

SUMMARY
Accelerated glycolysis is the main metabolic change observed in cancer, but the underlying molecular mechanisms and their role in cancer progression remain poorly understood. Here we show that deletion of the long noncoding RNA (lncRNA) Neat1 in MMTV-PyVT mice profoundly impairs tumor initiation, growth and metastasis, while specifically switching off the penultimate step of glycolysis. Mechanistically, NEAT1 directly binds and forms a scaffold bridge for the assembly of PGK1/PGAM1/ENO1 complexes, and thereby promotes substrate channeling for high and efficient glycolysis. Notably, NEAT1 is upregulated in cancer patients and correlates with high levels of these complexes, and genetic and pharmacological blockade of penultimate glycolysis ablates NEAT1-dependent tumorigenesis. Finally, we demonstrate that Pinin mediates glucose-stimulated nuclear export of NEAT1, through which it exerts isoform-specific and paraspeckle-independent functions. These findings establish a direct role for NEAT1 in regulating tumor metabolism, provide new insights into the Warburg effect, and identify potential targets for therapy.
Keywords: Aerobic glycolysis, Breast cancer, ENO1, Long noncoding RNA, NEAT1, PGAM1, PGK1, Pinin (PNN), Tumor metabolism, Warburg effect
Graphical Abstract

eTOC Blurb:
Park et al. report on a role for NEAT1 in promoting a glycolytic state in breast cancer, highlighting how lncRNAs regulate cancer development at a metabolic level in vivo. NEAT1 facilitates assembly of PGK1/PGAM1/ENO1 glycolytic complexes and substrate channeling. Through isoform switching, they identify an isoform-specific, paraspeckle-independent role for NEAT1 in tumorigenesis.
INTRODUCTION
Aerobic glycolysis, also termed the Warburg effect, in which glucose is processed into lactate, is a common feature of glucose metabolism in cancer cells, (Hanahan and Weinberg, 2011; Vander Heiden et al., 2009). Although the enzymes that catalyze the three committed steps of glycolysis, specifically hexokinase (HK), phosphofructokinase (PFK), and pyruvate kinase (PK), are key control elements in the glycolytic pathway, the fundamentals of metabolic control analysis (Fell, 1992) have suggested that the control mechanisms of glycolytic flux may be different under Warburg effect conditions. In fact, non-rate-limiting glycolytic enzymes, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM), enolase (ENO), and lactate dehydrogenase (LDH), have been shown to contribute to aerobic glycolysis and biosynthesis in cancer cells (Hitosugi et al., 2012; Liberti et al., 2017; Lin et al., 2020).
Long noncoding RNAs (lncRNAs) can regulate glucose metabolism by controlling expression of metabolic enzymes and transporters, or by modulating signaling pathways. However, a direct link between lncRNA and tumor glycolysis has yet to be established. Nuclear enriched abundant transcript 1 (NEAT1) is a highly abundant lncRNA which was originally identified as a structural component of nuclear paraspeckles (Clemson et al., 2009; Sasaki et al., 2009). NEAT1 has since been indicated as a regulator of tumorigenesis by transfection or carcinogen studies and/or correlative expression analyses (Adriaens et al., 2016; Chakravarty et al., 2014; Li et al., 2017b; Mello et al., 2017); however, the molecular mechanisms by which NEAT1 contributes to glucose metabolism in cancer progression are not yet understood, and in vivo evaluation of NEAT1 function has been limited. Notably, two isoforms of NEAT1, short NEAT1_1 (~3.7 kb) and long NEAT1_2 (~23 kb), are produced from the same locus by alternative 3′ processing (Naganuma et al., 2012). While NEAT1_2 is essential for the formation of paraspeckle nuclear bodies, NEAT1_1 localizes prominently outside of paraspeckles and is dispensable for paraspeckle formation (Adriaens et al., 2019; Isobe et al., 2020; Li et al., 2017a; Yamazaki et al., 2018), implicating NEAT1_1 in paraspeckle-independent mechanisms. However, the precise physiological function of NEAT1_1 in tumorigenesis remains largely unknown.
In this study, we describe the impact of NEAT1 in promoting a glycolytic state in breast cancer that is associated with more invasive and higher-grade tumors. Specifically, NEAT1 provides a scaffold structure for PGK1/PGAM1/ENO1 glycolytic multienzyme complexes which contribute to increased flux through the penultimate step in glycolysis. By genetically forcing isoform switching, we also uncover an isoform-specific, paraspeckle-independent function for NEAT1 in tumorigenesis.
RESULTS
Ablation of Neat1 in mouse mammary tumors attenuates aggressive breast cancer progression and metastasis
To examine the role of NEAT1 in breast cancer development in vivo, we investigated the consequences of loss of Neat1 in an MMTV-PyVT (polyoma virus middle T antigen) transgenic mouse model for human luminal breast cancer. Genetic ablation of Neat1 profoundly delayed onset of PyVT-driven spontaneous mammary tumors (Figure 1A and Figure S1A). By 130 days of age, PyVT;Neat1+/+ mice had developed medium-sized to large mammary tumors (median weight, 1.57 g), whereas tumor numbers and weight were markedly reduced in PyVT;Neat1−/− mice (0.18 g) (Figure 1B and Figure S1B). Histological analysis by H&E staining revealed severe malignant progression in PyVT;Neat1+/+ mice, as indicated by the formation of poorly differentiated and aggressive adenocarcinomas (Figure 1C). In contrast, the mammary glands of age-matched PyVT;Neat1−/− mice displayed hyperplasia-like, non-malignant features. Neat1 ablation also diminished intra-tumoral vascularization in PyVT mammary tumors (Figure 1D), and the expression of genes involved in either neovascularization, such as Angpt1 and Kdr (or Flk1/Vegfr2), or its reflected endothelial cell numbers, such as Pecam1 (or Cd31) and Tie1, were significantly reduced in PyVT;Neat1−/− compared to PyVT;Neat1+/+ mammary tumors (Figure S1C). Furthermore, multiple tumor nodules that had metastasized to the lung surface were observed in PyVT;Neat1+/+ mice, but none were found in PyVT;Neat1−/− mice (Figure 1E). Complete loss of Neat1 resulted in decreased ductal elongation and branching morphogenesis in virgin mice (Figure S1D), which is consistent with Neat1 being essential for mammary gland development (Standaert et al., 2014). It is noteworthy, however, that although mice with heterozygous Neat1 deletion exhibited normal mammary gland development, PyVT;Neat1+/− mice showed delayed tumor onset and reduced tumor growth and metastasis, indicating that Neat1 is haplo-insufficient with respect to mammary tumorigenesis and cancer progression.
Figure 1. Neat1 knockout or overexpression represses and promotes, respectively, aggressive breast tumor formation.

(A) Tumor-free survival (TFS) analysis of PyVT;Neat1+/+ (n = 42, median TFS 70 days), PyVT;Neat1+/− (n = 18, median TFS 89 days) and PyVT;Neat1−/− (n = 10, median TFS 103 days) mice.
(B) Mammary tumor weight isolated from PyVT;Neat1+/+ (n = 20), PyVT;Neat1+/− (n = 9) and PyVT;Neat1−/− (n = 8) mice was quantified (left). Representative images of mammary tumors isolated from mice are shown in right.
(C) H&E and anti-Ki-67 stained sections of mammary tumors isolated from PyVT;Neat1+/+, PyVT;Neat1+/− and PyVT;Neat1−/− mice. Scale bars, 75 μm.
(D) Sections of mammary tumors isolated from 130 days old PyVT;Neat1+/+ (n = 13) and PyVT;Neat1−/− (n = 4) mice stained with anti-CD31 (left) and quantification of intra-tumoral vessel numbers (right). Scale bars, 75 μm.
(E) H&E and anti-PyV T antigen (Ag) stained sections of lungs isolated from 130 days old PyVT;Neat1+/+, PyVT;Neat1+/− and PyVT;Neat1−/− mice (left). The number of metastatic sites in lungs of 130 days old PyVT;Neat1+/+ (n = 18), PyVT;Neat1+/− (n =9) and PyVT;Neat1−/− (n = 10) mice was quantified (right). Arrowheads indicate clusters of metastatic cells in the lung. Scale bars, 75 μm.
(F) Mammary tumor volumes of syngeneic C57BL/6 mice orthotopically implanted with Neat1OE PyVT cells were measured at different day. n = 9.
(G) Representative images of mammary tumors isolated from (F) (top) and quantification of tumor weight (bottom). n = 8.
(H) Sections of mammary tumors isolated from (F) stained with anti-Ki-67 (left) and quantification of the proportion of tumor cells positive for Ki-67 (right). Scale bars, 75 μm. n = 6.
Error bars represent +/− SD. p value was determined by Log-rank (Mantel-Cox) test (A), ANOVA with Tukey’s multiple comparisons test (B and E) or Student’s t test (D, F, G and H) (n.s., non-significant; *p<0.05; **p<0.01; ***p<0.001). See also Figure S1.
We further investigated the pathological role of NEAT1 in breast cancer by using a NEAT1 overexpression mouse model. To selectively activate transcription of the Neat1 gene, we employed a CRISPR-mediated activation (CRISPRa) system. Upregulation of total Neat1 (Neat1_1 and Neat1_2) by CRISPRa in the PyVT mammary tumor-derived cell line Py8119 (hereafter referred to as Neat1OE) increased rates of cellular proliferation and invasion (Figures S1E–S1H). When these Neat1OE cells were implanted into the mammary fat pads of syngeneic C57BL/6 mice, mammary tumor growth (as determined by tumor volume, weight and Ki-67 positivity) was significantly induced compared to mice bearing control cells (Figures 1F–1H).
Our Neat1 whole-body knockout mouse model raises the possibility of non-cell autonomous effects, especially considering NEAT1 is a possible mediator of cytokine production (Zhang et al., 2019). However, Neat1 knockout or upregulation in PyVT mice did not alter expression of cytokines in mammary tumors or fat tissues close to malignant lesions (Figures S1I and S1J). Moreover, when Py8119 cells were orthotopically implanted into syngeneic Neat1+/+, Neat1+/− and Neat1−/− mice, no significant differences were observed in tumor growth rates (Figure S1K), suggesting that NEAT1 can induce tumorigenicity of breast cancer by cell-intrinsic mechanism(s). The significant difference in the aggressiveness of tumor formation between control and Neat1OE cells in Rag1-deficient mice, which lack mature T and B lymphocytes, supports our conclusion that NEAT1 is very likely to increase intrinsic cell proliferation (Figure S1L). NEAT1 was shown to protect preneoplastic cells from increased DNA damage during chemical carcinogen-induced mouse skin tumorigenesis (Adriaens et al., 2016). However, in our PyVT mouse breast cancer model, DNA damage response signaling was not a major contributing factor to Neat1-dependent breast tumorigenesis; if anything, Neat1 ablation slightly decreased DNA damage in PyVT tumors, as determined by phosphorylation of ATM/ATR substrates such as H2AX, p53, KAP1 and CHK1 (Figures S1M–S1O). Taken together, these results suggest that NEAT1 plays a role in promoting the progression of breast tumors in vivo.
NEAT1 modulates glycolytic metabolism in breast cancer
Metabolic reprogramming toward aerobic glycolysis (or the Warburg effect) accompanies tumorigenesis (Hanahan and Weinberg, 2011; Vander Heiden et al., 2009). We therefore sought to determine a potential role for NEAT1 in the altered glucose metabolism of breast cancer. RNA interference (RNAi) targeting both NEAT1 isoforms in the highly-glycolytic human breast cancer cell line BT-474 (Timmerman et al., 2013) decreased cellular glucose consumption and lactate production, a metabolic signature consistent with the so-called ‘anti-Warburg effect’; this effect was most likely independent of induced cell-cycle arrest and progression (Figures S2A and S2B). Conversely, upregulation of NEAT1 with overexpression constructs or by CRISPRa-based strategy led to increases in glucose consumption and lactate production (Figure S2C). Using a metabolomics approach based on capillary electrophoresis mass spectrometry (CE-MS) supported by validation of key metabolites, we examined metabolic pathways in mammary tumors of PyVT;Neat1+/+ and PyVT;Neat1−/− mice. Notably, levels of penultimate glycolytic metabolites, including 3-phosphoglycerate (3-PG)/2-phosphoglycerate (2-PG) and phosphoenolpyruvate (PEP), as well as pyruvate and lactate, were significantly lower in mammary tumors of PyVT;Neat1−/− mice relative to those of PyVT;Neat1+/+ mice (Figures 2A–2C and Figure S2D). In sharp contrast, tumoral glucose, glucose-6-phosphate (G-6-P)/fructose-6-phosphate (F-6-P), and glyceraldehyde-3-phosphate (GA-3-P) were markedly heightened by Neat1 loss in PyVT mice. Furthermore, CE-MS metabolomic profiling of mammary tumors of mice orthotopically implanted with Neat1OE cells demonstrated preferential accumulation of 3-PG/2-PG, PEP and pyruvate and dramatic reduction of G-6-P/F-6-P and GA-3-P (Figure 2D). Likewise knockdown of total NEAT1 with siRNA in BT-474 cells reduced cellular levels of 2-PG, PEP, and pyruvate, while increasing levels of G-6-P and GA-3-P, confirming the selective regulation of the penultimate step in glycolysis by NEAT1 (Figure 2E). In contrast, forced expression of 3.7-kb NEAT1_1 in relatively low glycolytic MCF7 and T47D cells (Timmerman et al., 2013) led to acceleration of penultimate glycolysis (Figure 2F). Notably, metabolic profiling of mammary tumors of PyVT;Neat1−/− mice also revealed decreased glutamine and glutamate contents, although Neat1OE tumors did not show the opposite (Figures 2A and S2E). The crosstalk between glycolysis and glutamine metabolism has been demonstrated previously; for instance, glycolysis has been shown to promote glutamine uptake and glutaminolysis in multiple types of cells (Wang et al., 2018; Wellen et al., 2010). However, ablation or upregulation of Neat1 in PyVT mammary tumors showed minimal effects on the expression of the genes encoding glutamine uptake and glutaminolysis enzymes, including ASCT2 (Slc1a5), GLS1 and GLUD1, as well as PCB, a key compensatory anaplerotic enzyme (Figures S2F–S2H).
Figure 2. NEAT1 controls the penultimate step of glycolysis in breast tumors.

(A) Representative heat map of metabolome profiles in PyVT;Neat1+/+ and PyVT;Neat1−/− mice was analyzed by hierarchical clustering analysis. Heat map colors represent relative metabolite levels determined by CE-MS as indicated in the color key. G-6-P, glucose-6-phosphate; F-6-P, fructose-6-phosphate; 3-PG, 3-phosphoglyceric acid; 2-PG, 2-phosphoglyceric acid; PEP, phosphoenolpyruvic acid; Gro-3-P, glycerol-3-phosphate. n = 3.
(B) Average absolute concentrations (pmol) of glycolytic metabolites, as indicated, per 106 cells isolated from PyVT;Neat1+/+ and PyVT;Neat1−/− mammary tumors were measured by metabolite assays. GA-3-P, glyceraldehyde-3-phosphate. n = 3~6.
(C) A simplified representation depicting the glycolytic pathway in NEAT1-deficient tumors. High and low levels of metabolites in PyVT;Neat1−/− tumors are shown as red and green color, respectively. HK, hexokinse; GPI, G-6-P isomerase; PFK, phosphofructokinase; ALDO, aldolase; PK, pyruvate kinase.
