

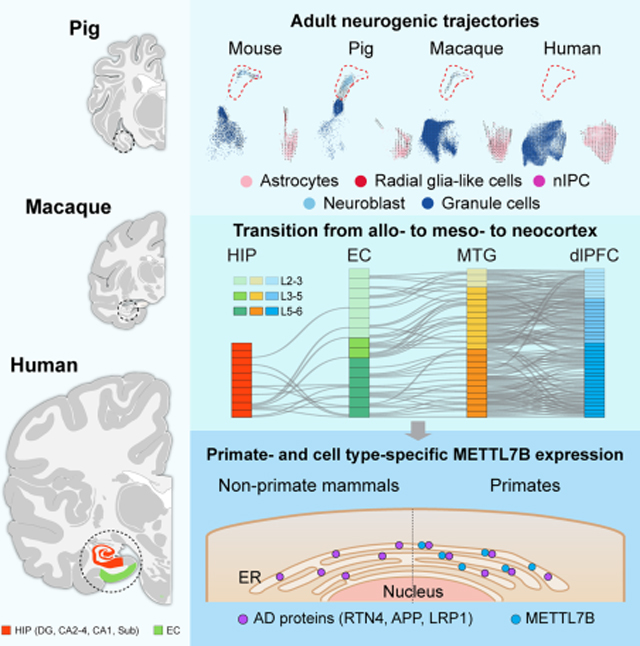
Summary
The hippocampal-entorhinal system supports cognitive functions, has lifelong neurogenic capabilities in many species, and is selectively vulnerable to Alzheimer’s disease. To investigate neurogenic potential and cellular diversity, we profiled single-nucleus transcriptomes in five hippocampal-entorhinal subregions in human, macaque, and pig. Integrated cross-species analysis revealed robust transcriptomic and histologic signatures of neurogenesis in adult mouse, pig and macaque, but not humans. Doublecortin (DCX), a widely accepted marker of newly generated granule cells, was detected in diverse human neurons, but it did not define immature neuron populations. To explore species differences in cellular diversity and implications for disease, we characterized subregion-specific transcriptomically-defined cell types and transitional changes from the three-layered archicortex to the six-layered neocortex. Notably, METTL7B defined subregion-specific excitatory neurons and astrocytes in primates, associated with endoplasmic reticulum and lipid droplet proteins, including Alzheimer’s disease-related proteins. Together this resource reveals cell-type- and species-specific properties shaping hippocampal-entorhinal neurogenesis and function.
Keywords: Single-cell, RNA-seq, hippocampus, entorhinal cortex, adult neurogenesis, neuroblast, immature neurons, doublecortin, evolution, neocortex, METTL7B, methylation, Alzheimer’s disease, aging
eTOC
Using snRNA-seq of the adult human, macaque and pig hippocampal-entorhinal system, Franjic et al. defined shared and divergent cell type features, like primate-specific expression of METTL7B in some excitatory neurons and astrocytes. They also identified robust transcriptomic and histologic signatures of neurogenesis in mouse, pig and macaque, but not human.
Graphical Abstract
Introduction
The hippocampal formation (HIP) and entorhinal cortex (EC) are critical components of a widespread neural network for memory and integration of space and time (Gloor, 1997; Andersen, 2007; Buzsaki and Moser, 2013) and are selectively vulnerable in Alzheimer disease (AD). Based on cytoarchitectonic, cellular, and circuit-related features, the hippocampal-entorhinal system can be divided into subregions that include the simpler three-layered allocortex of the dentate gyrus (DG), hippocampus proper (Cornu Ammonis, CA) and subiculum, and the more complex laminated periallocortex (mesocortex) of the pre- and parasubicular areas and the EC (Freund, 2002; Suzuki and Amaral, 2004; Klausberger and Somogyi, 2008). The molecular basis of cell-type diversity in these subregions and their homology with bordering neocortical cell types remains poorly understood (Kriegstein and Connors, 1986; Hoogland and Vermeulen-Vanderzee, 1989; Reiner, 1991; Ishizuka, 2001; Zeisel et al., 2015; Cembrowski et al., 2016b; Mercer and Thomson, 2017; Shepherd and Rowe, 2017). Laminar organization and cytoarchitecture changes gradually from allocortex to neocortex sectors across this region. Histological, physiological and connectional studies suggest that the allocortex is composed of glutamatergic excitatory projection neurons that resemble those in the deep layers of the mammalian neocortex (Kriegstein and Connors, 1986; Reiner, 1991; Ishizuka, 2001; Luzzati, 2015; Shepherd and Rowe, 2017).
Neurogenesis of granule cells in the adult DG has been documented across mammalian species (Patzke et al., 2015) and extensively studied in rodents, propelling a number of hypotheses about its functional role in cognitive processes and its potential for regenerative approaches. Adult neurogenesis also persists in non-human primates, though at substantially lower levels than in rodents (Gould et al., 1998; Kornack and Rakic, 1999; Ngwenya et al., 2006; Jabes et al., 2010; Kohler et al., 2011; Yuan et al., 2014). However, there is no consensus regarding the existence of significant neurogenesis in the adult human DG. Previous studies have provided evidence for the generation of granule cells in the adult and aged human DG through detection of cell proliferation (Eriksson et al., 1998; Spalding et al., 2013). Other studies have reported varied amounts of doublecortin (DCX; a widely adopted marker of neuroblasts and immature neurons) expressing cells in the adult human DG (Knoth et al., 2010; Boldrini et al., 2018; Moreno-Jimenez et al., 2019; Tobin et al., 2019). Similarly, bulk tissue RNA sequencing shows expression of DCX in the adult and aged human HIP, albeit dramatically lower than in the developing human or adult macaque HIP (Kang et al., 2011; Sousa et al., 2017; Zhu et al., 2018). Conversely, other studies have failed to identify neural progenitors or DCX-expressing neuroblasts after childhood in human DG samples (Dennis et al., 2016; Cipriani et al., 2018; Sorrells et al., 2018; Seki et al., 2019). Recently, single-cell RNA sequencing has been applied to characterize the process of DG neurogenesis at developmental and adult stages in mice, revealing the gene expression cascades along the granule cell lineage, from radial glia-like cells (RGLs) to neural intermediate progenitor cells (nIPCs), to neuroblast cells, to granule cells, (Hochgerner et al., 2018). Thus, this technique is well suited to bridge histologic and genetic analyses of cell lineage subtypes and possibly resolve the controversy surrounding human adult neurogenesis (Kempermann et al., 2018; Kuhn et al., 2018; Lee and Thuret, 2018; Paredes et al., 2018; Abbott and Nigussie, 2020).
Within the human hippocampal-entorhinal system, some cell types and circuits are selectively vulnerable to certain pathological processes including ischemia or Alzheimer disease’s pathology, and age-related neuronal loss (Schmidt-Kastner and Freund, 1991; Braak and Del Trecidi, 2015). Given this selectivity, a more detailed molecular and cellular profiling of this system will aid our understanding of the human brain and neuropsychiatric disease.
To investigate key cell type- and species-specific differences in gene expression, neurogenic capacity, and variable disease susceptibility, we performed high-coverage single-nucleus RNA sequencing (snRNA-seq) on five anatomically-defined subregions of the hippocampal-entorhinal system from adult human donors (DG, CA2–4, CA1, Sub and EC). We also profiled DG cell populations from adult rhesus macaques (Macaca mulatta), and all hippocampal fields from young-adult pigs (Sus scrofa). Similar to recent snRNA-seq studies of the postmortem adult human hippocampal-entorhinal system (Habib et al., 2017; Grubman et al., 2019; Ayhan et al., 2021; Leng et al., 2021), these findings identify a highly diverse cell populations with clear regional distinctions. Yet additionally, we investigated underlying species-level distinctions within this region by cross-species integrative comparisons with parallel samples from young-adult mice (Mus musculus) (Hochgerner et al., 2018), supporting higher-order inferences relative to evolution, behavior and disease. This resource is interactively accessible at http://resources.sestanlab.org/hippocampus.
Results
Transcriptomic diversity of adult human hippocampal and entorhinal cells
We used snRNA-seq to profile five subregions (DG, CA2–4, CA1, Sub and EC) microdissected from fresh frozen postmortem brains of clinically unremarkable adult human donors (age: 53 ± 5 years; postmortem interval (PMI): 15.6 ± 2.0 hours of mostly cold ischemic time; 2 females and 4 males, Fig. 1A–D; Table S1). Unbiased isolation of nuclei using our protocol (Li et al., 2018; Zhu et al., 2018) followed by snRNA barcoding, cDNA sequencing and quality filtering yielded 219,058 high-quality single-nucleus profiles (Fig. 1A–D, S1A–C). Expression patterns of major cell type markers identified 69,461 neurons (35.7 ± 4.1%) and 149,597 (64.3 ± 4.1%) non-neuronal cells (NNC) (Fig. 1B–D, S1D). Within neurons, there were 55,888 (77.8 ± 2.8% of all neurons) glutamatergic excitatory neurons (ExN) and 13,542 (22.1 ± 2.8%) GABAergic inhibitory neurons (InN), proportions that varied substantially between regions (Fig. S1C).
Figure 1. Cell type diversity in the human hippocampal-entorhinal system revealed by snRNA-seq.
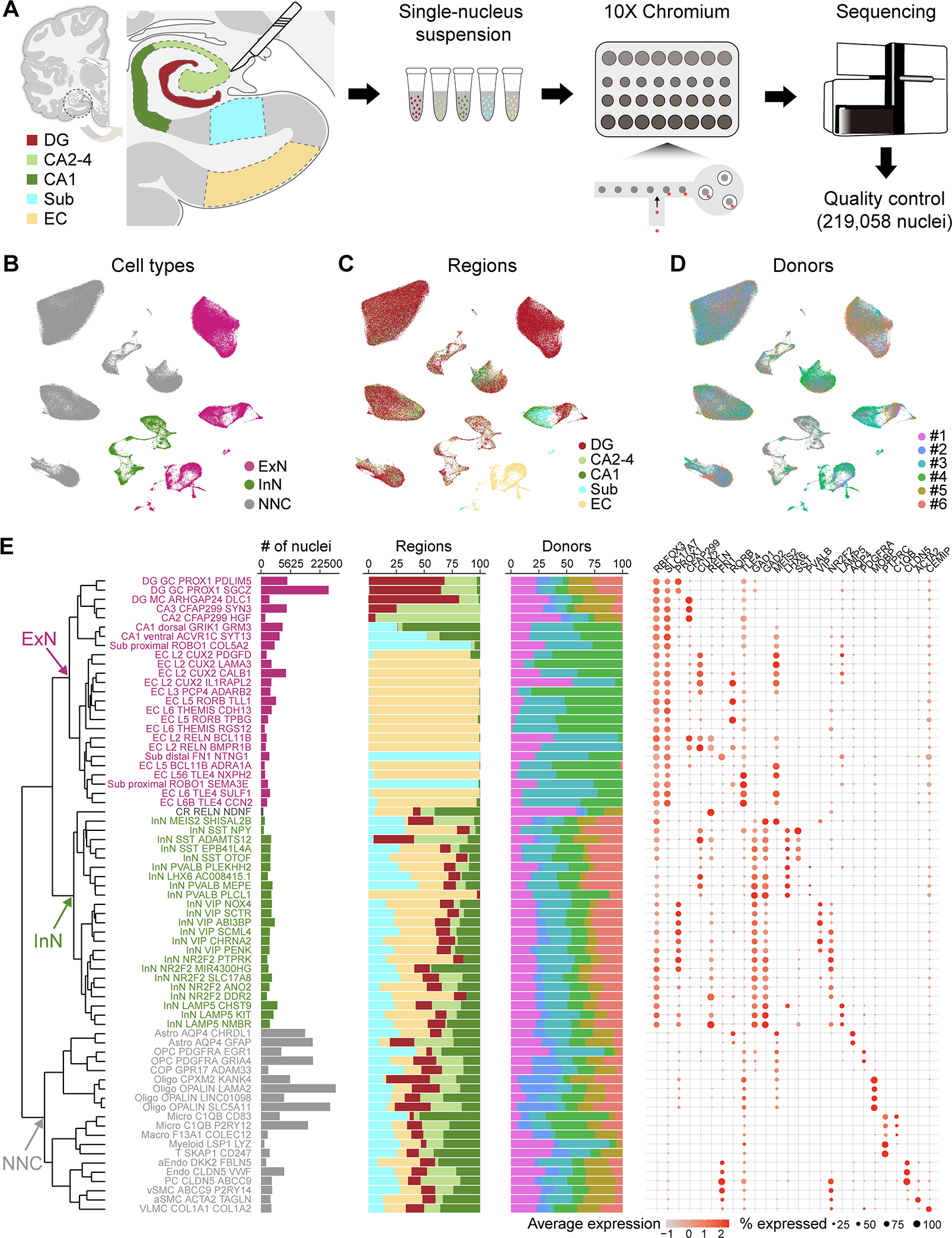
A, Schematic of the analytic workflow. B-D, UMAP visualization of all nuclei, colored by major cell types (B), subregions (C), and donors (D). E, Dendrogram depicting the hierarchical taxonomy across all cell subtypes. Bar plots show the number of nuclei, relative subregional and donor contributions, with coloring scheme conforming to panel B-D. Dot plot shows the expression of marker genes. GC, granule cell; MC, mossy cell; Astro, astrocyte; OPC, oligodendrocyte precursor cell; COP, committed OPC; Oligo, oligodendrocyte; Micro, microglia; Macro, macrophage; Myeloid, myeloid cell; T, T cell; aEndo, arterial endothelial cell; PC, pericyte; vSMC, venous smooth muscle cell; aSMC, arterial smooth muscle cell; VLMC, vascular and leptomeningeal cell. See also Figure S1.
Iterative clustering identified 69 transcriptomically distinct cell clusters across all donors (Fig. 1B–D) that were organized into a dendrogramatic taxonomy reflecting their unique gene expression patterns. This revealed 25 ExN subtypes, 23 InN subtypes, a Cajal Retzius-like cell type, and 20 NNC subtypes (Fig. 1E, S1E–F), which were all broadly mapped to those previously defined in adult human hippocampus (Fig. S1G–H), where subregions were not selectively dissected (Habib et al., 2017; Ayhan et al., 2021). Within ExN subtypes, apart from the expected transcriptomic diversity following the cytoarchitectonic organization of the HIP and EC (Fig. 1E), we found marked heterogeneity in the molecular profiles within regions, indicating a finer molecular subdivision than the apparent cytoarchitecture. For example, in the DG, we found two distinct subclusters of PROX1-expressing granule cells, characterized by the expression of PDLIM5 and SGCZ, respectively (Fig. S1F). Similar population diversity was identified in CA1, CA2–4 and Sub (Fig. 1E, S1F), matching those previously described (Nielsen et al., 2010; Slomianka et al., 2011; Cipriani et al., 2016; Cembrowski et al., 2018). Within the EC, ExN exhibited much more diversity than the expected division by laminar features, and we identified neuron subtypes marked by layer 2/3 markers (CUX2, RELN) or deep layer markers (TLE4, ADRA1A, and/or THEMIS).
In contrast to ExN, InN and NNC types were distributed more uniformly, without significant transcriptomic diversity across regions (Fig. 1E). InN subtypes were distinguished by major markers (SST, PVALB, VIP and LAMP5) and NNC populations included astrocytes, oligodendrocyte precursor cells, oligodendrocytes, microglia and vasculature cells.
These data therefore present high resolution cell populations extending previous findings outlining the functional cell diversity in the human hippocampal-entorhinal system (Freund, 2002; Suzuki and Amaral, 2004; Klausberger and Somogyi, 2008), which now enables detailed investigation of fundamental features of this system.
snRNA-seq reveals a neurogenic trajectory in macaque, pig and mouse DG that is virtually absent in humans
To ensure a robust analytic power to detect transcriptomic signatures of adult neurogenesis (Table S2), we collected 139,187 DG nuclei from 6 adult human donors (Fig. S2A; Table S1; STAR Methods), each with 1–8 technical replicates. We also generated snRNA-seq data from adult rhesus macaque as a reference for neurogenesis in the primate line, and from young adult pig (Table S1), as control for PMI effect, as it was analyzed at 30 minutes, 1 hour and 7 hours of warm ischemic PMI. To take full advantage of the inter-species information, we integrated our human, macaque and pig DG data with published single cell RNA-seq data from young adult mouse DG (Hochgerner et al., 2018), an established animal model with robust adult neurogenesis, to screen for neurogenic cells as well as DCX expression in these species. The integration revealed a broad cell type matching across species and showed that RGL cells clustered with astrocytes, as they share expression of multiple astrocyte markers (Fig. 2A, S2B–C) (Bonaguidi et al., 2011; Hochgerner et al., 2018; Arellano et al., 2021). Homologs of mouse nIPCs and neuroblasts were robustly observed in pig and macaque, but not in human, even though we profiled 25 times more cells in human and we were able to detect DCX transcripts in all human DG samples (Fig. 2A, S2A).
Figure 2. Cross-species analysis of transcriptomic signatures of adult neurogenic trajectories.

A-B, Seurat integration of all DG cells (A) or only astrocytes and the granule cell lineage (B) across species. In B, arrows indicate the direction and speed (arrow length) of the RNA velocity. C, Expression of cluster markers across species. The categories “progenitor” and “neuroblast” were manually annotated (Hochgerner et al., 2018; Berg et al., 2019). Middle: Dot plot depicting the expression of the markers with dots colored by species. Bottom: Marker expression in the 20 human cells residing in the nIPC and neuroblast domain as well as the randomly sampled human granule cells. The first two rows highlighted by arrows represent the two putative human neurogenic cells. RGL, radial-glia like cells; nIPC, neural intermediate progenitor cells; NB, neuroblasts. GC, granule cells; MC, mossy cells; CA2–4, CA2–4 ExN; CA1 Sub, CA1 and Sub ExN. See also Figure S2 and Tables S2.
Reintegration with only the granule cell lineage and astrocytes confirmed the alignment and unveiled a clearer trajectory from nIPC to neuroblast to granule cell in mouse, pig and macaque, but not in human (Fig. 2B). Those variations among species were recapitulated via RNA velocity (Fig. 2B), which infers cell differentiation lineage by leveraging splicing dynamics (Bergen et al., 2020). To more rigorously identify human cells matching homolog nIPCs and neuroblasts, we used Seurat and Harmony (Korsunsky et al., 2019; Stuart et al., 2019) to perform pairwise integration between human and each of other species. Summarizing all the integrations, we found a total of 20 cells located in the vicinity of homolog progenitors and neuroblast cells in human (Fig. 2C, S2D).
However, allocation in the domain of homolog progenitors and neuroblasts per se does not imply neurogenic identity, as multiple factors such as low cell quality, interspecies differences and method-specific bias may contribute to the misplacement. To assess the identity of these cells and the change of signatures delineating granule cell differentiation across species, we obtained the subtype marker genes in mouse, pig and macaque. Expression patterns of these markers confirmed the alignment of homolog progenitors and neuroblasts across species (Fig. S2E–F) and identified markers shared in at least two species. These include multiple cell cycle genes in nIPC (e.g., TOP2A, CENPF and MKI67) and some common neuroblast markers (DCX and CALB2) in neuroblast (red gene labels, Fig. 2C, S2G). The shared neuroblast markers also included ST8SIA2 (red gene labels, Fig. S2G), a gene encoding a polysialyltransferase that polysialylates NCAM to produce PSA-NCAM (Angata et al., 2002), which is also considered as a marker of neuroblasts and immature neurons (Seki et al., 2019). However, some shared markers showed distinct patterns in human with very high expression in mature granule cells (Fig. S2G). Moreover, multiple markers exhibited distinct patterns across species (e.g., NEUROD4 and DUSP14, blue gene labels, Fig. 2C), suggesting that transcriptomic neurogenic signatures are not fully preserved across species, and defining cell identity should be done with caution.
