

Abstract
Purpose:
Activation of Bruton’s tyrosine kinase (BTK) and phosphatidylinositol-3-kinase (PI3K) represent parallel, synergistic pathways in lymphoma pathogenesis. As predominant PI3Kδ inhibition is a possible mechanism of tumor escape, we proposed a clinical trial of dual BTK and pan-PI3K inhibition.
Patients and Methods:
We conducted a single-center phase I/Ib trial combining a BTK inhibitor (ibrutinib) and a pan-PI3K inhibitor (buparlisib) in 37 patients with relapsed/refractory (R/R) B-cell lymphoma. Buparlisib and ibrutinib were administered orally, once daily in 28 day cycles until progression or unacceptable toxicity. The clinical trial is registered with clinicaltrials.gov, NCT02756247.
Results:
Patients with mantle cell lymphoma (MCL) receiving the combination had a 94% overall response rate (ORR) and 33-month median progression-free survival; ORR of 31% and 20% were observed in patients with diffuse large B-cell lymphoma and follicular lymphoma, respectively. The maximum tolerated dose was ibrutinib 560 mg plus buparlisib 100 mg and the recommended phase II dose was ibrutinib 560 mg plus buparlisib 80 mg. The most common grade 3 adverse events were rash/pruritis/dermatitis (19%), diarrhea (11%), hyperglycemia (11%), and hypertension (11%). All grade mood disturbances ranging from anxiety, depression, to agitation were observed in 22% of patients. Results from serial monitoring of cell-free DNA samples corresponded to radiographic resolution of disease and tracked the emergence of mutations known to promote BTK inhibitor resistance.
Conclusions:
BTK and pan-PI3K inhibition in mantle cell lymphoma demonstrates a promising efficacy signal. Addition of BCL2 inhibitors to a BTK and pan-PI3K combination remain suitable for further development in mantle cell lymphoma.
Keywords: buparlisib, ibrutinib, lymphoma, refractory, relapsed
INTRODUCTION
Non-Hodgkin lymphomas (NHL) depend on multiple signaling pathways for cell proliferation, growth, and survival, including the phosphatidylinositol-3-kinase (PI3K) and B cell receptor (BCR) signaling pathways (1–3). In clinical trials, both PI3K and Bruton’s tyrosine kinase (BTK) inhibitors have demonstrated impressive single-agent activity across multiple types of lymphoma (4–6). In vitro combinatorial drug screening demonstrated synergistic tumoricidal activity between PI3K inhibitors and the BTK inhibitor, ibrutinib (7–9). Additional in vitro data suggest that selective inhibition of the PI3Kδ isoform is insufficient for tumor control because of a PI3Kɑ escape mechanism (10). Prior studies show tolerability and synergistic overall response rates for the combination of a PI3Kδ inhibitor with a BTK inhibitor in chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) (11–13). Therefore, we hypothesize that a pan-PI3K inhibitor may be better poised for synergistic activity with ibrutinib (7).
Clinical responses to single-agent targeted therapy with BTK or PI3K inhibitors in MCL, follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) are commonly partial responses with limited durability. Overall response rates (ORR) for ibrutinib in R/R MCL, FL, and DLBCL are 68% (5), 37% (14), and 22–40% (6), respectively. Median progression-free survival (PFS) for ibrutinib in the third line setting is 13.9 months (5) and 14 months (14) in patients with relapsed/refractory MCL and FL, respectively. In the second-line setting, ibrutinib had median PFS of 25 months while rituximab and ibrutinib showed a median PFS of 43 months in MCL (15,16).
Buparlisib is a pan-PI3K inhibitor with cross-tumor efficacy and tolerability. In vitro, buparlisib can decrease the phosphorylation status of downstream Akt effectors in DLBCL (17). A phase II study in lymphoma demonstrated modest activity for buparlisib with 12% ORR in 26 patients with DLBCL, including 1 complete response (CR) and 2 partial responses (PR) (18). Buparlisib penetrates the blood-brain barrier and, relative to other PI3K inhibitors, was observed to have higher incidences of reversible neuropsychiatric effects such as anxiety and depression observed across cancer types (19,20). In studies of buparlisib in lymphoma and breast cancer, neuropsychiatric effects included anxiety (18–25%), depression (18–29%), and agitation (7%) (18,19). Mood disorders were readily manageable with dose adjustments and/or treatment with appropriate concomitant medication. Other associated toxicities included reversible hyperglycemia, nausea, fatigue, and transaminitis (18).
Several studies have previously combined PI3K and BTK inhibitors. Umbralisib, a PI3Kδ and CK1ε inhibitor, was combined with ibrutinib in CLL and MCL with a reasonable toxicity profile (11). Two phase Ib clinical trials studying tirabrutinib in combination with idelalisib, a PI3Kδ inhibitor, in relapsed CLL and NHL, respectively, supported the combination as safe (12,13). In the NHL study, no serious adverse events of diarrhea, colitis, or transaminitis were observed (13). Based on the potential PI3Kɑ escape mechanism, we chose to investigate a pan-PI3K inhibitor with concurrent alpha, delta, and gamma inhibition which had not been previously studied in combination with BTK inhibition (10).
We hypothesized that combining the BTK inhibitor ibrutinib with the pan-PI3K inhibitor buparlisib would potentially increase duration of response to therapy and prevent development of resistant subclones. We designed this phase I clinical trial using ibrutinib and buparlisib to treat relapsed or refractory MCL, FL, and DLBCL to evaluate the safety and efficacy of the combination and follow lymphoma tumor mutational profiling in paired cell free DNA (cfDNA) and tumor tissue.
METHODS
Study design and oversight
In this phase I/Ib clinical trial, we enrolled adult patients with relapsed or refractory NHL. This combination study of ibrutinib and buparlisib had a standard 3+3 dose-escalation design followed by a dose-expansion cohort at the presumed MTD. Ibrutinib and buparlisib were given daily by mouth on a 28-day cycle, with dose reductions permitted after cycle 1. Drugs were administered once daily continuously until disease progression, intolerance, or 3 years since start of therapy. Patients were enrolled sequentially onto the dosing schedules as follows: buparlisib 80 mg and ibrutinib 420 mg, buparlisib 80 mg and ibrutinib 560 mg, buparlisib 100 mg and ibrutinib 560 mg in cohorts of 6 evaluable patients. This clinical trial was approved by the Memorial Sloan Kettering Cancer Center institutional review board, registered on clinicaltrials.gov as NCT02756247, and conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization guideline for Good Clinical Practice. Written informed consent was provided by all patients before screening and enrollment.
Patients
Patients were eligible if they were ≥18 years with histologically confirmed a) DLBCL who received ≥ 1 prior regimen and received, declined, or were ineligible for autologous or allogeneic stem cell transplant; b) FL who received ≥ 2 prior regimens; or c) MCL who received ≥ 1 prior regimen. Additional criteria included an Eastern Cooperative Oncology Group (ECOG) score ≤ 2 and adequate hematologic, renal, and hepatic function. Patients were excluded if they were previously treated with ibrutinib (other BTK inhibitors were allowed) or PI3K inhibitors. Additional inclusion and exclusion criteria are described in supplemental text.
Study assessments
The primary objectives were to assess safety and determine the MTD or recommended phase II dose of buparlisib and ibrutinib in combination. In addition to DLT events, laboratory values, and safety parameters in cycle 1, we selected the MTD based on continued assessment for adverse events beyond the DLT period. Secondary endpoints were overall response rate, duration of response, EFS, and PFS, defined as time from treatment start to progression or death, or censored at last follow-up. The early efficacy signal in patients with MCL prompted a protocol amendment to include formal phase II statistics with a target enrollment of 17 patients with MCL to the RP2D. In the expansion of the MCL cohort, the primary endpoint was the 6-month CR rate of ibrutinib and buparlisib, and the secondary endpoints were disease control rate, ORR, PFS, and EFS. Additional study assessments are detailed in the supplemental text.
Disease monitoring was evaluated with FDG-PET/CT or MRI of the chest, abdomen, and pelvis with contrast at baseline and repeated every 2 cycles until cycle 7, then every 3 cycles until cycle 12, and then every 4 cycles thereafter. Tumor response was based on a modified Lugano Classification, with complete responses requiring both FDG-PET resolution and at least a partial response by CT. RECIL response assessments were evaluated as a non-protocol-specified evaluation.
Exploratory assessments
Exploratory assessments included evaluation of tumor and ctDNA for next generation sequencing. Pre-treatment tumor biopsy samples were obtained for next-generation sequencing. Blood plasma samples for cfDNA were obtained at baseline and serial time points throughout the study to coincide with radiographic restaging. Baseline, cycle 2, and end of therapy timepoints were selected for analysis.
