

ABSTRACT
High-grade serous ovarian cancer (HGSOC) harbours aberrant epigenetic features, including DNA methylation. In this study we delineate pathways and networks altered by DNA methylation and associated with HGSOC initiation and progression to a platinum-resistant state. By including tumours from patients who had been treated with the hypomethylating agent (HMA) guadecitabine, we also addressed the role of HMAs in treatment of HGSOC. Tumours from patients with primary (platinum-naïve) HGSOC (n = 20) were compared to patients with recurrent platinum-resistant HGSOC and enrolled in a recently completed clinical trial (NCT01696032). Human ovarian surface epithelial cells (HOSE; n = 5 samples) served as normal controls. Genome-wide methylation profiles were determined. DNA methyltransferase (DNMT) expression levels were examined by immunohistochemistry and correlated with clinical outcomes. Cancer-related and tumorigenesis networks were enriched among differentially methylated genes (DMGs) in primary OC vs. HOSE. When comparing platinum-resistant and primary tumours, 452 CpG island (CGI)-containing gene promoters acquired DNA methylation; of those loci, decreased (P < 0.01) methylation after HMA treatment was observed in 42% (n = 189 CGI). Stem cell pluripotency and cytokine networks were enriched in recurrent platinum-resistant OC tumours, while drug metabolism and transport-related networks were downregulated in tumours from HMA-treated patients compared to HOSE. Lower DNMT1 and 3B protein levels in pre-treatment tumours were associated with improved progression-free survival. The findings provide important insight into the DNA methylation landscape of HGSOC tumorigenesis, platinum resistance and epigenetic resensitization. Epigenetic reprogramming plays an important role in HGSOC aetiology and contributes to clinical outcomes.
KEYWORDS: Ovarian cancer, epigenetics, methylation, platinum, resistance
Introduction
Epithelial ovarian cancer is the most lethal gynaecologic malignancy and with no adequate early detection screening, the majority of patients are diagnosed with advanced stage disease [1]. High-grade serous ovarian cancer (HGSOC), the most common OC histological type, initially responds to platinum-based therapy [2]. However, up to 75% of responding patients relapse and eventually develop platinum-resistant disease. HGSOC survival rates have remained essentially unchanged for decades [2].
It has been hypothesized that aberrant epigenetic processes, such as DNA methylation and histone modifications, play an important role in HGSOC initiation and progression to a platinum-resistant state [3–5]. Those epigenetic changes contribute to broad changes across the epigenome, including transcriptional inactivation of tumour suppressor genes (TSGs) [6]. A large number of TSGs have been identified as being hypermethylated and epigenetically silenced in OC [7–10]. Development-associated genes [11,12] have also been shown to be epigenetically silenced in advanced HGSOC. Furthermore, bench-to-clinic therapeutic interventions using tumours biopsied before and after hypomethylating agent (HMA) treatment identified demethylated TSGs whose increased expression was functionally linked to resensitization of ovarian tumours to platinum and associated with improved clinical outcomes [8,9,13–23]. Collectively, pre-clinical and clinical studies support the broad scale of epigenetic complexity in OC and a role for DNA methylation in platinum-resistant disease.
The overall objective of this study was to delineate pathways and networks altered by DNA methylation and associated with HGSOC initiation and progression to a platinum-resistant state. The effects of an HMA on methylated genes in platinum-resistant HGSOC were also measured. Towards this goal, we made multiple comparisons using bioinformatic analyses of differentially methylated genes (DMGs) generated from normal human ovarian surface epithelium cells (HOSE), primary (untreated) tumours, recurrent (platinum-resistant) tumours, and tumours from patients who had been treated with an HMA and platinum as part of a clinical trial (NCT01696032) [19]. By including tumours of HMA-treated-patients, we addressed the emerging role of HMAs in treatment of HGSOC. Pathways and networks associated with OC tumorigenesis and acquisition of platinum resistance were identified, including regulation of epithelial-mesenchymal transition, cancer metastasis signalling, and DNA damage-induced 14-3-3σ signalling. Functional validation of significant DMGs was performed. Further, expression of specific DNMT isoforms, the enzymes responsible for de novo and maintenance methylation in tumour samples was measured and correlated with clinical outcomes. The findings provide new insight into epigenetic reprogramming in HGSOC in general, and the DNA methylation landscape of platinum resistance and epigenetic resensitization in particular.
Materials and methods
Study samples/Patient
For this study, four groups of samples were analysed 1) Group 1: primary HGSOC (n = 20) obtained from the Indiana University Simon Comprehensive Cancer Centre Tissue Bank, according to institutional guidelines; 2) Group 2: recurrent platinum-resistant baseline HGSOC tumours from patients (n = 42) enrolled in a clinical trial (NCT01696032; study design and patient information were previously described [8]); 3) Group 3: HGSOC tumours from patients after treatment with the HMA guadecitabine + carboplatin (G + C) for two cycles (Cycle 2, Day 8, prior to the carboplatin infusion, n = 11; NCT01696032 [19]); 4) Group 4: human ovarian surface epithelial cells, HOSE (n = 5), served as normal epithelium controls for this study. Samples were tumour fragments (Group 1) or needle biopsies (Gropus 2 and 3) and were stored frozen in liquid nitrogen until use. HOSE cells were obtained from normal ovaries of five patients at Indiana University School of Medicine (Department of Obstetrics and Gynaecology) by scraping the ovarian surface epithelium and short-term culture. Cell culture conditions and procedures were described previously [24]. HOSE cells were placed in short-term culture and expanded (two to four passages). The purity of the HOSE cells was confirmed by keratin and vimentin immunostaining as described [24]. Normal fallopian tube epithelial cells were provided by Dr. Theresa Woodroff (Northwestern University, Chicago). All protocols and procedures for human subjects were approved by Indiana University School of Medicine.
Cell lines and culture conditions
OC cell lines OVCAR3, A2780 and SKOV3 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). OVCAR5 cell line was obtained from the Developmental Therapeutics Program at the National Cancer Institute. All cell lines were authenticated by short tandem repeat (STR) analysis and tested for mycoplasma contamination in 2017 (IDEXX BioAnalytics, Columbia, MO) and mycoplasma testing was thereafter performed twice yearly. Cell culture conditions, development of cisplatin-resistant cells and extraction of DNA and RNA are described in Supplemental Methods (SM).
