

Abstract
Interactions among risk factors for osteoarthritis (OA) are not well understood. We investigated the combined impact of two prevalent risk factors: mechanical loading and genetically abnormal cartilage tissue properties. We used cyclic tibial compression to simulate mechanical loading in the cho/+ (Col11a1 haploinsufficient) mouse, which has abnormal collagen fibrils in cartilage due to a point mutation in the Col11a1 gene. We hypothesized that the mutant collagen would not alter phenotypic bone properties and that cho/+ mice, which develop early onset OA, would develop enhanced load-induced cartilage damage compared to their littermates. To test our hypotheses, we applied cyclic compression to the left tibiae of 6-month-old cho/+ male mice and wild-type (WT) littermates for 1, 2, and 6 weeks at moderate (4.5N) and high (9.0N) peak load magnitudes. We then characterized load-induced cartilage and bone changes by histology, microcomputed tomography, and immunohistochemistry. Prior to loading, cho/+ mice had less dense, thinner cortical bone compared to WT littermates. In addition, in loaded and non-loaded limbs, cho/+ mice had thicker cartilage. With high loads, cho/+ mice experienced less load-induced cartilage damage at all time points and displayed decreased matrix metalloproteinase (MMP)-13 levels compared to WT littermates. The thinner, less dense cortical bone and thicker cartilage were unexpected and may have contributed to the reduced severity of load-induced cartilage damage in cho/+ mice. Furthermore, the spontaneous proteoglycan loss resulting from the mutant collagen XI was not additive to cartilage damage from mechanical loading, suggesting that these risk factors act through independent pathways.
Keywords: osteoarthritis, mechanical loading, Col11a1 haploinsufficiency, abnormal matrix properties, bone
Introduction
Osteoarthritis (OA) is a whole joint disease characterized by pain and stiffness due to articular cartilage degradation, subchondral bone changes, and osteophyte formation. OA is the leading cause of disability in the elderly and affects approximately 27 million people in the United States alone.1–5 The disease primarily affects articulating joints subjected to loading and motion such as the knees, fingers, and hips. OA has many risk factors, including aging, obesity, previous joint injury, joint malalignment, high levels of mechanical loading, and genetic abnormalities that affect cartilage integrity.6 Determining the interactions of these factors in influencing joint damage is critical to better understanding the disease and developing effective treatment options. In this study, we focused on the combined impact of mechanical loading and a genetically abnormal cartilage matrix in OA pathogenesis.
Mechanical loading and joint health have a unique relationship. Low levels of mechanical loading, such as mild exercise, can benefit cartilage by preventing injury-induced cartilage degradation,7,8 maintaining thicker cartilage,9 and increasing proteoglycan synthesis.10 However, higher levels of loading can lead to cartilage degradation by decreasing proteoglycan synthesis,11 inducing chondrocyte apoptosis,12 and rupturing critical stabilizing ligaments.13 To further elucidate the role of mechanical loading in OA, we and others developed a noninvasive preclinical model that applies controlled cyclic compression to mouse knee joints.14,15 We previously demonstrated that this model recapitulates OA symptoms in healthy mouse knees.14
Epidemiological studies of OA have estimated a heritability of 50% or more depending upon the affected joint, suggesting that half of the variation in disease susceptibility could be explained by genetic factors.16–19 Collagen XI is essential for collagen fibril formation in articular cartilage.20,21 Collagen fibrils, containing primarily type II collagen with the alpha 1 collagen XI [α1(XI)] chain incorporated, form a network in the cartilage extracellular matrix (ECM) that contributes to tensile strength and retention of proteoglycans in cartilage tissue. In this study, we focused on a mutation in the gene encoding α1(XI) that results in chondrodysplasia in humans and the development of secondary OA. Mice with the same point mutation in one allele of the Col11a1 gene (cho/+), display abnormally thick collagen fibrils in their cartilage ECM,22 accompanied by reduced tissue tensile properties and OA-like features starting at 3 months of age.22 The expression levels of both discoidin domain receptor (DDR)-2 and matrix metalloproteinase (MMP)-13 are elevated in the articular cartilage of cho/+ mice23 and contribute to degradation of the pericellular matrix.24 Although mice homozygous for the cho mutation display severe skeletal abnormalities resulting in perinatal lethality,20 cho/+ mice do not display overt skeletal or extra-skeletal25 developmental defects besides the aforementioned age-related changes in the cartilage matrix. Thus, cho/+ mice provide a model to study the contribution of a genetically abnormal cartilage matrix to load-induced OA in a well-controlled manner.
The influence of a genetically abnormal cartilage matrix on the severity of load-induced cartilage damage is unknown. Because cartilage damage was enhanced in mice overexpressing DDR-2 subjected to a surgically-induced posttraumatic model of OA,26 we predicted that cho/+ cartilage, with elevated levels of DDR-2, would experience exacerbated load-induced damage. Furthermore, the mechanical properties of the cartilage matrix are impaired in the cho/+ mouse, so we expected the cartilage to deform more under load, thereby increasing the strains on the chondrocytes, leading to cell death and cartilage degradation. However, the skeletal phenotype has not been carefully examined in adult cho/+ mice. Our previous RNAseq analyses on total RNA isolated from mouse bone showed that Col11a1 is among the most highly expressed genes in both cancellous and cortical bone.27 Altered subchondral bone properties in cho/+ mice could also influence the cartilage response to loading, and thus we could not accurately predict the overall response to load in the knee joints of the cho/+ mouse.
Therefore, we aimed to 1) determine whether the collagen XI mutation affects the intrinsic cortical and cancellous bone phenotype of cho/+ mice, and 2) examine the interaction between mechanical loading and genetically abnormal cartilage matrix properties in the onset and progression of OA. We subjected cho/+ mice and wild-type (WT) littermates to cyclic tibial compression in vivo and used radiographic, histological, and immunohistochemical techniques to assess the phenotypic bone and cartilage changes in the knee joint. We hypothesized that cartilage abnormalities due to the collagen XI mutation would exacerbate cartilage damage and subchondral bone adaptation associated with load-induced OA. However, we found that the development and progression of OA cartilage pathology was less severe in the mutant mice compared to their WT littermates. Our findings indicate that the altered bone and cartilage phenotypes may have contributed to these unexpected results and highlight the complex nature of the interactions between bone and cartilage properties in the evolution of load-induced OA.
Materials and Methods
Mouse Genotyping
Mouse genotyping was performed as described previously.20 Heterozygous cho/+ mice and WT littermates were housed together (2 to 4 mice per cage). Lighting was maintained at a 12-hours-on-12-hours-off schedule. Mice were given food and water ad libitum. Male 6-month-old cho/+ mice and WT littermates were used for the phenotype characterization and loading experiments. We previously confirmed the presence of cartilage damage in response to moderate and high loads in male 6-month-old C57BL/6 mice14 and the loss of superficial proteoglycan staining in non-loaded male cho/+ mice by 6 months of age, indicative of early OA-like symptoms.22
Mechanical Loading
We applied cyclic compression to the left tibiae of cho/+ mice20,22,23,28 and WT littermates at moderate (4.5N) and high (9.0N) peak load magnitudes for durations of 1, 2, and 6 weeks (n=6/group/genotype). Right limbs served as contralateral controls. With mice under general anesthesia (2% isoflurane, 1.0L/min, Webster), loading was applied to the left tibiae 5 days/week for 1200 cycles at a frequency of 4 Hz.29 Upon completion of loading, mice were euthanized. Knee joints were harvested and fixed in 4% paraformaldehyde overnight at 4°C. All experimental techniques were approved by the Cornell IACUC.
Cartilage and Bone Morphology Analyses
Microcomputed tomography (microCT) scans were used to assess phenotypic bone morphology and changes in response to loading. After fixation, tissues were transferred to 70% ethanol for short-term storage. Intact knee joints were scanned using microCT, with an isotropic voxel resolution of 10 μm (μCT35, Scanco, Bruttisellen, Switzerland; 55kVp, 145μA, 600ms integration time). The effects of beam hardening were reduced with a 0.5 mm aluminum filter. After scanning, knee joints were decalcified in formic acid and processed for paraffin embedding. Paraffin blocks were sectioned at a thickness of 6 μm from posterior to anterior using a rotary microtome (Leica RM2255, Wetzlar, Germany).
