

CASE PRESENTATION
A 55-year-old right-handed woman presented with a 3-year history of cognitive changes. Early symptoms included mild forgetfulness—for example, forgetting where she left her purse or failing to remember to retrieve a take-out order her family placed—and word-finding difficulties. Problems with depth perception affected her ability to back her car out of the driveway. When descending stairs, she had to locate her feet visually in order to place them correctly, such that when she carried her dog and her view was obscured, she had difficulty managing this activity. She struggled to execute relatively simple tasks, such as inserting a plug into an outlet. She lost the ability to type on a keyboard, despite being able to move her fingers quickly. Her symptoms worsened progressively for 3 years, over which time she developed a sad mood and anxiety. She was laid off from work as a nurse administrator. Her family members assumed responsibility for paying her bills, and she ceased driving.
Her past medical history included high blood pressure, Hashimoto’s thyroiditis with thyroid peroxidase antibodies, remote history of migraine, and anxiety. Medications included mirtazapine, levothyroxine, calcium, and vitamin D. She had no history of smoking, drinking alcohol, or recreational drug use. There was no known family history of neurologic diseases.
What Are Diagnostic Considerations Based on the History? How Might a Clinical Examination Help to Narrow the Differential Diagnosis?
Insidious onset and gradual progression of cognitive symptoms over the course of several years raise concern for a neurodegenerative disorder. It is helpful to consider whether or not the presentation fits with a recognized neurodegenerative clinical syndrome, a judgment based principally on familiarity with syndromes and pattern recognition. Onset of symptoms before age 65 should prompt consideration of syndromes in the spectrum of frontotemporal dementia (FTD) and atypical (nonamnesic) presentations of Alzheimer’s disease (AD) (1, 2). This patient’s symptoms reflect relatively prominent early dysfunction in visual-spatial processing and body schema, as might be observed in posterior cortical atrophy (PCA), although the history also includes mention of forgetfulness and word-retrieval difficulties. A chief goal of the cognitive examination would be to survey major domains of cognition—attention, executive functioning, memory, language, visual-spatial functioning, and higher somatosensory and motor functioning—to determine whether any domains stand out as more prominently affected. In addition to screening for evidence of focal signs, a neurological examination in this context should assess for evidence of parkinsonism or motor neuron disease, which can coexist with cognitive changes in neurodegenerative presentations.
The patient’s young age and history of Hashimoto’s thyroiditis might also prompt consideration of Hashimoto’s encephalopathy (HE; also known as steroid-responsive encephalopathy), associated with autoimmune thyroiditis. This syndrome is most likely attributable to an autoimmune or inflammatory process affecting the central nervous system. The time course of HE is usually more subacute and rapidly progressive or relapsing-remitting, as opposed to the gradual progression over months to years observed in the present case (3).
The patient’s mental status examination included the Montreal Cognitive Assessment (MoCA), a brief global screen of cognition (4), on which she scored 12/30. There was evidence of dysfunction across multiple cognitive domains (Figure 1). She was fully oriented to location, day, month, year, and exact date. When asked to describe a complex scene from a picture in a magazine, she had great difficulty doing so, focusing on different details but having trouble directing her saccades to pertinent visual information. She likewise had problems directing her gaze to specified objects in the room and problems reaching in front of her to touch target objects in either visual field. In terms of other symptoms of higher order motor and somatosensory functioning, she had difficulty demonstrating previously learned actions—for example, positioning her hand correctly to pantomime holding a brush and combing her hair. She was confused about which side of her body was the left and which was the right. She had difficulty with mental calculations, even relatively simple ones such as “18 minus 12.” In addition, she had problems writing a sentence in terms of both grammar and the appropriate spacing of words and letters on the page.
