

Abstract
Objective
Stromal interaction molecule 1 (STIM1) is a single-pass transmembrane endoplasmic/sarcoplasmic reticulum (E/SR) protein recognized for its role in a store operated Ca2+ entry (SOCE), an ancient and ubiquitous signaling pathway. Whereas STIM1 is known to be indispensable during development, its biological and metabolic functions in mature muscles remain unclear.
Methods
Conditional and tamoxifen inducible muscle STIM1 knock-out mouse models were coupled with multi-omics tools and comprehensive physiology to understand the role of STIM1 in regulating SOCE, mitochondrial quality and bioenergetics, and whole-body energy homeostasis.
Results
This study shows that STIM1 is abundant in adult skeletal muscle, upregulated by exercise, and is present at SR-mitochondria interfaces. Inducible tissue-specific deletion of STIM1 (iSTIM1 KO) in adult muscle led to diminished lean mass, reduced exercise capacity, and perturbed fuel selection in the settings of energetic stress, without affecting whole-body glucose tolerance. Proteomics and phospho-proteomics analyses of iSTIM1 KO muscles revealed molecular signatures of low-grade E/SR stress and broad activation of processes and signaling networks involved in proteostasis.
Conclusion
These results show that STIM1 regulates cellular and mitochondrial Ca2+ dynamics, energy metabolism and proteostasis in adult skeletal muscles. Furthermore, these findings provide insight into the pathophysiology of muscle diseases linked to disturbances in STIM1-dependent Ca2+ handling.
Keywords: Skeletal muscle, STIM1, Proteostasis, Energy metabolism, Exercise tolerance, Mitochondria, Calcium signaling, ER stress, Proteomics, Mass spectrometry
Graphical abstract
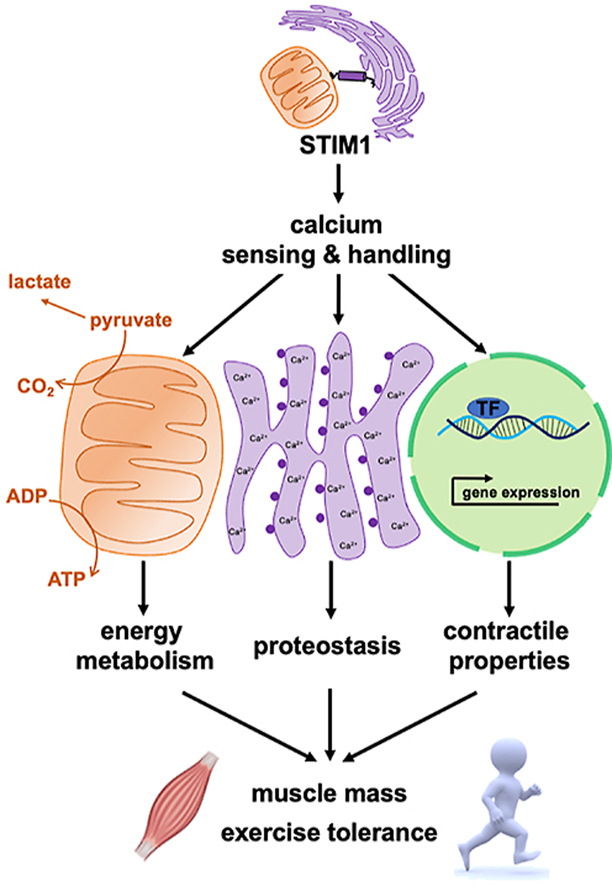
Highlights
-
•
STIM1 is a single-pass transmembrane E/SR protein recognized for its role in store operated Ca2+ entry.
-
•
The role of STIM1 in regulating mitochondrial function and energy homeostasis in adult muscle was investigated using a tissue-specific inducible knockout mouse line (iSTIM KO).
-
•
STIM1 is abundant in adult skeletal muscle, upregulated by exercise, and present at SR/mitochondria interfaces.
-
•
Deletion of STIM1 in adult muscle leads to diminished lean mass, reduced exercise capacity and altered mitochondrial metabolism.
-
•
Proteomics and phospho-proteomics analyses of iSTIM1 KO muscles revealed molecular signatures of low-grade E/SR stress and perturbed proteostasis.
1. Main
Decades of research have established a prominent role for calcium in mediating skeletal muscle excitation-contraction coupling (ECC), which requires release and reuptake of calcium from the sarcoplasmic reticulum (SR) in a process mediated by the ryanodine receptors and sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) pump activity, respectively [1]. By contrast, store-operated Ca2+ entry (SOCE) is an ancient and ubiquitous Ca2+ signaling pathway that is activated during cell stimulation to replenish exhausted internal stores, as well as promote intermediate and long-term Ca2+-dependent processes such as metabolism and gene expression. Evidence that SOCE plays a key role in skeletal muscle was revealed following the identification of patients with loss of function mutations in a Ca2+ sensor, known as stromal interaction molecule 1 (STIM1), as well as the plasma membrane-localized Ca2+ channel subunit, Ca2+ release-activated calcium modulator 1 (Orai1). Muscles of these patients exhibit delayed growth, accumulate lipid droplets, and have reduced content of oxidative fibers (type I and IIa). Gain of function mutations to STIM1 and Orai1 genes occur in patients with tubular aggregate myopathy, a disease wherein myofibers accumulate E/SR tubes causing muscle pain, cramping, and weakness. Thus, it is now clear that SOCE in skeletal muscle is distinguished as a Ca2+ signaling pathway distinct from its well-recognized and essential role in CARDIAC excitation-contraction coupling (ECC) [2].
STIM1 is a single-pass transmembrane E/SR protein that forms a complex with Orai1 and is best recognized for its role in activating the channel upon depletion of cellular calcium stores. STIM1 has also been implicated in the regulation of other channels, transporters, and signaling molecules [3]. Mice lacking STIM1, either globally (STIM1−/−) or selectively in skeletal muscle (mSTIM1−/−), display defects in skeletal muscle growth and reduced survival [4,5]. A similar reduction in muscle mass and weakness characterizes the phenotype of mice harboring either a dominant-negative mutation in the Orai1 (dn-Orai1) channel in skeletal muscle [6] or skeletal muscle-specific knockout (KO) of Orai1 [7]. However, the muscle phenotype of an inducible skeletal muscle Orai1 KO mouse model appears normal [7]. Notably, skeletal muscles from Orai1 loss-of-function mouse models exhibit normal mitochondrial morphology and function. By contrast, STIM1 loss of function mouse models exhibits prominent changes to mitochondrial ultrastructure [4].
Given the divergence in the muscle phenotypes resulting from the disruption of distinct components of SOCE, we considered the possibility that in a skeletal muscle STIM1 plays a broader biological role. Supporting this premise, recent studies have shown that STIM1 partitions into multiple domains of the SR, including the terminal cisternae near the T-tubule and the longitudinal SR near the Z-line [5,[8], [9], [10]]. Moreover, STIM1 is anchored at the myofiber Z-line by the intermediate filament, desmin, to promote SR Ca2+ refilling [11], which is unlikely to be required for STIM1-dependent activation of SOCE. We, therefore, posit that the STIM1-desmin domain mediates novel interactions between STIM1 and nearby membrane domains, including the mitochondria. Because calcium plays an essential role in activating several mitochondrial dehydrogenase enzyme complexes that participate in the tricarboxylic acid cycle (TCAC) [[12], [13], [14], [15]] and has been implicated in regulating ATP synthase (Complex V) [[16], [17], [18]], we further speculated that STIM1 might impact skeletal muscle energy metabolism and fuel selection, particularly in settings of energy stress such as exercise and acute starvation. Consistent with this notion, recent work involving studies in both human and rodent muscles identified and validated STIM1 as a direct substrate of the 5′AMP-activated kinase (AMPK), a master energetic sensor and metabolic regulator that phosphorylates its targets during physiological stresses such as high-intensity exercise [19].
To elucidate the interplay between STIM1, mitochondrial function, and energy homeostasis, we investigated the metabolic consequences of ablating the gene from adult skeletal muscle using an inducible KO mouse line (iSTIM−/-). Deletion of STIM1 from skeletal muscle of adult mice resulted in a substantial decline in muscle mass and reduced exercise capacity, which was attributable, in part, to disruptions in Ca2+-dependent regulation of oxidative metabolism and perturbed proteostasis. The results provide insight into the pathophysiology of muscle diseases linked to disturbances in STIM1-dependent calcium signaling.
2. Results
2.1. Muscle-specific loss of STIM1 alters mitochondrial quality
To elucidate the location of STIM1 in skeletal muscle, we performed immunohistochemistry of flexor digitorum brevis (FDB) muscles. Figure 1A shows that a subpopulation of STIM1 localizes near Cytochrome c oxidase IV(COX IV) and translocase of the outer membrane 20 (TOM20), inner and outer mitochondrial membrane proteins, respectfully. Subcellular distribution of STIM1 was further investigated using a STIM1-LacZ fusion protein and transmission electron microscopy (TEM) (Figure 1B). The dense material on TEM, which represents the aggregation of the β-galactose reaction product, reveals the location of STIM1-LacZ. The pattern depicted in the images reveals that a subpopulation of SR-localized STIM1 is juxtaposed to muscle mitochondria. The functional relevance of this observation was then investigated using a skeletal muscle-specific STIM1 knockout (KO) mouse model (mSTIM1−/−) [4]. As previously reported, this loss-of-function manipulation leads to profound growth delay, reduced myonuclear proliferation, and perinatal lethality. In this research, we show that muscles from the mSTIM1−/− mice studied at 1–2 weeks of age had reduced mitochondrial calcium levels in the basal state (Figure 1C) and response to electrical stimulation mimicking action potential (AP) (Figure 1D–E). Deletion of STIM1 also led to dysmorphic mitochondria, as assessed by TEM (Figure 1F). Moreover, mass spectrometry-based metabolite profiling revealed increased levels of lipid-derived acylcarnitines (Figure 1G), a class of oxidative intermediates commonly elevated in settings of mitochondrial dysfunction [20].
Figure 1.

Muscle-specific loss of STIM1 affects survival and mitochondrial quality. (A) Representative images and fluorescence intensity profiles of flexor digitorum brevis (FDB) fiber immunostained with STIM1, COXIV, and TOM20. (B) Distribution of STIM1-LacZ to subcellular localization with mitochondria (black arrow) in FDB muscle as determined by transmission electron microscopy (TEM). STIM1-LacZ expression is detected by staining of β-galactosidase activity. Myofibers were isolated from flexor digitorum brevis (FDB) muscles and loaded with Rhod-2 to determine mitochondrial Ca2+ content in the (C) basal state (n = 75–128 fibers) and in response to (D) electrical stimulation (n = 12–19). (E) Representative traces of mitochondrial Ca2+ transients in response to repeated bursts of electrical activity. (F) Representative transmission electron micrographs show (a) normal mitochondrial ultrastructure of STIM1fl/fl muscle and (b–f) abnormal mitochondrial size, shape, and cristae of mSTIM1−/− muscle. (G) Metabolomic analysis of acylcarnitines in gastrocnemius muscles at 2 weeks of age (n = 5–6). (H) STIM1 mRNA expression normalized to 18 S measured by RT-qPCR in tibialis anterior muscle after a 90-minute bout of treadmill running (n = 4). (I) Western blot and (J) quantification of STIM1 normalized total protein in gastrocnemius muscle from PGC1α transgenic (Tg) and non-transgenic (NT) littermates (n = 4). All data represent mean ± SEM. (C, D, G, and J) were analyzed using the Student's 2-tailed t-test. ∗p ≤ 0.05. (H) was analyzed by 1-way ANOVA with Dunnett's multiple comparisons test. ∗∗p≤ 0.01 and ∗∗∗p ≤ 0.001. n represents biological replicates.