Methylome analysis
DNA extracted from patient samples and HOSE cells was bisulphite converted and DNA methylation was assayed by using the Infinium Human Methylation 450 BeadChip (HM450; Illumina) at the University of Chicago Genomics Facility. Data preprocessing and analyses were conducted in the statistical programming environment R v3.1.2 with the package RnBeads v0.99, as previously described [8,15,25,26]. Normalization and background correction were applied to methylation data with manufacturer recommended algorithms implemented in methylumi package [27,28]. Methylation levels were averaged for the replicates for each biopsy or HOSE after normalization and the difference in methylation β-value between two groups or the mean of the pairwise difference for paired samples was calculated. To correct p-values for multiple hypothesis testing, false discovery rates (FDR) were calculated by using an improved Benjamini-Hochberg procedure [29], and the methylation changes in CpG sites/regions with FDR < 0.05 were considered statistically significant. Ingenuity Pathway Analysis (IPA) was used to identify functional interactions between differentially methylated genes. Average methylation signals on the CpG sites within each CpG island, gene body, and/or promoter region were hierarchically clustered with Pearson dissimilarity and average linkage as clustering parameters. Illumina HumanMethylation450 BeadChip results are available for download at Gene Expression Omnibus (GEO) data repository at the National Centre for Biotechnology Information (NCBI) under the accession number GSE102119.
Real-time RT-PCR
Expression (mRNA) levels of selected genes were measured in primary HGSOC tumours (n = 20), normal fallopian tube epithelium (FTE; n = 1), and cell lines by real-time RT-PCR (details are described in SM).
Immunohistochemistry (IHC)
Expression of DNA methyltransferases (DNMT1, DNMT3A and DNMT3B) was measured by IHC in archival paraffin-embedded HGSOC tumours obtained at time of diagnosis (before any therapy) from 32 patients included in Sample/Patient Group 2 (Recurrent). For IHC, tissue sections were treated with xylene to remove paraffin and then hydrated by immersion in decreasing concentrations of ethanol. Antigen retrieval was performed in 10 mM citrate buffer, pH 6.0, at 95°C for 30 minutes followed by removal of peroxidase activity using H2O2 for 15 minutes, blocking with 3% normal goat serum for 30 minutes at room temperature, and incubation with primary antibody at 4°C overnight. DNMT1 antibody (ab19905; Abcam,Cambridge, MA) was used at 10 µg/mL. Antibodies for DNMT3A (NB120-13,888, clone 64B1446) and DNMT3B (NB100-266; Novus Biologicals, Littleton, CO) were used at 5 µg/mL and 1/100 dilution, respectively. Tissue sections were incubated with secondary antibody using the avidin-biotin peroxidase technique with DAKO Detection Kit (DAKO North America, Carpinteria, CA). Staining was developed using Liquid DAB+ Substrate Chromogen System (DAKO North America, Carpinteria, CA). Staining intensity (0–3+ scale), and percentage of stained cells were quantified by a board-certified pathologist. IHC scores were calculated as the product of staining intensity by the percentage of stained cells.
Statistical analysis
Pearson correlation coefficients of DNMT expression levels and IHC scores were calculated using GraphPad Prism (San Diego, CA). Real-time RT-PCR data were analysed using Student t-test on Microsoft Excel for Windows. Values of P < 0.05 were considered significant. Associations between clinical outcomes (progression-free survival, PFS; overall survival, OS) with protein (IHC scores) and mRNA expression levels of DNMT1, DNMT3A and DNMT3B, were examined using proportional hazards regression in SAS V9.4 (SAS Institute, Cary NC). IHC scores and mRNA measurements were treated as categorical values with optimal cut point defined by the maximal chi-square method. All models accounted for early (Phase I or MTD groups) vs late (treatment of choice group that crossed over to G + C after progression) exposure in NCT01696032 [16,19]. For PFS, separate models were fitted that either included or did not include the subjects that crossed over to G + C after progression. Hazard ratios >1 mean that if the marker is above the defined cut point then the hazard of having the event are higher. Kaplan-Meier curves were generated for key results. Because of the small sample sizes, the size of the hazard ratio, rather than the p-value, was to guide interpretation of results. Hazard ratios >2 (or corresponding less than.5) are reported.
Results
Study population and workflow of data analysis
A total of 73 ovarian tumour samples were analysed, consisting of 20 primary untreated HGSOC tumours, 42 recurrent platinum-resistant HGSOC tumours, and 11 guadecitabine-treated platinum-resistant HGSOC tumours (Figure 1(a), data analysis workflow). Tumour biopsies from patients with platinum-resistant HGSOC enrolled on a clinical trial testing the combination of guadecitabine and carboplatin (NCT01696032) were collected on cycle 1 day 1 (C1D1; pre-guadecitabine), and guadecitabine-treated tumour biopsies were collected after two full cycles of daily X 5 doses of guadecitabine on day 8 (C2D8) (Supplementary Figure S1). Samples obtained from this clinical trial [8,19] were subjected to the analysis work flow (Figure 1(a)).
Figure 1.