To assess cartilage morphology, sections were stained with Safranin O/Fast Green at 90-μm intervals throughout the joint. Histological scoring was performed on these sections by two blinded researchers to examine cartilage damage in the medial and lateral tibial plateaus. Scores from all sections of each limb were averaged. In control limbs, a modified Mankin scoring system was used to characterize the articular cartilage phenotype of cho/+ and WT mice.22 Structural cartilage damage after loading was evaluated in all limbs using the OARSI scoring system.30 Cartilage thickness was measured in both the medial and lateral plateaus on three representative sections in the joint (posterior, middle, and anterior) as described previously.14,31
To assess osteophyte formation in response to loading, we examined Safranin O/Fast Green-stained histological sections for ectopic bone formation surrounding the joint. Osteophyte maturity was evaluated as described previously.32 Briefly, we identified the section from each joint that contained the largest portion of the medial tibial osteophyte. Using the representative slide, we scored osteophyte maturity: 0 for no osteophyte, 1 for a primarily cartilaginous osteophyte, 2 for a mixture of cartilage and bone, or 3 for primarily bony structure. We also measured the medial-lateral width of the osteophyte, defined as the distance between the medial end of the epiphysis and the end of the ectopic bone. Widths are reported as absolute values.
To assess peri-articular bone morphology, we examined cortical and cancellous bone in the epiphysis and metaphysis of the proximal tibia using microCT. We analyzed the subchondral cortical bone plate (SBP) and metaphyseal cortical shell for thickness and tissue mineral density (TMD). For the SBP, the volume of interest (VOI) encompassed all cortical bone beginning at the proximal end of the tibia extending distally until the epiphyseal cancellous bone was evident. The VOI for the metaphyseal cortical shell began distal to the growth plate and extended 10% of the tibial length, excluding cancellous bone. We analyzed isolated cancellous bone in the epiphysis and metaphysis for bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), and TMD. For the epiphysis, the VOI encompassed cancellous bone proximal to the growth plate and distal to the subchondral bone plate, excluding cortical bone. The VOI for metaphyseal cancellous bone encompassed the same region analyzed for the cortical shell, excluding the cortex.
Immunohistochemistry and TUNEL assay
We performed immunohistochemistry (IHC) to assess load-induced changes in cartilage proteins in cho/+ and WT mice. We evaluated markers of OA disease using specific antibodies against MMP-13 (Abcam, AB39012, Cambridge, MA) and DDR-2 (Abcam, AB5520, Cambridge, MA). We also assessed chondrocyte apoptosis using a TUNEL kit to detect DNA strand breaks (Sigma, 11684795910 Roche, Darmstadt, Germany). We analyzed the medial posterior tibial plateau at early time points under high loads, because the majority of load-induced damage occurs in this region,14 and a single session of 9N cyclic compression leads to progressive cell-mediated changes and cartilage degradation at 1 and 2 weeks.31
We stained one representative section from the posterior region of control and loaded limbs from animals loaded for 1 or 2 weeks at 9N. For MMP-13 and DDR-2 IHC, sections were deparaffinized, rehydrated, and incubated with 2.5% hyaluronidase at 37°C for antigen retrieval, and quenched for endogenous peroxidase activity. Sections for MMP-13 IHC were then incubated with 1% bovine serum albumin and 0.5% Triton X-100 for 1h. Sections for DDR-2 IHC were incubated with Dako protein block for 5min at room temperature. Then, the samples were incubated overnight at 4°C with specific antibodies against MMP-13 or DDR-2, followed by incubation with anti-rabbit horseradish peroxidase-conjugated secondary antibodies. Color development was performed using the peroxidase substrate DAB. Samples were dehydrated, coverslipped, and the percentage of positive immunostaining in the articular cartilage of the medial tibial plateau was calculated using ImageJ software (NIH) as described previously.33,34
For the TUNEL assay, sections were deparaffinized, rehydrated, and incubated with proteinase K at 37°C. The samples were then incubated with the TUNEL reaction mixture for 1h at 37°C. Finally, sections were coverslipped with antifade mounting media containing DAPI. TUNEL positive cells were measured in the articular cartilage of the medial tibial plateau, as described previously,33,34 and normalized to the total number of chondrocytes (DAPI+ signal) to account for potential changes in cellularity.
Statistical analyses
Bone parameters and Mankin scores of control limbs were compared between WT and cho/+ mice using a Student’s t-test. To minimize potential systemic effects of loading and aging, we analyzed control limbs only from the 1-week time point for our baseline bone phenotype comparisons. Baseline IHC parameters in control limbs were also compared between WT and cho/+ mice using a t-test.
To understand the effects of loading, we used a four-way ANOVA with load (loaded vs. control limb), magnitude (high vs. moderate), duration (1 vs. 2 vs. 6 weeks), and genotype (WT vs. cho/+) as variables, with animal as a random effect. We applied the ANOVA to assess OARSI histological scores, cartilage thickness, and all bone morphological parameters. To analyze the IHC and TUNEL results and osteophyte formation at high loads, we used a three-way ANOVA with load, genotype, and duration as variables, with animal as a random effect. Post-hoc testing was performed with Tukey’s tests for interaction effects and t-tests for individual effects.
Results
Bone and Cartilage Phenotypes
Cortical bone thickness and TMD were different between genotypes. The metaphyseal cortical shell was thinner (p=0.029) and had lower TMD (p=0.002) in cho/+ mice compared to WT littermates (Fig. 1A). Similarly, the medial subchondral bone plate was thinner (p=0.041) with a trend towards lower TMD (p=0.051) in cho/+ mice (Fig. 1B). Cancellous bone morphology in the proximal tibia was similar between WT and cho/+ mice. Specifically, cancellous BV/TV, Tb.Th, Tb.Sp, and TMD were not different in the epiphyseal and metaphyseal regions of either genotype (Figs. S-1A,S-2B). In addition, tibial length was not different between cho/+ and WT mice (17.70±0.32 vs. 17.75±0.34mm).
Figure 1.

Cortical bone phenotypes were different between cho/+ mice and WT littermates. Cortical bone in the (A) metaphyseal shell and (B) medial subchondral bone plate in cho/+ mice was thinner and less dense compared to WT littermates. Lines behind the dots show the mean ± SD of 24 mice (n=12/group). TMD = Tissue Mineral Density. *p<0.05 vs. WT by t-test.
Consistent with previous reports,22 loss of proteoglycan in the articular cartilage in cho/+ mice occurred spontaneously at 6 months of age, resulting in higher Mankin scores (p=0.011, Fig. S-2). Proteoglycan loss was localized to the superficial layer of the articular cartilage, as evident from the representative Safranin O/Fast Green images of control limbs (Fig. S-2). Cho/+ mice also had ~10% thicker cartilage than WT on the medial side of the joint in the posterior (78.49 vs. 71.28μm, p=0.028, Fig. 2C), middle (93.15 vs. 87.16μm, p=0.036), and anterior (93.07 vs. 84.97μm, p=0.071) regions. Cartilage in cho/+ mice was also thicker on the lateral side in the posterior region (75.94 vs. 68.63μm, p=0.012). In the medial tibial plateau, calcified cartilage thickness was not different between genotypes (data not shown), indicating that the thickened articular cartilage in cho/+ mice was due to thicker hyaline cartilage.
Figure 2.
