREM2 (red). (C) A cosedimentation assay to quantify sTREM2/fibrillar Aβ42 binding by incubating sTREM2 (10 nM) with various concentrations of fibrillar Aβ42. Quantification of fibril-bound sTREM2 using Western blot probed by anti–His-HRP conjugate antibody. (D) A binding curve obtained by fitting the data to a saturation binding model. The apparent binding constant is 2.6 ± 0.3 μM. (E) A cosedimentation assay to quantify sTREM2 and fibrillar Aβ40 binding by incubating 10 nM sTREM2 with various concentrations of fibrillar Aβ40. The Western blot was stained using anti–His-HRP conjugate antibody. (F) A binding curve obtained by fitting the data to a saturation binding model. The apparent binding constant for sTREM2 to Aβ40 is 2.3 ± 0.4 μM.
Next, we examined the binding of sTREM2 to fibrillar Aβ40 and Aβ42, using a sedimentation assay similar to that previously used for probing interactions of sTREM2 fused with the Fc domain of human IgG4 (30), albeit without reporting the binding affinity. We therefore performed a binding titration using a fixed concentration of sTREM2 (10 nM) with increasing concentrations of fibrillar Aβ40 and Aβ42, quantifying the amount of fibril-bound sTREM2 by Western blot (Fig. 2 C–F). The resulting binding curve is well described by a classic Langmuir isotherm with an apparent dissociation constant (Kapp) of 2.6 ± 0.3 µM for Aβ42 fibrils and 2.3 ± 0.4 µM for Aβ40 fibrils (Fig. 2 C–F).
R47H Has No Significant Effect on sTREM2 Binding to Fibrillar Aβ40 and Aβ42.
Using the cosedimentation assay, we also showed that the binding constants of sTREM2 WT and R47H mutant to fibrillar Aβ42 and fibrillar Aβ40 are the same within experimental error. Thus, this substitution did not cause a measurable change in the binding affinity to fibrillar Aβ42 and fibrillar Aβ40 (Fig. 3 C, D, G, and H). Interestingly, the R47H mutation is known to impair the binding of sTREM2 to other ligands, such as phospholipids (11). The lack of an effect of this mutation on the binding to fibrils indicates that sTREM2 has alternative binding surfaces for different ligands.
Fig. 3.

(A) The aggregation traces (dotted line) of 4.9 µM monomeric Aβ42 monitored by ThT fluorescence in the absence and presence of sTREM2 R47H. (B) The half-time (t1/2) derived from Aβ42 aggregation traces with sTREM2 WT or R47H. R47H displayed reduced inhibition. (C) Binding of fibrillar Aβ42 to sTREM2 R47H. (D) Binding curves of fibrillar Aβ42 to sTREM2 WT and R47H. Both WT and R47H showed similar binding affinity. (E) The aggregation traces (dotted line) of 5 µM monomeric Aβ40 monitored by ThT fluorescence in the absence and presence of sTREM2 R47H. (F) The half-time (t1/2) derived from Aβ40 aggregation traces with sTREM2 WT or R47H. R47H displayed reduced inhibition. (G) Binding of fibrillar Aβ40 to sTREM2 R47H. (H) Binding curves of fibrillar Aβ40 to sTREM2 WT and R47H. Both WT and R47H showed similar binding affinity. Replicates of aggregation kinetics for determination of t1/2 for sTREM2 WT and R47H are listed in SI Appendix, Table S1.
sTREM2 Inhibits the Fibrillization of Aβ40 and Aβ42 by Targeting the Secondary Nucleation Pathway of Fibril Formation.
TREM2-Fc dimeric constructs have been reported to interact with oligomeric and fibrillar Aβ, resembling several extensively studied molecular chaperones (36–41). We therefore asked whether monomeric sTREM2 can modulate the fibrillization of Aβ40 and Aβ42. For these studies, we used gel-filtered monomeric Aβ40 and Aβ42 (SI Appendix, Fig. S4), which were produced under carefully controlled conditions chosen to provide highly precise and reproducible kinetic measurements (42–45). We evaluated the fibrillization kinetics of Aβ42 (4.9 μM) and Aβ40 (5 μM) by monitoring the fluorescence of thioflavin T (ThT) under nonshaking conditions (43), in the absence and presence of different concentrations of sTREM2. Without sTREM2, Aβ42 aggregated rapidly with t1/2 of less than 1 h, consistent with a previous report (Fig. 4 A and B) (9, 43). However, the addition of a substoichiometric amount of sTREM2 attenuated the fibrillization of Aβ42 significantly (Fig. 4 A and B), as observed previously with a fluorescently tagged version of the peptide (29). A significant reduction was observed with concentrations of sTREM2 as low as 70 nM and a mole ratio of 0.015 ([sTREM2]/[Aβ42]). This reduction contrasts with observations for other chaperones, which typically require higher molar ratios for a large effect on the time course of fibrillization (42). Thus, sTREM2 is capable of inhibiting fibrillization at concentrations significantly below the Kdiss for binding to fibrils.