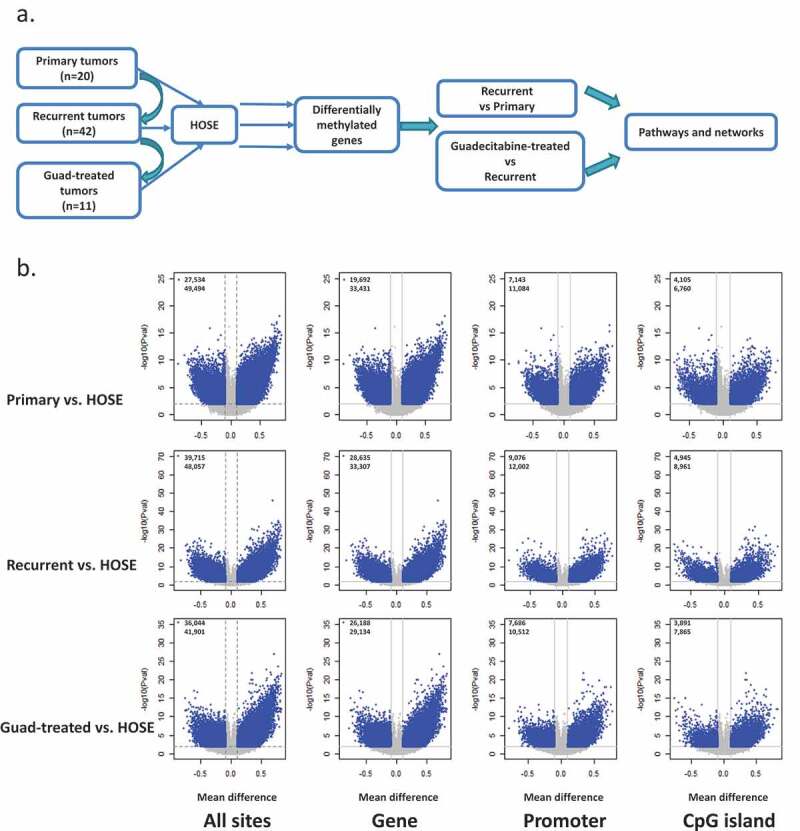
Differential methylation between each OC group and HOSE. (a) workflow of data analysis. (b) Volcano plots depicting differentially methylated CpG sites in different genomic regions (All CpG sites, Gene Body, Promoter, CpG island) of three OC groups compared to HOSE. Differentially methylated CpG sites are depicted as blue dots. The number of sites at different regions are indicated in each volcano plot. HOSE is human ovarian surface epithelium, which served as the normal control
Differential DNA methylation between tumours and HOSE
To begin investigating changes in DNA methylation in HGSOC tumours, we profiled methylated genes in HOSE, primary, recurrent, and guadecitabine-treated OC tumours using Infinium HumanMethylation 450 BeadChip arrays. Both fallopian tube and ovarian surface epithelia have been identified as cells of origin of HGSOC [30–33] and we used HOSE in the present study to define a normal baseline. Differential methylation between each tumour group (primary, recurrent, and HMA-treated) and HOSE was determined for all CpG sites, sites located within genes (promoter and body), promoter region, and CpG islands and shown as volcano plots in Figure 1(b). Methylation changes between groups were defined on the basis of CpG sites with a |Δβ| >0.1 and FDR<0.05. Applying these criteria to differential methylation at promoter region (tumour vs. normal), we identified 2647, 2642 and 2234 differentially methylated genes between primary tumours vs. HOSE, recurrent tumour vs. HOSE, and guadecitabine-treated tumour vs. HOSE, respectively (Table 1, Supplementary Tables S1-S3). Pathways enriched/altered by differentially methylated genes in these three comparisons were determined by using IPA. The most significantly enriched pathways common to all comparisons were FXR/RXR activation and LXR/RXR activation (Supplementary Figures S2a, S3a, S4a). Cancer-related and tumorigenesis networks were enriched in primary OC when compared to HOSE (Supplementary Figure S2b; red or green represents hypermethylated/inhibited or hypomethylated/activated molecules). DMGs in recurrent, platinum-resistant OC tumours compared to HOSE were enriched for networks related to stem cell pluripotency and cytokine metabolism (Supplementary Figure S3b). Drug metabolism and transport-related networks were significantly downregulated among DMGs from tumours collected from patients post-HMA treatment compared to HOSE (Supplementary Figure S4b). We observed similar trends in sites unrelated to an island, i.e., differentially methylated sites generally occurring within the gene body (Figure 1(b)). The entire list of networks enriched by DMGs in the OC groups compared to HOSE can be found in Supplementary Table S4.
Table 1.
Differentially methylated genes between different ovarian cancer groups and HOSE (FDR<0.05, |Δβ|>0.1)
| Numbers of Genes |
||
|---|---|---|
| Hypermethylated | Hypomethylated | |
| Primary OC vs. HOSE | 1810 | 837 |
| Recurrent OC vs. HOSE | 1541 | 1101 |
| Guadecitabine-treated OC vs. HOSE | 1364 | 870 |
Methylomic changes during acquired platinum resistance
To examine methylomic changes acquired during development of platinum resistance, we compared DMGs from primary vs. platinum-resistant tumours to HOSE cells. We considered methylation changes specific to development of cisplatin resistance as those observed in recurrent tumours but not in primary tumours. The analysis identified 452 hypermethylated genes specifically occurring in recurrent platinum-resistant cancer (Figure 2(a), blue ellipse, gene list in Supplementary Table S5). Biological pathways associated with those 452 genes, determined by IPA, included gene ontology (GO) terms related to migration and metastasis of OC cells, invasion of carcinoma cell lines, epithelial-mesenchymal transition (EMT), differentiation of mesenchymal stem cells, among others (Figure 2(b)). Furthermore, multiple pathways were represented by the 452 genes, including cancer metastasis signalling, DNA damage-induced 14-3-3σ signalling, and regulation of EMT (Figure 2(c)).
Figure 2.
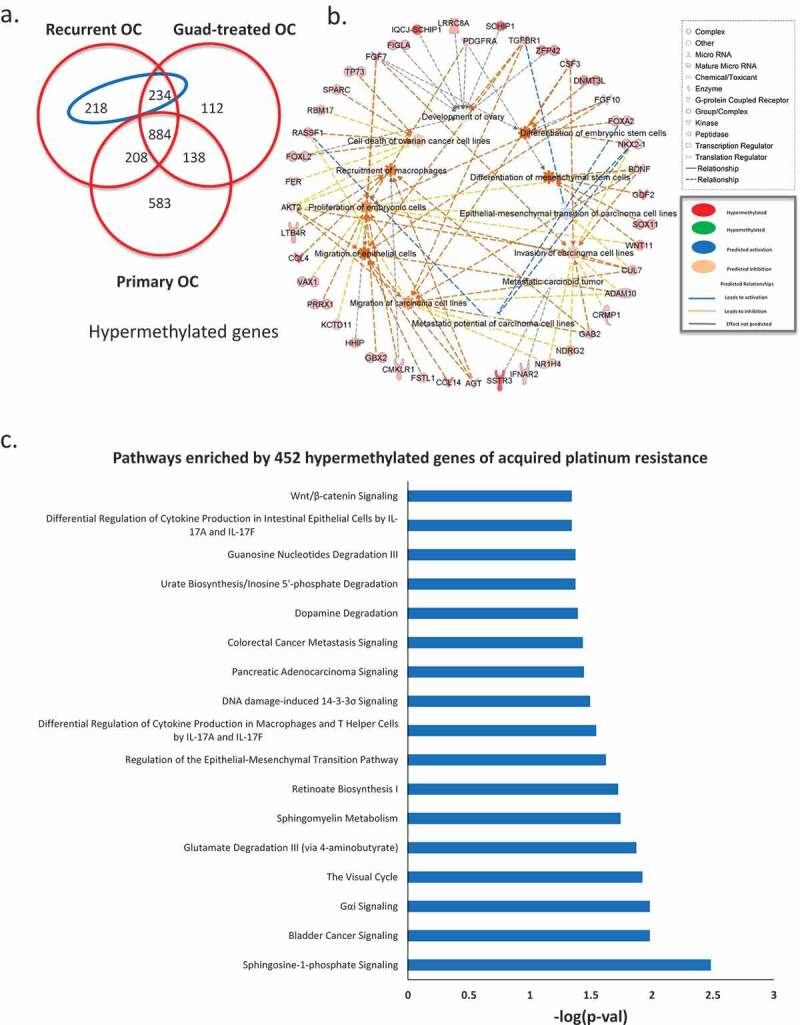
Methylomic changes during acquired platinum resistance. (a) Venn diagram shows unique and common hypermethylated genes in all three ovarian tumour groups, in the blue ellipse, 452 genes identified hypermethylated during acquired platinum resistance (hypermethylated in recurrent but not in primary OC). (b) Gene ontology analysis of the 452 genes hypermethylated in recurrent OC, different shapes on the out layer are genes from 452 gene list, whereas the ones in the middle are different gene ontology terms enriched by those genes from the outer layer. (c) Pathways enriched by 452 hypermethylated genes during acquired platinum resistance
Hypomethylating agent-induced methylomic changes
When we intersected hypomethylated genes for all three comparisons (primary HGSOC vs. HOSE, platinum-resistant HGSOC vs. HOSE, and guadecitabine-treated HGSOC vs.HOSE), we observed that guadecitabine-induced demethylation of 189 genes (Figure 3(a)), blue ellipse, gene list in Supplementary Table S6), which were hypermethylated in recurrent, platinum-resistant ovarian tumours compared to HOSE. Networks included EMT inhibition and reduced cell survival of cancer cells (Figure 3(b)). Pathway analysis showed that guadecitabine altered methylation of genes associated with cell cycle checkpoint regulation, vitamin-C transport and apoptosis signalling pathways (Figure 3(c)). These data offer support that cellular processes impacted by HMA treatment contribute to platinum re-sensitization.