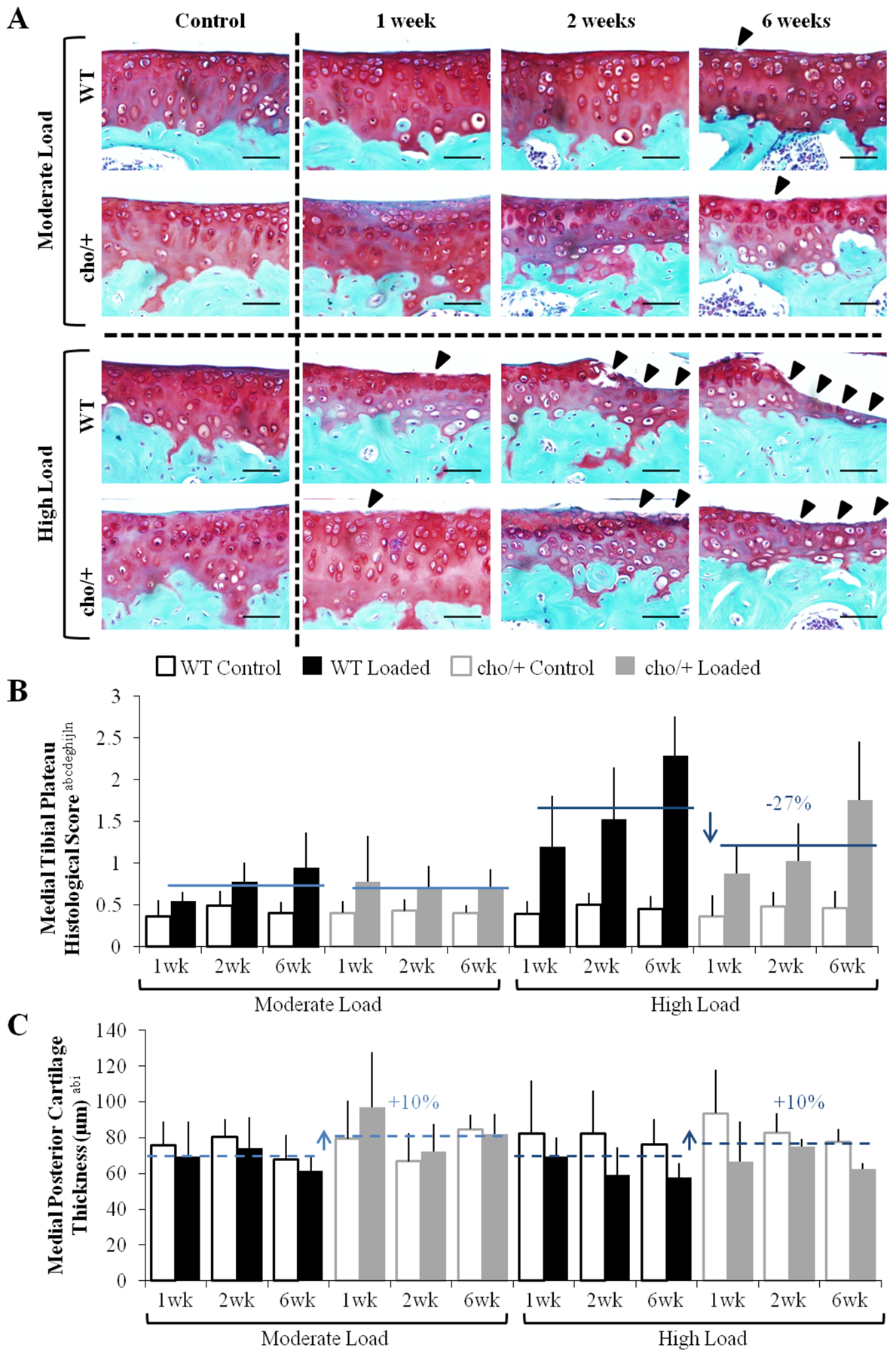
Cho/+ mice were less susceptible to load-induced cartilage degradation. (A) Moderate loading led to fibrillation after 6 weeks. High loads led to fibrillation after 1 week and progressed to erosion after 2 and 6 weeks. Damage is indicated by arrow heads. (B) Under high load magnitudes, cho/+ mice experienced less severe cartilage damage compared to WT mice, but moderate loads caused similar damage between genotypes. (C) High loads caused thinning in the posterior region of the tibial plateau. Cho/+ mice had approximately 10% thicker cartilage than WT mice. Scale Bars = 50 μm. Bars show the mean ± SD of 72 mice (n=6/group). Solid and dashed lines indicate post-hoc comparisons of the effect of genotype and genotype*load, respectively. p<0.05 by ANOVA indicated on vertical axis for effects of aGenotype, bLoad, cDuration, dMagnitude, eGenotype*Load, fGenotype*Duration, gGenotype*Magnitude, hLoad*Duration, iLoad*Magnitude, jDuration*Magnitude, kGenotype*Load*Duration, lGenotype*Load*Magnitude, mGenotype*Duration*Magnitude, nLoad*Duration*Magnitude, oGenotype*Load*Duration*Magnitude.
Load-Induced Articular Cartilage Morphological Changes
Mechanical loading induced articular cartilage degradation in the tibial plateaus of both WT and cho/+ mice (Fig. 2A). Moderate loading (4.5N) caused little damage after 1 and 2 weeks, but fibrillation of the articular surface was present after 6 weeks. Load-induced damage under moderate loads was similar in both genotypes. With high loads (9.0N), cartilage fibrillation occurred by 1 week. Erosion of the articular surface occurred after 2 weeks. At the 6-week time point, the articular cartilage was completely eroded to the tidemark, and in some cases, erosion occurred through the entire calcified cartilage layer, most often on the medial tibial plateau of WT mice. Under high loads, the medial tibial plateau of loaded limbs in cho/+ mice had significantly less cartilage damage than in WT (OARSI score 1.22 vs. 1.67, p=0.016, Fig. 2B).
The high load caused localized cartilage thinning in the posterior region of the tibial plateau (Fig. 2C). Cartilage thinning was also evident in cartilage in the middle region of the joint on the medial side with loading. The majority of thinning occurred in the hyaline cartilage (Fig. 2A). Load-induced cartilage thinning was similar in both genotypes. In contrast, moderate loading caused little to no thinning.
Osteophyte formation
Osteophyte formation was evident only in limbs loaded at high load magnitudes (Fig. 3A). After 1 week of high loads, small cartilaginous pre-osteophytes were present on the medial tibial plateau of all loaded joints. After 2 weeks, medial osteophytes grew significantly in size, but remained primarily cartilaginous with little to no mineralization. After 6 weeks of loading, osteophytes had undergone ossification and were predominantly bone. In the knees of a few animals, these bony osteophytes extended along the entire medial side of the joint. Osteophyte size and maturity was similar between genotypes (Figs. 3B,3C), with a slight trend towards smaller osteophytes in cho/+ mice (p=0.185, Fig. 3B).
Figure 3.
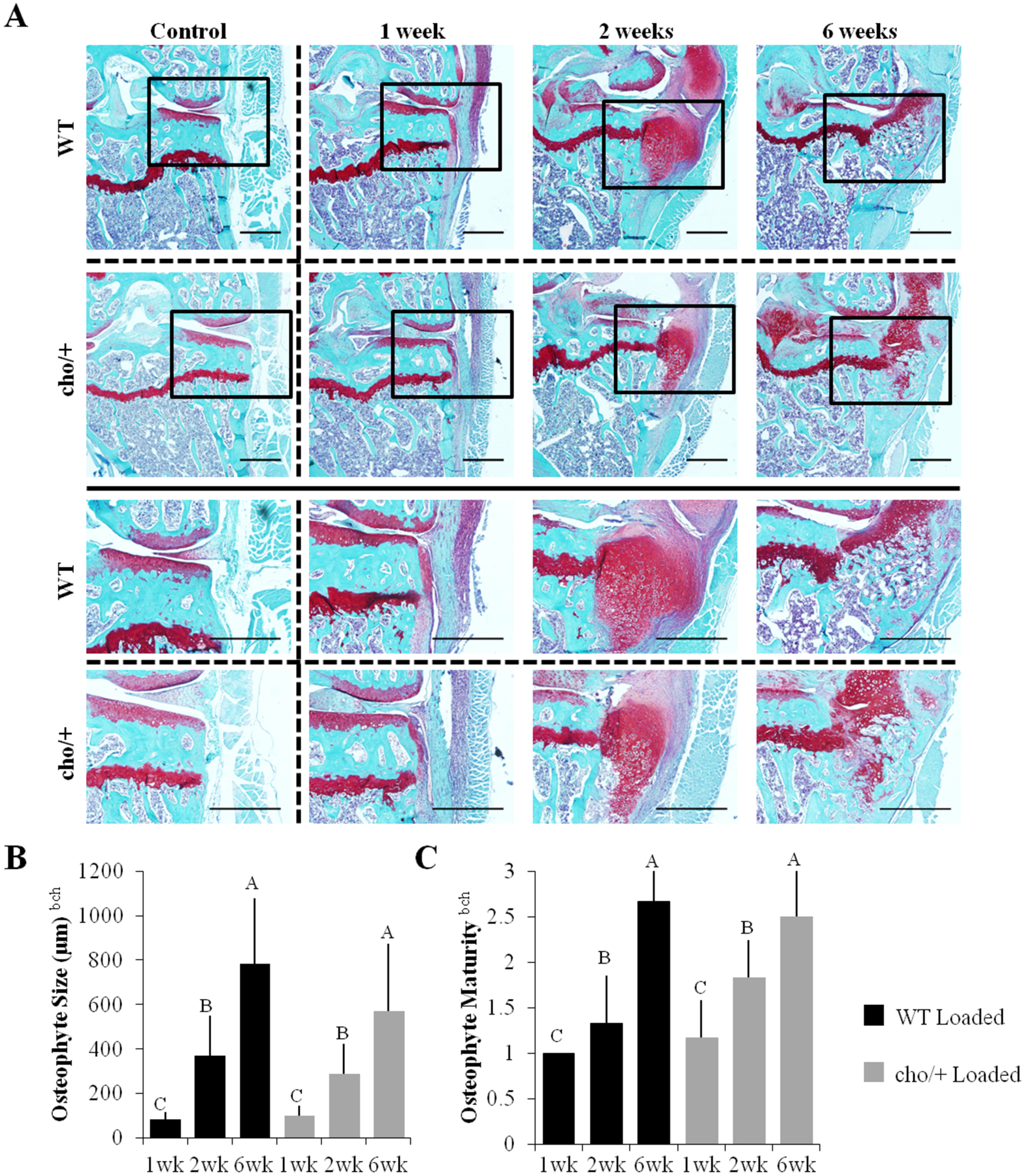
Osteophyte formation was similar between genotypes. (A) Osteophytes formed on the medial tibial plateau of loaded limbs. (B,C) After 1 week, osteophytes were small and primarily cartilagenous. After 6 weeks, osteophytes were larger and more mineralized. Scale Bars = 500 μm Bars show the mean ± SD of 36 mice (n=6/group). Same letters over the bars indicate similar mean values, and groupings with different letters indcate that the difference is significant by post-hoc comparisons of the effect of load*duration. p<0.05 by ANOVA indicated on vertical axis for effects of aGenotype, bLoad, cDuration, fGenotype*Duration, gGenotype*Load, hLoad*Duration, kGenotype*Load*Duration.
