

Abstract
Failure to establish immune tolerance leads to the development of autoimmune disease. The ability to regulate autoreactive T cells without inducing systemic immunosuppression represents a major challenge to develop new strategies to treat autoimmune disease. Here, we describe a translational method for bioengineering programmed death-ligand 1 and cluster of differentiation 86-functionalized mouse Schwann cells to prevent and ameliorate multiple sclerosis in established mouse models of chronic and relapsing-remitting experimental autoimmune encephalomyelitis (EAE). We show here that the intravenous administration of immune checkpoint ligand-functionalized mouse Schwann cells modifies the course of disease and ameliorates EAE. Further, we found that such bioengineered mouse Schwann cells inhibit the differentiation of myelin-specific helper T cells into pathogenic T helper type 1 and type 17 cells, promote the development of tolerogenic myelin-specific regulatory T cells and resolve inflammatory CNS microenvironments without inducing systemic immunosuppression.
Keywords: Experimental Autoimmune Encephalomyelitis, Immune Checkpoint, Metabolic Glycoengineering, Bioorthogonal Click Chemistry, Schwann Cells
A translational method for bioengineering programmed death-ligand 1 and cluster of differentiation 86-functionalized Schwann cells to prevent and ameliorate multiple sclerosis has been developed.
The immune system evolved robust immune responses against foreign antigens while tolerating self-antigens to avoid autoimmunity.[1, 2] Regulatory T (Treg) cells regulate homeostasis and maintain immunotolerance.[3] Failure to maintain immunotolerance leads to the development of autoimmune disease.[1, 4, 5] For example, autoreactive T cells attack the myelin in the central nervous system (CNS), causing the autoimmune neurological disorder multiple sclerosis (MS), which disrupts communication between the brain and peripheral system.[4, 6] At least 2.5 million people worldwide are affected by MS. Most patients initially experience episodes of reversible neurological deficits, followed by remission, before chronic neurological deterioration leads to severe, irreversible disabilities.[6] Unfortunately, MS cannot be completely cured, although available immunomodulatory therapies reduce the frequency and severity of MS relapses by inducing antigen-specific immunotolerance,[7] thus delaying the accumulation of disabilities. New treatment strategies involve the induction of antigen-specific Treg cells[8, 9] that suppress inflammatory pathogens and restore peripheral immunotolerance without causing systemic immunosuppression.
Immune checkpoints play key roles in maintaining immunotolerance.[10] For example, studies have found that coinhibitory immune checkpoint pathways such as programmed death 1 (PD1)-PD ligand 1 (PD-L1),[10, 11] and cytotoxic T lymphocyte associated protein 4 (CTLA-4)-cluster of differentiation 86 (CD86)[10, 12] directly regulate the development and maintenance of myelin-specific induced Treg cells.[13]. Here, we aimed to determine whether the intravenous (i.v.) administration of coinhibitory immune checkpoint ligand–bioengineered glia can be used as a prophylactic treatment to prevent the development of MS or use in therapeutic treatment to ameliorate active MS symptoms through the inhibition of pathogenic CD4+ lymphocyte T helper type 1 (Th1) and type 17 (Th17) cells and promote the development of myelin-specific Treg cells (Figure 1). Further, we investigated the possibility that creating a less proinflammatory CNS microenvironment through local cotreatment with an immunomodulatory drug (e.g., leflunomide (LEF)[14]) would confer the ability of oligodendrocytes (OLs) to repair myelin damage[15] and ameliorate MS symptoms (Figure 1). To achieve this, we utilized recent advances in bionanotechnology to bioengineer Schwann cells (SCs) (glial cells of the peripheral nervous system) with LEF-encapsulated nanoparticles (NPs) functionalized with PD-L1 and CD86 to upregulate the PD-1 and CTLA-4 signaling pathways in the engaged myelin-specific CD4+ T cells (Figure 1). We focused on SCs because they express diverse myelin-specific antigens such as myelin oligodendrocyte glycoprotein (MOG) and proteolipid protein (PLP) (Figure S1, Supporting Information). Furthermore, protocols were established to isolate SCs from the sural nerve and ex vivo proliferate for autologous SC transplant.[16]
Figure 1.
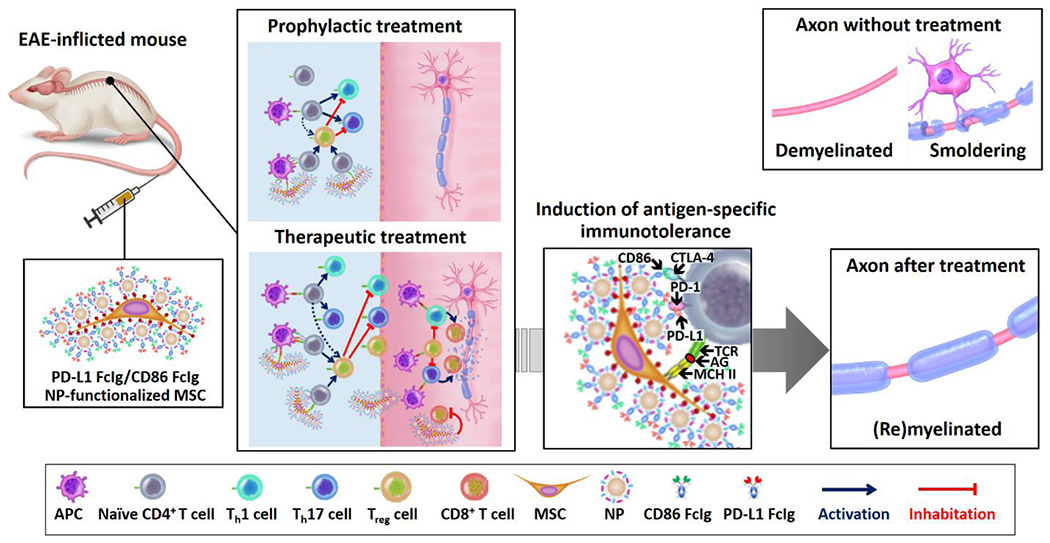
PD-L1- and CD86-functionalized MSCs prevent and ameliorate active EAE in the mouse. The scheme illustrates the mechanism of actions of drug-free and LEF-encapsulated PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs to prevent and treat EAE in the mouse. The myelin antigen-rich PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs can simultaneously present the myelin antigen to the myelin-specific CD4+ T cells and inhibit PD-1/PD-L1 and CTLA-4/CD86 immune checkpoint pathways. In prophylactic treatment, the i.v. administered functionalized MSCs inhibit the activation of myelin-specific CD4+ T cells and the subsequent differentiation into pathogenic Th1 and Th17 cells, and promote the development of myelin-specific Treg cells. In therapeutic treatment, the functionalized MSCs inhibit the activation of myelin-specific CD4+ T cells, reduce the pathogenic Th1 and Th17 cells, and promote the development of antigen-specific Treg cells. In addition, the induced Treg cells and i.v. administered MSCs can enter the CNS to inhibit the activation of pathogenic Th1 and Th17 cells and cytotoxic T cells. Furthermore, the encapsulated LEF release inside the CNS directly inhibits the proliferation of autoreactive CD4+ and CD8+ T cells and generates a less proinflammatory CNS microenvironment for the OL to repair the damaged myelin sheaths. The antigen-specific immunotherapy effectively prevents systemic immune suspension. (AG = antigen, TCR = T cell receptor, MCH II = major histocompatibility complex class II.)