Next, we sought to screen for the presence of these transcriptomically-defined markers along with other traditional progenitor and neuroblast markers in the adult human DG (Hochgerner et al., 2018; Berg et al., 2019). Among the 20 human cells clustered with homolog progenitors and neuroblast cells in the UMAP space, we observed extremely low expression for most of the markers and comparable expression to background granule cells for the rest (Fig. 2C). Notably, there is only one cell showing neuroblast features characterized by the co-expression of PROX1, DCX, CALB2, NEUROD6 and DPYSL3 (blue arrow in Fig. 2C). We also recognized one putative nIPC in human co-expressing PROX1 and several nIPC markers (cell indicated by red arrow in Fig. 2C), including TOP2A, CENPF and MKI67. Unbiasedly co-expression searching could only reveal one additional PROX1-expressing granule cell co-expressing these neuroblast markers (DCX, CALB2 and DPYSL3) in the human (Fig. S2G–H). Still, this gene profile was not specific enough to define putative neuroblasts, as high co-expression of these 3 putative neuroblast markers and PROX1 were observed in InN, especially in human.
As neuroblasts at later stages of maturation may possess a combination of progenitor and mature granule cell signatures reminiscent of the doublet features, we further incorporated the previously removed doublets into the human-mouse integrative analysis. Only a few human cells aligned with the mouse neuroblast subtype and their expression profiles were suggestive of glia, mature neuron or neuron/glia doublets, but not neurogenic cells (Fig. S2J). Considering that human prenatal and adult neuroblast cells may share transcriptomic similarity, as observed in mouse (Hochgerner et al., 2018), we further projected adult human DG data to fetal human DG data (Zhong et al., 2020). Once more, no clear granule cell trajectory or cells expressing nIPC or neuroblast markers were detected in adult humans (Fig. S2K–L). Taken together, our exhaustive integrative cross-species analysis identified clear and robust trajectories for adult neurogenesis in mouse, pig and macaque, but not in humans. We only identified one cell with transcriptomic profile consistent with neuronal progenitors and one cell with profile consistent with neuroblasts out of 139,187 all DG cells (0.0007% for each cell) and 32,067 granule cells (0.003% for each cell), a ratio substantially lower than previous estimations based on DCX protein expression and 14C incorporation analysis, which suggested a range of 28 to 1,218 neuroblasts for our sample size (see Table S2 for details).
DCX RNA is not a specific marker of neuroblasts or immature granule neurons
To further investigate the neurogenic potential, we profiled DCX RNA expression across species. Datasets from the four species were down-sampled to the same sequencing depth to have comparable metrics (STAR Methods). This revealed comparable magnitudes of DCX expression levels and similar expression patterns across species, but selective enrichment in mouse neuroblasts and, to a lesser extent, in pig and macaque neuroblasts (Fig. 3A). Importantly, DCX expression with at least one Unique Molecular Identifier (UMI) was found in granule cells across species with human showing the scarcest expression (Mouse – 3.21%, Pig – 14.88%, Rhesus – 3.08%, Human – 110/32067 or 0.34%) (Fig. 3A; Table S3). However, one UMI is a low baseline that could represent background and does not reliably confer cell identity (Fig. S1D). Accordingly, we compared DCX expression of at least 2 UMIs and found a similar pattern, still human showing the scarcest expression (Mouse – 0.51%, Pig – 1.97%, Rhesus – 0.2%, Human – 4/32067 or 0.01%, Table S3). Conversely, prominent DCX expression was detected in non- granule cells, especially in InN, where 7.28% expressed at least 2 DCX UMIs (Fig. 3A; Table S3), suggesting that low expression, rather than low detection, is the explanation for the scarce expression of DCX in human granule cells. Although human samples overall have a longer PMI (Table S1), this clear expression of DCX indicates that the long PMIs do not limit the detection of DCX transcripts. Nonetheless, we further evaluated the effect of PMI by performing snRNA-seq analysis in pig brains with PMI of 30 minutes, 1 hour and 7 hours. Pig brains were kept at room temperature (warm PMI), while human specimens used in our study spent typically less than 4 hours at room temperature (warm PMI) before they were refrigerated (cold PMI) to slow down tissue and cell deterioration until autopsy. The longer warm PMI in pig brains is likely to exacerbate the effect of the postmortem interval compared to cold preserved brains but the results showed comparable DCX expression and similar abundance of neural progenitors and neuroblasts in all three conditions, once again suggesting that PMI might not be a substantial factor influencing RNA preservation and/or detection (Fig. S3C). Similar to human, DCX expression outside the neuroblast/granule cell population was detected in all species analyzed, but it was more prominent in the pig and primates than in mice (Fig. 3B). These results suggest DCX expression per se is inadequate to define adult neurogenesis.
Figure 3. Hippocampal DCX expression across species.
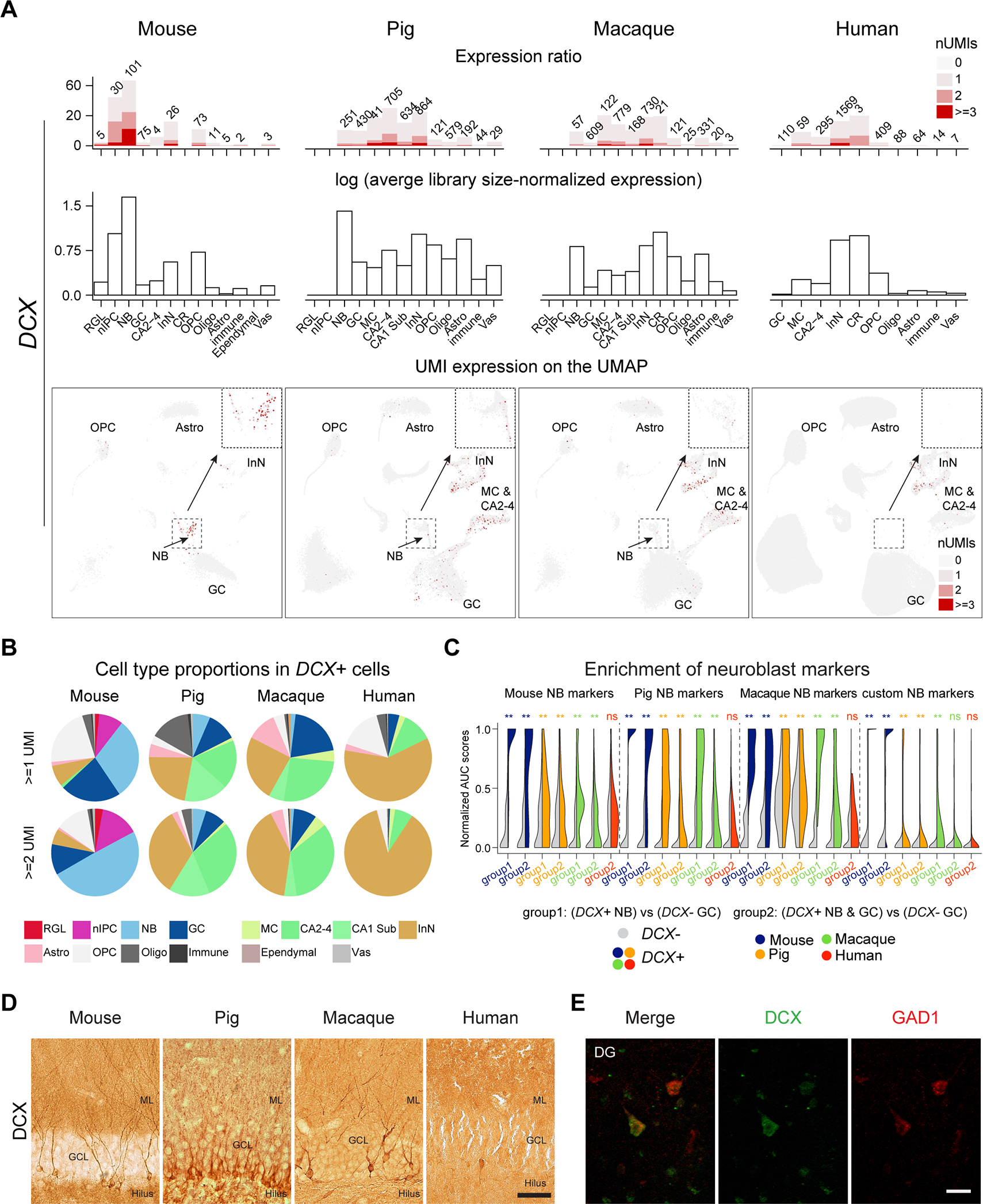
A, Top: The number (text label) and percentage (y axis) of cells expressing DCX. Middle: Average library size-normalized expression of DCX. Bottom: DCX expression on UMAP with insets highlighting the neuroblast domain. B, Cell type proportions of DCX-expressing cells across species. C, Enrichment of different set of neuroblast markers in DCX+ compared to DCX− cells. Significance was tested using one-tailed Wilcoxon rank sum test (**: p < 0.01, ns: not significant). D, Images of the mouse, pig, macaque and human DG immunolabeled against DCX. Scale bar represents 50 μm in mouse, pig and macaque and 75 μm in human. E, Colocalization of DCX and GAD1 in cells with InN morphology in the molecular layer of the human DG. Scale bar is 30 μm. GCL, granular cell layer; ML, molecular layer. Other abbreviations conform to Figure 2. See also Figure S3 and Table S3.
To further interrogate whether the 110 DCX-expressing human granule cells might represent neuroblasts, we tested if they were enriched in neuroblast markers compared to DCX-negative granule cells. Our results showed lack of enrichment in neuroblast markers (Fig. 3C), a pattern that persists in the original human data with higher depth prior to downsampling (Fig. S3B). This result was in stark contrast with mouse, pig and macaque, that showed conserved enrichment of neuroblasts markers in DCX-expressing granule cells, ratifying the robustness of the method to detect potential neuroblasts overriding species differences. This result further confirms the absence of detectable neurogenic trajectory in our human DG samples revealed by the integrative analysis, and suggests that mature granule cells express some degree of DCX in the adult human DG, similar to other mature neuronal populations.
We complemented these snRNA-seq analyses using immunohistochemistry with two different commonly used antibodies against DCX on young adult mouse and pig, and adult rhesus macaque and human medial temporal lobe. Mice, pig and macaque exhibited numerous DCX immunolabeled (DCX-IL) cells in the DG, with both immature and mature granule cell morphology (Fig. 3D), as previously reported (Guidi et al., 2011). In humans, we tested a cohort of 10 cases (Table S3) where the HIP, and EC or the amygdala were available. We screened the amygdala as an internal control for DCX detection, as it harbors a large population of cells strongly immunolabeled with both DCX antibodies in the paralaminar nucleus (Sorrells et al., 2019). To maximize detection, we used several protocols for antigen retrieval, including the one used in previous studies (Moreno-Jimenez et al., 2019; Flor-Garcia et al., 2020). However, we did not see significant differences between the protocols. With standard antigen retrieval citrate buffer, we could detect numerous reliably immunolabeled cells in the amygdala of all cases, plus occasional, scarcer DCX immunolabeled (DCX-IL) cells in the EC and perirhinal cortex, and rare DCX-IL cells in the Sub and CA fields of some cases (Table S3). While DCX-IL cells in the amygdala and EC showed strong labeling in the soma and processes, in the DG we only found some cells lightly immunolabeled by DCX located mostly in the molecular layer or in the SGZ and hilus, and occasionally, within the granule cell layer (Fig. S3D–G, M–Q). However, their morphology and localization were more consistent with GABAergic InN than with immature granule cells and, in fact, some of them were lightly labeled with GAD1, a marker of InNs (Fig. 3E and S3M–R). These results are consistent with our snRNA-seq analysis. Similar lightly labeled cells were found in other regions, even in pyramidal cells, which also suggests the possibility of background staining (Fig. S3D–G), though such labeling was not detected in controls lacking the primary antibody. Immunostaining against PSA-NCAM, a selective marker of neuroblasts and immature neurons in the DG of rodents (Seki, 2002) showed a completely different pattern of staining in the human, labeling numerous cells with InN morphology in the DG and hilar area (Fig. S3H), as previously shown (Mikkonen et al., 1998; Seki et al., 2019) that matched the predominant distribution outside the typical neurogenic lineage seen for DCX transcripts. We did not colocalize those markers, as anti-DCX antibodies require antigen retrieval, and anti-PSA-NCAM immunostaining does not tolerate the same treatment.
The possible effect of the PMI in the human samples did not preclude DCX detection, since we could detect DCX-IL cells in the amygdala and EC/perirhinal cortex in cases with up to 24 hours of mostly cold ischemic PMI. Additionally, we evaluated the effect of PMI in pig DG with 15 and 24 hours of cold ischemic PMI and in a macaque with a 16 hours cold ischemic PMI using immunohistochemistry (Fig. S3I–L; Table S3). In both cases there was a reduction in the number of DCX-IL cells, some of which exhibited varicose dendrites (Fig. 3D and S3I–L). However, these results indicate that long PMIs do not preclude detection of DCX-IL cells in the DG or adjacent cortex. Taken together, our integrated cross-species snRNA-seq analysis and DCX immunohistochemistry revealed clear and robust evidence for adult neurogenesis in our mouse, pig and macaque, but not in our human tissue samples.
Taxonomic relationship of neural cells across allo-, meso- and neo-cortex
The putative homology between neurons in the hippocampal-entorhinal system and neocortical neurons, and in particular the cytoarchitectonic and evolutionary transition between allo-, meso-, and neo-cortex, offers an opportunity to reveal organizational principles underlying the specialization and function of the cerebral cortex. Towards elucidating these principles, we compared cell profiles across hippocampal-entorhinal subregions and transcriptomically defined cell types within two human neocortical regions, the dorsolateral prefrontal cortex (dlPFC/DFC) (Li et al., 2018) and the middle temporal gyrus (MTG) (Hodge et al., 2019).
Aside from the marked heterogeneity we observed within each subfield of HIP (Fig. 1E), we also observed a clear distinction between ExN of the CA fields and Sub compared to those of EC (Fig.4A–B) as well as those of neocortical MTG and dlPFC. As expected from the laminar structure, we observed higher similarity between ExN of MTG and dlPFC, and to a lesser extent between ExN of the EC and those of MTG and dlPFC. (Fig. 4B). In total, we revealed 15 ExN subtypes with regional specificity (three in DG, two in CA2–4, two in CA1, two in Sub, five in EC, one in dlPFC; outlined in Fig. 4B).
Figure 4. Transcriptomic similarities and differences of hippocampal, entorhinal and neocortical cell types.
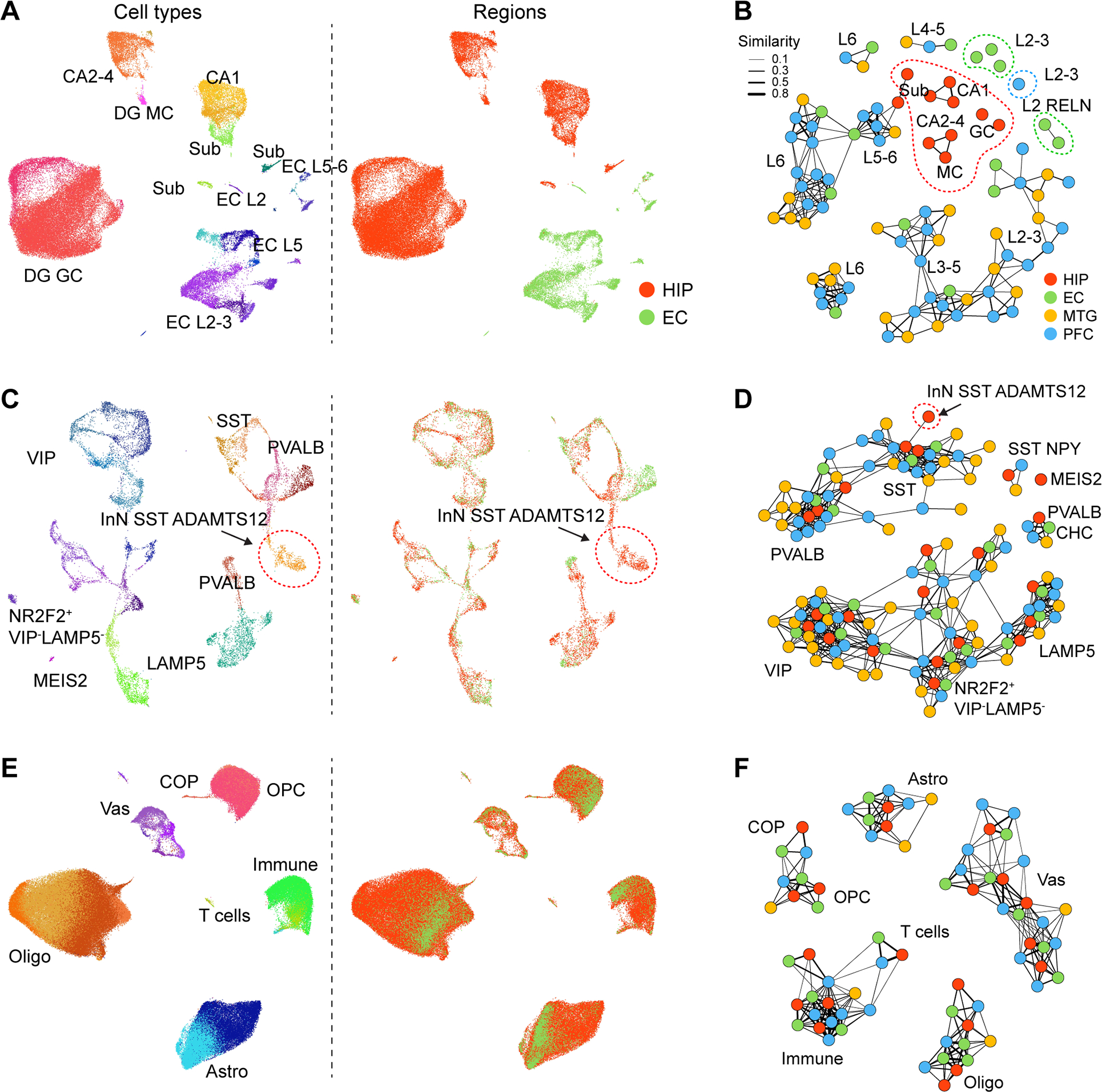
A, Left: UMAP showing all ExN nuclei colored by subtypes (left) or regions (right). B, Network demonstrating the extent of transcriptome similarities among ExN subtypes of HIP, EC, MTG (Hodge et al., 2019) and dlPFC (Li et al., 2018). Dots represent the subtypes within each brain region and the widths of lines represent the strength of similarity. Subtypes with regional-specificity were outlined in corresponding colors. C-F, As in panels A-B, for InN (C, D) and NNC (E, F). GC, granule cell; MC, mossy cell; CA2–4, CA2–4 ExN; CA1, CA1 ExN; Sub, Sub ExN; Astro, astrocyte; OPC, oligodendrocyte precursor cell; COP, committed OPC; Oligo, oligodendrocyte; Micro, microglia; Vas, vascular cells.
In particular, we found that deep-layer ExN subtypes in the neocortex were well-represented in the EC and to a lesser extent in the HIP, but upper-layer neuron subtypes were not well represented (Fig 5A–B, S4A). For example, we identified two EC subtypes characteristics of layer 2 RELN expression that, similar to a previous report (Witter et al., 2017), did not correspond closely to any ExN subtype detected in the neocortex (Fig. 4B). Consistent with this observation, all subtypes of HIP ExN showed higher expression of molecular markers for neocortical deep-layer ExN than for upper-layer ExN (Fig. S4A, S4B). Moreover, we observed lower expression of key molecular markers of intracerebral projection neurons in each of the HIP ExN subtypes as compared to other neocortical ExN populations (Fig. S4C), which likely reflects the restricted and largely ipsilateral telencephalic projections of the human hippocampal subfields to limbic areas (Cenquizca and Swanson, 2007). Nevertheless, there was one DG granule cell subtype transcriptomically resembling one upper layer ExN subtype in EC (Fig. 5A), which might recapitulate the similarities found between hippocampal formation and neocortex in mouse (Yao et al., 2021). We next identified several genes which underlie the molecular specificity of ExNs within the HIP, including CHRNA1, METTL7B and P2RX2. To gain insight into their potential roles in hippocampal development and maturation, we then examined their temporal expression and found mixed patterns of up and down regulation (Fig. 5C). This suggests the molecular coordination of hippocampal specification occurs at multiple time points using multiple processes.