Load-Induced Peri-Articular Bone Morphological Changes
High loads had no effect on medial subchondral cortical bone plate thickness (Fig. 4A), but decreased TMD (Fig. 4B). Moderate loads did not change SBP thickness (Fig. S-3A) or TMD (Fig. S-3B). Load-induced changes in the SBP were similar between the genotypes.
Figure 4.
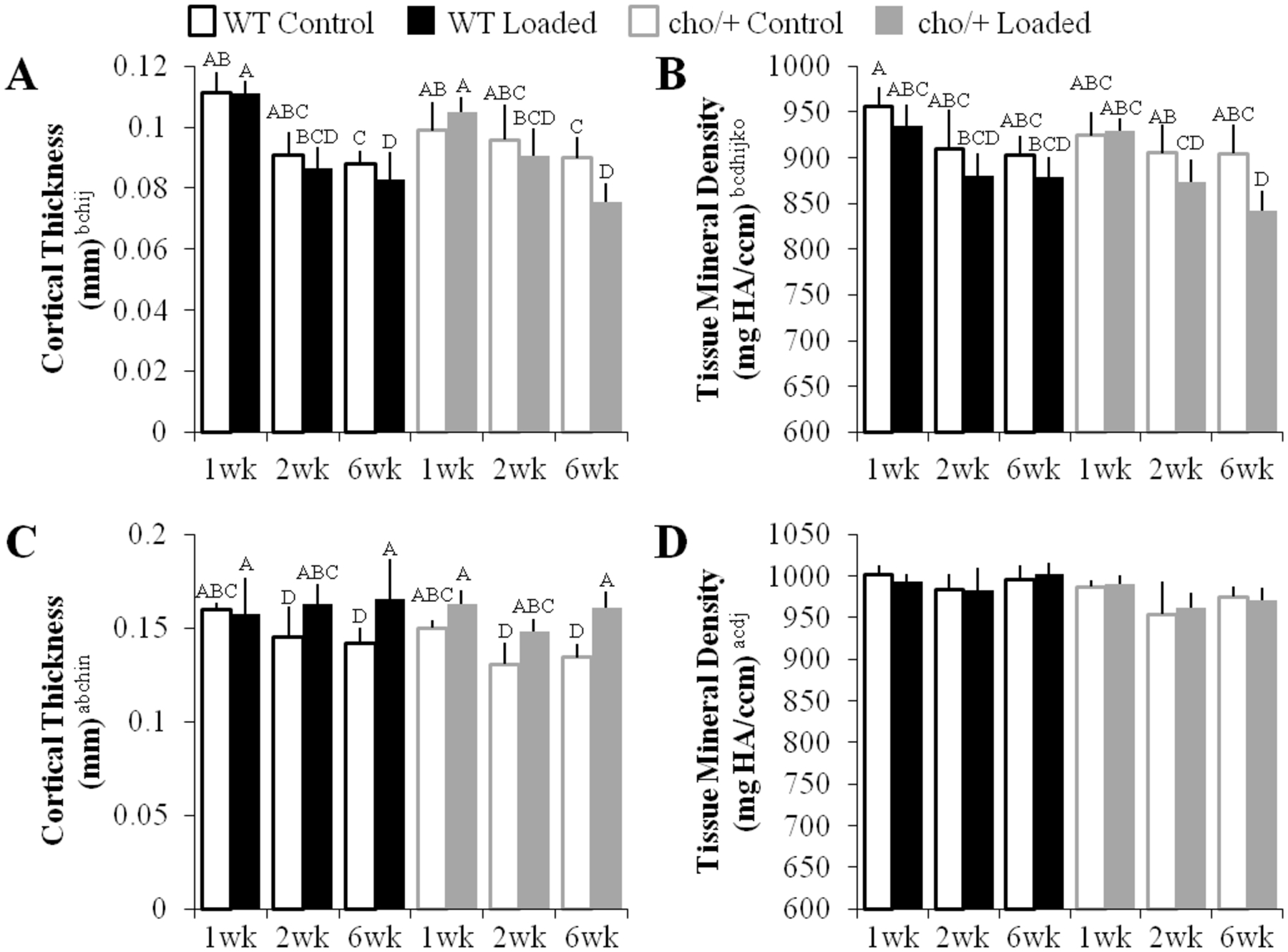
Cortical bone in the subchondral bone plate and metaphyseal cortical shell responded differently to high loads. (A,B) The subchondral cortical bone plate thinned and became less dense with loading. (C,D) The metaphyseal cortical shell maintained thickness and its density remained the same with loading. Bars show the mean ± SD of 36 mice (n=6/group). Same letters over the bars indicate similar mean values, and groupings with different letters indcate that the difference is significant by post-hoc comparisons of the effect of (A) load*duration, (B) genotype*load*duration*magnitude, and (C) load*duration*magnitude. p<0.05 by ANOVA indicated on vertical axis for effects of aGenotype, bLoad, cDuration, dMagnitude, eGenotype*Load, fGenotype*Duration, gGenotype*Magnitude, hLoad*Duration, iLoad*Magnitude, jDuration*Magnitude, kGenotype*Load*Duration, lGenotype*Load*Magnitude, mGenotype*Duration*Magnitude, nLoad*Duration*Magnitude, oGenotype*Load*Duration*Magnitude.
High loads maintained thickness in the metaphyseal cortical shell (Fig. 4C), whereas moderate loads had no effect on metaphyseal cortical thickness (Fig. S-3C). TMD was not affected by either load level (Figs. 4D, S-3D). The cortical shell changed similarly in response to loading in both genotypes. When pooled by genotype, cho/+ mice had thinner (p=0.002) and less dense (p<0.0001) cortical bone in the metaphyseal region.
Loading led to epiphyseal cancellous bone loss in both cho/+ and WT mice (Fig. 5A). Bone loss in the epiphysis was primarily trabecular thinning (p=0.038) with a trend towards greater separation (p=0.054, Fig. 5B) as a result of both high and moderate loads (Figs. S-4A, S-4B). Load-induced epiphyseal cancellous bone changes were similar in both genotypes. When pooled by genotype, cho/+ mice had a trend towards thinner trabeculae compared to WT (0.053 vs. 0.054mm, p=0.095).
Figure 5.