Potential links between STIM1 and skeletal muscle metabolism were further vetted by examining gene expression responses to an acute bout of exercise. As compared with the resting state, STIM1 mRNA levels measured immediately after a 90-minute graded treadmill exercise increased 6-fold and remained elevated for up to 24 h (Figure 1H). Interestingly, this mRNA expression pattern closely resembles that of The peroxisome proliferator-activated receptor co-activator-1α (PGC1α), a prominent transcriptional co-activator that functions as a master regulator of metabolism and mitochondrial biogenesis [21]. Likewise, STIM1 protein levels are elevated 2-fold in gastrocnemius muscles from MCK-PGC1α mice (Figure 1I–J), a well-established model of enhanced oxidative capacity and exercise tolerance. Taken together, these findings show that muscle-specific deficiency of STIM1 compromises tissue function during early development while also establishing a delicate connection between STIM1 and mitochondrial quality.
2.2. Conditional STIM1 deficiency in adult mice diminishes muscle mass
The foregoing observations implied a key role for STIM1 in regulating muscle metabolism and/or mitochondrial function during development. To assess the role of STIM1 during adulthood, we proceeded to generate a conditional KO model (iSTIM1−/−) (Figure 2A) wherein skeletal muscle-specific deletion of STIM1 can be induced upon treatment with tamoxifen (TMX). The model was validated via Western blot analysis of gastrocnemius muscles (Figure 2B), which confirmed an 80%–90% reduction of the STIM1 short isoform within 4 weeks of giving TMX injections (Figure 2C). The long isoform declined more gradually, reaching 60% and 75% reductions at 4 weeks and 18 weeks post-TMX, respectively (Figure 2D). As anticipated, Ca2+ entry following depletion of ryanodine receptor 1 (RYR-1) stores by caffeine and SERCA1 inhibition with cyclopiazonic acid (CPA) was absent in the Fura-2 loaded FDB muscle fibers from iSTIM1−/− mice (Figure 2E). To evaluate a broader role for STIM1 and SOCE in Ca2+ signaling, Fura-4-loaded FDB fibers were subjected to electrical field stimulation (EFS, 50 Hz) and Ca2+ transients were assessed in STIM1fl/fl and iSTIM−/− fibers (Figure 2F). Action potentials delivered to naïve iSTIM−/− fibers generated Ca2+ transients with a smaller amplitude than STIM1fl/fl fibers, although this difference was insignificant (data not shown). With repetitive 50-Hz trains, Ca2+ transients were readily sustained for the STIM1fl/fl fibers, whereas the peak amplitude of the Ca2+ transients from the same stimulus was significantly reduced for iSTIM1−/− fibers (Figure 2G), indicating that STIM1 senses and responds to RYR1 Ca2+ release in order to preserve EC coupling. Notably, unlike for STIM1fl/fl fibers, the post-stimulation cytosolic Ca2+ progressively increased in the iSTIM1−/− fibers (Figure 2H). Possible mechanisms to explain these changes to the EFS-Ca2+ transients include excessive RYR1 leak or the inability to cycle Ca2+ back to the SR or mitochondria. Given the close relationship of STIM1 to mitochondria established in Figure 1, Rhod-2 imaging was performed to assess mitochondrial Ca2+ dynamics. In these studies, STIM1 depleted fibers exhibited dramatically reduced mitochondrial Ca2+ following SR Ca2+ mobilization by caffeine and CPA (Figure 2I). Moreover, peak mitochondrial Ca2+ uptake in response to Ca2+ re-addition (SOCE) was markedly blunted in iSTIM1−/− fibers (Figure 2J). These results confirm that STIM1-SOCE is activated rapidly during EFS to counterbalance the RYR1-Ca2+ flux across the T-tubule and mitochondrial membranes [2,11]. Overall, the findings indicate that single and repetitive Ca2+ transients activated by electrical stimulation are reduced in the iSTIM1−/− fibers. We interpret these results as evidence that SR and mitochondrial Ca2+ stores are diminished, due both to the loss of SOCE as well as impaired SERCA1 refilling.
Figure 2.

Conditional deletion of STIM1 in adult muscle abolishes SOCE. (A) Schematic depicting the breeding strategy to generate mice with inducible muscle-specific deletion of STIM1. Transgenic HSA-MerCreMer mice were crossed with mice in which exon 2 of the STIM1 gene is flanked by loxP sequences (STIM1fl/fl). Adult mice received tamoxifen (TMX) at ages of 12–13 weeks (B–D) or 8 weeks (E–J). TMX-treated transgenic HSA-MerCreMer:STIM1fl/fl are denoted as iSTIM1−/− throughout the manuscript. (B) Western blots and quantification of (C) STIM1S and (D) STIM1L abundance normalized to total protein in gastrocnemius muscles 4, 8, and 18 weeks after TMX (n = 5–8). (E) Representative traces of cytosolic Ca2+ in response to a standard SOCE protocol in FDB fibers loaded with Fura-2 (n = 4). (F) Representative traces of cytosolic Ca2+ transients generated with a single (2 s pulse [short bars]) or repeated bursts (2 s pulse at 50 HZ every 5 s [long bar]) of electrical activity in STIM1fl/f (left) and iSTIM1−/− (right) FDB myofibers loaded with Fura-4 (n = 4). The amplitude of cytosolic Ca2+ in Fura-4 loaded FDB fibers (G) during and (H) after each electrical stimulus (2 s at 50 HZ) (n = 17–23 fibers). (I) Representative traces and (J) quantification of mitochondrial Ca2+ uptake in response to a standard SOCE protocol in FDB fibers loaded with Rhod-2 (n = 12–13). Data in (C, D, and J) were analyzed using Student's two-tailed t-test. n represents biological replicates.
We next sought to determine whether deletion of STIM1 in adult skeletal muscles influences whole-body energy homeostasis. To this end, body weight and composition were measured longitudinally after TMX injections. Compared with STIM1fl/fl control mice, body weight (Figure 3A) and lean mass (Figure 3B) were consistently lower in the iSTIM1−/− group, which was detectable by 6 weeks after TMX injections. Fat mass trended slightly lower in iSTIM1−/− mice (Figure 3C). Whereas control mice continued to gain lean and fat mass between 0 and 12 weeks post-TMX, the iSTIM1−/− mice did not (Figure 3D). Similar results were observed in female iSTIM1−/− mice (S. Figure 1A–F). By contrast, tibia length, which remains constant after maturity, was identical between genotypes (Figure 3E). Thus, the impact of STIM1 deletion on lean mass appeared to stem from diminished muscle growth. Immunostaining of the oxidative enzyme, succinate dehydrogenase, in soleus muscle sections suggested that mitochondrial content is similar between genotypes (Figure 3F). However, the myofiber cross-sectional area of iSTIM1−/− muscles was reduced by 38% (Figure 3G). Notably, other histological metrics were largely unremarkable (Figure 3H); and assessment of several molecular markers of mitochondrial mass and dynamics produced no evidence of altered mitochondrial quality in the iSTIM1−/− muscles (Figure 3I,J).
Figure 3.

Conditional STIM1 deficiency in adult mice diminishes lean mass. (A) Body weight, (B) lean mass, and (C) fat mass before and 6 or 12 weeks after TMX administration at 11–13 weeks of age (n = 12). (D) Change in body weight, lean, and fat mass 12 weeks after TMX compared to pre-TMX (n = 12). (E) Tibia length measured 18 weeks after TMX (n = 6–7). (F) Representative images and (G) quantification of the cross-sectional area of succinate dehydrogenase immunostained soleus muscles 8 weeks after TMX administration at 8 weeks of age (n = 5). (H) Representative images of H&E-stained soleus cross-sections 8 weeks after TMX (n = 5). (I) Western blot and (J) quantification of mitochondrial protein expression normalized to total protein in gastrocnemius muscle 4 weeks after TMX (n = 5). All data shown represent mean ± SEM. Data in (A-E, G, and J) were analyzed using Student's two-tailed t-test. ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001. n represents biological replicates.
3. Conditional STIM1 deficiency alters fuel use and compromises exercise tolerance
The lower body weight and lean mass pointed to potential alterations in whole-body energy homeostasis. To explore this possibility, we used indirect calorimetry to assess energy balance and fuel use during a 48-h period. Although VO2 was reduced in iSTIM−/- mice relative to controls (Figure 1, Figure 4H), energy expenditure was similar between genotypes when assessed using linear regression analysis to account for differences in lean mass (Figure 4B) and total body weight (Figure 4C) [22]. Physical activity (Figure 4D) and food consumption (Figure 4E) were also similar between genotypes, whereas the whole-body respiratory exchange ratio (RER) was unchanged in females (S. Figure 1I) but trended higher in male iSTIM1−/− mice (Figure 4F). This was most pronounced during the dark cycle when food consumption increases, implying that iSTIM1−/− mice elicit a more robust substrate shift away from lipid-derived fuels upon feeding. These results led us to investigate whether the metabolic consequences of iSTIM1−/− deletion in adult mice might be exacerbated by moderate and/or intense exercise. To this point, when mice were challenged with a graded treadmill protocol, iSTIM1−/− mice exhausted earlier (Figure 4G), resulting in a 41% decrease in the running distance (Figure 4H). Also notable, the exercise-induced rise in the RER was more rapid and pronounced in the iSTIM1−/− mice as compared with their STIM1fl/fl counterparts (Figure 4I). Accordingly, in KO mice relative to controls, the ‘crossover’ point at which total body carbohydrate oxidation exceeds fat utilization occurred much sooner (Figure 4J,K), and blood lactate levels measured immediately after exercise were 62% higher (Figure 4L). Taken together, the results imply that iSTIM1−/− mice have a lower lactate threshold, which in turn alters blood chemistry and ventilation [23], thereby driving a higher RER during transitions from low to high exercise intensity. These genotype-related shifts in exercise tolerance prompted analysis of metabolic signaling events known to be influenced by nutritional and physiological manipulations, such as phosphorylation of AMPK and pyruvate dehydrogenase (PDH). Using a separate cohort of mice subjected to the same treadmill test, gastrocnemius muscles were collected after 15 min of exercise. In this experiment, STIM1 deficiency was accompanied by increased pAMPK T172, consistent with more severe energetic stress (Figure 4M and 4N), whereas phosphorylated PDH (pPDH) was unaffected by genotype at the time point tested.
Figure 4.

Conditional STIM1 deficiency alters fuel use and compromises exercise tolerance. (A) Whole-body oxygen consumption, the relationship of energy expenditure to (B) lean mass and (C) whole body weight as well as (D), total activity, (E) food consumption, and (F) respiratory exchange ratio assessed over 48 h in STIM1−/− v. STIM1fl/fl fed a chow diet after receiving TMX at 12–15 weeks of age (n = 9–10). (G) Time, and (H) distance to exhaustion, and (I) respiratory exchange ratio in response to a high-intensity exercise test (n = 5–6). (J) Traces of substrate utilization by STIM1fl/fl (left) and iSTIM1−/− (right) mice and (K) a bar graph depicting the time to the crossover point where carbohydrate utilization exceeds fat utilization during exercise (n = 5–7). (L) Blood lactate prior to and immediately after 15′ of exercise (n = 5–7). (M) Western blots and (N) quantification of phosphoproteins normalized to total protein in gastrocnemius muscles immediately after 15′ of exercise (n = 5–7). All data represent mean ± SEM. Data in (A, D-I, K, L, and N) were analyzed using Student's two-tailed t-test. ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001. n represents biological replicates.