FIGURE 1.
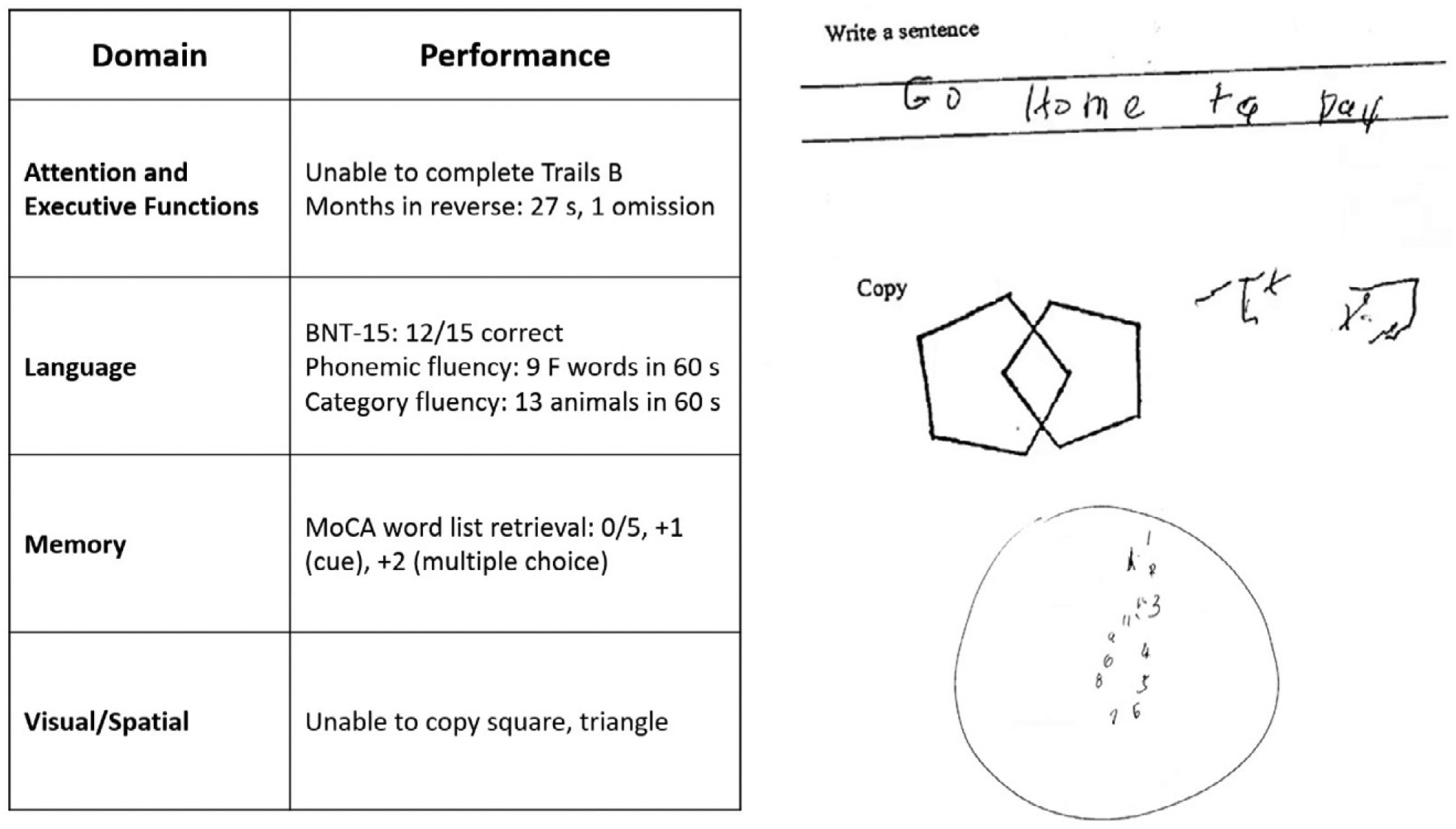
Selected elements of a 55-year-old patient’s cognitive examination at presentationa
a BNT-15=Boston Naming Test (15-Item); MoCA=Montreal Cognitive Assessment.
On elementary neurologic examination she had symmetrically brisk reflexes, with spread. She walked steadily with a narrow base, but when asked to pass through a doorway she had difficulty finding her way through it and bumped into the door jamb. Her elemental neurological examination was otherwise normal, including but not limited to brisk, full-amplitude vertical eye movements, normal visual fields, no evidence of peripheral neuropathy, and no parkinsonian signs such as slowness of movement, tremor, or rigidity.
How Does the Examination Contribute to Our Understanding of Diagnostic Considerations? What Additional Tests or Studies Are Indicated?
The most prominent early symptoms and signs localize predominantly to the parietal association cortex: The patient has impairments in visual construction, ability to judge spatial relationships, ability to synthesize component parts of a visual scene into a coherent whole (simultanagnosia or asimultagnosia), impaired visually guided reaching for objects (optic ataxia), and most likely impaired ability to shift her visual attention so as to direct saccades to targets in her field of view (oculomotor apraxia or ocular apraxia). The last three signs constitute Bálint syndrome, which localizes to disruption of dorsal visual networks (i.e., dorsal stream) with key nodes in the posterior parietal and prefrontal cortices bilaterally (5). She has additional salient symptoms and signs suggesting left inferior parietal dysfunction, including ideomotor limb apraxia and elements of Gerstmann syndrome, which comprises dysgraphia, acalculia, left-right confusion, and finger agnosia (6). Information was not included about whether she was explicitly examined for finger agnosia, but elements of her presentation suggested a more generalized disruption of body schema (i.e., her representation of the position and configuration of her body in space). Her less prominent impairment in lexical-semantic retrieval evidenced by impaired confrontation naming and category fluency likely localizes to the language network in the left hemisphere. Her impairments in attention and executive functions have less localizing value but would plausibly arise in the context of frontoparietal network dysfunction. At this point, it is unclear whether her impairment in episodic memory mostly reflects encoding and activation versus a rapid rate of forgetting (storage), as occurs in temporolimbic amnesia. Regardless, it does not appear to be the most salient feature of her presentation.
This localization, presenting with insidious onset and gradual progression, is characteristic of a PCA syndrome. If we apply consensus clinical diagnostic criteria proposed by a working group of experts, we find that our patient has many of the representative features of early disturbance of visual functions plus or minus other cognitive functions mediated by the posterior cerebral cortex (Table 1) (7). Some functions such as limb apraxia also occur in corticobasal syndrome (CBS), a clinical syndrome defined initially in association with corticobasal degeneration (CBD) neuropathology, a 4-repeat tauopathy characterized by achromatic ballooned neurons, neuropil threads, and astrocytic plaques. However, our patient lacks other suggestive features of CBS, including extrapyramidal motor dysfunction (e.g., limb rigidity, bradykinesia, dystonia), myoclonus, and alien limb phenomenon (Table 1) (8).
TABLE 1.