Targeted Next-Generation Sequencing
Baseline tumor tissue samples were analyzed by next-generation sequencing assays developed at MSK: MSK-IMPACT and MSK-IMPACT-Heme (21,22). For cfDNA samples, sequencing libraries were generated using the HEMEPACT (version 4) assay, similar to the MSK-IMPACT assay (23) with a different but overlapping 500 gene set targeting hematologic malignancies. For each patient, matched normal samples were obtained, when possible, from buffy coats, whole blood, nail, and/or saliva. Sequencing data were aligned and variant calling was done using the MSK ROSLIN pipeline (24). Mutation identification was performed first using the matched normal samples as controls to identify alterations only detected in the tumor, and the analysis was repeated using unmatched controls to identify alterations that may be present in the white blood cells as circulating disease. Two methods were used to identify mutations in cfDNA: genotyping known tumor mutations (tumor-guided analysis) and identification of mutations without prior knowledge (tumor independent analysis). We reported mutations found at a VAF of >1% for mutations previously identified in the tumor of the same patient, and >5% VAF for those identified through tumor independent analysis.
18F-FDG PET/CT Imaging and Analysis
18F-FDG PET/CT (PET/CT) imaging from baseline and after 2 and 4 cycles of therapy were blinded and interpreted by 2 independent nuclear medicine physicians. In case of discordant interpretation, a consensus was reached. In the PET/CT analysis, all lesions were visually identified, and all measurable lesions were selected for the analysis.
Statistical Analysis
We performed a 3+3 dose-escalation study with 3–6 patients enrolled per dose level, followed by a dose-expansion cohort of 12 patients at the RP2D for DLBCL and FL, and 17 patients at RP2D for MCL. The small sample size precluded formal efficacy hypothesis testing; however, the statistical goal was for the combination to improve the ORR from 25% to 40% in FL, and 30% to 50% in DLBCL. For the MCL dose expansion, a Simon mini-max two-stage design was applied with the goal of increasing 6-month CR rate from 20% to 50%. The one-sided type I error rate was 0.068 and the power was 0.92. We planned a sample size of 17 patients for the phase II dose expansion. Drug support was prematurely withdrawn and the clinical trial was closed to accrual.
Analysis of adverse events included treatment-related and -unrelated adverse events. Adverse events were counted once per patient by NCI-CTCAE v4.03 preferred terms at the worst severity and strongest causality. Treatment-related adverse events were those reported as possibly, probably, or definitely related to treatment. Attribution to each drug was ascribed by treating physicians and reviewed.
Analyses of safety, objective response, and survival using Kaplan-Meier curves were performed using R version 4.0.0. The MSK institutional data monitoring committee oversaw the study.
RESULTS
Patient Characteristics
Between May 13, 2016 and December 4, 2017, 37 patients were enrolled and received therapy with buparlisib and ibrutinib (Fig. 1). Eighteen patients had MCL, 5 FL, and 14 DLBCL (Table 1). Twenty-three patients (62%) had 2 or more prior lines of therapy. Median number of prior lines of systemic therapy was 1 for MCL (range, 1–3), 2 for FL (all had 2 prior regimens), and 3 for DLBCL (range, 1–7). Fifteen patients (41%) had abnormal levels of lactase dehydrogenase (LDH), and 14 patients (38%) had lesions ≥ 5 cm. Mutations or deletions in TP53 as assessed by a next-generation sequencing gene panel were observed in 3 of 9 patients with MCL, 1 of 4 patients with FL, and none of the 11 patients with DLBCL with baseline tumor sequencing (3rd row in Fig. 2A). ctDNA analysis found TP53 mutations in one additional patient with DLBCL and two patients with MCL who did not have baseline next generation targeted tumor sequencing (Fig. 2A).
Figure 1.
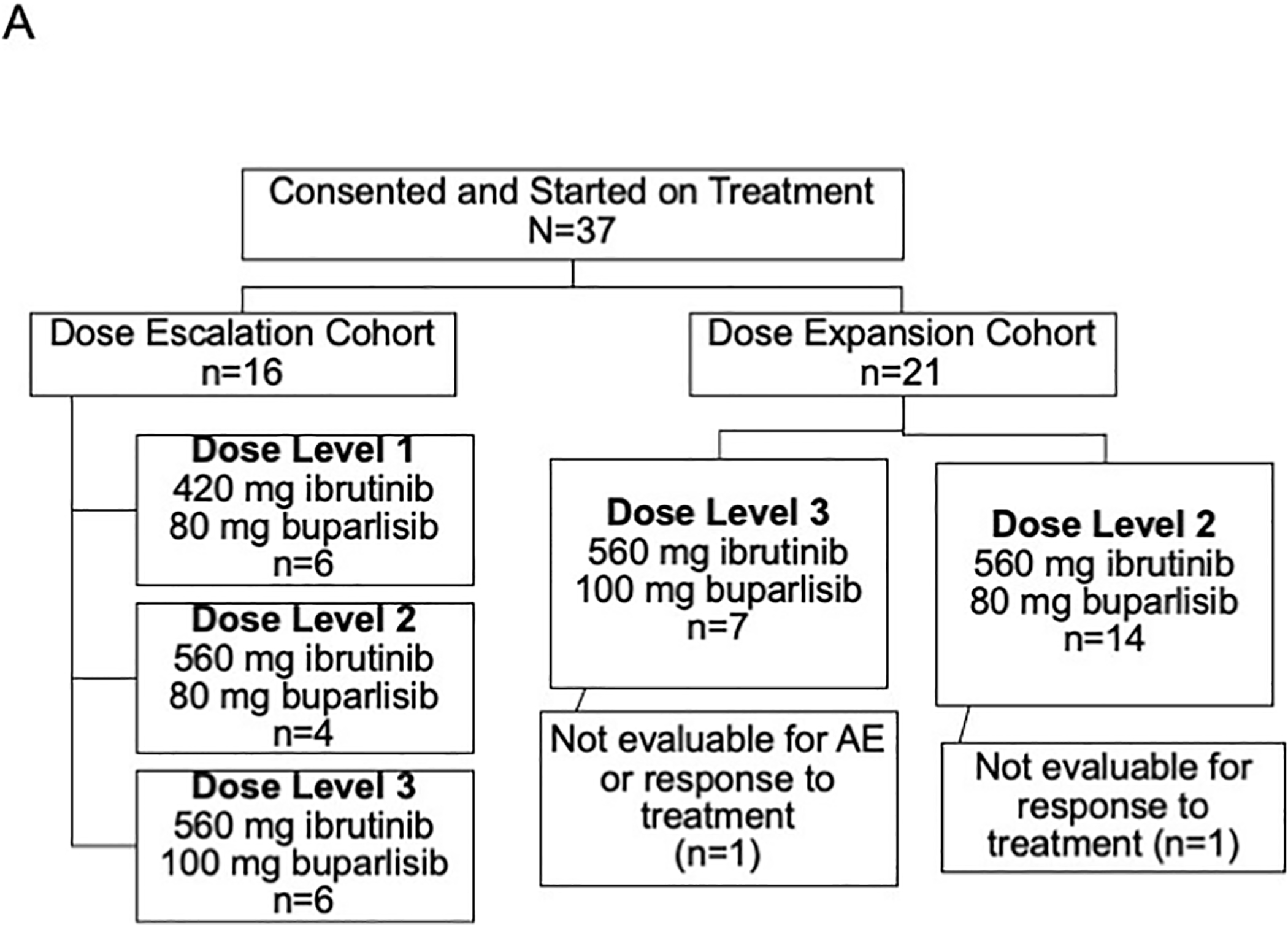
CONSORT diagram. Of 38 consented patients, 37 were started on treatment, 36 were evaluable for adverse events, and 35 were evaluable for clinical response.
Table 1.
