

Abstract
Biomolecular structure drives function, and computational capabilities have progressed such that the prediction and computational design of biomolecular structures is increasingly feasible. Because computational biophysics attracts students from many different backgrounds and with different levels of resources, teaching the subject can be challenging. One strategy to teach diverse learners is with interactive multimedia material that promotes self-paced, active learning. We have created a hands-on education strategy with a set of sixteen modules that teach topics in biomolecular structure and design, from fundamentals of conformational sampling and energy evaluation to applications like protein docking, antibody design, and RNA structure prediction. Our modules are based on PyRosetta, a Python library that encapsulates all computational modules and methods in the Rosetta software package. The workshop-style modules are implemented as Jupyter Notebooks that can be executed in the Google Colaboratory, allowing learners access with just a web browser. The digital format of Jupyter Notebooks allows us to embed images, molecular visualization movies, and interactive coding exercises. This multimodal approach may better reach students from different disciplines and experience levels as well as attract more researchers from smaller labs and cognate backgrounds to leverage PyRosetta in their science and engineering research. All materials are freely available at https://github.com/RosettaCommons/PyRosetta.notebooks.
Introduction
Structural models of proteins and other biomolecules help explain their functions and properties. Methods for computational structure prediction (i.e. protein folding and docking, as well as interactions with nucleic acids, carbohydrates, and other biomolecules) have been successful in many cases and certainly useful to drive structural and functional research hypotheses (1). Design of biomolecules (i.e. protein design, prediction of mutational effects, and molecular complex design) has also exhibited many successes, with potential impacts in medicine, biology, biotechnology, materials, and chemistry (2). Thus, there is a need to disseminate these interdisciplinary methods to a broader audience. Here, we present a set of workshops for teaching or self-study of biomolecular structure prediction and design.
Scientific and Pedagogical Background
Computational methods are a relatively inexpensive way to predict and manipulate biomolecular structures, especially when experimental methods prove difficult. There is a long history in biophysics of using computational modeling to better understand structure, dynamics, and function. In fact, the 2013 Nobel Prize in Chemistry was awarded for the pioneering contributions in quantum and molecular mechanics of complex chemical systems (3). There are now many available dynamic simulation tools for observing the behavior of biomolecules over time and predicting thermodynamic and kinetic properties from estimates of the system’s partition function. Some of these tools include CHARMM, Schrödinger software suite, MOE, NAMD, Amber, and Gromacs (4–9). A complementary approach to model biomolecules is with so-called structure prediction approaches. Instead of seeking a full description of all the states and kinetic rates of the system, these approaches seek the dominant, low-energy conformational state(s) that is (are) most relevant at biological conditions (10). These methods often accelerate calculations with approximations, such as constant bond lengths and angles, implicit solvent models, and empirically tuned energy functions. In exchange for these approximations, structure prediction approaches can capture the structure of large biomolecules in equilibrium without necessitating simulations over long timescales. These approaches are fundamentally based on optimization of an energy function in a very large conformational space. The same algorithmic components can then be used in reverse to design biomolecules by optimizing the energy function across different biomolecular sequences.
One leading structure prediction and design software suite is Rosetta, a collection of algorithms for protein structure prediction, docking, and design (10–13) as well as protein interactions with small molecules (14), nucleic acids (15), and carbohydrates in solution or in a lipid bilayer (16). Rosetta has been a scientific leader in several blind structure prediction challenges (17–21) and has shown proof-of-principle for many design goals, including de novo folds (22–24), loop design, interface design (25–28), symmetric assembly (29, 30), and mineral binding (31, 32). In addition to its success in science and engineering, Rosetta is suited for teaching structure prediction and design for several reasons. The Rosetta methods are available as a Python library called PyRosetta (33), which makes them easier to learn and combine with other scientific code libraries. PyRosetta allows access to low-level data and has a range of pre-built protocols for many tasks in biophysical research. Students can measure and manipulate protein conformations, dock proteins and small molecules, run folding algorithms, and explore other emerging topics in biomolecular structure prediction and design, such as RNA modeling and non-canonical amino acids. Furthermore, students can learn how to use these tools by creating and testing their own algorithms.
For about a decade now, structure prediction and design has been taught with PyRosetta, primarily through the use of a set of workshops that are available both as a printed book (34) and as downloadable PDF files (35). These workshops have been used to teach a course for undergraduate and graduate students at Johns Hopkins University for over ten years and intermittently at other schools, including the Massachusetts Institute of Technology, Stanford University, the University of Kansas, and the University of North Carolina. Workshops have been downloaded over 120,000 times (several tutorials over 1,000 times per year), and a complementary set of online lecture videos has registered over 14,000 views, reflecting a fast-growing interest in biomolecular structure prediction and design. In addition, these workshops have been an important resource for the Rosetta community, with the workshops being the primary learning tool for many now-senior core developers.
Despite the strong demand for educational resources, there have been several challenges in teaching with these materials. One problem in this interdisciplinary field has been how to train students from all levels and different skill sets. To address this challenge, the Rosetta Commons has established several programs and resources for students and researchers who are interested in Rosetta and PyRosetta, such as the PyRosetta and C++ Code Academies and the Rosetta Research Experience for Undergraduates (REU) (36). Other salient resources from the Rosetta community include: XML documentation (37), the Rosetta User Guide (38), Code Manual (39), and the active, managed user forum (40). While these resources have helped expose the field to a broad audience, hurdles still remain. Learning new software can be challenging for beginners, and the available beginner academies have limits on their annual cohort size. In addition, most of the resources currently available are in the form of code documentation or static text, which lack the interactive components that would enable active learning. Multimodal environments (e.g. including visualizations in addition to text) enhance students’ mental representations of fundamental concepts (41, 42), and in at least one coding class, interactive web-based content increased student time engaging with material and improved their quiz scores (43).
In addition, there are technological challenges that pose problems for new learners. Because the capabilities of Rosetta are constantly changing and expanding, as methods are modified and new algorithms added, educational resources need to evolve in parallel with the main Rosetta software. Since the original PyRosetta workshops, some commands and protocols have been deprecated or replaced, and many new frameworks have become standard. Static text workshops have been difficult to maintain because they require manual testing and updating. A related challenge is that PyRosetta is difficult to configure on Windows, making the barrier of entry for some beginners and self-learners prohibitively high.
In this work, we describe our latest contribution to address the pedagogical and technological limitations of previous educational resources by creating an accessible, multimedia platform for teaching biomolecular structure prediction and design methods with PyRosetta. Our solution combines the accessibility of Jupyter Notebooks, a shareable web application that supports live code, equations, visualization, and text (44), with the free computing power of Google Colaboratory (45) to develop a way for students of all experience levels to use PyRosetta on the cloud. Starting with our existing static workshops, we created a new, expanded set of interactive, multimedia Jupyter Notebooks with coding examples and conceptual questions that engage students with the material and let them test their understanding. We discuss below how this approach may improve engagement and retention, and how the technical implementation removes barriers to entry and enables the materials to stay current with emerging Rosetta methods.
Results
A. PyRosetta workshops cover a broad range of basic and advanced topics
To make a broad range of topics in the field accessible to the public, we have created a diverse set of PyRosetta workshops within Jupyter Notebooks (i.e., PyRosetta notebooks) and shared them in a public, open-source GitHub repository (https://github.com/RosettaCommons/PyRosetta.notebooks). These notebooks aim to teach both the fundamentals as well as the applications of biomolecular structure prediction and design. The set of notebooks currently includes 16 modules and is split into two parts. Part I introduces the basics of PyRosetta (Chapters 2–9), and Part II explores advanced applications (Chapters 10–16), such as antibody design and membrane protein modeling (Fig. 1). Chapter 1 walks students through the process of setting up PyRosetta in Google Colaboratory with step-by-step instructions and guiding screenshots.