(D) The levels of glycolytic metabolites (nmol), as indicated, per 1 mg of protein of mammary tumors isolated from syngeneic C57BL/6 mice orthotopically implanted with Neat1OE PyVT cells were determined by CE-MS. n = 7.
(E, F) Intracellular concentrations (nmol) of glycolytic metabolites, as indicated, per 1 mg of protein of BT-474 cells expressing NEAT1 siRNA targeting both the NEAT1_1 and NEAT1_2 isoforms together (E) and MCF7 cells overexpressing NEAT1_1 (F) were measured by CE-MS based and colorimetric assays. n = 3~4.
(G) Total RNAs from paired normal and breast cancer tissue samples from patients were subjected to RT-qPCR for total NEAT1 and NEAT1_2. n = 20. BRCA, breast carcinoma.
(H) Intracellular concentrations (nmol) of glycolytic metabolites, as indicated, per 1 mg of protein of (G) were measured by CE-MS based and colorimetric assays. n = 16.
(I) Correlation curves showing the positive relationship between NEAT1 and 2-PG, PEP and pyruvate, but not GA-3-P, levels. n = 16. The correlation coefficients were calculated by the Pearson’s Chi-Square test.
Error bars represent +/− SD. p value was determined by Student’s t test (n.s., non-significant; *p<0.05; **p<0.01; ***p<0.001). See also Figure S2.
To extend our observations to clinicopathologically relevant settings, we collected tumor samples paired with adjacent normal mammary tissues from breast cancer patients, and performed RT-qPCR to analyze total NEAT1 (NEAT1_1 and NEAT1_2) and NEAT1_2 expressions along with metabolism assays to detect glycolytic metabolites. The results showed significant upregulation of total NEAT1 (but not NEAT1_2) and high production rates of 2-PG, PEP and pyruvate, but not GA-3-P, in human breast cancer samples relative to normal tissues (Figures 2G and 2H). Importantly, when the levels of metabolites from the penultimate step of glycolysis were plotted against levels of NEAT1, significant positive correlations were seen between NEAT1 expression and 2-PG, PEP and pyruvate levels, but not GA-3-P levels (Figure 2I). NEAT1 has been shown to regulate genes at the transcriptional and post-transcriptional levels (Li et al., 2017b; West et al., 2014). However, Neat1 deletion or overexpression did not lead to marked alterations in penultimate glycolytic gene expression in PyVT mice (Figures S2I and S2J). Likewise, in our study population and publicly available datasets, high NEAT1 expression in breast cancer patients did not correlate with PGK1, PGAM1 or ENO1 expression levels (Figures S2K–S2M). Collectively, these data suggest that NEAT1 promotes metabolic reprogramming in breast cancer with acceleration of the penultimate step of glycolysis.
NEAT1 directly binds the penultimate enzymes of glycolysis
LncRNAs can act as biological signal transducers through specific interactions with signaling effector proteins (Wang and Chang, 2011). We therefore decided to explore the possible binding capability of NEAT1 to the enzymes involved in the penultimate step of glycolysis. To our surprise, we found that NEAT1, but not its adjacent lncRNA NEAT2 (also known as MALAT1) (Hutchinson et al., 2007), was highly enriched in the immunoprecipitates of endogenous PGK1, PGAM1 and ENO1 (Figure 3A and Figure S3A). This data was corroborated by the physical interactions between in vitro-transcribed NEAT1_1 and PGK1, PGAM1 and ENO1 recombinant proteins (Figure S3B). To further confirm the interactions of NEAT1 with PGK1, PGAM1 and ENO1, we developed a fluorescence polarization assay. For the fluorescein-labeled NEAT1_1, the dissociation constants (Kd) of 9.7 ± 3.5 nM, 19.6 ± 2.3 nM and 25.8 ± 2.5 nM were determined for the recombinant proteins PGK1, PGAM1 and ENO1, respectively (Figure S3C), suggesting that NEAT1 may exhibit preferential binding for three penultimate glycolytic enzymes: PGK1>PGAM1>ENO1. In agreement with our observations, a recent proteomic analysis with capture hybridization analysis of RNA targets (CHART)–enriched materials of NEAT1 in human breast cancer identified PGK1, PGAM5 and ENO1 as putative NEAT1-interacting proteins (West et al., 2014). It is worth noting that PGK1, PGAM1 and ENO1 have been described as RNA-binding proteins (RBPs) (Castello et al., 2012; Hernandez-Perez et al., 2011; Ho et al., 2010), and do not significantly regulate the stability of NEAT1 (Figure S3D).
Figure 3. NEAT1 interacts with PGK1, PGAM1 and ENO1.

(A) Lysates from MCF7 cells were subjected to NEAT1 RNA immunoprecipitation (RIP) with IgG and anti-PGK1, –PGAM1, –ENO1 or –GAPDH.
(B) Lysates from MCF7 cells were subjected to CLIP-qPCR for NEAT1_1 segments, as shown in top diagram, with IgG and anti-PGK1, –PGAM1 or –ENO1. A secondary structure model of NEAT1 (Lin et al., 2018) in bottom right. The four structural domains are highlighted with different colors, and the NEAT1_1 sequence motifs that are recognized by PGK1, PGAM1 and ENO1 are drawn as dashed lines.
(C, F) Generation of MCF7 cell lines lacking the NEAT1 sequence motifs that are recognized by PGAM1 and ENO1 (Δ1033~2115 bp; Δ1~2.1k) (C) or PGK1 (Δ2116~2805 bp; Δ2.1~2.8k) (F) using CRISPR/Cas9 technology with a series of sgRNAs (left). Lysates from wild-type (WT) and Δ1~2.1k or Δ2.1~2.8k cells were subjected to NEAT1 RIP with IgG and anti-PGK1, –PGAM1 or –ENO1 (right). n = 3~4.
(D, G) Glucose consumption (left) and lactate production (right) by WT and Δ1~2.1k (D) or Δ2.1~2.8k (G) cells were determined by enzymatic assays. n = 3.
(E, H) Intracellular concentrations (nmol) of glycolytic metabolites, as indicated, per 1 mg of protein of WT and Δ1~2.1k (E) or Δ2.1~2.8k (H) cells were measured by CE-MS based and colorimetric assays. n = 3~4.
(I, L, O) Lysates from MCF7 cells expressing PGK1 (I), PGAM1 (L) or ENO1 (O) shRNA together with exogenous WT or K/R-to-A mutants, as indicated, PGK1, PGAM1 or ENO1 from an ORF transcript lacking the 3’-UTR sequences targeted by shRNA were subjected to RIP-qPCR for NEAT1 with IgG and anti-PGK1, –PGAM1 or –ENO1 (left). Structure-based prediction of NEAT1 and PGK1, PGAM1 or ENO1 interaction (right). Basic residues are displayed in blue and acidic residues are shown in red. The PGK1, PGAM1 and ENO1 protein structures were visualized with Swiss-PdbViewer (PDBID:3C39, PDBID:4GPI and PDBID:3B97, respectively).
(J, M, P) Glucose consumption (μmol/106 cells) of (top) and lactate production (μM) (bottom) by MCF7 cells expressing PGK1 (J), PGAM1 (M) or ENO1 (P) shRNA together with exogenous WT or K/R-to-A mutants, as indicated, PGK1, PGAM1 or ENO1 were determined by enzymatic assays. n = 3~4.
(K, N, Q) Intracellular concentrations (nmol) of glycolytic metabolites, as indicated, per 1 mg of protein of MCF7 cells expressing PGK1 (K), PGAM1 (N) or ENO1 (Q) shRNA together with exogenous WT or K/R-to-A mutants, as indicated, PGK1, PGAM1 or ENO1 were determined by colorimetric assays. n = 3.
Error bars represent +/− SD. p value was determined by Student’s t test (n.s., non-significant; *p<0.05; **p<0.01; ***p<0.001). See also Figure S3.
To map the NEAT1 sequence motifs responsible for PGK1, PGAM1, and ENO1 bindings, we performed a cross-linking immunoprecipitation–qPCR (CLIP-qPCR) analysis. The short motifs of 3.7-kb NEAT1_1 bound by PGK1, PGAM1 and ENO1 were found to encompass 2764~2827 bp, 1183~1302 bp, and 1361~1539 bp (Figure 3B), suggesting that NEAT1 binds to PGK1, PGAM1 and ENO1 via three distinct regions. Deletions of the motif sequences 1033~2115 bp and 2116~2805 bp of NEAT1 using CRISPR/Cas9 technology with different sgRNAs (hereafter referred to as Δ1~2.1k and Δ2.1~2.8k, respectively) (Figures S3E and S3F) impeded its interaction with PGAM1/ENO1 and PGK1, respectively (Figures 3C and 3F). Furthermore, Δ1~2.1k and Δ2.1~2.8k in MCF7 cells decreased rates of glucose consumption and lactate production (Figures 3D and 3G), likely due to marked reductions of cellular PEP/pyruvate (with aberrant accumulation of GA-3-P/2-PG) and 2-PG/PEP/pyruvate (with aberrant GA-3-P accumulation), respectively (Figures 3E and 3H); these perturbations were associated with diminished cellular growth and invasion potentials (Figures S3E and S3F).
Next, we assessed the functional significance of the protein domains of PGK1, PGAM1 and ENO1 that bind to NEAT1. Predictive models and analysis using BindUP (Paz et al., 2016) and RBPpred (Zhang et al., 2010) have indicated that human PGK1 (PDBID:3C39), PGAM1 (PDBID:4GPI) and ENO1 (PDBID:3B97) respectively harbor two, one and four positive-electrostatic patches that may be involved in protein-RNA interaction. Indeed, the lysine (K)/arginine (R) sites, including R39, K141, R171 and R192 of PGK1, K5, R21 and R117 of PGAM1, and K5, K193 and R400 of ENO1, were found to be important for mediating NEAT1 binding (Figures 3I, 3L and 3O and Figure S3G); notably, the candidate K/R residues proximal to the catalytic sites of the three enzymes had been excluded to avoid competition between their substrates or co-factors and NEAT1. Given that human ENO1 can form a homodimer facing each other in an antiparallel fashion (Diaz-Ramos et al., 2012) (Figure S3H), it is conceivable that NEAT1 can also bind to the dimeric forms of ENO1 (Figure S3I). Critically, rates of glucose consumption and lactate production were markedly decreased by reconstitution with exogenous R39A, K141A, R171A and R192A mutant PGK1, K5A, R21A and R117A mutant PGAM1, and K5A, K193A and R400A mutant ENO1 from PGK1, PGAM1, and ENO1 ORF transcripts lacking the 3’-UTR sequences targeted by respective shRNAs (Figures 3J, 3M and 3P), and these effects were likely associated with slower 1,3-BPG→3-PG→2-PG→PEP conversion (Figures 3K, 3N and 3Q).
NEAT1 facilitates assembly of PGK1/PGAM1/ENO1 glycolytic complexes
Mounting evidence supports the idea that lncRNAs serve as central platforms upon which relevant molecular components can be assembled (Wang and Chang, 2011), and this feature may be fundamental to the precise control of the specificity and dynamics of intermolecular interactions and biological signaling processes. We therefore hypothesized that NEAT1 may serve as a molecular scaffold that dynamically modulates the penultimate step of glycolysis in cancer. To test this, we performed in vitro pull-down assays with PGK1, PGAM1 and ENO1 recombinant proteins in the presence of in vitro-transcribed 3.7-kb NEAT1_1 transcripts. Remarkably, NEAT1 sense transcripts induced complex formation with PGK1, PGAM1 and ENO1 in a dosage-dependent manner, while these components did not assemble in the presence of NEAT1 anti-sense transcripts (Figure 4A). Endogenous PGK1/PGAM1/ENO1 complexes were clearly purified from PyVT;Neat1+/+ mammary tumors (Figure 4B); however, the formation of these complexes was markedly attenuated by Neat1 ablation. Conversely, mammary tumors of mice bearing Neat1OE cells exhibited increased PGK1/PGAM1/ENO1 association (Figure 4C). Notably, the COOH-terminal domains of PGK1 (residues 101~417), PGAM1 (139~254), and ENO1 (135~434) were found to be essential for NEAT1-centered PGK1/PGAM1/ENO1 complex formation (Figure 4D). Although NEAT1-PGK1/PGAM1/ENO1 binding was likely independent of induced cell-cycle arrest and progression (Figure S4A), it was markedly reduced by Δ1~2.1k and Δ2.1~2.8k (Figures 4E and 4F). We further explored whether the NEAT1 scaffolding mechanism was directed specifically towards PGK1/PGAM1/ENO1. Co-immunoprecipitation analyses revealed that NEAT1-dependent association of metabolic enzymes into complexes does not occur in other steps of glycolysis catalyzed by hexokinase, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), aldolase or GAPDH (Figures S4B and S4C), suggesting that NEAT1 provides a scaffold structure specifically for PGK1/PGAM1/ENO1 in the glycolytic pathway (Figure 4G).
Figure 4. NEAT1 bridges PGK1/PGAM1/ENO1 complexes for high glycolysis.

(A) Recombinant PGK1, PGAM1 and ENO1 proteins incubated with in vitro-transcribed NEAT1_1 sense or anti-sense transcript (left) were immunoprecipitated (IP) with anti-PGK1 then immunoblotted (right). * and # indicate the heavy chain and light chain, respectively, of IgG. $ indicates non-specific band.
(B) Lysates from PyVT;Neat1+/+ and PyVT;Neat1−/− mammary tumors were IP with anti-PGK1 then immunoblotted as indicated.
(C) Lysates from mammary tumors of syngeneic C57BL/6 mice orthotopically implanted with Neat1OE PyVT cells were IP with anti-PGK1 then immunoblotted as indicated.
(D) Lysates from MCF7 cells expressing NEAT1_1 together with the GFP-tagged NH2- and COOH–terminal domains, as indicated, of PGK1 (left), PGAM1 (middle) or ENO1 (right) were IP with anti-GFP then immunoblotted as indicated. * indicates the heavy chain of IgG.
(E, F) Lysates from Δ1~2.1k (E) or Δ2.1~2.8k (F) MCF7 cells were IP with anti-PGK1 then immunoblotted as indicated. * indicates the heavy chain of IgG.
(G) A proposed model for the role of NEAT1 to constitute a scaffold bridge in the assembly of the PGK1/PGAM1/ENO1 multienzyme complexes for high glycolysis in breast cancer (created with BioRender.com).
(H–J) Lysates from MCF7 cells expressing PGK1 (H), PGAM1 (I) or ENO1 (J) shRNA together with exogenous WT or K/R-to-A mutants, as indicated, PGK1, PGAM1 or ENO1 from an ORF transcript lacking the 3’-UTR sequences targeted by shRNA (left) were IP with IgG (middle) and anti-PGK1, –PGAM1 or –ENO1 (right) then immunoblotted as indicated. * and # indicate the heavy chain and light chain, respectively, of IgG.
See also Figure S4.
Furthermore, various K/R-to-A mutants PGK1 (R39A, K141A, R171A and R192A), PGAM1 (R21A and R117A), and ENO1 (K5A, K193A and R400A), which had failed to interact with NEAT1, dramatically attenuated the formation of PGK1/PGAM1/ENO1 complexes (Figures 4H–4J). Notably, we found no significant difference in levels of NEAT1-PGK1/PGAM1/ENO1 association after supplementation with ubiquitin/MG132, sodium acetate or sodium succinate (Figures S4D and S4E), suggesting that protein modifications, including ubiquitination, acetylation or succinylation, on K sites of PGK1, PGAM1 and ENO1 (Hitosugi and Chen, 2014) are unlikely to operate or restrict this association. Taken together, these results suggest that NEAT1 forms a scaffold bridge for the assembly of PGK1/PGAM1/ENO1 complexes, and this multienzyme complex can accelerate processing of glycolytic intermediates.