Immune checkpoint ligand-functionalized MSCs were bioengineered via metabolic glycoengineering followed by the bioorthogonal click reaction.[17, 18] We evaluated direct bioconjugation (Figure 2a) and NP pre-anchoring conjugation (Figure 2b–c) strategies to functionalize the MSCs. These strategies employed azide-modified MSCs obtained by culturing MSCs with a subcytotoxic concentration of N-azidoacetylmannosamine tetraacylated (Ac4MaNAz; Figure S2, Supporting Information).[18] MSCs take up the ManNAz and convert it to azide-sialic acid derivatives to achieve N-linked glycosylate of cell surface proteins.[17] These azide-sialic acid derivatives on the surface of the glia provide sites for bioorthogonal strain-promoted azide-alkyne cycloaddition (SPAAC; Figure 2a(i)).[17] In the direct functionalization method, dibenzocyclooctyne (DBCO)-functionalized PD-L1 Fc-fusion proteins (PD-L1 FcIg) and CD86 Fc-fusion proteins (CD86 FcIg)[19, 20] (Figure S3a–c, Supporting Information) were directly conjugated to azide-modified MSCs through SPAAC[17, 18] at a target degree of conjugation of 5 μg of fusion protein per one million cells (Figue 2a). The NP pre-anchoring conjugation strategy involved the preparation of drug-free and LEF-encapsulated DBCO- and methyltetrazine (MTZ)-functionalized NPs (DBCO/MTZ NPs) via the nanoprecipitation method (Figure 2b).[20] The LEF-encapsulated DBCO/MTZ NPs (LEF NPs) were encapsulated with 3.3 wt/wt% of LEF, as determined by fluorescence spectroscopy.[21] By using fluorescence spectroscopy method[21] to quantify the amount of LEF retained in the NPs, we determined that the encapsulated LEF controlled release under physiological conditions (half-life 15.0 ± 0.3 h) (Figure 2b). We next conjugated DBCO/MTZ NPs to azide-modified MSCs via SPAAC at a target degree of conjugation of 500 μg NPs per one million cells (Figure 2c). We then conjugated TCO-functionalized PD-L1 FcIg and CD86 FcIg to the NP-functionalized MSCs through the inverse electron-demand Diels-Alder (IEDDA) reaction[22] with the same target degree of functionalization as for the directly functionalized MSCs (Figure 2c; Figure S3d, Supporting Information). Neither bioconjugation strategy did not significantly affected the size (Figure 2a–c) or viability of the MSCs (Figure S2, Supporting Information).
Figure 2.
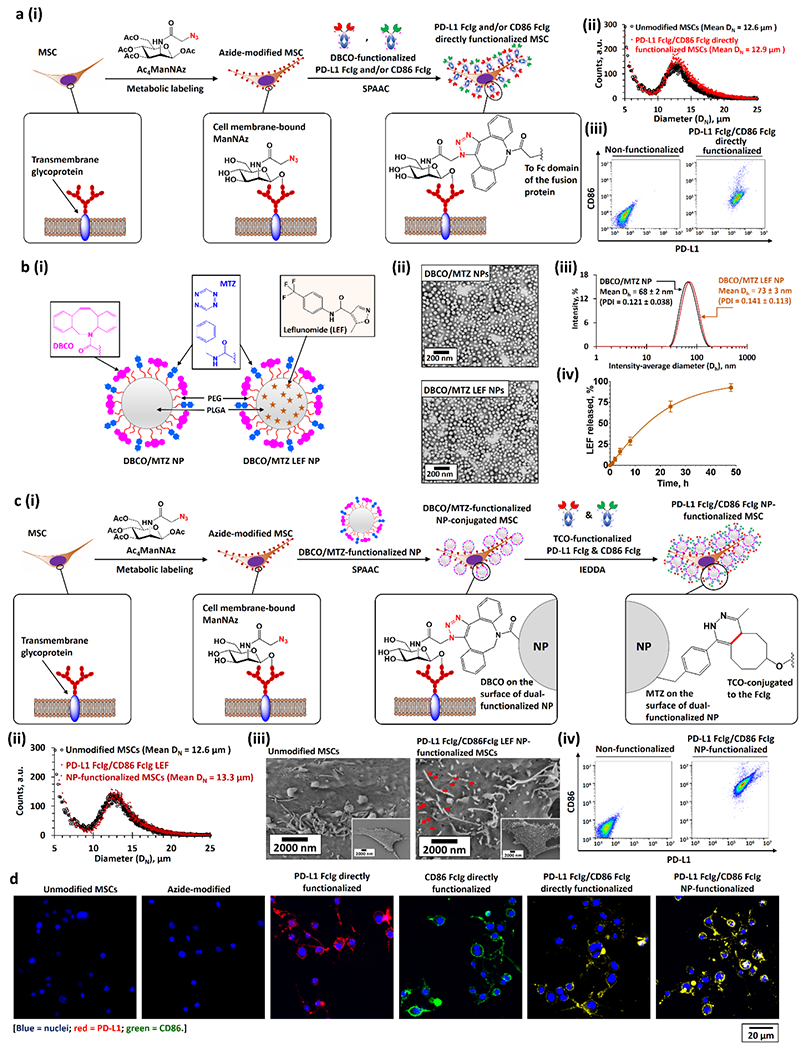
Bioengineering of PD-L1 and CD86 functionalized MSCs. a)(i) Bioengineering PD-L1 Fc-Ig and CD86 Fc-Ig directly functionalized MSCs through metabolic glycoengineering followed by SPAAC with DBCO-functionalized PD-L1 Fc-Ig and CD86 Fc-Ig. (ii, iii) Size distributions (ii), PD-L1, and CD86 expressions (iii) of unmodified and PD-L1 Fc-Ig and CD86 Fc-Ig directly functionalized MSCs. b)(i) Structures of drug-free DBCO and MTZ dual-functionalized PEG-PLGA NPs (DBCO/MTZ NPs) and LEF-encapsulated DBCO/MTZ NPs (DBCO/MTZ LEF NPs). (ii, iii) TEM images (ii), and intensity-average diameter (Dh) distributions (iii) of drug-free and LEF-encapsulated DBCO and MTZ dual-functionalized NPs. (iv) Drug-release profile of LEF-encapsulated DBCO and MTZ dual-functionalized NPs at physiological conditions in the presence of large excess of PBS. c)(i) Bioengineering of PD-L1 Fc-Ig and CD86 Fc-Ig NP-dual-functionalized MSCs. The dual-functionalized MSCs were engineered via 3 steps: first, metabolic labeling of Ac4ManNAZ gave azide-modified MSCs; second, the conjugation of DBCO/MTZ NPs (or DBCO/MTZ LEF NPs) onto the azide-modified MSCs through SPAAC at the physiological conditions; and finally, the bioconjugation of TCO-functionalized PD-L1 Fc-Ig and CD86 Fc-Ig onto the DBCO/MTZ NP-functionalized MSCs via IEDDA at the physiological conditions. (ii–iv) Size-distributions (ii), scanning electron microscopy (SEM) images (iii), and PD-L1 and CD86 expressions (iv) of unmodified and PD-L1 Fc-Ig and CD86 Fc-Ig NP-functionalized MSCs. Pseudopodia can be identified from the SME images of both unmodified and functionalized MSCs. The red arrows in the SEM images highlighted the PD-L1 FcIg/CD86 FcIg LEF NPs grafted on the surface of the MSCs. d) Representative CLSM images of different as-functionalized MSCs.
When we used A488-labeled PD-L1 FcIg and Texas Red–labeled CD86 FcIg (Figure S4, Supporting Information) for the bioconjugation, between 68% and 72% of the DBCO-functionalized fusion proteins were directly conjugated to the azide-modified MSCs (Figure S5, Supporting Information). When we functionalized using Cy5-labeled DBCO/MTZ NPs, 35 ± 5 μg of the NPs were conjugated to one million of the MSCs (and thus 1.16 μg of encapsulated LEF for the LEF NP-functionalized MSCs; Figure S6, Supporting Information), which allowed a quantitative conjugation of TCO-functionalized fusion proteins (i.e., 5 μg of TCO-functionalized fusion protein per million cells). Fluorescence-activated cell sorting (FACS) assay further confirmed that PD-L1 FcIg and CD86 FcIg were conjugated to the MSCs (Figure 2a,c; Figure S7, Supporting Information). The levels of PD-L1 and CD86 expressed by the directly functionalized MSCs declined much faster those functionalized through the NP pre-anchoring strategy because of cell proliferation and metabolic clearance (Figure S7,S8, Supporting Information).[17] The functionalization of MSCs was further confirmed by confocal laser scanning microscopy (CLSM) staining with A488-labeled anti-PD-L1 and phycoerythrin (PE)-labeled anti-CD86 antibodies (Figure 2d; Figure S9, Supporiting Information). Further, scanning electron microscopy indicated equal distribution of the conjugated PD-L1 FcIg/CD86 FcIg LEF NPs on the surface of the MSCs (Figure 2c(iii)).