Figure 3.
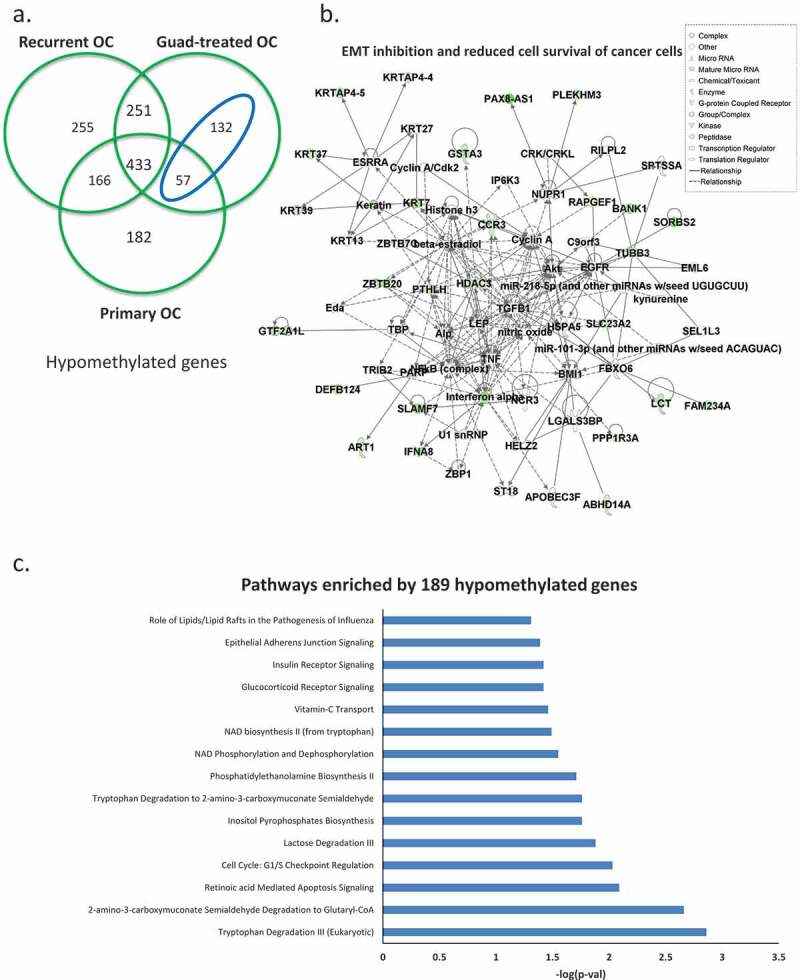
Hypomethylated genes induced by the HMA guadecitabine. (a) Venn diagram shows unique and common hypomethylated genes in all three ovarian tumour groups, in the blue ellipse, 189 genes identified as hypomethylated and induced by guadecitabine (genes hypomethylated in guadecitabine-treated but not in recurrent OC). Networks of epithelial-mesenchymal transition (EMT) inhibition and reduced cell survival of cancer cells (b) and pathways (c) enriched by the189 guadecitabine-induced hypomethylated genes
Functional validation
To validate our findings, expression levels of specific genes in response to treatment with an HMA were validated in preclinical models of platinum-resistance developed by us (Supplementary Figure 5). Gene selection was based on the bioinformatics analyses performed in this study and on previously recognized positive correlations between methylated transcripts and OC prognosis/outcome in the existing literature [34]. For example, FXYD domain containing ion transport regulator 6 (FXYD6), pyruvate dehydrogenase complex component X (PDHX), and ubiquitination factor E4A (UBE4A) were previously identified by us to be silenced via methylation during acquired platinum resistance [31]. In addition, interferon regulatory factor 9 (IRF9), ecotropic viral integration site 2A (EVI2A) and C-C motif chemokine ligand 19 (CCL19) had been found to be demethylated and re-expressed in ovarian tumour biopsies from HMA-treated patients [8]. Platinum-sensitive/parental (wild-type) and -resistant EOC cell lines OVCAR5, SKOV3 and A2780 were utilized (untreated or treated with vehicle (control) vs. guadecitabine). It is worth noting that recent genomic studies indicated that these cell lines may not have originated from HGSOC [32,33]. Basal expression level of FXYD6 was significantly lower in resistant OVCAR5 and resistant A2780 cells, while UBE4A was downregulated in resistant OVCAR5 cells relative to parental cells (Figure 4(a–c)). Although guadecitabine treatment upregulated FXYD6, PDHX, UBE4A, IRF9, EVI2A and CCL19 in the majority of EOC cell lines, the platinum-resistant cell lines were overall more responsive to the HMA (Figure 4(d–f)). Increased expression after guadecitabine treatment was observed for 5 of 6 genes in OVCAR3 cells (Figure 4(g)).
Figure 4.