Blockade of penultimate glycolysis impedes NEAT1-driven tumorigenesis
To determine whether glycolysis impacts NEAT1-dependent cancer cell growth, we cultured Neat1OE cells in a glucose-depleted medium. Glucose starvation resulted in a marked reduction in the proliferation ability of Neat1OE cells compared to that of control cells (Figure S5A); however, glutamine deprivation retarded the proliferation of both cells equally. Genetic blockade of glycolysis by RNAi for Pgk1, Pgam1 and Eno1 dramatically impeded increased growth of Neat1OE cells (Figure 5A). Likewise knockdown of PGK1, PGAM1 and ENO1 notably reversed the Warburg phenotypes observed in NEAT1-overexpressing T47D cells (Figures S5B and S5C), and attenuated NEAT1-induced promotion of cell proliferation and invasion (Figures S5D and S5E). Furthermore, in PyVT and T47D cells overexpressing NEAT1, pharmacological blockade of glycolysis with the PGK1 inhibitor NG52 (Wang et al., 2020), the PGAM1 inhibitor PGMI-004A (Hitosugi et al., 2012), and the ENO1 inhibitor POMHEX (Lin et al., 2020) [but not the GAPDH inhibitor koningic acid (Liberti et al., 2017)] resulted in far greater reductions in cell growth rates than was observed in control cells (Figure 5B and Figure S5F). Conversely, the anti-cancer effects of these agents were diminished in NEAT1-silenced cells (Figure S5G).
Figure 5. Blockade of penultimate glycolysis reduces NEAT1-dependent tumorigenesis.

(A) Growth curves of Neat1OE PyVT cells expressing Gapdh, Pgk1, Pgam1 or Eno1 siRNAs. n = 3. The inset indicates the RNAi mediated knockdown efficiency for the indicated genes.
(B) Growth curves of Neat1OE PyVT cells treated with the GAPDH inhibitor koningic acid (KA) (1 μM), the PGK1 inhibitor NG52 (10 μM), the PGAM1 inhibitor PGMI-004A (20 μM) or the ENO1 inhibitor POMHEX (1 μM). n = 3.
(C) Tumor-free survival (TFS) analysis of PyVT;Neat1+/+ and PyVT;Neat1−/− mice treated with vehicle (n = 21 for PyVT;Neat1+/+ mice, median TFS 70 days; n = 7 for PyVT;Neat1−/− mice, median TFS 111 days) or POMHEX (n = 9 for PyVT;Neat1+/+ mice, median TFS 96 days; n = 5 for PyVT;Neat1−/− mice, median TFS 112 days). TFS curves of vehicle-treated PyVT;Neat1+/− mice (n = 9, median TFS 91 days) are also shown.
(D) The weight of mammary tumor isolated from 130 days old vehicle-treated PyVT;Neat1+/+ (n = 15), POMHEX-treated PyVT;Neat1+/+ (n = 9) and vehicle-treated PyVT;Neat1+/− (n = 7) mice were quantified (left). Representative images of mammary tumors isolated from mice are shown in right.
(E) H&E and anti-Ki-67 stained sections of mammary tumors isolated from vehicle-treated PyVT;Neat1+/+, POMHEX-treated PyVT;Neat1+/+ and vehicle-treated PyVT;Neat1+/− mice. Scale bars, 75 μm.
(F) H&E and anti-PyVT Ag stained sections of lungs isolated from 130 days old vehicle-treated PyVT;Neat1+/+ (n = 11), POMHEX-treated PyVT;Neat1+/+ (n = 5) and vehicle-treated PyVT;Neat1+/− (n = 7) mice (left). Arrowheads indicate clusters of metastatic cells in the lung. The number of metastatic sites in lungs of mice was also quantified (right). Scale bars, 75 μm.
(G, H) Growth curves (G) and cell invasion assays (H) of BT-474 cells expressing NEAT1 siRNA targeting both NEAT1 isoforms treated with increasing concentrations (μg/ml), as indicated, of phosphoenolpyruvate (PEP). n = 4.
(I) TFS analysis of PyVT;Neat1+/+ and PyVT;Neat1+/− mice treated with vehicle (n = 20 for PyVT;Neat1+/+ mice, median TFS 87 days; n = 20 for PyVT;Neat1+/− mice, median TFS 99 days) or pyruvate (n = 12 for PyVT;Neat1+/+ mice, median TFS 80 days; n = 13 for PyVT;Neat1+/− mice, median TFS 82 days).
Error bars represent +/− SD. p value was determined by ANOVA with Tukey’s multiple comparisons test (A, B, D, F, G and H) or Log-rank (Mantel-Cox) test (C and I) (n.s., non-significant; *p<0.05; **p<0.01; ***p<0.001). See also Figure S5.
Next, to determine in vivo whether penultimate glycolysis serves as a promising therapeutic target in NEAT1-driven breast cancer, we administered intraperitoneally the ENO inhibitor, POMHEX (>98% purity) (Figure S5H), into PyVT;Neat1+/+ and PyVT;Neat1−/− mice; the in vivo specificity of POMHEX was defined as impaired conversion of 2-PG to PEP in PyVT mouse mammary tumors (Figure S5I). Critically, POMHEX treatment provided a significant survival benefit for PyVT;Neat1+/+ mice (median tumor onset at 96 days vs. 70 days for vehicle-treated mice, p = 0.0005) and delayed median tumor onset to a degree similar to that found in PyVT;Neat1+/− mice (91 days) (Figure 5C). In contrast, complete Neat1 loss abrogated the sensitivity of PyVT mice to POMHEX (113 days vs. 112 days for PyVT;Neat1−/− mice), supporting the existence of a link between NEAT1-positive breast cancer and vulnerability to inhibition of penultimate glycolysis. We also found that PyVT mammary tumor volume, weight, and histological aggressiveness were substantially reduced by treatment with POMHEX (Figures 5D and 5E). In contrast to multiple tumor nodules observed on the lung surface in vehicle-treated PyVT;Neat1+/+ mice at 130 days of age, no nodules were observed in age-matched POMHEX-treated PyVT;Neat1−/− mice (Figure 5F), indicating the robust antitumor effects of POMHEX in this PyVT mouse model of breast cancer. Given three types of ENO isoenzymes in mammals – ENO1 (or α-enolase), which is present in almost all mature tissues, neuronal ENO2 (γ-enolase), and muscle specific ENO3 (β-enolase), it is conceivable that PyVT mammary tumors, which show high levels of ENO1 in comparison to ENO2 and ENO3 (Figure S5J), are highly dependent on ENO1 to drive glycolysis and aggressive behavior, rendering these tumors exquisitely sensitive to POMHEX. Consistent with our observations, ENO1-deleted glioma has shown exceptional sensitivity to inhibition of neuronal ENO2 by POMHEX (Lin et al., 2020) in a therapeutic strategy recognized as ‘collateral lethality’ (Muller et al., 2012).
Finally, we sought to determine whether the impairment of breast tumorigenesis elicited by NEAT1-loss results from defective glycolysis. PEP, the penultimate product of glycolysis, markedly rescued the diminished rates of cellular proliferation and invasion in total NEAT1-silenced breast cancer cells when administered at increasing concentrations (Figures 5G and 5H). Most importantly, supplementation of pyruvate, the end product of glycolysis, significantly restored the delayed onset and slower growth of mammary tumors observed in PyVT;Neat1+/− mice (median tumor onset at 81 days vs. 104 days for vehicle-treated mice, p = 0.005) to a level similar to those observed in PyVT;Neat1+/+ mice (82 days) (Figure 5I). However, pyruvate supplement did not have a significant effect in PyVT;Neat1+/+ mice. Taken together, these results suggest that NEAT1’s regulation of glycolysis is a critical mechanism for tumorigenesis.
Glucose-stimulated nuclear export of NEAT1 is mediated by Pinin
NEAT1 is mainly enriched in the nucleus but also found in the cytoplasm (van Heesch et al., 2014), and intriguingly, it can be released from the nucleus to the cytoplasm during inflammasome activation (Zhang et al., 2019). We reasoned therefore that NEAT1 may be actively translocated to the cytoplasm, where it can effectively assist glycolysis. Indeed, we found that in the human and mouse breast cancer cell lines tested, NEAT1 interacted with PGK1, PGAM1, and ENO1 exclusively in the cytoplasm (Figure 6A and not shown). Under starvation conditions, re-addition of glucose [but not 2-deoxyglucose (2-DG), a nonmetabolizable glucose analog that inhibits the glycolytic pathway], significantly increased the ability of NEAT1 to bind with PGK1, PGAM1 and ENO1 (Figure 6B). Similarly, the formation of NEAT1-centered PGK1/PGAM1/ENO1 complexes in the cytoplasm was augmented upon glucose stimulation (Figure 6C). To further understand how NEAT1 participates in the activation of glycolysis, we decided to determine the presence of NEAT1 in the cytoplasmic fraction of MCF7 cells cultured under high glucose treatment condition. Our RNA fluorescence in situ hybridization (RNA-FISH) analysis and copy number measurement revealed that a small but appreciable proportion of NEAT1 is distributed in the cytoplasm (Figures S6A and S6B). Furthermore, subcellular fractionation experiments showed that glucose, but not 2-DG, greatly stimulates NEAT1 translocation from the nucleus to the cytoplasm in a concentration-dependent manner (Figure 6D). These data suggest that NEAT1 can be exported actively to the cytoplasm to assist in glycolysis.
Figure 6. Glucose-stimulated nuclear export of NEAT1 is Pinin-dependent.
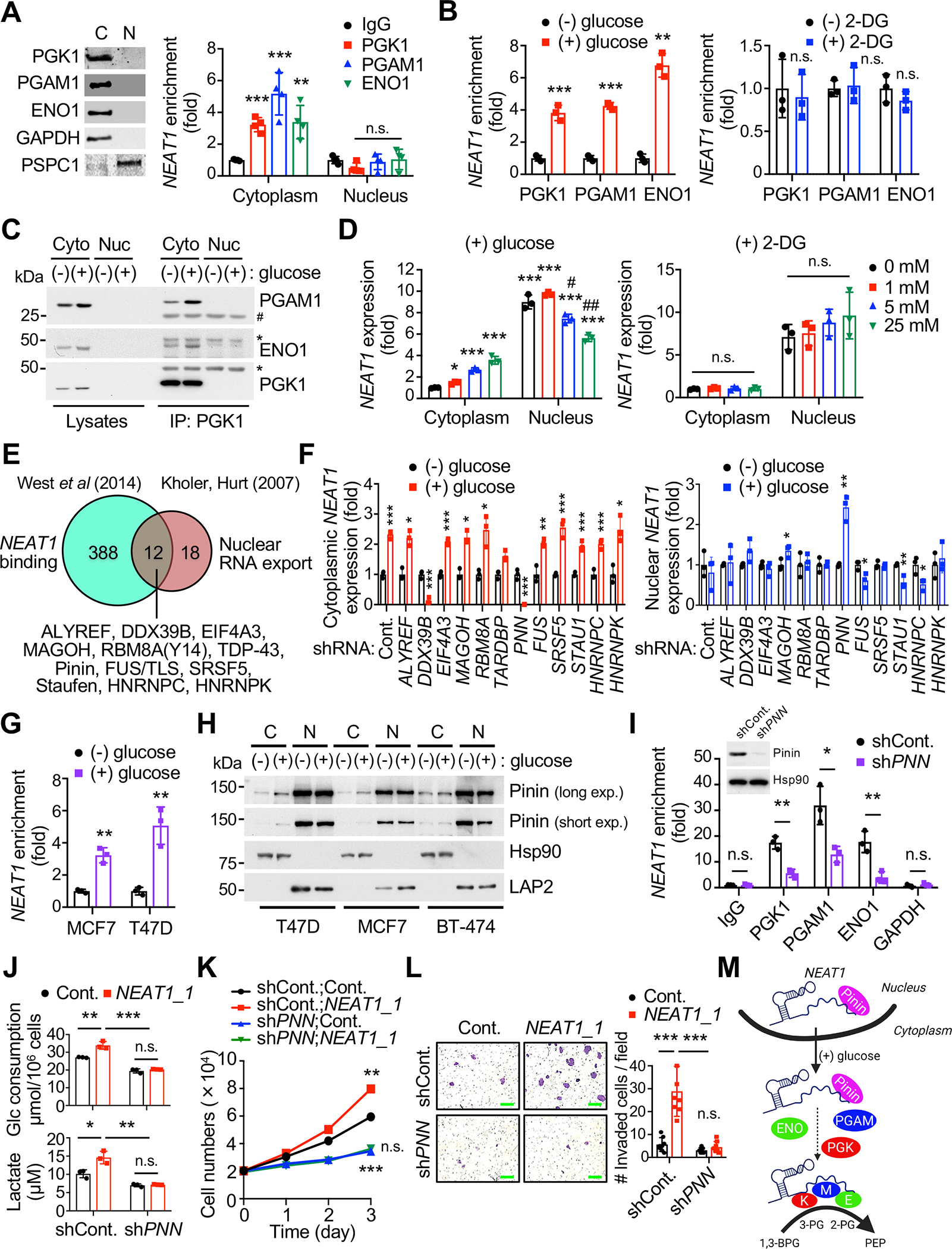
(A) Cytoplasmic (C) and nuclear (N) fractions from MCF7 cells (left) were subjected to NEAT1 RNA immunoprecipitation (RIP) with IgG and anti-PGK1, –PGAM1 or –ENO1 (right). n = 4. GAPDH and PSPC1 serve as controls.
(B) Cytoplasmic fraction from MCF7 cells cultured in glucose-free media for 16 hours [(−) glucose] and stimulated for 2 hours by re-addition of 5 mM glucose [(+) glucose] (left) or 2-deoxyglucose (2-DG) [(+) 2-DG] (right) were subjected to NEAT1 RIP with IgG and anti-PGK1, –PGAM1 or –ENO1. n = 3.
(C) Cytoplasmic and nuclear fractions from MCF7 cells grown in the indicated conditions were IP with anti-PGK1 then immunoblotted as indicated.
(D) Cytoplasmic and nuclear fractions from MCF7 cells cultured in glucose-free media and stimulated by re-addition of different amounts, as indicated, of glucose (left) or 2-DG (right) were subjected to RT-qPCR for NEAT1. n = 3.
(E) Bioinformatic analysis of public datasets for proteins with known roles in nuclear export of various classes of RNAs and proteomics with NEAT1 CHART-MS.
(F) Cytoplasmic (left) and nuclear (right) fractions from MCF7 cells expressing the indicated shRNAs grown in the indicated conditions were subjected to RT-qPCR. n = 3.
(G) Cytoplasmic fraction from MCF7 and T47D cells grown in the indicated conditions were subjected to NEAT1 RIP with IgG and anti-Pinin. n = 3.
(H) Cytoplasmic (C) and nuclear (N) fractions from T47D, MCF7 and BT-474 cells grown in the indicated conditions were immunoblotted as indicated. Hsp90 and LAP2 serve as controls.