Figure 1:
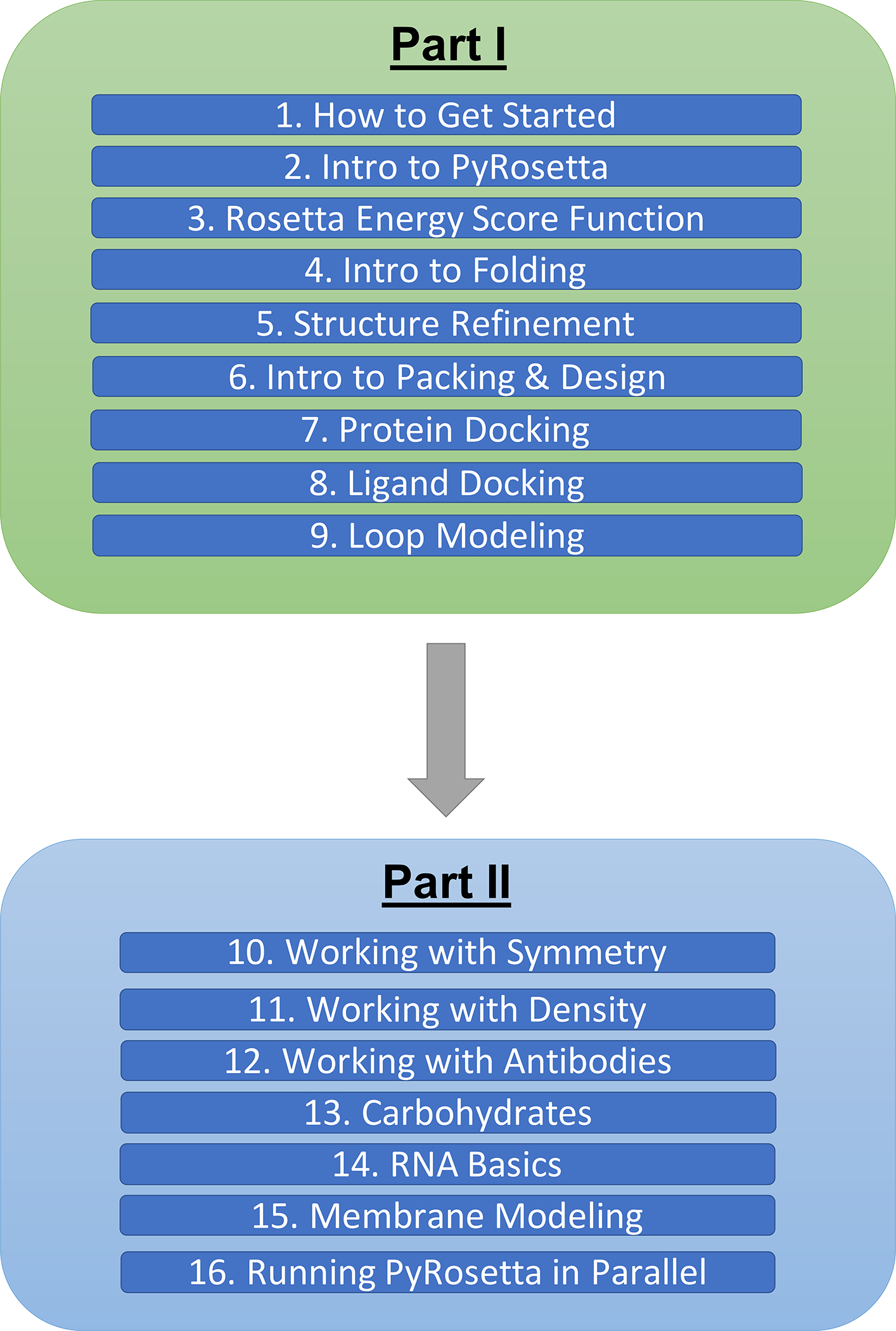
Map of general topics covered in the PyRosetta notebooks (as of October 2020).
Part I focuses on the two main scientific capabilities of Rosetta: sampling and scoring biomolecular conformations. The notebooks explain the technical basics of PyRosetta, starting with how to create a Pose object, which is the container that holds all atoms, molecules, coordinates, energies, and other details about the system. Next, students learn how to make a ScoreFunction object to approximate free energies and how to combine Mover objects to manipulate the Pose conformations. In addition to these technical skills, Part I builds the fundamental theoretical concepts that frame the challenges of sampling and scoring conformations. Students are introduced to Levinthal’s paradox, the idea that the conformational space available to proteins is exponentially large and thus impossible to search comprehensively (46). They also learn about Anfinsen’s Dogma, the idea that a folded protein is at a thermodynamic minimum free energy state (47). The workshops teach students how to employ various potential functions, which can be physics-based (van der Waals, Coulomb) or knowledge-based (hydrogen bonding, side-chain energies). Students learn that these functions can be empirically optimized for protein-scale phenomena, such as folding and design. Most exercises in Part I are short and provide detailed guidance. Moving GIF animations, schematics, and images of biomolecules (created in PyMOL software) are used to illustrate general concepts (48). For example, students are guided through the application of a TrialMover, which tests a conformational change, evaluates the new energy, and uses the Metropolis Monte Carlo criterion to either accept or reject the change (49). In addition to general concepts, some visualizations also depict expected outcomes, such as a PyMOL movie of a basic folding algorithm. The learning objectives for the workshops in Part I can be found in Table 1.
Table 1:
List of workshop topics and learning objectives in Part I.
| Current Topics | Students Will Be Able To |
|---|---|
|
1.00 How to Get Started 1.01 PyRosetta Google Drive Setup 1.02 PyRosetta Google Drive Usage Example 1.03 How to Install Local PyRosetta |
• Set up PyRosetta in Google Colab. • Set up PyRosetta on local computer (optional). |
|
2.00 Intro to PyRosetta 2.01 Pose Basics 2.02 Working with Pose Residues 2.03 Accessing PyRosetta Documentation 2.04 Getting Spatial Features from Pose 2.05 Protein Geometry 2.06 Visualization and PyMOL Mover 2.07 RosettaScripts in PyRosetta 2.08 Visualization and pyrosetta.distributed.viewer |
• Load a PDB structure. • Measure and alter protein structure (in internal or Cartesian coordinates). • Visualize macromolecules and PyRosetta ResidueSelectors within Jupyter Notebooks and through the PyMol-PyRosetta interface. • Run a RosettaScript from Python. • Instantiate and use individual configured components (objects) from a RosettaScript |
|
3.00 Rosetta Energy Score Functions 3.01 Score Function Basics 3.02 Analyzing Energy Between Residues 3.03 Energies and the PyMOL Mover |
• Test different score function components or weighted combinations. |
|
4.00 Intro to Folding 4.01 Basic Folding Algorithm 4.02 Low-Res Folding and Fragments |
• Explain the fundamental challenges of protein structure prediction. • Describe the use of protein fragments for building protein backbones. • Implement a Metropolis Monte Carlo search strategy. • Use standard PyRosetta protocols to optimize protein structure. |
|
5.00 Structure Refinement 5.01 High-Res Movers 5.02 Refinement Protocol |
• Implement a Monte Carlo-plus-minimization algorithm. • Use various standard PyRosetta Movers to manipulate protein structure. |
|
6.00 Intro to Packing and Design 6.01 Side-Chain Conformations and Dunbrack Energies 6.02 Packing Design Regional Relax 6.03 Design with a Resfile and Relax 6.04 Protein Design 2 6.05 HBnet Before Design 6.06 Intro to Parametric Backbone Design 6.07 Intro to de novo Protein Design 6.08 Point Mutation Scan |
• Optimize side-chain conformations for a set of specified residues using PyRosetta. • Write custom PyRosetta protocols to simultaneously optimize protein structure and sequence. • Integrate sidechain packing with small and shear moves and minimization in PyRosetta refinement protocols. • Precede sidechain packing with hydrogen bond network design • Design proteins using custom scorefunctions with non-pairwise decomposable scoreterms in PyRosetta. • Design symmetrical proteins using Parametric backbone design • Design families of proteins with regular arrangements of secondary-structure elements • Generate mutagenesis library for antigen-antibody binding with PyRosetta • Compare mutant and wild-type binding energy • Visualize mutagenesis results using a Python heatmap |
|
7.00 Protein Docking 7.01 Fast Fourier Transform Docking 7.02 Docking Moves in Rosetta |
• Describe the major approaches to docking (grid based, FFT, Monte Carlo) and their advantages and disadvantages. • Use the PyJobDistributor for job distribution |
|
8.00 Ligand Docking PyRosetta 8.01 Ligand Docking XMLObjects 8.02 Ligand Docking pyrosetta.distributed |
• Perform high-resolution protein-ligand refinement using the DockMCMProtocol mover. • Perform global ligand docking using XMLObjects. • Perform ligand docking with a genetic algorithm using pyrosetta.distributed. |
|
9.00 Loop Modeling 9.01 Using Gen KIC |
• Describe the loop modeling and loop closure problems. • Describe the cyclic coordinate descent and kinematic closure approach and identify their advantages and limitations. • Close loops using Gen KIC protocol. |
Part II guides learners through advanced applications of PyRosetta, relying on the basic skills and concepts introduced in Part I. Chapter 12, for example, explores how PyRosetta can also be used to model and design antibodies, which is an important challenge faced by pharmaceutical companies (50). In Chapter 14, students learn how to apply the same approaches to predict RNA structures, which are increasingly recognized for critical roles in catalysis and regulation (51). Chapter 15 explores the tools for investigating membrane proteins, which comprise approximately 60% of drug targets (52). A larger emphasis is placed on workshop exercises to introduce learners to a variety of questions and methods that are currently used in the field. For advanced students, Chapter 16 reviews more intensive tasks that can be executed outside of Google Colaboratory, such as parallelization with GNU and dask libraries (53). The learning objectives for the workshops in Part II can be found in Table 2.
