

ABSTRACT
Histone arginine methylation is a prevalent posttranslational modification (PTM) in eukaryotic cells and contributes to the histone codes for epigenetic regulation of gene transcription. In this study, we determined how local changes on adjacent residues in the histone H4 substrate regulate arginine asymmetric dimethylation and symmetric dimethylation catalysed by the major protein arginine methyltransferase (PRMT) enzymes PRMT1 and PRMT5, respectively. We found that phosphorylation at histone H4 Ser-1 site (H4S1) was inhibitory to activities of PRMT1 and PRMT5 in both monomethylating and dimethylating H4R3. Also, a positively charged H4K5 was important for PRMT1 catalysis because acetylation of H4K5 or the loss of the H4K5 ε-amine had a similar effect in reducing the catalytic efficiency of asymmetric dimethylation of H4R3. An opposite effect was observed in that acetylation of H4K5 or the loss of the H4K5 ε-amine enhanced PRMT5-mediated symmetric dimethylation of H4R3. Furthermore, we observed that N-terminal acetylation of H4 modestly decreased asymmetric dimethylation of H4R3 by PRMT1 and symmetric dimethylation of H4R3 by PRMT5. This work highlights the significance of local chemical changes in the substrate to regulating PRMT activity and unravels the pattern complexities and subtleties of histone codes.
KEYWORDS: PRMT, arginine methylation, ADMA, SDMA, acetylation, phosphorylation, histone, posttranslational modification, epigenetics
Introduction
Arginine methylation is a widespread posttranslational modification (PTM) in eukaryotic proteins that is comparable to serine phosphorylation and lysine ubiquitination [1]. The three main types of arginine methylation marks are NG-monomethylarginine (MMA), NG, NG-dimethylarginine (ADMA), and symmetric NG, N’G-dimethylarginine (SDMA). The protein arginine methyltransferases (PRMTs) are classified based on the types of methyl marks they produce: type I (MMA and ADMA), type II (MMA and SDMA), and type III (MMA) (Figure 1). The ADMA and SDMA marks in mammalian cells are mainly deposited by PRMT1 and PRMT5, respectively [2–4]. Knockout of PRMT1 in mouse embryonic fibroblast (MEF) cells reduces the ADMA level in proteins by about 58% [3], and knockout of PRMT5 in MEF cells reduces a prevalent loss of SDMA marks in proteins by approximately 95% [4]. Moreover, ADMA and SDMA modifications can signify divergent biological consequences. Free ADMA, outside the context of a protein (i.e., the free asymmetrically dimethylated arginine amino acid), can be found in systemic circulation, and the free ADMA concentration (not SDMA) is associated with high cardiovascular risks [5,6]. ADMA inhibits nitric oxide synthase (NOS), and administration of 3 mg/kg of ADMA over a course of 45 min in guinea pigs results in significantly increased blood pressure in comparison to a saline solution [7]. In the context of a protein sequence, the ADMA and SDMA marks on the nucleosomal histone tails also confer different functional consequences. Histone H4 methylation at Arg-3 (i.e., H4R3me) is a major epigenetic modification for regulation of gene transcription, as previously reviewed [8]. Symmetric dimethylation of H4R3 (H4R3me2s) by PRMT5 represses gene transcription by recruiting the DNA methyltransferase DNMT3A, which methylates CpG islands to silence gene transcription [9,10]. In contrast, asymmetric dimethylation of H4R3 (H4R3me2a) by PRMT1 recruits histones acetyltransferases p300 and PCAF that can acetylate the lysine residues in H3 and H4 histones and thereby promote gene transcription [11–13]. These and other examples of biological regulation owing to dimethylation of a shared site by the major PRMTs raises the mechanistic question of how ADMA and SDMA productions are regulated.
Figure 1.
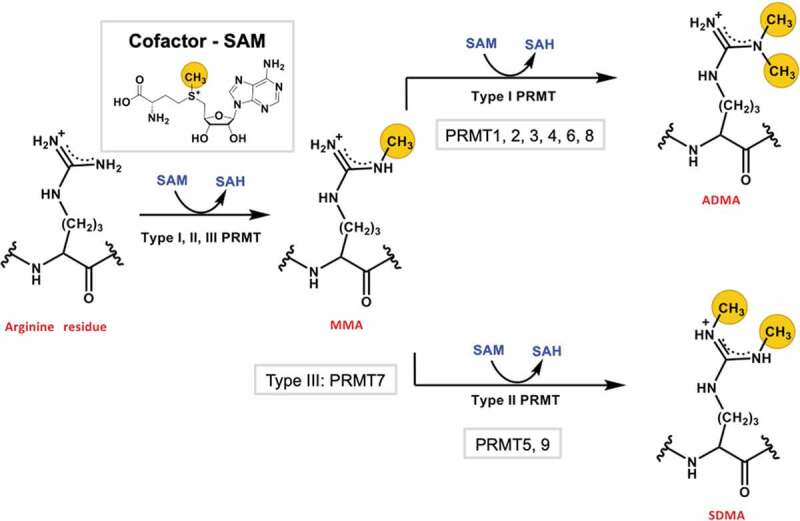
Protein arginine methylation catalysed by PRMTs. All types (I, II, and III) of PRMTs catalyse monomethylation of an arginine residue using the cosubstrate SAM. Type I and II PRMTs further catalyse a second methylation reaction that produce either ADMA or SDMA. The transferred methyl groups are highlighted with yellow circles.
The N-terminal tail of histone H4 is subject to multiple chemical modifications, including alpha-N-acetylation (Ac-H4), Arg-3 methylation (H4R3me), Ser-1 phosphorylation (H4S1ph), and various acylations of lysine residues 5, 8, 12, and 16, etc [8]. These PTMs are dynamic in nature and have distinct biological roles. For instance, H4S1ph and overall acetylation of lysine residues in the H4 N-terminal tail has been observed in the cytoplasm of duck erythroid cells, and translocation of the modified H4 histones to the nucleus results in deacetylation (except the N-terminus) and dephosphorylation of the H4 histones [14]. In previous studies, we have determined how some of the naturally occurring PTMs on the H4 N-terminal tail affect Arg-3 methylation activities of different PRMT enzymes (PRMT1, −3, −5, and −8) [15,16]. Those studies revealed a highly heterogeneous pattern of substrate PTM effects on H4 Arg-3 methylation. For instance, the impact of H4 lysine acetylation on Arg-3 methylation depends on the site of acetylation and the PRMT type. While H4 Lys-5 acetylation (H4K5ac) represses PRMT1-mediated Arg-3 methylation, H4K5ac enhances methylation by PRMT5. Also, Lys-16 acetylation (H4K16ac) subtly increases PRMT1 activity but decreases PRMT5 activity. Furthermore, we found that as the length of the short acyl side-chain modification at Lys-5 becomes longer from acetyl to propionyl to butyryl, an increasing inhibitory action was observed for all the PRMTs. Interestingly, the terminal alpha-N acetylation generally exhibits a slight inhibition on Arg-3 methylation by most PRMTs tested [15].
The arginine methylation process kinetically entails two chemical steps: monomethylation of the guanidine group followed by a second methylation. Our previous transient kinetic experiments reveal that arginine dimethylation is a distributive process in which, after the first turnover of methyl transfer, the monomethylated substrate is released out of the active pocket of PRMT and rebinds to the enzyme for the second methylation [8,17,18]. Our pre-steady-state kinetic results are in fairly good agreement with the steady-state experiments that advocated either fully or partially distributive kinetic behaviours of PRMT catalysis [19–23]. This kinetic model dictates that in the early stage of substrate methylation, the major product will be monomethylarginine, and only when the amount of monomethylarginine arises to a level comparable to the initial substrate, dimethylarginine product begins to appreciably accumulate [18]. In our previous reports on studying PTM crosstalks on the H4 tail, synthetic H4 peptides with an unmodified Arg-3 residue were used as PRMT substrates. Under the initial velocity condition, the products are primarily the monomethylarginine product H4R3me, with limited amounts of H4R3me2 formed. Because ADMA and SDMA have distinct biological consequences in the cell, herein we sought to determine how the formation of asymmetrically and symmetrically dimethylated H4R3 (i.e., H4R3me2a and H4R3me2s formation) was regulated by the adjacent PTMs on the H4 substrate, especially with regard to changes in local charges (e.g., Lys-5 acetylation and Ser-1 phosphorylation) (Figure 2). The results provide new molecular-level insights into the mechanisms of PRMT activity regulation as well as reveal the complex intercommunications between histone modification marks.
Figure 2.
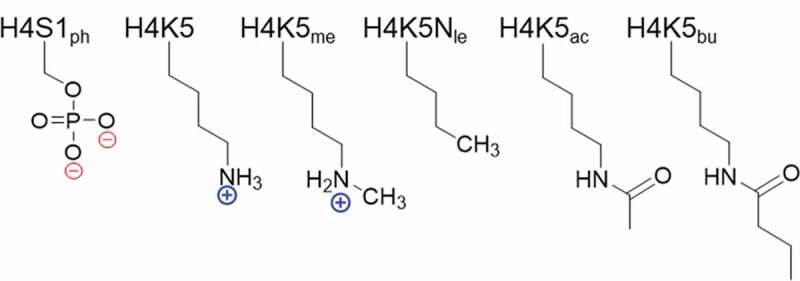
Chemical modifications at residues Ser-1 and Lys-5 on the H4 substrate. Modifications include phosphorylation (ph), methylation (me), norleucine substitution (Nle), acetylation (ac), and butyrylation (bu).
