

Abstract
Hyperlipidemia induces accelerated rejection of cardiac allografts and resistance to tolerance induction using costimulatory molecule blockade in mice due in part to anti-donor Th17 responses and reduced regulatory T cell function. Accelerated rejection in hyperlipidemic mice is also associated with increased serum levels of IL-6. Here, we examined the role of IL-6 in hyperlipidemia-induced accelerated rejection and resistance to tolerance. Genetic ablation of IL-6 prevented hyperlipidemia-induced accelerated cardiac allograft rejection. Using Th17-lineage fate tracking mice, we observed that IL-6 is required to promote the development of anti-donor Th17 lineage cells independently of antigen challenge. In contrast, the frequency of alloreactive T cells producing IL-2 or IFN-γ remained increased in hyperlipidemic IL-6-deficient mice. Ablation of IL-6 overcame hyperlipidemia-induced changes in Tregs, but was not sufficient to overcome resistance to costimulatory molecule blockade induced tolerance. We suggest that accelerated rejection in hyperlipidemic mice results from IL-6 driven anti-donor Th17 responses. While alterations in Tregs were overcome by ablation of IL-6, the reversal of hyperlipidemia-induced changes in Tregs was not sufficient to overcome increased Th1-type anti-donor T cell responses, suggesting that hyperlipidemia induced IL-6-independent effects on recipient immunity prevent tolerance induction.
Introduction
The canonical understanding of solid organ transplant rejection holds that transplant rejection is mediated by anti-donor T helper type 1 (Th1) CD4 and CD8 cytotoxic T cells (1–3). Operational tolerance to the transplant is thought to be mediated by host regulatory T cells (4, 5). However, this understanding has been gained largely through transplanting organs between young, healthy, animals. There has been relatively little work examining how co-morbidities present in the human transplant patient population affect the immunological response of patients to the transplanted organ. This is a clinically relevant issue because transplant patients frequently suffer from immunologically relevant co-morbidities like obesity, diabetes, or hyperlipidemia that lead to immune system dysfunction and systemic inflammation and can lead to disease associated metabolic disorders (6, 7). How these comorbidities affect the immunological response or the ability to induce operational tolerance to transplanted organs is not fully understood.
A major recipient characteristic associated with cardiac allograft rejection is a history of ischemic heart disease which can result from hyperlipidemia. Hyperlipidemia is a common co-morbidity among the heart transplant patient population, with nearly 90% of heart transplant patients becoming hyperlipidemic by 5 years after transplant (8), many of whom are resistant to clinically acceptable doses of cholesterol-lowering statin drugs (9). Hyperlipidemia leads to systemic inflammation (10). Hyperlipidemic humans and mice exhibit increased levels of inflammatory cytokines in their serum and increased inflammatory T cell responses (11–15). Given the systemic effects of hyperlipidemia that promote inflammation, we examined how hyperlipidemia affects transplant outcome. We have shown that hyperlipidemia promotes accelerated rejection of vascularized cardiac allografts (16–18). Hyperlipidemia-induced accelerated rejection is associated with an increase in anti-donor Th1 and Th17 responses as well as changes in Tregs and increases in serum concentrations of inflammatory cytokines (16).
Systemic changes induced by hyperlipidemia include increased serum levels of IL-6 (19–21). We observed that serum levels of IL-6 are increased in hyperlipidemic mice receiving cardiac allografts (16). IL-6 is a pleiotropic cytokine that has effects on many cell types (22), and is known to promote pro-inflammatory responses. IL-6 plays a critical role in the generation of Th17 cells, while IL-23 serves to maintain these cells (23–25). IL-6 also negatively regulates the TGF-β mediated generation of regulatory T cells, further increasing its pro-inflammatory effects (26). It has been previously shown that high lipid levels can stimulate macrophages to produce large amounts of IL-6 (27) and recognition of oxidized LDL by macrophages induces IL-6 secretion (28). Therefore, the connection between lipid levels and pro-inflammatory IL-6 production is clear and clinically relevant, yet the downstream effects of IL-6 on transplanted organs in hyperlipidemic recipients are not fully understood.
Here we show that IL-6 plays a central role in promoting Th17 cell mediated accelerated rejection in hyperlipidemic mice. Ablation of IL-6 prevented Th17 cell mediated accelerated rejection, and was sufficient to overcome alterations in Tregs observed in hyperlipidemic mice. In contrast, ablation of IL-6 had no effect on the ability of hyperlipidemia to induce an increase in the frequency of donor reactive T cells producing typical Th1-type cytokines. While ablation of IL-6 overcame accelerated rejection and alterations in Tregs in hyperlipidemic mice, these changes were not sufficient to overcome resistance to tolerance induced using CTLA4-Ig and anti-CD154. We suggest that hyperlipidemia alters rejection through IL-6 dependent mechanisms involving anti-donor Th17 responses and alterations in Tregs and IL-6 independent mechanisms that affect tolerance.
Materials and Methods
Mice.
IL-6−/−, ApoE−/−, Ai9 mice (29), B6(C)-H2-Ab1bm12/KhEgJ mice (bm12) and BALB/cJ (BALB/c) mice were obtained from Jackson Laboratories (Bar Harbor, ME). Mice expressing cre under the control of the IL-17 promoter were the generous gift of Dr. Brigitta Stockinger. All procedures involving mice were approved by our Institutional Animal Care and Use Committee in accordance with NIH guidelines.
Heterotopic heart transplantation.
Heterotopic cardiac transplants were performed and function assessed as previously described (30). All transplant recipients were between 10 and 17 weeks of age.
Costimulatory molecule blockade.
Co-stimulatory molecule blockade with anti-CD154 and CTLA-4Ig was performed as previously described (18, 31). In brief, mice were treated with 500 micrograms each CTLA-4Ig and MR1 (BioXcell, Lebanon NH) intraperitoneally (IP) on day zero relative to transplantation, followed by repeat injections consisting of 250 micrograms of each CTLA-4Ig and MR1 on days 2,4 and 6 relative to transplantation.
Flow Cytometry.
Cell surface staining was performed using standard methods. The following antibodies were used: Anti-CD4 (GK1.5, Biolegend, San Diego, CA), anti-CD3 (2C11, Biolegend), anti-CD8α (53–6.7, Biolegend), anti-FoxP3 antibody (FJK16A, eBioscience), anti-Akt pS473 (M89–61, BD Pharmingen), anti-human CD2 (hCD2- PE), anti-CD44 (IM7, Biolegend), anti-CD62L (MeL14, Biolegend), anti-Pten (BD Pharmingen), anti-PD1 (J43, BD Pharmingen).