To evaluate the effects of MSC-conjugated PD-L1, CD86, and encapsulated LEF on antigen-specific CD4+ T cell activation, we cultured mono- and dual-functionalized MSCs with MOG-specific CD4+ T cells isolated from 2D2 mice (2D2 cells)[23, 24] and quantified the PD-1 and CTLA-4 levels expressed by the 2D2 cells. Both types of directly monofunctionalized MSCs effectively upregulated the corresponding immune checkpoint pathway (Figure 3–b; Figure S10, Supporting Information). A 1:1 combination of both monofunctionalized MSCs and dual-functionalized MSCs concurrently upregulated both immune checkpoint pathways in 2D2 cells (Figure 3a–b; Figure S10, Supporting Information), but the upregulations were less effective than they were with the same amount of monofunctionalized MSCs. The drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs were as effective as the combination of two directly functionalized MSCs to upregulate the PD-1 and CTLA-4 expressions of 2D2 cells (Figure 3a–b; Figure S10, Supporting Information). Similar to the results of previous study,[25] small-molecule LEF upregulated CD86 expression in the MSCs and therefore increased the expression of CTLA-4 in the co-cultured 2D2 cells (Figure 3b; Figure S10, Supporting Information). Thus, PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs were more effective than drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs and dual directly functionalized MSCs in upregulating the CTLA-4 pathway (Figure 3b; Figure S10, Supporting Information). The remarkable upregulation of both inhibitory immune checkpoint pathways by the cocultured PD-L1 and CD86 dual-functionalized MSCs significantly reduced the level of effector molecules when evaluating the interferon gamma (IFN-γ, secreted from Th1 cells)[24, 26] and interleukin 17A (IL-17A, secreted from Th17 cells)[24, 27] secreted by the 2D2 cells through enzyme-linked immunosorbent assay (Figure 3c–d).
Figure 3.
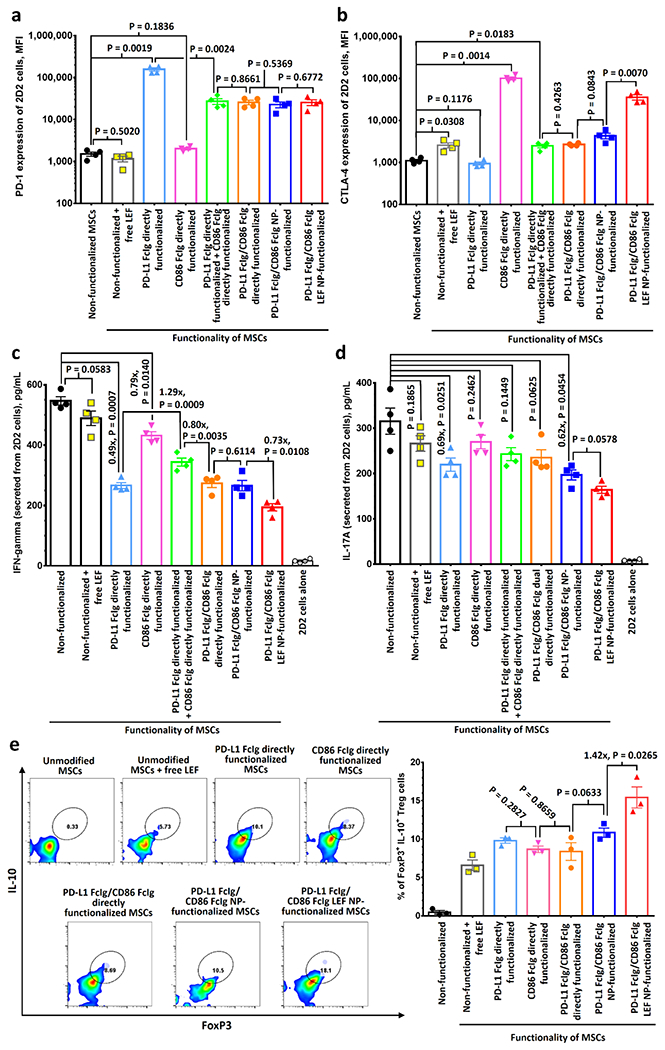
PD-L1- and CD86-functionalized MSCs upregulate PD-1 and CTLA-4 pathways in myelin-specific T cells, downregulate T cell activation and promote the development of induced regulatory T cells in vitro. a,b) PD-1 (a), and CTLA-4 (b) expressions of myelin-specific 2D2 T cells after incubated with different types of PD-L1 FcIg- and/or CD86 FcIg-functionalized MSCs for 48 h, as determined by FACS assay. Cells were initially gated at CD3+ cells. (n = 4) c,d) ELISA analysis of INF-gamma (c) and IL-17A (d) secreted from 2D2 CD4+ T cells after incubated with different functionalized MSCs. Supernatants were collected 48 h post-incubation for the ELISA analysis. (n = 4) e) Quantification of IL-10+ and FoxP3+ population in 2D2 CD4+ T cells after incubated with different functionalized MSCs for 48 h via FACS. Cells were initially gated at CD3+ cells. (n = 3) Statistical significance between two control/experimental groups were accessed by two-way ANOVA with Tukey correction. Data represent mean ± s.e.m.
To determine whether PD-L1- and CD86-functionalized MSCs can promote the development of antigen-specific induced Treg cells, we quantified the population of FoxP3+ and IL10+ CD4+ T cells after culturing the 2D2 cells with different functionalized MSCs for 72 h.[13, 24] Incubation with unmodified MSCs in the presence of small-molecule LEF induced approximately 6% of 2D2 cells to develop into Treg cells (Figure 3e; Figure S11, Supporting Information). All directly functionalized MSCs promoted the development of induced Treg cells, as indicated by finding that 8-10% of the CD4+ expressed cells were FoxP3+ and IL10+ (Figure 3e; Figure S11, Supporting Information). The drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs were as effective as the directly dual-functionalized MSCs in promoting native 2D2 cells to develop into induced Treg cells. In contrast, the LEF-encapsulated NP-functionalized MSCs were 42% more effective than those of the drug-free NP-functionalized MSCs in their ability to transform native 2D2 cells into induced Treg cells (Figure 3e; Figure S11, Supporting Information).
To demonstrate that PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs can directly inhibit the activation of CD8+ T cells and thus reduce inflammation in the CNS, we performed a carboxyfluorescein succinimidyl ester (CFSE) assay to quantify the proliferation of stimulated CD8+ T cells (isolated from wild-type C57BL/6 mice) after culturing them with drug-free and LEF-encapsulated PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (Figure S12, Supporting Information). The mean fluorescence intensity (MFI) of CFSE-labeled CD8+ T cells cocultured with PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs was 5.6 times higher than compared with that of these cells cultured with the unmodified MSCs (Figure S12, Supporting Information). These findings indicate that conjugated PD-L1[28] and CD86[29] effectively inhibited the proliferation of stimulated CD8+ T cells, independent of the antigen. The MFI of CD8+ T cells cocultured with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs was 4.5 times higher than compared with that of the MFI of cells cultured with drug-free functionalized MSCs (Figure S12, Supporting Information). These findings show that the encapsulated LEF released from the NPs inhibited the proliferation of activated CD8+ T cells in vitro.
To determine whether i.v. administration of PD-L1- and CD86-functionalized MSCs can ameliorate animal model of autoimmune inflammatory disease of the CNS, we used a monophasic chronic MOG35-55-induced EAE model because it is the best-characterized model to develop therapies for MS.[30] When we performed an in vivo toxicity study, we found that i.v. administration of unmodified and PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (2×106 cells per mouse, the maxinium number of cells that can be administrated for each i.v. injection[31]) did not induce detectable hepatotoxicity, and nephrotoxicity in healthy C57BL/6 mice (Figure S13, Supporting Information). In addition, we did not observe any breathing problems or weight loss after the i.v. administration of MSCs. All healthy mice survived the in vivo toxicity study.