Table 2:
List of workshop topics and learning objectives in Part II.
| 10.00 Working with Symmetry | • Create crystallographic symmetry files • Load proteins with symmetric components • Convert a monomer into a symmetric assembly. • Learn how to use common Rosetta protocols with symmetry enabled |
| 11.00 Working with Density | • Convert PDB density files into Rosetta-readable files • Load density files into Rosetta • Use RosettaDensity to score a structure and use density to guide modeling |
|
12.00 Working with Antibodies 12.01 Rosetta Antibody Framework and Simple Metrics 12.02 Rosetta Antibody Design |
• Load antibody structures into the RosettaAntibody framework • Retrieve antibody specific information such as CDR loop regions, clusters, etc. for use in custom protocols • Set antibody-specific residue selectors and configure task operations for use in modeling and design • Design new antibodies with the RosettaAntibodyDesign protocol |
|
13.00 Carbohydrates 13.01 Glycan Trees, Selectors and Movers 13.02 Glycan Modeling and Design |
• Load an oligosaccharide or a glycoprotein. • Use RosettaCarbohydrates to add glycans conjugated to proteins. • Evaluate sugar-sugar linkage energies. • Select carbohydrates and get carbohydrate chemical and connectivity information • Optimize carbohydrate structure through linkage torsions, ring conformers, and sidechain conformers. Design carbohydrate recognition motifs (sequons) for designing glycans into proteins |
| 14.00 RNA Basics | • Load nucleic acids and identify nucleic acid residues in poses. • Identify canonical and non-canonical base pairs in RNA structures. • View and manipulate nucleic acid torsion angles. • Evaluate nucleic acid energies using RNA-specific low- and high-resolution score functions. Isolate RNA-specific score terms (e.g. stacking energies, base pairing potential). • Decompose RNA structures into 3D RNA motifs. • Use idealized torsion angles for RNA residues to generate an idealized A-form helix. • Replace RNA residues with a new sequence for homology modeling. • Use RNA fragments when building an RNA backbone and use minimization to refine resulting structures. Build a Monte Carlo search strategy using these approaches. • Use the FARFAR protocol for sampling RNA structures, which combines fragment assembly and high-resolution minimization moves. |
|
15.00 Modeling Membrane Proteins 15.01 Accounting for the Lipid Bilayer 15.02 Membrane Protein ddG of mutation |
• Use membrane tools to orient a protein in the lipid bilayer. • Calculate the lowest energy orientation for a membrane protein. • Identify membrane protein pores and cavities. • Interpret model quality using terms from franklin2019, the membrane energy function. • Compute the ΔΔG of mutation |
|
16.00 Running PyRosetta in Parallel 16.01 PyData, DDGs, and PSSMs 16.02 PyData Miniprotein Design 16.03 GNU parallel 16.04 dask.delayed via SLURM 16.05 Ligand Docking dask |
• Parallelize macromolecule modeling tasks using distributed computing, elastic cloud computing, and high-performance computing infrastructures. • Parallelize PyRosetta jobs using GNU parallel and the SLURM job scheduling system. • Visualize and execute PyRosetta job parallelization with the dask module. • Analyze outputs from parallelized PyRosetta jobs in real-time as completed. |
B. Students can access the multimedia PyRosetta workshops on the Google Colaboratory platform
Google Colaboratory is an online web environment for Jupyter Notebooks on a cloud-based virtual machine accessible with any browser. Google Colaboratory provides students with powerful computational resources, including 13 GB of RAM, 33 GB of disk space, 2.30 GHz of CPU, and continuous sessions of up to 12 hours (45). While Jupyter Notebooks have been used for engineering education (54, 55), Google Colaboratory offers a few advantages for studying biomolecular modeling, starting with the free in-the-cloud computing power. Students can complete most of the PyRosetta Notebooks in the Google Colaboratory environment (Fig. 2). They can open notebook files and store different versions directly in their Google Drive. The initial configuration of the PyRosetta software package in Google Colaboratory is automated and takes approximately 10 minutes. Afterwards, students simply import the supporting pip package pyrosettacolabsetup (56) and the configured PyRosetta package. Students can complete the provided exercises to build their own solutions and modify any line of code in the workshops, which pair introductory passages, concepts, and exercises with supporting PyMOL images, movies, and diagrams (Fig. 3).
Figure 2:
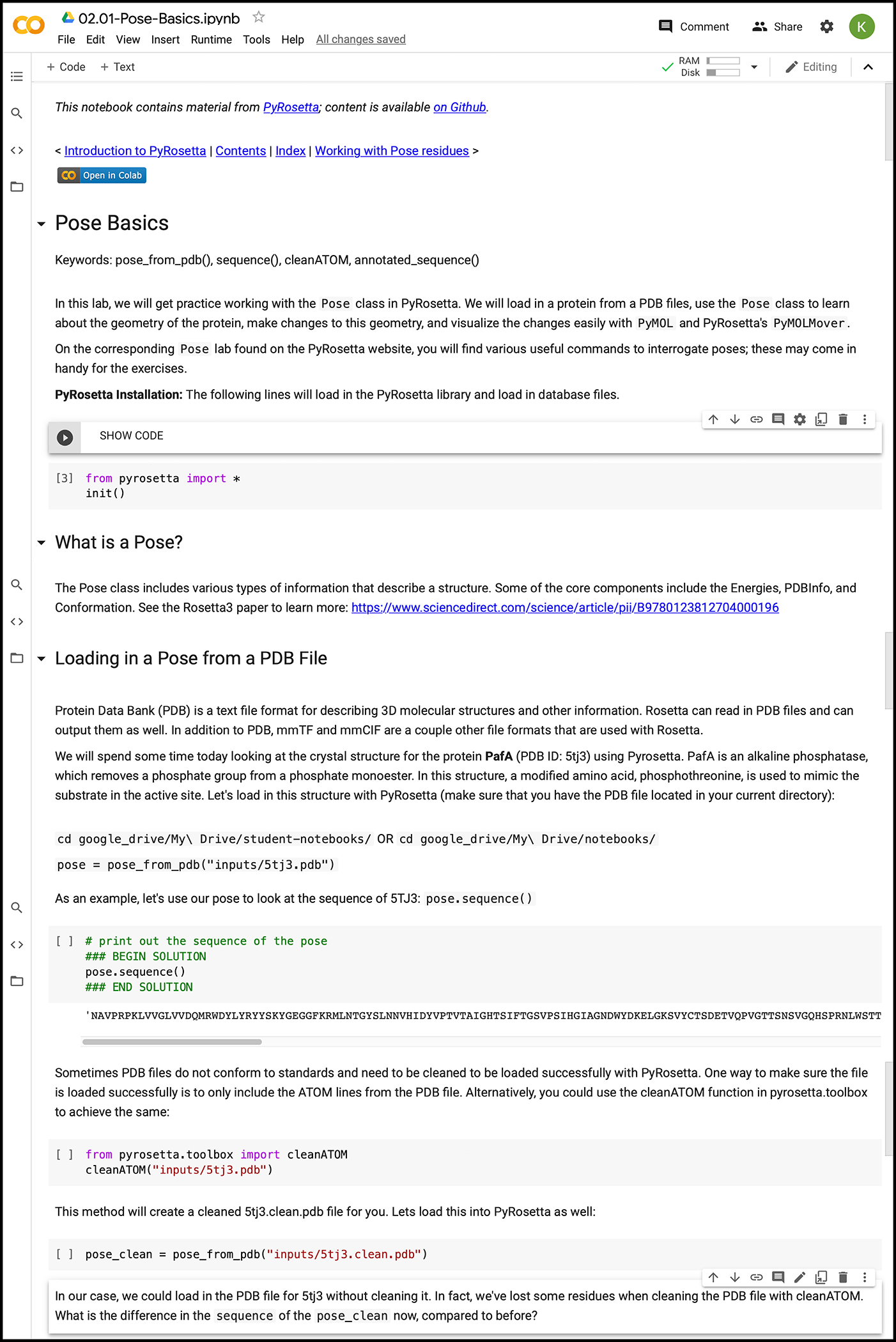
Screenshot of the Pose Basics workshop module in Google Colaboratory.