Figure 5. Taxonomic relationships of cell types across allo-, meso- and neo-cortex.
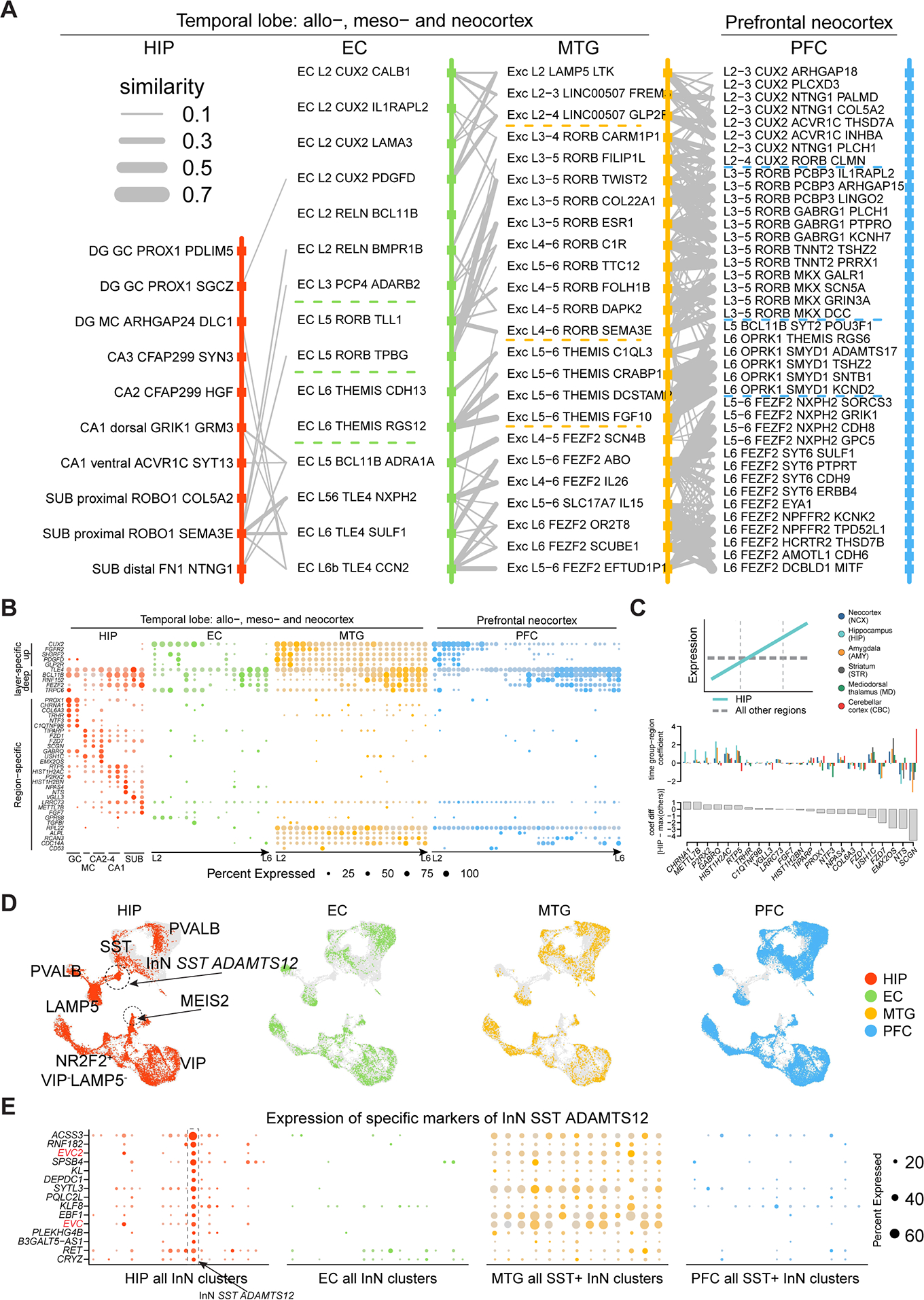
A, Transcriptomic relations across subtypes of pairwise regions organized according to layer distributions. Broad layer distinction was marked by dotted lines. B, Expression of neocortical upper-layer and deep-layer markers, as well as region-specific genes. C, Rank of the hippocampus-specific genes based on their temporal specificity in adult hippocampus using PsychENCODE data (Li et al., 2018). Top: Coefficients of time group-region with large positive values indicating upregulation along development (illustrated in the diagram). Bottom: Differences of the time group-region coefficients between HIP and the maximum of other regions. D, Integration of InN from 4 regions. Grey dots denote cells from other regions. E, Expression of the exclusive markers (rows) of the cluster ‘InN SST ADAMTS12’ across all InN subtypes (columns) in HIP and EC, and all SST+ InN subtypes (columns) in MTG and dlPFC. See also Figure S4.
In contrast to the observed patterns for ExN, InN did not exhibit an obvious transition between allo-, meso-, and neo-cortex, with just two HIP InN clusters (InN MEIS2 SHISAL2B and InN SST ADAMTS12) lacking a clear counterpart in EC, MTG, and dlPFC (Fig. 4C–D, 5D). The former matched to a white matter InN subtype (Frazer et al., 2017; Tasic et al., 2018) and the cell population variations could actually reflect tissue dissection differences. The other cell type, InN SST ADAMTS12, was marked by the expression of two EvC Ciliary Complex genes, EVC and EVC2 (Fig. 5E), involved in hippocampal ciliary sonic hedgehog signaling (Breunig et al., 2008; Rhee et al., 2016; Park et al., 2019). Lastly, NNC types constituted the most transcriptomically conserved populations across the allo-, meso-, and neocortical taxonomy, with a high similarity observed in each subtype across all regions (Fig. 4E–F). Notably, the putative interlaminar astrocytes (Astro AQP4 GFAP, layer 1) and protoplasmic astrocytes (Astro AQP4 CHRDL1, layer 2 to 6) (Oberheim et al., 2009; Hodge et al., 2019) were present in all the four regions (Fig. S4D), which points to the possibility that astrocyte lamination was not immediately a result of the six-layered neocortex in mammals but may be an ancient feature. Taken together, these findings indicate that ExNs exhibit the most prominent differences across allo-, meso-, and neo-cortex, including the increased prevalence of intracerebral projection neurons in the neocortex as compared to allocortex.
Primate, age and cell type-specific METTL7B expression
Among the genes specific to hippocampal ExN in the transition from allo- to neo-cortex, we identified CHRNA1 and METTL7B as two genes showing temporal specificity in adult hippocampus compared to other brain areas along development (Fig. 5C). METTL7B has been described to be predominantly expressed in enzymatically- and metabolically-active cells in the liver (Uhlen et al., 2015) and has not been studied in the vertebrate brain, so we decided to further investigate its possible role in hippocampus biology. METTL7B encodes a membrane protein associated with endoplasmic reticulum (ER) and lipid droplets (LD), and, by amino acid sequence homology, is predicted to belong to the protein methyltransferase superfamily (Turro et al., 2006; Thomas et al., 2013).
We mapped the cell type expression of METTL7B across all the species analyzed, and found METTL7B only expressed in human and macaque, but not in pig or mouse (Fig. 6A). These results were confirmed with bulk tissue RNA-seq, quantitative PCR, western blot and also using a lacZ reporter in the mouse (Fig. 6B–F, S5A). In humans, we observed the highest expression in ExN especially in the DG, followed by CA2–4 and then Sub and moderate expression in astrocytes (Fig. 6A). The same pattern of expression was found in macaque, but with higher expression in astrocytes. Immunolabeling of human and macaque hippocampal tissue confirmed these findings (Fig. 6G, S5B). Given that there was trace expression in human MTG (Fig. 5B), we surveyed 11 areas of the human neocortex and found high levels in the large pyramidal neurons of layer 5B (Fig. S5C–D), such as Betz and Meynert cells in M1C and V1C, respectively. Similar staining patterns were observed in macaque, with very little expression of METTL7B in cortico-cortical pyramidal neurons of neocortical and entorhinal layer 5A and upper layers (L2–4). Using immuno-electron microscopy, we confirmed and extended beyond previous reports, showing that METTL7B is localized to the ER and LD in macaque and human hippocampal neurons and astrocytes (Fig. 6H–I).
Figure 6. METTL7B defines subregion-specific excitatory neurons and astrocytes in primates.
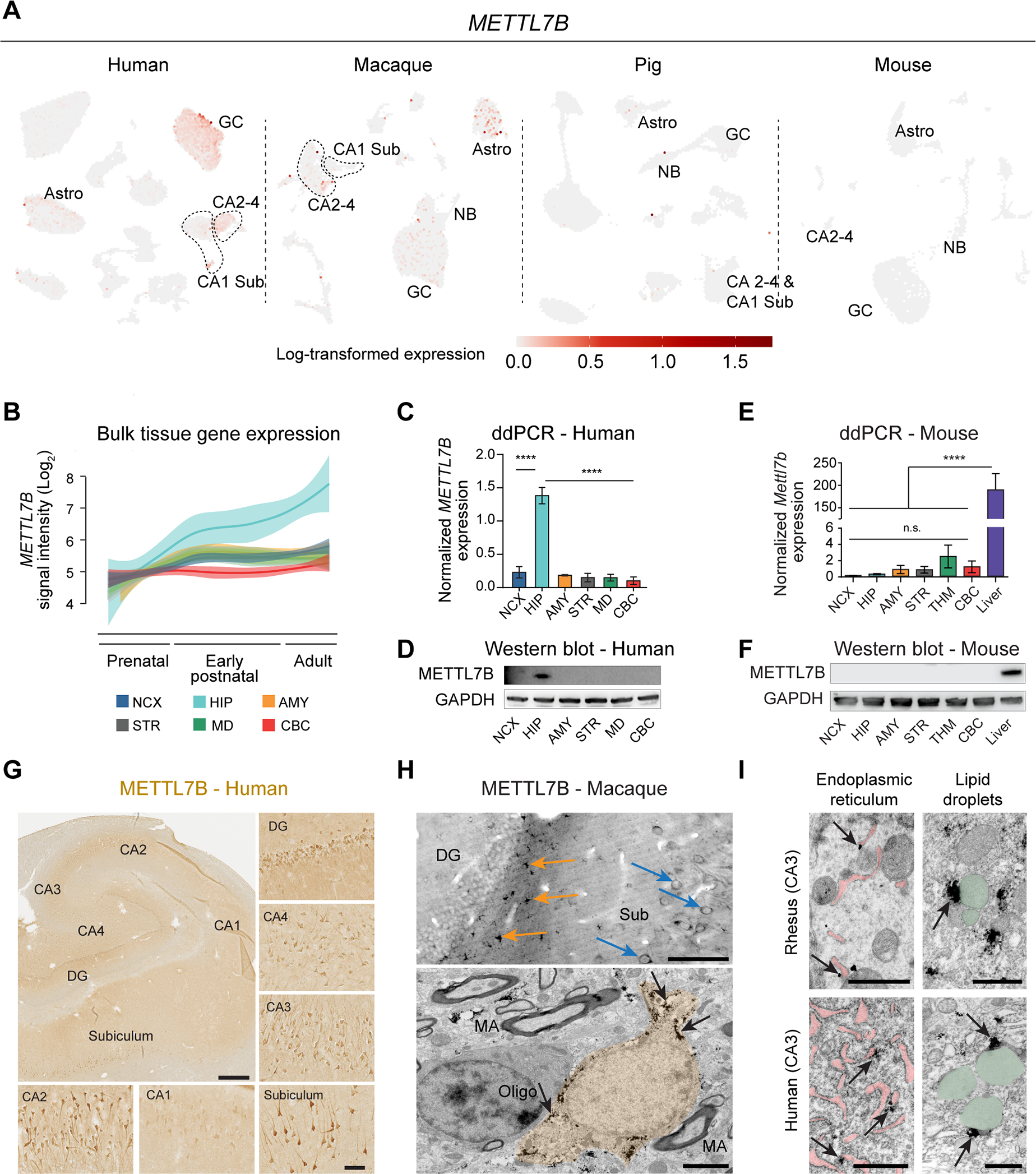
A, METTL7B expression in adult human HIP-EC, macaque DG, pig HIP and mouse DG. B, Expression of METTL7B showing temporal specificity in adult human hippocampus (Kang et al., 2011). C-D, Droplet digital PCR and immunoblot validation in six regions of adult human brain. One-way ANOVA with post-hoc Dunnett’s adjustment (****P<0.0001), N=3 per group. E-F, Same as (C) and (D) using mouse tissues, including liver as a positive control. G, METTL7B immunostaining of adult human hippocampus. Scale bars = 1 mm; insets = 100 μm; immunofluorescence = 10 μm. H, Upper panel: Numerous METTL7B Immunopositive astrocytes (orange arrows) and neurons (blue arrows). Bottom panel: Immunoelectron microscopy of astrocytes (orange; pointed with arrows). Scale bar is 100 μm (upper) and 2 μm (bottom). MA, myelinated axon. I, Immuno-electron microscopy CA3 hippocampal pyramidal neurons in rhesus macaque and human. Notice METTL7B labeling (arrows) on the outer surface of ER cisterns (pink) and in contact with LDs (green). Scale bar is 1μm for each panel. See also Figure S5.
This preferential expression of METTL7B in human and macaque prompted us to broaden our analysis and include in our study another primate, analyzing METTL7B in 16 homologous brain regions in human, chimpanzee and rhesus macaque (Sousa et al., 2017; Zhu et al., 2018). METTL7B expression in the chimpanzee brain is not distinct to humans, while it was more broadly upregulated throughout the cerebrum in macaque brain (Fig. S5E), possibly attributed to the elevated expression in astrocytes (Fig. 6A, S5B). Using published datasets (Cardoso-Moreira et al., 2019), we found METTL7B expression was enriched in the human, chimpanzee, and macaque cerebrum, but not in the cerebrum of mouse, rat, rabbit, and opossum (Fig. S5F), suggesting that the expression, and biological consequences thereof, are not conserved across mammals, and likely are primate-specific.
METTL7B interacts with proteins associated with endoplasmic reticulum, lipid droplet and Alzheimer’s disease
To gain more insights into the possible function of METTL7B in the primate hippocampus, we sought to identify METTL7B-interacting proteins by performing unbiased proteomic analysis with two different affinity-based approaches: HaloTag that has scarce non-specific binding (Hook, 2014) and BioID that is able to capture weak or transient interactions (Roux et al., 2012) (Fig. S6A–B, G–H). Using Significance Analysis of INTeractome (SAINT) (Choi et al., 2011), we identified 275 METTL7B interactors in HaloTag and 1804 interactors in BioID (Fig. S6D, J; Table S5; STAR Methods). Notably, both methods showed significant enrichment in ER- and LD-associated proteins (Fig. S6E, K), also confirmed by co-immunofluorescence (Fig. S6C, I). KEGG pathway enrichment analysis revealed potential relevance to protein processing in the ER, oxidative phosphorylation, endocytosis and neurodegenerative diseases including AD (Fig. S6F, L).
Intersecting the lists of METTL7B interacting proteins from both strategies, we found 110 high-confidence proteins, with the most enriched gene ontology term being protein processing in ER (Fig. 7A–B). We observed that many of those high confidence proteins overlapped with the KEGG AD Pathway (Fig. 7C, S6F), including amyloid precursor protein (APP), inhibition of γ-secretase (RTN3 and RTN4/NOGO), and amyloid binding (NAE1, LRP1, APBB1). We confirmed using our snRNA-seq dataset that many of these genes were extensively co-expressed with METTL7B in several hippocampal populations (Fig. S7A) and subsequent immunoblotting confirmed that candidate proteins RTN4, APP, and LRP1 were specific to METTL7B sample eluates. RTN3 was not detected in any of the samples, possibly due to low pull-down amounts (Fig. 7D–E). Additionally, using multiple independent methods including bulk tissue RNA sequencing of multiple brain regions, snRNA-seq of dlPFC (Mathys et al., 2019) and MTG (STAR Methods), and immunohistochemistry (Fig. S7B–D), we found selective enrichment of METTL7B-expressing astrocytes in AD brains compared to control brains, suggesting that METTL7B expression may participate in the glial response to the neuronal damage as AD progresses.
Figure 7. METTL7B-interacting proteins are enriched in the endoplasmic reticulum and lipid droplets.
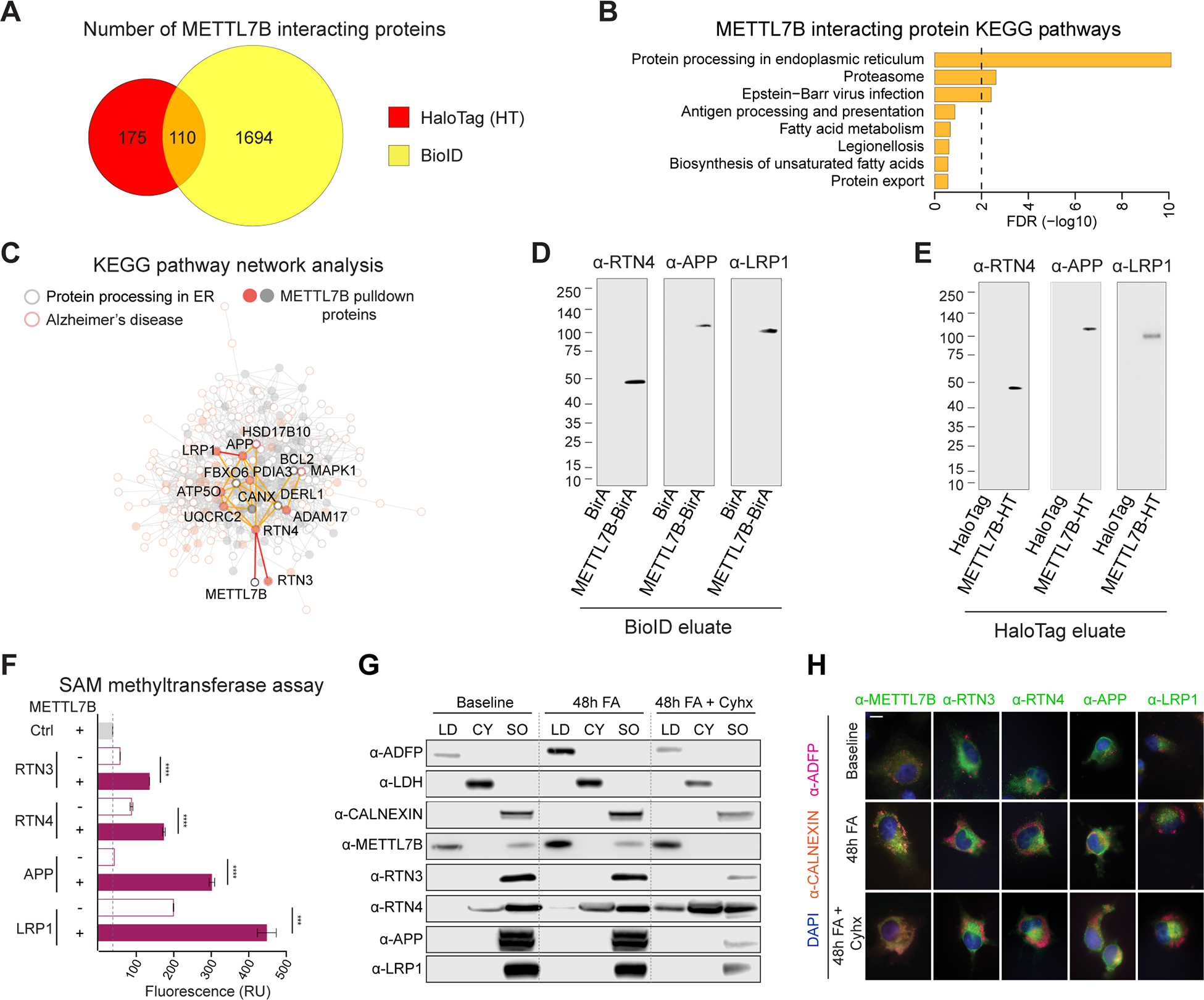
A, Venn diagram of high-confidence METTL7B interacting proteins revealed by HaloTag and BioID. B, KEGG enrichment of METTL7B interacting proteins from the intersection of HaloTag and BioID. C, Interaction network with proteins in KEGG Protein Processing in the ER pathway (grey) and Alzheimer’s disease pathway (orange). METTL7B interactors are shown as filled circles. D-E, Immunoblot confirmation of top interacting candidates. The molecular weight of the RTN4-immunoreactive band is consistent with a known proteolytic fragment of RTN4A or RTN4B (Kim et al., 2003; Sekine et al., 2020). F, SAM methyltransferase activity assay showing an increased reactivity in the presence of METTL7B. P-values calculated by unpaired two-tailed Student’s t test, N=3. G-H, Immunoanalysis of METTL7B translocation. Increased fatty acid (FA) load leads to a shift of METTL7B from ER to lipid droplets (LDs), while high confidence interactors remain unaffected. Blocking translation of new proteins with cycloheximide (Cyhx) suggests a complete shift of METTL7B. Scale bar = 10 μm. CY = cytosol; SO = sedimented organelle (containing the ER). All data are mean ± SEM. ****P< 0.0001, ***P< 0.001. See also Figures S6, S7, and Tables S4.