ELISPOT.
ELISpot assays were performed as previously described (32). Capture antibodies were: IL-2 (JES6–1A12), IL-4 (11B11), IFNγ (AN-18), and IL-17A (TC11–18H10.1). Biotinylated detection antibodies were: IL-2 (JES6–5H4), IL-4 (BVD6–24G2), IFNγ (R4–6A2), and IL-17A (TC11–8H4).
Serum lipid measurement.
Serum was analyzed for lipid levels using a cholesterol quantitation kit (Millipore Sigma).
Histology.
Hearts were fixed in 10% Buffered Formalin, embedded in paraffin and sections stained with H&E or Masson’s Trichrome.
Immunohistochemistry for CD4.
Formalin-fixed paraffin embedded sections underwent heat induced epitope retrieval. Rat anti mouse CD4 (4SM95, ThermoFisher) was used as a primary antibody followed by biotinylated goat anti-rat (Vector, Burlingame, CA) and ABC-HRP. Staining was developed using DAB/peroxide solution (ThermoFisher). Slides were counterstained with 50% Gill’s hematoxylin diluted in with water and then cleared. For quantification, 4 sections from each graft were obtained, at 100 μm intervals. FIJI color deconvolution and thresholding techniques were used to isolate DAB signal, and FIJI particle analysis was used to quantitate CD4 cells.
Statistics.
For survival curves, significance was determined using log-rank (Mantel-Cox) tests. For pathologic analysis and ELISPOT experiments, significance was determined using nested t-Tests. For all other analysis, significance was determined by Student’s unpaired parametric t test. Error bars represent standard deviation. For experiments determining if ablation of IL-6 abrogates effects observed in IL-6 sufficient mice, we performed power calculations using https://clincalc.com/stats/Power.aspx to ensure all experiments were powered >80%.
Results
Accelerated rejection in hyperlipidemic mice is IL-6 dependent.
To examine the role of IL-6 in hyperlipidemia-induced accelerated rejection, we utilized apolipoprotein E knockout mice (ApoE−/− mice) (33). ApoE−/− mice are perhaps the best-characterized model used study the effects of hyperlipidemia on immunity (34). Serum cholesterol, LDL, VLDL, and triglyceride levels are elevated in ApoE−/− mice and can be further increased by feeding a high-fat diet (HFD). While ApoE−/− mice become hyperlipidemic they do not rapidly become obese or hyperglycemic when fed a high-fat diet allowing us to eliminate these comorbidities as confounding factors. C57BL/6 mice fed a high-fat diet also become hyperlipidemic, although less degree than ApoE−/− mice and humans. However, C57BL/6 mice maintained on a high-fat diet also become hyperglycemic and rapidly become obese.
We bred ApoE−/− and IL-6−/− mice to generate ApoE−/−IL-6−/− double knockout mice. Groups of ApoE−/−IL-6−/− mice and IL-6 sufficient controls (ApoE−/−IL-6+/+) were fed normal chow or a high-fat diet for a minimum of four weeks. IL-6 deficiency had no effect on serum lipid levels in either ApoE−/− or ApoE+/+ controls maintained on normal chow. The degree of hyperlipidemia induced in ApoE−/− mice fed a high-fat diet was the same in the presence or absence of IL-6 (Fig. S1). Mice in each group then received MHC class II mismatched C57BL/6.C-H2bm12 (bm12) heterotopic cardiac transplants. As expected, bm12 hearts survived longer than 100 days when transplanted into normolipidemic C57BL/6 controls (Fig. 1, MST > 100 days). As previously shown (16, 18), allogeneic bm12 hearts transplanted into ApoE−/−IL-6+/+ mice fed a high-fat diet were rapidly rejected (Fig. 1, MST = 48 days, P = 0.01 relative to ApoE+/+IL-6+/+ controls fed normal chow). In contrast, bm12 hearts survived long-term when transplanted into IL-6 deficient ApoE−/−IL-6−/− mice fed a high-fat diet (Fig. 1, MST > 100 days, P = 0.0039 vs ApoE−/−IL-6+/+ mice fed a high-fat diet). Thus, hyperlipidemia-induced accelerated rejection is IL-6 dependent.
Figure 1. IL-6 deficiency prevents accelerated cardiac allograft rejection in hyperlipidemic recipients.
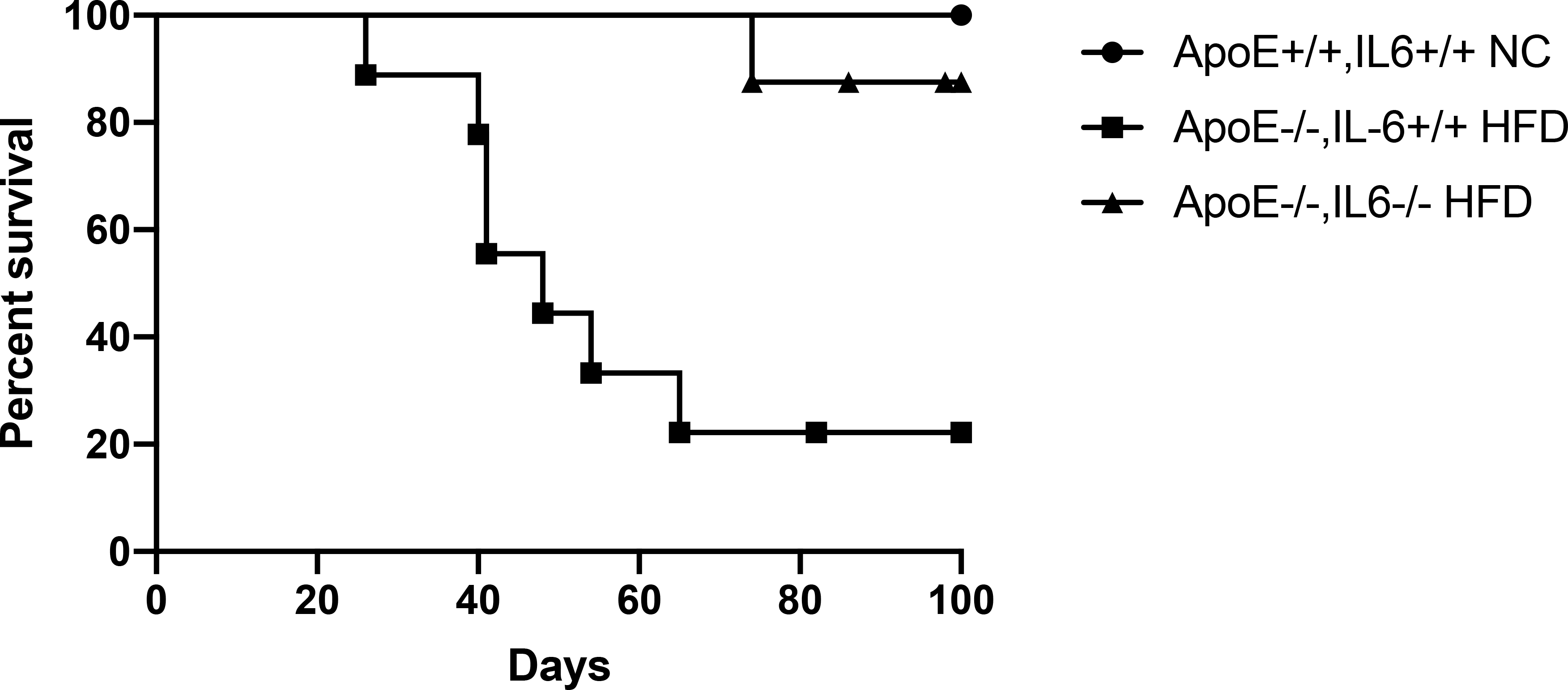
Groups of mice were fed either high fat diet (HFD) or normal chow (NC) for a minimum of four weeks prior to transplantation with allogeneic bm12 cardiac grafts. Shown is graft survival in ApoE−/−IL-6+/+ recipient mice fed HFD (squares, MST = 48 days, n = 9), ApoE+/+IL-6+/+ recipient mice fed NC (circles, MST > 100 days, n = 5), and ApoE−/−IL-6−/− recipient mice fed HFD (upright triangles, MST > 100 days, n = 8, P = 0.004 vs ApoE−/− HFD recipients). Significance was determined by log-rank (Mantel-Cox) test.