Results
Effects of H4S1 phosphorylation and H4K5 modifications on arginine methylation activity by PRMT1
To determine how chemical modifications with positive, negative, and neutral properties in the substrate may regulate PRMT activity on the shared H4R3 site, we performed filter-based radioactive methylation assays with [14C]SAM as a cosubstrate, either PRMT1 or PRMT5, and a small panel of synthetic histone H4 peptides (residues 1–20) with chemical modifications at H4S1 or H4K5 sites (Table 1). The methylation reactions were quenched with isopropanol, loaded onto P81 filter paper, and washed with sodium bicarbonate buffer (pH 9). The filter paper was allowed to air dry before immersion in Ultima Gold scintillation cocktail to prepare samples for scintillation counting. Measured counts per minute (CPM) values were converted to concentration of methylated product based on the known concentration of a [14C]SAM internal standard as previously described [15].
Table 1.
Sequences and molecular weights of synthetic H4 peptides
| Substrate | Sequence | Theoretical MW | Measured MW |
|---|---|---|---|
| Ac-H4(1–20) | Ac-SGRGKGGKGLGKGGAKRHRK | 2034.37 | 2034.9 |
| Ac-H4(1–20)S1ph | Ac-SphGRGKGGKGLGKGGAKRHRK | 2113.15 | 2114.3 |
| Ac-H4(1–20)K5me | Ac-SGRGKmeGGKGLGKGGAKRHRK | 2048.40 | 2048.2 |
| Ac-H4(1–20)K5Nle | Ac-SGRGNleGGKGLGKGGAKRHRK | 2019.35 | 2019.6 |
| Ac-H4(1–20)K5ac | Ac-SGRGKacGGKGLGKGGAKRHRK | 2076.41 | 2076.4 |
| Ac-H4(1–20)K5bu | Ac-SGRGKbuGGKGLGKGGAKRHRK | 2104.46 | 2104.3 |
| Ac-H4(1–20)R3me | Ac-SGRmeGKGGKGLGKGGAKRHRK | 2048.40 | 2048.3 |
| NH2-H4(1–20)R3me | NH2-SGRmeGKGGKGLGKGGAKRHRK | 2006.36 | 2007.2 |
| Ac-H4(1–20)S1phR3me | Ac-SphGRmeGKGGKGLGKGGAKRHRK | 2128.37 | 2128.0 |
| Ac-H4(1–20)R3meK5me | Ac-SGRmeGKmeGGKGLGKGGAKRHRK | 2062.42 | 2062.2 |
| Ac-H4(1–20)R3meK5Nle | Ac-SGRmeGNleGGKGLGKGGAKRHRK | 2033.38 | 2033.7 |
| Ac-H4(1–20)R3meK5ac | Ac-SGRmeGKacGGKGLGKGGAKRHRK | 2090.43 | 2090.6 |
| Ac-H4(1–20)R3meK5bu | Ac-SGRmeGKbuGGKGLGKGGAKRHRK | 2118.49 | 2118.0 |
To understand the overall arginine methylation by PRMT1, we first performed the radioactive methylation assay with recombinant human PRMT1 enzyme and substrates Ac-H4(1–20), Ac-H4(1–20)S1ph, Ac-H4(1–20)K5me, Ac-H4(1–20)K5ac, Ac-H4(1–20)K5Nle (Nle is norleucine), and Ac-H4(1–20)K5bu. We observed that phosphorylation of H4S1 reduced the rate of arginine methylation by approximately 2-fold [0.50 ± 0.020 min−1 with Ac-H4(1–20) vs. 0.22 ± 0.029 min−1 with Ac-H4(1–20)S1ph] (Figure 3(a)). The H4S1ph mark was reported to coexist with monomethylated, asymmetrically dimethylated, and symmetrically dimethylated H4R3 during early development of X. laevis[24]. We were curious to know if the inhibitory effect of H4S1ph on arginine methylation was present with other PRMTs that methylate H4. Using the radioactive methylation assay with two synthetic substrates, Ac-H4(1–20) and Ac-H4(1–20)S1ph, we observed that phosphorylation reduced arginine methylation by several other PRMT members (Fig. S1). The rate of arginine methylation was reduced by 3-fold for PRMT1, 8-fold for PRMT3, 5-fold for PRMT8, and 3-fold for PRMT5. The inhibitory effect of H4S1ph on arginine methylation by PRMT5 is consistent with previous observations [25]. To determine the underlying mechanism for the reduction in arginine methylation by H4S1ph, we characterized the steady-state kinetics of these enzymatic methylations by measuring PRMT1 activity as a function of peptide substrate concentrations. The Hill equation was applied to fit the concentration-rate curves as previously described [15] to determine kcat, K0.5, and n parameters (Table 2, Figure 3(b), and Fig. S2), wherein kcat was the turnover number, K0.5 was the substrate concentration at the half-maximal velocity (equivalent to Km in the Michaelis-Mention equation), and n was the Hill coefficient. In comparison to Ac-H4(1–20), H4S1 phosphorylation reduced kcat by 2-fold (0.26 ± 0.0070 min−1) and increased K0.5 by over 11-fold (5.2 ± 0.36 µM). These results suggest that the phospho group on Ser-1 mainly affected substrate binding with PRMT1, with a minor influence on the turnover.
Figure 3.
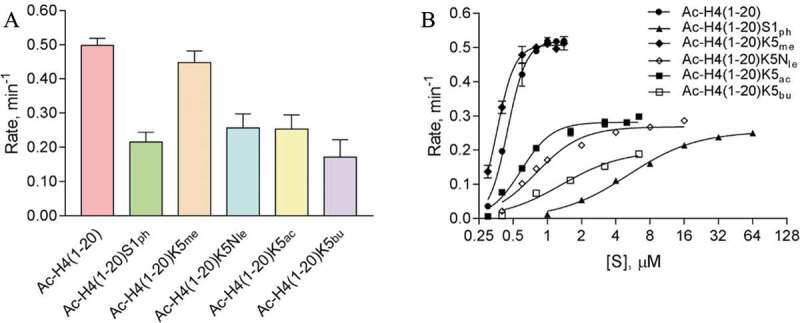
Arginine methylation of H4 peptides catalysed by PRMT1. (a) Single-point radioactive methylation assay of PRMT1 with H4 peptides. Reactions proceeded for 10 min at 30°C with 0.05 µM PRMT1, 15 µM [14C]SAM, and 15 µM of peptide substrate. (b) PRMT1 steady-state kinetics with H4K5 modified H4 peptides. Reactions proceeded for 15 min at 30°C with 0.05 µM PRMT1 and 15 µM [14C]SAM. With substrates Ac-H4(1–20) and Ac-H4(1–20)K5me, reactions were held for 8 min. Error bars represent standard deviation.
Table 2.
Steady-state kinetic parameters for H4R3 methylation by PRMT1
| Substrate | kcat (min−1) | K0.5 (µM) | n |
kcat/K0.5 (min−1 µM−1) |
|---|---|---|---|---|
| Ac-H4(1–20) | 0.52 ± 0.0078 | 0.45 ± 0.0085 | 5.3 ± 0.43 | 1.2 ± 0.028 |
| Ac-H4(1–20)S1ph | 0.26 ± 0.0070 | 5.2 ± 0.36 | 1.4 ± 0.12 | 0.050 ± 0.0037 |
| Ac-H4(1–20)K5me | 0.51 ± 0.0033 | 0.36 ± 0.0029 | 5.5 ± 0.26 | 1.4 ± 0.015 |
| Ac-H4(1–20)K5Nle | 0.27 ± 0.010 | 0.71 ± 0.047 | 2.8 ± 0.60 | 0.38 ± 0.029 |
| Ac-H4(1–20)K5ac | 0.28 ± 0.0088 | 0.59 ± 0.032 | 3.2 ± 0.52 | 0.47 ± 0.030 |
| Ac-H4(1–20)K5bu | 0.20 ± 0.031 | 1.3 ± 0.39 | 1.6 ± 0.57 | 0.15 ± 0.052 |
| Ac-H4(1–20)R3me | 0.40 ± 0.0062 | 0.58 ± 0.020 | 4.5 ± 0.62 | 0.69 ± 0.026 |
| NH2-H4(1–20)R3me | 0.17 ± 0.0075 | 0.42 ± 0.022 | 4.4 ± 0.92 | 0.40 ± 0.028 |
| Ac-H4(1–20)S1phR3me | 0.092 ± 0.0025 | 1.5 ± 0.12 | 1.0 ± 0.12 | 0.061 ± 0.0052 |
| Ac-H4(1–20)R3meK5me | 0.30 ± 0.0034 | 0.52 ± 0.014 | 3.3 ± 0.25 | 0.58 ± 0.017 |
| Ac-H4(1–20)R3meK5Nle | 0.20 ± 0.0054 | 0.59 ± 0.028 | 3.3 ± 0.51 | 0.34 ± 0.019 |
| Ac-H4(1–20)R3meK5ac | 0.17 ± 0.0052 | 0.57 ± 0.0045 | 2.5 ± 0.43 | 0.30 ± 0.0094 |
| Ac-H4(1–20)R3meK5bu | 0.076 ± 0.0052 | 0.87 ± 0.16 | 1.3 ± 0.26 | 0.087 ± 0.017 |
All kinetic parameters were determined using 15 µM [14C]SAM, 0.05 µM PRMT1, and varying concentrations of H4 peptide. Reactions with Ac-H4(1–20) and Ac-H4(1–20)K5me were held for 8 min at 30°C, while all other reactions were held for 15 min at 30°C.