To demonstrate a prophylactic effect, we i.v. administered MSCs 24h post-immunization (p.i.) with MOG35-55. The administration of unmodified MSCs did not significantly affect disease progression or severity (Figure 4a). Tail and hindlimb paralysis (EAE score ≥ 2.5) were observed between 18 and 22 days p.i. Prophylactic treatments with PD-L1 FcIg or CD86 FcIg directly mono-functionalized MSCs did not significantly delay disease onset, though both treatments reduced severity as indicated by maximum EAE scores p.i. and cumulative EAE scores by 60% and 40% (Figure 4b–c; Figure S14a–b, Supporting Information), respectively. Although prophylactic treatment with dual-functionalized MSCs did not completely prevent the development of EAE, its severity was significantly reduced (only 1 of 9 treated mice experienced partial hindlimb paralysis, EAE score ≥ 2.0) (Figure 4b–c; Figure S14a–b, Supporting Information). Spinal inflammation and demyelination in mice with EAE are markers of severity of clinical signs.[32] Histological studies (Figure 4d–e; Figure S15–S16, Supporting Information) through hematoxylin and eosin (H&E) stain and Luxol fast blue stain (LFB stain, stains for myelin[33]) revealed that prophylactic treatment with PD-L1 FcIg/CD86 FcIg directly dual-functionalized MSCs reduced spinal inflammation by an average of 81% and demyelination by 76% compared with untreated mice at the study endpoint (36 or 37 days p.i.).
Figure 4.
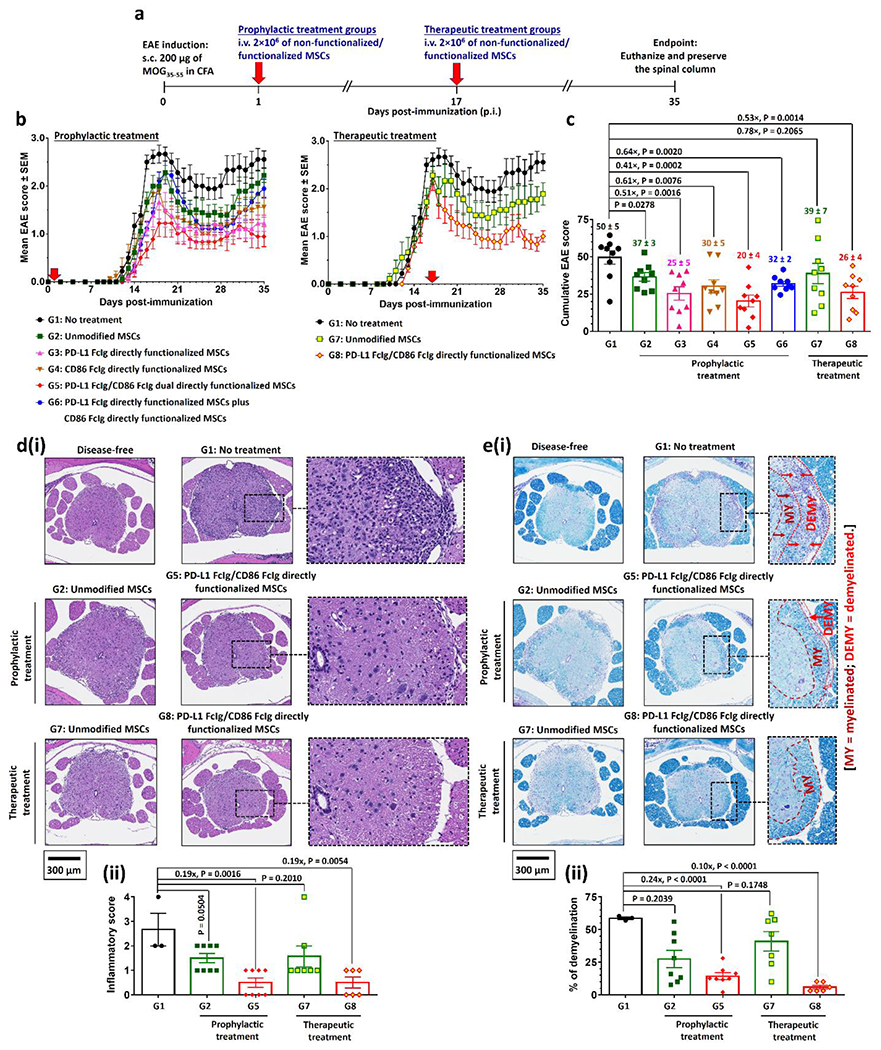
PD-L1 FcIg and CD86 FcIg directly functionalized MSCs prophylactically and therapeutically suppress MOG35-55-induced EAE in vivo. a) Prophylactic and therapeutic treatment schedules after immunization with MOG35-55 peptide. 2×106 of unmodified or functionalized MSCs were i.v. administrated 1 day (prophylactic treatment) or 17 days (therapeutic treatment) post-immunization (p.i.). Body conditions were monitor daily until day 35 p.i. Mice were euthanized day 36 or 37 p.i. The spinal columns were preserved for further histopathological studies. b) Time-dependent mean clinical scores of EAE inflicted mice after received different prophylactic or therapeutic treatments. In the absence of treatment, EAE progress from partial tail paresis (score 0.5), complete tail paresis (score 1.0), limp tail and hind leg inhibition (score 1.5), limp tail and weakness of hind legs (score 2.0), limp tail and no movement in one leg (score 2.5), to complete hind limb paralysis (score 3.0). (n = 9 mice per group.) c) Cumulative EAE scores of EAE inflicted mice after received different treatments. d)(i) Representative hematoxylin and eosin (H&E)-stained spinal cord sections preserved from healthy disease-free mouse and EAE-inflicted mice after received different prophylactic and therapeutic treatments with directly functionalized MSCs. (ii) Quantification of spinal inflammation from the H&E-stained images of spinal cords. (n = 3 for the non-treatment group; n = 8 for both prophylactic treatment groups; n = 7 for therapeutic treatment group treated with the non-functionalized MSCs; n = 6 for the therapeutic treatment group treated with the functionalized MSCs.) e)(i) Representative Luxol fast blue (LFB)-stained spinal cord sections preserved from healthy disease-free mouse and EAE-inflicted mice after received different prophylactic and therapeutic treatments with directly functionalized MSCs. Myelin fibers and phospholipids appear blue to green, neuropil appears pink, and nerve cells appear purple. (ii) Quantification of demyelination from the LFB-stained images of spinal cords. (n = 3 for the non-treatment group; n = 8 for both prophylactic treatment groups; n = 7 for therapeutic treatment group treated with the non-functionalized MSCs; n = 6 for the therapeutic treatment group treated with the functionalized MSCs.)
We therefore investigated the effects of treating EAE mice with PD-L1 and CD86 dual-functionalized MSCs after disease onset (Figure 4b–c; Figure S14c–d, Supporting Information). Therapeutic treatment with PD-L1 FcIg/CD86 FcIg directly functionalized MSCs significantly reduced cumulative EAE scores by 50% (Figure 4b–c; Figure S14c–d, Supporting Information), whereas the administration of unmodified MSCs only slighly reduced the cumulative EAE score (P = 0.2065, Figure 4c). At the study endpoint (35 days p.i.), 7 of 9 treated mice no longer suffered detectable hindlimb weakness, whereas at least one hindlimb of the untreated mice was completely paralyzed (EAE score ≥ 2.5; Figure 4b–c; Figure S14c–d, Supporting Information). Histological studies showed that therapeutic treatment with dual-functionalized MSCs reduced spinal inflammation and demyelination by 81% and 90%, respectively, compared with those of untreated EAE mice 36 or 37 days p.i. (Figure 4d–e; Figure S15–S16, Supporitng Information).