Fig. 4.

(A) The aggregation traces (dotted line) of 4.9 µM monomeric Aβ42 monitored by ThT fluorescence in the absence and presence of sTREM2. The solid lines are fits obtained using the secondary nucleation dominated, unseeded model with both knk+ and k2k+ as free fitting parameters. Fits with only knk+ (C) or k2k+ (D) as the free-fitting parameter. (B) Longer half-time (t1/2) with increasing amount of sTREM2. Relative rate constants knk+ (E) and k2k+ (F) from A. An asterisk (*) denotes rate constants from Aβ42 aggregation in the absence of sTREM2. (G) The aggregation traces (dotted line) of 5 µM monomeric Aβ40 monitored by ThT fluorescence in the absence and presence of sTREM2. The solid lines are fits using the multistep secondary nucleation dominated, unseeded model with both knk+ and k2k+ as free-fitting parameters. (H) Longer half-time (t1/2) with increasing amount of sTREM2. Replicates for aggregation and fitting kinetics with SD for sTREM2 WT are listed in SI Appendix, Table S1.
We observed similar effects for monomeric Aβ40, where the gel-purified monomeric peptide aggregated in the absence of sTREM2, as previously reported (44, 45). Addition of substoichiometric amounts of sTREM2 significantly attenuated the fibrillization of Aβ40 (Fig. 4 G and H), even at a concentration of 80 nM, as observed for Aβ42. This finding suggests that sTREM2 is a potent inhibitor of an intermediate in the amyloid fibrillization process.
To gain further molecular insight into the mechanism of inhibition, we performed a kinetic analysis using a well-established model of Aβ42, the more toxic species among the two forms of Aβ (43). In this model, the aggregation of Aβ42 can be described by two pathways. The primary pathway involves the nucleation of monomers into aggregation-competent oligomers with the rate constant of primary nucleation (kn) and elongation (k+). The secondary pathway involves the fibrillar surface of fibrillar Aβ42 that further catalyzes the formation of oligomers with k2 as the rate constant of secondary nucleation. We then used the Amylofit (44) server to analyze the data and fit kinetic parameters for the combined rate constants associated with both the primary pathway (knk+) and secondary pathway (k2k+). When both knk+ and k2k+ were set as free-fitting parameters, the fitted curve described the data well (Fig. 4A). sTREM2 affected mainly k2k+ (Fig. 4 E and F), which describes the secondary pathway of aggregation. However, the effect on the primary pathway is much less significant. To further support this result, we performed additional fitting by using k2k+ as the only free parameter, while constraining knk+ as the global fitting parameter. Changing k2k+ alone can explain the aggregation behavior (Fig. 4D). However, using knk+ of the primary pathway as the only free-fitting parameter resulted in a poor fit (Fig. 4C). Together with previous studies (37, 38, 46), this kinetic analysis supports an inhibition mechanism in which sTREM2 interacts with oligomeric intermediates to block the secondary pathway of aggregation.
AD-Associated R47H Substitution Has a Small but Significant Effect on Its Ability to Inhibit Fibril Formation.
The R47H mutant of TREM2, which is known to cause a threefold higher risk for AD (13, 16), has been suggested to have reduced binding to TREM2 ligands and incomplete TREM2 activation by its ligands (7, 8, 11, 12, 27, 47). However, the estimates of the extent to which the R47H substitution diminishes the interaction of sTREM2 constructs with oligomeric Aβ42 have varied across different studies and techniques (7, 29–31). We therefore examined the effect of sTREM2 R47H on the fibrillization kinetics of Aβ40 and Aβ42. The addition of sTREM2 R47H delayed the fibrillization of Aβ42, but the extent of inhibition is slightly weaker than WT sTREM2 (Fig. 3 A and B). Specifically, R47H leads to about a 25% reduction in t1/2 relative to WT over a wide range of protein concentrations. While this effect is relatively modest, it is highly reproducible (SI Appendix, Fig. S5 and Tables S1 and S2). Similarly, the R47H mutation showed a small decrease in t1/2 relative to WT for inhibition of Aβ40 fibrillization (Fig. 3 E and F), as observed for Aβ42. Thus, the R47H substitution has only a subtle effect on the inhibition of fibrillization of Aβ40 by sTREM2 WT.