Clinical features and neuropathological associations of posterior cortical atrophy and corticobasal syndromea
| Feature | Posterior cortical atrophy | Corticobasal syndrome |
|---|---|---|
| Cognitive and motor features | Visual-perceptual: space perception deficit, simultanagnosia, object perception deficit, environmental agnosia, alexia, apperceptive prosopagnosia, and homonymous visual field defect | Motor: limb rigidity or akinesia, limb dystonia, and limb myoclonus |
| Visual-motor: constructional dyspraxia, oculomotor apraxia, optic ataxia, and dressing apraxia | ||
| Other: left/right disorientation, acalculia, limb apraxia, agraphia, and finger agnosia | Higher cortical features: limb or orobuccal apraxia, cortical sensory deficit, and alien limb phenomena | |
| Imaging features (MRI, FDG-PET, SPECT) | Predominant occipito-parietal or occipito-temporal atrophy, and hypometabolism or hypoperfusion | Asymmetric perirolandic, posterior frontal, parietal atrophy, and hypometabolism or hypoperfusion |
| Neuropathological associations | AD>CBD, LBD, TDP, JCD | CBD>PSP, AD, TDP |
Consensus diagnostic criteria for posterior cortical atrophy per Crutch et al. (7) require at least three cognitive features and relative sparing of anterograde memory, speech-nonvisual language functions, executive functions, behavior, and personality. Diagnostic criteria for probable corticobasal syndrome per Armstrong et al. (8) require asymmetric presentation of at least two motor features and at least two higher cortical features. AD=Alzheimer’s disease; CBD=corticobasal degeneration; FDG-PET=[18]F-fluorodexoxyglucose positron emission tomography; JCD=Jakob-Creutzfeldt disease; LBD=Lewy body disease; PSP=progressive supranuclear palsy; SPECT=single-photon emission computed tomography; TDP=TDP–43 proteinopathy.
In addition to a standard laboratory work-up for cognitive impairment, it is important to determine whether imaging of the brain provides evidence of neurodegeneration in a topographical distribution consistent with the clinical presentation. A first step in most cases would be to obtain an MRI of the brain that includes a high-resolution T1-weighted MRI sequence to assess potential atrophy, a T2/fluid-attenuated inversion recovery (FLAIR) sequence to assess the burden of vascular disease and rule out less likely etiological considerations (e.g., infection, autoimmune-inflammatory, neoplasm), a diffusion-weighted sequence to rule out subacute infarcts and prion disease (more pertinent to subacute or rapidly progressive cases), and a T2*-gradient echo or susceptibility weighted sequence to examine for microhemorrhages and superficial siderosis.
A lumbar puncture would serve two purposes. First, it would allow for the assessment of inflammation that might occur in HE, as approximately 80% of cases have some abnormality of CSF (i.e., elevated protein, lymphocytic pleiocytosis, or oligoclonal bands) (9). Second, in selected circumstances—particularly in cases with atypical nonamnesic clinical presentations or early-onset dementia in which AD is in the neuropathological differential diagnosis—we frequently pursue AD biomarkers of molecular neuropathology (10, 11). This is most frequently accomplished with CSF analysis of amyloid-β-42, total tau, and phosphorylated tau levels. Amyloid positron emission tomography (PET) imaging, and most recently tau PET imaging, represent additional options that are approved by the U.S. Food and Drug Administration for clinical use. However, insurance often does not cover amyloid PET and currently does not reimburse tau PET imaging. [18]-F-fluorodeoxyglucose (FDG) PET and perfusion single-photon emission computed tomography imaging may provide indirect evidence for AD neuropathology via a pattern of hypometabolism or hypoperfusion involving the temporoparietal and posterior cingulate regions, though without molecular specificity. Pertinent to this case, a syndromic diagnosis of PCA is most commonly associated with underlying AD neuropathology—that is, plaques containing amyloid-β and neurofibrillary tangles containing tau (12–15).
The patient underwent MRI, demonstrating a minimal burden of T2/FLAIR hyperintensities and some degree of bilateral parietal volume loss with a left greater than right predominance (Figure 2A). There was relatively minimal medial temporal volume loss. Her basic laboratory work-up, including thyroid function, vitamin B12 level, and treponemal antibody, was normal. She underwent a lumbar puncture; CSF studies revealed normal cell counts, protein, and glucose levels and low amyloid-β-42 levels at 165.9 pg/ml [>500 pg/ml] and elevated total and phosphorylated tau levels at 1,553 pg/ml [<350 pg/ml] and 200.4 pg/ml [<61 pg/ml], respectively.