To determine whether the annotated methyltransferase domain of METTL7B exhibits methyltransferase activity, we incubated purified recombinant proteins (RTN4, APP, LRP1 and RTN3) with recombinant METTL7B in a continuous enzyme-coupled S-adenosylmethionine (SAM) methyltransferase assay. All four assayed samples produced a significant increase in signal compared to candidate proteins incubated alone (Fig. 7F), suggesting that METTL7B uses SAM as a methyl donor, and that METTL7B has enzymatic activity. We further showed that this METTL7B-mediated methylation could be limited in conditions with high levels of lipids, as METTL7B was translocated from ER to LD in this condition but these METTL7B interacting proteins remained in ER (Fig. 7G–H). Taken together, our cross-species transcriptomic analysis suggests a region and cell type-specific protein methylation mechanism that seems to be restricted to primates.
Discussion
We report an extensive single-cell transcriptomic analysis of several anatomically-defined cell populations in the adult human, macaque and pig hippocampal-entorhinal system. Our findings reveal fundamental species differences in adult hippocampal neurogenesis and delineate the molecular diversity of the cytoarchitectural transition from allo- to neocortex. These results also outline genes that are selectively enriched in certain species and cell types that may have a role in the specific biology and/or pathology of the hippocampal-entorhinal system
Unlike recent studies which mostly rely on one or two key markers (e.g., progenitors - nestin; neuroblasts and immature granule cells - DCX) (Boldrini et al., 2018; Moreno-Jimenez et al., 2019; Tobin et al., 2019), single cell RNA-seq studies are much more robust, since they leverage combinatorial gene expression profiles to identify cell populations more robustly (Hochgerner et al., 2018). This approach also allows for cross species analysis amplifying rare signals within a single species that may be masked when analyzed separately. Our cross-species analysis allowed identification of the neurogenic lineage in mouse, pig and macaque, that was virtually absent in the human. We only detected one cell with the transcriptomic profile characteristic of nIPC and one with putative neuroblast profile out of 32067 granule cells (0.003%) in our adult human DG samples, a proportion considerably lower than the expected 0.09–3.8% neuroblasts according to previous DCX immunostaining or 14C incorporation studies of the adult human HIP (see Table S2 for data and relevant studies).
The same analytic strategy detected much higher proportions of neuroblasts in the other species analyzed (mouse - 6.6%, pig - 55.6%, macaque - 2.0%) (Fig. 2B; Table S3). These proportions were higher than those previously estimated based on progenitor proliferation and identification of neuroblasts markers such as DCX (Table S2), suggesting that more studies are needed to fine tune the detection of these neurogenic populations. However, this apparently lax detection protocol confirms that our parameters are unlikely to have missed any appreciable neuroblast populations amongst the large pool of human DG granule cells surveyed, even if they might exhibit an ambiguous profile.
Alternative confounding of our cross-species integrative analysis from possible human-specific transcriptomic changes was ruled-out when human UMAP layouts did not include any clustering of neurogenic cells adjacent to the mature granule cell cluster. Likewise, the possibility that human neuroblasts exist in our samples, but their transcriptomic profile differs from other species and blends with related cell populations, is lessened by findings that all neurogenic lineages preceding mature granule cells were absent in human DG samples (Table S2–3).
We also extended our findings to existing snRNA-seq data of adult human HIP. We reappraised the identity of a recently reported neural progenitor cluster (Ayhan et al., 2021) marked by LPAR1, a gene reported to mark mouse DG neural progenitors (Walker et al., 2016; Hochgerner et al., 2018). Our analyses indicated that this cluster actually represented doublets formed by oligodendrocytes and granule cells (Fig. S3S). In addition, reanalysis of the pioneer HIP data (Habib et al., 2017) by Sorrells and coworkers (Sorrells et al., 2021) showed that the cell cluster labeled as neural stem cells was actually characteristic of ependymal cells.
Analysis of DCX transcripts in all species analyzed showed expression in mature neurons, mostly in InN, and in glial cells, indicating that DCX expression is not exclusive of DG neuroblasts (Fig. 3A–B). This pattern is in agreement with the reanalysis of Habib et al. 2017 data (Sorrells et al., 2021). Taken together, all transcriptomic analyses performed so far suggest the lack of neurogenic cell populations in adult human DG.
At the protein level, DCX was, with a few exceptions (Fig. S3L), present exclusively in the DG cells resembling neuroblasts and immature granule cells in all non-human species analyzed. Also, cells with immature morphology could be detected in other areas such as the EC of the macaque or the pyriform cortex of the mouse as previously described (Gomez-Climent et al., 2008; Zhang et al., 2009). In humans, there is intense controversy regarding DCX immunostaining in the human DG, with some reports showing negative results (Dennis et al., 2016; Cipriani et al., 2018; Sorrells et al., 2018; Sorrells et al., 2021) and others describing DCX-IL cells (Knoth et al., 2010; Epp et al., 2013; Boldrini et al., 2018; Le Maitre et al., 2018; Moreno-Jimenez et al., 2019; Tobin et al., 2019; Moreno-Jimenez et al., 2021). We detected clear DCX-IL cells in the amygdala and occasionally in the EC, but we could not find DCX-IL cells resembling neuroblasts in the DG in the same tissue sections. These inconsistencies in detecting DCX-IL cells in adult human DG cannot be fully attributed to postmortem denaturation and degradation of DCX protein, as DCX-IL cells were clearly detected in samples with prolonged PMIs (Fig. S3D–E and S3I–L). Moreno-Jimenez et al., 2019 reported an intensive protocol for antigen retrieval as a necessary step to label DCX cells in the human DG. However, they reported no positive cells in the EC, a relatively common finding in our study (Fig. S3D) and others (Sorrells et al., 2021) using conventional antigen retrieval. Since our analysis did not reveal neuroblasts both at the RNA or protein level (using diverse antigen-retrieval methods), the question remains about what those previously reported cells could be. Apart from underappreciated non-specific and off-target effects (Sorrells et al., 2021), those studies could label mature granule cells and InN that might contain low levels of DCX protein that was detected specially after a multi-step antigen retrieval. In support of this hypothesis is the fact that the faintly immnolabeled cells we detected, mostly in the vicinity of granule cell layer, exhibited the morphology of mature InN and co-labeled with antibodies against GAD1, a marker of InN (Fig. 3E, S3M–Q). This faint staining is far from the strong staining and well-defined morphology of soma and dendrites revealed in the EC and in the amygdala (Fig. S3D–E) and is similar to the light DCX immunostaining reported previously (Seki et al., 2019). Thus, our conclusion is that DCX protein might be expressed at very low levels in InN or in some mature granule cells that can be lightly immunolabeled under normal antigen retrieval, but that can show more intense and widespread staining under more elaborated tissue treatments and stringent conditions of antigen retrieval. In fact, Fig. 2I from Moreno-Jimenez et al., 2019 showed that around 75% of the DCX-IL cells were colocalized with NeuN (RBFOX3, 75%), a marker of mature granule cells, and 91% of the DCX-IL cells were also positive for Prospero homeobox1 (PROX1), a transcription factor expressed by granule cells that is also expressed by InN generated in the caudal ganglionic eminence (Ma et al., 2013; Laclef and Metin, 2018), supporting the possibility that most DCX-IL cells might actually represent mature granule cells or InN.
Although the PMI in human was longer than other species analyzed, human brains were kept at 4°C for most of the PMI period, while the pigs used as controls for PMI were kept at room temperature. This warm PMI will likely exacerbate the postmortem effects, but those conditions were not an obstacle to detect the neurogenic pathway in this species. It could be argued that the neurogenic pathway in the human DG is not detected because our snRNA-seq strategy might preferentially exclude neurogenic cells in human. However, it seems extremely unlikely that it will affect all cell types in the neurogenic lineage, from progenitors to neuroblasts, and only in the human. Overall, the most parsimonious interpretation of the combined results from our RNA transcript analysis and the DCX protein study is that, contrary to the other mammals analyzed, ongoing baseline neurogenesis does not occur, or is extremely rare in the adult human DG.
Similar species-related and cell-specific transcriptomic profiling that characterizes neurogenic potential also outlines the transition from allocortical to neocortical domains in the hippocampal-entorhinal system and shows that ExN are the main drivers of the differences between subfields (Fig. 4), which evidence a richer complement of ExNs than traditional descriptions based on cytoarchitecture. Our analysis provides a primer to further study these populations and characterize the possible implications for hippocampal-entorhinal physiology. These data refine our understanding of the evolution of allo-, meso-, and neo-cortex. The transcriptomic signatures we developed strongly suggest homology between mammalian allocortex and specifically deep layers of the EC and neocortex.
Among the genes contributing to the layer transition, we identified METTL7B to be important in hippocampus physiology and functions. We found METTL7B, equipped with methyltransferase activity, interacts with important AD-related proteins (e.g., APP, LRP1, RTN3, and RTN4). Importantly, our results suggest that these functional interactions in a subset of ExNs and astrocytes seem to be phylogenetically specific to Old World monkeys and apes (parvorder Catarrhini), species that show more marked signs of pathology related to aging such as AD than other species (Perez et al., 2013; Finch and Austad, 2015; Edler et al., 2017; Paspalas et al., 2018). Overall, our analyses provided multiple vignettes of how this resource can be used to identify cell types and genes that might be functionally relevant for the biology of the hippocampus, allowing for inter-species comparisons.
STAR Methods
Resource availability
Lead contact
Requests for further information, resources and reagents should be directed to and will be fulfilled by the lead contact, Nenad Sestan (nenad.sestan@yale.edu).
Materials availability
All plasmids and the lentivirus generated in this study are available from the Lead Contact without restriction. The Mettl7b mutant mice line generated is preserved as frozen sperm and will be available upon request. Reagents used in the study were of general use and from commercial sources.
Data and code availability
Supplement contains transcriptome analysis and proteomic data with analysis. RNA-seq data is deposited at http://www.psychencode.org/, https://biccn.org/data and NCBI GEO: GSE186538. The data can also be interactively visualized at: http://resources.sestanlab.org/hippocampus.
All scripts are available at Github repository https://github.com/sestanlab/Hippocampus.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request
Experimental model and subject details
Human, rhesus macaque and pig postmortem tissue
Human samples were obtained from the collections of the Sestan and Rakic laboratories and from Javier DeFelipe’s collection in the Instituto Cajal in Madrid (Spain). Rhesus macaque and pig brain specimens were obtained from the tissue collection of the Sestan and Rakic laboratories. All clinical histories, tissue specimens, and histological sections were evaluated to assess for signs of disease, injury, and gross anatomical and histological alterations.
Fresh tissue specimens for histology were fixed with 4% paraformaldehyde/PBS followed by 30% sucrose/PBS. No obvious signs of neuropathological alterations were observed in any of the human, macaque or pig specimens analyzed in this study. The postmortem interval (PMI) was defined as hours between time of death and time when tissue samples were fresh frozen or started to undergo fixation process.
Frozen archival tissue human specimens were used for snRNA-seq. No obvious signs of neuropathological alterations were observed in any of the specimens considered and analyzed in this study. For all other specimens, regions of interest were sampled from frozen tissue slabs or whole specimens stored at −80 °C. To ensure consistency between specimens, all dissections from the same species were performed by the same person. Frozen tissue slabs were kept on a chilled aluminum plate during dissections. EC and four hippocampal subregions (DG, CA 2–4, CA1, and Sub) were microdissected as previously reported (Kang et al., 2011) from fresh frozen post-mortem human brains previously cut into 1-cm thick serial, coronal sections, and snap frozen in isopentane (J. T. Baker).
All human (Homo sapiens) brain specimens used for snRNA-seq transcriptome and DCX immunostaining (Table S1 and S3) were de-identified and collected from clinically unremarkable donors and one case that died in status epilepticus. Tissue was collected following the guidelines provided by the Yale Human Investigation Committee (HIC) for the Sestan and Rakic collection or by the European Union for DeFelipe’s samples from Spain. Tissue was collected and handled in accordance with ethical guidelines and regulations for the research use of human brain tissue set forth by the NIH (http://bioethics.od.nih.gov/humantissue.html) and the WMA Declaration of Helsinki (http://www.wma.net/en/30publications/10policies/b3/index.html). Appropriate informed consent was obtained and all available non-identifying information was recorded for each specimen.
The brain tissue samples of Alzheimer disease were sourced from 4 biobanks, with Braak stage II-VI and/or CERAD confirmed neuropathologic diagnosis and the PMI span 8–28 hours (Table S7).
All studies using non-human primates and pigs were carried out in accordance with a protocol approved by Yale University’s Committee on Animal Research and NIH guidelines. Rhesus macaque (Macaca mulatta) brain samples were collected postmortem from 7 adult specimens (Table S1 and S3). Pig brain samples were collected postmortem from 18 young adult specimens (Table S1 and S3).
Method details
Anatomical definition of sampled subregions of the hippocampal formation and entorhinal cortex
The dentate gyrus (DG) was sampled from the posterior part of the anterior third of the hippocampal formation. It included all three layers: molecular, granular, and polymorphic. The deeper part of the hilus of the DG was dissected as part of the proximal portion (nearer DG) of the CA2–4 region.
Cornu Ammonis (CA) 2–4 region was sampled after DG was dissected and contained the remaining hilus containing CA4 and the proximal hippocampal fields CA3 and CA2 (until approximately the CA1 region), including all three layers: molecular, pyramidal and stratum oriens.
CA1 region (Sommer’s sector) was sampled from approximately the border of CA2 to the subiculum, comprising the most distal (from the DG) portion of cornu Ammonis. The border between CA1 and CA2 is difficult to reliably identify and thus small pieces of the neighboring CA2 and, vice versa, could have been occasionally present in the samples.
The subiculum (Sub) is part of the subicular complex (subiculum, presubiculum and parasubiculum) located between the hippocampus and EC. Our sample was taken adjacent to CA1, corresponding to the subiculum, and was composed of the molecular, pyramidal and polymorphic layers and the superficial region of underlying white matter.
The entorhinal cortex (EC) spreads over both the gyrus ambiens and a considerable part of the parahippocampal gyrus. The EC samples were collected from the middle portion of the parahippocampal gyrus of the same tissue slab used to dissect the subregions of the hippocampal formation, corresponding to the proper entorhinal subregion and Brodmann area 28. The EC was also defined by presence of numerous wart-like elevations (verrucae hippocampi) on the surface of the gyrus. Samples contained all cortical layers and the superficial region of underlying white matter.
Brain cell nuclei isolation
The brain cell nuclei were isolated according to our previous protocol (Li et al., 2018; Zhu et al., 2018) with some modifications. Hippocampal regions (DG, CA1, CA2–4, Sub) and adjacent entorhinal cortex were dissected from three frozen adult human brains (Table S1). In order to avoid experimental bias and evenly dissociate the tissue for cell nuclei isolation, whole tissue was finely pulverized to powder in liquid nitrogen with mortar and pestle (Coorstek #60316, #60317). All buffers were ice cold and all reagents used for consequent nuclear isolation were molecular biology grade unless stated otherwise. 5 – 10 mg of pulverized tissue was added into 5 ml of ice-cold lysis buffer consisting of 320 mM sucrose (Sigma #S0389), 5 mM CaCl2 (Sigma #21115), 3 mM Mg(Ace)2 (Sigma #63052), 10mM Tris-HCl (pH 8) (AmericanBio #AB14043), protease inhibitors w/o EDTA (Roche #11836170001), 0.1 mM EDTA (AmericanBio #AB00502), RNAse inhibitor (80U/ml) (Roche #03335402001), 1mM DTT (Sigma #43186), and 0.1% TX-100 (v/v) (Sigma#T8787). DTT, RNAse Protector, protease inhibitors, and TX-100 were added immediately before use. The suspension was transferred to Dounce tissue grinder (15ml volume, Wheaton #357544; autoclaved, RNAse free, ice-cold) and homogenized with loose and tight pestles, 30 cycles each, with constant pressure and without introduction of air. The homogenate was strained through 40 um tube top cell strainer (Corning #352340) which was pre-wetted with 1ml wash buffer: (250 mM sucrose (Sigma #S0389), 25 mM KCl (Sigma #60142), 5mM MgCl2 (Sigma #M1028), 20mM Tris-HCl (pH 7.5) (AmericanBio #AB14043; Sigma #T2413), protease inhibitors w/o EDTA (Roche #11836170001), RNAse inhibitor (80U/ml) (Roche #03335402001), 1mM DTT (Sigma #43186)). Additional 4 ml of wash buffer was added to wash the strainer. Final 10 ml of solution was mixed with 10 ml of 50% Optiprep (Axis-Shield# 1114542) solution (50% iodixanol (v/v), 250 mM sucrose (Sigma #S0389), 25 mM KCl (Sigma #60142), 5mM MgCl2 (Sigma #M1028), 20mM Tris-HCl (pH 7.5) (AmericanBio #AB14043; Sigma #T2413), protease inhibitors w/o EDTA (Roche #11836170001), RNAse inhibitor (80U/ml) (Roche #03335402001), 1mM DTT (Sigma #43186)) by inverting the tube 10x and carefully pipetted into 2 centrifuge tubes (Corning #430791). The tubes were centrifuged at 1000g, for 30 min at 4 °C on centrifuge (Eppendorf #5804R) and rotor (Eppendorf #S-4–72). Upon end of centrifugation, the supernatant was carefully and completely removed and total of 5 ml of resuspension buffer (250 mM sucrose (Sigma #S0389), 25 mM KCl (Sigma #60142), 5mM MgCl2 (Sigma #M1028), 20mM Tris-HCl (pH 7.5) (AmericanBio #AB14043; Sigma #T2413), protease inhibitors w/o EDTA (Roche #11836170001), RNAse inhibitor (80U/ml) (Roche #03335402001), 1mM DTT (Sigma #43186)) was added carefully on the pellets in tubes and centrifuged at 1000g, for 10 min at 4 °C on the same centrifuge and rotor. Supernatants were then carefully and completely removed, pellets were gently dissolved by adding 100 ul of resuspension buffer (see above) and pipetting 30x with 1ml pipette tip, pooled and filtered through 35 um tube top cell strainer (Corning #352340). Finally, nuclei were counted on hemocytometer and diluted to 1 million/ml with sample-run buffer: 0.1% BSA (Gemini Bio-Products #700–106P), RNAse inhibitor (80U/ml) (Roche#03335402001), 1mM DTT (Sigma #43186) in DPBS (Gibco #14190). Some DG nuclei samples (HSB179, HSB181, HSB282 and RMB3, Table S1) were fixed with methanol (American Bio AB#09110). At the end of nuclei isolation, four volumes of methanol (−20 °C) were added dropwise, while mixing the nuclei suspension (final concentration: 80% methanol). The methanol-fixed nuclei were kept on ice for 15 min and then stored at −80 °C. For rehydration nuclei were placed on ice, centrifuged on the same centrifuge and rotor as above - at 3000g, 10 min at 4 °C, resuspended in modified sample-run buffer (1% BSA), centrifuged at 1000g, for 10 min at 4 °C, resuspended in sample-run buffer, and prepared for 10x Genomics assay as indicated above.