(I) Cytoplasmic fraction from T47D cells expressing PNN shRNA grown in the indicated conditions were subjected to NEAT1 RIP with IgG and anti-PGK1, –PGAM1, –ENO1 or –GAPDH. n = 3. The inset indicates the RNAi mediated knockdown efficiency for PNN.
(J) Glucose consumption (top) and lactate production (bottom) by T47D cells expressing PNN shRNA together with NEAT1_1 as indicated were determined by enzymatic assays. n = 3.
(K, L) Growth curves (K) and cell invasion assays (L) of T47D cells expressing PNN shRNA and NEAT1_1 as indicated. n = 4.
(M) A proposed model for the role of Pinin for glucose-stimulated nuclear export of NEAT1 enabling PGK1/PGAM1/ENO1 complex formation (created with BioRender.com).
Error bars represent +/− SD. p value was determined by Student’s t test (A, B, D, F, G and I) or ANOVA with Tukey’s multiple comparisons test (J–L) (n.s., non-significant; *,#p<0.05; **,##p<0.01; ***p<0.001). See also Figure S6.
The recruitment of nuclear export factors to many types of RNAs, including messenger RNAs (mRNAs) and lncRNAs, determines their nuclear retention and export (Chen, 2016; Kohler and Hurt, 2007). Intriguingly, a recent CHART-MS assay has presented evidence for the binding of 12 putative nuclear export factors to NEAT1 (Kohler and Hurt, 2007; West et al., 2014) (Figure 6E). Our RNAi screen of 12 genes whose loss-of-function could impair nuclear export of NEAT1 in T47D cells subsequently identified the nuclear speckle-associated protein Pinin (PNN) (Alpatov et al., 2004). Depletion of Pinin led to the cytoplasmic depletion and aberrant nuclear accumulation of NEAT1 even upon glucose stimulation (Figure 6F), and this phenotype was not associated with changes in NEAT1 expression (Figure S6C). NEAT1 was also found to be greatly enriched in the immunoprecipitates of endogenous Pinin during glycolysis activation (Figure 6G). Next, we examined whether Pinin could be exported to the cytoplasm in response to glucose stimulation, and found glucose stimulation did indeed lead to the translocation of a portion of Pinin to the cytoplasm in a dose-dependent manner (Figure 6H and Figure S6D); however, Pinin was not released to the cytoplasm upon glutamine stimulation (Figure S6E). Consistently, the interactions of NEAT1 with PGK1, PGAM1, and ENO1 were significantly reduced in Pinin-silenced cells compared to control cells (Figure 6I). Knockdown of Pinin reversed the Warburg phenotypes observed in NEAT1_1-overexpressing cells (Figure 6J) and diminished the pro-growth and –invasive effects of NEAT1 (Figures 6K and 6L). Analysis of publicly available datasets (Liu et al., 2014; Tang et al., 2018) also revealed that Pinin-high tumors are significantly associated with poor prognosis in breast cancer patients (Figure S6F), and notably, PNN expression levels were strongly correlated with NEAT1 upregulation in patients across breast tumor intrinsic subtypes (Cancer Genome Atlas, 2012; Curtis et al., 2012) (Figure S6G). Taken together, these results imply a mechanism for glucose-stimulated NEAT1 nuclear export in which the binding of Pinin plays a key role (Figure 6M).
NEAT1 drives glycolysis and cancer progression in an isoform-specific manner
The human NEAT1 gene produces two transcripts, NEAT1_1 (~3.7 kb) and NEAT1_2 (~23 kb); of which only the latter is indispensable for de novo paraspeckle construction (Naganuma et al., 2012; Yamazaki et al., 2018). While NEAT1_1 is polyadenylated, NEAT1_2 lacks a polyadenylation signal (PAS) and instead contains a triple helix at its 3’ terminus (Sunwoo et al., 2009; Wilusz et al., 2012) (Figure 7A). To quantify NEAT1_1 and NEAT1_2 specifically, RNA templates for RT-qPCR analysis were therefore selectively reverse-transcribed using oligo(dT) primers and random hexamer primers, respectively. The resulting data showed that NEAT1_1, but not NEAT1_2, is specifically enriched in the immunoprecipitates of PGK1, PGAM1 and ENO1 (Figure 7A) and efficiently translocated to the cytoplasm in response to glucose stimulation (Figure 7B), suggesting that NEAT1_1 (but not NEAT1_2) transported to the cytoplasm may exert a paraspeckle-independent function in glycolysis.
Figure 7. NEAT1_1 isoform is essential for glycolysis and progression of breast cancer.
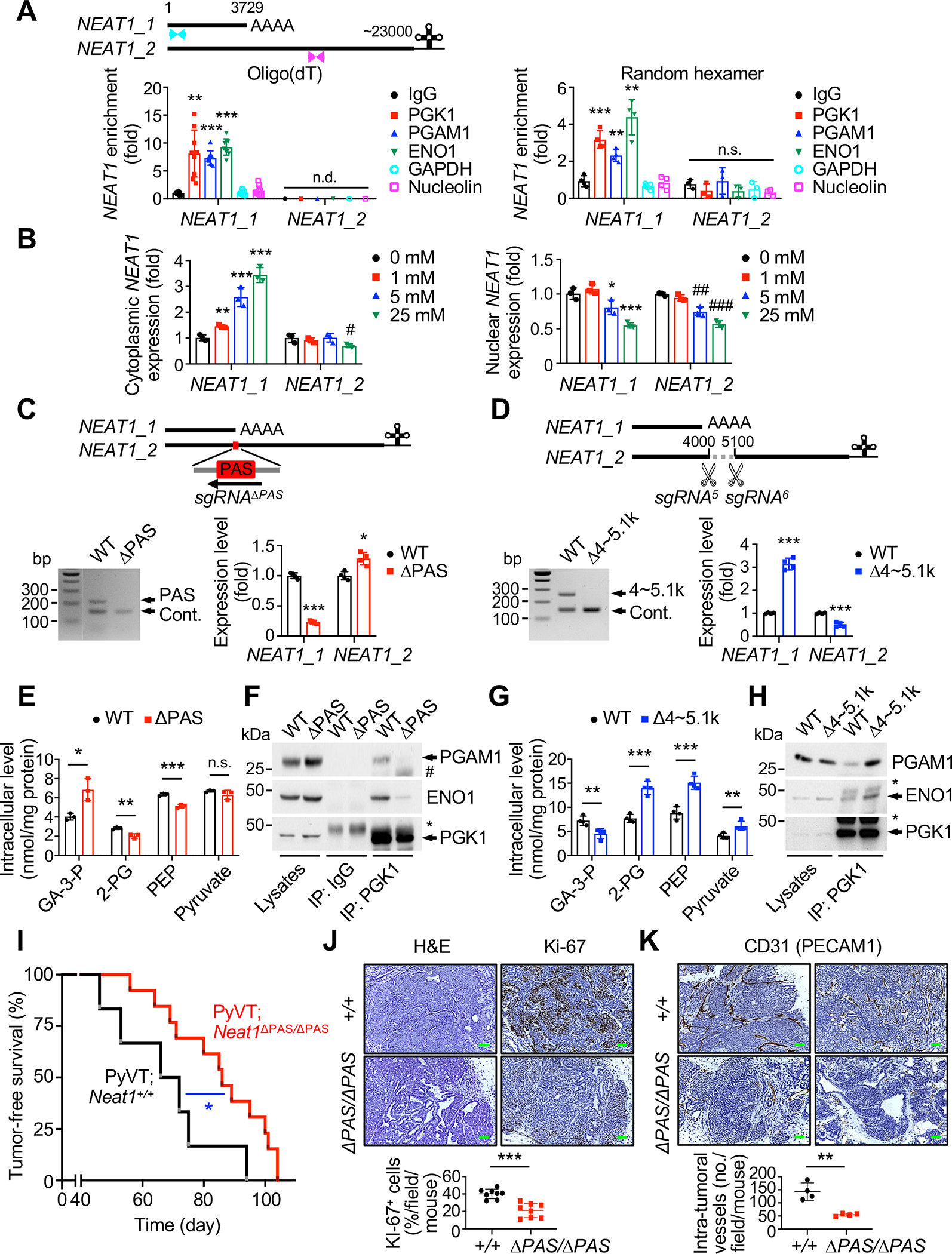
(A) Schematic representation of human NEAT1 isoforms (top). Blue and red arrowheads indicate RT-qPCR primers that specifically detect total NEAT1 and NEAT1_2, respectively. Lysates from MCF7 cells were subjected to NEAT1_1 and NEAT1_2 RNA immunoprecipitation (RIP) with IgG and anti-PGK1, –PGAM1 or –ENO1 and RT-qPCR with oligo(dT) priming (bottom left) or random hexamer priming (bottom right), respectively. Anti-GAPDH and –nucleolin serve as controls. n = 3~7. n.d., not determined.
(B) Cytoplasmic (left) and nuclear (right) fractions from MCF7 cells grown in the indicated conditions were subjected to RT-qPCR for NEAT1_1 and NEAT1_2. n = 3.
(C, D) Generation of MCF7 cell lines lacking the NEAT1 sequence motifs that are essential for NEAT1_1 (Δ3699~3786 bp; ΔPAS) (C) or NEAT1_2 (Δ4054~5116 bp; Δ4~5.1k) (D) using CRISPR/Cas9 technology with a series of sgRNAs (top). The resulting PCR genotyping (bottom left) and expression analysis by RT-qPCR (bottom right) in ΔPAS (C) and Δ4~5.1k (D) cells. n = 4.
(E, G) Intracellular concentrations (nmol) of glycolytic metabolites, as indicated, per 1 mg of protein of wild-type (WT) and ΔPAS (E) or Δ4~5.1k (G) cells were measured by CE-MS based and colorimetric assays. n = 3.
(F, H) Lysates from WT and ΔPAS (F) or Δ4~5.1k (H) cells were immunoprecipitated (IP) with IgG and anti-PGK1 then immunoblotted as indicated. * and # indicate the heavy and light chain of IgG.
(I) Tumor-free survival (TFS) analysis of the PyVT;Neat1+/+ (n = 7, median TFS 67 days) and PyVT;Neat1ΔPAS/ΔPAS (n = 13, median TFS 85 days) mice.
(J) H&E and anti-Ki-67 stained sections of mammary tumors isolated from 134~139 days old PyVT;Neat1+/+ and PyVT;Neat1ΔPAS/ΔPAS mice (top) and quantification of the proportion of tumor cells positive for Ki-67 (bottom). Scale bars, 75 μm. n = 4.
(K) Sections of mammary tumors isolated from 134~139 days old PyVT;Neat1+/+ and PyVT;Neat1ΔPAS/ΔPAS mice stained with anti-CD31 (top) and quantification of intra-tumoral vessel numbers (bottom). Scale bars, 75 μm. n = 4.
Error bars represent +/− SD. p value was determined by ANOVA with Tukey’s multiple comparisons test (A and B), Student’s t test (C–E, G, J and K) or Log-rank (Mantel-Cox) test (I) (n.s., non-significant; *,#p<0.05; **,##p<0.01; ***,###p<0.001). See also Figure S7.
In order to precisely define the isoform-specific roles for NEAT1 in glycolysis and tumorigenesis, we established two MCF7 clonal cell lines that carried: (1) deletion of the PAS essential for the production of NEAT1_1 using CRISPR/Cas9-mediated genome editing (this deletion hereafter referred to as ΔPAS), which strongly reduced NEAT1_1 expression (Figure 7C); and (2) deletion of the motif sequence 4054~5116 bp of NEAT1, which is involved in the alternative 3’-end processing that can facilitate NEAT1_2 synthesis (Yamazaki et al., 2018) (hereafter referred to as Δ4~5.1k). In this Δ4~5.1k clone, the expression levels of NEAT1_2 were significantly lower than in wild-type cells, whereas NEAT1_1 levels were ~3-fold higher (Figure 7D). Critically, the forced isoform switching of NEAT1_1 to NEAT1_2 by PAS deletion led to markedly decreased cellular proliferation and invasion in ΔPAS cells (Figure S7A), concomitant with significant reductions in levels of penultimate glycolysis (Figure 7E) and PGK1/PGAM1/ENO1 association (Figure 7F). In sharp contrast, Δ4~5.1k cells exhibited increased rates of cell proliferation/invasion (Figure S7B), processing of penultimate glycolytic intermediates (Figure 7G), and PGK1/PGAM1/ENO1 complex formation (Figure 7H). Together these data suggest that NEAT1_1 plays a distinct and essential role in promoting growth, invasion and glycolysis of breast cancer cells.
Finally, we sought to determine the physiological contributions of NEAT1_1–specific and paraspeckle-independent function to the pro-tumorigenic and glycolytic phenotypes observed in NEAT1-dependent tumors in vivo. To this end we generated female MMTV-PyVT mice lacking the PAS (Isobe et al., 2020) (hereafter referred to as PyVT;Neat1ΔPAS/ΔPAS) (Figures S7C and S7D), and analyzed mammary tumor development in PyVT;Neat1ΔPAS/ΔPAS mice. Genetic inactivation of Neat1_1 (by deletion of the Neat1 PAS) significantly delayed overall onset of PyVT-induced mammary tumors (median tumor onset 85 days vs. 67 days for PyVT;Neat1+/+ mice, p = 0.035; Figure 7I). Histological analysis further indicated that PyVT;Neat1ΔPAS/ΔPAS mice have a significantly lower rate of developing aggressive mammary tumors, which are poorly differentiated, highly vascularized, and highly glycolytic compared to those in PyVT;Neat1+/+ littermate controls (Figures 7J and 7K and Figure S7E). Notably, these phenotypes were not associated with mammary gland development aberration, because Neat1ΔPAS/ΔPAS mice displayed normal mammary gland development (Figure S7F). Taken together, our results demonstrate, through in vivo isoform switching transgenic strategies, that the NEAT1_1 isoform is a potent inducer of mammary tumor growth and glycolysis (Figure S7G).
DISCUSSION
Our investigation of how long noncoding RNAs (lncRNAs) could regulate cancer development in vivo through metabolic mechanisms has led us to several relevant conclusions. First, we have identified the lncRNA NEAT1-regulated PGK1/PGAM1/ENO1 multienzyme complex as a previously unappreciated ‘metabolon’ functioning at the penultimate step of glycolysis to elevate its speed and efficiency via substrate channeling, which offers new insights into the Warburg effect in cancer cells. Steadily accumulating evidence suggests that assemblies of consecutive enzymes are a cardinal feature of metabolic systems across all domains of life, and substrate channeling occurs in these complexes in several organisms as a means of metabolic regulation (Castellana et al., 2014; Srere, 1987). NEAT1 is a nuclear lncRNA entity, while assembly of PGK1/PGAM1/ENO1 proteins occurs in the cytoplasm. Interestingly, we found that NEAT1 levels are significantly elevated in the cytoplasm of cells in response to high glucose. Thus, the increased proportion of cytosolic NEAT1 in response to glycolysis activation could ultimately result in higher PGK1/PGAM1/ENO1 glycolytic scaffold complex formation. However, it remains to be studied how these limited cytosolic NEAT1 molecules manipulate a cloud of glycolytic enzymes during highly active tumorous glycolysis. This limitation may warrant future development of new methods to accurately determine the stoichiometry between cytosolic NEAT1 and PGK1/PGAM1/ENO1.