Considering the improved abilities of NP-functionalized MSCs to suppress pathogenic CD4+ T cell activation and to facilitate the development of antigen-specific Treg cells in vitro (Figure 3), we further investigated the abilities of drug-free and LEF-encapsulated PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs to prevent the development and serve as a treatment for mice with EAE (Figure 5a). Similar to unmodified MSCs alone (Figure 4b–c; Figure S14, Supporting Information), prophylactic treatment with PD-L1 FcIg and CD86 FcIg followed by unmodified MSCs (20 min apart) did not prevent or change the course of disease compared with untreated mice (Figure 5b–c; Figure S17, Supporting Information). Prophylactic treatment with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs did not completely prevent the onset of disease, although such treatment was 12% more effective than PD-L1 FcIg/CD86 FcIg directly functionalized MSCs for reducing cumulative EAE scores upon completion of the study (4 of 8 treated mice suffered partial tail paresis (EAE score = 0.5)) (Figure 5b–c; Figure S17a–b, Supporting Information). However, prophylactic treatment with LEF-encapsulated PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs did not further reduce the severity of EAE symptoms than drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (Figure S18, Supporting Information). Control prophylactic studies indicated that i.v. administration of small-molecule LEF with unconjugated PD-L1 FcIg and CD86 FcIg, or PD-L1 FcIg/CD86 FcIg LEF NPs followed by unmodified MSCs in treated mice did not inhibit the development of EAE or reduce the severity of the disease compared with untreated mice (Figure 5b–c; Figure S17a–b, Supporitng Information). Similarly, histological analysis showed that treatment with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs was as effective as treatment with dual-functionalized MSCs, reducing spinal inflammation by 87% and demyelination by 89% compared with the results for untreated mice (Figure 5d–e; Figure S19–S20, Supporting Information).
Figure 5.
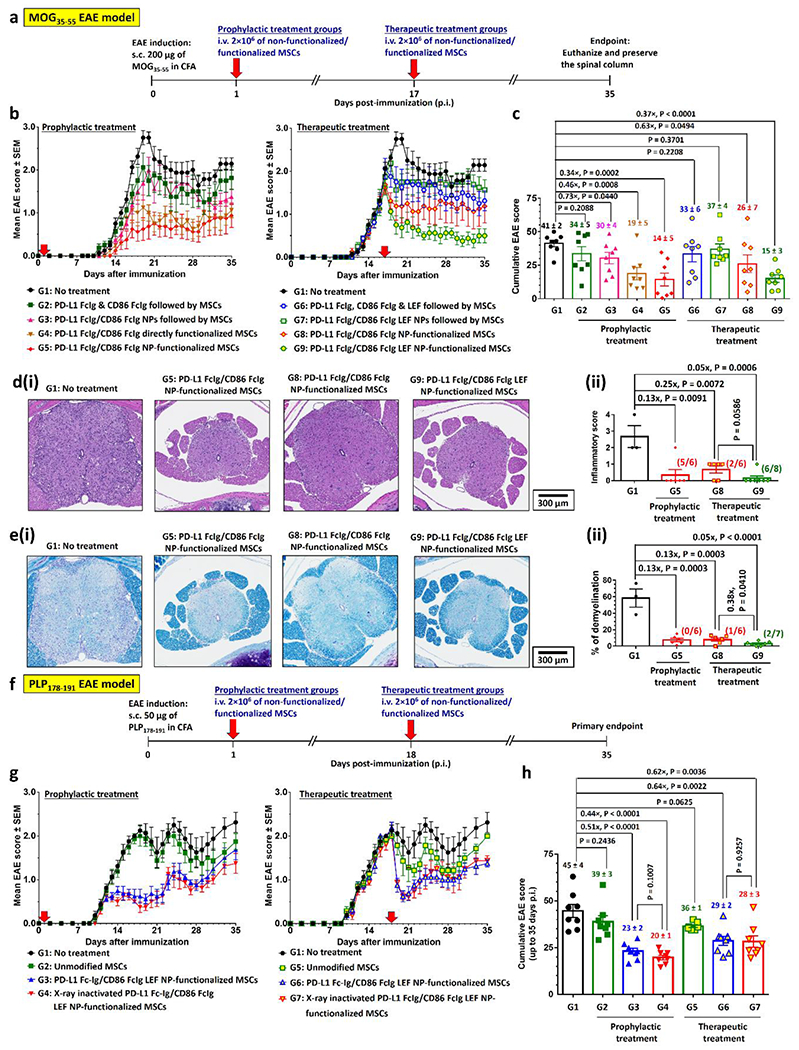
PD-L1- and CD86-conjugated NP-functionalized MSCs effectively suppress progressive chronic MOG35-55-EAE model and relapsing-remitting PLP178-191-EAE model in vivo, prophylactically, and therapeutically. a) Prophylactic and therapeutic treatment schedules with PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs in C57BL/6 mice after immunization with MOG35-55 peptide. Unmodified or functionalized MSCs were i.v. administrated 1 day (prophylactic treatment) or 17 days (therapeutic treatment) p.i.. Body conditions were monitor daily until 35 days p.i. Mice were euthanized 36 or 37 days p.i., spinal columns were preserved for further histopathological studies. In control treatment groups 2, 3, 6, and 7, free or NP conjugated PD-L1 Fc-Ig, and CD86 Fc-Ig (plus unencapsulated LEF) were i.v. administrated 20 min before the non-functionalized MSCs. b) Time-dependent mean clinical scores of MOG35-55-induced EAE inflicted mice after received different prophylactic and therapeutic treatments. (n = 8 mice per group; one non-treatment group mouse was found dead 28 days p.i.) c) Cumulative EAE scores of MOG35-55-EAE inflicted mice after received different treatments. d)(i) Representative H&E-stained spinal cord sections preserved from EAE-inflicted mice after received different prophylactic and therapeutic treatments with drug-free/LEF-encapsulated PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs. (ii) Quantification of spinal inflammation from the H&E-stained images of spinal cords. (n = 3 for the non-treatment group; n = 6 for the prophylactic treatment group and therapeutic treatment group treated with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs; n = 7 for the therapeutic treatment group treated with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs.) e)(i) Representative LFB-stained spinal cord sections preserved from EAE-inflicted mice after received different prophylactic and therapeutic treatments with drug-free/LEF-encapsulated PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs. (ii) Quantification of demyelination from the LFB-stained images of spinal cords. (n = 3 for the non-treatment group; n = 6 for the prophylactic treatment group and therapeutic treatment group treated with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs; n = 7 for the therapeutic treatment group treated with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs.) f) Prophylactic and therapeutic treatment schedules with PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs in C57BL/6 mice after immunization with PLP178-191 peptide. Unmodified or functionalized MSCs were i.v. administrated 1 day (prophylactic treatment) or 18 days (therapeutic treatment) p.i.. Body conditions were monitor until 35 days p.i. g) Time-dependent mean clinical scores of MOG35-55-induced EAE inflicted mice after received different prophylactic and therapeutic treatments. (n = 8 mice per group, except n = 7 for the therapeutic treatment group with unmodified MSCs.) h) Cumulative EAE scores of PLP178-191-EAE inflicted mice after received different treatments.
Similar to the results of the prophylactic study, therapeutic treatment with PD-L1 FcIg, CD86 FcIg and small-molecule LEF followed by unmodified MSCs (20 min apart) did not prevent or change the course of disease compared with untreated mice (Figure 5b–c; Figure S17, Supporting Information).The therapeutic treatment with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs was as effective as treatment with directly functionalized MSCs in inhibiting the progression of EAE and reversing certain associated symptoms (Figure 5b–c; Figure S17, Supporting Information). In contrast, the PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs were 29% more effective than the drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs in reducing cumulative EAE scores (Figure 5b–c; Figure S17c–d, Supporting Information). At 35 days p.i., all mice treated with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs regained hindlimb strength (EAE score ≤ 2.0; Figure 5b–c; Figure S17c–d, Supporintg Information), and 3 of 9 treated mice were symptom-free. This improved therapeutic efficiency shows that encapsulated LEF is required to control the proliferation of autoreactive T cells in the CNS. Consistent with the prophylactic study, treatment with small-molecule LEF, unconjugated PD-L1 FcIg, and CD86 FcIg or PD-L1 FcIg/CD86 FcIg LEF NPs followed by unmodified MSCs did not achieve significant therapeutic effects compared the result for untreated mice.