Baseline and clinical characteristics of treated patients (n=37)
| Characteristic | Total N = 37 |
MCL N = 18 |
FL N = 5 |
DLBCL N = 14 |
|---|---|---|---|---|
| Age, years, median (range) | 70 (48–84) | 71 (50–84) | 65 (56–84) | 72 (48–78) |
| Sex, n (%) | ||||
| F | 10 (27) | 2 (11) | 1 (20) | 7 (50) |
| M | 27 (73) | 16 (89) | 4 (80) | 7 (50) |
| Stage, n (%) | ||||
| II | 2 (5.4) | 1 (5.6) | 0 (0) | 1 (7.1) |
| III | 8 (22) | 0 (0) | 4 (80) | 4 (29) |
| IV | 27 (73) | 17 (94) | 1 (20) | 9 (64) |
| Prior lines of therapy, n (%) | ||||
| 1 | 14 (38) | 12 (67) | 0 (0) | 2 (14) |
| 2 | 15 (41) | 5 (28) | 5 (100) | 5 (36) |
| ≥ 3 | 8 (22) | 1 (5.6) | 0 (0) | 7 (50) |
| Prior stem cell transplant, n (%) | ||||
| Allogeneic | 4 (11) | 1 (5.6) | 2 (40) | 1 (7.1) |
| Autologous | 10 (27) | 6 (33) | 0 (0) | 4 (29) |
| None | 23 (62) | 11 (61) ` | 3 (60) | 9 (64) |
| Ki-67, n (%) | ||||
| < 30% | 10 (29) | 5 (31) | 4 (80) | 1 (7.1) |
| ≥ 30% | 25 (71) | 11 (69) | 1 (20) | 13 (93) |
| Unknown | 2 | 2 | 0 | 0 |
| Abnormal LDH (above ULN), n (%) | 15 (41) | 4 (22) | 1 (20) | 10 (71) |
| Lesion ≥ 5 cm, n (%) | 14 (38) | 8 (44) | 3 (60) | 3 (21) |
| Number extranodal sites, n (%) | ||||
| < 2 | 22 (59) | 10 (56) | 4 (80) | 8 (57) |
| ≥ 2 | 15 (41) | 8 (44) | 1 (20) | 6 (43) |
| Baseline SUVmax, median (range) | 13 (4–46) | 9 (4–16) | 16 (8–29) | 18 (8–46) |
| Total lesion glycolysis, median (range) | 804 (78–20,806) | 895 (78–20,806) | 928 (555–3,187) | 540 (88–7,146) |
| Tumor metabolic volume, cm3, median (range) | 148 (12–5,854) | 195 (12–5,854) | 152 (72–458) | 50 (12–762) |
Abbreviations: LDH, lactate dehydrogenase; SUVmax, maximum standardized uptake value; ULN, upper limit of normal.
Figure 2.
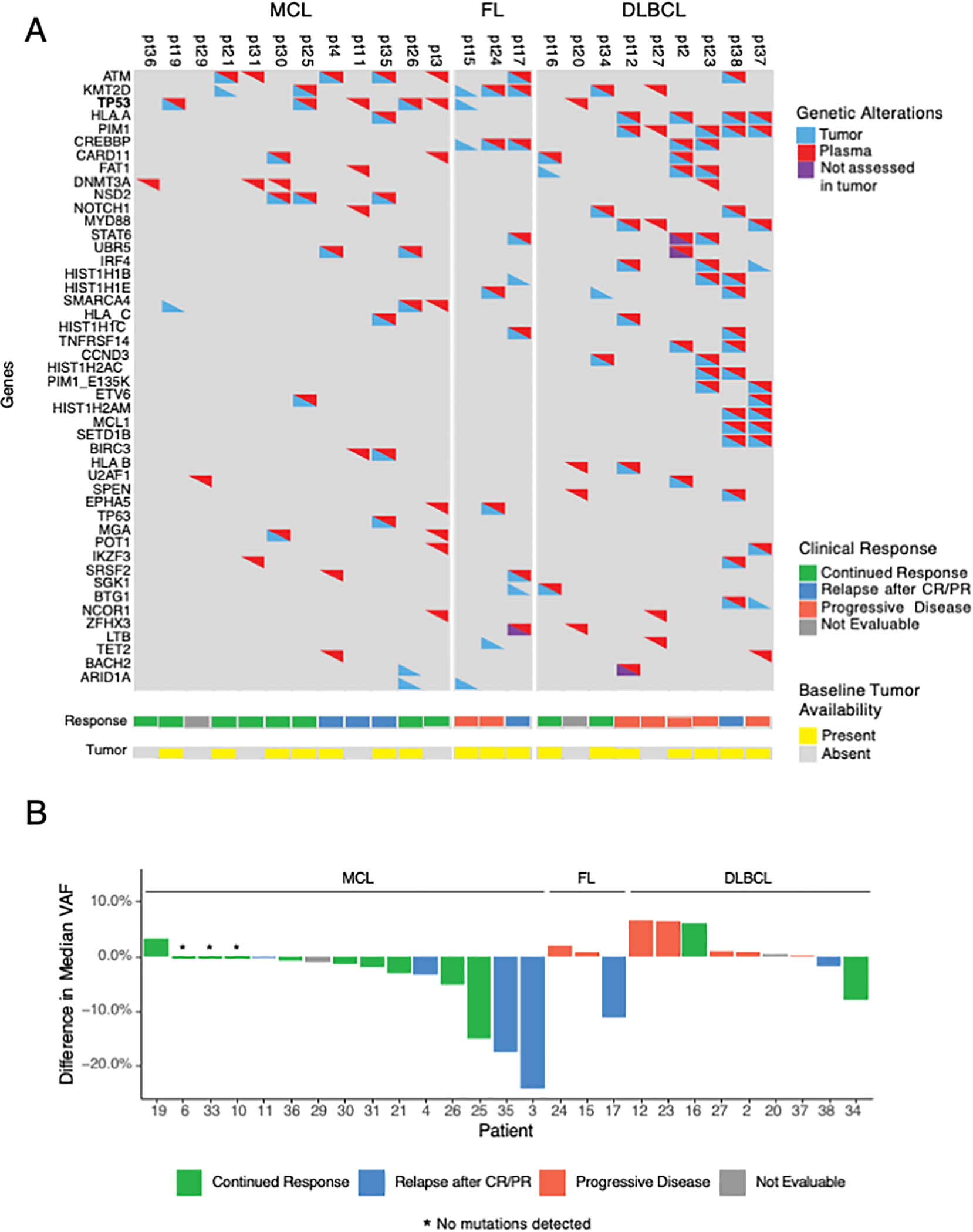
Association of change in cell-free DNA variant allele frequency (VAF) with clinical outcomes. A, Heatmap of mutations observed at least twice across 24 patients with detectable ctDNA. Not assessed in tumor refers to genes that were not assessed in the baseline tumor sequencing due to differences in the gene panels used – some patients had baseline sequencing with the MSK-IMPACT assay and others with the MSK-IMPACT-Heme assay. High concordance was observed between plasma and tumor sequencing. TP53 mutations were described in baseline tumor samples in 3 MCL patients and 1 FL patient. Plasma sequencing identified an additional 2 patients with MCL and 1 DLBCL patient with TP53 mutations. B, Waterfall plot of ctDNA difference (Δ) in median VAF at cycle 3 from baseline, as plotted against patient outcomes. Decreases in median VAF in MCL correlated with initial response but could not differentiate between patients who eventually relapsed.
Selection of Recommended Phase 2 Dose
In the dose escalation phase, patients received daily ibrutinib and buparlisib in 3 dose levels: dose level 1, 420 mg ibrutinib and 80 mg buparlisib; dose level 2, 560 mg ibrutinib and 80 mg buparlisib; and dose level 3, 560 mg ibrutinib and 100 mg buparlisib (Fig. 1). The dose-escalation phase enrolled 16 patients, 6, 4 and 6 respectively at dose levels 1, 2 and 3 (4 patients enrolled at dose level 2 because two patients consented concurrently on the same day). All 16 patients from the dose-escalation phase were evaluable for dose-limiting toxicities (DLT), as they received at least 50% of the intended doses in cycle 1 (Supplemental Table S1). Two of these patients experienced 1 DLT each: one patient on dose level 1 was hospitalized for grade 3 anorexia related to poor oral tolerance of the drugs, and one patient at dose level 3 reported headaches which upon further investigation subacute blood products were present in region of a pre-existing pituitary adenoma and event was classified as a grade 2 pituitary infarct. We selected dose level 3, ibrutinib 560 mg and buparlisib 100 mg, as the maximum tolerated dose (MTD) based on the observed events in the dose-escalation cohort.
Dose-expansion cohorts treated 21 additional patients, 7 at dose level 3 and 14 at dose level 2. The initial 4 of 7 patients in the dose level 3 expansion cohort experienced grade 2–3 toxicities, most notably rash and diarrhea which required dose adjustments (Supplemental Fig. S1A–C). Based on the related adverse events in cycle 1 of the expansion cohort, the study team proceeded with dose level 2, ibrutinib 560 mg and buparlisib 80 mg, as the recommended phase II dose (RP2D) to minimize toxicity of the combination. All patients on study at that time were at or dose reduced to dose level 2. The dose level 2 expansion cohort enrolled 14 additional patients, with 1 of 14 patients experiencing a grade 3 rash in cycle 1.