Second, we propose a new model for NEAT1 nuclear export in which glucose stimulates Pinin loading onto NEAT1 to favor its translocation to the cytoplasm, which enables NEAT1 to play a key role outside of paraspeckles. Pinin (PNN) was originally identified as a nuclear and desmosome-associated protein (Ouyang and Sugrue, 1992), and has since been found to participate in the regulation of pre-mRNA splicing and export from the nucleus (Alpatov et al., 2004; Murachelli et al., 2012). Although Pinin plays a role during mouse development (Leu et al., 2012), the precise role of Pinin in cancer progression have so far remained unknown. Our results are the first to indicate that Pinin can promote breast cancer cell glycolysis and subsequent growth and invasion, at least in part, by facilitating NEAT1 trafficking. Consequently, the notion that metabolic phenotypes and aggressive tumor behaviors are at least partially dependent on NEAT1 localization implies that other signaling pathways capable of translocating NEAT1 may drive tumor growth and glycolysis.
Third, we demonstrate that NEAT1_1 but not NEAT1_2 is a potent inducer of mammary tumor growth and glycolysis, which to our knowledge provides the first evidence for such isoform-specific and paraspeckle-independent functions. These and other studies show that Neat1_1–specific deficient (Neat1ΔPAS/ΔPAS) mice lack the overt developmental phenotypes observed in Neat1−/− mice (Adriaens et al., 2019; Isobe et al., 2020), suggesting that Neat1_1 may not play an essential role during normal development. Despite previous work showing that Neat1_1 is not required for carcinogen-induced skin cancer formation (Adriaens et al., 2019), we have found that Neat1_1 makes an important contribution to breast tumorigenesis and cancer progression. Thus the extent of NEAT1 isoform-specific functions is variable in different tissues, and may involve distinct pathophysiological features. Notably, NEAT1_1 serves as an estrogen receptor (ER)–inducible target and is most highly expressed in ER+ breast cancers (Chakravarty et al., 2014; Li et al., 2017b), whereas NEAT1_2 expression is associated with human epidermal growth factor receptor 2 (HER2)–enriched breast cancer subtypes (Kukharsky et al., 2020). Unlike NEAT1_2, NEAT1_1 localizes prominently outside of paraspeckles and is dispensable for paraspeckle formation (Adriaens et al., 2019; Isobe et al., 2020; Li et al., 2017a; Yamazaki et al., 2018), implicating NEAT1_1 in paraspeckle-independent functions. Despite the overlapping regions of both isoforms, the essentiality of NEAT1_1 for assembly of PGK1/PGAM1/ENO1 is likely due to its cytoplasmic translocation during glycolysis activation; it is thus possible that NEAT1_2–containing paraspeckles are not disrupted in this setting, which is the release prerequisite for NEAT1_2 (Zhang et al., 2019). While our studies have firmly linked NEAT1_1 loss (or gain) to impairment (or enhancement) of malignant progression and glycolysis of breast cancer in ΔPAS (or Δ4~5.1k), it cannot be excluded that upregulation (or downregulation) of NEAT1_2 and the subsequent hyper- (or hypo–) formation of paraspeckles by forced isoform switching (Isobe et al., 2020; Yamazaki et al., 2018) may oppose (or favor) tumorigenesis and glycolysis. Clearly, it remains a challenge to fully understand the context-dependent roles of NEAT1_1 and NEAT1_2.
Finally, from a clinical perspective, our studies indicate for the first time that high NEAT1 expression is associated with a significant acceleration of glycolytic intermediate processing as a direct consequence of multienzyme complex formation in patients. Moreover, our findings should prove highly relevant to the development of anticancer therapies designed to target penultimate glycolysis, epitomized here by treatment with POMHEX, for NEAT1-dependent breast cancer. Given the recent surge of interest in RNA-based therapies and antisense therapeutics (Winkle et al., 2021), combinatorial targeting of NEAT1 and the glycolytic pathway may represent a unique therapeutic regimen for patients with advanced breast cancer.
Limitations of study
This study defines the impact of the lncRNA NEAT1 in promoting a glycolytic state in breast cancer that is associated with more invasive and higher-grade tumors. Specifically, NEAT1 enables PGK1/PGAM1/ENO1 glycolytic scaffold complexes that optimize glycolysis, and genetic and pharmacologic rescue experiments support this mechanism. Two overlapping NEAT1 isoforms, NEAT1_1 and NEAT1_2, are distinct entities that are differentially regulated and functionally distinct. Our initial observations were mainly made using a mouse model lacking both Neat1_1 and Neat1_2 isoforms. To address isoform-specific role for NEAT1 in tumorigenesis, we employed in vivo isoform switching transgenic strategies. The genetic and functional analyses presented here support a pro-tumorigenic role for NEAT1_1 in a MMTV-PyVT-driven mouse breast cancer model. However, with this approach, we cannot rule out the possibility that the observed phenotype may result from the actions of deregulated NEAT1_2 and paraspeckles. Further explorations of the isoform-specific knockout of NEAT1 will help elucidate the full biological activities of this molecule, although technical challenges still remain for establishing a Neat1_2 specific knockout mouse model.
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Min Sup Song (msong1@mdanderson.org).
Materials availability
Unique reagents generated in this study will be made available upon reasonable request to the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
All the original images have been deposited on Mendeley at http://dx.doi.org/10.17632/nyhjzwtscc.1.
No new code has been generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Models and Subject Details
Mice
Mice were housed in a specific pathogen free (SPF) facility in groups (4~5 mice per cage) on a 12 h light/dark cycle, at room temperature of 22°C and with ad-libitum access to autoclaved water and irradiated standard chow diet (RM3-P, Special Diets Services). The health status of mice was daily monitored and only mice that displayed good general health were used for the in-house breeding colonies. Ages of mice at the time of experiment are indicated in the corresponding figures legends. The Neat1+/− and Neat1−/− (Nakagawa et al., 2011) and Neat1ΔPAS (Isobe et al., 2020) mice were generated as described previously and maintained on the C57BL/6 genetic background. Mice were genotyped by PCR with the following primers. WT-forward: 5’-CTAGTGGTGGGGAGGCAGT-3’; WT-reverse: 5’-AGCAGGGATAGCCTGGTCTT-3’; KO-reverse: 5’-GCACATCTGAACTTCAGC-3’; ΔPAS-forward: 5’-TGGGGAAATGTGAAGAAAGC-3’; ΔPAS-reverse: 5’-CTTCCCTCCCAGAGAGTTGA-3’. To generate PyVT;Neat1+/−, PyVT;Neat1−/− and PyVT;Neat1ΔPAS/ΔPAS compound mice, Neat1−/− and Neat1ΔPAS/ΔPAS mice were crossed with MMTV-PyVT transgenic mice (Guy et al., 1992) (Jackson Laboratory). Primers for genotyping are MMTV-PyVT: 5’-GGAAGCAAGTACTTCACAAGGG-3’ (forward), 5’-GGAAAGTCACTAGGAGCAGGG-3’ (reverse). Palpable mammary tumors were checked every other day for tumor-free survival curve. Experimental mice were euthanized at 130~139 days. At sacrifice, mammary tumors were removed and tumor weights were measured. Mammary tumors and lungs were fixed with 4% paraformaldehyde. For POMHEX and pyruvate experiments, at 40 or 80 days (POMHEX) and 30 days (pyruvate) after the birth, mice were treated daily with intraperitoneal injection of vehicle, POMHEX in ~2% DMSO/PBS (30 mg/kg) (Lin et al., 2020) or pyruvate in PBS (1 g/kg). Orthotopic injection models were established as previously described (Song et al., 2013). Briefly, 6~8 weeks old female Neat1+/+, Neat1+/− and Neat1−/− mice and Rag1+/+ and Rag1−/− mice (Mombaerts et al., 1992) (Jackson Laboratory), were anaesthetized with isoflurane. A small incision between the fourth nipple and the midline was made to expose the mammary fat pad. Then, control or Neat1OE PyVT cells (1 × 106) in 100 μl growth medium were injected into the mammary fat pad. The recipient mice were monitored and euthanized when the palpable mammary tumors reached 2 cm in diameter. All animal experiments in this study were approved by and adhered to the guidelines of the MD Anderson Cancer Center Animal Care and Use Committee.
Human tumor materials
Fresh frozen primary tumors and paired adjacent noncancerous breast tissues were obtained from individuals with breast cancer diagnosed at MD Anderson Cancer Center (MDACC). All patients were American female, and the median age was 56 years (range 36~80). All biopsies were evaluated at MDACC, and the clinicopathological diagnosis was based on established criteria: 16 invasive ductal carcinomas (pathological type); 1 T1N0M0, 2 T1N1M0, 1 T1N2M0, 11 T2N0M0, and 1 T2N1M0 (TNM stage); 13 ER+PR+HER2− and 3 ER−PR−HER2+ (molecular subtype). Four triple-negative breast cancer (TNBC) (ER−PR−HER2−) and adjacent normal tissue samples were previously described (Setijono et al., 2018). The biopsies were snap-frozen immediately after excision during the surgical procedure and further stored in liquid nitrogen until subsequent analyses. The tissue viability and optical metabolic imaging (OMI) endpoints resulting from this tissue handling protocol were evaluated at the MDACC Biobank. This work was performed in accordance with the Institutional Review Board (IRB) approval at MDACC. All tissue samples were collected in compliance with informed consent policy.
Cells
293T, MCF7, T47D, BT-474 and MMTV-PyVT mouse mammary tumor derived cell line Py8119 were obtained from ATCC (Manassas, VA). 293T and BT-474 cells were cultured in DMEM medium (11965118, Gibco) supplemented with 10% FBS (16000044, Gibco) and 1% penicillin/streptomycin (15070063, Gibco). MCF7 and T47D cells were cultured in DMEM medium supplemented with 10% FBS, 1% penicillin/streptomycin and 5 μg/ml insulin (I9278, Sigma). Py8119 cells were cultured in DMEM/Ham’s F-12 (1:1) medium (DFL13, Caisson Labs) supplemented with 5% FBS and 1% penicillin/streptomycin. Cells were maintained in a humidified, 5% CO2 atmosphere at 37°C. All cells were independently validated by short tandem repeat DNA fingerprinting and chromosomal analysis by the Characterized Cell Line Facility and the Molecular Cytogenetics Core at MDACC.
Method Details
Plasmids
Human NEAT1 was amplified from pCRII_TOPO-hNEAT1 (Addgene) by primers (5’-ATCAGGTACCGGAGTTAGCGACAGGGAGGG-3’, 5’-TACAGCGGCCGCTGAGTTTAGAACTCAAACTT-3’). The resulting PCR products were sub-cloned into the KpnI and NotI sites of the pcDNA3 plasmid (Invitrogen). Lentiviral ENO1 (NM_001428.30), PGK1 (NM_000291.3) and PGAM1 (NM_002629.3) were obtained from Genecopoeia. pCMV3-C-HA-PGK1, pCMV3-N-Myc-PGAM1 and pCMV3-C-Flag-ENO1 were from Sino Biologicals. pEGFP-C2-PGK1-N term (residues 1~100), pEGFP-C2-PGK1-C term (101~417); pEGFP-C2-PGAM1-N term (1~138), pEGFP-C2-PGAM1-C term (139~254); and pEGFP-C2-ENO1-N term (1~134), pEGFP-C2-ENO1-C term (135~434) were generated by PCR based cloning. The mutant constructs of R39A, K41A, K131A, K141A, R171A and R192A for PGK1; K5A, R21A, R116A, R117A and K222A for PGAM1; K5A, R179A, K193A and R400A for ENO1 were generated by site-directed mutagenesis using the Quikchanges II Site-Directed Mutagenesis kit (Agilent Technologies).
Genome editing using CRISPR/Cas9
The PX330-B/B plasmids bearing both U6-driven sgRNA expression cassette and Cas9, as previously described (Yamazaki et al., 2018), were used for genome editing. sgRNAs targeting NEAT1 (Δ1033~2115 bp for Δ1~2.1k; Δ2116~2805 bp for Δ2.1~2.8k; Δ4054~5116 bp for Δ4~5.1k; Δ3699~3786 for ΔPAS) and sgRNAs upregulating Neat1 in the CRISPRa system were designed using the CRISPR design website (crispr.mit.edu) (Table S1). To delete the different portions of NEAT1, the PX330-B/B plasmids (2 μg) containing two sgRNAs were co-transfected with pcDNA6/TR plasmids (0.2 μg) containing the blasticidin resistance gene (Invitrogen) into MCF7 cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were treated with 20 μg/ml blasticidin (Millipore) for 3 days, starting 1 day after transfection. Cells were then diluted into 96-well plates for selection of single clones. Genomic DNAs from the selected clones were extracted using the PureLink genomic DNA purification kit (Invitrogen) according to the manufacturer’s instructions. The resulting genomic DNAs were subjected to PCR genotyping analysis with genotype-specific primers (Table S1) to detect deletions. For CRISPRa-mediated Neat1 upregulation, 3 × 106 293T cells were plated per 10-cm culture dish and lenti dCAS-VP64_Blast and lenti MS2-P65-HSF1_Hygro plasmids (Konermann et al., 2015) (Addgene) were transfected with Lipofectamine 2000. For infection, Py8119 cells were plated at a density of 6 × 105 cells per 10-cm culture dish and infected by virus from 293T cells 48 h after transfection. After selection in blasticidin (20 μg/ml) and hygromycin (75 μg/ml, Invitrogen), cells were re-infected virus from 293T cells expressing control or five different Neat1 sgRNAs containing lenti sgRNA(MS2)_puro plasmids (Addgene). After selection in puromycin (2 μg/ml, Sigma), the upregulation of Neat1 in the resulting cells was determined by RT-qPCR analysis.
siRNAs and shRNAs
All the sources of siRNA duplexes and shRNA constructs used are shown in Table S1. To prepare retroviral particles, 3 × 106 293T cells were plated per 10-cm culture dish and GFP control, PGK1, PGAM1 or ENO1 shRNA was transfected with Lipofectamine 2000. For infection, BT-474, MCF7 or T47D cells were plated at a density of 6 × 105 cells per 10-cm culture dish and infected by virus from 293T cells 48 h after transfection. After selection in puromycin (2 μg/ml), cells were transfected with pcDNA3-NEAT1 or wild-type or K/R-to-A mutants PGK1, PGAM1 or ENO1.