IL-6 independent tissue destruction in cardiac allografts in hyperlipidemic mice.
Chronic rejection of bm12 cardiac allografts in normolipidemic C57BL/6 mice is characterized by neointimal proliferation and occlusion of donor blood vessels that leads to graft loss at greater than 100 days. Perivascular lymphocytic infiltration may be apparent, but cardiac muscle is largely spared (35, 36). In contrast, rejection of bm12 hearts transplanted into hyperlipidemic ApoE−/− recipients is characterized by significant lymphocytic infiltration and extensive destruction of graft tissue (16). As expected, bm12 hearts rejected by ApoE−/−IL-6+/+ mice fed a high-fat diet were characterized by significant lymphocytic infiltrate and large areas of necrosis and fibrosis throughout the transplanted heart (Fig. 2A). Bm12 hearts transplanted into ApoE sufficient controls maintained on normal chow showed areas of significant neointimal proliferation and vasculopathy consistent with chronic rejection (Fig. 2B), again consistent with previous reports. Bm12 heart transplants harvested from normolipidemic ApoE+/+IL-6−/− recipients maintained on normal chow also showed tissue damage consistent with chronic rejection including neointimal proliferation, vasculopathy and largely undamaged cardiac muscle (Fig. 2C). In contrast, bm12 hearts transplanted into ApoE−/−IL-6−/− mice fed a high-fat diet showed significant areas of lymphocytic infiltrate and tissue destruction (Fig. 2D). Thus, hyperlipidemia promotes greater apparent tissue damage in rejecting hearts in the absence of IL-6.
Figure 2. IL-6 independent tissue destruction in cardiac allografts in hyperlipidemic mice.
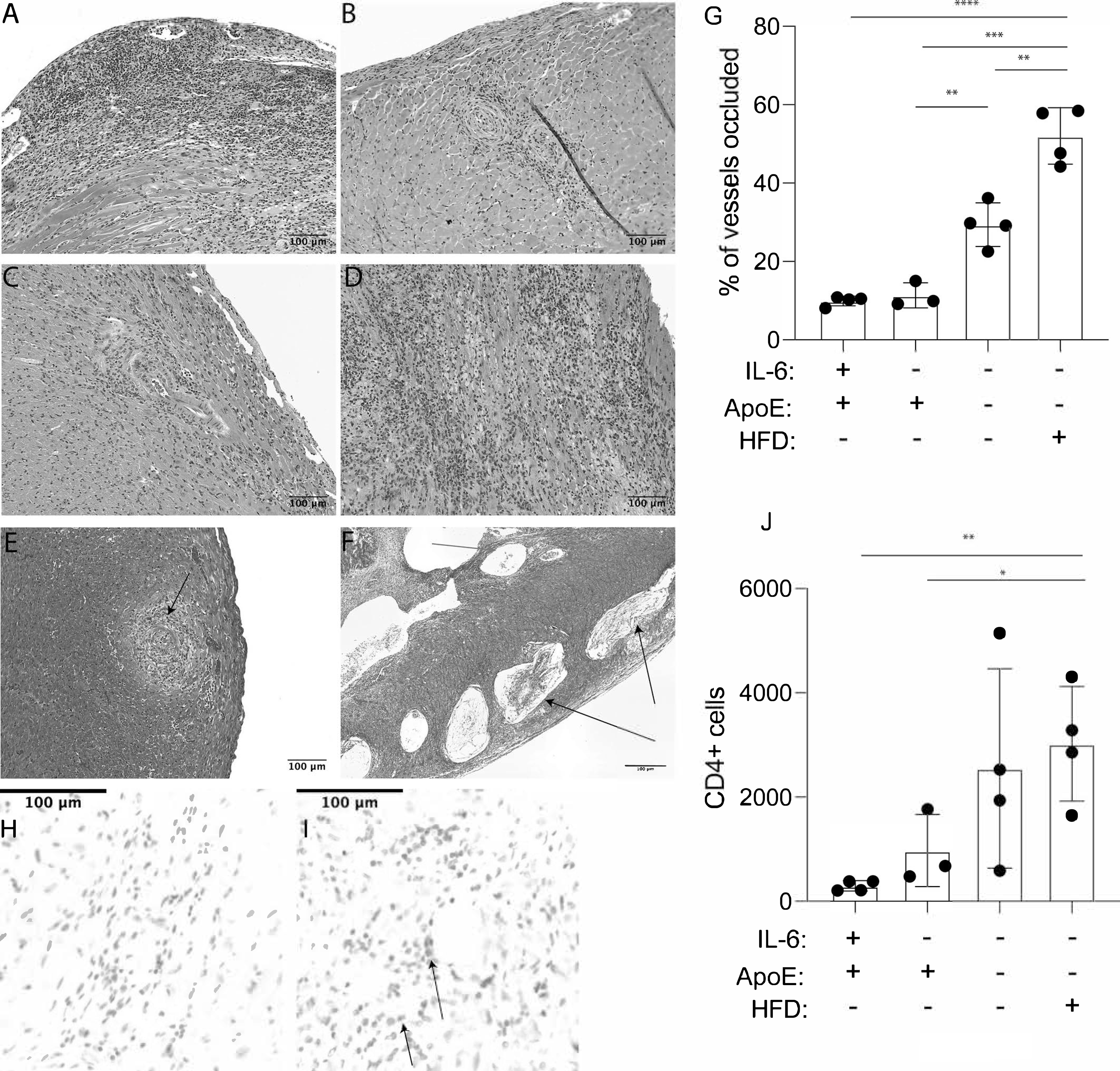
Age matched groups of mice were fed high fat diet (HFD) or normal chow (NC) for a minimum of four weeks prior to transplant with bm12 cardiac allografts. Grafts were harvested after 100 days, or at the time of rejection, and tissue sections were prepared. Shown are representative sections stained with hematoxylin and eosin (H&E). (A) ApoE−/−IL-6+/+ recipient mice fed HFD (B) ApoE+/+IL-6+/+ recipient mice fed NC (C) ApoE+/+IL-6−/− recipient mice fed NC (D) ApoE−/−IL-6−/− recipient mice fed HFD. Slides were scanned and individual pictures were captured at 15X magnification with CaseViewer software. A representative section showing Masson’s Trichrome staining of occluded vessels and unoccluded vessels in a section from a bm12 graft isolated from (E) ApoE+/+IL-6−/− recipient mice fed NC and (F) ApoE−/−IL-6−/− recipient mice fed HFD. (G) Quantification of occluded vessels in bm12 grafts harvested from recipient animals after 100 days. Tissue sections from 3–4 transplants were analyzed. Statistical significance was determined with a nested t-test. Representative immunohistochemical staining for CD4 in bm12 grafts harvested from (H) ApoE+/+IL-6−/− NC-fed recipient mice (I) ApoE−/−IL-6−/− HFD-fed recipient mice. (J) Quantitation of CD4 cells infiltrating grafts. Tissue sections from 3–4 transplants were analyzed. Error bars represent standard deviation. Statistical analysis was performed using a nested t-Test. **** = P < 0.0001,*** = P < 0.0005, ** = P < 0.005, * = P < 0.05.
We next quantitated the frequency of occluded vessels in rejecting hearts at 100 days after transplant. Occluded vessels in normolipidemic recipients showed significant neointimal proliferation without inclusion of lipids or cholesterol clefts (Fig. 2E) while in hyperlipidemic recipients, vessel occlusion included lipid deposits and cholesterol clefts (Fig. 2F). The frequency of occluded vessels in bm12 hearts harvested from hyperlipidemic ApoE−/−IL-6−/− mice fed normal chow was significantly increased compared to the frequency of occluded vessels in bm12 hearts harvested from normolipidemic ApoE+/+IL-6−/− mice fed normal chow (Fig. 2G). This frequency was further increased in ApoE−/−IL-6−/− mice fed a high-fat diet.