Tolerability of Ibrutinib and Buparlisib Over Treatment Course
Treatment-related adverse events (AE) were reported in all 36 AE-evaluable patients (Table 2). The 37th patient was not DLT-evaluable because of early progression and completion of less than 14 days of the intended cycle 1 dose. Table 2 describes related adverse events by grade occurring in more than 10% of patients. Adverse events were generally grade 1–2 and resolved with standard therapeutic interventions, dose interruptions, or dose reductions. The most common related adverse events of all grades were diarrhea (64%), thrombocytopenia (53%), fatigue (50%), hyperglycemia (39%), and rash/pruritis/dermatitis (39%). The most common grade 3 or higher related adverse events were rash/pruritis/dermatitis (19%), diarrhea (11%), hyperglycemia (11%), and hypertension (11%). Neurologic sequelae from mood disturbances (8 patients) and cognitive impairment (7 patients) were also noted and attributed to buparlisib. The cognitive impairment events were primarily anxiety, depression, and increased forgetfulness all of which were reversible except for one patient with existing dementia who continued to have neurologic decline thought unrelated to buparlisib. The patient had baseline Alzheimer dementia which had progressed in the 7.8 months he was on study, of which 6.2 months included buparlisib therapy. Grade 3 or higher non-treatment-related adverse events included hypertension (8%) and rash (6%) which were associated with patients’ baseline co-morbidities (Supplemental Table S2, Supplemental Fig. S2). The unrelated hypertension occurred when patients were off PI3K inhibition and were transient in nature. Similarly, the unrelated treatment emergent rashes were events of contact dermatitis or other dermatologic events clearly independent of therapy. No deaths were attributed to adverse events.
Table 2.
Treatment-related adverse events occurring in >10% of 36 AE-evaluable patients
| Adverse Event, n (%) | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total no. patients with any grade |
|---|---|---|---|---|---|
| Diarrhea | 14 (39%) | 5 (14%) | 4 (11%) | 0 (0%) | 23 (64%) |
| Thrombocytopenia | 9 (25%) | 9 (25%) | 1 (3%) | 0 (0%) | 19 (53%) |
| Fatigue | 13 (36%) | 4 (11%) | 1 (3%) | 0 (0%) | 18 (50%) |
| Hyperglycemia | 3 (8%) | 7 (19%) | 4 (11%) | 0 (0%) | 14 (39%) |
| Rash/Pruritis/Dermatitis | 4 (11%) | 3 (8%) | 7 (19%) | 0 (0%) | 14 (39%) |
| Dyspepsia/GERD | 9 (25%) | 3 (8%) | 1 (3%) | 0 (0%) | 13 (36%) |
| Anorexia | 4 (11%) | 5 (14%) | 2 (6%) | 0 (0%) | 11 (31%) |
| Nausea | 7 (19%) | 2 (6%) | 1 (3%) | 0 (0%) | 10 (28%) |
| Bruising | 8 (22%) | 1 (3%) | 0 (0%) | 0 (0%) | 9 (25%) |
| Elevated Bilirubin | 2 (6%) | 6 (17%) | 1 (3%) | 0 (0%) | 9 (25%) |
| Muscle Cramping | 5 (14%) | 4 (11%) | 0 (0%) | 0 (0%) | 9 (25%) |
| ALT Elevated | 6 (17%) | 0 (0%) | 2 (6%) | 0 (0%) | 8 (22%) |
| Mood Disturbances | 1 (3%) | 4 (11%) | 3 (8%) | 0 (0%) | 8 (22%) |
| AST Elevated | 4 (11%) | 2 (6%) | 1 (3%) | 0 (0%) | 7 (19%) |
| Cognitive Impairment | 2 (6%) | 4 (11%) | 0 (0%) | 1 (3%) | 7 (19%) |
| Hypertension | 1 (3%) | 2 (6%) | 4 (11%) | 0 (0%) | 7 (19%) |
| Mucositis | 4 (11%) | 2 (6%) | 1 (3%) | 0 (0%) | 7 (19%) |
| Cough | 4 (11%) | 2 (6%) | 0 (0%) | 0 (0%) | 6 (17%) |
| Dizziness/Lightheaded | 5 (14%) | 1 (3%) | 0 (0%) | 0 (0%) | 6 (17%) |
| Dry Skin | 6 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 6 (17%) |
| Myalgia | 4 (11%) | 2 (6%) | 0 (0%) | 0 (0%) | 6 (17%) |
| Arthralgia | 3 (8%) | 1 (3%) | 0 (0%) | 0 (0%) | 4 (11%) |
| CMV Reactivation | 3 (8%) | 1 (3%) | 0 (0%) | 0 (0%) | 4 (11%) |
| Headache | 2 (6%) | 1 (3%) | 1 (3%) | 0 (0%) | 4 (11%) |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CMV, cytomegalovirus; GERD, gastroesophageal reflux disease.
Dose interruptions of ibrutinib and buparlisib were allowed on protocol beyond cycle 1. The median duration of treatment for the combination of ibrutinib and buparlisib was 16 weeks (range 1–146 weeks). Graphical representations of dose modifications are provided in Supplemental Fig. S1A–C. Dose adjustments were made to one or both medications based on the adverse event. Treatment-related adverse events led to dose reductions of ibrutinib and buparlisib in 14 (38%) and 12 (32%) patients, respectively. The median percentage of protocol-specified dose taken by patients was 97% (range 46–100%) for ibrutinib and 91% (range 4–100%) for buparlisib. The most common cause of ibrutinib dose reduction was recurrent grade 1–2 muscle cramping (n=5), rash/pruritis/dermatitis (n=4: 2 grade 1 and 2 grade 3), and grade 2 myalgia (n=2). Buparlisib was dose-reduced in 12 patients, with the most common causes being grade 3 rash (n=3), grade 1–2 elevated bilirubin (n=3), and grade 1–2 fatigue (n=2). Thirteen patients (33%) stopped buparlisib while on study: 4 patients for cognitive impairment (1 grade 4, 1 grade 2, and 2 grade 1), 3 patients for grade 3 mood disturbances, 2 patients for grade 2 anorexia, 2 patients for AST elevation (1 grade 2, 1 grade 3) and 2 patients for edema (1 grade 2, 1 grade 3). Patients taken off buparlisib for adverse events were in a complete remission and remained on study while continuing ibrutinib monotherapy.
Efficacy
We evaluated all treated 37 patients for response (Table 3). Three patients demonstrated clinical progression in absence of radiographic staging. The median time to first response was 7 weeks (range 7–51 weeks) across all patients. Assessment by Lugano (25) and Response Evaluation Criteria in Lymphoma (RECIL) criteria (26) yielded similar results further validating the use of unidimensional measurements in clinical trials (Fig. 3B, Supplemental Fig. S3A). The overall median follow-up was 3.8 years (range 1–51 months).
Table 3.
Clinical response to ibrutinib and buparlisib combination in 37 patients, of whom 35 were evaluable for response
| Total (N=37) |
MCL (n=18) |
FL (n=5) |
DLBCL (n=14) |
|
|---|---|---|---|---|
| Evaluable for overall response | 35 | 17 | 5 | 13 |
| Best response by Lugano criteria, n (%) | ||||
| Overall response | 21 (60) | 16 (94) | 1 (20) | 4 (31) |
| Complete response | 17 (49) | 13 (76) | 1 (20) | 3 (23) |
| Partial response | 4 (11) | 3 (18) | - | 1 (8) |
| Stable disease | 4 (11) | - | 4 (80) | - |
| Progressive disease | 11 (31) | 1 (6) | - | 10 (77) |
| Treatment duration, weeks, median (range)b | 16 (1–146) | 39 (1–146) | 16 (8–79) | 7 (2–145) |
The 2 patients not evaluable for overall response were 1 patient with MCL who was off treatment for toxicity prior to response assessment and 1 patient with DLBCL who had a sudden death not attributed to treatment without formal radiographic assessment.
Ongoing as of March 31, 2021.
Figure 3.