3.1. Mitochondrial bioenergetics and energy transduction at 4 and 8 weeks are unaffected by STIM1 deficiency
We next questioned whether the foregoing deficits in energy homeostasis might be related to changes in mitochondrial respiratory function, an outcome that was predicted based on findings detailed in Figure 1. Here, we leveraged a recently developed respiratory diagnostics platform that permits a comprehensive assessment of mitochondrial energy transduction in the context of dynamic energy demands maintained within physiological ranges using a creatine kinase (CK) energetic clamp technique [24]. In these experiments, isolated mitochondria are exposed to graded adjustments in the ATP: ADP ratio in a manner that mimics shifts in energy demand occurring during transitions between rest and exercise. Measurements of steady-state oxygen flux (JO2), membrane potential (ΔΨm), NAD(P)H/NAD(P)+ redox state, and H2O2 production are performed in parallel to estimate respiratory conductance and efficiency [25]. In the current study, respiratory diagnostics were performed at 4 (S. Figure 2) and 8 (S. Figure 3) weeks after TMX injection. Freshly isolated mitochondria were exposed to three distinct substrate conditions; pyruvate/malate (PM), α-ketoglutarate (αKG), and palmitoyl-carnitine/malate (PcM), to evaluate energy fluxes dependent on PDH, αKGDH, and beta-oxidation, respectively. Surprisingly, we found no overt evidence of respiratory dysfunction in muscle mitochondria from iSTIM−/- mice as compared to controls. These results suggest exercise intolerance assessed in relatively young adult iSTIM1−/− mice were not attributable to significant remodeling and/or diminished quality of muscle mitochondria, but was a direct consequence of impaired Ca2+ signaling leading to perturbations in muscle contractile function and/or contraction-induced regulation of energy fluxes.
3.2. Deletion of STIM1 alters muscle pyruvate partitioning but not whole-body glucose tolerance
The exercise-intolerant phenotype of iSTIM1−/− mice combined with the negative results of the mitochondrial diagnostics assays prompted us to assess glucose flux in intact muscles. Therefore, mass spectrometry-based metabolic flux analysis was applied to isolated soleus and extensor digitorum longus (EDL) (S. Figure 4) muscles incubated with [U–13C] glucose and unlabeled palmitate (Figure 5A) ± insulin. In general, these experiments revealed a tendency for iSTIM1−/− muscles to accumulate more 13C labels in glycolytic intermediates and fewer labels in TCAC intermediates, particularly upon insulin stimulation. Diminished glucose flux into soleus muscle TCAC intermediates was most pronounced at 4 weeks (Figure 5B); whereas at 8 weeks post-TMX, genotype-related shifts in 13C enrichments were evident in the glycolytic intermediates (Figure 5C) and were accompanied by increased lactate efflux (Figure 5D). Notably, at 4 weeks post-TMX, the ratio of M5 citrate to M3 aspartate, which provides an estimation of PDH contribution to acetyl-CoA [26], was lower in iSTIM1−/− soleus muscles upon insulin stimulation (Figure 5E), suggestive of perturbed glucose flux into the TCAC. In sum, these findings are consistent with the notion that STIM1 deficiency results in reduced PDH flux. Further evidence to support this interpretation came from immunoblot assays of pPDH S300 and S293, both of which inhibit PDH activity [27,28] and were more abundant in gastrocnemius muscles from iSTIM1−/− mice at the 4-week timepoint (Figure 5F,G). Despite evidence suggesting that STIM1 deficiency alters PDH phosphorylation and/or pyruvate flux, whole-body glucose tolerance (Figure 5H,I and S. Figure 1G) and plasma insulin levels (Figure 5J) were similar between genotypes. Together, these findings imply that disruptions in mitochondrial Ca2+ signaling caused by STIM1 deletion resulted in a shift in pyruvate flux away from the TCAC and towards alternative metabolic fates. We surmise that the overall impact on glucose flux is more pronounced in vivo, particularly during muscle contraction, as strongly implied by the results of the treadmill test (Figure 4).
Figure 5.

Deletion of STIM1 alters muscle pyruvate partitioning but not whole-body glucose tolerance. (A) Scheme of isotopologue enrichment depicting labeling of glycolytic intermediates, TCAC intermediates, and TCAC-derived intermediates by [U–13C6]-glucose through the first turn of the TCAC via pyruvate dehydrogenase (black circles) and pyruvate carboxylase (blue circles). Muscles were incubated with 10 mM [U–13C]-glucose + 200 μM palmitate with or without 100 nM of insulin. Average 13C-labeling of glycolytic and TCAC intermediates in soleus muscles (B) 4 and (C) 8 weeks after TMX administered at 12–15 weeks of age (n = 4–6). (D) Lactate efflux from soleus during 90′ incubation (n = 4–6). (E) Ratio of M5 citrate to M3 pyruvate labeling as an estimation of the contribution of pyruvate to the acetyl-CoA pool through PDH (n = 4–6). (F) Western blots and quantification of phospho to total protein levels in gastrocnemius muscles 4 and 8 weeks after TMX (n = 5–8). (G) Blood glucose and (H) plasma insulin during an oral glucose tolerance test 6 weeks after TMX (n = 6–8). All data represent mean ± SEM. Data in (B-E, G-J) were analyzed using Student's two-tailed t-test. ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001. Outliers identified by Grubb's test were removed to reach statistical significance. n represents biological replicates.
3.3. Metabolomics and proteomics analysis of iSTIM−/- muscles reveals adaptive proteostasis
To further assess the impact of STIM1 deficiency on intermediary metabolism, targeted metabolomics was performed on the extracts of gastrocnemius muscles harvested 4 and 8 weeks post-TMX and after an overnight (16 h) fast (Figure 6A). Whereas small shifts in metabolite levels were detected at both time points, the most prominent and consistent signature associated with STIM1 deficiency was an increase in the abundance of a subset of amino acids and amino acid-derived acyl carnitines (e.g., arginine, proline, serine, glutamate/glutamine, and acylcarnitine C4-DC/Ci4-DC and C5OH). Additionally, levels of citrate as well as the C2 and C4OH acylcarnitine species, which can be derived from multiple fuel sources, trended higher in iSTIM1−/− muscles. Suspected perturbations in amino acid catabolism were supported by the finding that inhibitory phosphorylation of the enzyme that catalyzes the rate-limiting step in branched-chain amino acid oxidation (pBCKDH S293) was blunted 18% (S. Figure 5A,B), thereby favoring the branched-chain amino acids (BCAAs) catabolism. Moreover, several molecular markers that have been linked to regulation of autophagy and autophagic capacity were differentially regulated in STIM1−/− muscles, including phosphorylation of Unc-51 like autophagy activating kinase 1 (ULK1) at activating and inhibitory residues Ser555 and Ser757, respectively, and an elevated content of LC3-I and LC3I + II (S. Figure 5C,D).
Figure 6.

Metabolomics and proteomics analysis of iSTIM1−/− muscles reveals adaptive proteostasis. Metabolomic and proteomic analysis in gastrocnemius muscles from iSTIM1−/− v. STIM1fl/fl mice a 16 h fast. (A) Volcano plots depicting the relative abundance of organic acids, amino acids, and acylcarnitines identified 4 (left) and 8 weeks (right) after TMX administration at 12–13 weeks of age (n = 8). Gastrocnemius muscles collected 8 weeks after TMX and 16 h of fasting were used to evaluate protein and phospho-protein abundance by MS/MS proteomics and western blotting (n = 5). (B) Volcano plots depicting relative abundance (downregulated in blue and upregulated in red) of proteins (left) and phosphopeptides (right). (C) Principal component analysis of proteins (left) and phosphopeptides (right). (D) Heat map of a subset of differentially regulated proteins involved in proteostasis and contraction identified by pathway analysis. (E) Output of kinase-substrate enrichment analysis (KSEA) wherein at least 2 phospho-peptides with substrate sequence were identified; numbers indicate substrates previously linked to each kinase. (F) Western blot and (G) quantification of phosphoprotein expression normalized to total protein. Data in (A and G) were analyzed using Student's two-tailed t-test ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001. n represents biological replicates.
Broader insights into signaling pathways and biological processes influenced by STIM1 deficiency were gained using quantitative mass spectrometry-based proteomics and phosphoproteomics, performed on gastrocnemius muscle specimens harvested 8 weeks post-TMX following an overnight fast. These assays quantified 3,539 proteins and 12,442 phospho-peptides (S. Tables 1 and 2). Among these, 54 proteins and 49 phospho-peptides exhibited statistically significant changes by genotype at a stringent 10% false discovery rate (FDR). As anticipated, STIM1 emerged as the most downregulated protein and phosphopeptide (Figure 6B). Also downregulated were several contractile proteins characteristic of a slow/oxidative fiber type. Among the most upregulated proteins in STIM1 KO muscles were the Calcyclin-binding protein (CacyBP), which is involved in Ca2+-dependent ubiquitination and proteasomal degradation of target proteins [29], and IGFBP5, a secreted peptide that has been implicated as a negative regulator of muscle mass [30,31]. The list of upregulated proteins also included several known proteins to be involved in proteostasis (S. Table 1). Also noteworthy is that three members of the Ca2+/calmodulin-dependent (CaM) family of protein kinases, CAMK2A, CAMK2B, and CAMPK2G, were identified among the most downregulated phosphorylated proteins in iSTIM1−/− muscles (S. Table 2). Fittingly, CAMK2 is a well-recognized regulator of muscle metabolism [[32], [33], [34], [35], [36]] and hypertrophic signaling [37,38]. Upon filtering for proteins and phospho-peptides that met a less stringent cutoff of p < 0.01, principal components analysis (PCA) (Figure 6C) and hierarchical clustering (S. Figure 6A) revealed a clear separation between genotypes. The entire proteomics data set was used to perform pathway enrichment analysis using PathfindR (S. Figure 6B). This analysis identified ER protein processing, chaperones involved in autophagy, ribosomes, adrenergic signaling and muscle contraction as the top pathways affected by the loss of STIM1 (S. Table 1 and Figure 6D). Next, computational analysis of known phospho-signaling networks was performed using kinase-substrate enrichment analysis (KSEA) [39,40]. This algorithm predicts signaling cascades that might be affected by a given manipulation based on net changes in an identified set of phospho-peptide substrates that have previously been linked to a specific kinase. Results of this analysis suggested that signaling cascades mediated by Glycogen synthase kinase 3 beta (GSK3β), Protein kinase C delta (PKCD), MAPK-activated protein kinase 2 (MAPKAPK2), and Extracellular signal-regulated kinase 2 (ERK2) might be upregulated in STIM1−/− muscles, whereas DAPK3, AKT1 and CAMK2A were identified as suspected downregulated networks (Figure 6E). Many of these signaling proteins have established connections to Ca2+ homeostasis [35,41,42], proteostasis [[43], [44], [45], [46]], and/or muscle growth [36,38,[47], [48], [49], [50]]. Notably, predicted changes in CAMPK2A (pCAMPK2A T286) and ribosomal protein S6 (pS6 S235/236) were confirmed by Western blot analysis (Figure 6F,G). In aggregate, the results imply that STIM1 insufficiency in adult muscle results in wide-ranging perturbations in ER protein processing, ribosomal function, muscle fiber type, and growth signaling.