Single nucleus microfluidic capture and cDNA synthesis
The nuclei samples were placed on ice and taken either to Yale Center for Genome Analysis core facility or processed in the laboratory within 15 minutes for snRNA-seq with targeted nuclei recovery of 10000 nuclei, respectively, on microfluidic Chromium System (10x Genomics) by following the manufacturer’s protocol (10x Genomics, CG000183_Rev_A), with Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3, (10x Genomics #PN-1000075) and Chromium Single Cell B Chip Kit (10x Genomics #PN-1000074), Chromium i7 Multiplex Kit (10x Genomics #PN-120262) on Chromium Controller (10x Genomics). Due to limitations imposed by source RNA quantity, cDNA from nuclei was amplified for 14 cycles.
Single nucleus RNA-seq library preparation
Post cDNA amplification cleanup and construction of sample-indexed libraries and their amplification followed manufacturer’s directions (10x Genomics, CG000183_Rev_A), with the amplification step directly dependent on the quantity of input cDNA.
Sequencing of libraries
In order to reach sequencing depth of 20000 raw reads per nucleus, single nucleus libraries were run using paired end sequencing with single indexing on the HiSeq 4000 platform (Illumina) by following manufacturer’s instructions (Illumina; 10x Genomics, CG000183_Rev_A). To avoid lane bias, multiple uniquely indexed samples were mixed and distributed over several lanes.
Single nuclei expression quantification and quality control
We quantified the expression levels of genes in each potential nucleus represented by a cellular barcode using the 10X Genomics CellRanger pipeline (version 3.0.2). For the human samples, reads were mapped to human reference genome GRCh38 (Ensembl release 98) and quantified in units of Unique Molecular Identifiers (UMIs) based on the combined exon-intron human annotation. Same strategies were applied to macaque and pig except that genome assembly Mmul10 and susScr11 was used for rhesus macaque and pig, respectively. Associated NCBI RefSeq gene annotations of pig and rhesus macaque were downloaded from UCSC genome browser. We took advantage of the enhanced cell-calling methodology in CellRanger to distinguish true cells from damaged or empty droplets. Specifically, RNA content distribution of each barcode was compared to the background concentration which was generalized from extremely low RNA-containing barcodes, and was subsequently classified as damaged if comparable profiles were seen. To further rule out low-quality cells, we excluded nuclei with mitochondrial content greater than 10%. This loose criterion was set as we aimed to incorporate certain cell types into analyses such as endothelial cells which were shown to be prone to high mitochondrial content (Velmeshev et al., 2019). Additional filtering procedure was performed after clustering and low-dimensional embedding (see below) to eliminate cell clusters collectively displaying elevated mitochondrial and ribosomal gene expression and showing no signals of reasonable cell types.
Normalization, dimensionality reduction and clustering
We normalized the raw UMI counts using the ‘NormalizeData’ function in the R package Seurat with the scaling factor equal to 10,000 (Butler et al., 2018). To position all nuclei in a two-dimensional representation reflecting their transcriptomic similarities (Fig. 1B–1D), the top 2,000 highly variable genes were obtained by the Seurat function ‘FindVariableFeatures’ with the default variance stabilizing process. We further integrated nuclei from a given species on the basis of the summarized anchor features via the function ‘IntegrateData’ and embedded ensuing nuclei in the PCA dimensions followed by Uniform Manifold Approximation and Projection (UMAP) visualization (Becht et al., 2019). To cluster nuclei according to their nearest transcriptomic neighbors, we searched for shared nearest neighbors (SNN) in the PCA space with the neighbor number being 25 and optimized the graph modularity using the Seurat function ‘FindClusters’. In general, we performed an iterative removal-clustering approach to remove nuclei with high mitochondrial or ribosomal contents and without clear cluster-related markers followed by re-clustering of the remaining nuclei. Moreover, cells and clusters co-expressing combinatory of major cell-type (ExN, InN, Astro, OPC, Oligo, immune and Vas) signatures were manually marked as doublets and excluded from the downstream analytical flow. Lastly, we re-embedded cell types of interest (i.e., ExN, InN and NNC) in the PCA space and re-clustered them using the same procedure as mentioned above, as this would offer finer details into the cell types we sought to probe into.
Tree construction
To explore the taxonomic relationships among all cell subtypes, we constructed a hierarchical tree by first averaging the gene expression levels across cells of the same subtype. The derived expression was standardized to mean of zero and variance of one within each subtype across the anchor genes selected in the previous integration step. Following this step, we calculated the Euclidean distances between pairwise subtypes, and clustered these subtypes in a structured tree (Fig. 1E) by the ‘hclust’ function in R with the method set to ‘ward.D2’.
Relative cell cluster contribution from subregions and donors
Because of the absolute ratio of donors or subregions in each cluster can be biased by the differences of sample size as well as the subregions dissected in each donor, we used relative ratio instead to measure the contribution of donors or subregions to cells clusters. Specifically, we calculated the absolute ratio of a given cluster in each donor or subregion and divided this ratio by the sum of ratios across all subregions or donors. Results are visualized in Fig. 1E.
Global across-dataset comparison
We performed global comparisons with two previous human HIP single nuclei RNA-seq datasets (Habib et al., 2017; Ayhan et al., 2021). We calculated the average log-transformed expression of the highly variable genes across all clusters and then performed Pearson correlation to demonstrate the subtype-subtype similarity across datasets, which were further displayed in gradient heat maps (Fig. S1G–H). Because the annotated neural stem cell cluster in the pioneer HIP data actually represents an ependymal cell cluster (Sorrells et al., 2021), we updated the cluster label accordingly.
Classification of cell subtypes in human
We grouped cell clusters with strong signals of SLC17A7 expression into ExN. Furthermore, we categorized them into different subtypes through marker gene expression and comparisons with published datasets (Fig. S1G–H) (Cembrowski et al., 2016a; Cembrowski et al., 2016b; Habib et al., 2017; Cembrowski et al., 2018; Li et al., 2018; Hodge et al., 2019; Ayhan et al., 2021). Specifically, granule cells were characterized by the predominant composition of DG nuclei and prominent expression of PROX1. Mossy cells were described by the principal origin from DG and exclusive expression of ADCYAP1. We initially identified three granule cell subtypes characterized by the high expression of SGCZ, PDLIM5 and EGR1, respectively. Given that the EGR1-expressing subtype is solely contributed by one donor, which are most likely caused by batch effects rather than true biological variations, it was merged to the most similar cluster, SGCZ-expressing subtype. ExN from CA fields were arranged mainly according to subfields: CA3 pyramidal neurons (co-expression of CFAP299 and SYN3), CA2 pyramidal neurons (co-expression of CFAP299 and HGF), dorsal CA1 pyramidal neurons (co-expression of GRIK1 and GRM3), and ventral CA1 pyramidal neurons (co-expression of ACVR1C and SYT13). For the Sub ExN, we categorized them into three subtypes: one distal (away from CA1) (FN1+) subtype and two proximal ones (ROBO1+). Of note, the spatial registrations of CA and Sub cell subtypes were achieved on the basis of previous transcriptomic studies of hippocampal pyramidal neurons (Cembrowski et al., 2016a; Cembrowski et al., 2016b; Cembrowski et al., 2018). With regards to entorhinal ExN, we classified them by two means. First, we aligned them with ExN from single nucleus data of human MTG using the same procedure as described above. Second, we examined the subtype-specific marker genes in both our ExN and related literature reports. Specifically, two layer 2 subtypes were classified as RELN+ and one as CALB1+ (Witter et al., 2017). Other upper-layer subtypes were depicted based on marker gene expression of LAMA3, PDGFD, IL1RAPL2, and PCP4 (Ramsden et al., 2015; Tang et al., 2015; Ohara et al., 2018). The middle-to-deep layer subtypes were delineated by the specific gene expression of RORB, THEMIS, ADRA1A, and TLE4.
Cell clusters showing high GAD1 expression were then assigned as InN. InN clusters were first classified to major groups based on the expression of three canonical function markers (PVALB, SST, VIP) as well as LAMP5, a marker mostly representing a group of neurogliaform InN and recently being adopted as a major InN marker (Tasic et al., 2018; Hodge et al., 2019). For a given cluster expressing two markers simultaneously (e.g., InN LAMP5 NMBR cluster expresses both SST and LAMP5), it was assigned to the same major group of the neighboring cluster in the hierarchical tree. Additionally, we used LHX6 (a medial ganglionic eminence marker) and NR2F2 (a caudal ganglionic eminence marker) to classify the rest of the InN clusters which do not express these markers. Finally, each InN cluster was named after the combination of major group marker (eg. SST, VIP) and one top subtype marker (eg. ANO2). Apart from these InN clusters, we also identified a MEIS2-expressing InN cluster corresponding to the white-matter residing InN type described before (Frazer et al., 2017; Tasic et al., 2018) and a neuron cluster co-clustered with InN showing strong signals of RELN, NDNF, highly indicative of Cajal Retzius cells.
The remaining nuclei were collectively referred to as NNC. We classified these nuclei into four big groups based on marker gene expression of SOX10 (oligodendrocyte lineage-related cells), AQP4 (astrocytes), PTPRC (immune cells) and RGS5 (endothelial cells) (Fig. S2E, S2F). The first group was further subdivided by the expression of PDGFRA (oligodendrocyte precursor cells, OPCs), GPR17 (committed oligodendrocyte precursor cells, COPs), and MOBP (oligodendrocytes). We additionally grouped OPCs and oligodendrocytes into specific subtypes according to the high expression of specific genes: EGR1 and GRIA4 for OPCs; CPXM2, SLC5A11, LINC01098 and LAMA2 for oligodendrocytes. For astrocyte subtype specification, we classified them by the laminar distribution: GFAP+ ones located in deep layers and CHRDL1+ ones in upper layers (Lanjakornsiripan et al., 2018). Regarding immune cells, we used marker genes C1QB, F13A1, LYZ and SKAP1 to deconstruct them into microglia, macrophages, myeloid cells and T cells, respectively. Microglia were further subdivided via specific gene expression of P2RY12 and CD83. In terms of vasculature lineage, we employed combinational expression of genes to sort them into arterial endothelial cells (DKK2+), endothelial cells (CLDN5+ and VWF+), pericytes (CLDN5+ and ABCC9+), venous smooth muscle cells (ABCC9+ and P2RY14+), arterial smooth muscle cells (ACTA2+ and TAGLN+) and vascular and leptomeningeal cells (COL1A2+ and COL1A1+) (Vanlandewijck et al., 2018).
Most of clusters identified are shared across donors while certain exhibited minimal or even no representation in some of the donors (Fig. 1E). Among those disproportionally distributed clusters, two clusters, CR RELN NDNF and InN SST NPY, only account for 0.01%−0.02% of the cell population and were more prone to show disproportional distribution. Another interneuron subtype “InN PVALB PLCL1”, which exhibits certain level of depletion in HIP as compared to EC (Fig. 1E), is also absent in a donor where only DG region was dissected. All the EC ExN subtypes were exclusively contributed by EC and were missing in the donors where only DG regions were dissected. Additionally, we observed one cluster “T SKAP1 CD247” absent in one donor, probably reflecting variations of immune response across donors.
Classification of cell types in pig and rhesus macaque
The cell identity classification of pig and macaque were carried out using the same procedures as described above with a few exceptions. The annotation of nIPC and neuroblast was based on two criteria, expression of canonical cell type markers (nIPC: MKI67, CENPF, TOP2A; neuroblast: DCX, CALB2, PROX1) and clustering with mouse progenitors and neuroblast cells when integrated with mouse data. Due to the scarcity of RGL cells in pig and rhesus and their transcriptomic similarity to astrocytes, we classified those pig and macaque cells co-clustered with mouse RGL cluster as RGL cells. In total, we identified 8 and 7 RGL cells in pig (30 minutes PMI) and macaque, respectively.
Classification of cell types in fetal human hippocampus
Fetal human hippocampus cells (Zhong et al., 2020) were further subclustered using the same procedure described above to separate granule cell and pyramidal neuron differentiation lineages. Neural intermediate progenitor cells (nIPCs) were classified as SOX2+EOMES+NEUROG1+ and radial glia cells were annotated as SOX2+PAX6+VIMhighOLIG2lowEOMES−. Neuroblast cells were identified via the combinatory expression of DCX and NHLH1. Separation of DG versus non-DG ExN lineage was based on the expression of MEIS2 (non-DG lineage) and PROX1 (DG lineage).
Integrate dentate gyrus data across species and developmental stages
We used the same Seurat integration pipelines to integrate the DG data from mouse (Hochgerner et al., 2018), pig, rhesus macaque and human. Young adult mouse data referred to P120-P132 period of the dataset C in the original data and juvenile mouse data (P12-P35) referred to the dataset A (Hochgerner et al., 2018). Importantly, variable features were first selected via the Seurat function ‘FindVariableFeatures’ with the default variance stabilizing process for each sample and the union of highly variable genes were set as the anchor features for data integration. To more rigorously identify putative human nIPCs and neuroblasts, we applied pairwise integration between human and each of other species using both Seurat (Stuart et al., 2019) and Harmony (Korsunsky et al., 2019) harnessing the union of highly variable genes of each species pair. Here, for simplicity, we only used pig hippocampus data at 30 minutes postmortem interval for the four-species integration (Fig. 2A–B and S2D). Same integration pipelines were applied for the integration including human doublets (Fig. S2I–J) as well as the integration between fetal and adult human data (Fig. S2K–L).
RNA velocity analysis for mouse, pig, rhesus macaque and human
We first applied velocyto (La Manno et al., 2018) to count the abundances of un-spliced and spliced transcripts using the bam output of CellRanger in pig, rhesus macaque and human. With regard to the mouse data (Hochgerner et al., 2018), because of the incompatibility of public sequencing files with Cellranger input, we reperformed the read alignment and UMI counting using STARsolo (Dobin et al., 2013), a tool performing similar preprocessing analysis to CellRanger, and passed the bam files to velocyto package. We then applied scVelo (Bergen et al., 2020) to find variable genes, calculated RNA velocities via dynamical models and visualized the velocities on the UMAP embeddings where four species were integrated together using the Seurat pipelines described above (Fig. 2B).
Comparison of subtype markers across species and developmental stages
Subtype marker gene calculation was performed separately in each dataset using “FindMarkers” function in Seurat. We used the following strategy to minimize the marker set size bias and extrapolate the subtype similarity. Specifically, for each cluster, we checked the percentage of the top 75 markers (ranked by average fold changes) of species A present in species B markers and the percentage calculated in the reverse direction, which were then averaged to indicate the subtype similarity of the cluster between species A and B (Fig. S2C).
To get cell-type specific markers that are only enriched in a given cell type, we further retained marker genes with fold changes of expression ratio no less than 1.2 and adjusted p value (Bonferroni correction) no more than 0.01. The top 20 specific markers of each subtype were then visualized in dot plots (Fig. 2C). As there are insufficient RGL cells in pig and rhesus and limited nIPCs in pig, these clusters were not included in the marker analysis.
Expression profiling of DCX across species and regions
In order to compare the DCX expression across species, we down-sampled all the datasets to a comparable level. Specifically, we calculated the median of the total UMIs of the granule cell subtype in each species and computed a scaling factor using dataset with the lowest depth. We reasoned that granule cell cluster is the best anchor given that it presents in all species with high abundance and it is a crucial part of the granule cell lineage. Then, the UMIs of each cell were subsampled to the level equal to multiplying the original library size by the scaling factor and the generated down-sampled datasets were used for the comparative analysis including DCX expression (Fig. 3A–B and Table S3) and enrichment analysis (Fig. 3C).
Enrichment of neurogenic marker sets in DCX-expressing cells
To test whether DCX-expressing cells show enriched expression of these neuroblast markers, we compared the area under the curve (AUC) scores of these marker sets (Aibar et al., 2017) in DCX-expressing and DCX-negative cells using Wilcoxon rank sum test (one-tailed test, Fig. 3C). Gene expression ranking was first performed in each cell followed by calculation of the enrichment of the given marker set using AUC scores. Because expression ranking rather than expression level was used, the calculation was less vulnerable to expression units. We also used the down-sampled datasets to further minimize sequencing depth bias. We removed DCX gene from each of these marker sets prior to AUC score calculation as the presence of DCX in these markers could increase the AUC scores for DCX-expressing cells and bias the analysis.
Reanalysis of data from Ayhan et al., 2021
We extracted the relevant cell types from the study, which includes all the neurons, astrocytes and oligodendrocytes. Then we used the same Seurat integration pipeline described above to integrate the data from multiple batches and visualized the cell on the UMAP layout (Fig. S3S). We were not able to access the cell annotations for the original Gra.Neu.5 cluster, but we found a group of granule cells corresponding to that cluster clearly marked by LPAR1, a marker used to label cluster Gra.Neu.5. Since we observed strong expression of all the top oligodendrocyte markers (calculated by FindMarkers function in Seurat) in this cluster (one representative marker is shown in Fig. S3S), we then used AUCell (Aibar et al., 2017) to calculate the AUROC scores of oligodendrocyte markers to test their enrichment in this cluster. Doublet scores were calculated using Scrublet package (Wolock et al., 2019).
Cell subtype comparisons among HIP, EC, MTG and dlPFC
To explore the transcriptomic divergence across HIP, EC, MTG and dlPFC for all cell subtypes, we constructed a network demonstrating the relationships among the subtypes in the four brain regions based on the extent of overlap of their specific marker genes. In detail, in each region we first determined the marker genes of each subtype using the ‘FindAllMarkers’ function in Seurat. Subsequently, we generated a similarity matrix representing the overlap between marker genes of pairwise subtypes across all regions, followed by the visualization of this matrix in the form of a network via the R package ‘igraph’ through the force-directed graphopt algorithm (Fig. 4B, D and F). Especially, for ExN types we displayed their connections in a between-region manner (HIP and EC, EC and MTG, and MTG and dlPFC). To further examine the cell subtype connections across different regions, in each brain region we focused on marker genes detected in at least one subtype and assessed their expression across all subtypes of remaining brain regions visualized in heat maps (Fig. S4A). Additionally, given the upper- and deep-layer marker genes identified in MTG, we calculated the percentages of genes in each subtype of each region where expression was greater than the expression constraint of 40% quantile across all expression values (Fig S4B). Furthermore, we evaluated the expression of marker genes from intratelencephalic/intracerebral (IT) neurons and non-IT neurons of MTG in all subtypes of the four regions through first averaging the expression of each gene across cells of the same subtype and then displaying the average values across IT markers/Non-IT markers in scatter plots (Fig. S4C).
Identifying genes specific to ExN of different regions
In order to identify a list of genes that exhibit enriched expression to a specific region, we first calculated the expression ratio of all the genes across all the ExN subtypes. Stringent criteria were applied to minimize the influence of technical differences across datasets. Specifically, we required the gene to have a maximum expression ratio of 0.3 across all the ExN subtypes in that region and have a minimum of fold change of 2.2 compared to the expression ratios in other regions. We also filtered genes that were prominently expressed in other regions. The region-specific genes were visualized in Fig. 5B and the hippocampus ExN-specific genes were passed to the below analysis to survey their temporal specificity.
Temporal specificity of the HIP ExN-specific genes in bulk tissue transcriptomic datasets
Gene expression analysis was performed on the PsychENCODE RNA-seq datasets (Li et al., 2018). Time periods 3–15 were collapsed into three time groups: prenatal (periods 3–7), early postnatal (periods 8–12), and adult (periods 13–14). We used limma (Smyth et al., 2005) to run a regression that included the time group and brain region, as well as the region-group interactions, as factors. Genes were then ranked by the region-group coefficient differences between HIP and the maximum of other regions (Fig. 5C).