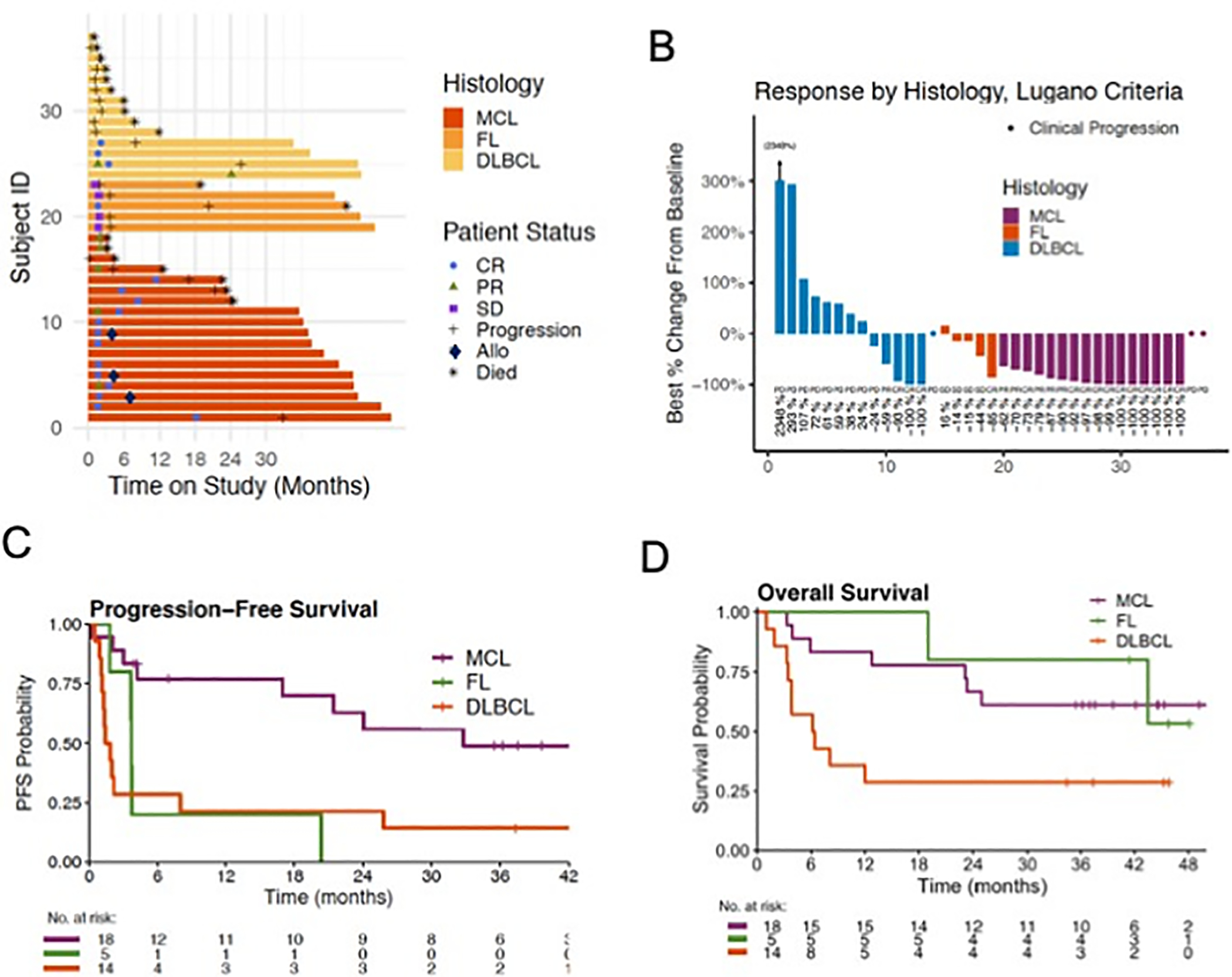
Clinical outcomes of patients treated with ibrutinib and buparlisib. A, Swim plot of 37 patients, 35 of whom were evaluable for response. Three patients with MCL proceeded to an allogeneic stem cell transplant while in remission. B, Waterfall plot using Lugano criteria. C, Progression-free survival in each disease cohort, showing median PFS of 33 months in MCL, 1.6 months in DLBCL, and 3.7 months in FL. D, Overall survival in each disease cohort. Median OS was 6.3 months in DLBCL, and not reached in MCL and FL.
In the MCL cohort, 18 patients were evaluable for PFS, EFS, and OS, while ORR was evaluable in 17 patients. One patient with MCL came off concurrent treatment with buparlisib and ibrutinib for toxicity prior to a radiographic restaging. The objective response rate was 94% and the CR rate 76% (Table 3). Three patients who achieved remission on therapy after multiply refractory MCL proceeded to an allogeneic stem cell transplant (Fig. 3A). The median time on treatment was 39 weeks (1–146 weeks). Two patients displaying PR converted to CR at 3.5 and 5.2 months, respectively. Median duration of response in the MCL cohort was 31 months (95% CI, 16 months – not reached [NR]), and median PFS was 33 months (21 - NR) based on median follow-up of 3.5 years (Fig. 3C). The median event-free survival (EFS) was 8.9 months (3.6 - NR) (Supplemental Fig. S3B). Patients discontinued therapy for adverse events but continued to have sustained lymphoma responses. Median overall survival was not reached (23 months - NR) (Fig. 3D).
In the FL cohort, an overall response was observed in 1 of 5 (20%) patients (Table 3). Median time on treatment was 16 weeks (8–79 weeks). The median PFS was 3.7 months (3.7 - NR) with median follow-up of 3.8 years (Fig. 3C). Median EFS was 3.7 months (3.7 - NR) (Supplemental Fig. S3B) and median OS was NR (43 - NR) (Fig. 3D).
In the DLBCL cohort, overall response was assessable in 13 patients while PFS, EFS and OS was evaluated in 14 patients. One DLBCL patient suffered a sudden death from treatment-unrelated causes and based on physical exam demonstrated a clinical response that was not verified with radiographic imaging. At the time, patient had 12 days of combination therapy and was undergoing a treatment holiday for diarrhea and rash. Overall responses were observed in 4 of 13 (31%) patients (Table 3). Median time on treatment was 7 weeks (2–145 weeks). Two of 8 patients with non-germinal center B-cell-like DLBCL had a CR, and 2 of 5 evaluable patients with germinal center B-cell-like DLBCL had a response (1 CR, 1 PR) (Fig. 3B). One patient with DLBCL converted from a partial to complete response at 15 weeks (Fig. 3A). Patients with DLBCL with a response experienced sustained responses evidenced by a median duration of response of 30 months (95% CI, 5.9 - NR). The median PFS was 1.6 months (1.3 - NR) with median follow-up of 3.4 years (Fig. 3C). The median EFS was 1.6 months (1.3–30) (Supplemental Fig. S3B) and median OS was 6.3 months (3.8 - NR) (Fig. 3D).
Dynamics of Circulating Tumor DNA (ctDNA) for Disease Monitoring
Plasma ctDNA mutations were highly concordant with tumor mutations.
Plasma sequencing of ctDNA identified mutations in 24/27 (88.9%) patients. In total, 225 mutations were found in the plasma; 64% were tumor-derived, while 36% were identified only in plasma, attributed to clonal progression during treatment or lacked baseline tumor for comparison. Across the whole cohort, an average of 8.3 mutations were identified in the plasma per patient and the number of tumor mutations correlated with the number of plasma mutations (R2 = 0.8433).
In 17 patients with tumor sequencing prior to therapy, 15 (88.2%) had detectable ctDNA. Circulating tumor DNA sequencing identified an additional 7 mutations in 4 of these patients from their baseline plasma. For example, patient 4 (MCL) harbored MSI1 N247S and TET2 H877L mutations in the baseline plasma sample which were not found in the tumor sample (Fig. 4D). In this case, the lack of synchronous tumor and plasma profiling precludes ability to discern tumor heterogeneity or clonal progression in the period between the tumor biopsy and plasma sampling (27,28). The median time between tumor biopsy and first plasma collection was 45 days (range −7 to 993 days, Supplemental Table S3). For synchronous plasma and tumor sampling (+/− 16 days, n = 6), 43 out of 57 tumor mutation calls were observed in plasma (75.4%) (Fig 2A).
Figure 4.