Effects of sTREM2 on Aβ Uptake.
We further examined the uptake of sTREM2–Aβ mixtures using human neuroglioma cells (H4 cell line) and microglial cells (HMC3 cell line). First, we established a cytotoxic dose–response curve using the MTT assay at various concentrations of sTREM2 and Aβ40 fibrils or Aβ42 oligomers on H4 cells in the presence and absence of WT and mutant sTREM2 (SI Appendix, Fig. S5 A–D). In this short-term in vitro assay, we found no significant effect of either sTREM2 or the mutant R47H on the toxicity of either Aβ species.
We next determined the cellular uptake of sTREM2 WT versus R47H in the presence of Aβ40 fibrils. H4 or HMC3 cells were treated with fibrillar Aβ40 (2.5 μM) and increasing concentrations of sTREM2 WT or R47H, and the amount associated with the cell fraction was determined using ELISA for sTREM2 and homogeneous time-resolved fluorescence (HTRF) for Aβ40 (Fig. 5). We observed a dose-dependent association of sTREM2, which was decreased approximately fourfold for the R47H mutant (Fig. 5 A and C). We confirmed the TREM2 ELISA kit equally measured recombinant sTREM2 WT and R47H in cell-free conditions (SI Appendix, Fig. S5G). These findings show that the R47H substitution greatly decreases the uptake of sTREM2. Similar results were also observed for sTREM2 uptake when mixed with Aβ42 oligomers (Fig. 5 C and G).
Fig. 5.

Aβ fibrils or oligomers (at 2.5 µM) were incubated with various concentrations of sTREM2 WT or R47H (10, 5, 2.5, and 1.25 µM) before adding to H4 cells represented as Aβ + sTREM2. The numerical values denote the concentration of sTREM2 either for WT or R47H. Control samples for sTREM2 (without the addition of Aβ) are represented as WT C and R47 C, added at a constant concentration of 2.5 µM. (A) ELISA to determine the amount of sTREM2 in the cell lysates of H4 cells treated with Aβ40 fibrils + sTREM2. (B) HTRF assay to determine the amount of Aβ40 in cell lysates of H4 cells treated Aβ40 fibrils + sTREM2. (C) ELISA to determine the amount of sTREM2 in the cell lysates of H4 cells treated with Aβ42 oligomers + sTREM2. (D) HTRF assay to determine the amount of Aβ42 in cell lysates of H4 cells treated Aβ42 oligomers + sTREM2. (E–H are identical to A–D, but with HMC3 cells.) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
sTREM2 also enhanced the uptake of Aβ42 fibrils. As the concentration of sTREM2 increased from 1.25 to 10 µM, the extent of uptake of Aβ42 increased markedly in a dose-dependent manner, reaching an approximately threefold increase at 10 µM sTREM2 (Fig. 5 D and H). The R47H mutant had a decreased ability to stimulate the uptake of Aβ42 fibrils. Over the concentration range examined, approximately twofold higher concentrations of R47H than WT were required to achieve a similar degree of stimulation. Thus, sTREM2 stimulates the uptake of Aβ fibrils while the R47H mutation decreasing the extent of this stimulation.
To determine if sTREM2 WT or R47H complexed with Aβ induces distinct functional responses in microglia (HMC3 cells), we examined the differential expression profile of 1,321 genes related to neuroinflammation and myeloid cell innate immunity using the Nanostring platform. Agglomerative clustering of gene expression across all the concentrations of sTREM2 WT and sTREM2 R47H identified a major gene cluster for cells treated with sTREM2 WT only and cells treated with sTREM2 WT mixed with Aβ40 fibrils, which segregated from the gene cluster for sTREM2 R47H, sTREM2 R47H mixed with Aβ40 fibrils, and Aβ40 fibril only treatment (Fig. 6 A and B). Notably, we observed a sTREM2 WT dose-dependent up-regulation of several of genes involved in microglial activation such as C3, CXCL1, RELB (a component of the NF-κB complex) (Fig. 6 C and D and SI Appendix, Figs. S8 and S9 and Table S3). In contrast, sTREM2 R47H induced little to no functional response and showed an expression profile similar to the Aβ fibril-only treatment. To further elucidate the functional pathways and microglial functions induced by the sTREM2–Aβ complex, we performed Ingenuity Pathway Analysis on the Nanostring dataset. Ingenuity Pathway Analysis identified the involvement of several canonical microglial activation pathways including chemokine, cytokine, interleukin (IL)-1, NF-κB, and p38 MAPK signaling in HMC3 cells upon induction with sTREM2 WT complexed with Aβ (SI Appendix, Table S4). Our data suggest that sTREM2 in complex with Aβ fibrils can mediate downstream signaling of genes for microglial activation, and the R47 site regulates this function.