FIGURE 2.
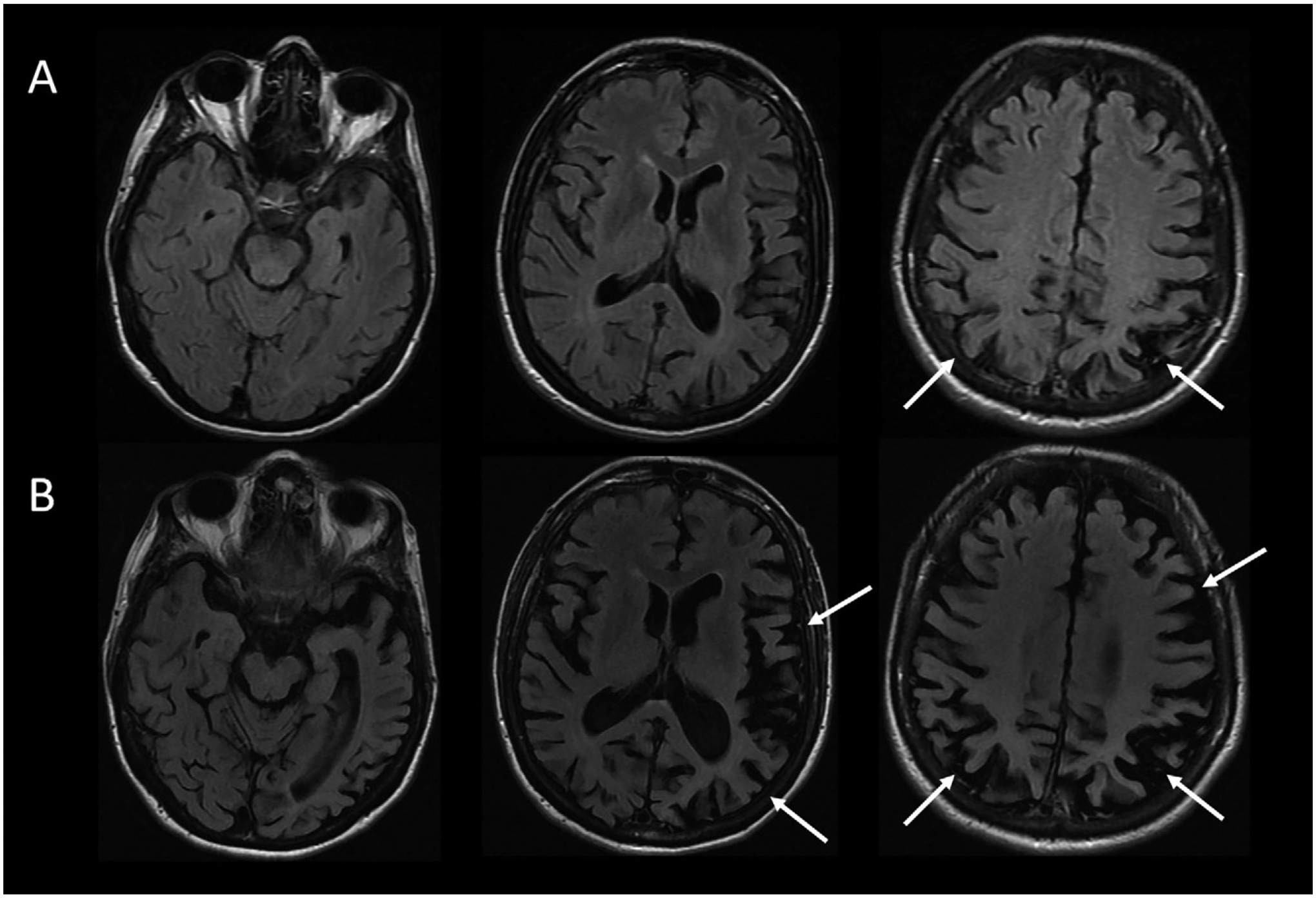
MRI brain scan of the patient at presentation and 4 years latera
a Arrows denote regions of significant atrophy.
Considering This Additional Data, What Would Be an Appropriate Diagnostic Formulation?
For optimal clarity, we aim to provide a three-tiered approach to diagnosis comprising neurodegenerative clinical syndrome (e.g., primary amnesic, mixed amnesic and dysexecutive, primary progressive aphasia), level of severity (i.e., mild cognitive impairment; mild, moderate or severe dementia), and predicted underlying neuropathology (e.g., AD, Lewy body disease [LBD], frontotemporal lobar degeneration) (16). This approach avoids problematic conflations that cause confusion, for example when people equate AD with memory loss or dementia, whereas AD most strictly describes the neuropathology of plaques and tangles, regardless of the patient’s clinical symptoms and severity. This framework is important because there is never an exclusive, one-to-one correspondence between syndromic and neuropathological diagnosis. Syndromes arise from neurodegeneration that starts focally and progresses along the anatomical lines of large-scale brain networks that can be defined on the basis of both structural and functional connectivity, a concept detailed in the network degeneration hypothesis (17). It is important to note that neuropathologies defined on the basis of specific misfolded protein inclusions can target more than one large-scale network, and any given large-scale network can degenerate in association with more than one neuropathology.
The MRI results in this case support a syndromic diagnosis of PCA, with a posteriorly predominant pattern of atrophy. Given the patient’s loss of independent functioning in instrumental activities of daily living (ADLs), including driving and managing her finances, the patient would be characterized as having a dementia (also known as major neurocognitive disorder). The preservation of basic ADLs would suggest that the dementia was of mild severity. The CSF results provide supportive evidence for AD amyloid plaque and tau neurofibrillary tangle (NFT) neuropathology over other pathologies potentially associated with PCA syndrome (i.e., CBD, LBD, TDP-43 proteinopathy, and Jakob-Creutzfeldt disease) (13, 14). The patient’s formulation would thus be best summarized as PCA at a level of mild dementia, likely associated with underlying AD neuropathology.
The patient’s symptoms progressed. One year after initial presentation, she had difficulty locating the buttons on her clothing or the food on her plate. Her word-finding difficulties worsened. Others observed stiffness of her right arm, a new symptom that was not present initially. She also had decreased ability using her right hand to hold everyday objects such as a comb, a brush, or a pen. On exam, she was noted to have rigidity of her right arm, impaired dexterity with her right hand for fine motor tasks, and a symmetrical tremor of the arms, apparent when holding objects or reaching. Her right hand would also intermittently assume a flexed, dystonic posture and would sometime move in complex ways without her having a sense of volitional control.
Four to 5 years after initial presentation, her functional status declined to the point where she was unable to feed, bathe, or dress herself. She was unable to follow simple instructions. She developed neuropsychiatric symptoms, including compulsive behaviors, anxiety, and apathy. Her right-sided motor symptoms progressed; she spent much of the time with her right arm flexed in abnormal postures or moving abnormally. She developed myoclonus of both arms. Her speech became slurred and monosyllabic. Her gait became less steady. She underwent a second MRI of the brain, demonstrating progressive bilateral atrophy involving the frontal and occipital lobes in addition to the parietal lobes and with more left > right asymmetry than was previously apparent (Figure 2B). Over time, she exhibited increasing weight loss. She was enrolled in hospice and ultimately passed away 8 years from the onset of symptoms.
Does Information About the Longitudinal Course of Her Illness Alter the Formulation About the Most Likely Underlying Neuropathological Process?