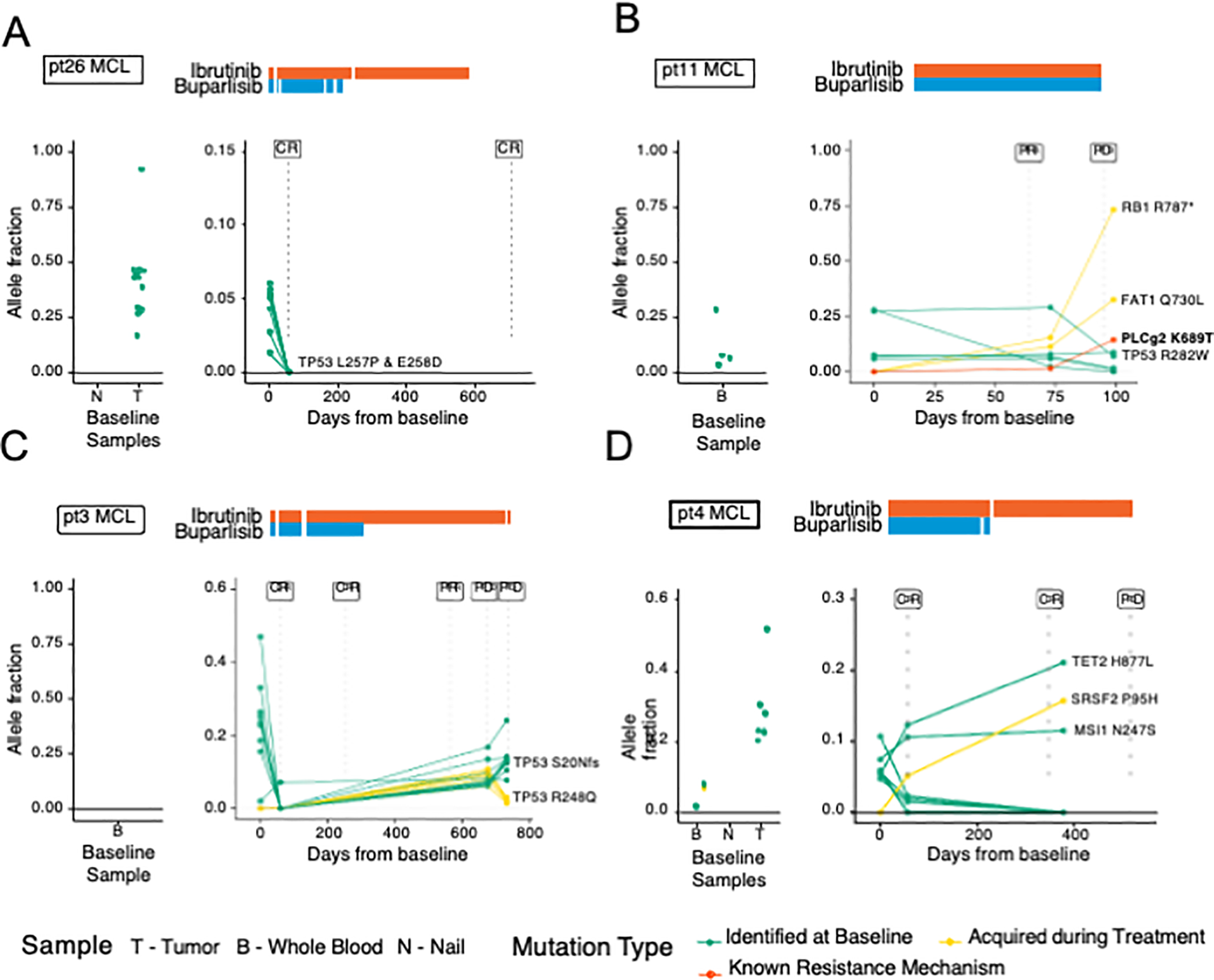
Cell-free DNA and baseline sample profiles of MCL patients. Patient cfDNA samples were serially assessed using HEMEPACT. Patterns of ctDNA responses emerge corresponding to radiographic imaging. A, In a patient with early CR, the VAFs all reduced to below detectable levels. B, Detection of new mutation PLCg2 K689T with known resistance mechanism to BTK therapy in patient with partial response and subsequent progression. C, Early CR with later progression tracked by increased VAF of pre-existing mutations. An existing TP53 mutation in R248Q mutation and a newly detected more dominant TP53 S20Nfs both increased in VAF with divergent clonal representation. D, Early CR with later progression including discordant ctDNA dynamics and a potential resistance mutation. Mutations TET2 and SRSF2 known to be relevant in clonal hematopoiesis were observed without clinical association with myelodysplastic syndrome during protocol period. T, tumor; B, whole blood; N, nail; BC, buffy coat; S, saliva
Clinically relevant ctDNA mutations can also be identified in the absence of paired tumor sequencing. In 9 patients without baseline tumor sequencing, mutations were identified in 7 of 10 (70%) patients (Fig. 2A). We identified 3 patients with TP53 mutations: 2 patients with MCL and TP53 R282W, S20Nfs*20 mutations, respectively, and 1 patient with DLBCL and a TP53 F113S mutation. In one MCL patient with tumor sequencing (patient 3), an additional mutation in TP53 at R248Q was identified as a treatment-acquired mutation (Fig. 4C). In total, 8 TP53 mutations were identified across 6 patients via plasma ctDNA, including those with matched tumor sequencing.
Patterns of ctDNA changes correspond to radiographic imaging and disease status.
We tracked the magnitude of variant allele frequency (VAF) changes relative to response (Fig. 2B). The median difference in VAF of non-relapsing and relapsing patients was −1.1% and −7.35%, respectively. Interestingly, the magnitude of the ctDNA change did not differentiate between patients achieving CR from those who eventually relapsed.
Twenty-four patients had ctDNA detected, of whom 22 patients had serial radiographic imaging available for analysis. Focusing on the 18 patients with MCL, 15 had detectable ctDNA sequenced and 3 had no detectable ctDNA at any timepoint. Of 15 patients with MCL with detectable ctDNA, 11 patients responded to therapy. Six patients with MCL (3 CR, 3 PR) had decreases in ctDNA VAF similar to patient 26 (Fig. 4A). In these patients, the pattern of ctDNA followed radiographic responses. Of 6 patients with MCL who had a CR or PR and decreases in ctDNA, only 2 relapsed. Both relapsed patients were identified by increases in ctDNA prior to radiographic relapses as in patient 3 (Fig. 4C). However, radiographic status of disease was not always concordant with ctDNA. Patients 11 and 4), as expected, developed a radiographic progression shortly after the increases in ctDNA (Fig. 4B, 4D). In contrast, patient 19 (Fig. 2B) had a detectable and rising TP53 Y220C mutation despite achieving a durable CR. Shifting of tumor clonality was also observed as in patient 3 who had an existing TP53 mutation (R248Q) with decreased VAF during period of clinical response with subsequent increase of VAF at relapse, and presence of a newly detected TP53 mutation (S20Nfs) at relapse. The dynamics in each tumors clonal populations may be a result of clonal selection under therapeutic pressure. We also observed emergence of mutations consistent with clonal hematopoiesis such as TET2 H877L and SRSF2 P95H (pt 4, Fig. 4D), however patient had no clinical signs of myelodysplastic syndrome throughout treatment.
Plasma ctDNA reveals emergence of potential resistance mutations.
Three patients in our cohort, 2 with MCL (patient 4 and patient 11; Fig. 4B, 4D) and 1 patient with DLBCL (patient 38) acquired new mutations while on therapy. Patient 11 acquired mutations in RB1, BIRC3 and PLCG2 which were observed as early as cycle 3. The known resistance mutation of PLGC2 K689T was not observed at time of consent but clonally expanded to 1.5% at cycle 3, and 14.4% at time of progression after cycle 4 (Fig. 4B). In this case, the resistant mutation was detected 1 cycle prior to relapse. Patient 4 acquired a mutation in SRSF2 P95H where VAF of this mutation increased from 0% at baseline, to 5.1% at cycle 3, and 15.7% at time of relapse in cycle 14 (Fig. 4D). This pattern was also observed in patient 38 with DLBCL where mutations in BCR T1018A and LMO1 X122 splice variant were acquired at cycle 9 and expanded at relapse. With more sensitive plasma sequencing, mutations suspicious for early resistance may be identified months prior to radiographic relapse.
DISCUSSION
High-throughput strategies using unbiased small molecule combination screening have found multiple cooperative interactions between ibrutinib and inhibitors of the PI3K pathway (7). In particular, a pan-PI3K which provides PI3Kα inhibition was suggested to suppress resistance mechanisms. The published clinical studies of PI3K and BTK inhibitors have used selective PI3K inhibitors (11–13); this study to our knowledge represents the first pan-PI3K inhibitor combination with a BTK inhibitor.
In our study, buparlisib plus ibrutinib demonstrated promising clinical activity in patients with relapsed MCL. The overall response rate was 94%, with 76% of patients achieving a complete response. The combination of buparlisib and ibrutinib in MCL resulted in a median PFS of 33 months. Three patients with multiply refractory disease achieved a complete remission while on combination therapy, permitting them to safely undergo an allogeneic stem cell transplant for disease consolidation. In our population of MCL patients, 67% were enrolled with 1 prior line of therapy. Despite the relatively low treatment exposures, the patients with MCL had high risk factors, with > 69% of patients having Ki-67 ≥ 30% and 45% (5/11) with TP53 aberration.
The overall response rate of 94% and 33 mo PFS of buparlisib and ibrutinib in MCL compare favorably to ibrutinib single agent in the second line and other BTK and PI3K combinations. Barring cross trial comparisons, rituximab-ibrutinib demonstrated a lower CR of 58% and a superior PFS of 43 mo (16). The patients with durations of responses <24 months who were treated with rituximab-ibrutinib had intermediate or high risk MIPI, blastoid disease, or high Ki-67. In the current study, 94% patients had intermediate to high risk MCL, and 67% patients had elevated Ki-67≥30%. In a series of 99 patients with MCL treated with ibrutinib in the second-line setting, median PFS was 25 months and ORR was 78% with a CR of 37%(15). Other published studies of combination BTK and PI3K inhibition in the relapsed setting also improve the CR rate compared to BTK treatment alone. Combination of ibrutinib and umbralisib, a preferential PI3Kδ inhibitor, showed ORR 67%, CR 19%, and PFS 10.5 months in MCL (11). Similarly, the combination of tirabrutinib and idelalisib demonstrated ORR of 93%, CR 7%, and PR 79% in 14 CLL patients, with a 27-month median duration of response and 32-month median PFS (12). Tirabrutinib and idelalisib showed ORR of 24% in DLBCL, 20% in FL, and 1 MCL patient had a complete response (13). While it is challenging to compare across studies in a small and variable patient population the combination of a pan-PI3K and BTK inhibitor may differ from combinations of PI3Kδ and BTK blockade by reducing an PI3Kα mediated resistance (10) and offer an oral regimen with high rates of complete responses.