Consistent with our previous observation[15], methylation of H4K5 did not affect the rate of arginine methylation by PRMT1 [0.45 ± 0.034 min−1 for Ac-H4(1–20)K5me] (Figure 3(a)). In the absence of the H4K5 ε-amine (norleucine substitution) or upon acetylation of H4K5, the rates of arginine methylation by PRMT1 decreased by 2-fold [0.26 ± 0.042 min−1 for Ac-H4(1–20)K5Nle and 0.25 ± 0.041 min−1 for Ac-H4(1–20)K5ac] as compared to the wild-type Ac-H4(1–20) substrate. Introducing a bulkier modification, butyrylated H4K5 further reduced the rate of arginine methylation to 0.17 ± 0.051 min−1. From our steady-state kinetics characterization (Table 2, Figure 3(b), and Fig. S2), the kcat values for H4K5 unmethylated and methylated substrates were similar (0.52 ± 0.0078 min−1 vs. 0.51 ± 0.0033 min−1, respectively). In contrast, there was a nearly 2-fold reduction in kcat upon acetylation of H4K5 (0.28 ± 0.0088 min−1) or loss of the ε-amine (0.27 ± 0.010 min−1). Also, the K0.5 values for Ac-H4(1–20)K5ac (0.59 ± 0.032 µM) and Ac-H4(1–20)K5Nle (0.71 ± 0.047 µM) were greater in comparison to the unmodified substrate, and this contributed to the overall reduction in catalytic efficiency of 0.47 ± 0.030 and 0.38 ± 0.029 min−1 µM−1 for these modifications, respectively. Moreover, the bulkier butyrylation of H4K5 resulted in a nearly 3-fold increase in K0.5 (1.3 ± 0.39 µM) and > 2-fold reduction in kcat (0.20 ± 0.031 min−1) when compared to Ac-H4(1–20). Hence, while the bulkier modification does appear to impair substrate recognition, maintaining the charge on H4K5 is important for PRMT1 catalysis.
Effects of H4S1 phosphorylation and H4K5 modifications on H4R3 methylation activity by PRMT5
For comparison with PRMT1, we also studied the effect of local modifications on the H4R3 methylation activity of the major type-II enzyme PRMT5. First, a single-point radioactive methylation assay was performed with recombinant human PRMT5-MEP50 complex and several modified H4 substrates (Figure 4). Methylation of H4K5 did not appear to significantly affect arginine methylation by PRMT5 (Figure 4(a)), consistent with previous results[15]. Upon acetylation or loss of the H4K5 ε-amine, there was a mild increase in the rate of arginine methylation. This is an indication that the absence of the H4K5 ε-amine increased the rate of arginine methylation, though acetylation appeared to have the strongest impact on increasing the rate of arginine methylation by PRMT5. Upon determining the steady-state kinetic parameters of PRMT5 with the H4K5 modified substrates, a similar trend appeared that the kcat values increased upon acetylation (0.12 ± 0.0026 min−1) or the absence of the H4K5 ε-amine (0.10 ± 0.0033 min−1) in comparison to the unmodified (0.090 ± 0.0039 min−1) or methylated H4K5 (0.066 ± 0.0016 min−1) (Table 3, Figure 4(b), and Fig. S3). Interestingly, the K0.5 value for H4K5me substrate was the lowest (0.25 ± 0.012 µM) and the Ac-H4(1–20)K5Nle substrate had the highest K0.5 (0.48 ± 0.011 µM). In contrast, the catalytic efficiency between Ac-H4(1–20)K5 and Ac-H4(1–20)K5me was about the same (0.26 min−1 µM−1). Ac-H4(1–20)K5Nle had the lowest catalytic efficiency (0.21 ± 0.084 min−1 µM−1), while Ac-H4(1–20)K5ac had the highest (0.40 ± 0.013 min−1 µM−1). As mentioned above, H4S1ph inhibited arginine methylation by PRMT5 (Figure 4(a)). We attempted to determine the steady-state kinetic parameters of PRMT5 for the Ac-H4(1–20)S1ph substrate, but the CPM values were barely above the background to provide reliable rate-concentration curves for (Fig. S4). Overall, we conclude that the absence of the positively charged H4K5 ε-amine was beneficial for PRMT5 turnover, yet further modification of H4K5 to have an acetyl group appeared to have an additive effect in increasing the rate of arginine methylation and substrate binding, thus enhancing the overall catalytic efficiency of PRMT5.
Figure 4.
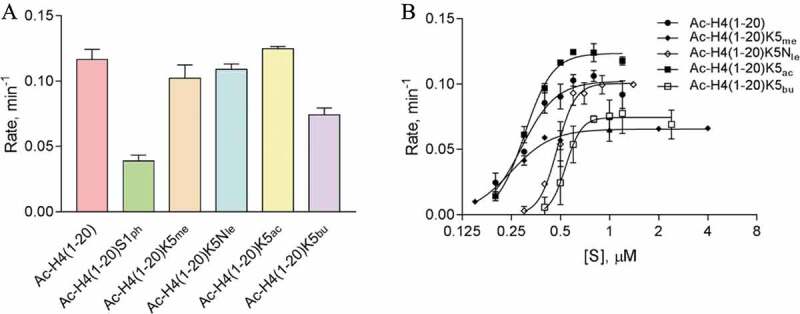
Arginine methylation of H4 peptides catalysed by PRMT5. (a) Single-point radioactive methylation assay of PRMT5 with H4 peptides. Reactions proceeded for 10 min at 30°C with 0.25 µM PRMT5, 15 µM [14C]SAM, and 15 µM of peptide substrate. (b) PRMT5 steady-state kinetics with H4 peptides. Reactions proceeded for 15 min at 30°C with 0.05 µM PRMT5 and 15 µM [14C]SAM. Error bars represent standard deviation.
Table 3.
Steady-state kinetic parameters for H4R3 methylation by PRMT5
| Substrate | kcat (min−1) | K0.5 (µM) | n |
kcat/K0.5 (min−1 µM−1) |
|---|---|---|---|---|
| Ac-H4(1–20) | 0.090 ± 0.0039 | 0.35 ± 0.014 | 7.6 ± 1.8 | 0.26 ± 0.015 |
| Ac-H4(1–20)S1ph | ND | ND | ND | ND |
| Ac-H4(1–20)K5me | 0.066 ± 0.0016 | 0.25 ± 0.012 | 3.5 ± 0.45 | 0.26 ± 0.014 |
| Ac-H4(1–20)K5Nle | 0.10 ± 0.0033 | 0.48 ± 0.011 | 7.7 ± 1.2 | 0.21 ± 0.084 |
| Ac-H4(1–20)K5ac | 0.12 ± 0.0026 | 0.30 ± 0.0073 | 5.1 ± 0.57 | 0.40 ± 0.013 |
| Ac-H4(1–20)K5bu | 0.074 ± 0.0018 | 0.54 ± 0.0095 | 9.0 ± 1.4 | 0.14 ± 0.0041 |
All kinetic parameters were determined using 15 μM [14C]SAM, 0.05 μM PRMT5, and varying concentrations of H4 peptide. All reactions were conducted at 30°C for 15 min. ND indicates that the parameters were not determined due to a lack of detectable changes in signal of methylated peptide product as the peptide concentration increased.
Effects of H4S1 phosphorylation and H4K5 modifications on the asymmetric dimethylation activity by PRMT1 (ADMA formation)
A substrate arginine residue can be methylated by PRMTs (except type III enzyme PRMT7) into monomethyl and dimethyl products. Upon examination of the major products by mass spectrometry with the H4R3 peptide used as substrate, we found that the major product was monomethylated arginine for PRMT1 while the methylated products of PRMT3, −5, and −8 were undetectable by mass spectrometry due to very low yields (Fig. S5). These results coincide with our kinetic model that arginine dimethylation in PRMT catalysis is a highly distributive process[18]. Therefore, the measured PRMT activities described earlier are largely a reflection on the MMA activity, that is, the first-step of H4R3 methylation. To examine how H4S1 phosphorylation and H4K5 modifications affect the ADMA activity of PRMT1, we made several H4 peptides that contained H4R3 in the monomethylated state (H4R3me) (Table 1). Radiometric biochemical tests were conducted to measure PRMT1 activity in methylating these H4R3me substrates. As shown in Figure 5(a), in comparison to the Ac-H4(1–20)R3me control (0.36 ± 0.0052 min−1), we observed that phosphorylation of H4S1 led to a > 3-fold reduction in the rate (0.11 ± 0.0035 min−1) of asymmetric arginine methylation by PRMT1. In contrast, methylation of H4K5 did not appear to affect the rate of asymmetric dimethylation (0.38 ± 0.019 min−1). Interestingly, the removal of the ε-amine by substituting the H4K5 with norleucine or acetylation of H4K5 resulted in an approximately 2-fold reduced rates in asymmetric dimethylation by PRMT1 in comparison to the positive control [0.19 ± 0.0034 min−1 for Ac-H4(1–20)R3meK5Nle and 0.20 ± 0.013 min−1 for Ac-H4(1–20)R3meK5ac]. Adding the bulky butyryl group to H4K5 further reduced the rate of asymmetric dimethylation to 0.10 ± 0.0046 min−1.
Figure 5.
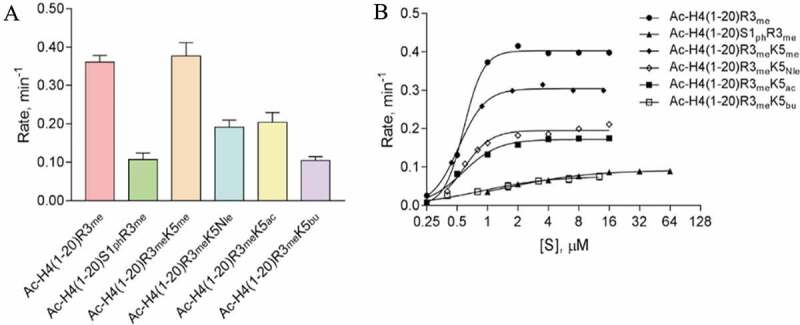
Impact of chemical modifications to H4S1 and H4K5 on PRMT1-mediated methylation of H4R3me substrate (ADMA formation). (a) Single-point radioactive methylation assay performed with 0.05 µM PRMT1, 15 µM [14C]SAM, and 15 µM of peptide substrate at 30°C for 10 min. (b) Overlay of PRMT1 steady-state kinetics with substrates Ac-H4(1–20)R3me, Ac-H4(1–20)S1phR3me, Ac-H4(1–20)R3meK5me, Ac-H4(1–20)R3meK5Nle, Ac-H4(1–20)R3meK5ac, or Ac-H4(1–20)R3meK5bu. Reactions were held for 15 min at 30°C with 0.05 µM PRMT1 and 15 µM [14C]SAM. Error bars represent the standard deviation.