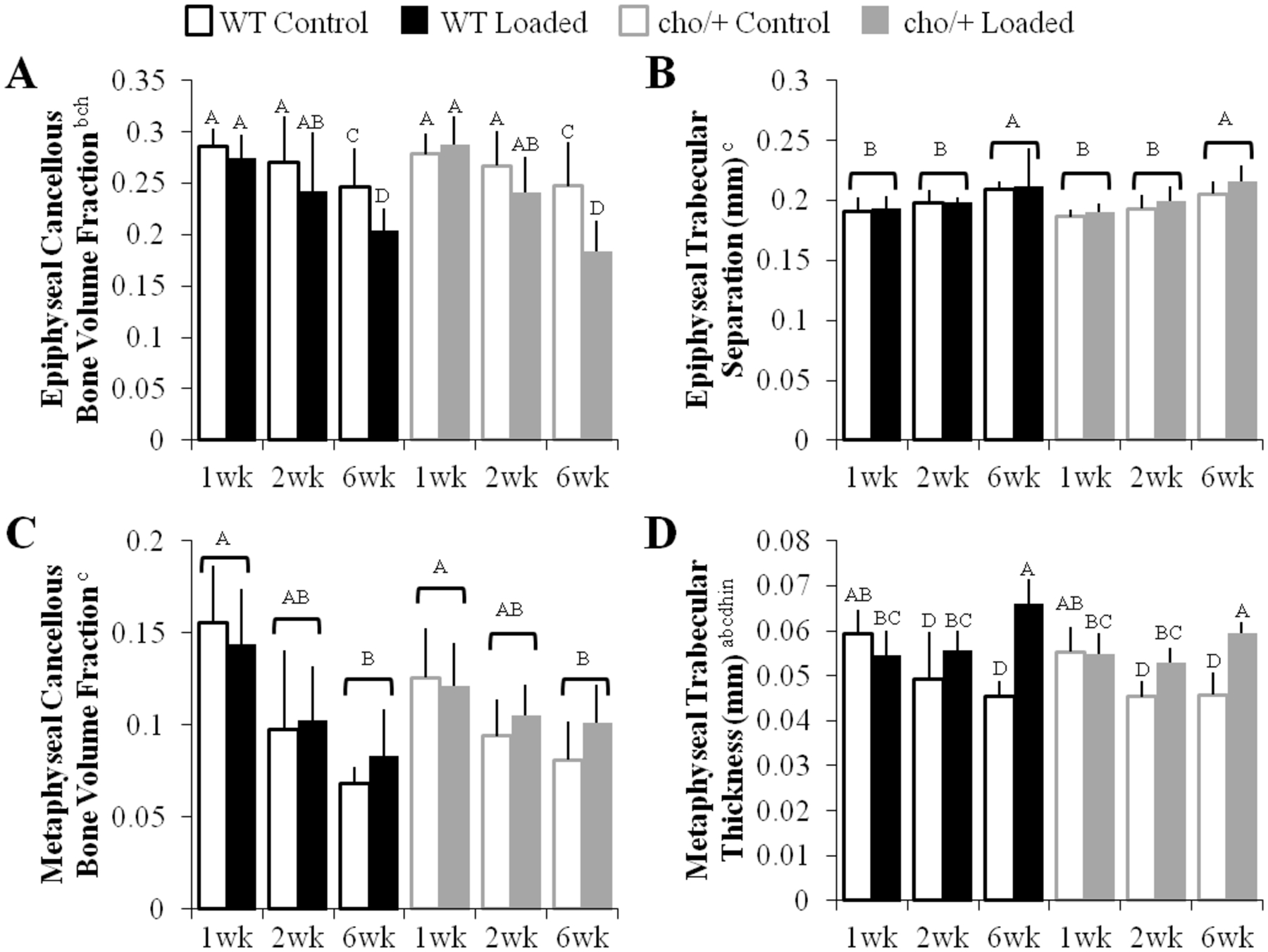
Cancellous bone in the epiphysis and metaphysis responded differently to high loads. (A) Epiphyseal cancellous bone decreased in bone volume fraction with high load magnitudes. (B) Epiphyseal trabecular separation increased with duration. (C) Cancellous bone in the metaphysis decreased with duration, but (D) trabecular thickness increased with loading. Bars show the mean ± SD of 36 mice (n=6/group). Same letters over the bars, or pooled bars, indicate similar mean values, and groupings with different letters indcate that the difference is significant by post-hoc comparisons of the effect of (A) load*duration, (B,C) duration, and (D) load*duration*magnitude. p<0.05 by ANOVA indicated on vertical axis for effects of aGenotype, bLoad, cDuration, dMagnitude, eGenotype*Load, fGenotype*Duration, gGenotype*Magnitude, hLoad*Duration, iLoad*Magnitude, jDuration*Magnitude, kGenotype*Load*Duration, lGenotype*Load*Magnitude, mGenotype*Duration*Magnitude, nLoad*Duration*Magnitude, oGenotype*Load*Duration*Magnitude.
Unlike the epiphysis, metaphyseal cancellous BV/TV was unaffected by loading in either genotype (Fig. 5C). However, loading increased Tb.Th and TMD (Fig. 5D). Tb.Sp did not change with loading in the metaphysis. These changes were not different between high and moderate loads (Figs. S-4C, S-4D). Load-induced metaphyseal cancellous bone changes were similar between genotypes. When pooled by genotype, metaphyseal trabeculae were thinner in cho/+ than WT mice (0.051 vs. 0.054mm, p=0.014), similar to the epiphysis.
Bone loss occurred over time in all four regions of bone we analyzed. Specifically, cortical bone thickness in the subchondral bone plate decreased over time in control and loaded limbs, and metaphyseal cortical shell thickness decreased in control limbs. In addition, epiphyseal and metaphyseal cancellous BV/TV decreased over time in both control and loaded limbs.
Immunohistochemical and TUNEL analyses
Control cho/+ limbs had high MMP-13 levels (p=0.031) and a trend towards increased DDR-2 immunostaining (p=0.111) compared to WT littermates, consistent with previous reports.22 Loading induced a trend towards decreased MMP-13 (1.3 vs. 1.9%) and DDR-2 (1.3 vs. 2.9%) immunostaining in cho/+ mice, whereas MMP-13 and DDR-2 levels were not altered with loading in WT littermates (Fig. 6). In addition, loading decreased cellularity in both genotypes (data not shown), and high loads also increased chondrocyte apoptosis in both genotypes (p=0.009), with a greater percentage of TUNEL+ cells in cho/+ mice compared to WT littermates after 2 weeks of loading (1.8 vs. 0.8μm2/cell #, Fig. 6).
Figure 6.
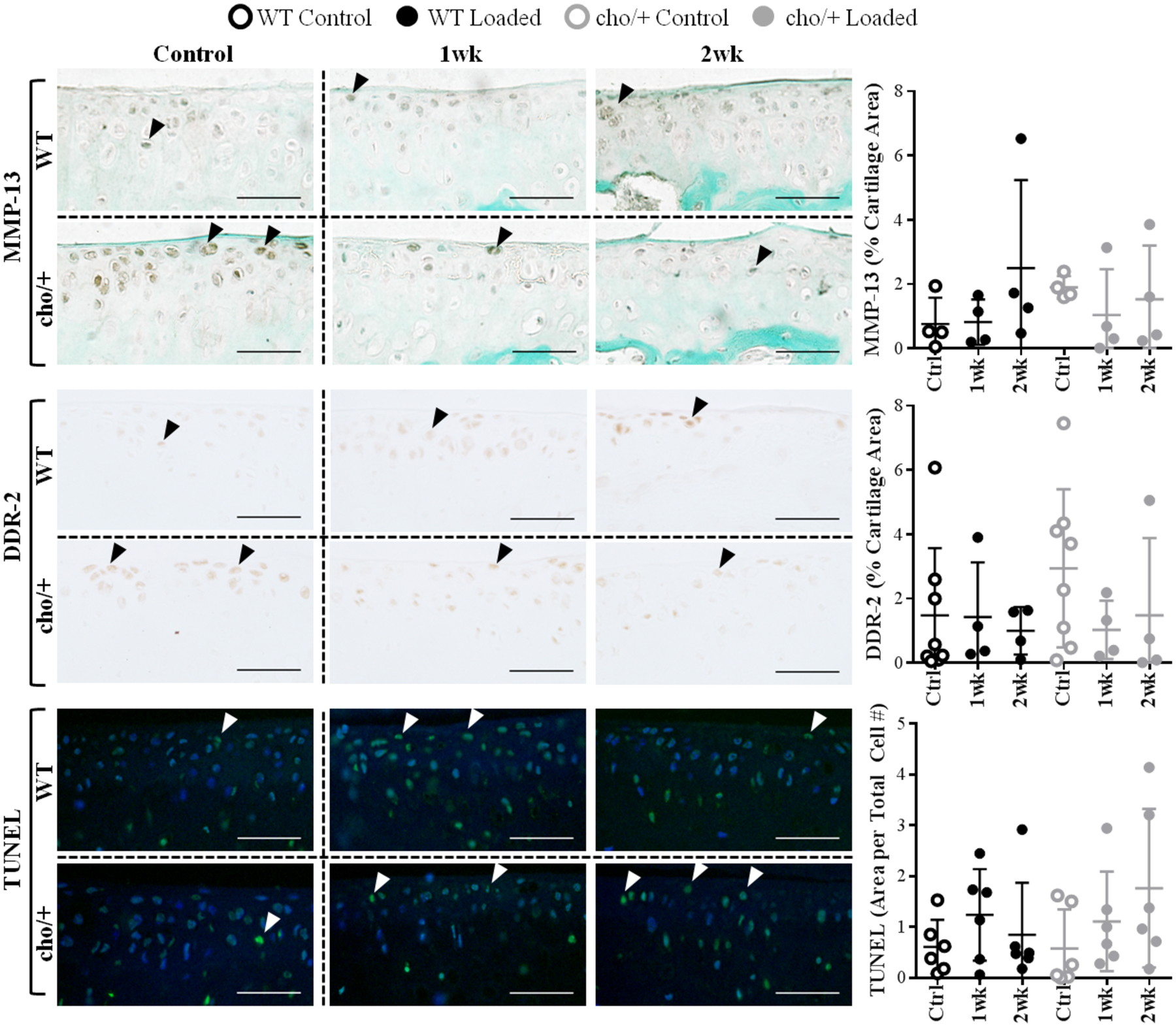
MMP-13 and DDR-2 levels were higher in control limbs of cho/+ mice compared to WT littermates. Loading induced a trend towards decreased MMP-13 and DDR-2 immunostaining in cho/+ mice, whereas it did not alter MMP-13 and DDR-2 levels in WT littermates. Apoptosis increased in loaded limbs, with slightly higher levels of apoptosis in cho/+ mice after 2 weeks of loading. Scale Bars = 50 μm. Lines behind the dots show the mean ± SD of 16–24 mice (n=4–8/group).