Rejection of bm12 hearts by C57BL/6 mice is CD4 T cell dependent (37). We therefore quantified graft infiltrating CD4 T cells to assess cell-mediated rejection. Allogeneic bm12 hearts transplanted into ApoE−/−IL-6−/− mice fed a high-fat diet were harvested after 100 days and tissue sections obtained were stained with anti-CD4 antibodies. We observed that these sections had an increased number of graft-infiltrating CD4+ cells when compared to sections from bm12 grafts placed in ApoE+/+IL-6−/− mice maintained on normal chow also harvested at 100 days (Fig. 2H–J). Together, these data suggest that even though bm12 grafts placed into IL-6-deficient hyperlipidemic animals survived longer than bm12 grafts placed into IL-6-replete hyperlipidemic recipients, hyperlipidemia is sufficient to induce IL-6-independent anti-donor responses that worsen rejection as observed by graft damage, vessel occlusion, and infiltrating CD4 cells.
IL-6 production is required for increases in Th17 cells in hyperlipidemic mice.
Hyperlipidemia promotes anti-donor Th17 responses based on an increased frequency of IL-17 producing anti-donor reactive CD4 T cells, and that IL-17 production plays a role in accelerated rejection (16). IL-6 plays an important role in Th17 cell differentiation (38, 39). We therefore examined whether the effect of IL-6 on hyperlipidemia induced accelerated rejection was the result of Th17 lineage anti-donor responses. To this end, we used Th17 lineage fate-mapping mice expressing cre recombinase under the control of the endogenous IL-17A promoter (40). These mice were bred to mice containing a loxP-flanked STOP sequence up-stream of the tdTomato gene which was knocked into the ROSA26 locus (Ai9 mice (29)) to generate IL-17cre/tdTomato mice in which transcription of the il17a locus leads to expression of cre recombinase and permanent expression of tdTomato by cells that have expressed IL-17A at any point in their history, even if they are not producing IL-17A at the time of analysis (40). IL-17cre/tdTomato mice were bred to ApoE−/− mice and ApoE−/−IL-6−/− mice to generate ApoE−/−IL-17cre/tdTomatoIL-6−/− and ApoE−/−IL-17cre/tdTomatoIL-6+/+ mice. Groups of mice were fed a high fat diet or normal chow for four weeks, and then spleens were examined by flow cytometry. Representative flow plots showing the percentage of tdTomato+ cells among CD3+CD4+ Lymphocytes are shown in Fig. 3A. Gating strategy is shown in Fig. S2. The frequency and absolute number of all CD3+ T cells expressing tdTomato in the spleen of naïve ApoE−/−IL-17cre/tdTomatoIL-6+/+ mice fed a high fat diet was significantly increased when compared with the frequency and absolute number in ApoE+/+IL-17cre/tdTomatoIL-6+/+ controls fed normal chow (Fig. 3B and 3C). In contrast, ApoE−/−IL-17cre/tdTomatoIL-6−/− mice fed a high-fat diet contained significantly lower numbers of tdTomato expressing CD3+ T cells when compared to IL-6 sufficient controls (Fig. 3B and 3C, P = 0.04). When CD4 T cells were specifically examined, the frequency and number of tdTomato+CD3+CD4+ Th17 lineage cells in IL-6 sufficient ApoE−/−IL-17cre/tdTomato mice fed a high-fat diet was significantly higher than in controls and ApoE−/−IL-17cre/tdTomato IL-6−/− mice fed a normal chow diet (Fig. 3D and 3E). The frequency (Fig. 3D, P = 0.02) and absolute number (Fig. 3E, P = 0.005) of tdTomato+ Th17 cells was significantly reduced in ApoE−/−IL-17cre/tdTomatoIL-6−/− mice fed a high-fat diet when compared with ApoE−/−IL-17cre/tdTomatoIL-6+/+ controls fed a high-fat diet. Very few tdTomato+ CD8 cells were observed in all groups and the frequency and absolute number was the same (Fig. S3A and B).