To understand the underlying mechanism for the changes in rates, we determined the steady-state kinetic parameters for PRMT1 with the same H4R3me substrates containing H4S1 or H4K5 chemical modifications (Figure 5(b), Table 2, and Fig. S6). The results were gained by fitting the rate-concentration data curves with the Hill equation. For Ac-H4(1–20)R3me, we determined a kcat of 0.40 ± 0.0062 min−1, a K0.5 of 0.58 ± 0.020 μM, a Hill coefficient of 4.5 ± 0.62 (potentially positive cooperativity), and a catalytic efficiency (kcat/K0.5) of 0.69 ± 0.026 min−1 μM−1 (Table 2). Upon phosphorylation of H4S1, the kcat was reduced approximately 4-fold while the K0.5 was increased approximately 2.6-fold, which contributed to the overall ~11-fold reduction in the catalytic efficiency for PRMT1. Interestingly, the Hill coefficient was 1.0 ± 0.12 for the phosphorylated substrate, indicating no apparent cooperativity. Methylation of H4K5 reduced the turnover number to 0.30 ± 0.0034 min−1 and the K0.5 to 0.52 ± 0.014 μM, and this resulted in a lower catalytic efficiency of 0.58 ± 0.017 min−1 μM−1 in comparison to the control, Ac-H4(1–20)R3me. Substrates with acetylation of H4K5 or substitution of the Lys-5 with norleucine had similar kinetic parameters. In comparison to the H4R3me control substrate, the kcat values were reduced by at least 2-fold to 0.20 ± 0.0054 min−1 with Ac-H4(1–20)R3meK5Nle and 0.17 ± 0.0052 min−1 with Ac-H4(1–20)R3meK5ac. Interestingly, the K0.5 values for the Ac-H4(1–20)R3meK5Nle (0.59 ± 0.028 μM) and Ac-H4(1–20)R3meK5ac (0.57 ± 0.0045 μM) were close to the Ac-H4(1–20)R3me control substrate (0.58 ± 0.020 μM). Nonetheless, the reduced kcat values with substrates Ac-H4(1–20)R3meK5Nle and Ac-H4(1–20)R3meK5ac contributed to the > 2-fold reduction in catalytic efficiency in comparison to the H4R3me control substrate. Altogether, these results supported that phosphorylation of H4S1 was inhibitory to PRMT1 catalysis of asymmetric dimethylation and hinders substrate recognition. Methylation of H4K5 appeared to have a greater impact on PRMT1 catalysis than substrate binding. Lastly, neutralization of the H4K5 charge by acetylation or removal of the ε-amine resulted in a similar inhibition of PRMT1 catalysis and little effect on substrate recognition by PRMT1. Thus, retaining the positive charge on H4K5 is important for PRMT1 catalysis of asymmetric dimethylation of arginine.
Effects of H4S1 phosphorylation and H4K5 modifications on the symmetric dimethylation activity by PRMT5 (SDMA formation)
To examine how positive, negative, or neutral chemical modifications affect symmetric dimethylation of H4R3 by PRMT5, we performed a single-point radioactive methylation assay on the relevant H4 peptides containing the R3me mark (Figure 6). Overall, phosphorylation of H4S1 reduced the rate of symmetric dimethylation just over 2-fold (0.0098 ± 0.00089 min−1) in comparison to the H4R3me control substrate (0.025 ± 0.0010 min−1). Also, methylation of H4K5 was modestly inhibitory (0.019 ± 0.0013 min−1) to H4R3me methylation. The loss of the ε-amine or acetylation of H4K5 led to similar enhanced rates of symmetric dimethylation by PRMT5 (0.034 ± 0.00067 min−1 and 0.035 ± 0.0016 min−1, respectively). Moreover, using a longer acyl modification, butyrylated H4K5, modestly increased the rate of symmetric dimethylation by PRMT5 to 0.031 ± 0.0035 min−1. We attempted to determine the steady-state kinetic parameters of PRMT5 with these various H4 substrates, but we could not detect meaningful changes in the methylated products at 0.05 μM PRMT5 condition, due to weak signals of the methylation reaction (Fig. S4). Nonetheless, these results support that the presence of the positive charge on H4K5 or adding a negatively charged phosphate group on H4S1 does not favour symmetric dimethylation by PRMT5. Neutralizing the charge of H4K5 by acetylation or removing the ε-amine favours symmetric dimethylation by PRMT5. Plus, PRMT5 did not appear to be hindered from methylating H4R3me in the presence of butyrylated H4K5, which was different from its modest inhibitory effect on the H4R3 monomethylation reaction (Figure 4).
Figure 6.
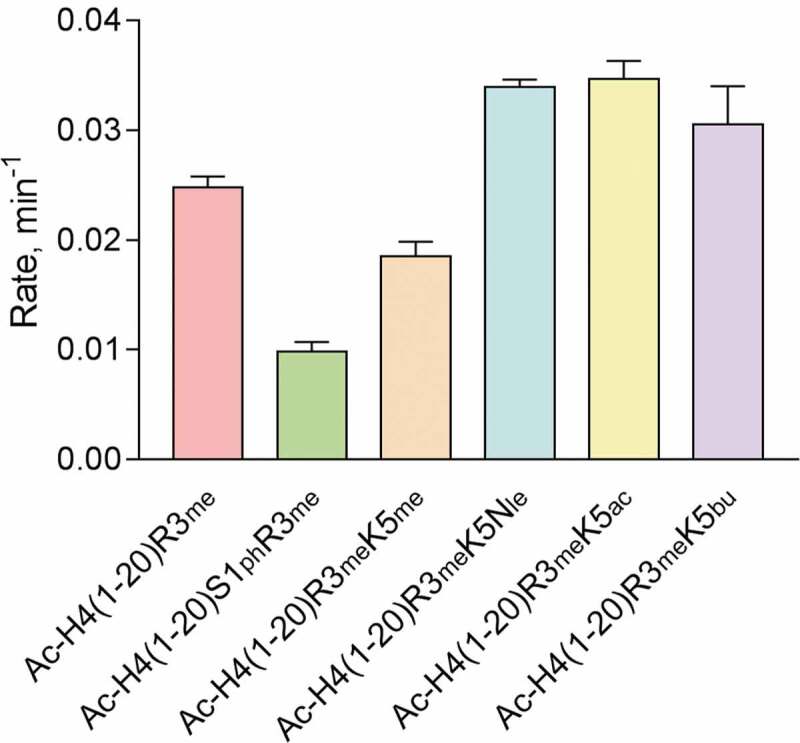
Impact of chemical modifications to H4S1 and H4K5 on PRMT5-mediated methylation of H4R3me substrate (SDMA formation). Single-point radioactive methylation assay performed with 0.25 µM PRMT5, 15 µM [14C]SAM, and 15 µM of peptide substrate at 30°C for 10 min. Error bars represent the standard deviation.
Effects of H4 N-terminal acetylation on ADMA activity of PRMT1 and SDMA activity of PRMT5
The N-terminal alpha-amino acetylation is an evolutionarily conserved modification and highly prevalent in human proteins (84%) [26]. Nα-acetyltransferase Nat4 in yeast and Naa40p (NatD) in humans were reported to acetylate the N-terminus of H4 [27,28]. While H4 N-terminal acetylation is inhibitory to H4R3 methylation by the yeast arginine methyltransferase hnRNP arginine N-methyl methyltransferase 1(Hmt1) and supports ribosomal DNA expression [29], it remains understudied as to how this modification impacts human PRMTs, particularly the effects on ADMA and SDMA production. To determine how the N-terminal acetylation of H4 affects type I and II PRMT activities, we performed a single-point radioactive methylation assay with PRMT1 and PRMT5 using Ac-H4(1–20)R3me and NH2-H4(1–20)R3me as substrates. For PRMT1, the absence of the N-terminal acetyl group modestly enhanced the rate of ADMA production [0.21 ± 0.00051 min−1 for Ac-H4(1–20)R3me vs. 0.24 ± 0.011 min−1 for NH2-H4(1–20)R3me] (Figure 7(a)). For PRMT5, the absence of the N-terminal acetyl group also seemed to enhance the rate of SDMA production [0.017 ± 0.0012 min−1 for Ac-H4(1–20)R3me vs. 0.021 ± 0.0032 min−1 for NH2-H4(1–20)R3me] (Figure 7(b)). To better understand the mechanism, we proceeded to determine the steady-state kinetic parameters for PRMT1 with NH2-H4(1–20)R3me. The absence of N-terminal acetylation resulted in an approximately 2-fold reduction in kcat (0.17 ± 0.0075 min−1) yet moderately improved K0.5 (0.42 ± 0.022 μM) in comparison to the Ac-H4(1–20)R3me control (Table 2 and Figure 7(c)). Overall, the data here demonstrated that the N-terminal acetylation of H4 slightly reduced PRMT1 and PRMT5 activity on Arg-3 dimethylation, which appeared to be in accord with the inhibitory effect of H4 N-terminal acetylation on Hmt1 activity in yeast [29].
Figure 7.