Exclusive markers of cluster InN SST ADAMTS12
To find hippocampus-specific transcriptome features in the cluster InN SST ADAMTS12, we first sought to confirm the enrichment of this cluster in hippocampus by integrating InN from HIP, EC, MTG and dlPFC using the ‘RunHarmony’ function in the Harmony R package (Fig. 5C) (Korsunsky et al., 2019). Following the integration, we identified a set of markers exclusively expressed in this cluster as compared to other interneuron clusters in hippocampus and SST-expressing interneuron clusters in MTG or dlPFC. To do so, we first calculated the markers of InN SST ADAMTS12 in the hippocampal-entorhinal dataset using “FindMarkers” function in Seurat and removed those identified as marker genes in SST subtypes in MTG and dlPFC.
Generation of knockout mice and tissue processing
All experiments with mice were performed in accordance with a protocol approved by Yale University’s Committee on Animal Research. Targeted embryonic stem (ES) cells (Mettl7btm1(KOMP)Vlcg) were obtained from Knockout Mouse Project (KOMP) repository. Chimeric mice were generated by blastocyst injection of ES cells at Yale Genome Editing Center (YGEC). Mice were bred for germline transmission to generate gene knockout mice. Genotyping was performed using the TUF/TUR primer set (145 bp) for the wild-type allele and the NeoFwd/SD primer set (351 bp) for the Mettl7b deletion allele.
Both wild type and Mettl7b mutant mice were reared in group housing in a 12h light:12h dark cycle and provided food and water ad libitum with veterinary care provided by Yale Animal Resource Center. Only mice in good, healthy condition, as approved by Yale Animal Resource Center, were used for breeding and experimentation. Multiple breeding pairs were maintained and siblings were never mated to increase genetic diversity, and prevent unintended selection for features that could affect results. Both sexes were used and randomly assigned for all experiments. Adult mice were anesthetized and intracardially perfused with ice-cold PBS and 4% PFA. All mouse brain tissue specimens were fixed by immersion in 4% PFA overnight at 4 °C and sectioned at 50 μm using a vibratome (Leica).
In situ hybridization
Human brain tissue samples were fixed in 4% PFA overnight at 4 °C and sectioned at 30 μm using a Leica VT1000 S vibratome. The RNA probes complementary to human METTL7B cDNA (NM_152637.2) were labeled with digoxigenin-UTP (Roche). After acetylation, sections were hybridized with the probes at 63 °C for 16 hours. Following hybridization, the riboprobes were immunolabeled with anti-digoxigenin-AP conjugate and the signal was developed with NBT/BCIP overnight in dark.
Immunolabeling and histology
For METTL7B immunohistochemistry (IHC), tissue sections were pretreated with antigen retrieval with citrate buffer pH 6 at 95C for 20 mins, incubated with anti-Mettl7b antibody raised in rabbit (Atlas antibodies HPA038644; RRID:AB_2676130; 1:500) followed by ImmPRES Excel Amplified HRP Polymer Staining Kit (Anti-Rabbit IgG, MP-7601–15, Vector Laboratories) per manufacturer’s protocol and using standard biotinylated secondary antibodies followed by Vectastain ABC-AP kit (AK-5000, Vector Labs) and developed with ImmPACT-DAB (SK-4105, Vector labs). For mouse α-β-galactosidase (lacZ) stain, tissue sections were blocked with blocking solution (5% normal donkey serum, 1% BSA, 0.1% glycine, 0.1% lysine, and 0.3% Triton X-100 in PBS) for 1 hour and incubated with primary antibodies and biotinylated secondary antibodies. The signal was amplified with Vectastain ABC-AP kit and developed with Vector Blue AP kit (SL-5300, Vector Labs) per manufacturer’s protocol. DCX IHC was performed with anti-DCX antibodies raised in guinea pig (EMD Millipore AB2253; RRID:AB_1586992; 1:4000) and antibodies raised in mouse (Santa Cruz sc-271390; RRID:AB_10610966; 1:500). Immunohistochemistry for GAD1 was performed with anti-GAD1 antibody raised in goat (R&D AF2086; RRID:AB_2107724; 1:200) and for PSA-NCAM with antibodies raised in mouse (5A5-s Hybridoma Bank; RRID:AB_528392; 1:500). All antibodies were incubated in 3% normal donkey serum, 0.25% Triton X-100 in PBS). Antigen retrieval (20 mins in citrate buffer pH 6 at 95C) was required for optimal results with DCX and GAD1 antibodies, but not in IHC for PSA-NCAM, as it precludes it to work. Chromogenic antibody detection was achieved with biotinylated secondary antibodies, followed by ABC-AP kit and ImmPACT-DAB as described for Mettl7b. DCX controls were performed in the same way, except the primary antibody was omitted. For colocalization of DCX and GAD1, anti-guinea pig biotinylated secondary antibodies followed by Streptavidin conjugated (Jackson Immunoresearch) antibodies were used for DCX and anti-goat secondary antibodies (Jackson Immunoresearch) for GAD1. DAPI was used for nuclear staining. All histology samples were imaged on Aperio ScanScope system, Leica microscope, Zeiss Axio Observer with an Apotome 2 system or on a Zeiss LSM 510 confocal microscope. Cell culture samples were fixed with ice-cold 4% paraformaldehyde (PFA) for 10 minutes at room temperature, blocked for 30 minutes at RT with blocking solution (5% normal donkey serum, 1% BSA, 0.1% glycine, 0.1% lysine, and 0.3% saponin in PBS), incubated with primary and appropriate Alexa Flour-conjugated secondary antibodies, and imaged on Zeiss LSM 510 confocal microscope.
IHC labeling for electron microscopy
Rhesus macaque brain (N=3) was fixed with intracardial perfusion of 4% paraformaldehyde and 0.05% glutaraldehyde mixture. Postmortem human brain (N=3) was fixed with immersion in same fixative. For antigen retrieval, vibratome 40-mm-thick slices from the hippocampus and entorhinal cortex were immersed in citrate buffer pH6 at 60°C during 20 min. Then, slices were blocked in 5% bovine albumin and incubated in rabbit METTL7B (1:500) polyclonal antibodies overnight at room temperature. For immunoperoxidase labeling, the slices were immersed in solution of biotinylated goat anti-rabbit antibodies (Jackson Immunoresearch Inc., West Grove, PA; 1:300) and developed by the Elite ABC kit (Vector Laboratories, Burlingame, CA) with Ni-intensified 3,3’-diaminobenzidine-4HCl as a chromogen and post-fixed with 1% OsO4. For immunogold labeling, after primary antibodies, slices were blocked in the mixture of 0.8% bovine albumin and 0.1% cold water fish skin gelatin (Aurion, Wageningen, The Netherlands). Then, slices were incubated with goat anti-rabbit IgGs conjugated with 1-nm gold particles (1:80) overnight at 4°C with subsequent silver intensification in R-Gent SE-LM kit (all from Aurion) and post-fixed with 0.5% OsO4. Slices were dehydrated and embedded in Durcupan (ACM; Fluka, Buchs, Switzerland) on microscope slides. For electron microscopic investigations, fragments from identified hippocampal zones were re-embedded into Durcupan blocks and cut by Leica UC7 ultramicrotome into 60-nm-thick sections. Ultrathin sections were collected on one-slot grids covered with Butvar B-98 films (EMS, Hatfield, PA), stained with lead citrate, and evaluated in Talos L120C electron microscope.
Plasmids
For expression of METTL7B, full length cDNA (NM_152637.2) was inserted into pCAGIG (a gift from Connie Cepko, Addgene #11159) (Matsuda and Cepko, 2004). For lentiviral generation, pFUGW (a gift from David Baltimore, Addgene #14883) (Lois et al., 2002) was digested with PacI, 3’ overhangs removed with Klenow (NEB) to form blunt ends, and additionally digested with BsrGI to release hUBC promoter and EGFP. The CAG-IRES-EGFP was removed from pCAGIG and ligated into pFUGW. For protein pulldown experiments, BirA-HA and HaloTag constructs were PCR-amplified from pcDNA3.1-MCS-BirA(R118G)-HA (a gift from Kyle Roux, Addgene #36047) (Roux et al., 2012) and pHTC-CMVneo-HaloTag (G7711, Promega), respectively, and ligated into pFUGW-CAG.
Lentiviral purification and generation of stable cell lines
Ten 15-cm dishes of sub-confluent Lenti-X 293T cells (Clontech) were used for each purification. pFUGW-CAG specific plasmids (BirA, METTL7B-BirA, HaloTag, METTL7B-HaloTag) along with pMD2.G, pRSVrev and pMDLg/pRRE (a gift from Didier Trono, Addgene #12259, #12253, #12251) (Dull et al., 1998) were transfected at 1:1:1:1 molar ratio using PolyJet (SignaGen). Cell culture media containing lentiviral particles (LVP) was collected at 48- and 60-hours post-transfection and filtered through 0.2 μm filter to remove cellular debris. Filtered supernatants were centrifuged at 100,000g for 2 hours. One milliliter of PBS was laid over LVP pellet and left overnight at 4 °C. Next day, resuspended pellets were centrifuged through 30% sucrose gradient to further purify the virus. Lentiviral titers were determined by transducing Lenti-X 293T cells and calculating titer from FACS data between 1–10% infection rate using formula: Titer (IU/ml) = (# cells seeded × dilution factor × % GFP-positive cells) / (volume of virus solution added).
For pulldown experiments, 50,000 ReNcell CX (EMD Millipore) cells were plated on a laminin coated 24-well plate in triplicate wells. Cells were transduced with lentiviral particles at MOI of 10 in a 150 μL of cell culture media supplemented with 10 μg/mL of protamine sulfate (#02194729, MP Biomedicals) and saved as ReN-CAG-BirA, ReN-CAG-METTL7B-BirA, ReN-CAG-HaloTag, and ReN-CAG-METTL7B-HaloTag stable cell lines.
Affinity capture of proteins
For BioID and HaloTag experiments, two million cells (ReN-CAG-BirA, ReN-CAG-METTL7B-Bira, ReN-CAG-HaloTag, ReN-CAG-METTL7B-HaloTag) were plated on four laminin coated 10-cm dishes. BioID pulldown was performed per protocol (Roux et al., 2013). At near confluency, cell culture media was supplemented with 50 μM biotin (B4639, Sigma-Aldrich). The next day, cells were rinsed twice with PBS, detached with Accutase (Millipore) for 10 minutes at 37 °C, centrifuged at 200 g for 3 minutes, rinsed with PBS, and centrifuged again. Bead-protein conjugates were resuspended in 50 mM ammonium bicarbonate. HaloTag pulldown was performed per manufacturer’s protocol (G6500, Promega). Proteins were eluted by resuspending HaloTag resin in 50 μL of 8 M urea prepared in 50 mM ammonium bicarbonate and shaking for 30 minutes at room temperature. Ten percent fractions of BioID and HaloTag eluates were saved for immunoblot and silver stain analysis.
Mass spectrometry and proteomic data analysis
BioID and HaloTag tryptic digestion was performed using the optimized method from the original published method (Kim et al., 2014). Proteins were reduced by adding 2 μl of 0.5M Tris (2-carboxyethyl) phosphine (TCEP) at 30 °C for 60 min. The reaction was cooled to room temperature (RT) and proteins were alkylated in the dark for 30 min by adding 4 μl of 0.5M Iodoacetamide. Sample volume was adjusted by adding 350 μl of 50 mM Ammonium Bicarbonate to dilute the 8M urea to 1M before trypsin digestion. Mass spectrometry grade trypsin (Promega) was added for overnight digestion at 30°C using Eppendorf Thermomixer at 700 rpm. Formic acid was added to the peptide solution (to 2%), followed by desalting by C18 TopTip (TT10C18.96, PolyLC) and finally dried on a SpeedVac. Tryptic peptides were resuspended in 100 μl of 2% Acetonitrile in 0.1% formic acid. Ten microliters of total tryptic peptides were used in triplicate runs for the 1D LC-MS/MS analysis, consisting of an EASY-nLC 1000 HPLC Acclaim PepMap peptide trap with a 25 cm- 2μm Easy-Spray C18 column, Easy Spray Source, and a Q Exactive Plus mass spectrometer (all from Thermo Fisher Scientific). A 230-min gradient consisting of 5–16%B (100% acetonitrile) in 140 min, 16–28% in 70 min, 28–38% in 10 min, 38–85% in 10 min was used to separate the peptides. The total LC time was 250 min. The Q Exactive Plus was set to scan precursors at 70,000 resolution followed by data-dependent MS/MS at 17,500 resolution of the top 12 precursors.
Protein identification and data analysis
The LC-MS/MS raw data of two technical replicates was combined and submitted to Sorcerer Enterprise v.3.5 release (Sage-N Research Inc.) with SEQUEST algorithm as the search program for peptide/protein identification. SEQUEST was set up to search the target-decoy UniProt Human Reviewed (v. March 2015) protein fasta database using trypsin for the enzyme and with the allowance of up to 2 missed cleavages, semi tryptic search, fixed modification of 57 Da for cysteines to account for carboxyamidomethylation and precursor mass tolerance of 50 ppm. Differential search included 226 Da on lysine for biotinylation (BioID samples), 16 Da for methionine oxidation, and 14, 28 and 42 Da on lysine for mono-, di- and tri- methylayion. The search results were viewed, sorted, filtered, and statically analyzed by using comprehensive proteomics data analysis software, Peptide/Protein prophet v.4.02 (ISB) (Nesvizhskii et al., 2003). The minimum trans-proteomic pipeline (TPP) probability score for proteins was set to 0.9 to assure very low error (less than FDR 2%) with good sensitivity. The differential spectral count analysis was done by QTools, an open source SBP in-house developed tool for automated differential peptide/protein spectral count analysis (Brill et al., 2009) and the protein prophet peptide report was utilized to report biotinylated peptides. The LC-MS/MS raw data were also submitted to Integrated Proteomics Pipelines (IP2) Version IP2 v.3 (Integrated Proteomics Applications, Inc.) with ProLuCID algorithm as the search program (Xu et al., 2006) for peptide/protein identification. ProLuCID search parameters were set up to search the UniProt Human Reviewed (v. March 2015) protein fasta database including reversed protein sequences using trypsin for enzyme with the allowance of up to 2 missed cleavages, semi tryptic search, fixed modification of 57 Da for cysteines to account for carboxyamidomethylation and precursor mass tolerance of 50 ppm. Differential search included 226 Da on lysine for biotinylation (for BioID samples), 16 Da for methionine oxidation, and 14, 28 and 42 Da on lysine for mono-, di- and tri- methylayion. The search results were viewed, sorted, filtered, and statically analyzed by using DTASelect for proteins to have protein FDR rate of less than 2.5% (Tabb et al., 2002). Differential label-free proteomics data analysis was done by IP2-Census, Protein Identification STAT COMPARE (Park et al., 2008) using two technical replicates. This result was a label-free quantification analysis, of duplicate technical data for each sample; using spectral count analysis with t-test and Gene Ontology analysis (Robinson et al., 2004).
Identification of true pulldown proteins based on mass spectrometry spectral counting data
We discriminated true prey-bait interactions from false interactions in the Halotag and BioID pulldowns by using Significance Analysis of INTeractome (SAINT) method (Choi et al., 2011; Teo et al., 2014). Briefly, the SAINT method utilizes MS/MS spectral counting data and models true and false prey-bait interactions as separate Poisson distributions to obtain the probability of a true protein-protein interaction based on Bayesian statistical inference. The estimated probability provides a quantitative measure of the confidence of prey-bait interactions such that false interactions can be filtered out in a statistically-controlled manner. Upon applying the SAINT method to MS/MS spectral count data available from each pulldown experiment system, we identified 275 (out of 3 cases and 3 controls) and 1795 (3 cases and 3 controls) proteins as true METTL7B interactors from Halotag and BioID pulldowns, respectively, at Bayesian False Discovery Rate (BFDR) of 5%.
Subcellular localization of METTL7B
To characterize subcellular localization of the true METTL7B interactors, we performed fold-enrichment test for major subcellular compartments cataloged in the Human Protein Atlas database (Uhlen et al., 2015) and mammalian lipid droplet proteomes (Hodges and Wu, 2010). Human Protein Atlas provides genome-wide analysis of major subcellular localization information of human proteins based on immunofluorescent stained cells. It consists of 20 main subcellular compartments and 10,003 proteins (www.proteinatlas.org). To make the fold-enrichment test comparable across Human Protein Atlas and the mammalian lipid droplet proteome datasets, we merged the mammalian lipid droplet protein list to Human Protein Atlas dataset as a separate subcellular localization category and used the entire Human Protein Atlas subcellular localization records uniformly as a null (background) set. We found that 73.8% (203/275) and 77.7% (1384/1795) of true METTL7B interactors from HaloTag and BioID pulldown experiments had matching HGNC gene symbols in Human Protein Atlas. Of the 152 mammalian cytoplasmic lipid proteins(Hodges and Wu, 2010), 80 proteins had matching HGNC gene symbols in the Human Protein Atlas. Twenty-three (HaloTag) and 37 (BioID) true METTL7B interactors were identified to be among 80 lipid droplet proteins in the Human Protein Atlas database.
Validation of pulldown experiments
We evaluated the performance of SAINT method by benchmarking the true METTL7B interactors against non-redundant physical BioGRID protein-protein interaction network (Stark et al., 2006). We computed the significance of interactions between proteins from the true METTL7B interactor set and the rest of the proteins (background set) in the protein-protein interaction (PPI) network by using binomial proportions test Z-score as follows (Abul-Husn et al., 2009):
| (Eq. 1) |
where
p1 : number of true METTL7B interactors among the adjacent PPI network neighbors of a given protein,
p2 : number of all the adjacent PPI network neighbors of a given protein,
N1 : number of the true METTL7B interactors present in the PPI network,
N2 : number of the all PPI network proteins, and
The Z-score thus provides an approximate quantitative measure of how significantly a given protein in the PPI network interacts with the true METTL7B interactors in the immediate neighborhood of the protein-protein interaction network compared to the background proteins in the protein-protein interaction network. We found that the true METTL7B interactors tend to interact much more significantly to each other than to the rest of proteins in the protein-protein interaction network (Wilcoxon rank sum test p-value < 2e-16, data not shown). This indicates that the true METTL7B interactors are significantly clustered and proximal to each other in the protein-protein interaction network as expected.
KEGG pathway enrichment analysis
Towards the assessment of KEGG pathway enrichment, only these 110 high-confidence METTL7B interacting proteins reported by both strategies, were submitted to the online software, i.e., DAVID Bioinformatics Resources 6.8 (Huang da et al., 2009), with the selection of “Homo sapiens” as species background and “KEGG_PATHWAY” as the targeted functional term.
Immunoblotting and silver stain
Tissue sample preparation:
Tissue was lysed in PBS with 0.01% Tween-20 and protease inhibitor cocktail (P-2714, Sigma-Aldrich), and sonicated in two sessions (30 pulses at an output level of 3 using a Microson Ultrasonic Cell Disruptor [Misonix]) with 1-minute rest on ice between sessions. Samples were centrifuged at 14 000 g for 10 minutes at 4 °C. Total protein concentrations were measured by the Bradford assay (#23246, Pierce).
Immunoblotting:
Samples were mixed with NuPAGE LDS Loading Buffer (NP0007) supplemented with 50 mM DTT, incubated at 72 °C for 10 minutes, and loaded on 4–12% Bis-Tris gel (NP0321, Thermo Fisher Scientific). Proteins were transferred to a 0.2 μm PVDF membrane (#162–0218, Bio-Rad), blocked with 5% non-fat milk or BSA in 1% TBST buffer, and blotted with appropriate primary and secondary HRP-conjugated antibodies. The signal was developed with SuperSignal West Pico Plus Chemiluminescent Substrate (#34577, Pierce) and visualized on G:BOX Chemi XRQ (Syngene) system.
Silver stain:
5% of HaloTag eluates were prepared as above and electrophoresed on 4–12% Bis-Tris gel. Gel was processed using Silver Stain for Mass Spectrometry kit per manufacturer’s instructions (#24600, Pierce).
SAM assay
Custom made recombinant METTL7B was expressed in E. Coli ArcticExpress and purified from inclusion bodies by GenScript. Recombinant RTN3, RTN4, LRP1, and APP peptide were purchased directly from vendors. SAMfluoro Methyltransferase Assay (786–431, G-Biosciences) was performed per manufacturer’s instructions using ~2 μg of METTL7B and ~1 μg of substrate protein. Recombinant proteins were incubated with or without METTL7B in triplicate wells. Assay was performed at 37 °C and resorufin fluorescence was measured on GloMax Multi Detection System (Promega) plate reader with an excitation wavelength of 530–540 nm and an emission wavelength of 585–595 nm.