3.4. Chronic deletion of STIM1 diminishes mitochondrial respiratory efficiency without exacerbating diet-induced glucose intolerance
The foregoing results revealed that STIM1 is essential for maintaining energy metabolism and proteostasis in adult mice. Because dysregulation of muscle energy homeostasis and elevated ER stress have been linked to obesity and type 2 diabetes, we sought to test the hypothesis of whether STIM1 deficiency might increase susceptibility to diet-induced weight gain and/or glucose intolerance. To this end, iSTIM1−/− and control littermates were fed an obesogenic high-fat diet (HFD) beginning at 10 weeks post-TMX. Mice were then monitored longitudinally for 20 weeks and compared against a parallel cohort of animals fed a standard chow (SC) diet. Primary endpoints included energy balance, whole-body glucose tolerance, and mitochondrial function.
The amount of body weight gained (Figure 7A) and fat amassed (Figure 7B) in response to the HFD was similar between genotypes (Figure 7C). Although total body lean mass was lower in iSTIM1−/− mice regardless of diet (Figure 7D), muscle weights normalized to tibia length were consistently lower in iSTIM1−/− mice fed SC, though this effect appeared to be partially offset by HF feeding (Figure 6E). HFD led to a predictable decline in whole body respiratory exchange ratio (RER), reflecting increased fat oxidation (Figure 7F). When analyzed as a function of lean mass or body weight, energy expenditure was higher in HFD-fed mice, regardless of genotype. However, the relationship between body weight and energy expenditure was unaffected by genotype (Figure 7G,H). Evidence of genotype-specific metabolic responses emerged from an acute fasting/refeeding challenge, whereas the RER of iSTIM1−/− mice rose gradually during the fasting period, indicative of a fuel shift away from fat-derived metabolites (Figure 7I,J). Additionally, the relative increase in whole-body glucose oxidation that occurred upon oral glucose administration and refeeding was blunted in the KO mice, regardless of the diet, consistent with impaired metabolic flexibility (Figure 7K).
Figure 7.

Chronic deletion of STIM1 alters whole-body fuel selection. (A) Body weight, (B) fat mass (C) percentage body fat, and (D) lean mass of iSTIM1−/− and STIM1fl/fl mice fed standard chow (SC) or a high-fat diet (HFD) (n = 6–8). (E) Wet weight of skeletal muscle normalized to tibia length after 20 weeks of diet and 30 weeks after TMX administered at 12–15 weeks of age (n = 6). (F) Respiratory exchange ratio over a 24 h light/dark period with ad libitum access to food after 8 weeks on diet (n = 5–8). Relationship between energy expenditure to (G) lean mass, and (H) body weight (n = 5–8). (I) Respiratory exchange ratio during 16 h of fasting, administration oral glucose bolus (3 g/kg), and a 5 h refeed after 13 weeks on diet, and (J) relative frequency distribution of RER during 16-h fasting period (n = 5–8). (K) Metabolic flexibility defined as the difference in RER in response to the 3 g/kg oral glucose bolus and the last hour of fasting (n = 5–8). Data in (A-F, I, and J) were analyzed using Student's two-tailed t-test. ∗p ≤ 0.05, ∗∗p ≤ 0.01 and ∗∗∗p ≤ 0.001. Outlier identified by Grubb's test was removed to reach statistical significance and is represented by a gray open circle. n represents biological replicates.
These observations prompted us to investigate the impact of chronic STIM1 deficiency on mitochondrial function and diet-induced adaptations in oxidative metabolism. These experiments revealed a respiratory phenotype in the SC group that was not evident at the earlier time points (S. Figures 2 and 3). Thus, chronic ablation of STIM1 resulted in a dramatic leftward shift in both the PM and αKG respiratory efficiency plots (Figure 7A), which depicts the relationship between JO2 (S. Figure 7A) and ΔΨm (S. Figure 7B). The leftward shift reflects a lower (less polarized) ΔΨm for any given rate of oxygen consumption; and occurs due to lower ΔΨm and modest alterations in mitochondrial JO2, and NAD(P)H/NAD(P)+ redox potential (S. Figure 7C), without any detectable shift in H2O2 emission (S. Figure 7D) or muscle abundance of canonical OxPhos subunits (Figure 8C,D). Notably, respiratory inefficiency was observed only in the presence of Complex I-linked substrates (PM and αKG) and not during fatty acid (PcM)-supported respiration. These findings suggest that the phenotype of iSTIM1−/− mitochondria is mediated by specific metabolic enzymes and/or pathways and not a result of generalized mitochondrial dysfunction. Remarkably, this pronounced phenotype was prevented in the cohort of iSTIM1−/− mice fed an HFD (Figure 8B,S. Figure 7, Figure 8). As shown previously [26], high-fat feeding enhances fatty acid-supported respiratory capacity, which was evident regardless of genotype (S. Figure 7D–G). Additionally, only in mitochondria extracted from iSTIM1−/− mice, chronic exposure to an HFD was accompanied by an increase in ΔΨm (S. Figure 8B,E), regardless of substrate, resulting in a rightward shift in the corresponding efficiency plots (S. Figure 8C,G).
Figure 8.

Chronic deletion of STIM1 diminishes mitochondrial respiratory efficiency without exacerbating diet-induced glucose intolerance. Respiratory efficiency evaluated by plotting JO2 vs. ΔΨm of mitochondria fueled with pyruvate/malate (PM), α-ketoglutarate (αKG), or palmitoylcarnitine/malate (PcM) isolated from skeletal muscle of mice fed a (A) standard chow (SC) (B) or 20-week high fat (HF) diet 30 weeks after TMX administration at 12–15 weeks of age (n = 4–6). (C) Western blot and (D) quantification of canonical OXPhos subunits normalized total protein (n = 6). (E) Oral glucose tolerance test after 10 weeks on diet (n = 6–8). (J) Western blot and quantification of STIM1 expression normalized to total protein in gastrocnemius muscle after 20 weeks of diet and 30 weeks after TMX (n = 6). All data represent mean ± SEM. Data in (A and B) were analyzed using 2-way ANOVA (S. Figure 6). Data in (D-F and H) were analyzed using Student's two-tailed t-test to test for statistical significance. Outlier identified by Grubb's test was removed to reach statistical significance. n represents biological replicates.
Despite their proclivity for reduced muscle mass, heightened ER stress, and disturbances in oxidative metabolism, iSTIM1−/− mice were not more susceptible to diet-induced glucose intolerance (Figure 8E,F). Lastly, Western blot analyses of muscles harvested at 30 weeks post-TMX confirmed that knockout of STIM1 had been maintained and that both isoforms of the protein were reduced by approximately 90% (Figure 8G,H).
4. Discussion
Though previous investigations have established STIM1 as an ER-localized Ca2+ sensor that activates Orai channels to promote SOCE during critical stages of development, its metabolic functions in adult skeletal muscle were not investigated. The present work provides strong evidence that STIM1 regulates Ca2+ handling in adult myofibers, and thus impacts a broad set of Ca2+-dependent signaling cascades and biological processes that influence energy metabolism and protein homeostasis. Prominent among these is mitochondrial Ca2+ loading in response to SR-Ca2+ release. Disruption of this process in iSTIM1−/− mice contributed to increased lactate production and early exhaustion during exercise. Moreover, we identified profound perturbations in signaling networks linked to ER stress and proteostasis, which were accompanied by diminished myofiber size and a substantial reduction in whole-body lean mass. Lastly, chronic deletion of STIM1 was associated with shifts in fuel preference during fasting and diminished respiratory efficiency evidenced in isolated mitochondria, but without affecting whole-body glucose tolerance. These results establish new roles for STIM1 protein in maintaining skeletal muscle health and function during early and middle adulthood.
Two working hypotheses have been offered to elucidate the function of SOCE in skeletal muscle. First, SOCE is slowly activated with store depletion, whether global or local, to refill stores and sustain Ca2+-dependent signaling during muscle development. Alternatively, SOCE is activated with each action potential in response to local RYR1 depletion, thereby acting as a countercurrent to the plasma membrane Na+/Ca2+ exchanger isoform 1 (NCX1) in order to prevent Ca2+ overload of the T-tubules. The findings reported herein support the latter model where Ca2+ released from the SR fails to refill the mitochondria. By sensing and responding to repetitive Ca2+ transients in skeletal muscle, STIM1 can adjust critical features of this tissue, including fuel choice, fiber type, gene expression, and protein synthesis, to optimize metabolic efficiency. Aligned with this premise, ablation of STIM1 in adult muscles severely abrogated the transfer of Ca2+ from the E/SR to mitochondria, in association with reduced exercise tolerance during a graded treadmill test. Because reduced muscle mass, weakness, and altered metabolism were not identified in similar studies with Orai1-deficient mice, we consider the STIM1-mitochondria Ca2+ signaling independent of SOCE. Moreover, we recently reported that STIM1 interacts with desmin filaments at the Z-lines of mouse muscles [11]. The STIM1-desmin interaction was found to be important for refilling SR and mitochondria Ca2+ stores whereas Orai1 has no overlapping function. Previous work demonstrated that exercise-induced phosphorylation of STIM1 via AMPK negatively regulates SOCE and that loss of STIM1 function improves exercise tolerance in Drosophila [19]. The investigators speculated that during high-intensity muscle contraction, SOCE-mediated Ca2+ entry contributes minimally to SR Ca2+ refilling and that excessive SOCE might lead to mitochondrial Ca2+ overload and early fatigue. Taken together with the current results, the two studies support a model wherein STIM1 acts as a Ca2+ rheostat to fine-tune Ca2+-dependent regulation of energy metabolism and cell signaling. It is important to point out that desmin, a STIM1 binding partner, is found only in invertebrates which may reconcile differences in exercise tolerance in STIM1 loss of function experiments for flies and mice.
Early exhaustion in iSTIM1−/− mice was accompanied by a striking leftward shift in the substrate crossover point – the exercise intensity corresponding with an accelerated rise in glucose use and lactate production. This phenomenon, also termed the lactate threshold, has been linked to limitations in the electron transport chain, beta-oxidation, and/or pyruvate flux through the PDH complex. Thus, comprehensive functional analysis of mitochondria isolated 4 and 8 weeks post-TMX provided no evidence of metabolic reprogramming, mitochondrial remodeling, and/or overt respiratory dysfunction in iSTIM1−/− mice. We, therefore, surmise that the rapid transition to glycolytic metabolism in these mice was caused by limitations in carbon flux through dehydrogenase enzymes that are activated upon mitochondrial Ca2+ loadings, such as PDH and alpha-ketoglutarate dehydrogenase (α-KGDH). For example, reversible phosphorylation of the E1α subunit (Ser-232, 293, or 300) of PDH is central to the regulation of enzymatic activity [51,52]. Phosphorylation by PDH specific kinases (PDK1-4) inhibits complex activity, whereas dephosphorylation by PDH phosphatases (PDP1/2) augments activity [53]. PDP1, the muscle-specific phosphatase that activates PDH-E1α, has a canonical Ca2+ binding domain that promotes catalysis [54]. Similarly, the enzymatic activities of NADP-isocitrate DH (IDH2), αKGDH, and glycerol-3 phosphate DH are also sensitive to Ca2+ [12,55,56]. Like PDP1, subunits of these enzymes contain Ca2+-binding EF-hand motifs that regulate steady-state activity and Vmax [55,57,58].