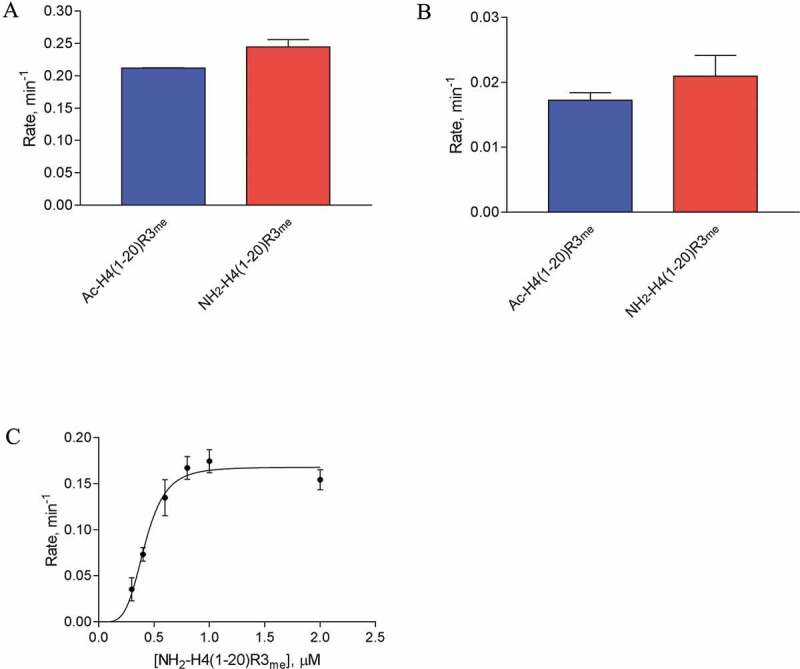
Impact of the N-terminal acetylation on H4R3me methylation by PRMT1 and PRMT5. (a) Single-point radioactive methylation assay performed with 0.05 µM PRMT1, 15 µM [14C]SAM, and 15 µM of peptide substrate at 30°C for 10 min. (b) Single-point radioactive methylation assay performed with 0.25 µM PRMT5, 15 µM [14C]SAM, and 15 µM of peptide substrate at 30°C for 10 min. (c) PRMT1 kinetics with substrate NH2-H4(1–20)R3me. Reactions were held for 15 min at 30°C with 0.05 µM PRMT1 and 15 µM [14C]SAM. All error bars represent standard deviation.
Discussion
The nucleosomal core histones are a hotbed of PTMs, especially in the N-terminal tail region, whose stochastic combination and intercommunication generate a herculean number of ‘histone codes’ for epigenetic regulation of genomic function [30,31]. An interesting observation is that different PRMTs may methylate the same arginine residues in a protein sequence and the type of methylation marks can result in divergent outcomes. Some notable examples include histone sites H3R2 and H4R3. Asymmetric dimethylation of H3R2 by PRMT6 represses transcription by antagonizing the deposit of a transcription activating mark H3K4me3 by the MLL1 complex [32–34]. In contrast, symmetric dimethylation of H3R2 by PRMT5 favours mono- and dimethylation of H3K4 by the MLL complex and H3R2me2s has been observed to coexist with H3K4me3 at highly transcribed genes [35,36]. For the H4R3 site, asymmetric dimethylation by PRMT1 is associated with active gene expression while symmetric dimethylation by PRMT5 represses gene transcription [8]. Previously, we studied how H4K5 acylations influence Arg-3 methylation by PRMT1 and PRMT5 [15,16]. Our data showed that H4 Lys-5 acetylation (H4K5ac) represses PRMT1-mediated Arg-3 methylation, H4K5ac enhances methylation by PRMT5. As the length of the short acyl side-chain modification at Lys-5 becomes longer from acetyl to propionyl to butyryl, an increasing inhibitory action was observed for all the PRMTs. A limitation of these previous studies was that the experiments were performed under initial velocity condition, and thus the products mainly contained the monomethylated arginine (H4R3me1), with limited amounts of H4R3me2 formed. Therefore, the PTM cross-talk effects observed before mainly reflected how the PTMs affected the MMA activity of PRMTs. In this work, we made an in-depth dive into the effects of the various histone PTMs on the H4R3 dimethylation activities of PRMT1 and PRMT5. Also, we investigated how Ser-1 phosphorylation impacted on H4R3 methylation.
The regulatory mechanisms of PRMT activity are not well studied. We and others have proposed that local chemical changes on the substrates caused by different PTM enzymes may form an important factor in regulating PRMT activity and specificity [8,25,37]. The major epigenetic mark H4R3 is a shared substrate for several PRMT enzymes, including PRMT1, −3, −5, −6, and −8 [21,33,38–42], with PRMT1 and PRMT5 being the dominant enzymes that catalyse ADMA and SDMA of H4R3 in vivo, respectively [2,9,38,43,44]. Notably, the histone H4 N-terminus nearby the Arg-3 site harbours multiple PTM marks such as Lys-5 methylation, acetylation, propionylation, crotonylation, butyrylation, 2-hydroxyisobutyrylation, citrullination, succinylation, formylation, and Ser-1 phosphorylation [45]. Understanding how the chemical properties of neighbouring PTMs modulate H4R3 methylation not only is of significance in cracking the histone codes but also offers molecular-scale insights into the regulatory mechanism of PRMT activity at the substrate level.
Our biochemical results show that different PTM groups, as well as the absence of the positively charged H4K5 ε-amine, can differentially affect the rates of ADMA production by PRMT1 and SDMA production by PRMT5 on H4R3. H4K5me does not affect the overall rate of H4R3 methylation by PRMT1, consistent with our previous observation [15]; and we observed in this work that H4K5me also does not strongly affect PRMT1-catalysed ADMA formation from H4R3me either (Figure 5(a)). In contrast, acetylation and other short chain acylation greatly reduced PRMT1 activity either on H4R3 or H4R3me substrate. Removal of the H4K5 ε-amino group led to the same reduction effect. The average pKa for a Lys side chain is 10.5 [46], and possibly lower to 8.2 in the context of the histone H4 sequence [47]. Physiological pH is reported to be approximately 7.2 for the cytosol and nucleus [48], and our reaction conditions were performed in a HEPES buffer system at pH 8. Either in cells or in our reactions, we would expect that the side chain of H4K5 to be protonated. Also, there are negative electrostatic regions just outside the active site of PRMT1 that may support the interaction with the positively charged H4K5 (Figure 8(a)). These data suggest that maintaining the positive charge of H4K5 was important for PRMT1 catalysis. Similar to PRMT1, H4K5me did not strongly affect monomethylation of H4R3 by PRMT5 (Figure 4 and ref. [15]); however, we did observe that H4K5me reduced PRMT5-catalysed symmetric dimethylation of H4R3me (Figure 6). In the PRMT5/MEP50 crystal structure with the H4 peptide and a SAM analogue, H4K5 forms H-bonds with Ser-310 in PRMT5, though these interactions appear to be between the backbone of the peptide and protein sequence [23]. In an electrostatic potential map, the area that Lys-5 occupies appears to be in a region of PRMT5 that is rich in negative electrostatic potential (Figure 8(b)). In this region, it appears difficult to explain why the loss of the Lys-5 ε-amine would be favourable for PRMT5 catalysis without experimental structural data to examine the molecular interactions.
Figure 8.
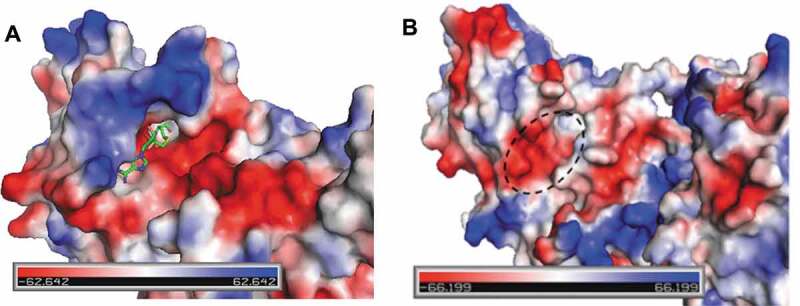
Electrostatic potential of PRMT1 and PRMT5. (a) A view of the active site of rat PRMT1 complexed with SAH (PDB ID 1ORI). SAH is coloured by element with green (carbon), blue (nitrogen), red (oxygen), and yellow (sulphur). (b) Electrostatic potential of human PRMT5 (PDB ID 5FA5) with the expected site for histone H4K5 to bind (dashed circle). The electrostatic potential range is depicted from red (electronegative) to blue (electropositive). The images were rendered with PyMOL.
We observed that H4S1 phosphorylation is inhibitory to arginine methylation of H4R3 by PRMT1, −3, −5, and −8 (Fig. S1). Ho and colleagues also observed that H4S1ph was inhibitory to PRMT5-catalysed arginine methylation of the H4(1–20) substrate, and this inhibitory effect appeared to dominate any other PTMs that could enhance arginine methylation when H4S1ph was absent (e.g., H4K5ac and H4K20me3) [25]. All PRMTs have a pair of conserved glutamate (EE) residues known as the ‘double-E loop’ (e.g., Glu-435 and Glu-444 in PRMT5) within the active site that is important for forming hydrogen bonds with the guanidino nitrogens of the substrate arginine [49]. Given the close proximity of the H4S1ph to H4R3, the negatively charged phosphate group on H4S1 may form an intramolecular salt bridge with the positively charged guanidine group of H4R3, and this may hinder substrate recognition of PRMT1 and PRMT5 (Fig. S7). Another possibility is that the protein surface of PRMT1 and PRMT5 may be a hindrance for accommodating a negatively charged phosphoserine. The area just outside the active site of PRMT1 appears to be hydrophobic and electronegative (Figure 8(a)), which would be unfavourable for the potentially −2 charged phosphoserine. For PRMT5, the region that H4S1 is expected to bind in the 4GQB crystal structure appears mostly hydrophobic (Phe-300, Tyr-304, Phe-580, Gln-309), and this would be unfavourable either for the highly, negatively charged phosphoserine (Figure 8(b)). An X-ray crystal structure would be of value to better understand how these chemical modifications regulate PRMT catalysis of the histone H4 substrate.