Nonetheless, the challenges of this combination are the neuropsychiatric toxicities attributable to buparlisib as well as toxicities arising from cumulative exposure to treatment. The combination of ibrutinib and buparlisib resulted in dose reductions, modifications and buparlisib was discontinued in 33% of the patients. The side effects leading to dose modification of ibrutinib were related to recurrent muscle cramping, rash or myalgias. However, other PI3K associated events such as colitis and pneumonitis and BTK associated cardiac, or bleeding events were rare. BTK and pan-PI3K combinations with a more favorable toxicity profile may be beneficial in future targeting of these pathways. In particular, regimens of combination targeted therapy may be suitable for patients requiring bridging treatment to CAR T therapy whose disease biology or clinical status preclude use of chemotherapy.
This study also compared next-generation sequencing from tumor biopsies and peripheral blood cfDNA and monitored the ctDNA dynamics throughout targeted combination therapy. Previous reports have demonstrated the potential of tracking circulating tumor DNA (ctDNA), the component of cfDNA that is derived specifically from the tumor, to monitor disease dynamics in MCL (29) and DLBCL (27,30), demonstrating applicability of this technique in lymphoma. Multiple studies have demonstrated that ctDNA can recapitulate next-generation sequencing of tumor samples. Our study further supports that and provides additional evidence to show that ctDNA profiling from peripheral blood can identify tumor mutations without previous knowledge of the tumor mutations. While ctDNA provided dynamic measurements with VAFs that correlated with radiographic outcomes, the magnitude of the ctDNA change did not segregate patients who achieve long term remission from those who later progress. Importantly, ctDNA profiling was also able to identify resistant mutations. Eight of 10 patients with sequencing performed at 3 or more timepoints had molecular progression identified prior to progression on radiographic scans. The sequencing technology employed in this study can detect mutations without prior knowledge down to a VAF of 2–5%. The dynamics of clonal and subclonal populations are observed but more robust and high depth sequencing strategies are needed to characterize tumor evolution. Sequencing methods with higher sensitivity such as those using unique molecular identifiers and deeper coverage (31) may allow for detection of resistant mutations at earlier timepoints and potentially guide timely change of therapy prior to radiographic progression. Studies involving this type of intervention after positive ctDNA are more advanced in other cancer types, for instance in non-small cell lung cancer where epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors have been prescribed following identification of EGFR mutations in plasma ctDNA (32,33) and in breast cancer where targeted therapies have been prescribed following identification of mutations in HER2 and ATK1 in ctDNA (34). Our ctDNA analysis is limited by a relatively small sample size and utilization of a suboptimal assay for ctDNA where detection of lower frequency VAFs may have been missed.
A limitation in our study is the relatively small population of patients enrolled in our study. The early efficacy signal in the MCL cohort may provide rationale to develop a larger study of a pan-PI3K and BTK combination in earlier lines of therapy. Additionally, our dedicated cfDNA detection panel is awaiting validation with an orthogonal method and other robust sequencing methods may be employed to detect status of minimal residual disease and emergence of resistance clones at higher sensitivity (35).
In conclusion, we report that the combination of buparlisib and ibrutinib for patients with MCL produced high overall response and complete response rates with a promising durability of response. The combination was associated with rashes, diarrhea, transient hyperglycemia, transient hypertension, and neuropsychiatric symptoms attributed to both the cumulative toxicity of extended oral treatment and the distinct side effect profile of buparlisib. Further investigation is warranted using BTK and PI3K inhibitors with a more favorable toxicity profile. BCL2 inhibition added to this doublet therapy may be considered for a multi-agent targeted therapy combination in MCL. Refinement of ctDNA monitoring technologies may better identify patients with enduring responses who benefit from early cessation of therapy.
Supplementary Material
Statement of translational relevance:
Previous studies reported that BTK inhibitors are clinically relevant for several B cell lymphomas and in particular mantle cell lymphoma. Combinations of targeted therapy such as BTK and BCL2 inhibitors were showed to be clinically active. Similarly, combinations of BTK and PI3Kδ inhibitors in B cell lymphoma have been studied with some success. The utilization of a pan-PI3K inhibitor is postulated to suppress a mechanism of resistance in this combination. In this phase I study, we evaluated the tolerability and efficacy of buparlisib, a pan-PI3K inhibitor, and ibrutinib, a BTK inhibitor in mantle cell, follicular and diffuse large B cell lymphoma. The combination produced high complete response rates particularly in mantle cell lymphoma however administration is limited by previously known neuropsychological toxicity profile of buparlisib. The study provides rationale for continued development of doublet or triplet combination targeted therapy in mantle cell lymphoma.
Acknowledgments
This clinical trial was sponsored by Memorial Sloan Kettering Cancer Center (MSK), with additional monetary and therapeutic drug support from Novartis and Janssen. This research was also funded in part by the NIH/NCI Cancer Center Support Grant (P30 CA008748) to MSK. Dr. Batlevi’s work has been supported by an ASCO Young Investigator Award, an ASH Clinical Scholar Award, and the Lymphoma Research Foundation Clinical Research Mentoring Program.
We thank the investigators and patients of this trial for their participation. We thank the clinical trial teams including Matthew Ho, Devin Callan, Ashley Ames, Shelley Levi, Salma Ahsanuddin, Robert Porzia, Amanda Copeland, Joanna Dicostanza, and Jenny Soiffer for assistance in managing patients and managing regulatory and data aspects of the clinical trial. Our clinical trials would not be able to function without the dedicated support of our clinical research team members.
Role of Funding Sources
This investigator-initiated clinical trial was sponsored by MSK with additional monetary and therapeutic drug support from Novartis and Janssen. The authors designed the clinical trial and interpreted the data in collaboration with the corporate study sponsors. The authors collected and analyzed the data independently, prior to review by the corporate sponsors. All authors had access to the raw data upon request and agree with the accuracy of this report. The corresponding authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Conflict of Interests
C. M. Stewart: None
L. Michaud: None
K. Whiting: None
R. Nakajima: None
C. Nichols: None
S. De Frank: None
P.A. Hamlin: research support from Portola Pharmaceuticals, Molecular Templates, Incyte, and J&J Pharmaceuticals; and has a consultancy role with Portola Pharmaceuticals, Celgene, Karyopharm Therapeutics, and Juno Therapeutics.
M.J. Matasar: research support from Genentech, Roche, GlaxoSmithKline, Bayer, Pharmacyclics, Janssen, Rocket Medical, and Seattle Genetics; has received honoraria from Genentech, Roche, Bayer, Pharmacyclics, Janssen, Seattle Genetics, and GlaxoSmithKline; and has a consultancy role with Genentech, Bayer, Merck, Juno, Roche, Teva, Rocket Medical, and Seattle Genetics.
J.F. Gerecitano: employment at Janssen.
P. Drullinsky: None
A. Hamilton: None
D. Straus: advisory role with Seattle Genetics.
S.M. Horwitz: research support from ADC Therapeutics, Aileron Therapeutics, Celgene, Forty Seven, Infinity Pharmaceuticals/Verastem Oncology, Kyowa Hakko Kirin, Millennium Pharmaceuticals/Takeda Oncology, Seattle Genetics, and Trillium; and has a consultancy role with ADC Therapeutics, Aileron, Corvus Pharmaceuticals, Forty Seven, Innate Pharma, Kyowa Hakko Kirin, Millennium/Takeda, Mundipharma, Portola, and Seattle Genetics.
A. Kumar: research support from AbbVie, Adaptive Biotechnologies, Celgene, Pharmacyclics, and Seattle Genetics; and has an advisory role with Celgene.
C.H. Moskowitz: research support from Bristol-Myers Squibb, Merck, and Seattle Genetics; has a consultancy role with AstraZeneca, Bristol-Myers Squibb, Karyopharm Therapeutics, Merck, Seattle Genetics, Takeda, and Vaniam Group; and has an advisory role with AstraZeneca, Karyopharm Therapeutics, Merck, Seattle Genetics, Takeda, and Vaniam Group.
A. Moskowitz: research support from Incyte, Seattle Genetics, Bristol-Myers Squibb, and Merck; and has a consultancy role with Kyowa Hakko Kirin Pharma, Miragen Therapeutics, Takeda Pharmaceuticals, ADC Therapeutics, Seattle Genetics, Cell Medica, Bristol-Myers Squibb, and Erytech Pharma.
A.D. Zelenetz: research support from MEI Pharma, MorphoSys, Sandoz, Celgene, Roche, and Gilead Sciences; has a consultancy role with Genentech/Roche, Gilead, Celgene, Janssen, Amgen, Novartis, and Adaptive Biotechnologies; and serves on the board of directors (Data Monitoring Committee Chair) for BeiGene.