Figure 3: Notebook multimedia examples.

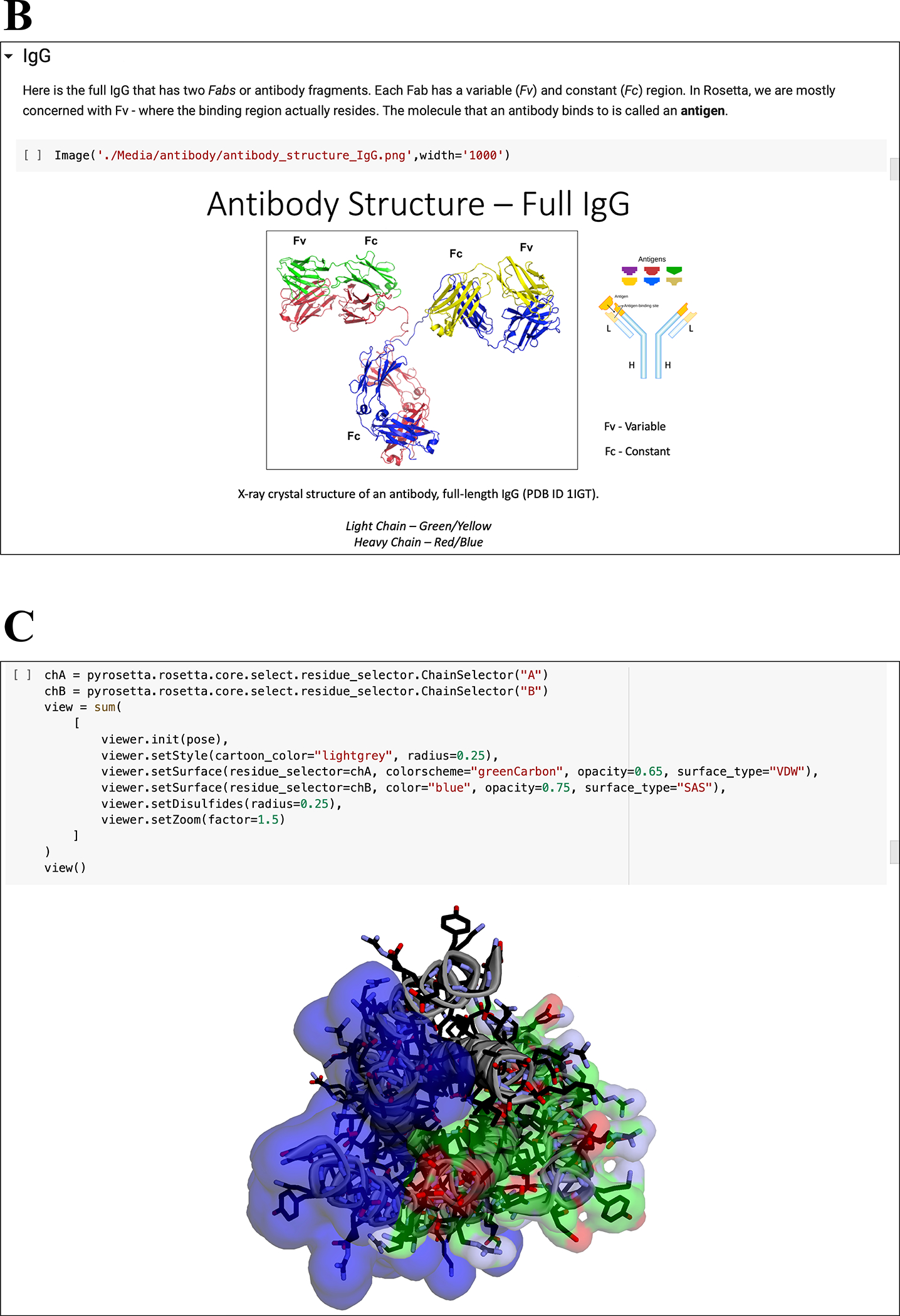
(A) Moving GIFs of small and shear movers from “05.01 High Res Movers.” (B) Images and diagrams of antibody structures from “12.00 Working with Antibodies.” (C) Integration of PyRosetta with py3Dmol for interactive macromolecular visualization in Jupyter Notebooks.
C. Jupyter Notebooks enable features for students and instructors
To create both student and instructor versions of assignments in the notebooks, we incorporated nbgrader (57). The nbgrader module enables developers and instructors to create and maintain a single master copy of each workshop. The master copy includes solutions to all exercises, and the student version of the workshop is automatically generated without selected solutions (Fig. 4). Thus, developers can write PyRosetta coding examples and problems for students to attempt on their own. To help students locate examples of specific concepts and commands, we also incorporated nbpages (58), which enables the automatic generation of the Table of Contents and a searchable Keyword Index in notebook and markdown form (Fig. 5). These tools are activated by the provided make-student-nb.bash script, which developers can use to update the student notebooks, Table of Contents, and Keywords Index with a single command.
Figure 4: Example of student exercises.