Ser-1 phosphorylation and Arg-3 methylation on H4 and H2A were reported previously to coexist in vivo at high levels during the later embryonic stages of X. laevis[24]. H4S1ph has been observed in newly synthesized histones [14,50], in condensed chromatin during mitosis[50], in D. melanogaster and mouse spermatogenesis [51], in S. cerevisiae sporulation [51], and at sites of DNA double-strand breaks (DSB) that are repaired by nonhomologous end joining (NHEJ) [52]. Recombinant human casein kinase II (CK2) was shown to phosphorylate H4S1 in vitro, and deletion of CK2 in yeast reduced H4S1ph levels in vivo[52]. The activity of CK2α in Ser-1 phosphorylation seems to favour for an H4 peptide without N-terminal acetylation [53]. Possibly, CK2α activity in H4S1 phosphorylation is jointly regulated by N-terminal acetylation and Arg-3 methylation. A further detailed inspection on this interrelationship is warranted in the future, especially in the cellular context. Yet we observed that H4S1ph is inhibitory to Arg-3 methylation by all the examined PRMTs (Fig. S1), supporting that the absence of H4S1ph would promote arginine-3 methylation. We posited that the H4R3 methylation mark is likely introduced by PRMTs prior to the setup of Ser-1 phosophorylation[8]. Except histone H4, there are other proteins in which serine phosphorylation and arginine methylation intercommunicates [54,55]. For instance, some consensus site motifs in kinase Akt substrates contain both phosphorylated serine and methylated arginine, and serine phosphorylation and arginine methylation are mutually exclusive and have opposing effects in generating neurotoxicity [56]. Smith et al. [57] recently found that the presence of phosphorylation in an SRGG motif in yeast proteins Npl3p and Nop1p dramatically decreases arginine methylation mediated by the methyltransferase Hmt1p. It appears to be a general rule that serine phosphorylation proximal to the substrate arginine is inhibitory to arginine methylation.
Previous proteomic studies have shown that H4R3me1 is more abundant than H4R3me2 [58,59], which coincide with our data that the rates of asymmetric and symmetric dimethylation of H4R3 by PRMT1 and PRMT5 are slower in comparison to their monomethylation activity. Specifically, the turnover number of arginine monomethylation by PRMT1 with the Ac-H4(1–20) substrate was 0.52 ± 0.0078 min−1 while that of the Ac-H4(1–20)R3me substrate was 0.40 ± 0.0062 min−1 (Table 2). Our results are consistent with previous reports that also observed a reduced rate in asymmetric dimethylation by PRMT1 compared to monomethylation [15,18,22]. Similar to PRMT1, PRMT5-catalysed SDMA formation from a pre-monomethylated H4R3 peptide also exhibited a reduced rate in comparison to the unmethylated H4R3 substrate, conforming to an earlier study [25]. Furthermore, consistent with previous reports [15,60], we observed that PRMT1 and PRMT5 kinetics often exhibited a sigmoidal shape, and hence, we applied the Hill equation to determine the kinetic parameters. In all cases for PRMT1, except with the Ac-H4(1–20)S1ph substrate, we observed Hill coefficients > 2 which suggests positive cooperativity. Future studies would be needed to determine if the underlying mechanism for the positive cooperativity is thermodynamically or kinetically driven.
Taken together, we sought to understand the regulatory mechanisms of substrate recognition and catalysis by PRMTs, particularly with respect to arginine dimethylation. We have examined how the chemical properties on the substrate, specifically the importance of local charges, regulate the substrate recognition and catalysis by the major type I and type II PRMTs, PRMT1 and PRMT5. We observed similarities and yet differences depending on the local changes in charged residues as well as the type of chemical modification. The absence of the positively charged H4K5 supports SDMA production by PRMT5 yet hinders ADMA production by PRMT1 (Figure 9). H4S1 phosphorylation is inhibitory to arginine methylation, both monomethylation and dimethylation, by PRMT1 and PRMT5. This work expands our fundamental understanding of arginine methylation and its regulation. In the future, it would be highly interesting to determine whether the patterns and mechanisms of histone PTMs regulating PRMT-catalysed ADMA and SDMA formation persist in the context of the nucleosomes. This information would be of importance for elucidating the dynamics and consequences of complex histone code establishment.
Figure 9.
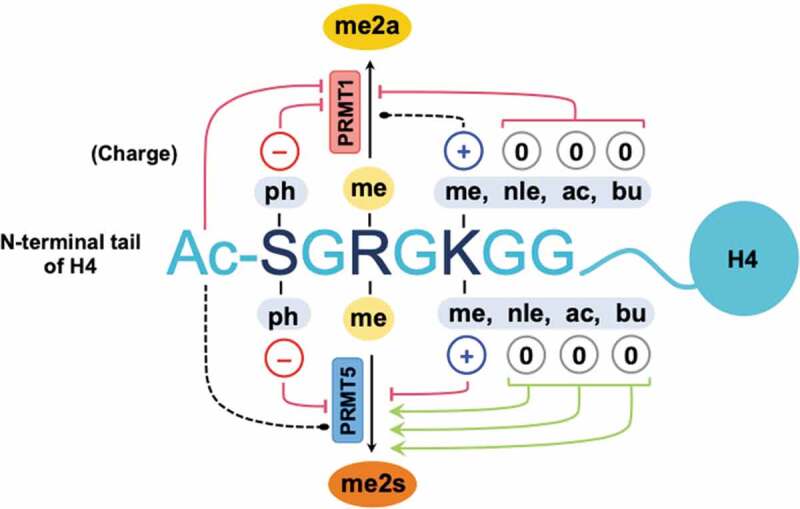
Summary of the impacts of chemical modifications to histone H4S1 and H4K5 on asymmetric and symmetric dimethylation of H4R3 by PRMT1 and PRMT5. Phosphorylation of H4S1 inhibits ADMA and SDMA production by PRMT1 and PRMT5, respectively. H4K5 methylation does not affect asymmetric dimethylation by PRMT1, yet it is inhibitory to PRMT5. Maintaining the positively charged H4K5 ε-amine is important for ADMA production by PRMT1. Acetylation, the loss of the ε-amine (norleucine), or butyrylation of H4K5 supports symmetric dimethylation of arginine by PRMT5. Red line with blunt end = inhibiting, green arrow = activating, and black dotted line = little or no effect.
Materials and methods
Chemicals and reagents
The N-α-Fmoc-protected amino acids were purchased from either Novabiochem or ChemPep Inc. High-performance liquid chromatography (HPLC) grade methanol and acetonitrile were purchased from either Sigma-Aldrich or British Drug Houses (BDH). Phenylmethanesulfonyl fluoride (PMSF) was purchased from either Gold Biotechnology or Sigma-Aldrich. Kanamycin, ampicillin, and isopropyl β-D-1-thiogalactopyranoside (IPTG) were purchased from Gold Biotechnology. Unless otherwise stated, the remaining chemical reagents described were purchased from Fisher Scientific, Acros Organics, Sigma-Aldrich, Alfa Aesar, BDH, Research Products International Corp., Macron Fine Chemicals, Bio-Rad, or J. T. Baker.