J. Rademaker: None
G. Salles: consultancy role with Abbvie, Bayer, Beigene, BMS/Celgene, Debiopharm, Epizyme, Genentech/Roche, Genmab, Incyte, Ipsen, Janssen, Kite/Gilead, Loxo, Milteniy, Morphosys, Novartis, Rapt, Regeneron, Takeda, Velosbio
V. Seshan: None
H. Schoder: None
A. Younes: employment at AstraZeneca.
D.W.Y. Tsui: received honoraria from Nanodigmbio, Cowen, and BoA Merrill Lynch; research funding from ThermoFisher Scientific, Epic Sciences, Prostate Cancer Foundation; travel, accommodations, expenses from Nanodigmbio at the time of the study. DWY Tsui is currently an employee of PetDx, Inc.
C.L. Batlevi: research support from Janssen, Novartis, Epizyme, Autolus, Roche, and Bayer; has received honoraria from Dava Oncology; and has a consultancy role with Skipta, Kite Pharma, MorphoSys, Bristol-Myers Squibb, Karyopharm Therapeutics, Genentech, and TG Therapeutics.
REFERENCES
- 1.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010;463(7277):88–92 doi 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wullenkord R, Friedrichs B, Erdmann T, Lenz G. Therapeutic potential of PI3K signaling in distinct entities of B-cell lymphoma. Expert Rev Hematol 2019;12(12):1053–62 doi 10.1080/17474086.2019.1676716. [DOI] [PubMed] [Google Scholar]
- 3.Blachly JS, Baiocchi RA. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br J Haematol 2014;167(1):19–32 doi 10.1111/bjh.13065. [DOI] [PubMed] [Google Scholar]
- 4.Flinn IW, Kahl BS, Leonard JP, Furman RR, Brown JR, Byrd JC, et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-delta, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood 2014;123(22):3406–13 doi 10.1182/blood-2013-11-538546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369(6):507–16 doi 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med 2015;21(8):922–6 doi 10.1038/nm.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. PNAS 2014;111(6):2349–54 doi 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul J, Soujon M, Wengner AM, Zitzmann-Kolbe S, Sturz A, Haike K, et al. Simultaneous Inhibition of PI3Kdelta and PI3Kalpha Induces ABC-DLBCL Regression by Blocking BCR-Dependent and -Independent Activation of NF-kappaB and AKT. Cancer Cell 2017;31(1):64–78 doi 10.1016/j.ccell.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Psyrri A, Papageorgiou S, Liakata E, Scorilas A, Rontogianni D, Kontos CK, et al. Phosphatidylinositol 3’-kinase catalytic subunit alpha gene amplification contributes to the pathogenesis of mantle cell lymphoma. Clin Cancer Res 2009;15(18):5724–32 doi 10.1158/1078-0432.CCR-08-3215. [DOI] [PubMed] [Google Scholar]
- 10.Iyengar S, Clear A, Bödör C, Maharaj L, Lee A, Calaminici M, et al. P110α-mediated constitutive PI3K signaling limits the efficacy of p110δ-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood 2013;121(12):2274–84 doi 10.1182/blood-2012-10-460832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman JM, et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: a multicentre phase 1–1b study. Lancet Haematol 2019;6(1):e38–e47 doi 10.1016/S2352-3026(18)30196–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danilov AV, Herbaux C, Walter HS, Hillmen P, Rule SA, Kio EA, et al. Phase Ib Study of Tirabrutinib in Combination with Idelalisib or Entospletinib in Previously Treated Chronic Lymphocytic Leukemia. Clin Cancer Res 2020;26(12):2810–8 doi 10.1158/1078-0432.CCR-19-3504. [DOI] [PubMed] [Google Scholar]
- 13.Morschhauser F, Dyer MJS, Walter HS, Danilov AV, Ysebaert L, Hodson DJ, et al. Phase 1b study of tirabrutinib in combination with idelalisib or entospletinib in previously treated B-cell lymphoma. doi: 10.1038/s41375-020-01108-x [epub ahead of print Dec 17, 2020]. Leukemia 2020. doi 10.1038/s41375-020-01108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: a phase 2 consortium trial. Blood 2018;131(2):182–90 doi 10.1182/blood-2017-09-804641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rule S, Dreyling M, Goy A, Hess G, Auer R, Kahl B, et al. Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: extended 3.5-year follow up from a pooled analysis. Haematologica 2019;104(5):e211–e4 doi 10.3324/haematol.2018.205229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain P, Romaguera J, Srour SA, Lee HJ, Hagemeister F, Westin J, et al. Four-year follow-up of a single arm, phase II clinical trial of ibrutinib with rituximab (IR) in patients with relapsed/refractory mantle cell lymphoma (MCL). Br J Haematol 2018;182(3):404–11 doi 10.1111/bjh.15411. [DOI] [PubMed] [Google Scholar]
- 17.Zang C, Eucker J, Liu H, Coordes A, Lenarz M, Possinger K, et al. Inhibition of pan-class I phosphatidyl-inositol-3-kinase by NVP-BKM120 effectively blocks proliferation and induces cell death in diffuse large B-cell lymphoma. Leuk Lymphoma 2014;55(2):425–34 doi 10.3109/10428194.2013.806800. [DOI] [PubMed] [Google Scholar]
- 18.Younes A, Salles G, Martinelli G, Bociek RG, Barrigon DC, Barca EG, et al. Pan-phosphatidylinositol 3-kinase inhibition with buparlisib in patients with relapsed or refractory non-Hodgkin lymphoma. Haematologica 2017;102(12):2104–12 doi 10.3324/haematol.2017.169656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrido-Castro AC, Saura C, Barroso-Sousa R, Guo H, Ciruelos E, Bermejo B, et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res 2020;22(1):120 doi 10.1186/s13058-020-01354-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Gooijer MC, Zhang P, Buil LCM, Citirikkaya CH, Thota N, Beijnen JH, et al. Buparlisib is a brain penetrable pan-PI3K inhibitor. Sci Rep 2018;8(1):10784 doi 10.1038/s41598-018-29062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ptashkin RN, Benayed R, Ziegler J, Rema AB, Sadowska J, Kiecka I, et al. MSK-IMPACT Heme: validation and clinical experience of a comprehensive molecular profiling platform for hematologic malignancies. American Association for Cancer Research annual meeting; March 29-April 3, 2019; Atlanta, GA. Abstract 3409; doi: 10.1158/1538-7445.AM2019-3409. Cancer Res 2019;79 (13 suppl)(13 suppl) doi 10.1158/1538-7445.AM2019-3409. [DOI] [Google Scholar]
- 22.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–13 doi 10.1038/nm.4333 http://www.nature.com/nm/journal/vaop/ncurrent/abs/nm.4333.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Molec Diagn 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Z, Kumar N, Bolipata A, Song T, Kandoth C, Liu F, et al. A portable bioinformatics pipeline for the FDA authorized IMPACT DNAseq assay. AACR Annual Meeting; March 29-April 3, 2019; Atlanta, GA. Abstract 2482; doi: 10.1158/1538-7445.AM2019-2482. Cancer Res 2019;79 (13 suppl)(13 suppl) doi 10.1158/1538-7445.AM2019-2482. [DOI] [Google Scholar]
- 25.Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32(27):3059–68 doi 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Younes A, Hilden P, Coiffier B, Hagenbeek A, Salles G, Wilson W, et al. International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann Oncol 2017;28(7):1436–47 doi 10.1093/annonc/mdx097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bohers E, Viailly P-J, Becker S, Marchand V, Ruminy P, Maingonnat C, et al. Non-invasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: analysis of a prospective cohort. Blood Cancer J 2018;8:74 doi 10.1038/s41408-018-0111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AFM, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med 2016;8(364):364ra155 doi 10.1126/scitranslmed.aai8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agarwal R, Chan Y-C, Tam CS, Hunter T, Vassiliadis D, Teh CE, et al. Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med 2019;25:119–29 doi 10.1038/s41591-018-0243-z. [DOI] [PubMed] [Google Scholar]
- 30.Camus V, Sarafan-Vasseur N, Bohers E, Dubois S, Mareschal S, Bertrand P, et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leuk Lymphoma 2016;57(9):2171–9 doi 10.3109/10428194.2016.1139703. [DOI] [PubMed] [Google Scholar]
- 31.Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 2012;9:72–4 doi 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 32.Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. J Clin Oncol 2016;34(28):3375–82 doi 10.1200/jco.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Cheng Y, An T, Gao H, Wang K, Zhou Q, et al. Detection of EGFR mutations in plasma circulating tumour DNA as a selection criterion for first-line gefitinib treatment in patients with advanced lung adenocarcinoma (BENEFIT): a phase 2, single-arm, multicentre clinical trial. Lancet Respir Med 2018;6(9):681–90 doi 10.1016/S2213-2600(18)30264-9. [DOI] [PubMed] [Google Scholar]
- 34.Turner NC, Kingston B, Kilburn LS, Kernaghan S, Wardley AM, Macpherson IR, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol 2020;21(10):1296–308 doi 10.1016/S1470-2045(20)30444-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun 2021;12(1):3770 doi 10.1038/s41467-021-24109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.