RNA isolation and digital droplet PCR
Total RNA was extracted from human and mouse brain tissue samples, or cultured cells, using RNAeasy Plus Mini Kit (#74134, Qiagen) per manufacturer’s protocol. RNA concentrations and quality were determined using R6K ScreenTape (#5067–5576, Agilent) and TapeStation analyzer (Agilent). cDNA was synthesized from 1 μg of total RNA using SuperScript III First-Strand Synthesis kit (#18080051, Invitrogen) and random primers. Digital droplet PCR was performed using QX200 Droplet Digital PCR (Bio-Rad) and data was normalized to TBP expression. PCR amplification was performed using primer sets and probes listed in Table S6.
snRNA-seq profiling of human Alzheimer’s disease brain middle temporal gyrus
To understand the cellular heterogeneity and disease-associated cellular changes in human AD brain, we performed unbiased massively parallel snRNA-seq with post-mortem frozen human brain tissues of middle temporal gyrus (MTG), a brain cortical region strongly affected by AD. The collection and characteristics of the AD and neurologically intact control brain samples has been described previously (Kostylev et al., 2015; Kostylev et al., 2018). From 12 individuals with and without AD, we isolated brain nuclei by sucrose gradient ultracentrifugation, generated single nucleus libraries with 10x Chromium platform (10x Genomics), and sequenced on NovaSeq S4 sequencer (Illumina). We integrated snRNA-seq data of human brains from these 12 individuals of both AD (Braak Stage V/VI, n = 6) and age-matched normal controls (Ctrl, Braak Stage I/II, n = 6) by single nucleus analysis using Seurat (Stuart et al., 2019). After quality control filtering, we profiled and analyzed 64,845 single nucleus transcriptomes, clustered all the cells jointly across the 12 donors that include 6 females and 6 males, and identified and annotated the major cell types of the human brain by interrogating the expression patterns of known gene markers, including neurons (GRIN1), excitatory neurons (ExN, SLC17A7), inhibitory neurons (InN, GAD1), astrocytes (Astro, AQP4), microglia (Micro, ITGAM), oligodendrocytes (Oligo, MBP), oligodendrocyte precursor cells (OPC, PDGFRA), and endothelial cells (Endo, CLDN5). Full analysis and data will be reported elsewhere (Zhang and Strittmatter, unpublished communication).
Quantification and statistical analysis
Differential gene expression tests, as well as the differential enrichment test of neuroblast markers, were carried out using the wilcox.test function implemented in R. This method does not require the assumption of normal distribution, which is appropriate for single cell data, that there is no consensus distribution model established. The related results are shown in Fig. 2C, 3C, 4B, 4E, S2G, S3B. The regression of gene expression with factors including time group, brain region, as well as the region-group interactions, were performed via the R package limma (Smyth et al., 2005). The results are shown in Fig. 4C. The differential test of METTL7B expression in AD brains versus control brains was performed using two-tailed Fisher’s exact test (Fig. S7C–D)
In the proteomic data analysis, we identified true pulldown proteins via Significance Analysis of INTeractome (SAINT) (Choi et al., 2011), and benchmarked its acquisition using binomial proportions test detailed in a previous study (Abul-Husn et al., 2009). Gene ontology enrichment tests were performed using an online software, DAVID Bioinformatics Resources (Huang da et al., 2009).
Additional statistical tests were applied to test the significance of signal differences in certain biochemical assays using the following methods: differential gene expression tests in ddPCR were carried out using one-way ANOVA with post-hoc Dunnett’s adjustment (Fig. 6C, E); the signal differences in the SAM methyltransferase assay were tested using two-tailed t-test (Fig. 7F);
Supplementary Material
Table S1 (related to Figure 1) | Human, macaque and pig specimens used for snRNA-seq.
Table S4 (related to Figure 7) | LC-MS/MS data from BioID and HaloTag experiments.
Table S5 (related to Figure 7) | SAINT analysis of LC-MS/MS data from BioID and HaloTag experiments.
Table S6 (related to STAR methods) | Primer and probe sequences.
Table S7 (related to Figure S7) | Human AD tissues used for METTL7B immunostaining.
Table S3 (related to Figure 3) | DCX expression in IHC samples and transcriptome data.
Table S2 (related to Figure 2) | Quantitative data on neurogenic rates estimated by DCX immunostaining and 14C incorporation in mouse, macaque and human DG from the literature.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| a-Streptavidin-Cy3 (1:1000) | BioLegend | Cat#405215 |
| a-Streptavidin-HRP, high sensitivity (1:40,000) | Pierce | Cat#21130 |
| Chicken a-ADFP (1:1000) | Abcam | Cat#ab37516; RRID:AB_722641 |
| Chicken a-BirA (1:1000) | Abcam | Cat#ab14002; RRID:AB_300830 |
| Donkey a-Goat | Jackson ImmunoResearch | Cat#705-225-147; RRID:AB_2307341 |
| Donkey a-Guinea pig IgG (H+L), biotin | Jackson ImmunoResearch | Cat#706-065-148; RRID:AB_2340451 |
| Donkey a-Mouse, Alexa Fluor 555 | Invitrogen | Cat#A-31570; RRID:AB_2536180 |
| Donkey a-Rabbit IgG (H+L), biotin | Jackson ImmunoResearch | Cat#711-065-152; RRID:AB_2340593 |
| Donkey a-Rabbit IgG, Alexa Fluor 488 | Invitrogen | Cat#A-21206; RRID:AB_2535792 |
| Donkey streptavidin conjugated | Jackson ImmunoResearch | Cat#016-160-084; RRID:AB_2337244 |
| Goat a-Chicken IgY H&L (HRP) | Abcam | Cat#ab97150 RRID:AB_10679811 |
| Goat a-Chicken IgY, Alexa Fluor 647 | Invitrogen | Cat#A-21449; RRID:AB_2535866 |
| Goat a-GAD1 (1:200) | R&D | Cat#AF2086; RRID:AB_2107724 |
| Goat a-Rabbit IgG H&L (HRP) | Abcam | Cat#ab97080; RRID:AB_10679808 |
| Guinea pig a-DCX (1:4000) | EMD Millipore | Cat#AB2253; RRID:AB_1586992 |
| Mouse a-DCX (1:500) | Santa Cruz | Cat#sc-271390; RRID:AB_10610966 |
| Mouse a-PSA-NCAM (1:500) | DBSH | Cat#5A5-s; RRID:AB_528392 |
| Mouse a-CALNEXIN (1:50) | Santa Cruz | Cat#sc-23954; RRID:AB_626783 |
| Mouse a-GAPDH (1:2500) | Invitrogen | Cat#MA5-15738-HRP; RRID:AB_2537659 |
| Mouse a-HaloTag (1:1000) | Promega | Cat#G9211; RRID:AB_2688011 |
| Rabbit a-APP (Y188) (1:200 IF, 1:10,000 WB) | Abcam | Cat#ab32136; RRID:AB_2289606 |
| Rabbit a-b-galactosidase (1:500) | Invitrogen | Cat#A-11132; RRID:AB_22153 |
| Rabbit a-CALNEXIN (1:1000) | Cell Signaling | Cat#2679; RRID:AB_2228381 |
| Rabbit a-Lactate Dehydrogenase (1:5000) | Abcam | Cat#ab52488; RRID:AB_2134961 |
| Rabbit a-LRP1 (1:200 IF, 1:1000 WB) | Abcam | Cat#ab92544; RRID:AB_2234877 |
| Rabbit a-METTL7B (1:500 IHC, 1:1000 WB) | Atlas Antibodies | Cat#HPA038644; RRID:AB_2676130 |
| Rabbit a-RTN3 (1:50 IF, 1:1000 WB) | Protein Tech | Cat# 12055-2-AP; RRID:AB_2301357 |
| Rabbit a-RTN4 (NOGO A+B) (1:200 IF, 1:2000 WB) | Abcam | Cat#ab47085; RRID:AB_881718 |
| Vectastain ABC-AP kit | Vector Labs | Cat#AK-5000 |
| Vector Blue AP kit | Vector Labs | Cat#SL-5300 |
| Vectastain Elite ABC-HRP kit | Vector Labs | Cat#PK-6100 |
| CFWS Gelatin | Aurion | Cat#900.033 |
| ImmPRES Excel Amplified HRP Polymer Staining Kit | Vector Labs | Cat#MP-7601-15 |
| R-Gent SE-LM | Aurion | Cat#500.011 |
| Goat-anti-Rabbit IgG (H&L) (gold particles conjugated) | Aurion | Cat#806.011 |
| Durcupan™ ACM | Sigma | Cat#44610 |
| Anti-Digoxigenin-AP, Fab fragments | Roche | Cat#11093274910 |
| Chemicals, peptides, and recombinant proteins | ||
| APP (peptide) | rPeptide | Cat#A-1203-1 |
| LRP1 | Abnova | Cat#H00004035-G01 |
| METTL7B (24-244 aa) | GenScript | This paper |
| RTN3 | Antibodies-Online | Cat#ABIN3111137 |
| RTN4 | Sino Biological | Cat#13030-H09E |
| Digoxigenin-UTP | Roche | Cat#11209256910 |
| BsrGI | New England BioLabs | Cat#R0575L |
| PacI | New England BioLabs | Cat#R0547L |
| Klenow | New England BioLabs | Cat#M0210M |
| Trypsin Gold, Mass Spectrometry Grade | Promega | Cat#V5280 |
| PolyJet | SignaGen | Cat#SL100688 |
| Protamine sulfate | MP Biomedicals | Cat#02194729 |
| NBT/BCIP Stock Solution | Roche | Cat# 11681451001 |
| Protector RNase Inhibitor | Roche | Cat#03335402001 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#11836170001 |
| Optiprep | Axis-Shield | Cat#1114542 |
| Bovine Serum Albumin (BSA), Fraction V—Molecular Biology Grade | Gemini Bio-Products | Cat#700-106P |
| Critical commercial assays | ||
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10x Genomics | Cat#PN-1000075 |
| TMRDirect (1:1000) | Promega | Cat#G2991 |
| HaloTag | Promega | Cat#G6500 |
| C18 TopTip | PolyLC | Cat#TT10C18.96 |
| Chromium Single Cell B Chip Kit | 10x Genomics | Cat#PN-1000074 |
| Chromium i7 Multiplex Kit (10x Genomics #PN-120262) | 10x Genomics | Cat#PN-120262 |
| Deposited data | ||
| Human adult hippocampus snRNA-seq | This paper | GSE186538 |
| GRCh38 (Ensembl release 98) | Ensembl, GENCODE | https://www.gencodegenes.org/human/# |
| Mmul10 | UCSC, RefSeq | https://hgdownload.soe.ucsc.edu/goldenPath/rheMac10/bigZips/ |
| susScr11 | UCSC, RefSeq | https://hgdownload.soe.ucsc.edu/goldenPath/susScr11/bigZips/ |
| PsychENCODE RNA-seq data | Li et al., 2018 | http://www.development.psychencode.org/ |
| Developmental human brain exon array data | Kang et al., 2011 | https://hbatlas.org/ |
| Human, chimpanzee and macaque RNA-seq data | Zhu et al., 2018 | https://evolution.psychencode.org/ |
| Mammalian brain development RNA-seq data | Cardoso-Moreira et al., 2019 | https://apps.kaessmannlab.org/evodevoapp/ |
| Human fetal hippocampus scRNA-seq data | Zhong et al., 2020 | GSE131258 |
| Mouse adult hippocampus scRNA-seq data | Hochgerner et al., 2018 | GSE95753 |
| Human hippocampus DroNc-seq data | Habib et al., 2017 | https://portals.broadinstitute.org/single_cell |
| Axis-specific human hippocampus snRNA-seq data | Ayhan et al., 2021 | https://cells.ucsc.edu/?ds=human-hippo-axis |
| snRNA-seq data of human Alzheimer’s disease brain middle temporal gyrus | NCBI GEO | GSE188545 |
| snRNA-seq data of human Alzheimer’s disease brain prefrontal cortex | Mathys et al., 2019 | https://www.synapse.org/#!Synapse:syn18485175 |
| Bulk tissue RNA-seq data of Alzheimer’s disease brains | Swarup Lab | http://swaruplab.bio.uci.edu:3838/bulkRNA/ |
| Experimental models: Cell lines | ||
| Targeted embryonic stem (ES) cells Mettl7btm1(KOMP)Vlcg | Knockout Mouse Project (KOMP) repository | https://www.komp.org/redirect.html |
| ReN-CAG-BirA | This paper | N/A |
| ReN-CAG-METTL7B-BirA | This paper | N/A |
| ReN-CAG-HaloTag | This paper | N/A |
| ReN-CAG-METTL7B-HaloTag | This paper | N/A |
| Lenti-X 293T cells | Clontech | Cat#632180 |
| ReNcell CX | EMD Millipore | Cat#SCC007 RRID:CVCL_E922 |
| Experimental models: Organisms/strains | ||
| Mettl7btm1(KOMP)Vlcg Chimeric Mice | Yale Genome Editing Center | https://medicine.yale.edu/compmed/ags/ |
| Oligonucleotides | ||
| Primers for genotyping Mettl7btm1(KOMP)Vlcg Chimeric Mice | This paper | See Table S6 |
| asMTfwd 5’-ATGGACATCCTGGTCCCACT-3’ | This paper | See Table S6 |
| asMTrev 5’-GCAATTTAATACGACTCACTATAGGGAGATTTGACAGCCTTTCCCATGATGT-3’ | This paper | See Table S6 |
| Human METTL7B IDT (Hs.Pt.58.39517850) | This paper | See Table S6 |
| Human TBP IDT (Hs.PT.58v.39858774) | This paper | See Table S6 |
| Mouse Mettl7b-fwd 5’-GGTCAGGTAAAGCATGAGAGAG-3’ | This paper | See Table S6 |
| Mouse Mettl7b-probe 5’-/56-FAM/CGCTGCAG/ZEN/GGTGATCATTCATCA/3IABkFQ/-3’ | This paper | See Table S6 |
| Recombinant DNA | ||
| METTL7B, cDNA (NM_152637.2) | This paper | Integrated DNA Technologies |
| pHTC-CMVneo-HaloTag | Promega | Cat#G7711 |
| pMD2.G | Dull et al., 1998 | Addgene #12259 |
| pRSVrev | Dull et al., 1998 | Addgene #12253 |
| pMDLg/pRRE | Dull et al., 1998 | Addgene #12251 |
| pDTET-METTL7B | This paper | N/A |
| hPKG promoter (M60581.1) | This paper | Integrated DNA Technologies |
| pCAGIG | Matsuda and Cepko, 2004 | Addgene #11159 |
| pFUGW | Lois et al., 2002 | Addgene #14883 |
| pcDNA3.1-MCS-BirA(R118G)-HA | Roux et al., 2012 | Addgene #36047 |
| pCW57.1 | David Root | Addgene #41393 |
| Software and algorithms | ||
| CellRanger v3.0.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| R version 3.6.1 | R project | https://www.r-project.org/ |
| Seurat v3 | Stuart et al., 2019 | https://satijalab.org/seurat/index.html |
| Harmony | Korsunsky et al., 2019 | https://github.com/immunogenomics/harmony |
| scrublet | Wolock et al., 2019 | https://github.com/swolock/scrublet |
| AUCell | Aibar et al., 2017 | https://github.com/aertslab/AUCell |
| UMAP | Becht et al., 2018 | https://github.com/lmcinnes/umap |
| limma | Smyth et al., 2005 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| scVelo | Bergen et al., 2020 | https://scvelo.readthedocs.io/ |
| velocyto | La Manno et al., 2018 | http://velocyto.org/velocyto.py/index.html# |
| STARsolo | Dobin et al., 2013 | https://github.com/alexdobin/STAR/blob/master/docs/STARsolo.md |
| Shiny | Rstudio | https://www.shinyapps.io/ |
| SEQUEST | Sage-N Research Inc. | http://proteomicsresource.washington.edu/protocols06/sequest.php |
| Peptide/Protein prophet v.4.02 | Nesvizhskii et al., 2003 | http://proteinprophet.sourceforge.net/index.html |
| QTools | Brill et al., 2009 | N/A |
| ProLuCID | Xu et al., 2006 | https://www.manula.com/manuals/ip2/ip2/1/en/topic/7-1-prolucid-search-engine |
| DTASelect | Tabb et al., 2002 | https://www.scripps.edu/cravatt/protomap/dtaselect_instructions.html |
| Census | Park et al., 2008 | http://fields.scripps.edu/yates/wp/?page_id=824 |
| Ontologizer | Robinson et al., 2004 | http://ontologizer.de/ |
| SAINT | Choi et al., 2011; Teo et al., 2014 | http://saint-apms.sourceforge.net/Main.html |
| DAVID | Huang et al., 2009 | https://david.ncifcrf.gov/ |
| Other | ||
| UC7 ultramicrotome | Leica | N/A |
| Talos L120C TEM | Thermo Fisher Scientific | N/A |
| EASY-nLC 1000 Liquid Chromatograph | Thermo Fisher Scientific | Cat#LC120 |
| Acclaim™ PepMap™ 100 C18 HPLC Columns | Thermo Fisher Scientific | Cat#164941 |
| Exactive™ Plus Orbitrap Mass Spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAPFALGMBCA |
Highlights.
Single-nucleus RNA-seq of adult hippocampal-entorhinal cells in human, monkey and pig
Transcriptomic signatures of adult neurogenesis in mouse, pig and monkey, but not human
Excitatory neuron diversification delineates transitions from 3- to 6-layered cortex
METTL7B defines subregion specific excitatory neurons and astrocytes in primates
Acknowledgments:
We thank K. Meyer for help with gene expression analysis, A. Rosa Campos and K. Motamedchaboki (SBP Medical Discovery Institute, La Jolla, CA) for help with generating proteomics data, the University of Maryland Brain and Tissue Bank, M. Horn, M. Pletikos, and S. Wilson for assistance with tissue acquisition and processing, J. DeFelipe and the NIH NeuroBioBank for providing human tissue, and K. Gobeske and other lab colleagues for their comments. We also thank A. Duque for using equipment from MacBrainResource (NIH/NIMH MH113257). This work was supported by NIH grants MH116488, MH124619, RF1AG074341, and DA023999. The project that gave rise to these results received the support of a fellowship from “la Caixa” Foundation (ID 100010434). The fellowship code is LCF/BQ/PI19/11690010. G. Santpere is supported by grant MS20/00064 from Instituto de Salud Carlos III (Spain) and cofunded by the European Social Fund and grant PID2019–104700GA-I00 funded by AGENCIA ESTATAL DE INVESTIGACIÓN, Spain.