Histological analysis showed that therapeutic treatment with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs was as effective as treatment with dual-functionalized MSCs in reducing spinal cord inflammation by 75% and demyelination by 87% (compared with the results for untreated mice) at 36 or 37 days p.i. (Figure 5d–e; Figure S19–S20, Supporting Information). Treatment with LEF-encapsulated MSCs further reduced spinal inflammation by 95% (6 of 7 treated mice did not exhibit detectable spinal inflammation) and demyelination by 95% (2 of 7 treated mice did not exhibit detectable demyelination) compared with the results for untreated mice 36 or 37 days p.i. Although the degree of demyelination in EAE mice treated with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs was similar in mice treated with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (Figure S20, Supporting Information), LEF-encapsulated MSCs significantly reduced spinal inflammation (7 of 8 treated mice did not exhibit detectable inflammation) compared with the drug-free functionalized MSCs (3 of 8 treated mice did not exhibit detectable inflammation) (Figure S19, Supporting Information). Though treatment with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs also reduced EAE clinical signs, it less effectively reduced spinal cord inflammation and demyelination than with treatment with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs. These findings support our hypothesis that the functionalized MSCs served as a vehicle for the therapeutic delivery of LEF into the spinal cord, thereby reducing the proliferation of autoreactive T cells in the CNS.
Recognizing that not all the EAE mice were cured after the first therapeutic treatment, we administered a second dose of PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs to the EAE mice 35 days p.i. In a separate therapeutic treatment study (Figure S21, Supporting Information), 4 of 6 mice responded to the second treatment with the PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs. The average EAE score significantly decreased by 50% (from 0.8 to 0.4) after the second treatment, and 3 of 6 of those mice were symptom-free at the study endpoint (50 days p.i.; Figure S21, Supporting Information).
To demonstrate that PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs can treat relapsing-remitting MS, we used a PLP178-191-induced EAE model.[34] Unlike the MOG35-55-inflicted EAE model, the EAE symptoms of PLP178-191-inflicated mice partially recovered after the first phase of onset before the symptoms grew worse (Figure 5f). Although prophylactic treatment with PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs did not completely prevent the development of EAE symptoms in this model, it significantly ameliorated clinical symptoms as well as the cumulative EAE score (49% at up to 35 days p.i.) (Figure 5g–h; Figure S22, Supporting Information). Similar to the therapeutic effects of the MOG-induced model of EAE, therapeutic treatment with functionalized MSCs reduced the cumulative EAE score by 43% (Figure 5g–h; Figure S22, Supporting Information). Similar to the outcome of using the MOG35-55-immunized model, a second therapeutic treatment, administered 17 days after the first treatment, significantly reduced disease progression from 0.0402 day−1 to 0.0044 day−1 (89% decrease; Figure S23, Supporting Information). These findings support the conclusion that a booster dose further improved the efficiency of therapy.
To prove that i.v. administered MSCs did not directly involve remyelination, we administered 50 Gy X ray-irradiated PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs for prophylactic and therapeutic treatments. The dying X ray-irradiated MSCs (Figure S24, Supporting Information) were as effective as the non-irradiated MSCs in reducing clinical signs and cumulative EAE scores, which indicates that bioengineered MSCs are not directly involved in myelin repair (Figure 5g–h; Figure S22, Supporting Information).
We next performed an ex vivo imaging study in the MOG35-55-immunized EAE model to determine the biodistribution 48 h after the i.v. administration of VivoTag 680 (VT680)-labeled unmodified and PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (Figure S25–S26, Supporting Information). In the prophylactic imaging groups, the majority of the administered MSCs were accumulated in peripheral organs, with less than ≤ 0.2% of the injected dose (ID) of the MSCs detected in the CNS (Figure S25–S26, Supporting Information). This indicates that the observed prophylactic effect may involve the induction of immunosuppressive T cells outside the CNS. In contrast, approximately 1.75% ID and 0.75% ID of MSCs were accumulated in the brain and spinal cord in the therapeutic treatment groups (Figure S25–S26, Supporting Information), respectively. Although the majority of administered MSCs remained in peripheral organs to induced immunosuppressive immune cells outside the CNS, the CNS-infiltrating MSCs may be required to maintain CNS-specific immunotolerance for the therapeutic treatment in MOG35-55-immunized EAE mice. The lack of long-term in vivo toxicity observed in the healthy C57BL/6 mice (Figure S13, Supporting Information) suggesting the majority of MSCs that initially accumulated in the liver and lung were dead (due to lack of correct tissue microenvironments) and cleard by the mononuclear phagocytes (e.g., macrophages).
We next analyzed MOG35-55-specific CD4+ T cell populations 3 days after prophylactic and therapeutic treatments with i.v. administered drug-free and LEF-encapsulated PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (Figure S27, Supporting Information).[35] Prophylactic treatment with both functionalized MSCs were equally effective in promoting the development of MOG35-55-specific splenic Treg cells (approximately 70% of MOG35-55+ CD4+ cells being FoxP3+) and slightly reduced the numbers of splenic MOG35-55-specific Th1 and Th17 cells (Figure 6a; Figure S28, Supporting Information). Similarly, therapeutic treatment with both PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs was equally effective in promoting the development of MOG35-55-specific splenic Treg cells (with approximately 25% of the splenic MOG35-55+ CD4+ cells being FoxP3+) and slightly reducing the number of MOG-specific splenic Th1 and Th17 cells (Figure 6b; Figure S29, Supporting Information). In contrast, treatment with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs induced 62% more MOG35-55-specific spinal CD4+ Treg cells than with treatment with drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (Figure 6b; Figure S30, Supporting Information). Thus, 32.2 ± 7.6% of CD8+ T cells infiltrating the spinal cord expressed IFN-gamma (Figure 6b; Figure S30, Supporting Information), whereas 76.7 ± 2.8% and 67.2 ± 4.4% of the CD8+ T cells infiltrating the spinal cords of mice treated or not treated with the drug-free PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs expressed IFN-gamma, respectively. Moreover, PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs effectively inhibited the development of EAE and reversed certain early-onset symptoms by promoting the development of MOG35-55-specific Treg cells (Figure 6c; Figure S31, Supporting Information). Further, histopathological analysis of the spinal cord preserved 36 or 37 days p.i. revealed that prophylactic and therapeutic treatments with the PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs promoted the development of suppressive CD4+ FoxP3+ Treg cells in the spinal cord (Figure 6d).
Figure 6.