Antibodies
Antibodies against CD31 (1:200 for immunohistochemistry, 77699), ENO1 (1:1000 for immunoblotting, 3810), ENO2 (1:1000, 9536), GAPDH (1:2000, 2118), Aldolase A (1:1000, 8060), p-S345 Chk1 (1:1000, 2348), Chk1 (1:1000, 2360), p-S15 p53 (1:1000, 9284), p53 (1:1000, 2524), p-S139 H2A.X (1:200 for immunohistochemistry; 1:1000 for immunoblotting, 9718), acetyllysine (1:1000, 9441), cleaved PARP (1:1000, 9546), cleaved caspase 3 (1:1000, 9664), p-T389 S6K (1:1000, 9234), S6K (1:000, 2708), p-T172 AMPK (1:1000, 2535) and Myc-Tag (1:2000, 2276) were purchased from Cell Signaling. Antibodies against hexokinase II (1:1000, 22029), PFKFB3 (1:1000, 13763), ALYREF (1:1000, 16690), DDX39B (1:1000, 14798), EIF4A3 (1:1000, 17504), MAGOH (1:1000, 12347), RBM8A/Y14 (1:1000, 14958), TDP-43 (1:1000, 10782), Pinin (1:1000; 1:200 for RNA immunoprecipitation, 18266), FUS/TLS (1:1000, 11570), SRSF5 (1:1000, 16237), Staufen (1:1000, 14225), HNRNPC (1:1000, 11760) and HNRNPK (1:1000, 11426) were obtained from Proteintech. Antibodies against PGK1 (1:200 for RNA immunoprecipitation, ab38007), PGAM1 (1:200 for RNA immunoprecipitation, ab96622), ENO1 (1:200 for RNA immunoprecipitation, ab155102), GLS1 (1:1000, ab156876) and GLUD1 (1:1000, ab166618) were from Abcam. Anti-β-Actin (1:10000, A1978), –Flag (1:2000, F1804) and –Hsp90 (1:1000, H1775) antibodies were purchased from Sigma. Antibodies specific for PGK1 (1:1000, sc-48342), PGAM1 (1:1000, sc-130334), PSPC1 (1:1000, sc-374181), ASCT2 (1:1000, sc-99002) and PCB (1:1000, sc-67021) were purchased from Santa Cruz Biotechnology. Anti-KAP1 (1:1000, 610334) and anti-LAP2 (1:1000, 611000) were from BD Biosciences. Anti-GAPDH (1:500 for RNA immunoprecipitation, 2275-PC-100) antibody was from R&D Systems. Anti-p-S824 KAP1 (1:1000, A300–767A) antibody was from Bethyl Laboratories. Anti-mono/polyubiquitin (1:1000, BMLPW8810) was from Enzo Life Sciences. Anti-succinyllysine (1:1000, PTM401) was from PTM Biolabs. Anti-ENO3 (1:1000, MBS9407781) was from MyBiosource. Anti-Staufen (1:1000, GTX106566) was from GeneTex. Anti-GFP (1:1000, A11120) was from Invitrogen. Anti-Ki-67 (1:300 for immunohistochemistry, RM-9106) antibody was from Lab Vision. Anti-PyV T Ag (1:250 for immunohistochemistry, NB100–2749) antibody was from Novus Biologicals.
Reverse transcription-quantitative PCR
Total RNA was isolated with Trizol reagent (Invitrogen) and reverse-transcribed with the PrimeScript reverse transcriptase (Takara) or the Maxima reverse transcriptase (Thermo Scientific). Expression of specific mRNAs was determined with a LightCycler (Roche) or iCycler (Bio-Rad) using the Roche SYBR green PCR master (Roche) or the KAPA SYBR FAST qPCR kit (Kapa Biosystems). All the sources of real-time RT-qPCR primers used are listed in Table S1.
Immunohistochemistry
Mouse tissue paraffin sections were deparaffinized, rehydratred and subjected to heat-mediated antigen retrieval in Citrate buffer. After blocking the endogenous peroxidase by 3% H2O2, sections were blocked in 10% goat serum and incubated with appropriate antibodies. The signal was detected with biotinated horseradish peroxidase reagent (Vectastain ABC kit, Vector Labs) and DAB (Vector Labs) according to manufacturer’s instructions. The sections were counter-stained with hematoxylin. The images were acquired with a Leica DM1000 microscope. The stained slides were scanned on an Automated Cellular Image System III (Dako, Denmark) for quantification by digital image analysis. The quantification of staining density was performed using an ImagePro software (Media Cybernetics) and calculated based on the average staining intensity and the percentage of positively stained cells throughout different types of palpable lesions (premalignant and malignant lesions) from mouse mammary glands at about the same shape and size.
Whole-mount staining
The fourth inguinal glands of mice were dissected at the indicated ages and were spread on a glass slide. After fixation with acidic alcohol for 2 h, the tissues were hydrated and stained in Carmine alum for overnight as previously described (Song et al., 2013). Samples were then dehydrated, cleared with xylene and mounted. Ductal elongation and branching were quantified using an ImageJ software.
Metabolomics
Capillary electrophoresis mass spectrometry (CE-MS)–based metabolomics of mammary tumors of PyVT;Neat1+/+ and PyVT;Neat1−/− mice, as well as of syngeneic C57BL/6 mice orthotopically implanted with Neat1OE PyVT cells was carried out as previously described (Vila et al., 2017). 10 mg of a pool of different types of palpable lesions (premalignant and malignant lesions) from mouse mammary glands at about the same shape and size was used for the metabolic extraction. The extraction step involved the addition of 0.5 ml ice-cold methanol containing 50 μM methionine sulfone and camphor-10-sulfonic acid as internal standards, and the extracts were dried by vacuum centrifugation, dissolved with 50 μl ultrapure water and analyzed by CE-MS. CE-MS analysis was performed by an Agilent CE System combined with TOFMS (Agilent Technologies), an Agilent CE System and an Agilent 6460 TripleQuad LC/MS. Metabolites were separated through a fused silica capillary (50 μm i.d. × 80 cm) preconditioned with cation/anion buffer (HMT) and filled with 1 M formic acid (cation) or 50 mM ammonium acetate solution (pH 8.5) (anion) as electrolyte, and sheath liquid was delivered at a rate of 10 μl/min. Sample solution was injected at a pressure of 50 mbar for 10 s. The applied voltage was set at 27 kV. Electrospray ionization-mass spectrometry (ESI-MS) (cation) or Agilent 6460 TripleQuad LC/MS (anion) was conducted and the capillary voltage was set at 4000. A flow rate of heated dry nitrogen gas (300°C) was maintained at 5 psig. Exact mass data were acquired at the rate of 1.5 cycles/s over a 50~1000 m/z range. The data obtained by CE-TOFMS and CE-MS/MS analyses were preprocessed using an automatic integration software, MassHunter (Agilent Technologies). Each metabolite was identified and quantified based on the peak information including m/z, migration time, and peak area. The peak area was then converted to relative peak area by normalization with internal standard peak area and total cell number. The quantified data were evaluated for statistical significance by Welch’s t test.
Detection of glycolytic metabolites
Intracellular concentrations of glucose-6-phosphate (G-6-P), 2-phosphoglycerate (2-PG), phosphoenolpyruvate (PEP), and pyruvate were quantified using the glycolytic metabolite detection kits (K657, K778, K365, and K609, respectively; BioVision) according to the manufacturer’s specifications. Cellular glyceraldehyde-3-phosphate (GA-3-P) levels were measured by the Baylor School of Medicine Metabolomics Core as described previously (Dasgupta et al., 2018). Briefly, 2~5 × 106 cells were lysed with 750 μl ice-cold methanol:water (4:1) containing 20 μl spiked internal standard (L-Zeatin). A 450 μl ice-cold chloroform was added and vortex mixed. The aqueous and organic layers were separated, dried and resuspended with methanol:water (50:50). The extract was deproteinized using a 3 kDa molecular filter (Amicon ultracel-3K Membrane; Millipore) and the filtrate was dried. Prior to liquid chromatography-mass spectrometry (LC-MS), the dried extracts were re-suspended in identical volumes of injection solvent composed of water:methanol (50:50). LC-MS HPLC analysis was performed using an Agilent 1290 series HPLC system equipped with a degasser, binary pump, thermostatted auto sampler and column oven (Agilent Technologies). Cellular levels of GA-3-P were normalized to protein concentrations.
Measurement of glucose consumption and lactate production
Glucose consumption and lactate production were measured as previously described (Vila et al., 2017). Briefly, 1~2 × 105 cells were seeded into 6-well plate. 12 h later, the medium was replaced with 3 ml complete medium and supernatants were collected 24 h later and analyzed for glucose and lactate content using the Glucose Colorimetric Assay kit II (BioVision) and the Lactate Colorimetric Assay kit II (BioVision), respectively, according to the manufacturer’s instructions. Glucose consumption was extrapolated by subtracting the measured glucose concentrations in the medium from the original glucose concentration (25 mM). Both glucose consumption and lactate production were normalized to total cell number.
Cell proliferation and invasion assays
Typically, cell proliferation and invasion assays were performed as previously described (Park et al., 2019). Growth curves were generated by seeding 2 × 104 cells into 12-well plate. Plates were stained with crystal violet at each indicated time point. The dye was extracted with 10% acetic acid followed by plate reading at 595 nm in an iMark Plate Reader (Bio-Rad). Transwell invasion assay was performed using the BioCoat Matrigel invasion chamber with 8-μm polycarbonated filters (BD Biosciences) according to the manufacturer’s instructions. 5 × 104 cells were suspended in 0.5 ml serum-free medium and seeded in the upper chamber. 10% FBS was used as a chemoattractant in the lower wells. After 12 h-incubation in the presence of 2 h pre-treatment of mitomycin C (5 μg/ml) - no significant apoptosis was seen, cells that invaded to the bottom of the membrane were fixed with 10% formalin, stained with 0.1% crystal violet and counted on a Leica DMIIL inverted microscope.
Cell cycle analysis
Cells were synchronized by the double thymidine block protocol as previously described (Song et al., 2004). Briefly, cells synchronized and released from thymidine block were harvested at the indicated times. Then, cells were fixed in 70% ethanol, stained with propidium iodide (25 μg/ml) (Sigma) and incubated for 30 min at 37 °C with RNase A (20 μg/ml) (Roche). The DNA contents of the cells were evaluated by flow cytometry with a FACSCalibur instrument (BD Biosciences).
Cytoplasmic/nuclear fractionation
Cytoplasmic/nuclear fractionation was performed as previously described (Song et al., 2008). Briefly, 1~2 × 105 cells were seeded into 6-well plate. 12 h after seeding, cells were cultured in glucose-free medium (11966025, Gibco) containing 10% dialyzed FBS (26400044, Gibco), and 16 h later, the medium was then changed to glucose- or 2-deoxyglucose–containing culture medium at the indicated concentrations. 2 h after stimulation, cells were subjected to sequential fractionation and isolate cytoplasmic and nuclear RNAs and proteins using hypotonic and hypertonic lysis buffers or the NE-PER fractionation kit (Pierce) according to the manufacturer’s instructions.
RNA immunoprecipitation
RNA immunoprecipitation (RIP) analysis from whole cell extracts was performed as previously described (Min et al., 2017). Briefly, cells were lysed in buffer containing 20 mM Tris-HCl (pH7.5), 100 mM KCl, 5 mM MgCl2, 0.5% NP-40 and cleared by centrifugation. The lysates were incubated with protein A-Sepharose beads coated with antibodies against PGK1 (ab38007, Abcam), PGAM1 (ab96622), ENO1 (ab155102), GAPDH (2275-PC-100, R&D Systems), Pinin (18266, Proteintech; ab244250) or nucleolin (ab22758) with control IgG (Sigma) for 1 h at 4°C. After the beads were washed four times with NT2 buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40], the immunoprecipitates were treated with 20 units of RNase-free DNase I for 15 min at 37°C and proteinase K (0.5 mg/ml) in 0.1% SDS for 15 min at 55 °C to remove DNAs and proteins, respectively. The RNAs isolated from the IPs by acidic phenol extraction were then subjected to RT-qPCR using the primers listed in Table S1. The RIP results were normalized to 18S rRNA or GAPDH mRNA.
Cross-linking immunoprecipitation–qPCR
The NEAT1 short sequence motifs responsible for PGK1, PGAM1 and ENO1 bindings were purified by ultraviolet (UV) cross-linking immunoprecipitation (CLIP) followed by RT-qPCR (CLIP-qPCR) as previously described (Yoon et al., 2014). For our CLIP experiments with PGK1, PGAM1 and ENO1, growing MCF7 cells were washed once with cold PBS and irradiated for 400 mJ/cm2 in a Gene Linker chamber (Bio-Rad). Cells were lysed and the lysates were digested with RNase T1 (1 U/μl) for 5 min and subjected to RIP with anti-PGK1, –PGAM1 or –ENO1. Short segments of NEAT1 bound with PGK1, PGAM1 and ENO1, and thereby protected from RNase T1 digestion, were purified by RT-qPCR using the primers listed in Table S1.
Co-immunoprecipitation and in vitro pull-down assays
For extracts of PyVT mammary tumors, a pool of different types of palpable lesions (premalignant and malignant lesions) from mouse mammary glands at about the same shape and size were lysed in buffer containing 50 mM Tris-HCl (pH7.5), 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA and 1% Triton X-100 with homogenization and cleared by centrifugation. Co-immunoprecipitation with appropriate antibodies in cell and tissue lysates was performed as described (Park et al., 2019; Vila et al., 2017). For in vitro pull-down assay, purified PGK1 (MSB203356, MyBioSource) was incubated with protein A/G-agarose (Pierce) coated with anti-PGK1 (sc-48342, Santa Cruz Biotechnology) and the beads were washed three times with lysis buffer. NEAT1 was in vitro transcribed (IVT) from pCRII_TOPO-hNEAT1 using the MEGAscript T7 Transcription kit (Thermo Scientific) according to the manufacturer’s instructions. PGK1 beads were incubated with recombinant PGAM1 (MBS203464) and ENO1 (MBS203689) proteins in the presence of 0.5 or 2.0 μg of IVT sense or anti-sense NEAT1 for 2 h at 4°C and the reaction mixture was washed 5 times with lysis buffer and subjected to immunoblotting.
RNA fluorescence in situ hybridization (RNA-FISH)
For human NEAT1 detection, we used the Thermo Scientific probe as follows. Cells were grown in 24-well plate on 18 mm glass coverslips. They were fixed and permeabilized using the provided buffers in the ViewRNA Cell Plus Assay kit (Thermo Scientific) following the manufacturer’s instructions. Target probe hybridization and signal amplification steps were carried out according to the manufacturer’s provided protocol by utilizing the kit-supplied buffers and reagents. Coverslips were mounted using SlowFade Diamond Antifade Mountant (S36963, Thermo Scientific) and imaged 1~2 days following assay completion using a Leica SP5 confocal microscope. All slides were counterstained with DAPI. All samples were imaged under same settings: 63× oil, Line averaging: 4, All lasers 20% power, Sequential scanning for each of the 4 channels, Steps of 0.4 μm for an average of 8~10 stacks, Gain settings (DAPI: 794, Em. 488: 958, Em. 546: 1053, Em. 647: 670). The maximum intensity projections were merged to produce the multicolor merged images. All z slices of individual channels are saved as well. For mouse Neat1 detection, we followed the Stellaris procedure as follows. Cells were grown to ~80% confluence on 18 mm glass coverslips, fixed with 3.7% formaldehyde in 1X PBS, and then permeabilized with 0.1% Triton X-100 in 1X PBS according to the FISH in Adherent Cells protocol provided by the manufacturer (Biosearch Technologies) for Stellaris RNA FISH probes. All subsequent wash and hybridization steps followed manufacturer protocol and utilized Stellaris Wash Buffers A, B and Hybridization buffer prepared as described in the protocol. Cells were incubated with a 125 nM working probe solution at 37 °C for ~12 hr. Coverslips were mounted using SlowFade Diamond Antifade Mountant and imaged 1~2 days following assay completion using a Leica SP5 confocal microscope.