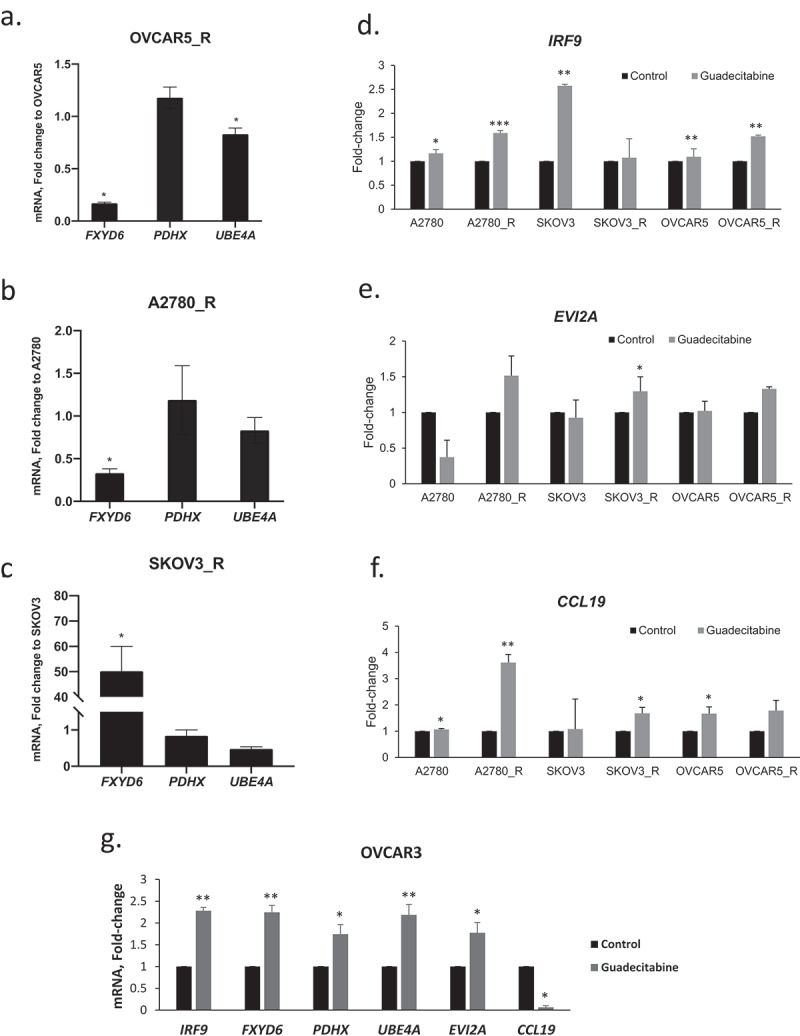
Effects of guadecitabine on expression of selected genes. (a–c) Basal mRNA expression levels of FXYD6, PDHX and UEB4A measured by real-time RT-PCR in platinum resistant (r) OVCAR5_R, A2780_R and SKOV3_R; (d–f) expression levels of IRF9, EVI2A and CCL19 on parental and cisplatin-resistant (R) A2780, SKOV3, and OVCAR5 treated with guadecitabine (100 nM) for 72 hours, and (g) OVCAR3 cells treated with the guadecitabine (100 nM) for 72 hours. Bars represent mean ± SD, n = 3 (* P < 0.05, ** P < 0.01)
Expression of DNMTs in primary OC tumours
The DNMTs (1, 3A and 3B) are the enzymes responsible for maintenance and de novo DNA methylation, respectively. Given our observations related to DNA methylation changes associated with HGSOC progression obtained through methylomic analyses, we sought to determine the expression levels of DNMTs and potential associations between expression and clinical outcomes. DNMT1, DNMT3A, and DNMT3B mRNA expression levels were examined in primary HGSOC tumours relative to FTE (Sample/Patient Group 1). DNMT1 and DNMT3B expression levels were similar, and expression of both was higher than DNMT3A (Figure 5(a)). A positive correlation between DNMT1and DNMT3B was observed (Figure 5(b)), whereas no correlation was seen between DNMT3A and DNMT1 or DNMT3B (Figure 5(c)). A positive correlation between DNMT3B and DNMT1 mRNA levels was also noted in a subgroup of recurrent HGSOC tumours of Sample/Patient Group 2 (Supplementary Figure S6a-b), further indicating that an association between de novo (DNMT3B) and maintenance (DNMT1) methylation [35] persisted through development of platinum resistance.
Figure 5.
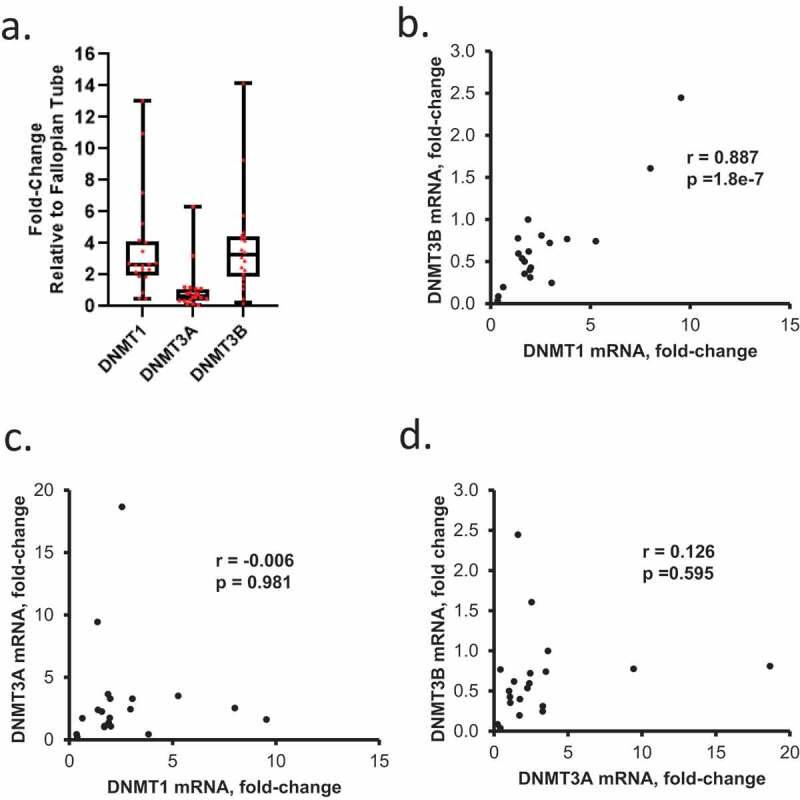
Expression levels of DNMT1, DNMT3A, and DNMT3B mRNAs in OC tumours. (a) DNMT1 and DNMT3B mRNA levels are positively correlated in HGSOC tumours. Box plots show medians and 25–75% quartiles of DNMT mRNAs levels measured by real-time RT-PCR in HGSOC primary tumours (n = 20, Sample/Patient Group 1) relative to fallopian tube epithelium. (b-d) Scatter plots and correlation coefficients between mRNA levels of DNMT1 and DNMT3B (b), DNMT1 and DNMT3A (c), or DNMT3A and DNMT3B (d) in HGSOC primary tumours (n = 20, Sample/Patient Group 1)
IHC was used to measure DNMT protein expression and localization in a subset of 32 HGSOC specimens obtained at time of diagnosis from patients included in Sample/Patient Group 2 (Figure 6(a); representative immunostaining). IHC scores for DNMT1 were greater (P < 0.05) than DNMT3A or 3B (Figure 6(b)). A positive correlation (P = 0.027) was observed between DNMT3A and DNMT3B IHC scores (Figure 6(c)). Although DNMT1 and DNMT3B mRNA levels were correlated (Figure 5(b)), the correlation between IHC scores of these DNMTs did not reach statistical significance (P = 0.07, Figure 6(c)), perhaps due to the limited number of specimens.