Evidence that STIM1 deletion does indeed alter pyruvate partitioning at PDH emerged from studies in which stable isotope tracing was applied to isolated soleus and extensor digitorum longus (EDL) muscles. These experiments revealed reduced coupling of glycolysis and pyruvate oxidation along with elevated lactate efflux. Moreover, decreased mitochondrial PDH flux was accompanied by increased pPDH, consistent with impaired dephosphorylation of the enzyme by the Ca2+-dependent PDP1. Although pPDH was unchanged in iSTIM1−/− muscles collected after 15 min of treadmill running, that the anticipated contrast between genotypes could possibly have surfaced at an earlier point in the treadmill test, closer to the time of substrate crossover in the KO mice. Notably, iSTIM1−/− mice did not exhibit increased whole-body lipid catabolism and/or enhanced mitochondrial fat oxidation, which conflicts with studies in mice with muscle-specific deletion of the mitochondrial Ca2+ uniporter (MCU). As compared with iSTIM1−/− mice, MCU-deficient muscles display a similar disruption in mitochondrial Ca2+ signaling and PDH activity [59] but adapt by increasing reliance on lipid fuels. Conversely, iSTIM1−/− mice maintained a higher RER during exercise and fasting, indicative of a lower rate of beta-oxidation. This distinction between models might be explained by reports showing that STIM1-mediated SOCE is required for lipolysis [60,61], which would affect the use of intramuscular triacylglycerol stores in iSTIM1−/− muscles.
In addition to the foregoing alterations in pyruvate flux, hints that amino acid catabolism might reciprocally be upregulated in iSTIM1−/− muscles emerged from mass spectrometry-based metabolic profiling and a corresponding reduction in inhibitory phosphorylation of branched-chain α-ketoacid dehydrogenase complex (BCKDC). A constitutive increase in amino acid oxidation would elevate fasting RER and blunt the typical feeding-induced rise in glucose oxidation, resulting in metabolic inflexibility–a phenotype observed in iSTIM1−/− mice. This interpretation fits with the finding that deletion of STIM1 led to a rapid divergence in total body lean mass due to diminished muscle growth. Skeletal muscle mass is regulated by a complex set of processes and systems that balance rates of protein synthesis and degradation while also managing proper folding, trafficking, and clearance of proteins. The cellular organelle primarily responsible for protein quality control is the E/SR, and Ca2+ plays a critical role in maintaining an ER environment that permits normal protein processing [62]. In the current study, proteomics analysis of iSTIM1−/− muscles revealed that pathways associated with protein processing in the ER, protein folding, and ribosomal protein synthesis are broadly upregulated in this model. These findings strongly suggest that STIM1 deficiency disrupts Ca2+ balance in the ER leading to perturbations in processing, quality, and/or turnover of muscle proteins. Further insights into potential molecular mechanisms responsible for the STIM1-related decline in muscle mass were gleaned from kinase-substrate enrichment analysis (KSEA) of the phosphoproteomics data. Leveraging known kinase–substrate relationships, this analysis identified DAPK3, CAMK2A, and AKT1 as potential downregulated signaling pathways, whereas GSK3β, PKCD, MAPKAPK2, and ERK2 emerged as signaling pathways that might be more active in iSTIM−/- muscles. Interestingly, DAPK3 has been implicated in regulating Ca2+ sensitization and muscle contractility [63], whereas CAMK2A is well-recognized as a molecular sensor that connects Ca2+ signals to transcriptional control of fiber type [35,36], mitochondrial biogenesis [34,64], and hypertrophy [37,38]. Also, AKT1 holds a prominent position as a central hub in growth signaling, whereas activated GSKβ antagonizes growth, partly through inhibition of AKT1. Lastly, the predicted activation of ERK2 signaling in KO muscles would favor increased protein translation, in part via its downstream targeting and phosphorylation of the S6 ribosomal protein [65,66], which was confirmed by immunoblot analysis. Taken together, these unbiased molecular profiles suggest that deletion of STIM1 resulted in the selective dampening of pathways linked to growth signaling, while ribosomal activation and presumably protein synthesis showed an increase. Integration of the proteomics, metabolomics, and physiologic assessments support a model wherein iSTIM1−/− muscles adapt to low-grade ER stress by slowing growth and augmenting protein quality control mechanisms. This apparent shift in adaptive proteostasis implies more protein synthesis and turnover in the context of a lower setpoint for muscle mass, which together serves to maintain resting energy homeostasis.
Although the proteomic analysis could not identify a clear mitochondrial signature, it is important to underscore that these assays were performed on muscles harvested at a relatively early (8 week) time point after TMX treatment. Whereas mitochondria from iSTIM1−/− muscles appeared functionally normal when assayed in vitro at 4 and 8 weeks post-TMX, subject mice studied after 30 weeks of STIM1 deficiency displayed an inefficient oxidative phenotype characterized by a leftward shift in the relationship between JO2 and ΔΨm. This phenotype was most pronounced when respiration was supported by Complex I-linked substrates (pyruvate and αKG) and was prevented by feeding the KO mice an HF diet for 20 weeks. These findings imply that the abnormalities in mitochondrial bioenergetics resulting from chronic STIM1 ablation are related to deficits in the flux of pyruvate and glutamate, the preferred substrates for mitochondria resident in muscles comprised predominately of white/glycolytic myofibers. When mice are fed an HFD, white muscles adapt by augmenting their beta-oxidation machinery [26], which enhances myofiber capacity to support energy demands, including those associated with mitochondrial quality control, using lipid substrates.
Lastly, it is noteworthy that STIM1 deficiency did not increase susceptibility to whole-body glucose intolerance. This result was surprising because chronic E/SR stress and subsequent activation of the unfolded protein response (UPR) is increasingly recognized as a pathogenetic mechanism linking obesity, insulin resistance, and type 2 diabetes [67,68]. The disconnect between E/SR stress and metabolic dysfunction in iSTIM1−/− might stem from counter-regulation of signaling networks and/or proteostatic processes that mitigate the unfolded stress response (UPR) and thus act to avert catastrophic disruption of protein quality. To this point, CAMK2 has been identified as a pathological mediator of E/SR stress, and its suppression prevents UPR-induced inhibition of insulin signaling [69,70]. In aggregate, the findings imply that low-grade E/SR stress alone is not sufficient to cause muscle insulin resistance.
In aggregate, this work establishes a critical role for STIM1 in regulating cellular and mitochondrial Ca2+ dynamics, energy metabolism, and proteostasis in adult skeletal muscle. Additionally, the iSTIM1−/− mice provide a new and intriguing model wherein perturbations in Ca2+ homeostasis and protein processing are accompanied by rapid resetting of muscle size and whole-body lean mass. These findings offer important insights into the biological mechanisms that connect genetic mutations in STIM1 to musculoskeletal and cardiometabolic diseases in humans [71] while having new implications for a potential role of STIM1 and Ca2+ dysregulation in age-related decline of skeletal muscle mass and function.
5. Limitations
The current study did not measure mitochondrial bioenergetics and/or substrate flux during contraction; therefore, it did not capture shifts in energy metabolism during physiological settings when Ca2+ loading of mitochondria increases dramatically. Additionally, although the proteomic analyses provide strong circumstantial evidence that muscle proteostasis is disrupted in the context of STIM1 deficiency, assays to directly measure rates of muscle protein synthesis, turnover, and/or autophagic flux are especially challenging and were not performed. The study raises new questions regarding the role of STIM1 in regulating mitochondria-SR contact sites and physical interactions between the two organelles. Furthermore, the specific functions of the L and S STIM1 isoforms remain unknown. These knowledge gaps are worthy of future investigation.
6. Contact for reagent and resource sharing
6.1. Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Deborah M. Muoio (debbie.muoio@duke.edu) and/or co-senior and co-corresponding author, Paul Rosenberg (paul.rosenberg@duke.edu)
6.2. Materials availability
This study did not generate new unique reagents. Mouse lines generated in this study will be distributed upon request to other investigators under a Material Transfer Agreement.
6.3. Data and code availability
All raw data from proteomics were submitted to ProteomeXchange.
No unpublished custom code, software, or algorithm was used in this study. All software packages and methods used in this study have been properly detailed and referenced under the Software and algorithms listed in reporting summary. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
7. Experimental model and subject details
All animal procedures were performed in accordance with the National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee at Duke University School of Medicine (Protocol A064-16-03). The mice were group-housed in light (12-h light/12-h dark intervals) and temperature (22 °C) controlled room and had ad libitum access to food and water unless noted otherwise. Male mice were used for most experiments and randomly assigned to experimental groups. A subset of experiments performed using female mice is reported in S. Figure 1. Mice with muscle-specific overexpression of PGC-1α (Tg) and non-transgenic (NT) littermates were provided by Dr. Bruce Spiegelman. STIM1-LacZ and STIM1fl/fl mice were generated as previously described [5]. Mice with skeletal muscle-specific deletion of STIM1 (mSTIM1−/−) were generated by crossing transgenic Myogenin-Cre mice with homozygous STIM1fl/fl mice as previously described [4]. Mice with inducible skeletal muscle-specific deletion of STIM1 (iSTIM1−/−) were generated by breeding transgenic HSA-MerCreMer mice [72] (purchased from Jackson Laboratories) with STIM1fl/fl mice. Tamoxifen (TMX) dissolved in corn oil was used to induce knock-out of the STIM1 gene. For early imaging experiments, TMX (225 mg/kg) was administered to 8-week-old mice by intraperitoneal injection for 5 days. In subsequent phenotyping experiments, TMX (80 mg/kg) was administered to 11-15-week-old mice by oral gavage for 3 days. Mice were fed a standard chow diet (Lab Diets PicoLab Rodent Diet 5053, Lab SupplyNorthlake, TX, USA) unless otherwise noted. For the diet study, the mice were fed a standard chow diet for 10 weeks after TMX injection, then divided into two experimental groups, and fed a standard chow diet or high fat (HF) diet with additional sucrose (Research Diets D03021303i; 25% sucrose, 45% fat) for 20–22 weeks. Unless otherwise stated, mice were anesthetized by intraperitoneal injection with Nembutal (100 mg/kg BW) prior to organ removal.
8. Method details
8.1. Chemicals and reagents
All chemicals were purchased from Sigma–Aldrich (St. Louis, MO, USA) unless otherwise stated. Creatine kinase from rabbit skeletal muscle was purchased from Roche Life Science (Penzberg, Germany). Tetramethylrhodamine methyl ester and Amplex Ultra Red were purchased from Thermo Fischer Scientific. d-Glucose (U-13C6) was purchased from Cambridge Isotope Labs (Tewksbury, MA, USA). Potassium pyruvate was purchased from Combi-Blocks (San Diego, CA, USA). Protease (Complete mini EDTA-free) and phosphatase (PhosStop) inhibitor tablets (Roche). 11-plex Tandem Mass Tags (TMT) were purchased from Thermo Fisher Scientific. Sequencing Grade Modified. Sequencing grade trypsin was purchased from Promega (Durham, NC, USA). Lysyl Endopeptidase (LysC) was purchased from Wako Chemicals (Richmond, VA, USA). tC18 SEP-PAK SPE columns were purchased from Waters (Milford, MA, USA). Ni-NTA Magnetic Agarose Beads were purchased from Qiagen (Germantown, MD, USA). Fura-2 AM, Rhod-2AM and Fura-4AM were purchased from Invitrogen (Waltham, MA, USA).