Fluorescence polarization assay
Fluorescein-modified NEAT1 was in vitro transcribed with fluorescein-12-UTP (Roche) using the MEGAscript T7 Transcription kit. To examine the RNA-binding affinity, recombinant PGK1, PGAM1 and ENO1 were incubated with 1 nM fluorescein-labeled NEAT1 in assay buffer (20 mM Tris-HCl pH 7.4, 0.5% glycerol, 150 mM NaCl, 4 mM MgCl2, 1 U/μL RiboLock RNase inhibitor or 20 mM Tris-HCl pH 7.4, 150 mM NaCl, 4 mM MgCl2, 10 mM DTT, 0.05% NP-40, 0.5% glycerol, 1 U/μL RiboLock RNase inhibitor) at room temperature. The assay was performed in 96-well non-binding black plate (Corning), with fluorescence polarization measured using a Spectramax iD5 microplate reader (Molecular Device) with the excitation and emission wavelengths 490 nm and 530 nm, respectively. Millipolarization units (mP) were used to express fluorescence polarization values defined by the equation mP = 1000 × [(IⅡ − GI⊥) / (IⅡ + GI⊥)], where IⅡ and I⊥ are parallel and perpendicular emission intensity measurements, respectively. The data was corrected for background (RNA sample alone), and G-factor, then fitted by a one-site binding model using the Equation, y = y0 + (ymax * x / KD + x), where y is the corrected fluorescence polarization, y0 is polarization value without enzyme, ymax is maximum binding fluorescence polarization signal, x is the enzyme concentration, and Kd is the dissociation constant. x and ymax were used as fitting parameters and nonlinear regression was performed using GraphPad Prism. Measurements were taken after 5 min incubation between enzyme and RNA at room temperature.
Copy number analysis
In vitro-transcribed NEAT1_1 at predetermined concentrations were used to generate linear standard curves dependent on the moles of template RNA for absolute quantification of NEAT1_1 RNA molecules. Total RNA was extracted using Trizol (Invitrogen) from either the cytoplasmic or the nuclear fraction, and 0.5 μg of RNA from each cellular compartment was reverse transcribed using the Maxima H Minus Reverse Transcriptase (Thermo Scientific). cDNA was diluted 1:10 in DEPC-treated water, and 2.5 μl was used for qPCR with 2.5 μl SYBR (Kapa Biosystems) and 5 μl forward and reverse primer mix. Average RNA copy number in the cytoplasm or the nucleus per cell were quantitated from the sample Ct values referenced against the linear regression of the standard curve followed by calculations to account for dilutions and cell numbers as previously described (Zealy et al., 2018).
Cancer biostatistical analysis
The correlation analysis between NEAT1 and PGK1, PGAM1, ENO1, GAPDH and PNN expressions in patients across breast tumor intrinsic subtypes was carried out using previously published RNA-seq database (Cancer Genome Atlas, 2012; Curtis et al., 2012) (n = 2610). Kaplan-Meier plotting and log-rank test of Pinin were done using a publicly accessible online tool KM Plotter (http://kmplot.com) (Liu et al., 2014; Tang et al., 2018) (n = 126 and 65, respectively). Patients were divided into two classes based on Pinin expression: the high- and low–Pinin groups were split based on the median value calculated across the entire dataset to generate two groups of equal size.
Quantification and Statistical Analysis
To assess significant differences, SPSS v24.0 and GraphPad Prism v8.2 were used. Data are expressed as the mean +/− SD of the values from the independent experiments performed, as indicated in the corresponding figures legends. The numbers of biological replicates, and what they represent, are indicated in each figure legend. For animal experiments, all the mice with the same genotypes were randomly assigned into each group. Comparisons between two groups were performed by an unpaired two-tailed Student’s t test. Multi-comparisons were performed by one-way analysis of variance (ANOVA) with Tukey’s post hoc test unless otherwise noted. Log-rank (Mantel-Cox) test was applied to test the incidence or survival distributions of two groups. The correlation coefficients were calculated by the Pearson’s Chi-Square test. Prior to analyzing the statistical significance of differences among groups, we tested whether the data were normally distributed at the 0.05 level and whether variance was similar among groups using Shapiro-Wilk normality test and F-test, respectively. p values below 0.05 were considered statistically significant.
Supplementary Material
Table S1. Oligonucleotide sequences used in this study, Related to STAR Methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-CD31 | Cell Signaling Technology | 77699; RRID:AB_2722705 |
| Rabbit polyclonal anti-ENO1 | Cell Signaling Technology | 3810; RRID:AB_2246524 |
| Rabbit polyclonal anti-ENO2 | Cell Signaling Technology | 9536; RRID:AB_2099308 |
| Rabbit monoclonal anti-GAPDH (For immunoblotting) | Cell Signaling Technology | 2118; RRID:AB_561053 |
| Rabbit monoclonal anti-Aldolase A | Cell Signaling Technology | 8060; RRID:AB_2797635 |
| Rabbit monoclonal anti- p-S345 Chk1 | Cell Signaling Technology | 2348; RRID:AB_331212 |
| Mouse monoclonal anti-Chk1 | Cell Signaling Technology | 2360; RRID:AB_2080320 |
| Rabbit polyclonal anti- p-S15 p53 | Cell Signaling Technology | 9284; RRID:AB_331464 |
| Mouse monoclonal anti-p53 | Cell Signaling Technology | 2524; RRID:AB_33174 |
| Rabbit monoclonal anti-p-S139 H2A.X | Cell Signaling Technology | 9718; RRID:AB_2118009 |
| Rabbit polyclonal anti-acetyllysine | Cell Signaling Technology | 9441; RRID:AB_331805 |
| Mouse monoclonal anti-cleaved PARP | Cell Signaling Technology | 9546; RRID:AB_2160593 |
| Rabbit monoclonal anti-cleaved caspase 3 | Cell Signaling Technology | 9664; RRID:AB_2070042 |
| Rabbit monoclonal anti-p-T389 S6K | Cell Signaling Technology | 9234; RRID:AB_2269803 |
| Rabbit monoclonal anti-S6K | Cell Signaling Technology | 2708; RRID:AB_390722 |
| Rabbit monoclonal anti-p-T172 AMPK | Cell Signaling Technology | 2535; RRID:AB_331250 |
| Mouse monoclonal anti-Myc Tag | Cell Signaling Technology | 2276; RRID:AB_331783 |
| Rabbit monoclonal anti-hexokinase II | Proteintech | 22029-1-AP; RRID:AB_11182717 |
| Rabbit polyclonal anti-PFKFB3 | Proteintech | 13763-1-AP; RRID:AB_2162854 |
| Rabbit polyclonal anti-ALYREF | Proteintech | 16690-1-AP; RRID:AB_2878300 |
| Rabbit polyclonal anti-DDX39B | Proteintech | 14798-1-AP; RRID:AB_2061854 |
| Rabbit polyclonal anti-EIF4A3 | Proteintech | 17504-1-AP; RRID:AB_2097393 |
| Rabbit polyclonal anti-MAGOH | Proteintech | 12347-1-AP; RRID:AB_2265988 |
| Rabbit polyclonal anti-RBM8A/Y14 | Proteintech | 14958-1-AP; RRID:AB_2178820 |
| Rabbit polyclonal anti-TDP-43 | Proteintech | 10782-2-AP; RRID:AB_615042 |
| Rabbit polyclonal anti-Pinin | Proteintech | 18266-1-AP; RRID:AB_10642138 |
| Rabbit polyclonal anti-FUS/TLS | Proteintech | 11570-1-AP; RRID:AB_2247082 |
| Rabbit polyclonal anti-SRSF5 | Proteintech | 16237-1-AP; RRID:AB_2185204 |
| Rabbit polyclonal anti-Staufen | Proteintech | 14225-1-AP; RRID:AB_2302744 |
| Rabbit polyclonal anti-HNRNPC | Proteintech | 11760-1-AP; RRID:AB_2117500 |
| Rabbit polyclonal anti-HNRNPK | Proteintech | 11426-1-AP; RRID:AB_2264314 |
| Rabbit polyclonal anti-Pinin | Proteintech | 18266-1-AP; RRID:AB_10642138 |
| Rabbit polyclonal anti-PGK1 (For RNA immunoprecipitation) | Abcam | ab38007; RRID:AB_2161220 |
| Rabbit polyclonal anti-PGAM1 (For RNA immunoprecipitation) | Abcam | ab96622; RRID:AB_10687155 |
| Rabbit monoclonal anti-ENO1 (For RNA immunoprecipitation) | Abcam | ab155102 |
| Rabbit monoclonal anti-GLS1 | Abcam | ab156876; RRID:AB_2721038 |
| Rabbit monoclonal anti-GLUD1 | Abcam | ab166618; RRID:AB_2815030 |
| Rabbit polyclonal anti-Pinin | Abcam | ab244250; RRID:AB_2868585 |
| Rabbit polyclonal anti-nucleolin | Abcam | ab22758, RRID:AB_776878 |
| Mouse monoclonal anti-LAP2 | BD Biosciences | 611000; RRID:AB_398313 |
| Mouse monoclonal anti-KAP1 | BD Biosciences | 610334; RRID:AB_397724 |
| Mouse monoclonal anti-Actin | Sigma | A2228; RRID:AB_476697 |
| Mouse monoclonal anti-Flag | Sigma | F1804; RRID:AB_262044 |
| Mouse monoclonal anti-Hsp90 | Sigma | H1775; RRID:AB_260026 |
| Mouse monoclonal anti-PGK1 (For immunoblotting/co-immunoprecipitation) | Santa Cruz Biotechnology | sc-48342; RRID:AB_628116 |
| Mouse monoclonal anti-PGAM1 (For immunoblotting) | Santa Cruz Biotechnology | sc-130334; RRID:AB_2160792 |
| Mouse monoclonal anti-PSPC1 | Santa Cruz Biotechnology | sc-374181; RRID:AB_10989076 |
| Mouse monoclonal anti-ASCT2 | Santa Cruz Biotechnology | sc-99002; RRID:AB_2239446 |
| Rabbit polyclonal anti-PCB | Santa Cruz Biotechnology | sc-67021; RRID:AB_2283532 |
| Rabbit polyclonal anti-GAPDH (For RNA immunoprecipitation) | R&D Systems | 2275-PC-100; RRID:AB_2107456 |
| Rabbit polyclonal anti-Anti-p-S824 KAP1 | Bethyl Laboratories | A300-767A; RRID:AB_669740 |
| Mouse monoclonal anti-Anti-mono/polyubiquitin | Enzo Life Sciences | BML-PW8810; RRID:AB_10541840 |
| Rabbit polyclonal anti-Anti-succinyllysine | PTM Biolabs | PTM-401; RRID:AB_2687628 |
| Rabbit polyclonal anti-Anti-ENO3 | MyBiosource | MBS9407781 |
| Rabbit polyclonal anti-Staufen | GeneTex | GTX106566; RRID:AB_1952044 |
| Mouse monoclonal anti-Anti-GFP | Invitrogen | A-11120; RRID:AB_221568 |
| Rabbit monoclonal anti-Ki-67 | Lab Vision | RM-9106; RRID:AB_2335745 |
| Rat monoclonal anti- PyVT Ag | Novus Biologicals | NB100-2749; RRID:AB_10001944 |
| Mouse IgG | Sigma | 12-371; RRID:AB_145840 |
| Rabbit IgG | Sigma | 12-370; RRID:AB_145841 |
| Biological Samples | ||
| Invasive ductal carcinomas and paired normal breast tissue cohort | MD Anderson Cancer Center (MDACC) | N/A |
| Triple-negative breast cancer (TNBC) | (Setijono et al., 2018) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 2000 | Thermo Scientific | 11668500 |
| Fetal Bovine Serum, dialyzed, US origin | Thermo Scientific | 26400044 |
| Puromycin | Sigma | P8833 |
| Blasticidin | Millipore | 203350 |
| Mitomycin C | Sigma | M4287 |
| Propidium iodide | Sigma | P4170 |
| ROCHE RNase A | Sigma | 10109142001 |
| Phosphoenolpyruvate | Sigma | 860077 |
| Sodium pyruvate | Sigma | P2256 |
| Carmine Alum | Sigma | C1022 |
| D-Glucose | Sigma | G8270 |
| 2-Deoxy-D-glucose | Sigma | D8375 |
| ROCHE Fluorescein-12-UTP | Sigma | 11427857910 |
| POMHEX | (Lin et al., 2020) | N/A |
| PGMI-004A | Glixx Laboratories | 1313738-90-7 |
| Koningic acid | Abcam | ab144269 |
| NG52 | MediChem | HY-15154 |
| Recombinant His-PGK1 | MyBioSource | MSB203356 |
| Recombinant His-PGAM1 | MyBioSource | MSB203464 |
| Recombinant His-ENO1 | MyBioSource | MSB203689 |
| Recombinant His-GAPDH | Sino Biological | 10094-H07E |
| Bacillus subtilis dapB, Type 1: Alexa Fluor 546 | Thermo Scientific | VF1-11712-VC |
| HU NEAT1, Type 1: Alexa Fluor 546 | Thermo Scientific | VA1-12621-VC |
| HU GAPDH, Type 4: Alexa Fluor 488 | Thermo Scientific | VA4-10641-VC |
| Stellaris® FISH Probes, Mouse Neat1 5’ Segment with Quasar® 570 Dye | Biosearch Technologies | SMF-3009-1 |
| Stellaris® FISH Probes, Mouse Neat1 Middle Segment with Quasar® 570 Dye | Biosearch Technologies | SMF-3010-1 |
| Critical Commercial Assays | ||
| Dharmafect I siRNA transfection reagent | Dharmacon | T-2001 |
| Vectastain ABC HRP kit | Vector Laboratories | PK-4000 |
| QIAquick PCR purification kit | Qiagen | 28104 |
| QuikChange II Site-Directed Mutagenesis kit | Agilent Technology | 200524 |
| PureLink™ Genomic DNA Mini Kit | Invitrogen | K182000 |
| PrimeScript 1st strand cDNA Synthesis kit | Takara | 6110A |
| Maxima 1st strand cDNA Synthesis kit | Thermo Scientific | K1672 |
| FastStart Essential DNA Green Master | Roche | 6402712001 |
| MEGAscript T7 Transcription kit | Thermo Scientific | AM 1334 |
| MEGAscript SP6 Transcription kit | Thermo Scientific | AM1330 |
| KAPA SYBR FAST qPCR kit | Kapa Biosystems | KK4606 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Pierce | 78833 |
| Glucose Colorimetric Assay kit II | Bio Vision | K686-100 |
| Lactate Colorimetric Assay kit II | Bio Vision | K627-100 |
| Glucose-6-phosphate Colorimetric Assay kit | Bio Vision | K657-100 |
| 2-Phosphoglycerate Colorimetric Assay kit | Bio Vision | K778-100 |
| Phosphoenolpyruvate Colorimetric Assay kit | Bio Vision | K365-100 |
| Pyruvate Colorimetric Assay kit | Bio Vision | K609-100 |
| ViewRNA Cell Plus Assay kit | Thermo Scientific | 88-19000-99 |
| Deposited Data | ||
| Correlation analysis between NEAT1 and PGK1, PGAM1, ENO1, GAPDH and PNN expressions in patients across breast tumor intrinsic subtypes | (Cancer Genome Atlas, 2012; Curtis et al., 2012) | n = 2610 |
| Kaplan-Meier plotting and log-rank test of Pinin | (Liu et al., 2014; Tang et al., 2018) | n = 126 and 65, respectively |
| Original images | Mendeley | http://dx.doi.org/10.17632/nyhjzwtscc.1 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL-3216 |
| MCF7 | ATCC | HTB-22 |
| T47D | ATCC | HTB-133 |
| BT-474 | ATCC | HTB-20 |
| Py8119 | ATCC | CRL-3278 |
| Experimental Models: Organisms/Strains | ||
| Neat1 knockout mice | (Nakagawa et al., 2011; Nakagawa et al., 2014) | N/A |
| MMTV-PyVT transgenic mice | Jackson Laboratory | 022974 |
| Rag1−/− mice | Jackson Laboratory | 002216 |
| Neat1 ΔPAS mice | (Isobe et al., 2020) | N/A |
| Oligonucleotides | ||
| Primers; see Table S1 | This paper | N/A |
| siRNAs; see Table S1 | (Clemson et al., 2009); Dharmacon | N/A |
| shRNAs; see Table S1 | Sigma; (Vila et al., 2017) | N/A |
| sgRNAs; see Table S1 | (Yamazaki et al., 2018); (Konermann et al., 2015) | N/A |
| Recombinant DNA | ||
| pCRII_TOPO-hNEAT1 | Addgene | 61518 |
| pcDNA3-hNEAT1 | This paper | N/A |
| Lv242-B PGK1 | Genecopeia | EX-C0528-Lv242-B |
| Lv242-B PGAM1 | Genecopeia | EX-C0543-Lv242-B |
| Lv242-B ENO1 | Genecopeia | EX-C0061-Lv242-B |
| pCMV3-C-HA-PGK1 | Sino Biologicals | HG11114-CY |
| pCMV3-N-Myc-PGAM1 | Sino Biologicals | HG16274-NM |
| pCMV3-C-Flag-ENO1 | Sino Biologicals | HG11554-CF |
| pEGFP-C2-PGK1-N term (residues 1∼100) | This paper | |
| pEGFP-C2-PGK1-C term (101∼417) | This paper | |
| pEGFP-C2-PGAM1-N term (1∼138) | This paper | |
| pEGFP-C2-PGAM1-C term (139∼254) | This paper | |
| pEGFP-C2-ENO1-N term (1∼134) | This paper | |
| pEGFP-C2-ENO1-C term (135∼434) | This paper | |
| pCMV3-C-HA-PGK1 R39A | This paper | N/A |
| pCMV3-C-HA-PGK1 K41A | This paper | N/A |
| pCMV3-C-HA-PGK1 K131A | This paper | N/A |
| pCMV3-C-HA-PGK1 K141A | This paper | N/A |
| pCMV3-C-HA-PGK1 R171A | This paper | N/A |
| pCMV3-C-HA-PGK1 R192A | This paper | N/A |
| pCMV3-N-Myc-PGAM1 K5A | This paper | N/A |
| pCMV3-N-Myc-PGAM1 R21A | This paper | N/A |
| pCMV3-N-Myc-PGAM1 R116A | This paper | N/A |
| pCMV3-N-Myc-PGAM1 R117A | This paper | N/A |
| pCMV3-N-Myc-PGAM1 K222A | This paper | N/A |
| pCMV3-C-Flag-ENO1 K5A | This paper | N/A |
| pCMV3-C-Flag-ENO1 R179A | This paper | N/A |
| pCMV3-C-Flag-ENO1 K193A | This paper | N/A |
| pCMV3-C-Flag-ENO1 R400A | This paper | N/A |
| PX330-B/B | Addgene | 42230 |
| PX330-B/B Δ1∼2.1k | This paper | N/A |
| lenti dCAS-VP64_Blast | Addgene | 61425 |
| lenti MS2-P65-HSF1_Hygro | Addgene | 61426 |
| lenti sgRNA(MS2)_puro | Addgene | 73795 |
| pcDNA6/TR | Thermo Scientific | V102520 |
| pLKO.1 GFP shRNA | (Vila et al., 2017) | N/A |
| Software and Algorithms | ||
| ImageJ 1.46r | NIH | https://imagej.nih.gov/ij/ |
| ImagePro software | Media Cybernetics | N/A |
| IBM SPSS Statistics 23 | IBM | v24.0 |
| GraphPad Prism | GraphPad Software | v8.2 |
HIGHLIGHTS:
NEAT1 plays a role in tumor initiation, growth and metastasis in breast cancer.