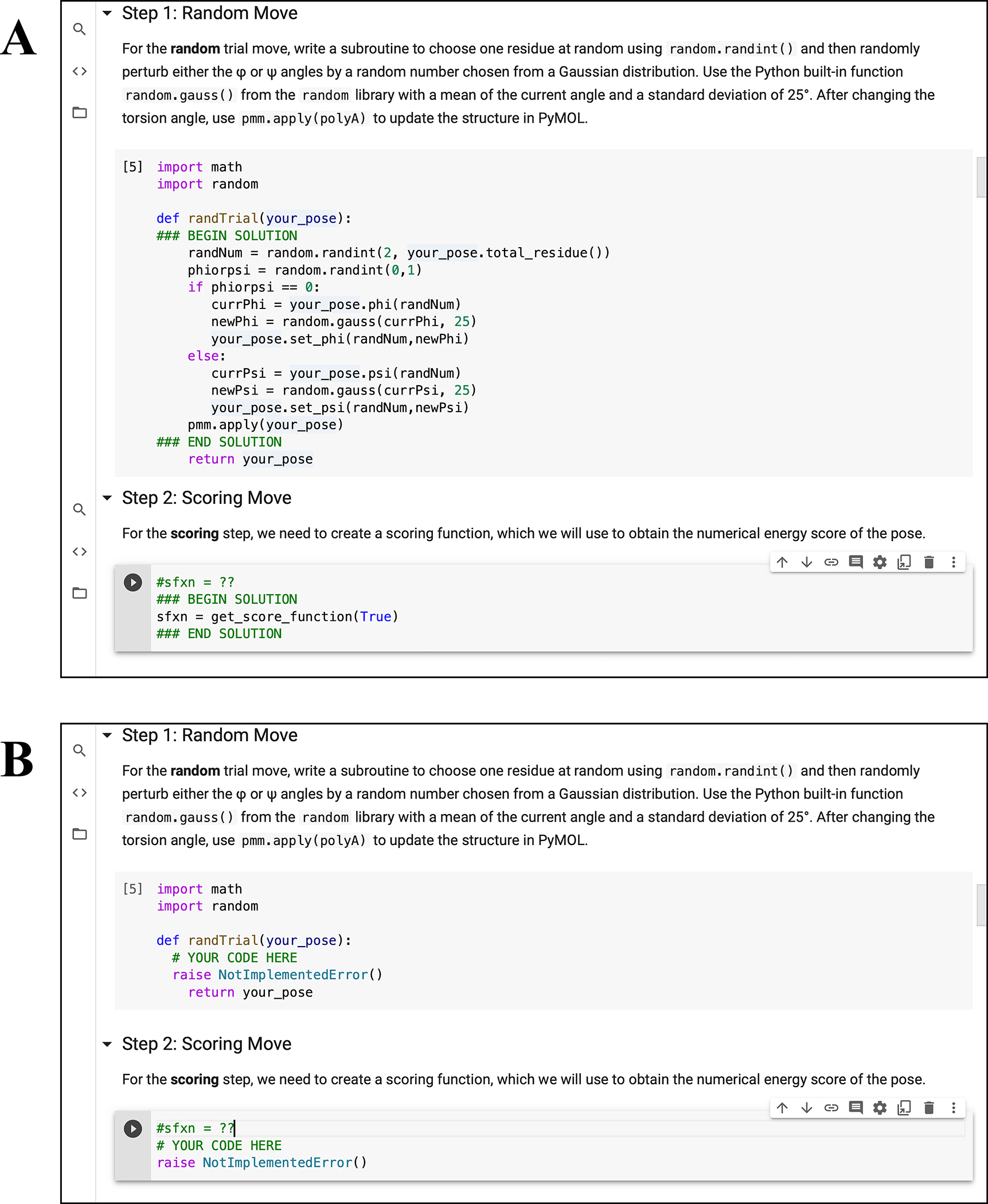
(A) Instructor version of “04.01 Basic Folding Algorithm” workshop with written solutions and (B) student version of the workshop with omitted solutions.
Figure 5:
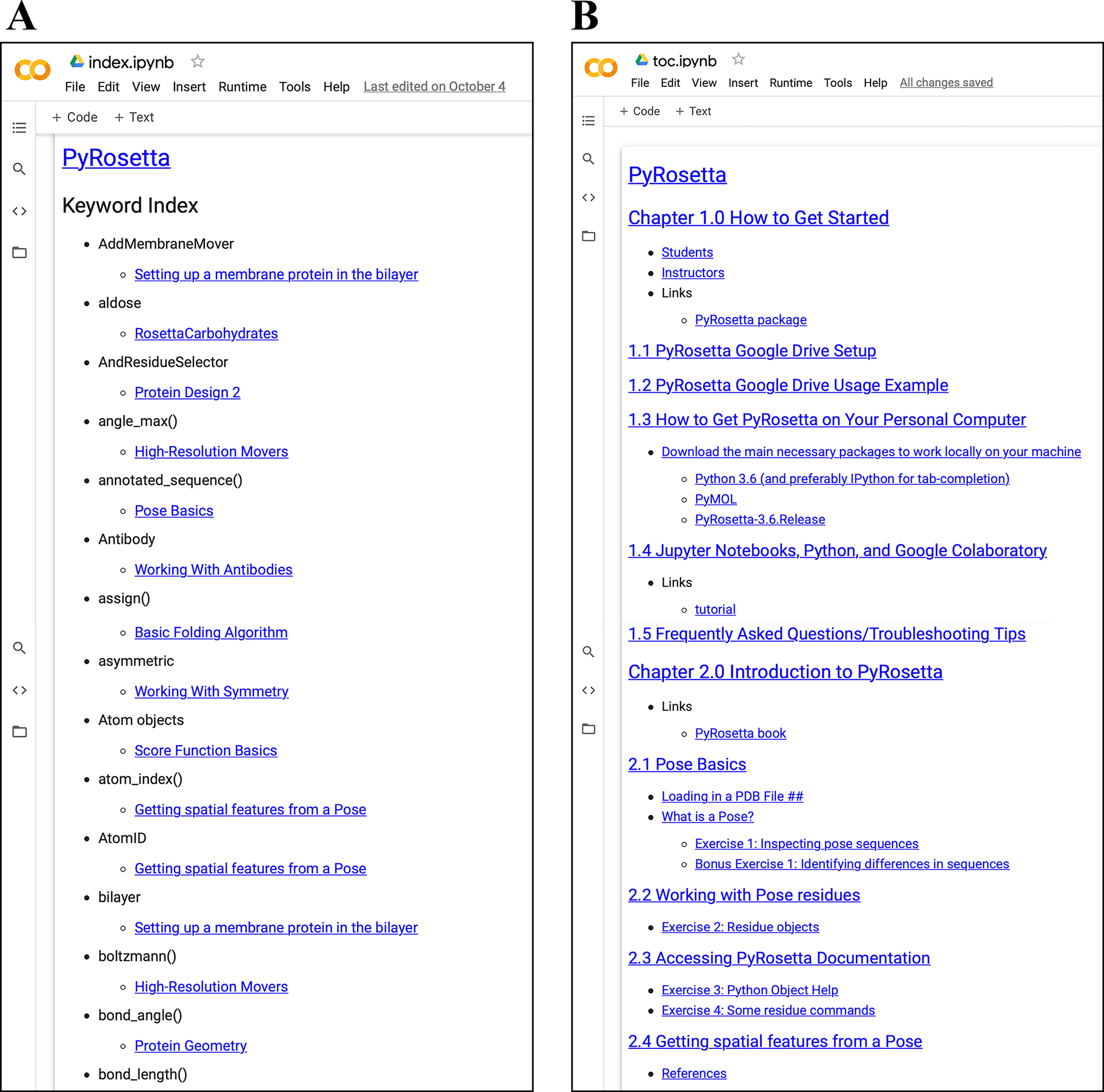
Automatically generated supporting material using nbpages.
(A) Keyword index notebook with links. (B) Table of contents notebook with links.
Instructors can make changes to the original set of Jupyter Notebook workshops by forking the main public repository (59). This allows instructors to tailor the workshops for specific curriculums. In addition, any changes to the repository files that would benefit the public can be incorporated directly into the main repository via GitHub pull requests, which can be reviewed and approved by a Rosetta Commons member. Fig. 6A shows a workflow for instructors who simply want to use the workshops in their courses, and Fig. 6B illustrates a workflow for instructors who wish to make changes to the material.
Figure 6: Instructor workflows.