Fig. 6.

Agglomerative clustering showing differential gene expression for (A) myeloid and (B) neuroinflammation pathways related genes using nSolver software. The data were analyzed in triplicate for each concentration of sTREM2 WT and sTREM2 R47H (10 µM, 5 µM, 2.5 µM, 1.25 µM) added with Aβ (2.5 µM) to HMC3 cells and for Aβ (2.5 µM) added alone. Volcano plot showing expression of genes in myeloid and neuroinflammation pathways for (C) 10 µM WT + 2.5 µM Aβ and (D) 10 µM R47H + 2.5 µM Aβ compared to cells treated only with 2.5 µM Aβ. All the genes with a significance threshold (gray line) of P < 0.05 and log2 (fold-change) values <−1 (green line) or >+1 (red line) are displayed on the plots. For exact values of log2 fold-change, refer to SI Appendix, Table S3.
Integrative Modeling of the Fibrillar Aβ40–sTREM2 Complex Based on Chemical Cross-Links Identified a Unique Binding Interface in sTREM2.
Our cell-based assay indicated that the R47H mutant influences its uptake but has no significant effect on the binding to Aβ fibrils (Figs. 3 D and H and 5 C, D, G, and H). In an attempt to rationalize these observations, we mapped the structure of the fibrillar Aβ–sTREM2 complex, by using integrative structure modeling (48, 49) based on XL-MS data (50). Aβ40 fibrils were formed using a procedure that produces relatively homogenous fibrils (32, 51) that are structurally similar to a published threefold symmetric solid NMR structure of Aβ40 (33). To avoid the complexity of posttranslational modifications of sTREM2 expressed in the mammalian system, we used sTREM2 obtained from bacterial expression system to form a complex with Aβ40 fibrils in vitro. This complex was chemically cross-linked using two cross-linkers, BS3 and NHSF. BS3 targets lysine sidechains and protein N termini, while NHSF links lysine sidechains or protein N termini to sidechains of several residues, including tyrosine, histidine, lysine, serine, and threonine (52). Western blot analysis showed successful cross-linking for sTREM2 and Aβ40 by both BS3 and NHSF (SI Appendix, Fig. S10). The number of cross-links identified by MS analysis was relatively modest, due to the paucity of Lys residue in both Aβ and sTREM2. Nevertheless, 15 intermolecular cross-links were observed (Fig. 7A).
Fig. 7.

Summary of the interprotein cross-links information used in integrative structure modeling. (A) A plot to represent the cross-links between Aβ40 fibril and sTREM2. Red and golden lines represent the BS3 and NHSF cross-links between Aβ40 fibril and sTREM2, respectively. (B) The representative structures and an indication of the cluster size are shown as ribbons and density maps, respectively, for the two largest of the five cluster solutions. Cluster I (Left) and cluster II (Right) represent 35% and 16% of the entire ensemble of models, respectively. R47 of TREM2 is denoted by red spheres. (C) Average contact maps between fibrillar Aβ40 and sTREM2. The x and y axes denote the residue numbers of sTREM2, and fifteen chains (C1, •••, C7, C, C8, •••, C14) of one of the protofilaments of Aβ40 fibril, respectively. Each bin of the map indicates the relative frequency of a contact between two residues in the cluster models, from 0 for red to 1 for blue; a contact between two residues is defined to occur when the distance between their Cα atoms is less than 12.0 Å.
Next, the structural model of the fibrillar Aβ40–sTREM2 complex was computed by integrative modeling (48, 53). The model was obtained by sampling configurations of the crystal structure of sTREM2 (PDB ID code 5UD7) (8) and the ssNMR structure of Aβ40 fibrils (PDB ID code 2LMP) (33) in order to find those configurations that are consistent with the chemical cross-links identified by XL-MS, symmetry considerations, and the excluded volume principle (SI Appendix). The modeling addressed the challenge of ambiguous assignment of cross-links to individual residues, arising from the arrangement of multiple copies of the Aβ40 monomer in the complex (48, 53). Five clusters of models were found that satisfy at least 80% of the cross-links. The structural variability within and among the clusters reflects both the actual heterogeneity of the samples used to obtain the data and the modeling uncertainty arising from limited input information and modeling assumptions; it is generally impossible to deconvolute them from each other and we do not attempt to do so here.