Figure 3. IL-6 deficiency decreases Th17-lineage frequency and absolute numbers in hyperlipidemic mice.

Groups of age matched ApoE+/+IL-17cre/tdTomato IL-6+/+, ApoE−/−IL-17cre/tdTomato IL-6+/+ and ApoE−/−IL-17cre/tdTomato IL-6−/− mice were maintained on normal chow or high fat diet for a minimum of four weeks. Splenocytes were isolated and stained with antibodies specific for murine CD3 and CD4. Cells were examined for expression of CD3, CD4 and tdTomato by flow cytometry. (A) Representative flow plots gated on live CD3+CD4+ splenocytes showing the percentage of tdTomato+ cells. (B) Combined frequencies of all CD3+ splenocytes expressing tdTomato. (C) Absolute number of all CD3+ splenocytes expressing tdTomato. Shown is the combined mean. (D) Combined frequencies of CD3+CD4+ splenocytes expressing tdTomato. (E) Absolute number of CD3+CD4+ splenocytes expressing tdTomato. Shown is the combined mean of ≥5 mice per group. Significance was determined using unpaired Students t-Test. Error bars represent standard deviation. *** = P < 0.0005, ** = P < 0.005, * = P < 0.05.
Hyperlipidemia induced alterations in regulatory T cells are dependent on IL-6.
Hyperlipidemia results in changes in Tregs that lead to reduced function including an increase in levels of phosphorylated Akt (18). More recently, we have observed that hyperlipidemia induced Akt phosphorylation independently decreases regulatory T cell function (JI personal observation). Therefore, we next examined the effect of IL-6 deficiency on Tregs from hyperlipidemic mice. Groups of IL-6−/− and ApoE−/−IL-6−/− mice were maintained on normal chow or fed a high-fat diet for four weeks prior to sacrifice. Tregs were then examined by flow cytometry to assess the extent to which hyperlipidemia-induced IL-6 production promoted alterations in Tregs. In contrast to our previous observations, the levels of p-Akt in Tregs from ApoE+/+IL-6−/− mice fed a high-fat diet were the same as observed in controls maintained on normal chow (Fig. 4A). The levels of p-Akt in Tregs derived from ApoE−/−IL-6−/− mice fed a high-fat diet were also the same as observed in controls maintained on normal chow, suggesting that the increase in Akt phosphorylation in Tregs from hyperlipidemic mice is IL-6 dependent.
Figure 4. Effects of IL-6 deficiency on regulatory T cell phenotype.

Groups of mice were fed high fat diet (HFD) or normal chow for a minimum of four weeks. Animals were sacrificed, and splenic Tregs examined by flow cytometry to assess Akt phosphorylation (pAkt). (A) Representative flow plots gated on live CD4+FoxP3+ splenocytes. (B) MFI of PD-1 relative to C57BL/6 controls in CD4+CD44hiCD62LloFoxP3+ splenocytes for indicated IL-6 replete groups of mice. Shown are the combined results of three independent experiments. (C) MFI of PD-1 relative to C57BL/6 controls in CD4+CD44hiCD62LloFoxP3+ splenocytes for indicated IL-6 deficient groups of mice. Shown are the combined results of three independent experiments. (D) Pten in IL-6 replete groups of hyperlipidemic mice. Shown is the MFI of Pten relative to C57BL/6 controls in CD4+FoxP3+ splenocytes. Combined results of three independent experiments. (E) Pten in IL-6 deficient groups of hyperlipidemic mice. Shown is the MFI of Pten relative to C57BL/6 controls in CD4+FoxP3+ splenocytes. Combined results of two independent experiments. (F) Frequency of CD4+CD44loCD62LhiFoxP3+ and CD4+CD44hiCD62LloFoxP3+ Tregs in indicated groups of IL-6 replete mice. Shown is combined results of three independent experiments. (G) Frequency of CD4+CD44loCD62LhiFoxP3+ and CD4+CD44hiCD62LloFoxP3+ Tregs in indicated groups of IL-6 deficient mice. Shown is combined results of three independent experiments. Statistical significance was determined by Student’s t-test. Asterisk indicates P < 0.05.
We observed that while the total frequency of Tregs expressing PD-1 on their surface was the same in hyperlipidemic and control mice, surface levels of PD-1 are reduced on eTregs from hyperlipidemic mice when compared with controls based on MFI (Fig. 4B). The decrease in surface expression of PD-1 correlated with levels of hyperlipidemia in that the reduction in surface PD-1 levels was greatest on Tregs from ApoE−/− mice fed a high-fat diet (Fig. 4B). In contrast, we did not observe a decrease in surface expression of PD-1 in ApoE+/+IL-6−/− mice fed a high-fat diet when compared with controls (Fig. 4C). Ligation of PD-1 on Tregs by its ligand PD-L1 results in the up-regulation of Pten, which negatively regulates PI3K and Akt. Tregs from ApoE+/+ mice fed a high-fat diet showed reduced levels of Pten when compared with controls maintained on normal chow, (Fig. 4D). Pten levels were further reduced in Tregs from ApoE−/− mice maintained on normal chow and greatest in ApoE−/− mice fed a high-fat diet (Fig. 4D). Similar results obtained by Western blot showed a trend toward a decrease in Pten levels in Tregs from hyperlipidemic mice (not shown). In the absence of IL-6, induction of hyperlipidemia had no effect of Pten levels (Fig. 4E).
We have previously shown that hyperlipidemia leads to a reduction in the frequency of CD62LhighCD44low Tregs and an increase in CD62LlowCD44high Tregs (18). While IL-6 deficiency was sufficient to overcome changes in Akt, PD1 and Pten, hyperlipidemia induced changes in CD62LhighCD44low and CD62LlowCD44high Treg frequencies remained apparent in hyperlipidemic IL-6 deficient mice. As observed previously in IL-6 sufficient mice, the frequency of CD62LhighCD44low Tregs in ApoE+/+IL-6−/− mice fed a high-fat diet was reduced when compared with controls maintained on normal chow (Fig. 4F). Similarly, the frequency of CD62LlowCD44high Tregs was increased in hyperlipidemic mice when compared with controls.