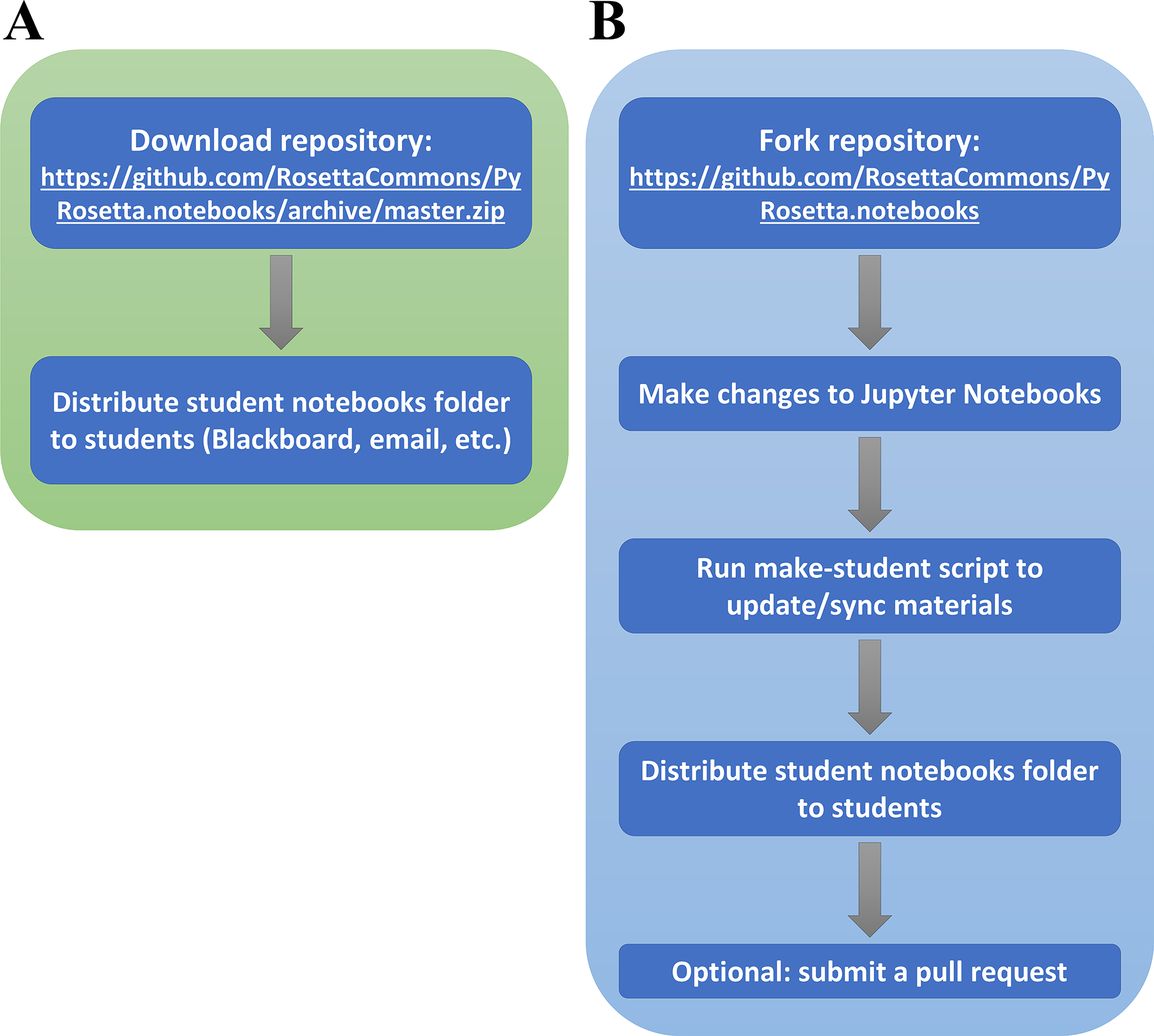
(A) Workflow without making changes (B) Workflow with changes to share with others via pull request to the RosettaCommons GitHub repository.
Further, in Chapter 2 we showcase the ability to visualize macromolecules directly within the Jupyter notebooks using py3Dmol, a web-based Jupyter widget encompassing an interactive 3Dmol.js molecular viewer (60). The py3Dmol bindings (in the pyrosetta.distributed.viewer namespace) facilitate on-the-fly, interactive visualization of PyRosetta ResidueSelector objects, which allow students to choose subsets of residues based on sequence, chemistry, or structural properties (Fig. 3C). For those who install PyRosetta on their local computer, the motions of a protein in a protocol can be watched in an external PyMOL window using the PyRosetta PyMOLObserver (61).
Chapter 16 demonstrates how to scale up simulations to high performance computing resources using the Slurm Workload Manager with GNU parallel, dask and distributed modules (53, 62). We additionally introduce the pyrosetta.distributed.dask namespace for PyRosetta integration with the dask-jobqueue module, providing a user-friendly interface for PyRosetta pre-initialization of worker machines allowing options-based configuration of macromolecular modeling tasks in distributed computing and cloud computing environments. These developments will enable future pedagogical programs to encompass advanced macromolecular modeling exercises and allow for additional educational content to be added with ease.
D. Learning outcomes
We piloted early versions of the notebooks in four separate teaching contexts. They were used in a formal university course in spring 2019 for a combined graduate/undergraduate elective course, in a Code Academy for new graduate students and postdocs in Rosetta Commons labs, and for a one-week code school for a class of undergraduate summer interns. Finally, we have shared the GitHub link with several individuals learning PyRosetta on their own.
In the spring of 2019, the formal university course, ChemBE414/614: Protein Structure Prediction and Design, enrolled 9 undergraduate students and 11 Masters/PhD students. Over the years, students who took this course have come from departments of chemical and biomolecular engineering, biomedical engineering, biophysics, chemistry, computer science, and applied math. In the pre-course survey, roughly half (45%) of the spring 2019 class indicated that they had “good” or “expert-level” familiarity with Python. Nearly all of these students had programming experience in some language prior to the course. However, their experience levels varied greatly in programming, biology, chemistry, and math. Following the course, students gave high reviews (quality of course 4.65/5.00 and teaching effectiveness 4.53/5.00). The biomolecular computation skills gained by students were evidenced by a range of successful course projects on topics including “Structure-based prediction of peptide-MHC binding,” “Finding the relationship between epistasis and score in sequentially mutated TEM-1 β-lactamase,” and, on the methodological side, “Comparison of optimization methods used in protein structure prediction.” This course used the Notebooks on local Linux or Mac installations, without the use of the Google Colaboratory platform. In the pilot, multiple students mentioned the technical challenges of using PyRosetta on their computer in course evaluation.
Like ChemBE414/614, Code Academy trainees varied in programming and scientific experience. In the pre-course survey, 7 out of 19 of trainees (37%) indicated that they had “very little to no programming experience”. After the course, trainees were asked to respond to a post-course survey. When asked about whether Jupyter Notebooks were an effective teaching tool, 13 out of 15 respondents selected “Agree” or “Strongly Agree.” Furthermore, all the respondents agreed or strongly agreed that the course gave them confidence to write more advanced protocols for their research. Code Academy trainees completed mini-projects such as “Antibody design for Ebola” and “Modeling intrinsically disordered proteins for cell signaling.”
The summer interns continued to complete successful research projects in ten different academic labs and one industry research site (36). Finally, some self-paced learners who tested the complete multimedia workshops shared comments including: “these notebooks make PyRosetta more approachable to non-experts”, “you can install PyRosetta in your Google Drive and use it from many different machines”, and “attempting problems myself allowed me to pinpoint gaps in my understanding.”
Discussion
Protein structure prediction and design tools are powerful and have the potential to impact biophysics and many cognate disciplines, but there are several challenges for students including access to the tools and the varied backgrounds of students. Here we have described a set of interactive notebooks for learning biomolecular structure prediction and design that can be used in a classroom context or for individual self-study. Educators within the Rosetta community have already used these notebooks extensively, and we hope that educators teaching high school, undergraduate, and graduate courses will also benefit from using and adapting these notebooks. A good starting point for new instructors to develop the necessary background to teach these workshops is a recent review article by Kuhlman and Bradley (63). Then, new instructors can complete the workshops themselves and read the associated primary literature linked within each notebook.
Students are advised to have some familiarity with basic Python capabilities, including creating/calling variables, functions, and classes. For a classroom setting, reviewing these skills prior to attempting the workshops may be beneficial. In ChemBE414/614 at Johns Hopkins, the instructor spent one week reviewing the necessary skills and assigned one homework assignment practicing Python.
One of the advantages of this platform is that it is free and publicly available on GitHub (https://github.com/RosettaCommons/PyRosetta.notebooks). Another advantage is that PyRosetta can be accessed through the Google Colaboratory online in a web browser, which requires no local computer installation and can quickly integrate open-source packages. In addition to its accessibility, Google Colaboratory provides students with free and powerful computational resources (45). These advantages address the current technological challenges with the current resources for PyRosetta. This online platform also provides an environment for multimodal learning material, such as molecular visualization movies and coding examples. With a broad scope of topics from contributors of different areas of expertise, students are also able to gain exposure to the different applications of PyRosetta and develop the skills to pursue more in-depth applications. For instructors, this set of modules can be easily adapted to a course syllabus by modifying workshops or adding relevant examples.