This patient developed clinical features characteristic of corticobasal syndrome over the longitudinal course of her disease. With time, it became apparent that she had lost volitional control over her right arm (characteristic of an alien limb phenomenon), and she developed signs more suggestive of basal ganglionic involvement (i.e., limb rigidity and possible dystonia). This presentation highlights the frequent overlap between neurodegenerative clinical syndromes; any given person may have elements of more than one syndrome, especially later in the course of a disease. In many instances, symptomatic features that are less prominent at presentation but evolve and progress can provide clues regarding the underlying neuropathological diagnosis. For example, a patient with primary progressive apraxia of speech or nonfluent-agrammatic primary progressive aphasia could develop the motor features of a progressive supranuclear palsy (PSP) clinical syndrome (e.g., supranuclear gaze impairment, axial rigidity, postural instability), which would suggest underlying PSP neuropathology (4-repeat tauopathy characterized by globose neurofibrillary tangles, tufted astrocytes, and oligodendroglial coiled bodies).
If CSF biomarker data were not suggestive of AD, the secondary elements of CBS would substantially increase the likelihood of underlying CBD neuropathology presenting with a PCA syndrome and evolving to a mixed PCA-CBS. But the CSF amyloid and tau levels are unambiguously suggestive of AD (i.e., very low amyloid-β-42 and very high p-tau levels), the neuropathology of which accounts for not only a vast majority of PCA presentations but also roughly a quarter of cases presenting with CBS (18, 19). Thus, underlying AD appears most likely.
NEUROPATHOLOGY
On gross examination, the brain weighed 1,150 g, slightly less than the lower end of normal at 1,200 g. External examination demonstrated mild cortical atrophy with widening of the sulci, relatively symmetrical and uniform throughout the brain (Figure 3A). There was no evidence of atrophy of the brainstem or cerebellum. On cut sections, the hippocampus was mildly atrophic. The substantia nigra in the midbrain was intact, showing appropriate dark pigmentation as would be seen in a relatively normal brain. The remainder of the gross examination was unremarkable.
FIGURE 3.
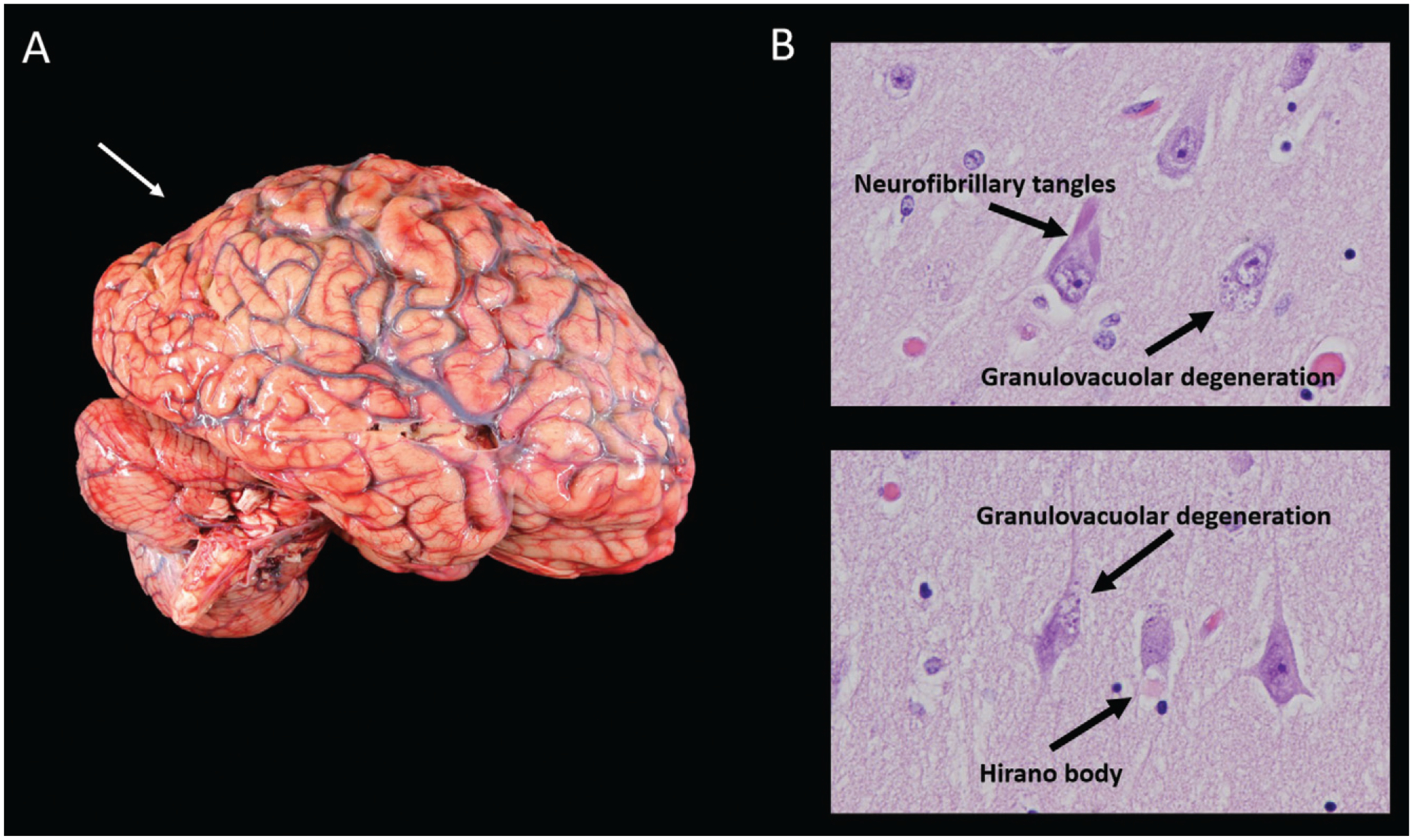
Mild cortical atrophy with posterior predominance and neurofibrillary tangles, granulovacuolar degeneration, and a Hirano bodya
a Panel A shows the gross view of the brain, demonstrating mild cortical atrophy with posterior predominance (arrow). Panel B shows the hematoxylin and eosin of the hippocampus at high power, demonstrating neurofibrillary tangles, granulovacuolar degeneration, and a Hirano body.