Figure 6.
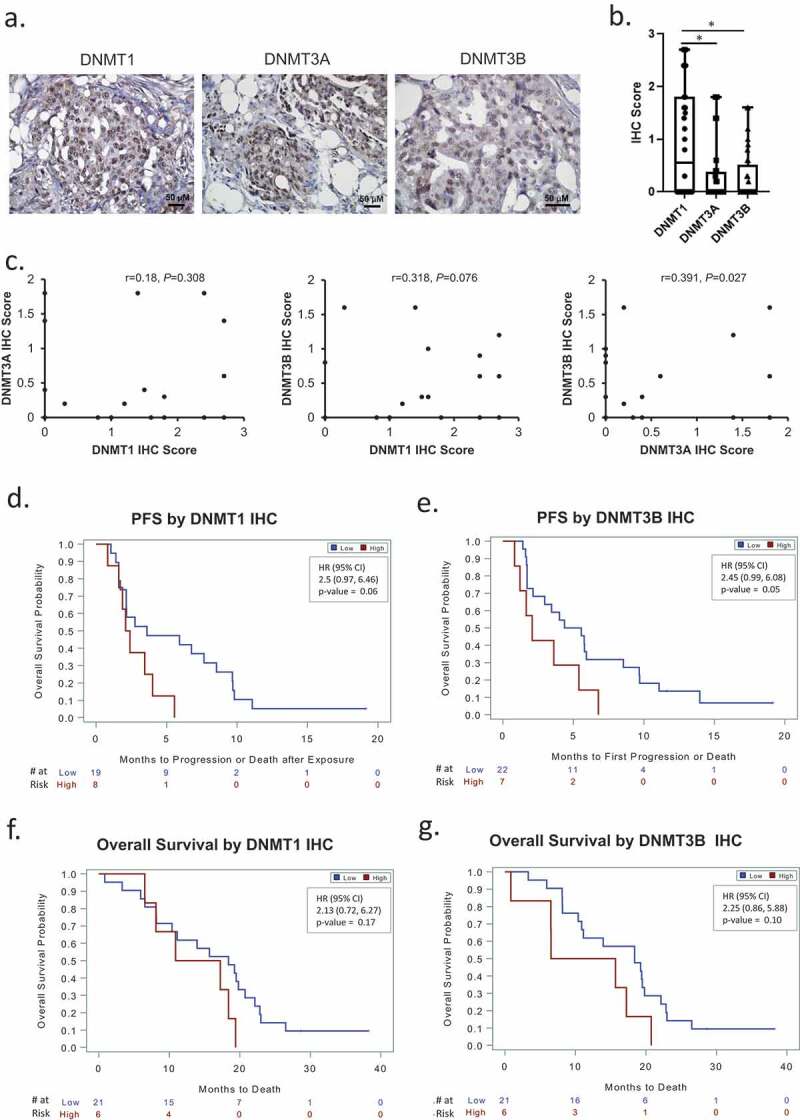
DNMT1 and DNMT3B are associated with progression-free (PFS) and overall survival (OS) of ovarian cancer patients. (a) Examples of DNMT1, DNMT3A and DNMT3B IHC immunostaining in sections from the same recurrent HGSOC tumour (original magnification: 200X). (b) Box plots show medians and 25–75 quartiles of DNMT1, DNMT3A, and DNMT3B IHC scores in recurrent HGSOC tumours (n = 32, Sample/Patient Group 2) before patients received guadecitabine treatment (*P < 0.05). (c) Scatter plots and correlation coefficients of IHC scores between DNMT1 and DNMT3A, DNMT1 and DNMT3B, or DNMT3A and DNMT3B in recurrent HGSOC tumours (n = 32, Sample/Patient Group 2). (d–g) Kaplan-Meier plots of progression-free survival (PFS) (d–e) and overall survival (f–g) by IHC score level (low or high) of DNMT1, or DNMT3B in recurrent HGSOC tumours from patients included in C above, and subsequently treated with a combination of carboplatin and guadecitabine. HR, hazard ratio
Finally, it was of interest to examine whether DNMT protein levels in the primary ovarian cancer tumours associated with clinical outcomes (PFS, OS). Intriguingly, hazard ratio values indicated that low IHC scores for DNMT1 or DNMT3B were associated with both longer PFS (Figure 6(d–e)) and OS (Figure 6(f–g)), inferring perhaps that lower levels of methylation in primary tumours may be associated with improved outcomes.
Discussion
It has been hypothesized that aberrant DNA methylation plays an important role in HGSOC, including in processes related to tumour initiation and progression to platinum-resistance, a common occurrence in the disease and a major cause of OC deaths. To address this important premise, we interrogated DNA methylomes from tumours collected before chemotherapy (treatment naïve), chemoresistant tumours (acquired, carboplatin) and chemo-resensitized tumours (by an HMA). By comparing epigenomic signatures from these patient cohorts to normal (HOSE) methylomes and integrating methylomic changes with transcriptomic data from these tumour cohorts, previously analysed [34], key genes and pathways contributing to OC tumorigenesis were identified and subsequently validated in OC cell lines. Several compelling findings of this study are highlighted.