In addition to the advantages, the platform has some limits. The Google Colaboratory platform complicates communication with the visualization software PyMOL, and we have so far been unable to make this connection simple. While the PyMOLObserver is the archetypal tool for real-time visualization of PyRosetta modeling trajectories (61), students with a local installation of PyRosetta are nevertheless able to view their algorithms in real-time on their local computer’s PyMOL. Within the Google Colaboratory, the pyrosetta.distributed.viewer (with py3Dmol bindings) currently supports dynamic visualization updates upon biomolecular conformational changes, which is convenient for viewing intermediate steps of biomolecular modeling tasks, such as between PyRosetta Movers, directly within Jupyter Notebooks. Although the pyrosetta.distributed.viewer mimics only a subset of PyMOL functionalities, it accepts ResidueSelector-based user inputs, thus allowing a more streamlined interface to interactive biomolecular modeling and design. However, because py3Dmol does not have multithreading or communication between threads, Google Colaboratory users cannot continuously update an instance of the 3Dmol.js molecular viewer as a PyRosetta protocol trajectory is calculated.
Our Notebooks may be compared with other educational materials for computational molecular biophysics. There are several textbook style software resources, such as the beginner’s guide to CHARMM (64) and the web-based lessons on CHARMM (65). Recently, MOE has been used for an integrated engineering curriculum (66). Additionally, FoldIt (67) and EteRNA (68) have been used in an interdisciplinary week-long program for undergraduate and high school students (69). Our contribution of PyRosetta notebooks is complementary to these and has advantages with its active involvement of students, multimedia integration, and engagement with viable and leading tools that can be used flexibly in new, innovative research. In addition, unlike many other electronic workshop materials, the PyRosetta notebooks are used to automatically test new versions of the PyRosetta software. Each Jupyter Notebook is converted into a simple Python script that is run continually on the community’s testing servers, and any malfunctions from new or modified code must be fixed before accepted into the main Rosetta repository. The automated notebook testing and GitHub pull request practices ensure that the workshops always remain functional for users.
Overall, the PyRosetta Notebooks are designed to be a gateway tool to introduce students to the fundamentals of biomolecular structure prediction and design. This platform could potentially be employed in high school lab courses, STEM summer programs, advanced undergraduate courses, as well as graduate courses. In addition, we encourage students to seek additional support from the distributed, collaborative network by posting on the Rosetta Commons forum (https://www.rosettacommons.org/forum). In addition, Google Colaboratory is compatible with TensorFlow and is heavily used for developing machine learning models because of the free GPU resources. In the future, machine learning could be incorporated with the capabilities of PyRosetta to explore new areas of research in the field (70). Furthermore, the GitHub archive is a living collection that will continually expand to include other applications of PyRosetta and aspects of macromolecular modeling to introduce a broader audience to the field.
Acknowledgements
Sid Chaudhury co-wrote the original set of (paper) workshops that grew over the years into this collection. Evan Baugh expanded these with a set of code-level tools into the first set of workshops posted online. JJG thanks the students of ChemBE 414/614 at Johns Hopkins University over the past decade who have completed the workshops as they have evolved and provided feedback each year. Funding provided from a Rosetta Commons mini-grant, a Johns Hopkins Center for Educational Resources Technology Fellowship to KHL, a Stanford School of Medicine TRS Teaching Innovation Grant to MAA, a Hertz Foundation Fellowship to RFA, NSF Graduate Research Fellowships to JCK, RR, and RFA, and grants NSF/DBI 1659649, NIH R01-GM127578, NIH R01-GM078221, NIH R01-GM73151, NIH R35 GM122517, NSF/CCI 1740549, and NSF CHE-1740549.
Footnotes
Use of Human Subjects
The use of student feedback and course evaluations in this study was reviewed and approved by the Homewood Institutional Review Board at Johns Hopkins University (HIRB #11185).
Conflict of Interest
The teaching tools described in this paper relate to the Rosetta software, and some of the funding for this study was provided by the Rosetta Commons. JJG and JBS are unpaid board members of the Rosetta Commons. Under institutional participation agreements between the University of Washington, acting on behalf of the Rosetta Commons, Johns Hopkins University and UC-Davis (as well as other Rosetta Commons institutions where work was performed on this report) may be entitled to a portion of revenue received on commercial licensing of Rosetta software. As a member of the Scientific Advisory Board, JJG has a financial interest in Cyrus Biotechnology. Cyrus Biotechnology distributes the Rosetta software, which may include methods described in this paper. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
References
- 1.Guzenko D, Lafita A, Monastyrskyy B, Kryshtafovych A, Duarte JM, Assessment of protein assembly prediction in CASP13. Proteins Struct. Funct. Bioinforma. (2019) 10.1002/prot.25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang PS, Boyken SE, Baker D, The coming of age of de novo protein design. Nature (2016) 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- 3.NobelPrize.org, The Nobel Prize in Chemistry 2013. Nobel Media AB 2019. [Google Scholar]
- 4.Brooks BR, et al. , CHARMM: The biomolecular simulation program. J. Comput. Chem. (2009) 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LLC, Schrodinger Software Suite, New York, NY, USA, 2011. [Google Scholar]
- 6.Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc., Montreal, QC, Canada, 2013. [Google Scholar]
- 7.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K, Scalable molecular dynamics with NAMD. J. Comput. Chem. (2005) 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, Ferguson D, Seibel G, Kollman P, AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. (1995) 10.1016/0010-4655(95)00041-D. [DOI] [Google Scholar]
- 9.Berendsen HJC, van der Spoel D, van Drunen R, GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. (1995) 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 10.Das R, Baker D, Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. (2008) 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 11.Leman JK, Weitzner BD, Lewis SM, Consortium R, Bonneau R, Macromolecular modeling and design in Rosetta: new methods and frameworks. Preprints, 1–14 (2019). [Google Scholar]
- 12.Leaver-Fay A, et al. , “Rosetta3: An object-oriented software suite for the simulation and design of macromolecules” in Methods in Enzymology, (2011) 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J, Practically useful: What the R osetta protein modeling suite can do for you. Biochemistry (2010) 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, Meiler J, Small-molecule ligand docking into comparative models with Rosetta. Nat. Protoc. (2013) 10.1038/nprot.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng CY, Chou FC, Das R, “Modeling complex RNA tertiary folds with Rosetta” in Methods in Enzymology, (2015) 10.1016/bs.mie.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 16.Alford RF, Koehler Leman J, Weitzner BD, Duran AM, Tilley DC, Elazar A, Gray JJ, An Integrated Framework Advancing Membrane Protein Modeling and Design. PLoS Comput. Biol. (2015) 10.1371/journal.pcbi.1004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ovchinnikov S, Park H, Kim DE, DiMaio F, Baker D, Protein structure prediction using Rosetta in CASP12. Proteins Struct. Funct. Bioinforma (2018) 10.1002/prot.25390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ, Blind prediction performance of RosettaAntibody 3.0: Grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins Struct. Funct. Bioinforma. (2014) 10.1002/prot.24534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burman SSR, Nance ML, Jeliazkov JR, Labonte JW, Lubin JH, Biswas N, Gray JJ, Novel sampling strategies and a coarse-grained score function for docking homomers, flexible heteromers, and oligosaccharides using Rosetta in CAPRI Rounds 37–45. bioRxiv Biophys., 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marcu O, Dodson EJ, Alam N, Sperber M, Kozakov D, Lensink MF, Schueler-Furman O, FlexPepDock lessons from CAPRI peptide–protein rounds and suggested new criteria for assessment of model quality and utility. Proteins Struct. Funct. Bioinforma. (2017) 10.1002/prot.25230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao Z, et al. , RNA-Puzzles Round III: 3D RNA structure prediction of five riboswitches and one ribozyme. RNA (2017) 10.1261/rna.060368.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D, Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science (80-. ) 302, 1364–1368 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D, Principles for designing ideal protein structures. Nature 491, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobs TM, Williams B, Williams T, Xu X, Eletsky A, Federizon JF, Szyperski T, Kuhlman B, Design of structurally distinct proteins using strategies inspired by evolution. Science 352, 687–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Humphris EL, Kortemme T, Prediction of Protein-Protein Interface Sequence Diversity Using Flexible Backbone Computational Protein Design. Structure 16, 1777–1788 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Guntas G, Purbeck C, Kuhlman B, Engineering a protein–protein interface using a computationally designed library. Proc. Natl. Acad. Sci. 107, 19296–19301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch E-M, Wilson IA, Baker D, Computational Design of Proteins Targeting the Conserved Stem Region of Influenza Hemagglutinin. Science (80-. ). 332, 816–821 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strauch E-M, et al. , Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 35, 667–671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO, Baker D, Accurate design of co-assembling multi-component protein nanomaterials. Nature 510, 103–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, André I, Gonen T, Yeates TO, Baker D, Computational Design of Self-Assembling Protein Nanomaterials with Atomic Level Accuracy. Science (80-. ). 336, 1171–1174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masica DL, Schrier SB, Specht EA, Gray JJ, De novo design of peptide-calcite biomineralization systems. J. Am. Chem. Soc. 132, 12252–12262 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Pyles H, Zhang S, De Yoreo JJ, Baker D, Controlling protein assembly on inorganic crystals through designed protein interfaces. Nature 571, 251–256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaudhury S, Lyskov S, Gray JJ, PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics (2010) 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray JJ, Chaudhury S, Lyskov S, Labonte JW, The PyRosetta interactive platform for protein structure prediction and design: A set of educational modules. 3rd ed. Createspace Independent Publishing Platform; (2017). [Google Scholar]
- 35.Gray JJ, Chaudhury S, Lyskov S, Labonte JW, PyRosetta Tutorials [accessed December 11, 2019]. http://www.pyrosetta.org/tutorials [Google Scholar]
- 36.Alford RF, Leaver-Fay A, Gonzales L, Dolan EL, Gray JJ, A cyber-linked undergraduate research experience in computational biomolecular structure prediction and design. PLoS Comput. Biol. (2017) 10.1371/journal.pcbi.1005837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D, Rosettascripts: A scripting language interface to the Rosetta Macromolecular modeling suite. PLoS One 6, 1–10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.RosettaCommons, What is Rosetta?, [accessed December 11, 2019]. https://www.rosettacommons.org/docs/latest/Home. [Google Scholar]
- 39.RosettaCommons, Rosetta Manuals, [accessed December 11, 2019]. https://www.rosettacommons.org/manuals/latest/.