Discussion
We sought to determine whether the mutant alpha XI collagen chain incorporated into the cartilage collagen fibrillar network and resultant changes in articular cartilage composition in cho/+ mice altered the responses in the cartilage and bone in knee joints subjected to cyclic tibial compression compared to WT littermates. We confirmed the presence of differences in cartilage morphology and the development of the spontaneous onset of OA-like features between 6-month-old cho/+ mice and WT littermates as previously reported.22 Unexpectedly, we found that prior to loading, the cortical bone in the subchondral bone plate and metaphysis was thinner and less dense in cho/+ mice compared to their WT littermates, whereas the collagen XI mutation was not associated with any change in cancellous bone structure.
We originally hypothesized that the collagen XI mutation would not affect bone phenotype based on collagen XI’s key role in fibril formation in cartilage.21,22,35 Our results, however, suggest that collagen XI plays a role in the formation and/or maintenance of cortical bone. Indeed, previous studies support our results that fibrillar collagen components play a role in bone morphogenesis and structure.20,36 Mice homozygous for the Col11a1 mutation (cho/cho) do not survive after birth because of improper skeletal formation, including a shortened spine and shortened bones in the appendicular and thoracic skeleton.20 Col11a1-deficient mice also have altered bone microarchitecture during embryonic development.37 In addition, mice with a deficiency in collagen IX, another key component of collagen fibrils in the cartilage ECM, have abnormal skeletal properties.36 Lastly, Col11a2 mutations lead to mild dwarfism in Labrador retrievers.38 Consistent with these previous findings, cho/+ mice had thinner and less dense cortical bone compared to WT littermates. These results emphasize the involvement of collagen XI in the development and potentially the composition and properties of bone in the mature skeleton.
Contrary to our hypothesis, cho/+ mice were less susceptible to load-induced cartilage damage compared to WT littermates, despite the spontaneous proteoglycan loss and increased DDR-2 and MMP-13 levels associated with the collagen XI mutation. One explanation for the less severe cartilage damage may be related to the differences in cartilage thickness between genotypes. Cho/+ mice had ~10% thicker cartilage in the medial tibial plateau compared to WT littermates. Cartilage swelling correlates with proteoglycan loss in early-stage experimental OA;39 thus, the cartilage in 6-month-old cho/+ mice may experience swelling concurrently with the associated spontaneous proteoglycan loss typical of the early OA-like phenotype of cho/+ mice at this age. Whereas load-induced cartilage thinning was not different between genotypes, the overall thicker cartilage in cho/+ mice may have reduced the load-induced structural cartilage stresses. Our observation is consistent with previous reports, also based on in vivo loading experiments, that showed thicker cartilage decreased the peak contact pressures and shear forces in the articular cartilage.40 Furthermore, mice susceptible to spontaneous cartilage lesions (Str/ort) were protected from load-induced mechanical trauma compared to control mice (CBA) because of thicker articular cartilage.40 In both studies, the decreased susceptibility to load-induced cartilage damage may be due, partly, to dissipation of mechanical stresses by the thicker articular cartilage.
The thinner, less dense cortical bone in the metaphyseal shell and medial subchondral bone plate in cho/+ mice may also have played a role in attenuating load-induced cartilage damage. Subchondral cortical bone stiffness is important in initiating OA.41–43 Clinically, individuals with extremely elevated bone density are more likely to develop OA in the hip and knee,44,45 and bone density and OA incidence may be inversely related.46 Thus, the thinner, less dense cortical bone in cho/+ mice could have contributed to the reduced severity of load-induced cartilage damage. Of interest, these results differ from previous findings showing enhanced cartilage damage in transgenic mice conditionally overexpressing DDR-2 in the articular cartilage subjected to the surgical destabilization of the medial meniscus (DMM) OA model.26 However, the bone properties of these transgenic mice prior to DMM surgery were not assessed and most likely differed from those of cho/+ mice, which could account for our different results.
Our findings confirm the mechanistic connection of DDR-2 and MMP-13 in cartilage degradation in cho/+ mice. DDR-2 is a cell surface receptor that drives MMP-13 expression and activity and is elevated in OA disease in both human articular cartilage and murine models, including the cho/+ mouse.22–24,47 We found that elevated DDR-2 and MMP-13 levels correlated with spontaneous proteoglycan loss in cho/+ mice, but were not associated with the OA-like changes induced by loading in either genotype. In loaded limbs, we observed trends towards decreased MMP-13 and DDR-2 levels in cho/+ mice, whereas loading did not alter MMP-13 or DDR-2 levels in WT littermates. Independent of the differences in DDR-2 and MMP-13 levels between genotypes, loading decreased cellularity and increased cell death similarly in cho/+ and WT cartilage, with only minor differences in apoptosis at 2 weeks post-loading, when cho/+ mice showed a trend towards increased numbers of TUNEL+ cells compared to WT. Taken together, differences in susceptibility to load-induced cartilage damage may be due to tissue-level characteristics rather than the cellular responses we examined. Specifically, the thicker cartilage and the thinner, less dense cortical bone in cho/+ mice were likely the driving factors in lowering the susceptibility of cho/+ mice to load-induced cartilage degradation, in agreement with previous findings.40
We clearly observed two distinct etiologies of OA and an unexpected interaction in the development of cartilage damage in our study. Cho/+ mice developed spontaneous OA from genetically weaker matrix properties that resulted in DDR-2-driven collagenase activity and gradual proteoglycan loss throughout their lifespan.22 On the other hand, cyclic tibial compression at high-load levels led to rapid structural changes in the cartilage matrix with little evidence of proteoglycan loss.14 The lack of correlation between loading and DDR2/MMP13 levels was unexpected, but was likely an example of the different mechanisms involved in load-induced versus genetic OA. We can only speculate that other receptors and cartilage-degrading enzymes (i.e., collagenases and aggrecanases) not analyzed in the context of this study contributed to load-induced cartilage degradation. Ultimately, the contribution of the genetic abnormalities of the cartilage in the cho/+ mouse was not additive to load-induced cartilage damage, suggesting that these two risk factors do not act synergistically or additively.48
Our study had several limitations, potential alternative strategies, and strengths. With our microCT analysis of subchondral bone, we analyzed the full volume of the subchondral cortical bone plate, but could not isolate specific regions within the subchondral bone. To examine location-specific changes, we measured localized subchondral bone thickness from histology slides in the anterior, middle, and posterior regions. The histology measurements matched our microCT findings. In particular, loading did not affect subchondral bone plate thickness in any of these three regions, and thickness decreased over time. Similarly, cortical and cancellous bone in the epiphysis and metaphysis decreased with time in this study. The changes in bone architecture over time can be attributed to aging, as rapid bone changes can occur in mice around 6 months of age.49
The mechanical loading regimen is not representative of all forms of joint loading. Cyclic tibial compression allows for precise control of the loading regimen, produces consistent cartilage damage, and does not depend on mouse activity levels or willingness to run on a treadmill or wheel. However, alternative techniques for applying elevated loads could be considered. For example, excessive treadmill running leads to OA in C57Bl/6 mice50 and could have produced a different response in cho/+ mice compared to cyclic tibial compression, but treadmill running also has a multitude of systemic effects.51 Cyclic tibial compression allowed us to focus solely on the specific characteristics of mechanical loading, including duration and magnitude, and their effects on the progression of load-induced OA.