Histological examination confirmed that the neurons in the substantia nigra were appropriately pigmented, with occasional extraneuronal neuromelanin and moderate neuronal loss. In the nucleus basalis of Meynert, NFTs were apparent on hematoxylin and eosin staining as dense fibrillar eosinophilic structures in the neuronal cytoplasm, confirmed by tau immunohistochemistry (IHC; Figure 4). Low-power examination of the hippocampus revealed neuronal loss in the subiculum and in Ammon’s horn, most pronounced in the cornu ammonis 1 (CA1) sub-field, with a relatively intact neuronal population in the dentate gyrus. Higher power examination with hematoxylin and eosin demonstrated numerous NFTs, neurons exhibiting granulovacuolar degeneration, and Hirano bodies (Figure 3B). Tau IHC confirmed numerous NFTs in the CA1 region and the subiculum. Amyloid-β IHC demonstrated occasional amyloid plaques in this region, less abundant than tau pathology. An α-synuclein stain revealed scattered Lewy bodies in the hippocampus and in the amygdala.
FIGURE 4.
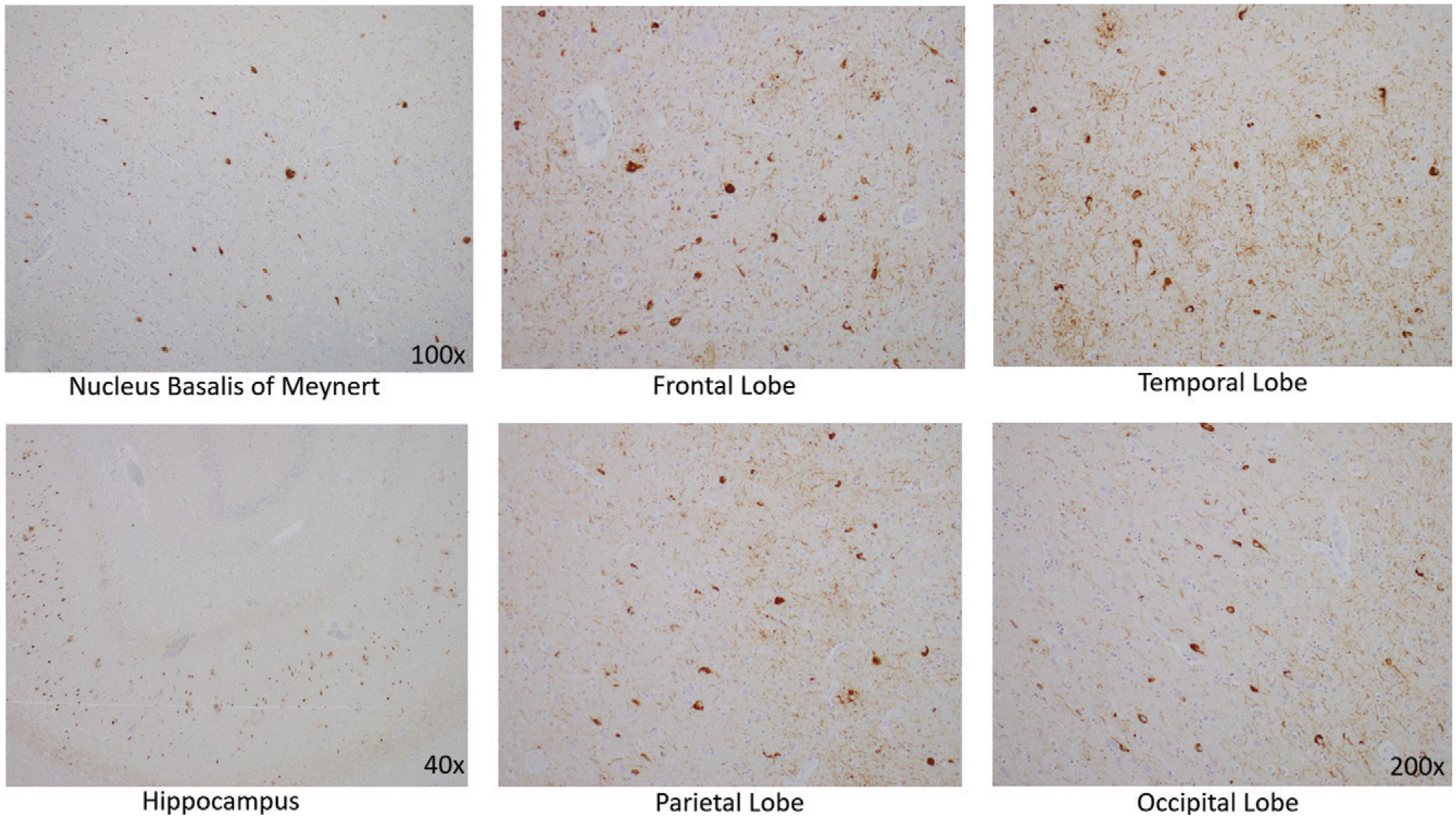
Tau immunohistochemistry demonstrating neurofibrillary tangles (staining brown) in the nucleus basalis of Meynert, in the hippocampus, and in the cerebral cortex of the frontal, temporal, parietal, and occipital lobes
In the neocortex, tau IHC highlighted the extent of the NFTs, which were very prominent in all of the lobes from which sections were taken: frontal, temporal, parietal and occipital. Numerous plaques on amyloid-β stain were likewise present in all cortical regions examined. The tau pathology was confined to the gray matter, sparing white matter. There were no ballooned neurons and no astrocytic plaques—two findings one would expect to see in CBD (Table 2).
TABLE 2.