8.2. Oral glucose tolerance test
Mice were singly housed and fasted on alpha-dry bedding for 5 h with free access to water. Glucose (1.5 g/kg body weight) was given orally and measured in whole blood obtained from the tail vein using a hand-held glucometer at 0, 10, 20, 30, 45, 60, and 90 min after glucose gavage. Blood for plasma insulin (oral glucose-stimulated insulin secretion) was collected from the tail vein 0, 10, and 30 min following glucose administration using heparin-coated capillary tubes (Sarstedt Microvette CB LH). Insulin was determined using ALPCO STELLUX Chemi Rodent Elisa Kit per manufacturer's instructions (ALPCO, Salem, NH, USA).
8.3. Body composition and muscle mass
Body weight and composition were measured longitudinally after TMX administration. Body composition was determined using a MiniSpec model mq10 nuclear magnetic resonance analyzer (Bruker Optics, Billerica, MA, USA). Soleus, plantaris, gastrocnemius, and tibialis anterior muscles were carefully excised from the hindlimb and weighed. Tibia length was determined using electronic calipers. Muscle weights were normalized to tibia length to account for any differences in body size.
8.4. Metabolic flexibility and locomotor activity
Indirect calorimetry was determined using the 8-chamber Comprehensive Lab Monitoring System (Columbus Instruments, Columbus, OH, USA) at the flow rate of 0.5 L/min. At the beginning of each run, the calorimeter O2 and CO2 sensors were calibrated using a reference gas containing N2 (79%), CO2 (0.5%), and O2 (20.5%). Oxygen consumption (VO2) and CO2 production (VCO2) were directly measured in series for 30 s intervals every 20 min over a period of 24–48 h. The total locomotor activity of mice moving freely in their cages was registered continuously by an infrared photocell beam grid. Mice had free access to food and water throughout the experiments unless otherwise noted. For the fasting-glucose challenge, food was removed after 3 h and mice fasted for 16 h then received glucose orally (3 g/kg body weight) 10 min prior to the subsequent cage measurement. Raw data files from Oxymax v.5.25.05 were uploaded to CalR for data analysis (https://calrapp.org) [22]. RER was calculated by VCO2 produced divided by VO2 consumed (RER = VCO2/VO2, where V is Volume). Energy expenditure (EE) was determined using the modified Weir equation wherein EE = (3.815 + 1.232 × RER) × VO2 [73]. Metabolic flexibility was calculated by subtracting the peak RER in response to glucose from the average RER of the last hour of fasting. Percent relative cumulative frequency of RER was calculated to determine how frequently specific RER values occur in each genotype [74].
8.5. Acute exercise
The acute exercise test was conducted as previously described [75]. After 3 days of habituation to the open-air Exer-3/6 treadmill equipped with a shock grid (Columbus Instruments), the exercise bout started at 5 m/min for 10 min to warm up, speed was increased to 10 m/min for 20 min, and then by 1 m/min every 2 min to a final rate of 15 m/min, which was maintained for 1 h. Control mice were habituated but not exercised. Tibialis anterior muscles were collected immediately post-exercise or after 3 or 24 h of recovery and immediately flash frozen.
8.6. Exhaustive exercise test
Mice were habituated to a single-line enclosed metabolic treadmill equipped with a shock grid (Columbus Instruments) for 3 days prior to experiments. The exhaustive exercise test began with a 10-min 0 m/min acclimation period followed by a speed of 6 m/min for 5 min, followed by increasing speed 3 m/min every 5 min. Once 20 m/min was reached, speed increased by 2 m/min every 2 min until exhaustion, defined as remaining on the shock grid for 10 s without engaging the treadmill. Oxygen consumption and carbon dioxide production were continually measured by indirect calorimetry and used to determine RER. RER values were used to calculate the crossover point; the point of the exercise at which energy derived from carbohydrates predominates over energy from lipids [23]. Time and distance to exhaustion were collected to measure exercise tolerance. To assess lactate and molecular signaling immediately after exercise mice were acclimatized to an open-air Exer-3/6 treadmill for 3 days. Basal blood lactate was measured from the tail vein (Nova Biomedical). The exercise was started at 6 m/min with increasing speed by 3 m/min every 5 min for 20 min, at which point post-exercise blood lactate was measured, and mice were euthanized with isoflurane prior to organ removal.
8.7. Mitochondrial isolation from skeletal muscle
Mitochondria were isolated from skeletal muscle by differential centrifugation using two buffer formulations. The buffers for Ca2+ and functional experiments are as follow: Buffer A (PBS with 10 mM EDTA, pH 7.4); Buffer B (50 mM MOPS, 100 mM KCl, 1 mM EGTA, 5 mM MgSO4, pH 7.1); and Buffer C (Buffer B supplemented with 2 g/L fatty-acid free BSA). Mice were fasted for 16 h with access to water then skeletal muscles (quadriceps and gastrocnemius) were excised and immediately placed in ice-cold buffer A. Skeletal muscles were minced, incubated on ice in Buffer A supplemented with 0.5% (w/v) trypsin for 5 min with vortexing every 30 s then centrifuged 200×g for 5 min at 4 °C. The supernatant was discarded and pellets resuspended in Buffer C, homogenized with a Teflon pestle and borosilicate glass vessel, and centrifuged at 800×g for 5 min at 4 °C. The supernatant was filtered through two layers of gauze and centrifuged 9,000×g for 10 min at 4 °C. The pellet was resuspended in Buffer B and centrifuged at 9,000×g for 3 min at 4 °C. The supernatant was aspirated from each tube and mitochondrial pellets were suspended in 100 μL Buffer B. Protein content was determined by Pierce BCA protein assay and mitochondria were diluted with Buffer B to a final concentration of 10 mg/mL for all functional and Ca2+ assays.
8.8. Intramitochondrial Ca2+
The o-Cresolphthalein Complexone (OCC) assay was used to determine mitochondrial [Ca2+] ([Ca2+]m) as previously described [76] with minor modifications. In brief, isolated mitochondria were centrifuged at 4 °C. The supernatant was then aspirated from each tube and mitochondrial pellets were resuspended in 0.6 N HCl and homogenized using a chilled Teflon pestle. The mitochondrial suspension was then sonicated with a probe sonicator (three 10 s bursts power setting of 4) boiled for 30 min at 95 °C, and centrifuged at 12,000×g for 10 min. The supernatants were loaded onto a 96-well plate with the OCC assay reagent (1.75 M 2-amino-2-methyl-1-propanol, 80 μM o-cresolphthalein complexone, 30 mM HCl, 3.45 mM 8-hydroxychloroquine, pH 10.7) and absorbance was measured at 575 nm.
8.9. Mitochondrial respiratory control
High-resolution respirometry was performed using OROBOROS Oxygraph-2k using a modified version of the creatine kinase (CK) bioenergetic clamp in the presence of excess CK, and known amounts of ATP, creatine (Cr), and phosphocreatine (PCr) as described previously [25]. The CK reaction couples the interconversion of ATP and ADP to PCr and Cr. All assays were conducted in 2 mLs of Buffer Z (105 mM MES potassium salt, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2) supplemented with Cr (5 mM), PCr (1.5 mM), CK (20 U/mL), fatty acid free BSA (2.5 mg/mL), and EGTA (1 mM; pH 7.2) at 37 °C with constant stirring. At the start of each assay, isolated mitochondria (0.025 mg/mL) were added to the assay buffer followed by the addition of respiratory substrates, then ATP (5 mM). Next, sequential additions of PCr were added to achieve the final concentrations of 3, 6, 9, 12, and 15 mM. The purpose of sequential additions is to progressively shift the CK equilibrium, altering energy demand (ATP/ADP ratio), and gradually lowering respiratory flux (JO2). The free energy of ATP hydrolysis (ΔGATP) after each PCr addition was calculated using the online tool (https://dmpio.github.io/bioenergetic-calculators/) which uses the equation ΔGATP = ΔGATP⁰ −2.3 RT∗K∗2.3∗log [(PCr]KCK/[Cr][Pi])], where ΔGATP⁰ is the standard ΔGATP (−7.592 kcal/mol), R is the gas constant (8.3145 calK−1mol−1), and T is the temperature (310.15 K). The slope of the linear portion represents the conductance of the overall system under specific substrate conditions. The following substrate combinations were used: 5 mM Pyruvate with 2.5 mM Malate (PM), α-ketogluterate (αKG; 5 mM), and 20 μM Palmitoyl-carnitine with 2.5 mM Malate (PcM).
8.10. Mitochondrial membrane potential and NAD(P)H/NAD(P)+ redox
Fluorescent determination of membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ were carried out simultaneously using QuantaMaster Spectrofluorometer. The ΔΨ was determined using the ratiometric fluorescent dye tetramethylrhodamine methyl ester (TMRM) wherein excitation/emission (ex/em) intensities at 572/590 nm and 551/590 nm were recorded. The ex/em parameters for NAD(P)H were 340/450 nm. All assays were conducted in 0.2 mL Buffer Z supplemented with Cr (5 mM), PCr (1.5 mM), CK (20 U/mL), and TMRM (0.2 μM) at 37 °C with constant stirring. At the start of each assay, isolated mitochondria (0.1 mg/mL) were added to the assay buffer followed by the addition of respiratory substrates (PM, αKG, and PcM), ATP (5 mM), and then sequential additions of PCr to achieve the final concentrations of 3, 6, 9, 12, and 15 mM. After the last PCr addition, potassium cyanide (4 mM) was added to induce a 100% reduction of the NAD(P)H/NAD(P)+ redox couple followed by alamethicin (12.5 μg/mL) to permeabilize the mitochondria and create a state of 0% reduction. NAD(P)H/NAD(P)+ throughout the experiment was expressed as % reduction based on the following equation: (F1–F0%)/(F100%-F0%). Mitochondrial ΔΨ was determined by converting the fluorescent 572/551 nm ratio to millivolts using a KCl standard curve generated in the presence of valinomycin.
8.11. Mitochondrial H2O2 emission
Rates of mitochondrial H2O2 emissions (JH2O2) were determined fluorometrically (QuantaMaster Spectrofluorometer; excitation/emission 565/600 nm) using Amplex Ultra Red (AUR)/horseradish peroxidase (HRP) detection system. All assays were conducted in 0.2 mL Buffer Z supplemented with Cr (5 mM), PCr (1.5 mM), CK (20 U/mL), AUR (10 μM), HRP (1 U/mL) and superoxide dismutase (20 U/mL) at 37 °C with constant stirring. At the start of each assay, isolated mitochondria (0.1 mg/mL) were added to the assay buffer, followed by respiratory substrates (PM, αKG, and PcM), ATP (5 mM), auranofin (AF; 0.1 μM), and then sequential additions of PCr to achieve the final concentrations of 6 and 15 mM. After the final PCr addition, 1-chloro-2,4-dinitrobenzene (CDNB) was added to approximate maximal JH2O2.