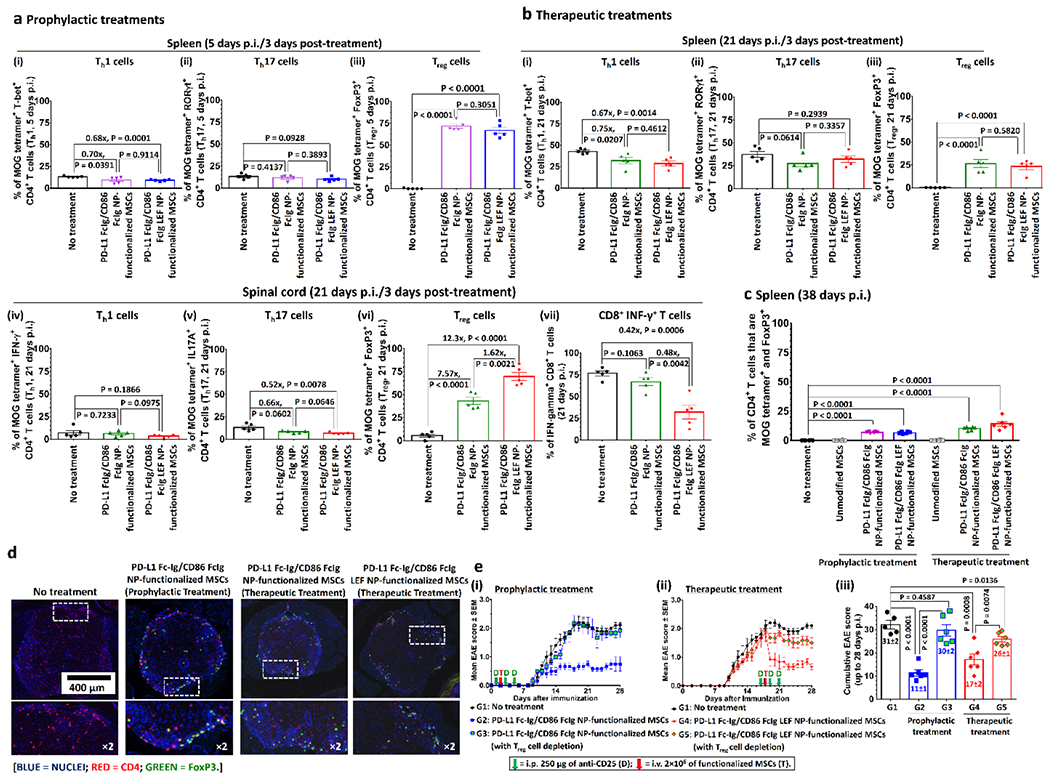
PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs effectively promote the development of MOG-specific Treg cells in the MOG35-55-EAE mouse model. a) Splenetic MOG-specific (i) Th1, (ii) Th17, and (iii) Treg cells populations in EAE-inflicted mice 3 days after different prophylactic treatments (5 days p.i.). (n = 5) b) Splenetic MOG-specific (i) Th1, (ii) Th17, and (iii) Treg cells populations in EAE-inflicted mice 3 days after different therapeutic treatments (5 days p.i.). MOG-specific (iv) Th1, (v) Th17 and (vi) Treg cells, and (vii) antigen non-specific INF-γ+ cytotoxic T cell populations in the spinal cord of EAE-inflicted mice 3 days after different therapeutic treatments (21 days p.i.). (n = 5) c) Splenetic MOG-specific Treg cells populations in EAE-inflicted mice 38 days p.i. after different prophylactic and therapeutic treatments. (n = 6) d) Representative anti-CD4- and anti-FoxP3-stained immunofluorescence images of spinal cord preserved from non-treated EAE-inflicted mice and different treated EAE-inflicted mice 38 days p.i. e) Prophylactic and therapeutic treatments with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs in MOG35-55-immunized mice with and without Treg cell depletion. Mice in Treg cell depletion groups received 3 intraperitoneal (i.p.) injections of anti-CD25 (200 μg per injection) before and after the treatments with the MSCs to achieve Treg cell depletion. (iii) Time-dependent mean clinical scores of MOG35-55-induced EAE inflicted mice after received different (i) prophylactic and (ii) therapeutic treatments. (iii) Cumulative EAE scores of MOG35-55-EAE inflicted mice after received different treatments with and without Treg cell depletion. (n = 6)
To confirm these findings, we performed Treg cell depletion studies with CD25-specific antibodies in MOG35-55-immunized mice (Figure 6e).[36] Similar to the result for untreated mice, Treg cell-depleted mice developed severe EAE symptoms after prophylactic treatment with PD-L1 FcIg/CD86 FcIg NP-functionalized MSCs (cumulative EAE score = 31 ± 2 versus 29 ± 2 in the non-treatment control group) (Figure 6e). The depletion of Treg cells before treatment with PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs significantly reduced the therapeutic efficiency of the functionalized MSCs and increased the cumulative EAE scores by 88% (Figure 6e). These findings indicate that Treg cells induced by PD-L1 FcIg/CD86 FcIg LEF NP-functionalized MSCs are required to maintain immunotolerance to MOG35-55-induced EAE.
The immune system evolved to elicit robust immune responses against foreign antigens while tolerating self-antigens to avoid autoimmunity.[1, 2] Failure to establish peripheral immune tolerance leads to the development of autoimmune diseases, ranging from type 1 diabetes to MS.[1, 2] Treg cells are required to maintain immune tolerance and homeostasis.[3] Numerous in vivo studies and clinical trials employed stimulated bulk Treg cells for the treatment of autoimmune diseases;[9, 37] however, the absence of antigen specificity increases the risk of systemic immunesuspension.[9, 37] Autoantigen-specific chimeric antigen receptor Treg cells are available to suppress MS,[8] although the clinical outcomes are disappointing because of the rapid mutation of autoantigens and insufficient long-term potency of the infused Treg cells.[9] Recent studies have focused on the administration of encephalitogenic peptide-conjugated microparticles,[38] blood-brain barrier permeable peptide-functionalized CTLA-4,[39] and encephalitogenic peptide-conjugated isologues leukocytes[40, 41] to induce antigen-specific immune tolerance through the reduction the population of pathogenic helper T cells and induction of antigen-specific Treg cells. However, clinical trials showed that only a small group of MS patients with human leukocyte antigen haplotypes DR2 or DR4 benefit from these treatments.[41] In addition, the long-term treatment response of these highly antigen-specific treatments is often compromised by the epitope shift and autoantigen mutation.[42]
In this study, we employed metabolic glycoengineering and bioorthogonal click chemistry to bioengineer PD-L1- and CD86-functionalized SCs to prevent and treat MS. Autologous SCs can be isolated from sural nerve, ex vivo bioengineer before administered back to the patient. In contrast to other antigen-specific MS treatment strategies, the functionalized SCs were designed to present a broad range of myelin antigens to engaged pathogenic helper T cells, to inhibit their activation, and to induce the development of myelin antigen-specific Treg cells to suppress the autoreactive immune cells. Comprehensive in vitro and in vivo studies show that immune checkpoint ligand–functionalized SCs effectively inhibited the differentiation of myelin-specific helper T cells into pathogenic Th1 and Th17 cells, promoted the development of antigen-specific Treg cells and resolved the inflammatory CNS microenvironment in established mouse EAE models. The less proinflammatory microenvironment allows the OLs to repair myelin damage and ameliorate EAE clinical signs. The facile bioorthogonal conjugation strategy reported here allows on-demand modular-based functionalization of SCs. This reversible bioconjugation strategy was associated with low toxicity and prevented potential irreversible adverse effects associated with inhibitory immune checkpoint pathways. However, the clear limitation of current treatment strategy is that only a small amount of i.v. administrated immune checkpoint-functionalized MSCs can enter the CNS and systemic circulation of these immunosuppressive MSCs may induce systemic immunosuppression. Further studies should focus on intraspinal administer the functionalized MSCs (or other glial cells) to facilitate the repairing of damaged myeline (re-myelination) and induce local antigen-specific immunotolerance. Nevertheless, the present study provides a new framework for treating MS and supports its further evaluation in other models of autoimmune disease.
Supplementary Material
Acknowledgements
We thank the Microscopy Service Laboratory Core, Animal Study Core, Small Animal Imaging Facility, Animal Clinical Laboratory, Animal Histopathology Core Facility, Translation Pathology Lab, UNC Flow Cytometry Core Facility, UNC Macromolecular Interactions Facility and UNC Michael Hooker Proteomics Centre in the School of Medicine, and Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) at the University of North Carolina at Chapel Hill for their assistance with procedures in this manuscript. UNC Lineberger Animal Studies Core is sipported in part by an NCI Center Core Support Grant (CA16086) to the UNC Lineberger Comprehensive Cancer Center. The UNC Flow Cytometry Core Facility is supported in part by P30CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. This work was supported by the University Cancer Research Fund from the University of North Carolina and R01CA178748 grant from the National Institutes of Health/National Cancer Institute. A.Z.W. was also supported by the National Institutes of Health Center for Nanotechnology Excellence Grant U54-CA151652. R.T. was supported by National Institutes of Health/NIAID grants R01AI139475 and R01AI141631.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Animal Welfare Statement
All procedures involving experimental animals were performed according to the protocol (protocol 20-261.0) approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee. Blood glucose, bodyweight, and body condition scores of NOD mice were monitored independently twice per week by the UNC Lineberger Animal Studies Core at the University of North Carolina at Chapel Hill.
Contributor Information
Kin Man Au, Laboratory of Nano- and Translational Medicine, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Radiation Oncology, University of Texas Southwestern Medical Center, Dallas, TX 75230, USA.