Increased T cell alloreactivity in hyperlipidemic mice is independent of IL-6 production.
Hyperlipidemia is sufficient to drive an increase in alloreactive T cells and does not require prior exposure to donor alloantigen (16). To determine the degree to which this increase is dependent on the production of IL-6, we quantitated the number of alloreactive CD4 T cells that respond to allogeneic splenocytes. Groups of ApoE+/+IL-6−/− and ApoE−/−IL-6−/− mice were fed either normal chow or a high-fat diet for four weeks. Mice were sacrificed, CD4 T cells were purified, and cytokine production after stimulation with fully allogeneic BALB/c splenocytes was examined by ELISpot assay. The frequency of both IL-2 and IFN-γ producing alloreactive CD4 T cells was significantly increased in hyperlipidemic IL-6−/− groups when compared to IL-6−/− controls with normal lipid levels (Fig. 5A). As expected from our previous result showing the effect of IL-6 deficiency on Th17 lineage cells, mice lacking IL-6 are deficient in IL-17 producing alloreactive cells (data not shown). The frequency of IL-17 producing alloreactive CD4 T cells was low and was not altered by lipid status in IL-6 deficient mice (Figure 5A). To further characterize the frequency of alloreactive cells in our model of MHC class II mismatched allograft rejection, we next tested the frequency of T cells responding to bm12 splenocytes, rather than fully allogeneic BALB/c splenocytes. The frequency of bm12 reactive IL-2 producing CD4 T cells increases in IL-6 deficient mice as they became hyperlipidemic (Figure 5B). The frequency of alloreactive CD4 cells that produced IFN-γ in response to bm12 target cells also significantly increased in hyperlipidemic ApoE−/−IL-6−/− mice fed high fat diet or normal chow (Figure 5B). However, as expected, the frequency of IL-17A-producing bm12-specific alloreactive CD4 T cells was significantly lower in all IL-6 deficient mice when compared with ApoE−/−IL-6+/+ mice fed a high fat diet (Fig. 5B).
Figure 5. Hyperlipidemia induces increases in the frequency of alloreactive cytokine-producing CD4 T cells in IL-6 deficient recipients.

Groups of age matched IL-6−/− mice and ApoE−/−IL-6−/− mice were fed either normal chow or high-fat diet for a minimum of 4 weeks. ELISpot assays were performed using splenic CD4+ T cells stimulated with (A) irradiated BALB/c splenocytes and assayed for production of IL-2 (left), IFN-γ (middle), and IL-17A (right) or (B) irradiated bm12 splenocytes and assayed for production of IL-2 (left), IFN-γ (middle) and IL-17A (right). Statistical significance was determined by nested t-Test * = P < 0.05, ** = P < 0.01, *** = P < 0.0005. Combined results of three or more independent experiments performed on pooled cells from 2–3 mice in each group per experiment.
IL-6 deficiency does not reverse resistance to costimulatory molecule-blockade induced tolerance.
Fully allogeneic BALB/c cardiac grafts are rapidly rejected when transplanted into wild-type C57BL/6 recipients. However, it is possible to induce long-term acceptance of fully allogeneic hearts by treating recipients with CTLA-4Ig and anti-CD154 MR1 to block costimulation through CD28 and CD154 respectively (41–45). In ApoE−/− mice fed a high fat diet costimulatory molecule blockade prolongs survival of fully allogenic hearts but does not lead to long-term acceptance or operational tolerance (18). Insofar as IL-6 ablation was sufficient to overcome hyperlipidemia-induced accelerated rejection of bm12 hearts, we examined whether resistance to costimulatory molecule blockade induced tolerance was also IL-6 dependent. While normolipidemic control mice treated with CTLA-4Ig and anti-CD154 accepted fully allogeneic hearts long term, ApoE−/−IL-6−/− mice fed a high-fat diet and treated with CTLA-4Ig and anti-CD154 rejected BALB/c hearts with the same kinetics as IL-6 sufficient ApoE−/− controls fed a high-fat diet (Fig. 6).
Figure 6. Resistance to costimulatory molecule-blockade induced tolerance to cardiac allografts is not overcome by IL-6 deficiency.

Groups of age-matched mice were placed on high fat diet (HFD) or maintained on normal chow (NC) for a minimum of four weeks, and then transplanted with allogeneic BALB/c cardiac grafts. Recipients were treated with 500 micrograms CTLA-4Ig and MR1 IP on day 0, followed by 250 micrograms CTLA-4Ig and MR1 IP on day 2,4 and 6 relative to transplant to induce tolerance as described. Graft survival in ApoE+/+IL-6+/+ maintained on normal chow (circles, MST > 100 days, n = 3), ApoE−/−IL-6+/+ maintained on high fat diet (squares, MST = 28 days, n = 3), and ApoE−/−IL-6−/− mice maintained on high fat diet (triangles, MST = 30 days, n = 3, P = 0.02 vs ApoE+/+IL-6−/− NC P = 0.8 vs ApoE−/−IL-6+/+ HFD recipients).
Discussion
The common co-morbidity of hyperlipidemia leads to accelerated rejection of cardiac allografts (16, 18). Hallmarks of hyperlipidemia-induced accelerated rejection include increases in the frequency of alloreactive T cells, effects on Tregs, and increases in anti-donor Th17 responses. These hyperlipidemia-induced changes occur without the need for prior exposure to donor antigen, suggesting that hyperlipidemia is sufficient to drive these effects (16, 18). We noted that hyperlipidemia leads to increases in serum levels of the pro-inflammatory cytokine IL-6. The role of IL-6 in rejection is poorly understood in terms of its effect on adaptive immune responses, although a role for IL-6 in donor and recipient has been suggested (46–48). The role IL-6 plays in rejection in the context of recipient hyperlipidemia is unknown. Here we set out to determine the role of IL-6 in hyperlipidemia-induced accelerated rejection.
Ablation of recipient IL-6 prevented hyperlipidemia-induced accelerated rejection of MHC class-II mismatched allogeneic heart transplants. Analysis of tissue sections from hearts transplanted into hyperlipidemic recipients revealed that the majority of pathology associated with accelerated rejection is dependent on recipient IL-6 production. We did not observe gross pathological changes in rejection due to IL-6 deficiency in the absence of hyperlipidemia. IL-6 was required to generate an increase in the frequency of anti-donor reactive Th17 cells in hyperlipidemic mice. We previously observed that IL-17 deficiency prolongs survival of allogenic hearts transplanted into hyperlipidemic recipients but does not prevent accelerated rejection (16). Using lineage tracking mice we show that hyperlipidemia-induced IL-6 production is required to generate anti-donor Th17 lineage cells and that in the absence of this effector population, accelerated rejection is prevented. Insofar as IL-17 ablation delayed but did not prevent acute rejection of hearts transplanted into hyperlipidemic mice (16), we suggest that in addition to effects on IL17 production, IL-6 has additional effects that drive accelerated rejection.