The structural variability in the most populated model cluster (the average Cα root-mean-square deviation between each model in a cluster and the cluster centroid) is ∼5.0 Å. This resolution is sufficient to define the orientation of sTREM2 relative to the fibril surface (Fig. 7). The two most populated clusters, accounting for most of the entire ensemble of models (Fig. 7B), differ by an approximate twofold rotation of the subunits about an axis orthogonal to the fibril axis. Both share the fibril interaction via surface 2 (Fig. 7A). Moreover, none of the other models showed an interaction between surface 1 and the fibrils. Thus, modeling suggests that the R47 site projects away from the fibril (Fig. 7 B and C), with surface 2 rather than surface 1 (that contains R47) forming primary interactions with the fibril. Thus, this model is fully consistent with the finding that the R47H substitution has little to no effect on the binding to Aβ fibrils or its fibrillization kinetics (Fig. 3).
Discussion
Aggregation of peptides and proteins into amyloid plaques is a key feature in AD. Aβ40 and Aβ42 found in plaques display markedly different aggregation behavior. Aβ40 is more abundant form, while Aβ42 is more aggregation prone (43). The major source of aggregates in the case of Aβ40 and Aβ42 is a fibril-catalyzed nucleation process (44, 45), which proceeds from monomeric Aβ via intermediate oligomeric forms that represent the primary nucleating species. A secondary pathway involves nucleation on the surface of preformed fibrils. This pathway is believed to be particularly significant, because it can generate toxic oligomers as intermediates in the secondary nucleation process (43, 54).
The kinetics of aggregation is complex as Aβ can act simultaneously as a reactant, intermediate, and a product. Moreover, the kinetics of Aβ aggregation is strongly affected by interactions with chaperone-like proteins that inhibit fibrillization. The Knowles group has previously described kinetic models for suppression of aggregation of amyloidogenic proteins in the presence of various chaperones, which inhibit a specific intermediate (37, 42). One such chaperone, Brichos, interacts with Aβ and selectively inhibits the secondary nucleation pathway. Brichos also can suppress the toxicity associated with aggregation by interacting with amyloid fibrils selectively to break the critical catalytic cycle through which toxic oligomers are generated. Using similar kinetic models, our results support inhibition of secondary nucleation pathways in aggregation of Aβ. Since the secondary nucleation is at the surface of the fibrils, and does not depend on the monomeric species, the inhibition we observed was at substochiometric ratios of sTREM2 to Aβ.
Our kinetic and binding studies, together with a low-resolution fibrillar Aβ–sTREM2 complex models, furthermore show that a patch of basic residues (containing residues R46, R47, K48, and R62) on sTREM2, which is altered in AD mutants, does not directly interact with fibrillar Aβ40. These results led us hypothesize that sTREM2 recognizes ligands using at least two distinct surfaces of its structure. Surface 1 is crucial for binding anionic ligands, such as cell-surface glycans or negatively charged membranes, as confirmed in crystallographic (8) and very recent molecular dynamics studies (34). These studies indicate that the AD-risk variant R47H induces structural instability in the complementarity determining region (CDR loop), which forms the hydrophobic patch near the top of surface 1 as viewed in Fig. 1. This structural instability is consistent with our results. In the WT TREM2 structure, the side chain of the residue R47H projects into the N-terminal region of the CDR loop, participating in extensive hydrogen bond networks with residues S65, T66, H67, and N68. However, this H-bonding is absent when the residue is mutated to histidine (R47H). Instead, the imidazole moiety of histidine (in R47H mutant) forms H-bond with T66 and π–π stacking interactions with the H67 completely changing the secondary architecture of the protein.
In contrast, we found that sTREM2 serves to enhance cellular uptake of fibrils in HMC3 cells, which have endogenous membrane tethered TREM2. This finding was somewhat surprising, as we expected that sTREM2 might compete with binding to membrane-bound TREM2, decreasing rather than increasing uptake. It is instead likely that TREM2’s extracellular domain acts similarly, irrespective of whether it is present in solution or tethered to the cell surface. In both cases, it would appear to act as a molecular glue, using one surface to bind to Aβ amyloid and the other to bind to acidic ligands on the cell surface (Fig. 8). This finding is also consistent with our observation of enhanced uptake in H4 cells, which lack TREM2. In support of this hypothesis, Song et al. (55) report that mice expressing the human TREM2 R47H variant do not exhibit sTREM2 binding to neuronal surfaces or amyloid plaques, as observed in mice expressing the common variant. Additionally, depletion of positive charge because of histidine mutation could potentially lead to effects on binding and uptake.
Fig. 8.