Roland Tisch, Department of Microbiology and Immunology School of Medicine, University of North Carolina at Chapel Hill, 27599, USA; Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Andrew Z. Wang, Laboratory of Nano- and Translational Medicine, Carolina Center for Cancer Nanotechnology Excellence, Carolina Institute of Nanomedicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Radiation Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA; Department of Radiation Oncology, University of Texas Southwestern Medical Center, Dallas, TX 75230, USA
References
- [1].Sinha AA, Lopez MT, McDevitt HO, Science 1990, 248, 1380. [DOI] [PubMed] [Google Scholar]
- [2].Sakaguchi S, Powrie F, Ransohoff RM, Nat Med 2012, 18, 54. [DOI] [PubMed] [Google Scholar]
- [3].Sakaguchi S, Yamaguchi T, Nomura T, Ono M, Cell 2008, 133, 775. [DOI] [PubMed] [Google Scholar]
- [4].Goverman JM, Immunol Rev 2011, 241, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vandenbark AA, Offner H, Immunology 2008, 125, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Goldenberg MM, P T 2012, 37, 175. [PMC free article] [PubMed] [Google Scholar]
- [7].Torkildsen O, Myhr KM, Bo L, Eur J Neurol 2016, 23 Suppl 1, 18; [DOI] [PMC free article] [PubMed] [Google Scholar]; Mendes A, Sa MJ, Arq Neuropsiquiatr 2011, 69, 536; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kammona O, Kiparissides C, Brain Sci 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jyothi MD, Flavell RA, Geiger TL, Nat Biotechnol 2002, 20, 1215. [DOI] [PubMed] [Google Scholar]
- [9].Duffy SS, Keating BA, Moalem-Taylor G, Front Neurosci 2019, 13, 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Joller N, Peters A, Anderson AC, Kuchroo VK, Immunol Rev 2012, 248, 122; [DOI] [PMC free article] [PubMed] [Google Scholar]; Chitnis T, Khoury SJ, J Allergy Clin Immunol 2003, 112, 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Trabattoni D, Saresella M, Pacei M, Marventano I, Mendozzi L, Rovaris M, Caputo D, Borelli M, Clerici M, J Immunol 2009, 183, 4984. [DOI] [PubMed] [Google Scholar]
- [12].Gerdes LA, Held K, Beltran E, Berking C, Prinz JC, Junker A, Tietze JK, Ertl-Wagner B, Straube A, Kumpfel T, Dornmair K, Hohlfeld R, Ann Neurol 2016, 80, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bilate AM, Lafaille JJ, Annu Rev Immunol 2012, 30, 733. [DOI] [PubMed] [Google Scholar]
- [14].Aly L, Hemmer B, Korn T, Curr Neuropharmacol 2017, 15, 874; [DOI] [PMC free article] [PubMed] [Google Scholar]; Klotz L, Eschborn M, Lindner M, Liebmann M, Herold M, Janoschka C, Torres Garrido B, Schulte-Mecklenbeck A, Gross CC, Breuer J, Hundehege P, Posevitz V, Pignolet B, Nebel G, Glander S, Freise N, Austermann J, Wirth T, Campbell GR, Schneider-Hohendorf T, Eveslage M, Brassat D, Schwab N, Loser K, Roth J, Busch KB, Stoll M, Mahad DJ, Meuth SG, Turner T, Bar-Or A, Wiendl H, Sci Transl Med 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Duncan ID, Radcliff AB, Heidari M, Kidd G, August BK, Wierenga LA, Proc Natl Acad Sci U S A 2018, 115, E11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mosahebi A, Fuller P, Wiberg M, Terenghi G, Exp Neurol 2002, 173, 213; [DOI] [PubMed] [Google Scholar]; Oudega M, Xu XM, J Neurotrauma 2006, 23, 453; [DOI] [PubMed] [Google Scholar]; Baron-Van Evercooren A, Avellana-Adalid V, Lachapelle F, Liblau R, Mult Scler 1997, 3, 157; [DOI] [PubMed] [Google Scholar]; Hood B, Levene HB, Levi AD, Neurosurg Focus 2009, 26, E4. [DOI] [PubMed] [Google Scholar]
- [17].Agatemor C, Buettner MJ, Ariss R, Muthiah K, Saeui CT, Yarema KJ, Nat Rev Chem 2019, 3, 605; [DOI] [PMC free article] [PubMed] [Google Scholar]; Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VD, Yarema KJ, Glycobiology 2009, 19, 1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Au KM, Medik Y, Ke Q, Tisch R, Wang AZ, Adv Mater 2021, e2101253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Au KM, Tripathy A, Lin CP, Wagner K, Hong S, Wang AZ, Park SI, ACS Nano 2018, 12, 1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Au KM, Wang AZ, Park SI, Sci Adv 2020, 6, eaaz9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sharma P, Gangopadhyay D, Mishra PC, Mishra H, Singh RK, J Med Chem 2016, 59, 3418. [DOI] [PubMed] [Google Scholar]
- [22].Oliveira BL, Guo Z, Bernardes GJL, Chem Soc Rev 2017, 46, 4895. [DOI] [PubMed] [Google Scholar]
- [23].Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK, J Exp Med 2003, 197, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Roberts RA, Eitas TK, Byrne JD, Johnson BM, Short PJ, McKinnon KP, Reisdorf S, Luft JC, DeSimone JM, Ting JP, Biomaterials 2015, 72, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Korn T, Magnus T, Toyka K, Jung S, J Leukoc Biol 2004, 76, 950. [DOI] [PubMed] [Google Scholar]
- [26].Bradley LM, Dalton DK, Croft M, J Immunol 1996, 157, 1350. [PubMed] [Google Scholar]
- [27].Tsai HC, Velichko S, Hung LY, Wu R, Clin Dev Immunol 2013, 2013, 267971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, Escors D, EMBO Mol Med 2011, 3, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jeannin P, Magistrelli G, Aubry JP, Caron G, Gauchat JF, Renno T, Herbault N, Goetsch L, Blaecke A, Dietrich PY, Bonnefoy JY, Delneste Y, Immunity 2000, 13, 303. [DOI] [PubMed] [Google Scholar]
- [30].Mendel I, Kerlero de Rosbo N, Ben-Nun A, Eur J Immunol 1995, 25, 1951. [DOI] [PubMed] [Google Scholar]
- [31].Ben Nasr M, Tezza S, D’Addio F, Mameli C, Usuelli V, Maestroni A, Corradi D, Belletti S, Albarello L, Becchi G, Fadini GP, Schuetz C, Markmann J, Wasserfall C, Zon L, Zuccotti GV, Fiorina P, Sci Transl Med 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Constantinescu CS, Farooqi N, O’Brien K, Gran B, Br J Pharmacol 2011, 164, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xiang Z, Nesterov EE, Skoch J, Lin T, Hyman BT, Swager TM, Bacskai BJ, Reeves SA, J Histochem Cytochem 2005, 53, 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tompkins SM, Padilla J, Dal Canto MC, Ting JP, Van Kaer L, Miller SD, J Immunol 2002, 168, 4173. [DOI] [PubMed] [Google Scholar]
- [35].Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, Akilli-Ozturk O, Kranz LM, Berger H, Petschenka J, Diken M, Kreiter S, Yogev N, Waisman A, Kariko K, Tureci O, Sahin U, Science 2021, 371, 145. [DOI] [PubMed] [Google Scholar]
- [36].Setiady YY, Coccia JA, Park PU, Eur J Immunol 2010, 40, 780. [DOI] [PubMed] [Google Scholar]
- [37].Arellano B, Graber DJ, Sentman CL, Discov Med 2016, 22, 73. [PMC free article] [PubMed] [Google Scholar]
- [38].Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD, Nat Biotechnol 2012, 30, 1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim GR, Kim WJ, Lim S, Lee HG, Koo JH, Nam KH, Kim SM, Park SD, Choi JM, Adv Sci (Weinh) 2021, 8, 2004973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, King NJ, Miller SD, J Immunol 2011, 187, 2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M, Martin R, Sci Transl Med 2013, 5, 188ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Smith CE, Eagar TN, Strominger JL, Miller SD, Proc Natl Acad Sci U S A 2005, 102, 9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Au KM, Park SI, Wang AZ, Science Advances 2020, 6, eaba8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li Y, Kurlander RJ, J Transl Med 2010, 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Miller SD, Karpus WJ, Curr Protoc Immunol 2007, Chapter 15, Unit 15 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Okuda Y, Okuda M, Bernard CC, J Neuroimmunol 2002, 131, 115. [DOI] [PubMed] [Google Scholar]
- [47].Chou WC, Guo Z, Guo H, Chen L, Zhang G, Liang K, Xie L, Tan X, Gibson SA, Rampanelli E, Wang Y, Montgomery SA, Brickey WJ, Deng M, Freeman L, Zhang S, Su MA, Chen X, Wan YY, Ting JP, Nature 2021, 591, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.