Recipient IL-6 deficiency did not alter the gross pathological appearance of chronic rejection in controls. IL-6 deficiency reduced the extent of tissue damage and lymphocytic infiltrate observed in hearts transplanted into hyperlipidemic mice, however the degree of damage observed remained greater than in IL-6 replete controls. Given that a role for donor-derived IL-6 has been postulated in the pathogenesis of chronic rejection, it remains to be determined if hyperlipidemia increases production of IL-6 by the graft, and if that production may lead to increased tissue destruction. While recipient IL-6 was required to generate Th17 cells, the frequency of IL-2 and IFN-γ producing alloreactive CD4 T cells was significantly increased in hyperlipidemic mice regardless of the presence or absence of IL-6. Thus, IL-6 deficiency did not lead to a global decrease in inflammatory cytokine production by alloreactive T cells, but specifically affected IL-17A producing cells. Thus, increases in alloreactive Th1-like cells producing IL-2 and IFN-γ are IL-6 independent.
We observed that several hyperlipidemia-induced changes in Tregs are IL-6 dependent including increases Akt activation in Tregs, and alterations in PD-1 and Pten expression levels. However, hyperlipidemia induced changes in the frequencies of CD62LhighCD44low and CD62LlowCD44high Tregs remained apparent in hyperlipidemic IL-6 deficient mice. These data suggest that hyperlipidemia induces IL-6 dependent as well as IL-6 independent effects on Tregs. Hyperlipidemia induced resistance to tolerance induction using CTLA-4Ig and anti-CD154 costimulatory molecule blockade (18). Hyperlipidemic IL-6 deficient mice treated with CTLA-4Ig and anti-CD154 rejected BALB/c hearts with the same kinetics as IL-6 sufficient controls. This was not related to an inability to induce tolerance in IL-6−/− mice (48). Thus, prevention of Th17 responses and restoration of Treg phenotype by IL-6 ablation in hyperlipidemic mice, while sufficient to overcome accelerated rejection, is not sufficient to overcome resistance to costimulatory molecule blockade induced tolerance.
Hyperlipidemia-induced changes in the frequencies of CD62LhighCD44low and CD62LlowCD44high Tregs remained apparent in hyperlipidemic IL-6 deficient mice. It has previously been reported that there is a dramatic shift in the ratio of CD62LhighCD44low to CD62LlowCD44high Tregs in mice exposed to LPS leading to an increase in CD62LlowCD44high Tregs (49). In hyperlipidemic mice this change appears to be IL-6 independent. Moreover, this alteration occurs in the absence of Th17 lineage cells. While the mechanisms leading to alterations in CD62LhighCD44low to CD62LlowCD44high ratios in hyperlipidemic mice remain unknown, based on published studies showing a role for LPS in altering these ratios it is tempting to speculate that hyperlipidemia may also lead to alterations in innate signaling perhaps through an TLR4 independent mechanism.
Insofar as IL-6 deficiency overcomes hyperlipidemia-induced anti-donor Th17 responses and defects in Tregs, it is interesting that in the absence of IL-6, resistance to tolerance induction persists. We suggest that resistance may reflect a significantly increased anti-donor Th1 like response combined with IL-6 independent defects in Tregs that contribute to the inability to induce tolerance. These data highlight the complexity of using costimulatory molecule blockade to induce tolerance in individuals with hyperlipidemia and highlight the importance of considering how concomitant health conditions in transplant patients affect outcomes. Moreover, our data demonstrate that targeting IL-6 dependent and independent responses may be required to overcome rejection or resistance to tolerance induction.
Supplementary Material
Acknowledgements
The authors thank members of the Iacomini laboratory for helpful discussions and critical review of the manuscript. We also thank Dr. Rex Neal Smith for help with pathology analysis. This work was supported by NIH grants R01AI116714, R56AI116714, R21AI136022 and Grant-in Aid 7-04-RA-45 from the American Heart Association. LW is supported by NIH Fellowship 1F30AI143006.
Abbreviations:
- H&E
Hematoxylin and Eosin
- Th1
T helper type 1
- HFD
high-fat diet
- LDL
low-density lipoprotein
- VLDL
very low-density lipoprotein
- MFI
median fluorescence intensity
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Strom TB, Roy-Chaudhury P, Manfro R, Zheng XX, Nickerson PW, Wood K et al. The Th1/Th2 paradigm and the allograft response. Current opinion in immunology 1996;8(5):688–693. [DOI] [PubMed] [Google Scholar]
- 2.Wood KJ, Goto R. Mechanisms of rejection: current perspectives. Transplantation 2012;93(1):1–10. [DOI] [PubMed] [Google Scholar]
- 3.Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev 2003;196:51–64. [DOI] [PubMed] [Google Scholar]
- 4.Yeung MY, Grimmig T, Sayegh MH. Costimulation Blockade in Transplantation. Adv Exp Med Biol 2019;1189:267–312. [DOI] [PubMed] [Google Scholar]
- 5.Lechler RI, Garden OA, Turka LA. The complementary roles of deletion and regulation in transplantation tolerance. Nat Rev Immunol 2003;3(2):147–158. [DOI] [PubMed] [Google Scholar]
- 6.Guarner V, Rubio-Ruiz ME. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip Top Gerontol 2015;40:99–106. [DOI] [PubMed] [Google Scholar]
- 7.Donath MY. Inflammation as a sensor of metabolic stress in obesity and type 2 diabetes. Endocrinology 2011;152(11):4005–4006. [DOI] [PubMed] [Google Scholar]
- 8.Lund LH, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dipchand AI et al. The registry of the International Society for Heart and Lung Transplantation: thirty-first official adult heart transplant report−−2014; focus theme: retransplantation. J Heart Lung Transplant 2014;33(10):996–1008. [DOI] [PubMed] [Google Scholar]
- 9.Zakliczynski M, Boguslawska J, Wojniak E, Zakliczynska H, Ciesla D, Nozynski J et al. In the era of the universal use of statins dyslipidemia’s are still common in heart transplant recipients: a cross-sectional study. Transplantation proceedings 2011;43(8):3071–3073. [DOI] [PubMed] [Google Scholar]
- 10.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473(7347):317–325. [DOI] [PubMed] [Google Scholar]
- 11.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005;352(16):1685–1695. [DOI] [PubMed] [Google Scholar]