Schematic representation showing binding surfaces of TREM2 in soluble and membrane-bound form. Aβ fibrils primarily bind along surface 2, which is away and distinct from surface 1 that houses the R47H mutation and plays a role in cellular binding and uptake. The membrane-bound form of TREM2 is attached to the membrane via a transmembrane helix and a flexible extracellular sequence. This membrane-bound form mediates signal transduction through the adapter protein DAP12 when TREM2 binds to polyvalent ligands that induce clustering of the DAP12 subunits.
The possibility of two distinct binding surfaces in TREM2’s extracellular domain may also have implications in understanding the role of membrane-tethered TREM2 in mediating the targeting and encapsulation of amyloid plaques by microglial processes, known as the microglia barrier (56, 57). The R47H mutation in humans and humanized knockin mouse models disrupts the microglial barrier formation around Aβ deposits leading to a more toxic plaque phenotype (19, 55, 58). Considering our findings here, the loss of microglia–amyloid interactions in carriers of mutant TREM2 might be due to diminished binding of membrane phospholipids from adjacent injured neurons or other disease-relevant biomolecules, such as ApoE, known to decorate the plaque surface (59–61), rather than Aβ itself. Consistent with this idea is the finding that mice deficient in ApoE exhibit a similar plaque phenotype as TREM2 deficiency (62), suggesting that ApoE is an important plaque component for attracting TREM2+ microglia processes.
sTREM2, produced by cleavage of the ectodomain of TREM2 or by alternative splicing, has ligand binding capacity, decreases Aβ plaques in AD brain, and serves as a biomarker that regulates the immune response in AD (56). Previous reports showed that the R47H variant causes loss-of-function in membrane-bound TREM2 and microglial activation impairment, and similar effect could also occur for soluble TREM2 (7). While R47H mutation did not change the sTREM2–Aβ binding capacity in vitro, it affected its uptake, thus reducing the intracellular signaling for microglial activation in response to fibrillar Aβ in microglial cells. Our results highlight the role of sTREM2 in AD, not only surrounding insoluble Aβ plaques but also favoring the clearance of diffusible Aβ by microglia and inducing the expression of genes required to trigger neuroinflammatory responses and microglial survival (63).
In conclusion, our results have helped define functional roles of sTREM2, which can bind with moderate affinity to fibrils and acts as a potent inhibitor of fibril formation. Moreover, the common variant of sTREM2, but not R47H, is readily taken up by both neuroglioma derived and microglial cell lines, and it can enhance uptake of fibrillar Aβ. These roles overlap with that of membrane bound TREM2, which mediates signaling when it binds multivalent ligands, that can either be Aβ aggregates, acidic phospholipids, ApoE, or a mixture of these ligands that are presented in polyvalent forms. These findings also provide insight into a possible chaperone-like role of sTREM2, which might have important implications for the initiation, processing, and propagation of plaques during the progression of AD.
Materials and Methods
Detailed experimental methods can be found in SI Appendix. The synthetic gene for sTREM2 WT and sTREM2 R47H mutant protein was obtained from Twist Biosciences and was cloned in vector pTwist Puro. The gene encoded the human TREM2 signal peptide and extracellular domain (residues 19 to 157) until the ADAM cleavage site (sequence in the SI Appendix). The glycosylation sites N20 and N79 were left unchanged, and a 6-His tag was added at the C terminus of the gene sequence to aid in protein purification, which is detailed in SI Appendix.
NMR Spectrometry.
15N HSQC experiments were conducted at University of California, San Francisco NMR facility on a Bruker Avance I 800 MHz spectrometer equipped with a 5-mm triple-resonance z-gradient TXI-Cryoprobe. Aβ samples were prepared following a previously reported method (35). Data were collected at 10 °C for 100 µM 15N Aβ with or without 100 µM sTREM2 in 20 mM sodium phosphate buffer (pH 7.0). NMR data were processed with NMRPipe (64) and visualized with Sparky (65).
Aggregation Kinetics of Aβ40 and Aβ42.
The aggregation of Aβ40 and Aβ42 was monitored following a previously published method (43). Briefly, Aβ40 and Aβ42 were first prepared in monomeric form using gel-filtration chromatography. The final assay solution contained 4.9 μM Aβ42 or 5 μM Aβ40, various concentrations of sTREM2 WT/R47H, and 6 μM ThT in a total volume of 100 μL per well in a 96-well plate (Corning 3881). Fluorescence signal (λex = 444, λem = 485) was monitored on a Spectramax M5 plate reader (Molecular Devices) using bottom read at 37 °C without shaking. The kinetic traces were first fitted using a sigmoidal function to derive t1/2. The global fitting of kinetic traces was done on Amylofit (https://amylofit.com/amylofitmain/login) using a “Secondary Nucleation Dominated, Unseeded” model (44).