Alternative forms of genetic OA also should be considered. Mice that are deficient in type IX collagen (Col9a1−/−) also develop abnormal collagen fibrils in the cartilage matrix leading to OA52 and show trabecular bone deterioration.53 Mice with mutations in other matrix-related genes, such as lubricin, have decreased cellularity in the superficial layer of their articular cartilage.54 Lastly, investigating the effects of cyclic tibial compression in older cho/+ mice at more advanced stages of OA22 may provide insights into the interaction between genetics and loading during the progression of OA.
To our knowledge, this study is the first to examine the cortical and cancellous bone properties in the cho/+ mouse strain. We demonstrated that mice with mutant collagen XI exhibit changes in cortical bone, with limited changes in cancellous bone. Our study provides evidence that thinner, less dense cortical bone and thicker cartilage may be associated with decreased severity of load-induced OA. Genetically abnormal cartilage matrix properties due to the collagen XI mutation were not additive to load-induced cartilage damage. The modes of damage involved in spontaneous OA in cho/+ mice and load-induced OA differed from each other at both the tissue and cellular levels. Our findings provide further insights into the complex nature of the interactions between bone and cartilage properties in the evolution of load-induced OA and demonstrate the utility of using mice with intrinsic alterations in cartilage and bone properties and defined loading conditions to elucidate the role of mechanobiological factors in OA development and progression.
Supplementary Material
Acknowledgments
We thank Lyudamila Lukashova of the Hospital for Special Surgery and the CARE staff at Cornell University. We would also like to thank our funding sources: NIH R21-AR064034, the Clark and Kirby Foundations, Office of Academic and Diversity Initiatives (NSA), and the Howard Hughes Medical Institute (AO).
References
- 1.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, Gabriel S, Hirsch R, Hochberg MC, Hunder GG, Jordan JM, Katz JN, Kremers HM, Wolfe F, National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turkiewicz A, Petersson IF, Björk J, Hawker G, Dahlberg LE, Lohmander LS, Englund M. Current and future impact of osteoarthritis on health care: a population-based study with projections to year 2032. Osteoarthr Cartil. 2014;22(11):1826–1832. doi: 10.1016/j.joca.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Felson DT. Osteoarthritis: Priorities for osteoarthritis research: much to be done. Nat Rev Rheumatol. 2014;10(8):447–448. doi: 10.1038/nrrheum.2014.76. [DOI] [PubMed] [Google Scholar]
- 4.Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol. 2014;10(7):437–441. doi: 10.1038/nrrheum.2014.44. [DOI] [PubMed] [Google Scholar]
- 5.Hootman JM, Helmick CG. Projections of US prevalence of arthritis and associated activity limitations. Arthritis Rheum. 2006;54(1):226–229. doi: 10.1002/art.21562. [DOI] [PubMed] [Google Scholar]
- 6.Felson DT. Risk Factors for Osteoarthritis. Clin Orthop Relat Res. 2004;427(427):S16–S21. doi: 10.1097/01.blo.0000144971.12731.a2. [DOI] [PubMed] [Google Scholar]
- 7.Hamamura K, Zhang P, Zhao L, Shim JW, Chen A, Dodge TR, Wan Q, Shih H, Na S, Lin C-C, Sun H Bin, Yokota H. Knee loading reduces MMP13 activity in the mouse cartilage. BMC Musculoskelet Disord. 2013;14(1):312. doi: 10.1186/1471-2474-14-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galois L, Etienne S, Grossin L, Watrin-Pinzano A, Cournil-Henrionnet C, Loeuille D, Netter P, Mainard D, Gillet P. Dose-response relationship for exercise on severity of experimental osteoarthritis in rats: a pilot study. Osteoarthritis Cartilage. 2004;12(10):779–786. doi: 10.1016/j.joca.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Hubbard-Turner T, Guderian S, Turner MJ. Lifelong physical activity and knee osteoarthritis development in mice. Int J Rheum Dis. 2015;18(1):33–39. doi: 10.1111/1756-185X.12291. [DOI] [PubMed] [Google Scholar]
- 10.Sah RL-Y, Kim Y-J, Doong J-YH, Grodzinsky AJ, Plass AHK, Sandy JD. Biosynthetic response of cartilage explants to dynamic compression. J Orthop Res. 1989;7(5):619–636. doi: 10.1002/jor.1100070502. [DOI] [PubMed] [Google Scholar]
- 11.Kurz B, Jin M, Patwari P, Cheng DM, Lark MW, Grodzinsky AJ. Biosynthetic response and mechanical properties of articular cartilage after injurious compression. J Orthop Res. 2001;19(6):1140–1146. doi: 10.1016/S0736-0266(01)00033-X. [DOI] [PubMed] [Google Scholar]
- 12.Stolberg-Stolberg JA, Furman BD, William Garrigues N, Lee J, Pisetsky DS, Stearns NA, DeFrate LE, Guilak F, Olson SA. Effects of cartilage impact with and without fracture on chondrocyte viability and the release of inflammatory markers. J Orthop Res. 2013;31(8):1283–1292. doi: 10.1002/jor.22348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christiansen B a., Anderson MJ, Lee C a., Williams JC, Yik JHN, Haudenschild DR. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthr Cartil. 2012;20(7):773–782. doi: 10.1016/j.joca.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Ko FC, Dragomir C, Plumb DA, Goldring SR, Wright TM, Goldring MB, van der Meulen MCH. In Vivo Cyclic Compression Causes Cartilage Degeneration and Subchondral Bone Changes in Mouse Tibiae. Arthritis Rheum. 2013;65(6):1569–1578. doi: 10.1002/art.37906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulet B, Hamilton RW, Shefelbine S, Pitsillides AA. Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum. 2011;63(1):137–147. doi: 10.1002/art.27765. [DOI] [PubMed] [Google Scholar]
- 16.Felson DT, Zhang Y. An update on the epidemiology of knee and hip osteoarthritis with a view to prevention. Arthritis Rheum. 1998;41(8):1343–1355. doi:. [DOI] [PubMed] [Google Scholar]
- 17.van Meurs JBJ. Osteoarthritis year in review 2016: genetics, genomics and epigenetics. Osteoarthr Cartil 2017;25(2):181–189. doi: 10.1016/j.joca.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Valdes AM, Spector TD. Genetic epidemiology of hip and knee osteoarthritis. Nat Rev Rheumatol. 2011;7(1):23–32. doi: 10.1038/nrrheum.2010.191. [DOI] [PubMed] [Google Scholar]
- 19.Spector TD, MacGregor AJ. Risk factors for osteoarthritis: genetics. Osteoarthr Cartil. 2004;12:39–44. doi: 10.1016/j.joca.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Lacerda D a, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, Oxford JT, Morris NP, Andrikopoulos K, Ramirez F. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995;80(3):423–430. [DOI] [PubMed] [Google Scholar]
- 21.Blaschke UK, Eikenberry EF, Hulmes DJS, Galla HJ, Bruckner P. Collagen XI nucleates self-assembly and limits lateral growth of cartilage fibrils. J Biol Chem. 2000;275(14):10370–10378. doi: 10.1074/jbc.275.14.10370. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, Flahiff CM, Waldman B a, Wu D, Olsen BR, Setton L a., Li Y Osteoarthritis-like changes and decreased mechanical function of articular cartilage in the joints of mice with the chondrodysplasia gene (cho). Arthritis Rheum. 2003;48(9):2509–2518. doi: 10.1002/art.11233. [DOI] [PubMed] [Google Scholar]
- 23.Lam NP, Li Y, Waldman AB, Brussiau J, Lee PL, Olsen BR, Xu L. Age-dependent increase of discoidin domain receptor 2 and matrix metalloproteinase 13 expression in temporomandibular joint cartilage of type IX and type XI collagen-deficient mice. Arch Oral Biol. 2007;52(6):579–584. doi: 10.1016/j.archoralbio.2006.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BE, Li Y. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J Biol Chem. 2005;280(1):548–555. doi: 10.1074/jbc.M411036200. [DOI] [PubMed] [Google Scholar]