Firstly, we observed that a remarkable number of gene promoters are silenced by DNA methylation in all three OC stages (primary, recurrent or HMA-treated vs. normal ovarian epithelium; Figure 1(b)), supporting the hypothesis that epigenetic reprogramming plays an important role in tumorigenesis. Network analysis highlights that both general processes of tumorigenesis as well as specific processes such as cell migration, cell invasion and EMT are altered by DNA methylation and potentially contribute to platinum resistance. Pathway analysis also showed that EMT, cancer metastasis signalling and DNA damage-induced 14-3-3σ signalling are altered during platinum resistance, in agreement with our previous work demonstrating that 14-3-3σ contributes to platinum sensitivity in OC [8]. These findings are congruent with other studies linking epigenetic abnormalities including DNA methylation with chronic DNA damage, tumour initiation and progression to platinum-resistance [36–40]. We recognize as a limitation the use of HOSE, as opposed to FTE, as the comparator in our study, as studies suggest that most epithelial OC originate in the fallopian tube [32,33]. However, very recent studies also support the ovarian surface epithelium as the origin of OC [30,31]. A recent methylomic analysis noted a higher resemblance between methylomic profiles of HGSOC and FTE than between HGSOC and HOSE [33]. Nevertheless, our results contribute to the body of evidence supporting that targeting epigenetic abnormalities may represent a possible treatment strategy for OC.
HMAs have been approved by the U.S. Food and Drug Administration for treating haematological malignancies (myelodysplastic syndrome and acute myeloid leukaemia) [41,42]. By reversing cancer-specific CpG island methylation, HMAs cause broad changes in gene expression [43]. Previous bench-to-clinic therapeutic interventions by us and others targeting aberrant DNA methylation in OC support that platinum re-sensitization can be an effective strategy [7–9,14–17,19,44,45]. In support of those reports, we show here that an HMA induces significant hypomethylation in tumours compared to normal HOSE. The 189 hypomethylated genes in Figure 3(a) (blue ellipse) are common to both primary tumours and tumours that had been treated by guadecitabine, suggesting that these genes were originally hypermethylated in the recurrent, platinum-resistant OC tumours. Pathway and network analyses demonstrate inhibition of EMT and reduced cell survival of cancer cells as key features of those 189 genes, along the compelling observation on the vitamin-C transport pathway (Figure 3(c)). A role for vitamin C in regulating the cancer epigenome via TET and Jumonji enzymes has recently been reported [46–49]. Vitamin C increases viral mimicry induced by 5-aza-2ʹ-deoxycytidine [50], and the findings here offer additional support for vitamin C in broadly reprogramming epigenomic medicines such as HMAs. In this regard, further assessment of both methylomic and transcriptomic data from our recent study [19] shows upregulation of TET3 and the viral defence gene interferon regulatory factor 9 (IRF9) after guadecitabine treatment ([8]; data not shown), supportive of the novel concept that epigenetic changes induced by an HMA-vitamin C combination have the potential to improve patient outcomes [51].
To validate the bioinformatic findings, we selected multiple genes based on functional relevance and utilized epithelial OC cell lines, including platinum-sensitive and acquired platinum-resistant cells (Figure 4). We demonstrate that several chemoresistance-associated genes, which were also reactivated in patient tumours by HMA, are upregulated in OC model systems and/or also inducible by guadecitabine in vitro. IRF9 is a viral defence gene, and CCL19 has been show to regulate EMT via ERK signalling pathway in OC patients [52]. EVI2A can interact with DOK2, a TSG and epigenetically regulated transcript in cancers [53]. PDHX is a structural component of the PDH complex that plays a critical role in cell metabolism, and suppression of PDHX leads to cancer cell proliferation [54]. These in vitro findings corroborate the tumour bioinformatics analysis and provide additional support for a role of methylation-mediated gene silencing in platinum-resistant OC.
Finally, we investigated DNMT expression in the patient samples. In primary OC tumours, DNMT1 and DNMT3B mRNA levels were found to be positively correlated. In this regard, studies evaluating DNMT1 mRNA as a prognostic indicator in other cancer types [55,56] support an important role for this enzyme in patient survival. We suggest that combined expression of DNMT1 and DNMT3B could serve as a predictor of response to epigenetic therapy in OC. In recurrent OC tumours, at the protein level, DNMT1 was higher than DNMT3A or DNMT3B, perhaps indicating a central role for DNA methylation maintenance over de novo DNA methylation in OC tumours with acquired platinum resistance. Further, based on an integrated data analysis with PFS or OS, low DNMT1 or DNMT3B IHC scores correlated with better prognosis/clinical outcome, warranting further investigation of these DNMTs as potential predictors of response of OC patients to HMA therapy. Validation of these findings in larger cohorts is necessary.
Overall, this study provides important insight into the DNA methylation landscape of HGSOC tumorigenesis, platinum resistance and epigenetic resensitization. By analysing methylomic changes in human ovarian tumours relative to clinical outcomes, we infer that key tumorigenicity-associated pathways can be altered through methylation gains and losses supporting future interventions targeting epigenetic events in this difficult to treat malignancy.
Supplementary Material
Acknowledgments
We thank the Genomics Facility of The University of Chicago for help with Illumina HumanMethylation450 BeadChip. We thank Dr. Theresa Woodruff (Northwestern University) for providing the normal fallopian tube epithelial cells. This work was funded in part by the National Cancer Institute Award CA182832-01 and a V Foundation Translational Grant (to DM and KPN).
Funding Statement
This work was supported by the National Cancer Institute Award CA182832-01 and the V- Foundation (to D.M. and K.P.N).
Authors’ contributions
HC and FF: acquisition, analysis and interpretation of the data, writing original draft. GJ: data curation, software, formal analysis. SMP: data curation, software, formal analysis. CZ: data curation, software, formal analysis. HNK: investigation, resources. YL: data curation, software, formal analysis. KNP and DM: conceived the idea, supervised research personnel and interpreted data. All authors read, critically reviewed and approved the final manuscript.
Disclosure statement
The authors declare that they have no competing interests.
Data availability statement
Illumina HumanMethylation450 BeadChip results are available for download at Gene Expression Omnibus (GEO) data repository at the National Center for Biotechnology Information (NCBI) under the accession number GSE102119.