Neuropathological features of this case compared with a case of corticobasal degenerationa
| Feature | Case of PCA/CBS due to AD | Exemplar case of CBD |
|---|---|---|
| Macroscopic findings | Cortical atrophy: symmetric, mild | Cortical atrophy: often asymmetric, predominantly affecting perirolandic cortex |
| Substantia nigra: appropriately pigmented | Substantia nigra: severely depigmented | |
| Microscopic findings | Tau neurofibrillary tangles and beta-amyloid plaques | Primary tauopathy |
| No tau pathology in white matter | Tau pathology involves white matter | |
| Hirano bodies, granulovacuolar degeneration | Ballooned neurons, astrocytic plaques, and oligodendroglial coiled bodies | |
| (Lewy bodies, limbic) |
AD=Alzheimer’s disease; CBD=corticobasal degeneration; CBS=corticobasal syndrome; PCA=posterior cortical atrophy.
The case was designated by the neuropathology division as Alzheimer’s-type pathology, Braak stage V–VI (of VI), due to the widespread neocortical tau pathology, with LBD primarily in the limbic areas.
COMMENTARY
Our patient had AD neuropathology presenting atypically with a young age at onset (52 years old) and a predominantly visual-spatial and corticobasal syndrome as opposed to prominent amnesia. Syndromic diversity is a well-recognized phenomenon in AD. Nonamnesic presentations include not only PCA and CBS but also the logopenic variant of primary progressive aphasia and a behavioral-dysexecutive syndrome (20). Converging lines of evidence link the topographical distribution of NFTs with syndromic presentations and the pattern of hypometabolism and cortical atrophy. Neuropathological case reports and case series suggest that atypical AD syndromes arise in the setting of higher than normal densities of NFTs in networks subserving the functions compromised, including visual association areas in PCA-AD (21), the language network in PPA-AD (22), and frontal regions in behavioral-dysexecutive AD (23). In a large sample of close to 900 cases of pathologically diagnosed AD employing quantitative assessment of NFT density and distribution in selected neocortical and hippocampal regions, 25% of cases did not conform to a typical distribution of NFTs characterized in the Braak staging scheme (24). A subset of cases classified as hippocampal sparing with higher density of NFTs in the neocortex and lower density of NFTs in the hippocampus had a younger mean age at onset, higher frequency of atypical (nonamnesic) presentations, and more rapid rate of longitudinal decline than subsets defined as typical or limbic-predominant.
Tau PET, which detects the spatial distribution of fibrillary tau present in NFTs, has corroborated postmortem work in demonstrating distinct patterns of tracer uptake in different subtypes of AD defined by clinical symptoms and topographical distributions of atrophy (25–28). Amyloid PET, which detects the spatial distribution of fibrillar amyloid- β found in amyloid plaques, does not distinguish between typical and atypical AD (29, 30). In a longitudinal study of 32 patients at early symptomatic stages of AD, the baseline topography of tau PET signal predicted subsequent atrophy on MRI at the single patient level, independent of baseline cortical thickness (31). This correlation was strongest in early-onset AD patients, who also tended to have higher tau signal and more rapid progression of atrophy than late-onset AD patients.
Differential vulnerability of selected large-scale brain networks in AD and in neurodegenerative disease more broadly remains poorly understood. There is evidence to support multiple mechanisms that are not mutually exclusive, including metabolic stress to key network nodes, trophic failure, transneuronal spread of pathological proteins (i.e., prion-like mechanisms), and shared vulnerability within network regions based on genetic or developmental factors (32). In the case of AD, cortical hub regions with high intrinsic functional connectivity to other regions across the brain appear to have high metabolic rates across the lifespan and to be foci of convergence of amyloid-β and tau accumulation (33, 34). Tau NFT pathology appears to spread temporally along connected networks within the brain (35). Patients with primary progressive aphasia are more likely to have a personal or family history of developmental language-based learning disability (36), and patients with PCA are more likely to have a personal history of mathematical or visuospatial learning disability (37).
This case highlights the symptomatic heterogeneity in AD and the value of a three-tiered approach to diagnostic formulation in neurodegenerative presentations. It is important to remember that not all AD presents with amnesia and that early-onset AD tends to be more atypical and to progress more rapidly than late-onset AD. Multiple lines of evidence support a relationship between the burden and topographical distribution of tau NFT neuropathology and clinical symptomatology in AD, instantiating network-based neurodegeneration via mechanisms under ongoing investigation.
Acknowledgments
Supported by NIH grants K08 AG065502 (to Dr. Miller) and T32 HL007627 (to Dr. Miller).
The authors have confirmed that details of the case have been disguised to protect patient privacy.
Footnotes
The authors report no financial relationships with commercial interests.