NEAT1 facilitates assembly of PGK1/PGAM1/ENO1 complexes and substrate channeling.
NEAT1’s regulation of glycolysis is a critical mechanism for tumorigenesis.
NEAT1 nuclear export underlies its isoform-specific role in tumorigenesis.
ACKNOWLEDGEMENTS
We thank D. Yu, A. Sahin and P. P. Pandolfi for sharing reagents and critical discussions. We thank J. M. Kang, J. H. Park, M. Chang, J. Huang, K. Misare, M. Maples, M. Ivan, Q. Hao and M. Liu for excellent technical assistance. We thank the Baylor School of Medicine Metabolomics Core, supported by CPRIT Proteomics and Metabolomics Core Facility (N. Putluri, V. Putluri) (RP170005), for their technical support in metabolome analysis. We are grateful to T. Garvey for critical editing of the manuscript. This work was supported by grants from the National Institutes of Health (NIH) (CA196740, CA258100, M.S.S.), the Department of Defense (W81XWH-20-1-0379, M.S.S.), and an American Cancer Society Institutional Research Grant awarded to the Hollings Cancer Center at the Medical University of South Carolina (J.-H.Y.). J.W.L., K.-W.M. (NRF-2019R1C1C1002886), H.M., S.J.S. (NRF-2018R1A2B6006630), and N.T.G., H.H.V., J.H.C. (NRF-2019R1A2C4069796) were supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning. J.-H.C. was supported by a Hollings Cancer Center’s Cancer Center Support Grant (CA138313) Fellowship from the Medical University of South Carolina, M.C.B. was supported by NIH training grants (TL1TR001451, UL1TR001450), and A.K. was supported by NIH grants (DK124553, CA246233, GM130457, DK123704).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adriaens C, Rambow F, Bervoets G, Silla T, Mito M, Chiba T, Asahara H, Hirose T, Nakagawa S, Jensen TH, et al. (2019). The long noncoding RNA NEAT1_1 is seemingly dispensable for normal tissue homeostasis and cancer cell growth. RNA 25, 1681–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriaens C, Standaert L, Barra J, Latil M, Verfaillie A, Kalev P, Boeckx B, Wijnhoven PW, Radaelli E, Vermi W, et al. (2016). p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med 22, 861–868. [DOI] [PubMed] [Google Scholar]
- Alpatov R, Munguba GC, Caton P, Joo JH, Shi Y, Shi Y, Hunt ME, and Sugrue SP (2004). Nuclear speckle-associated protein Pnn/DRS binds to the transcriptional corepressor CtBP and relieves CtBP-mediated repression of the E-cadherin gene. Mol Cell Biol 24, 10223–10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellana M, Wilson MZ, Xu Y, Joshi P, Cristea IM, Rabinowitz JD, Gitai Z, and Wingreen NS (2014). Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat Biotechnol 32, 1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406. [DOI] [PubMed] [Google Scholar]
- Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M, et al. (2014). The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun 5, 5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL (2016). Linking Long Noncoding RNA Localization and Function. Trends Biochem Sci 41, 761–772. [DOI] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, and Lawrence JB (2009). An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell 33, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, Choi JM, Jung SY, Coarfa C, Westbrook TF, et al. (2018). Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature 556, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Ramos A, Roig-Borrellas A, Garcia-Melero A, and Lopez-Alemany R (2012). alpha-Enolase, a multifunctional protein: its role on pathophysiological situations. J Biomed Biotechnol 2012, 156795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell DA (1992). Metabolic control analysis: a survey of its theoretical and experimental development. Biochem J 286 ( Pt 2), 313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, and Muller WJ (1992). Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hernandez-Perez L, Depardon F, Fernandez-Ramirez F, Sanchez-Trujillo A, Bermudez-Cruz RM, Dangott L, and Montanez C (2011). alpha-Enolase binds to RNA. Biochimie 93, 1520–1528. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, and Chen J (2014). Post-translational modifications and the Warburg effect. Oncogene 33, 4279–4285. [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, et al. (2012). Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MY, Tang SJ, Ng WV, Yang W, Leu SJ, Lin YC, Feng CK, Sung JS, and Sun KH (2010). Nucleotide-binding domain of phosphoglycerate kinase 1 reduces tumor growth by suppressing COX-2 expression. Cancer Sci 101, 2411–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, and Chess A (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe M, Toya H, Mito M, Chiba T, Asahara H, Hirose T, and Nakagawa S (2020). Forced isoform switching of Neat1_1 to Neat1_2 leads to the loss of Neat1_1 and the hyperformation of paraspeckles but does not affect the development and growth of mice. RNA 26, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler A, and Hurt E (2007). Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol 8, 761–773. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukharsky MS, Ninkina NN, An H, Telezhkin V, Wei W, Meritens CR, Cooper-Knock J, Nakagawa S, Hirose T, Buchman VL, et al. (2020). Long non-coding RNA Neat1 regulates adaptive behavioural response to stress in mice. Transl Psychiatry 10, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu S, Lin YM, Wu CH, and Ouyang P (2012). Loss of Pnn expression results in mouse early embryonic lethality and cellular apoptosis through SRSF1-mediated alternative expression of Bcl-xS and ICAD. J Cell Sci 125, 3164–3172. [DOI] [PubMed] [Google Scholar]
- Li R, Harvey AR, Hodgetts SI, and Fox AH (2017a). Functional dissection of NEAT1 using genome editing reveals substantial localization of the NEAT1_1 isoform outside paraspeckles. RNA 23, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang Z, Liu X, Cheng X, Zhang Y, Han X, Zhang Y, Liu S, Yang J, Xu B, et al. (2017b). The FOXN3-NEAT1-SIN3A repressor complex promotes progression of hormonally responsive breast cancer. J Clin Invest 127, 3421–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. (2017). A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab 26, 648–659.e648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Schmidt BF, Bruchez MP, and McManus CJ (2018). Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res 46, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Satani N, Hammoudi N, Yan VC, Barekatain Y, Khadka S, Ackroyd JJ, Georgiou DK, Pham CD, Arthur K, et al. (2020). An enolase inhibitor for the targeted treatment of ENO1-deleted cancers. Nat Metab 2, 1413–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NQ, Stingl C, Look MP, Smid M, Braakman RB, De Marchi T, Sieuwerts AM, Span PN, Sweep FC, Linderholm BK, et al. (2014). Comparative proteome analysis revealing an 11-protein signature for aggressive triple-negative breast cancer. J Natl Cancer Inst 106, djt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello SS, Sinow C, Raj N, Mazur PK, Bieging-Rolett K, Broz DK, Imam JFC, Vogel H, Wood LD, Sage J, et al. (2017). Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KW, Jo MH, Shin S, Davila S, Zealy RW, Kang SI, Lloyd LT, Hohng S, and Yoon JH (2017). AUF1 facilitates microRNA-mediated gene silencing. Nucleic Acids Res 45, 6064–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al. (2012). Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 488, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murachelli AG, Ebert J, Basquin C, Le Hir H, and Conti E (2012). The structure of the ASAP core complex reveals the existence of a Pinin-containing PSAP complex. Nat Struct Mol Biol 19, 378–386. [DOI] [PubMed] [Google Scholar]
- Naganuma T, Nakagawa S, Tanigawa A, Sasaki YF, Goshima N, and Hirose T (2012). Alternative 3’-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J 31, 4020–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Naganuma T, Shioi G, and Hirose T (2011). Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J Cell Biol 193, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang P, and Sugrue SP (1992). Identification of an epithelial protein related to the desmosome and intermediate filament network. J Cell Biol 118, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Yao Y, Xia W, Setijono SR, Kim JH, Vila IK, Chiu HH, Wu Y, Billalabeitia EG, Lee MG, et al. (2019). PTEN self-regulates through USP11 via the PI3K-FOXO pathway to stabilize tumor suppression. Nat Commun 10, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz I, Kligun E, Bengad B, and Mandel-Gutfreund Y (2016). BindUP: a web server for non-homology-based prediction of DNA and RNA binding proteins. Nucleic Acids Res 44, W568–W574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YT, Ideue T, Sano M, Mituyama T, and Hirose T (2009). MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci U S A 106, 2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setijono SR, Park M, Kim G, Kim Y, Cho KW, and Song SJ (2018). miR-218 and miR-129 regulate breast cancer progression by targeting Lamins. Biochem Biophys Res Commun 496, 826–833. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, and Pandolfi PP (2008). The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Song SJ, Ayad NG, Chang JS, Lee JH, Hong HK, Lee H, Choi N, Kim J, Kim H, et al. (2004). The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol 6, 129–137. [DOI] [PubMed] [Google Scholar]
- Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, Beringer G, Brikbak NJ, Yuan X, Cantley LC, et al. (2013). MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 154, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srere PA (1987). Complexes of sequential metabolic enzymes. Annu Rev Biochem 56, 89–124. [DOI] [PubMed] [Google Scholar]
- Standaert L, Adriaens C, Radaelli E, Van Keymeulen A, Blanpain C, Hirose T, Nakagawa S, and Marine JC (2014). The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA 20, 1844–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS, and Spector DL (2009). MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 19, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zhou M, Dorsey TH, Prieto DA, Wang XW, Ruppin E, Veenstra TD, and Ambs S (2018). Integrated proteotranscriptomics of breast cancer reveals globally increased protein-mRNA concordance associated with subtypes and survival. Genome Med 10, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ, et al. (2013). Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, Hao W, MacInnes AW, Cuppen E, and Simonis M (2014). Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol 15, R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila IK, Yao Y, Kim G, Xia W, Kim H, Kim SJ, Park MK, Hwang JP, Gonzalez-Billalabeitia E, Hung MC, et al. (2017). A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 31, 208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, and Chang HY (2011). Molecular mechanisms of long noncoding RNAs. Mol Cell 43, 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li JJ, Guo LY, Li P, Zhao Z, Zhou H, and Di LJ (2018). Molecular link between glucose and glutamine consumption in cancer cells mediated by CtBP and SIRT4. Oncogenesis 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WL, Jiang ZR, Hu C, Chen C, Hu ZQ, Wang AL, Wang L, Liu J, Wang WC, and Liu QS (2020). Pharmacologically inhibiting phosphoglycerate kinase 1 for glioma with NG52. Acta Pharmacol Sin 42, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, and Thompson CB (2010). The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24, 2784–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, and Kingston RE (2014). The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell 55, 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, and Sharp PA (2012). A triple helix stabilizes the 3’ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26, 2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkle M, El-Daly SM, Fabbri M, and Calin GA (2021). Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov 20, 629–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, and Hirose T (2018). Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol Cell 70, 1038–1053. [DOI] [PubMed] [Google Scholar]
- Yoon JH, De S, Srikantan S, Abdelmohsen K, Grammatikakis I, Kim J, Kim KM, Noh JH, White EJ, Martindale JL, et al. (2014). PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun 5, 5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zealy RW, Fomin M, Davila S, Makowsky D, Thigpen H, McDowell CH, Cummings JC, Lee ES, Kwon SH, Min KW, et al. (2018). Long noncoding RNA complementarity and target transcripts abundance. Biochim Biophys Acta 1861, 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Cao L, Zhou R, Yang X, and Wu M (2019). The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat Commun 10, 1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang H, Chen K, Ruan J, Shen S, and Kurgan L (2010). Analysis and prediction of RNA-binding residues using sequence, evolutionary conservation, and predicted secondary structure and solvent accessibility. Curr Protein Peptide Sci 11, 609–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligonucleotide sequences used in this study, Related to STAR Methods.
Data Availability Statement
All the original images have been deposited on Mendeley at http://dx.doi.org/10.17632/nyhjzwtscc.1.
No new code has been generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.