8.12. Western blot analysis
Frozen gastrocnemius muscles were powdered under liquid nitrogen and homogenized in 2x lysis buffer (4% SDS, 10 mM NaF, 1 mM EDTA, 20% glycerol, and 20 mM Tris; pH 6.8) supplemented with phosphatase inhibitor cocktails 2 and 3 and protease inhibitor using TissueLyser II bead mill (Qiagen) for 2 min at 30 Hz. Samples were removed, freeze-thawed three times, and sonicated with a probe sonicator (three 10 s bursts, power setting of 4). Samples were centrifuged at 10,000×g for 10 min at RT and the supernatant was transferred to a clean tube. Protein concentration was determined using Pierce BCA protein assay. Samples were diluted with 2x lysis buffer and mixed with an appropriate volume of 5x loading buffer. Thirty micrograms of protein per well were loaded onto a 12 + 2, 18, or 26 well 4%–15% Criterion TGX Stain-Free SDS-PAGE gel (Bio-Rad, Hercules, CA, USA), transferred a 0.2 μm Nitrocellulose membrane via the Bio-Rad TurboBlot Transfer system, and total protein was visualized using Pierce Reversible Stain Memcode Kit or Ponceau S solution. Membranes were cut horizontally into multiple sections to simultaneously probe for different proteins. Membranes were blocked in fish gelatin in phosphate-buffered saline for 1 h at room temperature and incubated with primary antibodies (1:1000) overnight at 4 °C with rocking. Membranes were washed with tris buffered saline with TWEEN (0.05%), incubated with appropriate secondary antibody (LiCor Biosciences) diluted in fish gelatin (1:10,000), washed, and imaged with Odyssey Imager. Band intensities were determined using ImageJ and normalized to total protein. The following primary antibodies were used: STIM1 (ab57834), VDAC1 (ab15895), PDHA (ab110330), PDHA Ser300 (AP1064), PDHA Ser293 (ABS204), AMPKα (CST 2531), AMPKα Tyr172 (CST 2532), CAMK2α (sc-13141), CAMK2 Thr286 (CST 3361), S6 ribosomal protein (CST 2217), S6 ribosomal protein Ser235/236 (CST 2211), Ulk1 (CST 8054), Ulk1 Ser757 (CST 6888), Ulk1 Ser555 (CST 8054), OPA1 (CST 80471), Drp1 (CST 8570), MFF (CST 84580), COXIV (CST 4850), SERCA1 (MA3-911), Total OXPHOS (MS604).
8.13. Gene expression
Total RNA was extracted from frozen tibialis anterior powdered under liquid nitrogen using Trizol/chloroform extraction method paired with RNeasy Mini spin columns (Qiagen) with on-column DNase treatment. RNA concentration was determined using a NanoDrop. cDNA was synthesized using the iScript cDNA synthesis kit (BioRad). Real-time quantitative PCR was performed using TaqMan Master Mix (STIM1 assay TaqMan#_Mm01158413) and Applied Biosystems 7000 Sequencing Detection System. The threshold count values were normalized to 18 S rRNA (18 S assay Applied Biosystems #4319413 E) to calculate fold change.
8.14. Isolation of myofibers for Ca2+ imaging
Myofibers from flexor digitorum brevis (FDB) muscles were isolated as previously described [77]. In brief, FDB muscles were carefully dissected, cleaned of connective tissue, and digested for 2.5 h at 37 °C in DMEM supplemented with 2% collagenase (Worthington). After digestion, muscles were transferred to a plate with Opti-MEM supplemented with 10% horse serum, the tendons were separated, and single fibers were liberated under a dissecting microscope by trituration. Myofibers recovered for 2 h prior to subsequent imaging experiments.
8.15. Ca2+ imaging of FDB myofibers
To measure mitochondrial Ca2+, Rhod-2 (2 μM) was loaded into FDB muscle fibers on ice for 30min. The Rhod-2 was subsequently washed out with Tyrode solution for 5–10 min at room temperature to sequester the Ca2+ indicator to mitochondria. To measure dynamic Ca2+ activities in response to store depletion and SOCE, epi-fluorescence of Rhod-2 was excited at 560 nm with a pE 340fura illumination system (cool Led) and epi-fluorescence at 580 nm were recorded with a pco edge 5.5 scientific CMOS digital camera. The illuminator and camera were controlled and data were acquired with Micro-Manager software. The data were further analyzed with ImageJ software. To measure Ca2+ uptake to the mitochondria in response to electrical field stimulation (EFS), myofibers were cultured in a perfusion trough with a 35-mm insert with a pair of electrodes. The electrical stimulus was generated by an A310 Accupulser (World Precision Instruments, Sarasota, CA, USA) and an A385 stimulus Isolator (World Precision Instruments) connected to the dish insert. Bursts of stimulation consisted of 1 ms current pulses (100 mA) applied at 50 Hz for the indicated stimulus duration. Trains of stimuli were generated by applying a 2 s stimulus every 5 s for the indicated train duration. Image processing and analysis were performed using Metafluor software (Molecular Devices). For analysis of mitochondrial Ca2+ levels in myofibers, regions of interest were made within the myofiber and nuclei excluded.
For cytosolic Ca2+ measurements, FDB muscle fibers were loaded with either Fura-2AM or Fura-4AM depending on the question to be addressed. Fura-2 (2 μM) was loaded into muscle fibers at 37 °C for 45min in zero Ca2+ Tyrode solution including (in mM) 145 NaCl, 5.4 KCl, 0.33 NaH2PO4, 1 MgCl2, 5 HEPES and 10 glucose, pH 7.4. 50 μM BTS was used to inhibit muscle contraction and reduce the motion artifact during Ca2+ imaging. FDB muscle fibers on a 35 mm MatTek dish were mounted on the motorized Nikon eclipse TE 2000-E microscope stage. The microscope was equipped with a 40× oil-based objective. The muscle fibers were continuously perfused with oxygenated experimental solutions. Epi-fluorescence of Fura-2 was excited by a pE340fura illumination system. Ca2+ images were recorded with a PCO edge version 5.5 digital camera. The imaging data were acquired in every 1s with Metafluor software (Molecular Device). After a stable baseline was recorded Tyrode solution that contains 30 μM CPA, 2–10 mM Ca2+ was applied to activate SOCE. SOCE was measured as the initial slope of fura-2 fluorescence. For E/SR Ca2+ release following EFS, a low-affinity Ca2+ indicator fura-4F was used to avoid possible saturation. 2 μM Fura-4F was loaded in normal Tyrode solution for 45 min at 37 °C. The microscope chamber was fitted with platinum wires that were attached to the field stimulation unit. After 2–5 min baseline recordings, fibers were stimulated at 50 Hz in a Tyrode solution. The imaging data were acquired every 1s with Metafluor software (Molecular Device). The amplitude of the [Ca2+]c following EFS for all fibers was determined as previously described (Li MCB 2012).
8.16. Transmission electron microscopy
Mice were anesthetized with sodium pentobarbital and perfuse fixed with 2% paraformaldehyde, 2% glutaraldehyde, and 0.1 M phosphate buffer, pH 7.4. For analysis of mitochondrial ultrastructure tibialis anterior muscles were subsequently dissected and cut into small pieces (∼11 mM) and postfixed in 2% paraformaldehyde, 2% glutaraldehyde, and 0.1 M phosphate buffer, pH 7.4 for 24 h then 1% osmium tetraoxide for 1 h. They were stained with 2% Uranyl Acetate, dehydrated in an Ethanol series, and embedded in an epoxy resin mixture. Ultrathin sections were cut at 60 nm mounted on copper grids and studied with an FEI Technai G2 Twin transmission electron microscope. For ultrastructural localization of STIM1-LacZ by TEM, FDB fibers were dissected and stained for LacZ in LacZ staining solution (5 mM K3Fe(CN)6, 5 mM K4FE(CN)6, 5 mM EGTA, 0.01% deoxycholate, 0.02% NP40, 2 mM MgCl2, 1 mg/mL X-gal) for 2 h. Stained tissue was post-fixed in 2% PFA and 2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, thereafter fixed in 1% osmium tetraoxide and stained en bloc with 1% uranyl acetate. The tissues were then dehydrated in a graded ethanol series, taken through a series of Spurr's resin/Ethanol washes and embedded in Spurr's resin. Thin sections were cut at 70 nm, mounted on copper grids, and counterstained with 2% uranyl acetate and lead citrate. Grids were viewed and photographed using an FEI Technai G2 Twin transmission electron microscope. All Images were acquired with a TIA CCD camera.
8.17. Histochemistry
Soleus muscles were carefully dissected, mounted in OCT with gum tragacanth and frozen in liquid nitrogen chilled isopentane. Samples were stored at −80 °C until sectioning. Cross-sections of the belly of the muscle were cut at a thickness of 14 μm. For succinate dehydrogenase (SDH) activity staining, unfixed slides were incubated in SDH staining solution (15 mg Nitro Blue Tetrazolium, 1x PBS, 0.1 M sodium succinate) for 15 min at room temperature then washed in PBS fixed in 4% paraformaldehyde and mounted in aquamount. For hematoxylin and eosin staining, the sections were fixed in 4% paraformaldehyde, stained with Gills 2 hematoxylin, washed in Scott's tap water, then stained with 0.1% Eosin/0.01% Phloxine, dehydrated, and then mounted in Cytoseal medium. Images were obtained on a Nikon Eclipse E400 microscope.
8.18. Immunohistochemistry
To isolate individual fibers, the FDB muscle was dissected from the foot carefully and immediately digested overnight at 37 °C in digestion medium (DMEM with 10% FBS, 1X Penstrep, and Collagenase A (Roche) at a concentration of 0.5–1 mg/mL). After digestion, the muscles were triturated in fiber medium (DMEM with 10% FBS and 1X Penstrep) until individual fibers separated. Fibers were plated onto glass-bottomed plated coated with 20ug/ml laminin. Fibers were allowed a period of 2 h to recover then fixed in 4% PFA for 5 min, washed in PBS, and blocked in 10% heat-inactivated goat serum. Then the fibers were incubated with primary antibody overnight at room temperature in fiber antibody solution (PBS, 2% heat-inactivated goat serum, and 0.3% Triton X-100). Primary antibodies used were STIM1 rabbit (Protein Tech 11565-1-AP), STIM1 Mouse (BD Biosciences 610,954), Tom20 Rabbit (Proteintech 11802-1-AP), and COX IV Mouse (ProteinTech Protein Tech 60251-1-Ig). After washing, fibers were incubated with secondary antibody for 1 h (Molecular Probes—Alexa Fluor 488 (green) and Alexa Fluor 561 (red) washed and mounted in VECTASHIELD Antifade Mounting Medium. Staining was analyzed on a Zeiss 510 confocal microscope.
8.19. Targeted metabolomics for amino acids, acylcarnitines, and organic acids
Frozen gastrocnemius muscles from 16 h fasted mice were powdered under liquid nitrogen and weighed. Samples were diluted to 50 mg/mL with ice-cold 50% acetonitrile containing 0.3% formic acid and disrupted with a TissueLyser II bead mill (Qiagen) for 2 min at 30 Hz. Samples were aliquoted for the determination of acylcarnitines, amino acids, or organic acids by mass spectrometry using stable isotope dilution techniques by Metabolomics Core at Duke Molecular Physiology Institute. Acylcarnitines and amino acids were measured by tandem-flow injection mass spectrometry as previously described [20] using a Quattro Micro system running 4.0 MassLynx software (Waters). Organic acids were quantified as described previously [78] using a Trace Ultra GC coupled to a Trace DSQ MS operating under Xcalibur 1.4 software (Thermo Fisher).