Contributor Information
Scott M. McGinnis, Department of Neurology, Division of Cognitive and Behavioral Neurology, Center for Brain/Mind Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston
Andrew M. Stern, Department of Neurology, Division of Cognitive and Behavioral Neurology, Center for Brain/Mind Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston
Jared K. Woods, Department of Pathology, Division of Neuropathology, Brigham and Women’s Hospital, Harvard Medical School, Boston
Matthew Torre, Department of Pathology, Division of Neuropathology, Brigham and Women’s Hospital, Harvard Medical School, Boston
Mel B. Feany, Department of Pathology, Division of Neuropathology, Brigham and Women’s Hospital, Harvard Medical School, Boston
Michael B. Miller, Department of Pathology, Division of Neuropathology, Brigham and Women’s Hospital, Harvard Medical School, Boston
David A. Silbersweig, Department of Psychiatry, Center for Brain/Mind Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston
Seth A. Gale, Department of Neurology, Division of Cognitive and Behavioral Neurology, Center for Brain/Mind Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston
Kirk R. Daffner, Department of Neurology, Division of Cognitive and Behavioral Neurology, Center for Brain/Mind Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston
REFERENCES
- 1.Balasa M, Gelpi E, Antonell A, et al. : Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology 2011; 76:1720–1725 [DOI] [PubMed] [Google Scholar]
- 2.Mercy L, Hodges JR, Dawson K, et al. : Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 2008; 71:1496–1499 [DOI] [PubMed] [Google Scholar]
- 3.Kothbauer-Margreiter I, Sturzenegger M, Komor J, et al. : Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment. J Neurol 1996; 243:585–593 [DOI] [PubMed] [Google Scholar]
- 4.Nasreddine ZS, Phillips NA, Bédirian V, et al. : The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005; 53:695–699 [DOI] [PubMed] [Google Scholar]
- 5.Rizzo M, Vecera SP: Psychoanatomical substrates of Bálint’s syndrome. J Neurol Neurosurg Psychiatry 2002; 72:162–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rusconi E: Gerstmann syndrome: historic and current perspectives. Handb Clin Neurol 2018; 151:395–411 [DOI] [PubMed] [Google Scholar]
- 7.Crutch SJ, Schott JM, Rabinovici GD, et al. : Consensus classification of posterior cortical atrophy. Alzheimers Dement 2017; 13: 870–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armstrong MJ, Litvan I, Lang AE, et al. : Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80:496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall GA, Doyle JJ: Long-term treatment of Hashimoto’s encephalopathy. J Neuropsychiatry Clin Neurosci 2006; 18:14–20 [DOI] [PubMed] [Google Scholar]
- 10.Johnson KA, Minoshima S, Bohnen NI, et al. : Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement 2013; 9:e-1–e-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw LM, Arias J, Blennow K, et al. : Appropriate use criteria for lumbar puncture and cerebrospinal fluid testing in the diagnosis of Alzheimer’s disease. Alzheimers Dement 2018; 14:1505–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alladi S, Xuereb J, Bak T, et al. : Focal cortical presentations of Alzheimer’s disease. Brain 2007; 130:2636–2645 [DOI] [PubMed] [Google Scholar]
- 13.Renner JA, Burns JM, Hou CE, et al. : Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 2004; 63: 1175–1180 [DOI] [PubMed] [Google Scholar]
- 14.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. : Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004; 63:1168–1174 [DOI] [PubMed] [Google Scholar]
- 15.Victoroff J, Ross GW, Benson DF, et al. : Posterior cortical atrophy: neuropathologic correlations. Arch Neurol 1994; 51:269–274 [DOI] [PubMed] [Google Scholar]
- 16.Dickerson BC, McGinnis SM, Xia C, et al. : Approach to atypical Alzheimer’s disease and case studies of the major subtypes. CNS Spectr 2017; 22:439–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seeley WW, Crawford RK, Zhou J, et al. : Neurodegenerative diseases target large-scale human brain networks. Neuron 2009; 62: 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SE, Rabinovici GD, Mayo MC, et al. : Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011; 70:327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitwell JL, Jack CR Jr, Boeve BF, et al. : Imaging correlates of pathology in corticobasal syndrome. Neurology 2010; 75:1879–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warren JD, Fletcher PD, Golden HL: The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol 2012; 8:451–464 [DOI] [PubMed] [Google Scholar]
- 21.Hof PR, Archin N, Osmand AP, et al. : Posterior cortical atrophy in Alzheimer’s disease: analysis of a new case and re-evaluation of a historical report. Acta Neuropathol 1993; 86:215–223 [DOI] [PubMed] [Google Scholar]
- 22.Mesulam MM, Weintraub S, Rogalski EJ, et al. : Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain 2014; 137:1176–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blennerhassett R, Lillo P, Halliday GM, et al. : Distribution of pathology in frontal variant Alzheimer’s disease. J Alzheimers Dis 2014; 39:63–70 [DOI] [PubMed] [Google Scholar]
- 24.Murray ME, Graff-Radford NR, Ross OA, et al. : Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011; 10: 785–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ossenkoppele R, Lyoo CH, Sudre CH, et al. : Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer’s disease. Alzheimers Dement 2020; 16:335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips JS, Das SR, McMillan CT, et al. : Tau PET imaging predicts cognition in atypical variants of Alzheimer’s disease. Hum Brain Mapp 2018; 39:691–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tetzloff KA, Graff-Radford J, Martin PR, et al. : Regional distribution, asymmetry, and clinical correlates of tau uptake on [18F]AV-1451 PET in atypical Alzheimer’s disease. J Alzheimers Dis 2018; 62:1713–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia C, Makaretz SJ, Caso C, et al. : Association of in vivo [18F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 2017; 74:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Formaglio M, Costes N, Seguin J, et al. : In vivo demonstration of amyloid burden in posterior cortical atrophy: a case series with PET and CSF findings. J Neurol 2011; 258:1841–1851 [DOI] [PubMed] [Google Scholar]
- 30.Lehmann M, Ghosh PM, Madison C, et al. : Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain 2013; 136:844–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.La Joie R, Visani AV, Baker SL, et al. : Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med 2020; 12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou J, Gennatas ED, Kramer JH, et al. : Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 2012; 73:1216–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buckner RL, Sepulcre J, Talukdar T, et al. : Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci 2009; 29: 1860–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoenig MC, Bischof GN, Seemiller J, et al. : Networks of tau distribution in Alzheimer’s disease. Brain 2018; 141:568–581 [DOI] [PubMed] [Google Scholar]
- 35.Liu L, Drouet V, Wu JW, et al. : Trans-synaptic spread of tau pathology in vivo. PLoS One 2012; 7:e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogalski E, Johnson N, Weintraub S, et al. : Increased frequency of learning disability in patients with primary progressive aphasia and their first-degree relatives. Arch Neurol 2008; 65: 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller ZA, Rosenberg L, Santos-Santos MA, et al. : Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol 2018; 75:728–737 [DOI] [PMC free article] [PubMed] [Google Scholar]