Footnotes
Declaration of interests:
Authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Abbott LC, and Nigussie F (2020). Adult neurogenesis in the mammalian dentate gyrus. Anat Histol Embryol 49, 3–16. [DOI] [PubMed] [Google Scholar]
- Abul-Husn NS, Bushlin I, Moron JA, Jenkins SL, Dolios G, Wang R, Iyengar R, Ma’ayan A, and Devi LA (2009). Systems approach to explore components and interactions in the presynapse. Proteomics 9, 3303–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen P (2007). The hippocampus book (Oxford; New York: Oxford University Press; ). [Google Scholar]
- Angata K, Suzuki M, and Fukuda M (2002). ST8Sia II and ST8Sia IV polysialyltransferases exhibit marked differences in utilizing various acceptors containing oligosialic acid and short polysialic acid. The basis for cooperative polysialylation by two enzymes. J Biol Chem 277, 36808–36817. [DOI] [PubMed] [Google Scholar]
- Arellano JI, Morozov YM, Micali N, and Rakic P (2021). Radial Glial Cells: New Views on Old Questions. Neurochem Res 46, 2512–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan F, Kulkarni A, Berto S, Sivaprakasam K, Douglas C, Lega BC, and Konopka G (2021). Resolving cellular and molecular diversity along the hippocampal anterior-to-posterior axis in humans. Neuron 109, 2091–2105 e2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology 37, 38-+. [DOI] [PubMed] [Google Scholar]
- Berg DA, Su Y, Jimenez-Cyrus D, Patel A, Huang N, Morizet D, Lee S, Shah R, Ringeling FR, Jain R, et al. (2019). A Common Embryonic Origin of Stem Cells Drives Developmental and Adult Neurogenesis. Cell 177, 654–668 e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, et al. (2018). Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 22, 589–599 e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming GL, and Song H (2011). In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145, 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Del Trecidi K (2015). Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv Anat Embryol Cell Biol 215, 1–162. [PubMed] [Google Scholar]
- Breunig JJ, Sarkisian MR, Arellano JI, Morozov YM, Ayoub AE, Sojitra S, Wang B, Flavell RA, Rakic P, and Town T (2008). Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A 105, 13127–13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill LM, Motamedchaboki K, Wu S, and Wolf DA (2009). Comprehensive proteomic analysis of Schizosaccharomyces pombe by two-dimensional HPLC-tandem mass spectrometry. Methods 48, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, and Moser EI (2013). Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci 16, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, Liechti A, Ascencao K, Rummel C, Ovchinnikova S, et al. (2019). Gene expression across mammalian organ development. Nature 571, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembrowski MS, Bachman JL, Wang L, Sugino K, Shields BC, and Spruston N (2016a). Spatial Gene-Expression Gradients Underlie Prominent Heterogeneity of CA1 Pyramidal Neurons. Neuron 89, 351–368. [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, Phillips MG, DiLisio SF, Shields BC, Winnubst J, Chandrashekar J, Bas E, and Spruston N (2018). Dissociable Structural and Functional Hippocampal Outputs via Distinct Subiculum Cell Classes. Cell 173, 1280–1292 e1218. [DOI] [PubMed] [Google Scholar]
- Cembrowski MS, Wang L, Sugino K, Shields BC, and Spruston N (2016b). Hipposeq: a comprehensive RNA-seq database of gene expression in hippocampal principal neurons. Elife 5, e14997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenquizca LA, and Swanson LW (2007). Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res Rev 56, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, and Nesvizhskii AI (2011). SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods 8, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani S, Ferrer I, Aronica E, Kovacs GG, Verney C, Nardelli J, Khung S, Delezoide AL, Milenkovic I, Rasika S, et al. (2018). Hippocampal Radial Glial Subtypes and Their Neurogenic Potential in Human Fetuses and Healthy and Alzheimer’s Disease Adults. Cereb Cortex 28, 2458–2478. [DOI] [PubMed] [Google Scholar]
- Cipriani S, Nardelli J, Verney C, Delezoide AL, Guimiot F, Gressens P, and Adle-Biassette H (2016). Dynamic Expression Patterns of Progenitor and Pyramidal Neuron Layer Markers in the Developing Human Hippocampus. Cereb Cortex 26, 1255–1271. [DOI] [PubMed] [Google Scholar]
- Dennis CV, Suh LS, Rodriguez ML, Kril JJ, and Sutherland GT (2016). Human adult neurogenesis across the ages: An immunohistochemical study. Neuropathol Appl Neurobiol 42, 621–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edler MK, Sherwood CC, Meindl RS, Hopkins WD, Ely JJ, Erwin JM, Mufson EJ, Hof PR, and Raghanti MA (2017). Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiol Aging 59, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp JR, Beasley CL, and Galea LA (2013). Increased hippocampal neurogenesis and p21 expression in depression: dependent on antidepressants, sex, age, and antipsychotic exposure. Neuropsychopharmacology 38, 2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, and Gage FH (1998). Neurogenesis in the adult human hippocampus. Nat Med 4, 1313–1317. [DOI] [PubMed] [Google Scholar]
- Finch CE, and Austad SN (2015). Commentary: is Alzheimer’s disease uniquely human? Neurobiol Aging 36, 553–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor-Garcia M, Terreros-Roncal J, Moreno-Jimenez EP, Avila J, Rabano A, and Llorens-Martin M (2020). Unraveling human adult hippocampal neurogenesis. Nat Protoc 15, 668–693. [DOI] [PubMed] [Google Scholar]
- Frazer S, Prados J, Niquille M, Cadilhac C, Markopoulos F, Gomez L, Tomasello U, Telley L, Holtmaat A, Jabaudon D, et al. (2017). Transcriptomic and anatomic parcellation of 5-HT3AR expressing cortical interneuron subtypes revealed by single-cell RNA sequencing. Nat Commun 8, 14219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF (2002). Changes in the views of neuronal connectivity and communication after Cajal: examples from the hippocampus. Prog Brain Res 136, 203–213. [DOI] [PubMed] [Google Scholar]
- Gloor P (1997). The temporal lobe and limbic system (New York: Oxford University Press; ). [Google Scholar]
- Gomez-Climent MA, Castillo-Gomez E, Varea E, Guirado R, Blasco-Ibanez JM, Crespo C, Martinez-Guijarro FJ, and Nacher J (2008). A population of prenatally generated cells in the rat paleocortex maintains an immature neuronal phenotype into adulthood. Cereb Cortex 18, 2229–2240. [DOI] [PubMed] [Google Scholar]
- Gould E, Tanapat P, McEwen BS, Flugge G, and Fuchs E (1998). Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci U S A 95, 3168–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Guidi S, Bianchi P, Alstrup AK, Henningsen K, Smith DF, and Bartesaghi R (2011). Postnatal neurogenesis in the hippocampal dentate gyrus and subventricular zone of the Gottingen minipig. Brain Res Bull 85, 169–179. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgerner H, Zeisel A, Lonnerberg P, and Linnarsson S (2018). Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neurosci 21, 290–299. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges BD, and Wu CC (2010). Proteomic insights into an expanded cellular role for cytoplasmic lipid droplets. J Lipid Res 51, 262–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland PV, and Vermeulen-Vanderzee E (1989). Efferent connections of the dorsal cortex of the lizard Gekko gecko studied with Phaseolus vulgaris-leucoagglutinin. J Comp Neurol 285, 289–303. [DOI] [PubMed] [Google Scholar]
- Hook B (2014). Cleaner Protein with HaloTag Purification Resins (Promega Corporation Web site.: Promega; ). [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ishizuka N (2001). Laminar organization of the pyramidal cell layer of the subiculum in the rat. J Comp Neurol 435, 89–110. [DOI] [PubMed] [Google Scholar]
- Jabes A, Lavenex PB, Amaral DG, and Lavenex P (2010). Quantitative analysis of postnatal neurogenesis and neuron number in the macaque monkey dentate gyrus. Eur J Neurosci 31, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gage FH, Aigner L, Song H, Curtis MA, Thuret S, Kuhn HG, Jessberger S, Frankland PW, Cameron HA, et al. (2018). Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 23, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Kc B, Zhu W, Motamedchaboki K, Doye V, and Roux KJ (2014). Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proceedings of the National Academy of Sciences 111, E2453 LP–E2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPre T, Qiu D, and Strittmatter SM (2003). Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38, 187–199. [DOI] [PubMed] [Google Scholar]
- Klausberger T, and Somogyi P (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoth R, Singec I, Ditter M, Pantazis G, Capetian P, Meyer RP, Horvat V, Volk B, and Kempermann G (2010). Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One 5, e8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler SJ, Williams NI, Stanton GB, Cameron JL, and Greenough WT (2011). Maturation time of new granule cells in the dentate gyrus of adult macaque monkeys exceeds six months. Proc Natl Acad Sci U S A 108, 10326–10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornack DR, and Rakic P (1999). Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc Natl Acad Sci U S A 96, 5768–5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, and Strittmatter SM (2015). Prion-Protein-interacting Amyloid-beta Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J Biol Chem 290, 17415–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostylev MA, Tuttle MD, Lee S, Klein LE, Takahashi H, Cox TO, Gunther EC, Zilm KW, and Strittmatter SM (2018). Liquid and Hydrogel Phases of PrP(C) Linked to Conformation Shifts and Triggered by Alzheimer’s Amyloid-beta Oligomers. Mol Cell 72, 426–443 e412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein AR, and Connors BW (1986). Cellular physiology of the turtle visual cortex: synaptic properties and intrinsic circuitry. J Neurosci 6, 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Toda T, and Gage FH (2018). Adult Hippocampal Neurogenesis: A Coming-of-Age Story. J Neurosci 38, 10401–10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laclef C, and Metin C (2018). Conserved rules in embryonic development of cortical interneurons. Semin Cell Dev Biol 76, 86–100. [DOI] [PubMed] [Google Scholar]
- Lanjakornsiripan D, Pior BJ, Kawaguchi D, Furutachi S, Tahara T, Katsuyama Y, Suzuki Y, Fukazawa Y, and Gotoh Y (2018). Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat Commun 9, 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Maitre TW, Dhanabalan G, Bogdanovic N, Alkass K, and Druid H (2018). Effects of Alcohol Abuse on Proliferating Cells, Stem/Progenitor Cells, and Immature Neurons in the Adult Human Hippocampus. Neuropsychopharmacology 43, 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, and Thuret S (2018). Adult Human Hippocampal Neurogenesis: Controversy and Evidence. Trends Mol Med 24, 521–522. [DOI] [PubMed] [Google Scholar]
- Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci 24, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872. [DOI] [PubMed] [Google Scholar]
- Luzzati F (2015). A hypothesis for the evolution of the upper layers of the neocortex through co-option of the olfactory cortex developmental program. Front Neurosci 9, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Wang C, Wang L, Zhou X, Tian M, Zhang Q, Zhang Y, Li J, Liu Z, Cai Y, et al. (2013). Subcortical origins of human and monkey neocortical interneurons. Nat Neurosci 16, 1588–1597. [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer A, and Thomson AM (2017). Cornu Ammonis Regions-Antecedents of Cortical Layers? Front Neuroanat 11, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkonen M, Soininen H, Kalvianen R, Tapiola T, Ylinen A, Vapalahti M, Paljarvi L, and Pitkanen A (1998). Remodeling of neuronal circuitries in human temporal lobe epilepsy: increased expression of highly polysialylated neural cell adhesion molecule in the hippocampus and the entorhinal cortex. Ann Neurol 44, 923–934. [DOI] [PubMed] [Google Scholar]
- Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, Avila J, and Llorens-Martin M (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25, 554–560. [DOI] [PubMed] [Google Scholar]
- Moreno-Jimenez EP, Terreros-Roncal J, Flor-Garcia M, Rabano A, and Llorens-Martin M (2021). Evidences for Adult Hippocampal Neurogenesis in Humans. J Neurosci 41, 2541–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Ngwenya LB, Peters A, and Rosene DL (2006). Maturational sequence of newly generated neurons in the dentate gyrus of the young adult rhesus monkey. J Comp Neurol 498, 204–216. [DOI] [PubMed] [Google Scholar]
- Nielsen JV, Blom JB, Noraberg J, and Jensen NA (2010). Zbtb20-induced CA1 pyramidal neuron development and area enlargement in the cerebral midline cortex of mice. Cereb Cortex 20, 1904–1914. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, et al. (2009). Uniquely hominid features of adult human astrocytes. J Neurosci 29, 3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara S, Onodera M, Simonsen OW, Yoshino R, Hioki H, Iijima T, Tsutsui KI, and Witter MP (2018). Intrinsic Projections of Layer Vb Neurons to Layers Va, III, and II in the Lateral and Medial Entorhinal Cortex of the Rat. Cell Rep 24, 107–116. [DOI] [PubMed] [Google Scholar]
- Paredes MF, Sorrells SF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, et al. (2018). Does Adult Neurogenesis Persist in the Human Hippocampus? Cell Stem Cell 23, 780–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, and Yates JR 3rd (2008). A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods 5, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Jang HJ, and Lee JH (2019). Roles of Primary Cilia in the Developing Brain. Front Cell Neurosci 13, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas CD, Carlyle BC, Leslie S, Preuss TM, Crimins JL, Huttner AJ, van Dyck CH, Rosene DL, Nairn AC, and Arnsten AFT (2018). The aged rhesus macaque manifests Braak stage III/IV Alzheimer’s-like pathology. Alzheimers Dement 14, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzke N, Spocter MA, Karlsson KAE, Bertelsen MF, Haagensen M, Chawana R, Streicher S, Kaswera C, Gilissen E, Alagaili AN, et al. (2015). In contrast to many other mammals, cetaceans have relatively small hippocampi that appear to lack adult neurogenesis. Brain Struct Funct 220, 361–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SE, Raghanti MA, Hof PR, Kramer L, Ikonomovic MD, Lacor PN, Erwin JM, Sherwood CC, and Mufson EJ (2013). Alzheimer’s disease pathology in the neocortex and hippocampus of the western lowland gorilla (Gorilla gorilla gorilla). J Comp Neurol 521, 4318–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden HL, Surmeli G, McDonagh SG, and Nolan MF (2015). Laminar and dorsoventral molecular organization of the medial entorhinal cortex revealed by large-scale anatomical analysis of gene expression. PLoS Comput Biol 11, e1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A (1991). A comparison of neurotransmitter-specific and neuropeptide-specific neuronal cell types present in the dorsal cortex in turtles with those present in the isocortex in mammals: implications for the evolution of isocortex. Brain Behav Evol 38, 53–91. [DOI] [PubMed] [Google Scholar]
- Rhee S, Kirschen GW, Gu Y, and Ge S (2016). Depletion of primary cilia from mature dentate granule cells impairs hippocampus-dependent contextual memory. Sci Rep 6, 34370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Wollstein A, Bohme U, and Beattie B (2004). Ontologizing gene-expression microarray data: characterizing clusters with Gene Ontology. Bioinformatics 20, 979–981. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, and Burke B (2013). BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci 74, 19 23 11–19 23 14. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, and Freund TF (1991). Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40, 599–636. [DOI] [PubMed] [Google Scholar]
- Seki T (2002). Expression patterns of immature neuronal markers PSA-NCAM, CRMP-4 and NeuroD in the hippocampus of young adult and aged rodents. J Neurosci Res 70, 327–334. [DOI] [PubMed] [Google Scholar]
- Seki T, Hori T, Miyata H, Maehara M, and Namba T (2019). Analysis of proliferating neuronal progenitors and immature neurons in the human hippocampus surgically removed from control and epileptic patients. Sci Rep 9, 18194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine Y, Lindborg JA, and Strittmatter SM (2020). A proteolytic C-terminal fragment of Nogo-A (reticulon-4A) is released in exosomes and potently inhibits axon regeneration. J Biol Chem 295, 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM, and Rowe TB (2017). Neocortical Lamination: Insights from Neuron Types and Evolutionary Precursors. Front Neuroanat 11, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomianka L, Amrein I, Knuesel I, Sorensen JC, and Wolfer DP (2011). Hippocampal pyramidal cells: the reemergence of cortical lamination. Brain Struct Funct 216, 301–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, and Scott HS (2005). Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21, 2067–2075. [DOI] [PubMed] [Google Scholar]
- Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, et al. (2018). Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Paredes MF, Velmeshev D, Herranz-Perez V, Sandoval K, Mayer S, Chang EF, Insausti R, Kriegstein AR, Rubenstein JL, et al. (2019). Immature excitatory neurons develop during adolescence in the human amygdala. Nat Commun 10, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Paredes MF, Zhang Z, Kang G, Pastor-Alonso O, Biagiotti S, Page CE, Sandoval K, Knox A, Connolly A, et al. (2021). Positive Controls in Adults and Children Support That Very Few, If Any, New Neurons Are Born in the Adult Human Hippocampus. J Neurosci 41, 2554–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AMM, Zhu Y, Raghanti MA, Kitchen RR, Onorati M, Tebbenkamp ATN, Stutz B, Meyer KA, Li M, Kawasawa YI, et al. (2017). Molecular and cellular reorganization of neural circuits in the human lineage. Science 358, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, and Tyers M (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34, D535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki WA, and Amaral DG (2004). Functional neuroanatomy of the medial temporal lobe memory system. Cortex 40, 220–222. [DOI] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, and Yates JR 3rd (2002). DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Ebbesen CL, Sanguinetti-Scheck JI, Preston-Ferrer P, Gundlfinger A, Winterer J, Beed P, Ray S, Naumann R, Schmitz D, et al. (2015). Anatomical Organization and Spatiotemporal Firing Patterns of Layer 3 Neurons in the Rat Medial Entorhinal Cortex. J Neurosci 35, 12346–12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, and Choi H (2014). SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics 100, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Klein MS, Stevens AP, Reinders Y, Hellerbrand C, Dettmer K, Gronwald W, Oefner PJ, and Reinders J (2013). Changes in the hepatic mitochondrial and membrane proteome in mice fed a non-alcoholic steatohepatitis inducing diet. J Proteomics 80, 107–122. [DOI] [PubMed] [Google Scholar]
- Tobin MK, Musaraca K, Disouky A, Shetti A, Bheri A, Honer WG, Kim N, Dawe RJ, Bennett DA, Arfanakis K, et al. (2019). Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 24, 974–982 e973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro S, Ingelmo-Torres M, Estanyol JM, Tebar F, Fernandez MA, Albor CV, Gaus K, Grewal T, Enrich C, and Pol A (2006). Identification and characterization of associated with lipid droplet protein 1: A novel membrane-associated protein that resides on hepatic lipid droplets. Traffic 7, 1254–1269. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker TL, Overall RW, Vogler S, Sykes AM, Ruhwald S, Lasse D, Ichwan M, Fabel K, and Kempermann G (2016). Lysophosphatidic Acid Receptor Is a Functional Marker of Adult Hippocampal Precursor Cells. Stem Cell Reports 6, 552–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Doan TP, Jacobsen B, Nilssen ES, and Ohara S (2017). Architecture of the Entorhinal Cortex A Review of Entorhinal Anatomy in Rodents with Some Comparative Notes. Front Syst Neurosci 11, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291 e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Venable JD, Park SK, Cociorva D, Lu B, Liao L, Wohlschlegel J, Hewel J, and Yates JR (2006). ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Molecular & cellular proteomics 5. [Google Scholar]
- Yao Z, van Velthoven CTJ, Nguyen TN, Goldy J, Sedeno-Cortes AE, Baftizadeh F, Bertagnolli D, Casper T, Chiang M, Crichton K, et al. (2021). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241 e3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TF, Li J, Ding F, and Arias-Carrion O (2014). Evidence of adult neurogenesis in non-human primates and human. Cell Tissue Res 358, 17–23. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Cai Y, Chu Y, Chen EY, Feng JC, Luo XG, Xiong K, Struble RG, Clough RW, Patrylo PR, et al. (2009). Doublecortin-expressing cells persist in the associative cerebral cortex and amygdala in aged nonhuman primates. Front Neuroanat 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Ding W, Sun L, Lu Y, Dong H, Fan X, Liu Z, Chen R, Zhang S, Ma Q, et al. (2020). Decoding the development of the human hippocampus. Nature 577, 531–536. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Sousa AMM, Gao T, Skarica M, Li M, Santpere G, Esteller-Cucala P, Juan D, Ferrandez-Peral L, Gulden FO, et al. (2018). Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (related to Figure 1) | Human, macaque and pig specimens used for snRNA-seq.
Table S4 (related to Figure 7) | LC-MS/MS data from BioID and HaloTag experiments.
Table S5 (related to Figure 7) | SAINT analysis of LC-MS/MS data from BioID and HaloTag experiments.
Table S6 (related to STAR methods) | Primer and probe sequences.
Table S7 (related to Figure S7) | Human AD tissues used for METTL7B immunostaining.
Table S3 (related to Figure 3) | DCX expression in IHC samples and transcriptome data.
Table S2 (related to Figure 2) | Quantitative data on neurogenic rates estimated by DCX immunostaining and 14C incorporation in mouse, macaque and human DG from the literature.
Data Availability Statement
Supplement contains transcriptome analysis and proteomic data with analysis. RNA-seq data is deposited at http://www.psychencode.org/, https://biccn.org/data and NCBI GEO: GSE186538. The data can also be interactively visualized at: http://resources.sestanlab.org/hippocampus.
All scripts are available at Github repository https://github.com/sestanlab/Hippocampus.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request