Protein expression
Details for recombinant protein expression of human PRMT1, PRMT3, and PRMT8 were described previously[15]. Recombinant 6xHis-tagged human PRMT5 was coexpressed with recombinant 6xHis-tagged human MEP50 using the Bac-to-Bac baculovirus expression system (Invitrogen, Life Technologies). In detail, pFB-LIC-Bse-PRMT5 and pFB-LIC-Bse-MEP50 were heat shock transformed in the DH10Bac E. coli strain according to the Bac-to-Bac Invitrogen protocol, and the recombinant bacmids were isolated with the PureLink HiPure Plasmid Miniprep Kit (Invitrogen, Thermo Scientific). Plasmid concentration and purity were determined using a Nanodrop. Sf9 cells cultured in Sf-900 II SFM + 100 U of penicillin/streptomycin (P/S) to a cell density of 1.5 × 106–2.5 × 106 cells/mL (>95% viability) before plating 2 mL of 4 × 105 cells/mL per well in a 6-well tissue culture plate. Cells were allowed at least 30 min to adhere to the plate before replacing the medium with 2.5 mL/well of Grace’s unsupplemented insect cell medium containing 15% Sf-900 II SFM (no antibiotics). The DNA-lipid complexes were prepared with PRMT5 or MEP50 recombinant bacmid, Cellfectin II, and Grace’s unsupplemented medium according to the Bac-to-Bac Invitrogen protocol, and each well received 2 µg total of PRMT5 or MEP50 recombinant bacmid. Transfection proceeded for 5 h at 27.5°C before replacing the transfection medium with 2 mL of Sf-900 II SFM + 100 U P/S per well. After 7 days of incubation at 27.5°C, the medium from each well as pooled and centrifuged (1000 rpm for 5 min) to collect the supernatant as the P1 viral stock. P1 viral stocks were stored at 4°C. Viral amplification of the P1 stock to achieve a higher titre P2 stock was performed on 6-well tissue culture plates. Each well contained 2 mL of 1 × 106 cells/mL. The volume of P1 inoculum was calculated based on a multiplicity of infection (MOI) of 0.1 per well (P1 target viral concentration 1 × 106–1 × 107 IFU/mL). Cells were incubated at 27.5°C for 30 min to allow the cells to attach to the plates before adding the calculated volume of inoculum and incubating for 7 days at 27.5°C. The P2 viral stock was harvested by transferring the medium from each well to 50 mL centrifuge tubes and centrifuged for 10 min at 1000 rpm (room temperature). The supernatant was collected as the P2 viral stock and stored at 4°C. The viral titre concentration (IFU/mL) was determined using a 96-well baculovirus plaque assay with a viral dilution range of 10−1 to 10−7. The target range for the P2 viral titre stocks was 1 × 107–1 × 108 IFU/mL. For protein expression, Sf9 insect cells were cultured in suspension at 27.5°C (130 rpm) in Expression Systems ESF 921 + 100 U P/S medium until the cell density was 1.5 × 106 cells/mL for a 200 mL culture. The 200 mL culture was then inoculated with P2 viral stocks of PRMT5 and MEP50 to achieve an MOI of 2. Cultures were incubated for 72 h at 27.5°C (130 rpm) before harvesting the cells by centrifugation (1000 rpm, 10 min) and discarding the supernatant. Cell pellets were flash frozen with liquid N2 and stored at −80°C until there was time for the purification process. Frozen cell pellets were thawed and disrupted twice at 100 psi in 20 mL of cell lysis buffer (50 mM Tris pH 8, 300 mM NaCl, 10% glycerol, 0.1% Triton X-100, 1 mM PMSF). Cell lysates were centrifuged for 60 min at 4°C (18000 rpm) in an Avanti J-26 XPI centrifuge with a JA-30.50 rotorhead. 10 mL of Ni-NTA (EMD Millipore) slurry was loaded into a 50 mL column and equilibrated with 4 × 15 mL of column equilibration/wash buffer (50 mM Tris pH 8, 250 mM NaCl, 30 mM Imidazole, 10% glycerol, 1 mM TCEP, 1 mM PMSF). The equilibrated nickel resin was rocked with the cell lysate supernatant on an orbital shaker for 1 h at 4°C. Afterwards, the resin was drained by gravity flow and washed with 4 × 25 mL of equilibration/wash buffer (50 mM Tris pH 8, 250 mM NaCl, 30 mM Imidazole, 10% glycerol, 1 mM TCEP, 1 mM PMSF). Proteins were eluted with 3 × 5 mL of elution buffer (50 mM Tris pH 8, 250 mM NaCl, 300 mM Imidazole, 10% glycerol, 1 mM TCEP, 1 mM PMSF). Elution samples were loaded into 10,000 MWCO SnakeSkin (Thermo Scientific) dialysis tubing and dialysed in storage buffer (50 mM Tris pH 8, 250 mM NaCl, 10% glycerol, 1 mM TCEP) with 3 buffer replacements over a course of 18 h at 4°C. Proteins were concentrated by centrifugation (6000 rcf, 4°C) in Vivaspin 20 ultrafiltration devices (10000 MWCO, GE Healthcare). Protein concentrations were determined with the Bradford Assay (Thermo Scientific), and protein purities were verified by SDS-PAGE (Fig. S8).
Peptide synthesis
Peptides were synthesized by Fmoc solid-phase peptide synthesis protocol on an AAPPTec Focus XC synthesizer using a Wang resin (Fmoc-Lys(Boc)-Wang, 100–200 mesh, ChemPep). Synthesis scale was either 50 or 100 µmole. Coupling of Fmoc-Ser(HPO3Bzl)-OH (ChemPep), Fmoc-Lys(me,Boc)-OH (ChemPep), or Fmoc-Lys(ac)-OH (ChemPep) utilized 5 eq amino acid (AA), 5 eq HCTU [O-(1 H-6-chlorobenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate], 5 eq of HOBt, and 15 eq of DIPEA in 6 mL DMF and mixed overnight. Coupling of Fmoc-Arg(me,Pbf)-OH (Novabiochem) utilized 5 eq AA, 5 eq HCTU, 5 eq of HOBt, and 15 eq of DIPEA in 6 mL NMP and mixed overnight. Coupling of Fmoc-Nle-OH (Chem-Impex) utilized 10 eq AA in NMP, 10 eq HCTU in DMF, 10 eq of HCTU in DMF, and 100 eq of NMM in DMF and mixed for 60 min. To butyrylate H4K5, Fmoc-Lys(DDE)-OH was double coupled using 10 eq of AA in NMP, 10 eq HCTU in DMF, and 40 eq NMM in DMF for 45–55 min at room temperature for the first coupling and then allowed to react overnight at room temperature during the second coupling. We proceeded to finish synthesizing the remaining residues in the H4 peptide and cap the N-terminus to avoid accidentally deprotecting the Fmoc group from Fmoc-Lys(DDE) during the removal of DDE with hydrazine. Lys(DDE) was deprotected with 2% hydrazine in DMF for 2 h at room temperature, drained, and repeated with fresh 2% hydrazine in DMF for 2 h at room temperature. After washing the resin with DMF, the butyrylation reaction was prepared with 10 eq of butyric acid in DMF, 10 eq HCTU in DMF, and 40 eq NMM in DMF for 1 h at room temperature and then repeated, allowing the reaction to proceed overnight. In general, the Fmoc deprotection reactions were performed with 20% (v/v) piperidine in dimethylformamide. The N-terminus of each peptide, unless stated otherwise, was acetylated by mixing the Fmoc deprotected peptides with 50 eq acetic anhydride, 50 eq DIPEA prepared in DMF (4:1, DMF:acetic anhydride) for 30 min. Peptides were cleaved from Wang resin with 2.5% ethanedithiol (EDT), 5% deionized water, 5% thioanisole, 5% phenol, 1% triisopropylsilane, and 81.5% trifluoroacetic acid (TFA). Peptides were precipitated in cold diethyl ether and pelleted by centrifugation (5 min, 2000 rpm). After centrifugation, the crude peptides were dissolved in water for lyophilization. Purification was performed on a Shimadzu reversed-phase high-performance liquid chromatography (RP-HPLC) system equipped with a Polaris 5 C18-A, 150 mm × 21.2 mm column (Agilent). Peptides were purified with a linear gradient using 0.05% TFA in water and 0.05% TFA in acetonitrile as the two mobile phases. The purified peptides were confirmed and characterized by MALDI (Table 1), and the peptide purity was checked by analytical HPLC. Peptide stock concentrations were calibrated using 1H NMR with an internal standard, 4,4-dimethyl-4-silapentane-1-sulphonic acid (DSS), as previously described [15] (Table S1 and Fig. S9).
Radioactive methylation assay (filter-based assay)
Reactions were performed as previously described [15]. For the reactions performed with PRMT5, the P81 filter paper dimensions were 1.1 × 2 cm and the filters were provided by the Protein Chemistry & Metabolism Unit at St. Vincent’s Institute of Medical Research, Australia. A comparison of the P81 filter paper manufactured by Whatman, Reaction Biology, and St. Vincent’s Institute of Medical Research is available in the Supporting Information (Fig. S10). Each methylation reaction was supplied with either [14C]-isotope-labelled SAM ([14C]-SAM 56.3 mCi/mmol or [14C]-SAM 58 mCi/mmol, catalogue No. NEC363050UC from PerkinElmer, Inc.). The reaction buffer contained 50 mM HEPES (pH 8), 10 mM NaCl, 0.5 mM EDTA, and 0.5 mM DTT. Each peptide was pre-mixed at room temperature with [14C]-SAM before initiating the reaction by adding the PRMT for a final concentration of 0.02 μM with the 15 min reactions or 0.05 μM with the 35 min reactions as indicated in the results. The reaction time points were chosen within the linear phase of the progress curves of PRMT1 and PRMT5 with various chemically modified H4 peptides (Fig. S11). Once initiated, all reactions were incubated at 30°C. Because we observed a low signal to background difference at 0.05 µM of PRMT5 (Fig. S4), we tried higher concentrations of PRMT5 at 0.25 µM and 0.5 µM to see if the rates were comparable and potentially further reduce the experimental error values. Consistently, in both cases at higher concentrations of PRMT5, methylation of H4K5 does not affect PRMT5-catalysed methylation (Fig. S12). Reactions were quenched by an equal volume of isopropanol, immediately vortexed, pulse spun down, and then loaded onto P81 Whatman filter paper (2.2 cm x 2 cm for each sample, Reaction Biology) to dry for 30 min at room temperature. Afterwards, the filter paper samples (except [14C]-SAM reference samples) were washed three times (20 min/wash) with 50 mM NaHCO3 (pH 9). All filter paper samples were allowed to dry for at least 3 h at room temperature. Each filter paper sample was immersed in 5 mL of scintillation cocktail (Ultima Gold mixture, PerkinElmer) and incubated for 30 min ― 1 h at room temperature in the dark before liquid scintillation counting on a Beckman Coulter LS 6500 Multi-Purpose Scintillation Counter (5 min/sample). The counts per minute (cpm) was measured by liquid scintillation counting and converted to the concentration of methylated products (P) based on the known concentration and cpm measured for the [14C]-SAM reference samples (Eq. 1). The experiments were performed, at minimum, in duplicate. Either KaleidaGraph (Version 4.03) or GraphPad Prism 7 was used to fit the kinetic data with equation 1 (Hill) to calculate the steady-state kinetic parameters kcat, K0.5, and n (Hill coefficient) based on our previous studies[15]. For consistency and comparison with other reported steady-state kinetic parameters in the PRMT field, the units are reported in minutes and µM.
(Eq. 1, Hill equation)
Supplementary Material
Acknowledgments
This work was funded by NIH grant R01GM126154 to Y.G.Z. We thank the Proteomics and Mass Spectrometry Facility and the Chemical Sciences Magnetic Resonance Facility of the University of Georgia for their assistance in collecting the MS and NMR data.
Funding Statement
This work was supported by the National Institutes of Health [R01GM126154].