Integrative Structure Modeling of Fibrillar Aβ40–sTREM2 Complex.
The structure of the fibrillar Aβ40–sTREM2 complex was generated by using an integrative approach. The structural models were determined by combining the structure of sTREM2 (PDB ID code 5UD7) (8), Aβ40 fibers (PDB ID code 2LMP) (33), and our XL-MS data. Four stages of modeling included: 1) gathering data, 2) representing system and translating input information into spatial restraints, 3) conformational sampling to generate an ensemble of models that satisfies the restraints, and 4) analyzing and validating the ensemble structures and data (48, 66, 67) (SI Appendix, Fig. S11). The integrative structure modeling protocol (stages 2, 3, and 4) was scripted using the Python Modeling Interface package (66), which is a library for modeling macromolecular assemblies based on our open-source Integrative Modeling Platform package; files containing input data, scripts, and output results are available at https://github.com/integrativemodeling/sTREM2-fAbeta_modeling. Given that many studies are conducted using mouse models and mouse TREM2, we also examined the surfaces of mouse TREM2, using AlphaFold (68) to create a molecular model (SI Appendix, Fig. S14 and Table S5). The surface is quite similar to that of WT human TREM2, so the modeling results should hold for the mouse as well as human species.
Supplementary Material
Acknowledgments
We thank John Nicoludis (University of California, San Francisco [UCSF]), Robert Newberry (UCSF), and Miranda Sullivan (UCSF) for their technical guidance; Shigenari Hayashi (UCSF) for running the mass spectrometer; and Stanley B. Prusiner, Director of the UCSF Institute for Neurodegenerative Diseases, for stimulating discussions, access to equipment, and technical resources critical for the completion of this study. This work was supported by the NIH Grants P30AG062422 (sub-award to C.C.), RF1AG061874 (to C.C. and W.F.D.), P01AG002132 (to A.S., C.C., and W.F.D.), R01GM083960 (to A.S.), and P41GM109824 (to A.S.).
Footnotes
Reviewers: A.D.S., Imperial College London; and T.H., Sanford Burnham Prebys Medical Discovery Institute.
The authors declare no competing interest.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2114486119/-/DCSupplemental.
Data Availability
All study data, and scripts are included in the main text and SI Appendix. Files containing input data, scripts, and output results of integrative modeling are available at GitHub, https://github.com/integrativemodeling/sTREM2-fAbeta_modeling. The mass spec raw data have been deposited to the ProteomeXchange Consortium via the iProX partner repository under Accession Number PXD030722 and Project ID IPX0003927000.
References
- 1.DeTure M. A., Dickson D. W., The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 14, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson P. T., Braak H., Markesbery W. R., Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 68, 1–14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe D. J., Hardy J., The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goedert M., Ghetti B., Spillantini M. G., Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006254 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoyagi A., et al. , Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med. 11, eaat8462 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulland T. K., Colonna M., TREM2–A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y., et al. , TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023–1031.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sudom A., et al. , Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J. Biol. Chem. 293, 12634–12646 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vilalta A., et al. , Wild-type sTREM2 blocks Aβ aggregation and neurotoxicity, but the Alzheimer’s R47H mutant increases Aβ aggregation. J. Biol. Chem. 296, 100631 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng Q., et al. , TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal. 3, ra38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., et al. , TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeh F. L., Wang Y., Tom I., Gonzalez L. C., Sheng M., TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Guerreiro R., et al. ; Alzheimer Genetic Analysis Group, TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin S. C., et al. , Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum. Mol. Genet. 23, 5838–5846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin S. C., et al. , TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol. Neurodegener. 10, 19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonsson T., et al. , Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meilandt W. J., et al. , Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42: Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 40, 1956–1974 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., et al. , TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 213, 667–675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan P., et al. , TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90, 724–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parhizkar S., et al. , Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 22, 191–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong L., et al. , Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 10, 1365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suárez-Calvet M., et al. ; Dominantly Inherited Alzheimer Network, Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 8, 369ra178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suárez-Calvet M., et al. ; Alzheimer’s Disease Neuroimaging Initiative, Early increase of CSF sTREM2 in Alzheimer’s disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol. Neurodegener. 14, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewers M., et al. ; Alzheimer’s Disease Neuroimaging Initiative (ADNI), Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. EMBO Mol. Med. 12, e12308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewers M., et al. ; Alzheimer’s Disease Neuroimaging Initiative, Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer’s disease. Sci. Transl. Med. 11, eaav6221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlepckow K., et al. , An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol. Med. 9, 1356–1365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atagi Y., et al. , Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey C. C., DeVaux L. B., Farzan M., The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J. Biol. Chem. 290, 26033–26042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kober D. L., et al. , Functional insights from biophysical study of TREM2 interactions with apoE and Aβ1‐42. Alzheimers Dement. 