- 25.Szymko-Bennett YM, Kurima K, Olsen B, Seegmiller R, Griffith AJ. Auditory function associated with Col11a1 haploinsufficiency in chondrodysplasia (cho) mice. Hear Res. 2003;175(1–2):178–182. doi: 10.1016/S0378-5955(02)00736-0. [DOI] [PubMed] [Google Scholar]
- 26.Xu L, Polur I, Servais JM, Hsieh S, Lee PL, Goldring MB, Li Y. Intact pericellular matrix of articular cartilage is required for unactivated discoidin domain receptor 2 in the mouse model. Am J Pathol. 2011;179(3):1338–1346. doi: 10.1016/j.ajpath.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly NH, Schimenti JC, Ross FP, Van Der Meulen MCH. Transcriptional profiling of cortical versus cancellous bone from mechanically-loaded murine tibiae reveals differential gene expression. 2016. doi: 10.1016/j.bone.2016.02.007. [DOI] [PMC free article] [PubMed]
- 28.Fernandes RJ, Weis M, Scott MA, Seegmiller RE, Eyre DR. Collagen XI chain misassembly in cartilage of the chondrodysplasia (cho) mouse. Matrix Biol. 2007;26(8):597–603. doi: 10.1016/j.matbio.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynch ME, Main RP, Xu Q, Walsh DJ, Schaffler MB, Wright TM, van der Meulen MCH. Cancellous bone adaptation to tibial compression is not sex dependent in growing mice. J Appl Physiol. 2010;109(3):685–691. doi: 10.1152/japplphysiol.00210.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18 Suppl 3:S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 31.Ko FC, Dragomir CL, Plumb DA, Hsia AW, Adebayo OO, Goldring SR, Wright TM, Goldring MB, van der Meulen MCH. Progressive cell-mediated changes in articular cartilage and bone in mice are initiated by a single session of controlled cyclic compressive loading. J Orthop Res. February 2016. doi: 10.1002/jor.23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, Shah M, Thompson EW. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60(12):3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girish V, Vijayalakshmi A. Affordable image analysis using NIH Image/ImageJ. Indian J Cancer. 41(1):47. http://www.ncbi.nlm.nih.gov/pubmed/15105580. Accessed January 24, 2017. [PubMed] [Google Scholar]
- 34.Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014;9(5):e96801. doi: 10.1371/journal.pone.0096801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez RR, Seegmiller RE, Stark MR, Bridgewater LC. A type XI collagen mutation leads to increased degradation of type II collagen in articular cartilage. Osteoarthr Cartil. 2004;12(4):314–320. doi: 10.1016/j.joca.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Kamper M, Hamann N, Prein C, Clausen-Schaumann H, Farkas Z, Aszodi A, Niehoff A, Paulsson M, Zaucke F. Early changes in morphology, bone mineral density and matrix composition of vertebrae lead to disc degeneration in aged collagen IX −/− mice. Matrix Biol. 2016;49(2016):132–143. doi: 10.1016/j.matbio.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Hafez A, Squires R, Pedracini A, Joshi A, Seegmiller RE, Oxford JT. Col11a1 Regulates Bone Microarchitecture during Embryonic Development HHS Public Access. J Dev Biol J Dev Biol. 2015;3(4):158–176. doi: 10.3390/jdb3040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frischknecht M, Niehof-Oellers H, Jagannathan V, Owczarek-Lipska M, Drögemüller C, Dietschi E, Dolf G, Tellhelm B, Lang J, Tiira K, Lohi H, Leeb T. A COL11A2 mutation in Labrador retrievers with mild disproportionate dwarfism. PLoS One. 2013;8(3):e60149. doi: 10.1371/journal.pone.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calvo E, Palacios I, Delgado E, Sánchez-Pernaute O, Largo R, Egido J, Herrero-Beaumont G. Histopathological correlation of cartilage swelling detected by magnetic resonance imaging in early experimental osteoarthritis. Osteoarthr Cartil. 2004;12(11):878–886. doi: 10.1016/j.joca.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Poulet B, Westerhof T a T, Hamilton RW, Shefelbine SJ, Pitsillides a. a. Spontaneous osteoarthritis in Str/ort mice is unlikely due to greater vulnerability to mechanical trauma. Osteoarthr Cartil. 2013;21(5):756–763. doi: 10.1016/j.joca.2013.02.652. [DOI] [PubMed] [Google Scholar]
- 41.Radin EL, Rose RM. Role of subchondral bone in the initiation and progression of cartilage damage. Clin Orthop Relat Res. 1986;(213):34–40. http://www.ncbi.nlm.nih.gov/pubmed/3780104. Accessed November 3, 2016. [PubMed] [Google Scholar]
- 42.Day JS, van der Linden JC, Bank RA, Ding M, Hvid I, Sumner DR, Weinans H. Adaptation of subchondral bone in osteoarthritis. Biorheology. 2004;41(3‐4):359–368. [PubMed] [Google Scholar]
- 43.Hayami T, Pickarski M, Wesolowski GA, Mclane J, Bone A, Destefano J, Rodan GA, Duong LT. The role of subchondral bone remodeling in osteoarthritis: Reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum. 2004;50(4):1193–1206. doi: 10.1002/art.20124. [DOI] [PubMed] [Google Scholar]
- 44.Hardcastle SA, Dieppe P, Gregson CL, Hunter D, Thomas GER, Arden NK, Spector TD, Hart DJ, Laugharne MJ, Clague GA, Edwards MH, Dennison EM, Cooper C, Williams M, Davey Smith G, Tobias JH. Prevalence of radiographic hip osteoarthritis is increased in high bone mass. Osteoarthr Cartil. 2014;22(8):1120–1128. doi: 10.1016/j.joca.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardcastle SA, Dieppe P, Gregson CL, Arden NK, Spector TD, Hart DJ, Edwards MH, Dennison EM, Cooper C, Sayers A, Williams M, Davey Smith G, Tobias JH. Individuals with high bone mass have an increased prevalence of radiographic knee osteoarthritis. Bone. 2015;71:171–179. doi: 10.1016/j.bone.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Im G-I, Kim M-K. The relationship between osteoarthritis and osteoporosis. J Bone Miner Metab. 2014;32(2):101–109. doi: 10.1007/s00774-013-0531-0. [DOI] [PubMed] [Google Scholar]
- 47.Xu L, Peng H, Glasson S, Lee PL, Hu K, Ijiri K, Olsen BR, Goldring MB, Li Y. Increased expression of the collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007;56(8):2663–2673. doi: 10.1002/art.22761. [DOI] [PubMed] [Google Scholar]
- 48.Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. April 2017. doi: 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
- 49.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22(8):1197–1207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- 50.Takasu N [Experimental study on the effect of forced running on occurrence of osteoarthritis in the knee of C 57 BL mice]. Nihon Seikeigeka Gakkai Zasshi. 1992;66(11):1165–1175. http://www.ncbi.nlm.nih.gov/pubmed/1484236. Accessed May 27, 2016. [PubMed] [Google Scholar]
- 51.Boveris A, Navarro A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. 2007. doi: 10.1016/j.freeradbiomed.2007.08.015. [DOI] [PubMed]
- 52.Hu K, Xu L, Cao L, Flahiff CM, Brussiau J, Ho K, Setton LA, Youn I, Guilak F, Olsen BR, Li Y. Pathogenesis of osteoarthritis-like changes in the joints of mice deficient in type IX collagen. Arthritis Rheum. 2006;54(9):2891–2900. doi: 10.1002/art.22040. [DOI] [PubMed] [Google Scholar]
- 53.Wang CJ, Iida K, Egusa H, Hokugo A, Jewett A, Nishimura I. Trabecular Bone Deterioration in col9a1+/− Mice Associated With Enlarged Osteoclasts Adhered to Collagen IX-Deficient Bone. J Bone Miner Res. 2008;23(6):837–849. doi: 10.1359/jbmr.080214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karamchedu NP, Tofte JN, Waller KA, Zhang LX, Patel TK, Jay GD. Superficial zone cellularity is deficient in mice lacking lubricin: a stereoscopic analysis. Arthritis Res Ther. 2016;18(1):64. doi: 10.1186/s13075-016-0967-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.