Disclosure statement
The authors do not have conflicts of interest to declare.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Larsen SC, Sylvestersen KB, Mund A, et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signaling. 2016;9:rs9. [DOI] [PubMed] [Google Scholar]
- [2].Tang J, Frankel A, Cook RJ, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–7730. [DOI] [PubMed] [Google Scholar]
- [3].Dhar S, Vemulapalli V, Patananan AN, et al. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci Rep. 2013;3:1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hadjikyriacou A, Yang Y, Espejo A, et al. Unique features of human protein arginine methyltransferase 9 (PRMT9) and its substrate RNA splicing factor SF3B2. J Biol Chem. 2015;290:16723–16743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bouras G, Deftereos S, Tousoulis D, et al. Asymmetric dimethylarginine (ADMA): a promising biomarker for cardiovascular disease? Curr Top Med Chem. 2013;13:180–200. [DOI] [PubMed] [Google Scholar]
- [6].Fulton MD, Brown T, Zheng YG.. The biological axis of protein arginine methylation and asymmetric dimethylarginine. Int J Mol Sci. 2019;20:3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vallance P, Leone A, Calver A, et al. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. [DOI] [PubMed] [Google Scholar]
- [8].Fulton MD, Brown T, Zheng YG.. Mechanisms and inhibitors of histone arginine methylation. Chem Rec. 2018;18:1792–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhao Q, Rank G, Tan YT, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16:304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Le Guezennec X, Vermeulen M, Brinkman AB, et al. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol. 2006;26:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li XG, Hu X, Patel B, et al. H4R3 methylation facilitates beta-globin transcription by regulating histone acetyltransferase binding and H3 acetylation. Blood. 2010;115:2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rezai-Zadeh N, Zhang X, Namour F, et al. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev. 2003;17:1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang SM, Litt M, Felsenfeld G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 2005;19:1885–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ruiz-Carrillo A, Wangh LJ, Allfrey VG. Processing of newly synthesized histone molecules. Science. 1975;190:117–128. [DOI] [PubMed] [Google Scholar]
- [15].Fulton MD, Zhang J, He M, et al. The intricate effects of alpha-amino and lysine modifications on arginine methylation on the N-terminal tail of histone H4. Biochemistry. 2017;56:3539–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Feng Y, Wang J, Asher S, et al. Histone H4 acetylation differentially modulates arginine methylation by an in Cis mechanism. J Biochem. 2011;286:20323–20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Feng Y, Xie N, Jin M, et al. Analysis of PRMT1 catalysis. Biochemistry. 2011;50:7033–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hu H, Luo C, Zheng YG. Transient kinetics define a complete kinetic model for protein arginine methyltransferase 1. J Biol Chem. 2016;291:26722–26738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lakowski T, Frankel A. Kinetic analysis of human protein arginine N-methyltransferase 2: formation of monomethyl-and asymmetric dimethyl-arginine residues on histone H4. Biochem J. 2009;421:253–261. [DOI] [PubMed] [Google Scholar]
- [20].Kolbel K, Ihling C, Bellmann-Sickert K, et al. Type I arginine methyltransferases PRMT1 and PRMT-3 act distributively. J Biol Chem. 2009;284:8274–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Osborne TC, Obianyo O, Zhang X, et al. Protein Arginine Methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry. 2007;46:13370–13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gui S, Wooderchak-Donahue WL, Zang T, et al. Substrate-induced control of product formation by protein arginine methyltransferase 1. Biochemistry. 2013;52:199–209. [DOI] [PubMed] [Google Scholar]
- [23].Antonysamy S, Bonday Z, Campbell RM, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A. 2012;109:17960–17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang WL, Anderson LC, Nicklay JJ, et al. Phosphorylation and arginine methylation mark histone H2A prior to deposition during Xenopus laevis development. Epigenet Chromatin. 2014;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ho MC, Wilczek C, Bonanno JB, et al. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One. 2013;8:e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Arnesen T, Van Damme P, Polevoda B, et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc Natl Acad Sci U S A. 2009;106:8157–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hole K, Van Damme P, Dalva M, et al. The human N-alpha-acetyltransferase 40 (hNaa40p/hNatD) is conserved from yeast and N-terminally acetylates histones H2A and H4. PLoS One. 2011;6:e24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Song OK, Wang X, Waterborg JH, et al. Nalpha-acetyltransferase responsible for acetylation of the N-terminal residues of histones H4 and H2A. J Biol Chem. 2003;278:38109–38112. [DOI] [PubMed] [Google Scholar]
- [29].Schiza V, Molina-Serrano D, Kyriakou D, et al. N-alpha-terminal acetylation of histone H4 regulates arginine methylation and ribosomal DNA silencing. PLoS Genet. 2013;9:e1003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yamamoto K, Chikaoka Y, Hayashi G, et al. Chemical proteomic approaches to reveal histone H4 modification dynamics in cell cycle: label-free semi-quantification of histone tail peptide modifications including phosphorylation and highly sensitive capture of histone PTM binding proteins using photo-reactive crosslinkers. Mass Spectrom (Tokyo). 2015;4:A0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Arnaudo AM, Garcia BA. Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin. 2013;6. DOI: 10.1186/1756-8935-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guccione E, Bassi C, Casadio F, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–937. [DOI] [PubMed] [Google Scholar]
- [33].Hyllus D, Stein C, Schnabel K, et al. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007;21:3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Iberg AN, Espejo A, Cheng D, et al. Arginine methylation of the histone H3 tail impedes effector binding. J Biol Chem. 2008;283:3006–3010. [DOI] [PubMed] [Google Scholar]
- [35].Migliori V, Muller J, Phalke S, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19:136–144. [DOI] [PubMed] [Google Scholar]
- [36].Yuan CC, Matthews AG, Jin Y, et al. Histone H3R2 symmetric dimethylation and histone H3K4 trimethylation are tightly correlated in eukaryotic genomes. Cell Rep. 2012;1:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jain K, Warmack RA, Debler EW, et al. Protein arginine methyltransferase product specificity is mediated by distinct active-site architectures. J Biol Chem. 2016;291:18299–18308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Strahl BD, Briggs SD, Brame CJ, et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11:996–1000. [DOI] [PubMed] [Google Scholar]
- [39].Allali-Hassani A, Wasney GA, Siarheyeva A, et al. Fluorescence-based methods for screening writers and readers of histone methyl marks. J Biomol Screen. 2012;17:71–84. [DOI] [PubMed] [Google Scholar]
- [40].Tweedie-Cullen RY, Brunner AM, Grossmann J, et al. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. Plos One. 2012;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wilczek C, Chitta R, Woo E, et al. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in xenopus laevis eggs. J Biol Chem. 2011;286:42221–42231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee J, Sayegh J, Daniel J, et al. PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J Biol Chem. 2005;280:32890–32896. [DOI] [PubMed] [Google Scholar]
- [43].Pawlak MR, Scherer CA, Chen J, et al. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000;20:4859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fabbrizio E, El Messaoudi S, Polanowska J, et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002;3:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang H, Sabari BR, Garcia BA, et al. SnapShot: histone modifications. Cell. 2014;159:458–458 e451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Grimsley GR, Scholtz JM, Pace CN. A summary of the measured pK values of the ionizable groups in folded proteins. Pro Sci. 2009;18:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Manning LR, Manning JM. Phosphorylation of serine induces lysine p Ka increases in histone N-termini and signaling for acetylation. transcription implications. Biochemistry. 2018;57:6816–6821. [DOI] [PubMed] [Google Scholar]
- [48].Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. [DOI] [PubMed] [Google Scholar]
- [49].Schapira M, Ferreira de Freitas R. Structural biology and chemistry of protein arginine methyltransferases. MedChemComm. 2014;5:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barber CM, Turner FB, Wang Y, et al. The enhancement of histone H4 and H2A serine 1 phosphorylation during mitosis and S-phase is evolutionarily conserved. Chromosoma. 2004;112:360–371. [DOI] [PubMed] [Google Scholar]
- [51].Krishnamoorthy T, Chen X, Govin J, et al. Phosphorylation of histone H4 Ser1 regulates sporulation in yeast and is conserved in fly and mouse spermatogenesis. Genes Dev. 2006;20:2580–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cheung WL, Turner FB, Krishnamoorthy T, et al. Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S-cerevisiae. Curr Biol. 2005;15:656–660. [DOI] [PubMed] [Google Scholar]
- [53].Ju J, Chen A, Deng Y, et al. NatD promotes lung cancer progression by preventing histone H4 serine phosphorylation to activate Slug expression. Nat Commun. 2017;8:928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lu Y, Ma W, Li Z, et al. The interplay between p16 serine phosphorylation and arginine methylation determines its function in modulating cellular apoptosis and senescence. Sci Rep. 2017;7:41390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen M, Qu X, Zhang Z, et al. Cross-talk between Arg methylation and Ser phosphorylation modulates apoptosis signal-regulating kinase 1 activation in endothelial cells. Mol Biol Cell. 2016;27:1358–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Basso M, Pennuto M. Serine phosphorylation and arginine methylation at the crossroads to neurodegeneration. Exp Neurol. 2015;271:77–83. [DOI] [PubMed] [Google Scholar]
- [57].Smith DL, Erce MA, Lai YW, et al. Crosstalk of phosphorylation and arginine methylation in disordered SRGG repeats of saccharomycescerevisiae fibrillarin and its association with nucleolar localization. J Mol Biol. 2020;432:448–466. [DOI] [PubMed] [Google Scholar]
- [58].Pesavento JJ, Bullock CR, LeDuc RD, et al. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J Biol Chem. 2008;283:14927–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tweedie-Cullen RY, Brunner AM, Grossmann J, et al. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS One. 2012;7:e36980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jain K, Jin CY, Clarke SG. Epigenetic control via allosteric regulation of mammalian protein arginine methyltransferases. Proc Natl Acad Sci U S A. 2017;114:10101–10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.