Ethics approval and consent to participate
All protocols and procedures for human subjects were approved by Indiana University School of Medicine.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vaughan S, Coward JI, Bast JRC, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–494. . [DOI] [PubMed] [Google Scholar]
- [4].Cancer Genome Atlas Research N . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baylin SB, Jones PA.. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016;8(9):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Balch C, Fang F, Matei DE, et al. Minireview: epigenetic changes in ovarian cancer. Endocrinology. 2009;150(9):4003–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fang F, Balch C, Schilder J, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116(17):4043–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fang F, Cardenas H, Huang H, et al. Genomic and epigenomic signatures in ovarian cancer associated with resensitization to platinum drugs. Cancer Res. 2018;78(3):631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fang F, Zuo Q, Pilrose J, et al. Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget. 2014;5(11):3579–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wei SH, Balch C, Paik HH, et al. Prognostic DNA methylation biomarkers in ovarian cancer. Clin Cancer Res. 2006;12(9):2788–2794. . [DOI] [PubMed] [Google Scholar]
- [11].Fiegl H, Windbichler G, Mueller-Holzner E, et al. HOXA11 DNA methylation–a novel prognostic biomarker in ovarian cancer. Int J Cancer. 2008;123(3):725–729. [DOI] [PubMed] [Google Scholar]
- [12].Ibanez de Caceres I, Battagli C, Esteller M, et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004;64(18):6476–6481. [DOI] [PubMed] [Google Scholar]
- [13].Fang F, Munck J, Tang J, et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin Cancer Res. 2014;20(24):6504–6516. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matei D, Ghamande S, Roman L, et al. A phase I clinical trial of guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer: clinical, pharmacokinetic, and pharmadynamic analysis. Clin Cancer Res. 2018;24(10):2285 - 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Matei D, Fang F, Shen C, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9):2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang Y, Cardenas H, Fang F, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74(17):4922–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zeller C, Dai W, Steele NL, et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene. 2012;31(42):4567–4576. [DOI] [PubMed] [Google Scholar]
- [18].Wang Y, Zong X, Mitra S, et al. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight. 2018;3(23):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oza AM, Matulonis UA, Alvarez Secord A, et al. A randomized phase ii trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer. Clin Cancer Res. 2020;26(5):1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang Y, Petropoulos S, Liu J, et al. The signature of liver cancer in immune cells DNA methylation. Clin Epigenetics. 2018;10(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Parashar S, Cheishvili D, Mahmood N, et al. DNA methylation signatures of breast cancer in peripheral T-cells. BMC Cancer. 2018;18(1):574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Flanagan JM, Wilhelm-Benartzi CS, Metcalf M, et al. Association of somatic DNA methylation variability with progression-free survival and toxicity in ovarian cancer patients. Ann Oncol. 2013;24(11):2813–2818. [DOI] [PubMed] [Google Scholar]
- [23].Flanagan JM, Wilson A, Koo C, et al. Platinum-based chemotherapy induces methylation changes in blood DNA associated with overall survival in patients with ovarian cancer. Clin Cancer Res. 2017;23(9):2213–2222. . [DOI] [PubMed] [Google Scholar]
- [24].Ahluwalia A, Hurteau JA, Bigsby RM. DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol Oncol. 2001;82(2):299–304. [DOI] [PubMed] [Google Scholar]
- [25].Assenov Y, Muller F, Lutsik P, et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11(11):1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cardenas H, Vieth E, Lee J, et al. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics. 2014;9(11):1461–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Du P, Zhang X, Huang CC, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11(1):587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Langmead B, Trapnell C, Pop M. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29(4):1165–1188. [Google Scholar]
- [30].Lohmussaar K, Kopper O, Korving J, et al. Assessing the origin of high-grade serous ovarian cancer using CRISPR-modification of mouse organoids. Nat Commun. 2020;11(1):2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang S, Dolgalev I, Zhang T, et al. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat Commun. 2019;10(1):5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hao D, Li J, Jia S, et al. Integrated analysis reveals tubal- and ovarian-originated serous ovarian cancer and predicts differential therapeutic responses. Clin Cancer Res. 2017;23(23):7400–7411. [DOI] [PubMed] [Google Scholar]
- [33].Klinkebiel D, Zhang W, Akers SN, et al. DNA methylome analyses implicate fallopian tube epithelia as the origin for high-grade serous ovarian cancer. Mol Cancer Res. 2016;14(9):787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li M, Balch C, Montgomery JS, et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics. 2009;2(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].He Y, Ecker JR. Non-CG methylation in the human genome. Annu Rev Genomics Hum Genet. 2015;16(1):55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Polo SE, Kaidi A, Baskcomb L, et al. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. Embo J. 2010;29(18):3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Smeenk G, Wiegant WW, Vrolijk H, et al. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190(5):741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chou DM, Adamson B, Dephoure NE, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107(43):18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4(8):e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].O’Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20(5):606–619. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. [DOI] [PubMed] [Google Scholar]
- [42].Scott LJ. Azacitidine: a review in myelodysplastic syndromes and acute myeloid leukaemia. Drugs. 2016;76(8):889–900. [DOI] [PubMed] [Google Scholar]
- [43].Yang X, Han H, De Carvalho DD, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fu S, Hu W, Iyer R, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer. 2011;117(8):1661–1669. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Matei D, Ghamande S, Roman L, et al. A phase i clinical trial of guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer: clinical, pharmacokinetic, and pharmacodynamic analyses. Clin Cancer Res. 2018;24(10):2285–2293. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–816. [DOI] [PubMed] [Google Scholar]
- [48].Gillberg L, Orskov AD, Liu M, et al. Gronbaek K: vitamin C - A new player in regulation of the cancer epigenome. Semin Cancer Biol. 2018;51:59–67. [DOI] [PubMed] [Google Scholar]
- [49].Blaschke K, Ebata KT, Karimi MM, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500(7461):222–226. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu M, Ohtani H, Zhou W, et al. Vitamin C increases viral mimicry induced by 5-aza-2ʹ-deoxycytidine. Proc Natl Acad Sci U S A. 2016;113(37):10238–10244. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gillberg L, Orskov AD, Nasif A, et al. Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: normalization of plasma vitamin C induces epigenetic changes. Clin Epigenetics. 2019;11(1):143. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cheng S, Guo J, Yang Q. Yang X: crk-like adapter protein regulates CCL19/CCR7-mediated epithelial-to-mesenchymal transition via ERK signaling pathway in epithelial ovarian carcinomas. Med Oncol. 2015;32(3):47. [DOI] [PubMed] [Google Scholar]
- [53].Poage GM, Houseman EA, Christensen BC, et al. Kelsey KT: global hypomethylation identifies Loci targeted for hypermethylation in head and neck cancer. Clin Cancer Res. 2011;17(11):3579–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Eastlack SC, Dong S, Ivan C. Suppression of PDHX by microRNA-27b deregulates cell metabolism and promotes growth in breast cancer. Mol Cancer. 2018;17(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Supic G, Kozomara R, Zeljic K, et al. Magic Z: prognostic value of the DNMTs mRNA expression and genetic polymorphisms on the clinical outcome in oral cancer patients. Clin Oral Investig. 2017;21(1):173–182. [DOI] [PubMed] [Google Scholar]
- [56].Piyathilake CJ, Badiga S, Borak SG, et al. Partridge EE: A higher degree of expression of DNA methyl transferase 1 in cervical cancer is associated with poor survival outcome. Int J Womens Health. 2017;9:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Illumina HumanMethylation450 BeadChip results are available for download at Gene Expression Omnibus (GEO) data repository at the National Center for Biotechnology Information (NCBI) under the accession number GSE102119.