- 40.RosettaCommons, Rosetta Forums, [accessed December 11, 2019]. https://www.rosettacommons.org/forum. [Google Scholar]
- 41.Freeman S, Eddy SL, McDonough M, Smith MK, Okoroafor N, Jordt H, Wenderoth MP, Active learning increases student performance in science, engineering, and mathematics. Proc. Natl. Acad. Sci. U. S. A. (2014) 10.1073/pnas.1319030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sankey M, Birch D, Gardiner M, Engaging students through multimodal learning environments: The journey continues in ASCILITE 2010 - The Australasian Society for Computers in Learning in Tertiary Education, (2010). [Google Scholar]
- 43.Edgcomb AD, Vahid F, Effectiveness of online textbooks vs. Interactive web-native content. ASEE Annu. Conf. Expo. Conf. Proc. (2014). [Google Scholar]
- 44.Kluyver T, Ragan-kelley B, Pérez F, Granger B, Bussonnier M, Frederic J, Kelley K, Hamrick J, Grout J, Corlay S, Ivanov P, Avila D, Abdalla S, Willing C, Jupyter Notebooks—a publishing format for reproducible computational workflows (2016) 10.3233/978-1-61499-649-1-87. [DOI]
- 45.Carneiro T, Da Nobrega RVM, Nepomuceno T, Bin Bian G, De Albuquerque VHC, Filho PPR, Performance Analysis of Google Colaboratory as a Tool for Accelerating Deep Learning Applications. IEEE Access (2018) 10.1109/ACCESS.2018.2874767. [DOI] [Google Scholar]
- 46.Karplus M, The Levinthal paradox: Yesterday and today. Fold. Des. 2, 69–75 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Anfinsen, Folding of Protein Chains. Science (80-. ). 181, 223–230 (1973). [DOI] [PubMed] [Google Scholar]
- 48.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. [Google Scholar]
- 49.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E, Equation of state calculations by fast computing machines. J. Chem. Phys. (1953) 10.1063/1.1699114. [DOI] [Google Scholar]
- 50.Weitzner BD, Jeliazkov JR, Lyskov S, Marze N, Kuroda D, Frick R, Adolf-Bryfogle J, Biswas N, Dunbrack RL, Gray JJ, Modeling and docking of antibody structures with Rosetta. Nat. Protoc. (2017) 10.1038/nprot.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das R, Karanicolas J, Baker D, Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods (2010) 10.1038/nmeth.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Overington JP, Al-Lazikani B, Hopkins AL, How many drug targets are there? Nat. Rev. Drug Discov. (2006) 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 53.Ford AS, Weitzner BD, Bahl CD, Integration of the Rosetta Suite with the Python Software Stack via reproducible packaging and core programming interfaces for distributed simulation. Protein Sci. (2019) 10.1002/pro.3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kantor J, Introduction to Chemical Engineering Analysis, [accessed December 15,0 2019]. http://jckantor.github.io/CBE20255/. [Google Scholar]
- 55.Marrugo A, Sensors and Actuators, [accessed December 15, 2019]. https://github.com/agmarrugo/sensors-actuators. [Google Scholar]
- 56.Le KH, pyrosettacolabsetup, https://pypi.org/project/pyrosettacolabsetup/.
- 57.Jupyter P, Blank D, Bourgin D, Brown A, Bussonnier M, Frederic J, Granger B, Griffiths T, Hamrick J, Kelley K, Pacer M, Page L, Pérez F, Ragan-Kelley B, Suchow J, Willing C, nbgrader: A Tool for Creating and Grading Assignments in the Jupyter Notebook. J. Open Source Educ. (2019) 10.21105/jose.00032. [DOI] [Google Scholar]
- 58.Kantor J, nbpages, https://pypi.org/project/nbpages/.
- 59.GitHub, About collaborative development models (2020), [accessed April 9, 2020]. https://help.github.com/en/github/collaborating-with-issues-and-pull-requests/about-collaborative-development-models.
- 60.Rego N, Koes D, 3Dmol.js: Molecular visualization with WebGL. Bioinformatics 31, 1322–1324 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baugh EH, Lyskov S, Weitzner BD, Gray JJ, Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS One (2011) 10.1371/journal.pone.0021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rocklin M, Dask: Parallel Computation with Blocked algorithms and Task Scheduling in Proceedings of the 14th Python in Science Conference, (2015) 10.25080/majora-7b98e3ed-013. [DOI] [Google Scholar]
- 63.Kuhlman B, Bradley P, Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. (2019) 10.1038/s41580-019-0163-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schleif R, A Concise Guide to CHARMM and the Analysis of Protein Structure and Function (2013), [accessed October 14, 2020]. https://pages.jh.edu/~rschlei1/Random_stuff/publications/charmmbook.pdf.
- 65.Miller BT, Singh RP, Schalk V, Pevzner Y, Sun J, Miller CS, Boresch S, Ichiye T, Brooks BR, Woodcock HL, Web-Based Computational Chemistry Education with CHARMMing I: Lessons and Tutorial. PLoS Comput. Biol. 10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ludovice P, MacNair D, Integrated Molecular Modeling Education in the Chemical Engineering Curriculum. AIChE 2019 Annual Meeting, Orlando, FL (2019, November 12). [Google Scholar]
- 67.Khatib F, Desfosses A, Koepnick B, Flatten J, Popović Z, Baker D, Cooper S, Gutsche I, Horowitz S, Building de novo cryo-electron microscopy structures collaboratively with citizen scientists. PLoS Biol. (2019) 10.1371/journal.pbio.3000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee J, Kladwang W, Lee M, Cantu D, Azizyan M, Kim H, Limpaecher A, Yoon S, Treuille A, Das R, RNA design rules from a massive open laboratory. Proc. Natl. Acad. Sci. (2014) 10.1073/pnas.1313039111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taly A, Nitti F, Baaden M, Pasquali S, Molecular modelling as the spark for active learning approaches for interdisciplinary biology teaching. Interface Focus 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kandathil SM, Greener JG, Jones DT, Recent developments in deep learning applied to protein structure prediction. Proteins Struct. Funct. Bioinforma. (2019) 10.1002/prot.25824. [DOI] [PMC free article] [PubMed] [Google Scholar]