- 12.Leonarduzzi G, Gamba P, Gargiulo S, Biasi F, Poli G. Inflammation-related gene expression by lipid oxidation-derived products in the progression of atherosclerosis. Free Radic Biol Med 2012;52(1):19–34. [DOI] [PubMed] [Google Scholar]
- 13.Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des 2012;18(28):4266–4288. [DOI] [PubMed] [Google Scholar]
- 14.Omoigui S The Interleukin-6 inflammation pathway from cholesterol to aging--role of statins, bisphosphonates and plant polyphenols in aging and age-related diseases. Immun Ageing 2007;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444(7121):860–867. [DOI] [PubMed] [Google Scholar]
- 16.Yuan J, Bagley J, Iacomini J. Hyperlipidemia Promotes Anti-Donor Th17 Responses That Accelerate Allograft Rejection. Am J Transplant 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bagley J, Yuan J, Iacomini J. Impact of hyperlipidemia on alloimmunity. Current opinion in organ transplantation 2017;22(1):14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagley J, Yuan J, Chandrakar A, Iacomini J. Hyperlipidemia Alters Regulatory T Cell Function and Promotes Resistance to Tolerance Induction Through Costimulatory Molecule Blockade. Am J Transplant 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryu H, Lim H, Choi G, Park Y-J, Cho M, Na H et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nature Immunology 2018;19(6):583–593. [DOI] [PubMed] [Google Scholar]
- 20.Roytblat L, Rachinsky M, Fisher A, Greemberg L, Shapira Y, Douvdevani A et al. Raised interleukin-6 levels in obese patients. Obes Res 2000;8(9):673–675. [DOI] [PubMed] [Google Scholar]
- 21.Eder K, Baffy N, Falus A, Fulop AK. The major inflammatory mediator interleukin-6 and obesity. Inflammation Research 2009;58(11):727. [DOI] [PubMed] [Google Scholar]
- 22.Kishimoto T Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther 2006;8 Suppl 2(Suppl 2):S2–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev 2014;13(6):668–677. [DOI] [PubMed] [Google Scholar]
- 24.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003;421(6924):744–748. [DOI] [PubMed] [Google Scholar]
- 25.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006;441(7090):231–234. [DOI] [PubMed] [Google Scholar]
- 26.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006;24(2):179–189. [DOI] [PubMed] [Google Scholar]
- 27.Frisdal E, Lesnik P, Olivier M, Robillard P, Chapman MJ, Huby T et al. Interleukin-6 protects human macrophages from cellular cholesterol accumulation and attenuates the proinflammatory response. J Biol Chem 2011;286(35):30926–30936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang R, Zhang Y, Xu L, Lin Y, Yang X, Bai L et al. Protein Inhibitor of Activated STAT3 Suppresses Oxidized LDL-induced Cell Responses during Atherosclerosis in Apolipoprotein E-deficient Mice. Sci Rep 2016;6:36790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010;13(1):133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation 1973;16:343. [DOI] [PubMed] [Google Scholar]
- 31.Yuan X, Ansari MJ, D’Addio F, Paez-Cortez J, Schmitt I, Donnarumma M et al. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci U S A 2009;106(26):10734–10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bagley J, Sawada T, Wu Y, Iacomini J. A critical role for interleukin 4 in activating alloreactive CD4 T cells. Nat Immunol 2000;1(3):257–261. [DOI] [PubMed] [Google Scholar]
- 33.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992;258(5081):468–471. [DOI] [PubMed] [Google Scholar]
- 34.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci U S A 1992;89(10):4471–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chong AS, Alegre M-L, Miller ML, Fairchild RL. Lessons and limits of mouse models. Cold Spring Harb Perspect Med 2013;3(12):a015495–a015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Win TS, Rehakova S, Negus MC, Saeb-Parsy K, Goddard M, Conlon TM et al. Donor CD4 T cells contribute to cardiac allograft vasculopathy by providing help for autoantibody production. Circ Heart Fail 2009;2(4):361–369. [DOI] [PubMed] [Google Scholar]
- 37.Fischbein MP, Ardehali A, Yun J, Schoenberger S, Laks H, Irie Y et al. CD40 signaling replaces CD4+ lymphocytes and its blocking prevents chronic rejection of heart transplants. J Immunol 2000;165(12):7316–7322. [DOI] [PubMed] [Google Scholar]
- 38.Hirota K, Ahlfors H, Duarte JH, Stockinger B. Regulation and function of innate and adaptive interleukin-17-producing cells. EMBO reports 2012;13(2):113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Current opinion in immunology 2007;19(3):281–286. [DOI] [PubMed] [Google Scholar]
- 40.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 2011;12(3):255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verbinnen B, Billiau AD, Vermeiren J, Galicia G, Bullens DM, Boon L et al. Contribution of regulatory T cells and effector T cell deletion in tolerance induction by costimulation blockade. J Immunol 2008;181(2):1034–1042. [DOI] [PubMed] [Google Scholar]
- 42.Vogel I, Verbinnen B, Maes W, Boon L, Van Gool SW, Ceuppens JL. Foxp3+ regulatory T cells are activated in spite of B7-CD28 and CD40-CD40L blockade. European journal of immunology 2013;43(4):1013–1023. [DOI] [PubMed] [Google Scholar]
- 43.Taylor PA, Noelle RJ, Blazar BR. CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med 2001;193(11):1311–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrer IR, Wagener ME, Song M, Kirk AD, Larsen CP, Ford ML. Antigen-specific induced Foxp3+ regulatory T cells are generated following CD40/CD154 blockade. Proceedings of the National Academy of Sciences 2011;108(51):20701–20706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. NatImmunol 2002;3(2):135. [DOI] [PubMed] [Google Scholar]
- 46.Shen H, Goldstein DR. IL-6 and TNF-alpha synergistically inhibit allograft acceptance. J Am Soc Nephrol 2009;20(5):1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diaz JA, Booth AJ, Lu G, Wood SC, Pinsky DJ, Bishop DK. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am J Transplant 2009;9(8):1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X, Boenisch O, Yeung M, Mfarrej B, Yang S, Turka LA et al. Critical role of proinflammatory cytokine IL-6 in allograft rejection and tolerance. Am J Transplant 2012;12(1):90–101. [DOI] [PubMed] [Google Scholar]
- 49.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 2014;211(1):121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.