17, 475–488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lessard C. B., et al. , High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol. Med. 10, e9027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong L., et al. , Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 13, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elkins M. R., et al. , Structural polymorphism of Alzheimer’s β-amyloid fibrils as controlled by an E22 switch: A solid-state NMR study. J. Am. Chem. Soc. 138, 9840–9852 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paravastu A. K., Leapman R. D., Yau W.-M., Tycko R., Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dean H. B., Roberson E. D., Song Y., Neurodegenerative disease—Associated variants in TREM2 destabilize the apical ligand-binding region of the immunoglobulin domain. Front. Neurol. 10, 1252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roche J., Shen Y., Lee J. H., Ying J., Bax A., Monomeric Aβ1–40 and Aβ1–42 peptides in solution adopt very similar Ramachandran map distributions that closely resemble random coil. Biochemistry 55, 762–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beeg M., et al. , Clusterin binds to Aβ1–42 oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J. Biol. Chem. 291, 6958–6966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen S. I. A., et al. , A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cristóvão J. S., et al. , The neuronal S100B protein is a calcium-tuned suppressor of amyloid-β aggregation. Sci. Adv. 4, eaaq1702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mainz A., et al. , The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 22, 898–905 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Mansson C., et al. , Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31066–31076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willander H., et al. , BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J. Biol. Chem. 287, 31608–31617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arosio P., et al. , Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen S. I., et al. , Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meisl G., et al. , Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Meisl G., et al. , Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. U.S.A. 111, 9384–9389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheidt T., et al. , Secondary nucleation and elongation occur at different sites on Alzheimer’s amyloid-β aggregates. Sci. Adv. 5, eaau3112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kober D. L., et al. , Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. eLife 5, e20391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rout M. P., Sali A., Principles for integrative structural biology studies. Cell 177, 1384–1403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sali A., From integrative structural biology to cell biology. J. Biol. Chem. 296, 100743 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young M. M., et al. , High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 97, 5802–5806 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang T., Jo H., DeGrado W. F., Hong M., Water distribution, dynamics, and interactions with Alzheimer’s β-amyloid fibrils investigated by solid-state NMR. J. Am. Chem. Soc. 139, 6242–6252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang B., et al. , Proximity-enhanced SuFEx chemical cross-linker for specific and multitargeting cross-linking mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 115, 11162–11167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viswanath S., Chemmama I. E., Cimermancic P., Sali A., Assessing exhaustiveness of stochastic sampling for integrative modeling of macromolecular structures. Biophys. J. 113, 2344–2353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee J., Culyba E. K., Powers E. T., Kelly J. W., Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song W. M., et al. , Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 215, 745–760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Condello C., Yuan P., Grutzendler J., Microglia-mediated neuroprotection, TREM2, and Alzheimer’s disease: Evidence from optical imaging. Biol. Psychiatry 83, 377–387 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Condello C., Yuan P., Schain A., Grutzendler J., Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 6, 6176 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leyns C. E. G., et al. , TREM2 function impedes tau seeding in neuritic plaques. Nat. Neurosci. 22, 1217–1222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Y., et al. , Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 22, 586–594 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Namba Y., Tomonaga M., Kawasaki H., Otomo E., Ikeda K., Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 541, 163–166 (1991). [DOI] [PubMed] [Google Scholar]
- 61.Navarro A., Del Valle E., Astudillo A., González del Rey C., Tolivia J., Immunohistochemical study of distribution of apolipoproteins E and D in human cerebral β amyloid deposits. Exp. Neurol. 184, 697–704 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Ulrich J. D., et al. , ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 215, 1047–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong L., et al. , DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.F. Delaglio et al., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 65.Lee W., Tonelli M., Markley J. L., NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Russel D., et al. , Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 10, e1001244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saltzberg D. J., et al. , Using integrative modeling platform to compute, validate, and archive a model of a protein complex structure. Protein Sci. 30, 250–261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jumper J.et al., . Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pettersen E. F., et al. , UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data, and scripts are included in the main text and SI Appendix. Files containing input data, scripts, and output results of integrative modeling are available at GitHub, https://github.com/integrativemodeling/sTREM2-fAbeta_modeling. The mass spec raw data have been deposited to the ProteomeXchange Consortium via the iProX partner repository under Accession Number PXD030722 and Project ID IPX0003927000.