8.20. Ex vivo muscle incubations, lactate efflux and 13C-glucose flux analysis
Soleus and EDL muscles were carefully dissected from 16 h fasted mice under anesthesia and placed in low calcium KHB buffer (118 mM NaCl, 4.73 mM KCL, 1.18 mM MgSO4, 0.47 mM CaCl2, 1.17 mM KH2PO4, 25 mM NaHCO3, 12.5 mM HEPES) supplemented with 0.5 mM glucose and 0.5 mM l-carnitine as previously described [26,79]. Muscles were incubated in a 29 °C water bath with gassing (5% CO2 and 95% O2) and shaking for 15 min. After 15 min, the temperature was raised to 37 °C, and muscles were incubated for an additional 15 min. After the 30-min incubation period, muscles were transferred to a tracer incubation plate containing [U–13C6]Glucose (10 mM) and a palmitate (200 μM) BSA (0.5%) mixture with or without insulin (100 nM). The plate was sealed with parafilm and incubated at 37 °C for 90 min with shaking. Muscles and incubation media were then flash-frozen in liquid nitrogen and stored at −80 °C for analysis. Frozen muscles were weighed and homogenized in a 1:1:1 mixture of methanol, chloroform, and water with norvaline standard (10 μ/mL) using a handheld rotary at 5.5 settings. Samples were centrifuged at maximum speed at 4 °C for 20 min. The aqueous phase was transferred to a clean tube and dried on 80 position MULTIVAP nitrogen with nitrogen flow at 37 °C. Upon dryness methyoxyamine (2% wt/vol) in pyridine was added and samples were heated at 40 °C for 90 min. MTBSTFA +1% TBDMCS was added and samples were incubated at 60 °C for 30 min. The samples were transferred to an injection vial and injected into the GC–MS as previously described [80] and metabolite enrichments were corrected for natural abundance [81]. Lactate concentration in the media was determined kinetically by following the reduction of NAD+ at 340 nm in assay buffer (175 mM hydrazine sulfate, 68 mM glycine, 2.9 mM EDTA, and 50 U/mL lactate dehydrogenase). Lactate efflux rate was calculated by dividing lactate concentration by muscle mass and experimental duration (1.5 h).
8.21. Phosphoproteomics data acquisition
Samples were prepared for phosphoproteomics based on a previously described workflow [82]. Briefly, approximately 15 mg of powdered gastrocnemius muscles were lysed in 8 M urea-containing buffer with a combination of tissue tearing, freeze-thaw, and sonication. Equal amounts of protein (400 μg) were reduced (5 mM DTT), alkylated (15 mM iodoacetamide), and digested with LysC (5 μg) and trypsin (10 μg). Peptides were desalted by solid-phase extraction (SPE) and each sample and a pooled mixture were labeled with a unique 11-plex Tandem Mass Tag (TMT) reagent (0.8 mg). All 11 samples were combined and separated into 12 fractions by high pH reversed-phase (HPRP) chromatography as described previously [83]. Each fraction was subjected to phosphopeptide enrichment via immobilized metal affinity chromatography (IMAC) as described previously [82], retaining 5% of each input fraction for assessment of unmodified peptides. All samples were subjected to nano LC-MS/MS analysis using an EASY-nLC 1200 ultra-performance liquid chromatography system (Thermo Fisher Scientific) coupled to a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific) via an EASY-Spray nanoelectrospray ionization source (Thermo Fisher Scientific) using previously described methods [26].
8.22. Proteomics data analysis
Data were searched against the UniProt mouse complete proteome database, which consisted of 55,485 sequences on the date of download (2/3/2021). Data analysis was performed using Proteome Discoverer 2.5 (PD2.5), using both Sequest HT and MS Amanda 2.0 with the following default parameters: oxidation (15.995 Da on M) as a variable modification and carbamidomethyl (57.021 Da on C) and TMT11plex (229.163 Da on-peptide N-term and K) as fixed modifications, and 2 missed cleavages (full trypsin specificity). Phosphopeptide runs included phosphorylation (79.966 Da on S, T, or Y) as a variable modification. PSMs were filtered to 1% FDR using Percolator [84] and PTM site localization was determined using ptmRS [85], with PSMs grouped to peptides at 1% FDR and a 90% PTM localization threshold. Peptides were grouped to proteins at 1% FDR, using the rules of strict parsimony. Protein and Peptide Isoforms tabs from the PD2.5 results were exported as tab-delimited .txt files and analyzed with in-house scripts based on a previously described workflow [86], modified to be conducted entirely in R (version 4.0.5) [87]. The R-package limma (3.46.0) was used to construct linear models for differential expression of peptides and proteins [88]. The eBayes function within limma was used to compute moderated statistics. Multiple hypothesis correction was performed using the Benjamini-Hochberg method with a significance threshold of ≤ 0.1. Phosphopeptide measurements were calculated alone (abundance) and with normalization to any change in the corresponding Master Protein (relative occupancy) by subtracting log2 Master Protein values from PTM-containing quantitation values on a sample-specific basis. KSEA analysis was performed using a slightly altered function from the KSEAapp package (0.99.0) [39,40]. Pathway analysis was processed using the pathfindR package (1.6.2) [89]. All plots were created in R using the ggplot2 (3.3.5) package. All proteomics raw data for proteomics were submitted to the Proteome Xchange Consortium, with accession number PXD030674.
8.23. Statistical analysis
Data are presented as mean ± SEM. Statistical analysis was performed in GraphPad Prism 9 (GraphPad Software, San Diego, CA) using 2-tailed unpaired student's t-tests, one-way ANOVA with Dunnet's post-hoc testing where appropriate, and two-way ANOVA. Figures were generated using GraphPad Prism 9.0.1. The level of significance was set at p ≤ 0.05 for all experiments unless stated otherwise.
Author contributions
Conceptualization, R.J. Wilson, D.M. Muoio, and P.B. Rosenberg; Methodology, S.P. Lyons, T.R. Koves, S.B. Crown, P.A. Grimsrud; Investigation, R.J. Wilson, S.P. Lyons, T.R. Koves, H. Zhang, V.G. Bryson, T. Li, and S.B. Crown; Writing-Original Draft, R.J. Wilson, and D.M. Muoio; Writing-Review and Editing, R.J. Wilson, D.M. Muoio, P.B. Rosenberg, S.P. Lyons, T.R. Koves, S.B. Crown, and P.A. Grimsrud; Funding acquisition, R.J. Wilson, D.M. Muoio, and P.B. Rosenberg; Resources, D.M. Muoio and P.B. Rosenberg.; Supervision, D.M. Muoio, P.B. Rosenberg, and P.A. Grimsrud.
Acknowledgements
The authors thank Greg Wait and Erik Soderblom of the Duke Proteomics and Metabolomics Shared Resource for performing peptide pre-fractionation for the proteomics experiments. This work was supported by the National Institutes of Health (T32HL00710 [R.J.W.], R01DK109911 [D.M.M. + P.B.R.]). We also thank the DMPI Metabolomics Core Lab (supported by Diabetes and Endocrine Research Center grant P30 DK124723) for assistance with the metabolomics assays.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101429.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Calderon J.C., Bolanos P., Caputo C. The excitation-contraction coupling mechanism in skeletal muscle. Biophysical Reviews. 2014;6(1):133–160. doi: 10.1007/s12551-013-0135-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koenig X., Choi R.H., Launikonis B.S. Store-operated Ca(2+) entry is activated by every action potential in skeletal muscle. Communications Biology. 2018;1:31. doi: 10.1038/s42003-018-0033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson H.A., Roe M.W. Molecular physiology and pathophysiology of stromal interaction molecules. Experimental Biology and Medicine (Maywood) 2018;243(5):451–472. doi: 10.1177/1535370218754524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li T., Finch E.A., Graham V., Zhang Z.S., Ding J.D., Burch J., et al. STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Molecular and Cellular Biology. 2012;32(15):3009–3017. doi: 10.1128/MCB.06599-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stiber J., Hawkins A., Zhang Z.S., Wang S., Burch J., Graham V., et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nature Cell Biology. 2008;10(6):688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei-Lapierre L., Carrell E.M., Boncompagni S., Protasi F., Dirksen R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nature Communications. 2013;4:2805. doi: 10.1038/ncomms3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrell E.M., Coppola A.R., McBride H.J., Dirksen R.T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated Ca2+ entry. The FASEB Journal. 2016;30(12):4109–4119. doi: 10.1096/fj.201600621R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boncompagni S., Michelucci A., Pietrangelo L., Dirksen R.T., Protasi F. Exercise-dependent formation of new junctions that promote STIM1-Orai1 assembly in skeletal muscle. Scientific Reports. 2017;7(1):14286. doi: 10.1038/s41598-017-14134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyfenko A.D., Dirksen R.T. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. Journal of Physiology. 2008;586(20):4815–4824. doi: 10.1113/jphysiol.2008.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurebayashi N., Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. Journal of Physiology. 2001;533(Pt 1):185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H., Bryson V.G., Wang C., Li T., Kerr J.P., Wilson R., et al. Desmin interacts with STIM1 and coordinates Ca2+ signaling in skeletal muscle. JCI Insight. 2021;6(17) doi: 10.1172/jci.insight.143472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCormack J.G., Denton R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochemical Journal. 1979;180(3):533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutter G.A., Burnett P., Rizzuto R., Brini M., Murgia M., Pozzan T., et al. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(11):5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rutter G.A., Denton R.M. The binding of Ca2+ ions to pig heart NAD+-isocitrate dehydrogenase and the 2-oxoglutarate dehydrogenase complex. Biochemical Journal. 1989;263(2):453–462. doi: 10.1042/bj2630453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutter G.A. Ca2(+)-binding to citrate cycle dehydrogenases. International Journal of Biochemistry. 1990;22(10):1081–1088. doi: 10.1016/0020-711x(90)90105-c. [DOI] [PubMed] [Google Scholar]
- 16.Scholz T.D., Balaban R.S. Mitochondrial F1-ATPase activity of canine myocardium: effects of hypoxia and stimulation. American Journal of Physiology. 1994;266(6 Pt 2):H2396–H2403. doi: 10.1152/ajpheart.1994.266.6.H2396. [DOI] [PubMed] [Google Scholar]
- 17.Boerries M., Most P., Gledhill J.R., Walker J.E., Katus H.A., Koch W.J., et al. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Molecular and Cellular Biology. 2007;27(12):4365–4373. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamada E.W., Huzel N.J. Ca2+-binding properties of a unique ATPase inhibitor protein isolated from mitochondria of bovine heart and rat skeletal muscle. Cell Calcium. 1985;6(6):469–479. doi: 10.1016/0143-4160(85)90022-3. [DOI] [PubMed] [Google Scholar]
- 19.Nelson M.E., Parker B.L., Burchfield J.G., Hoffman N.J., Needham E.J., Cooke K.C., et al. Phosphoproteomics reveals conserved exercise-stimulated signaling and AMPK regulation of store-operated calcium entry. The EMBO Journal. 2019;38(24) doi: 10.15252/embj.2019102578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O., et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabolism. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Koves T.R., Li P., An J., Akimoto T., Slentz D., Ilkayeva O., et al. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. Journal of Biological Chemistry. 2005;280(39):33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 22.Mina A.I., LeClair R.A., LeClair K.B., Cohen D.E., Lantier L., Banks A.S. CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metabolism. 2018;28(4):656–666 e651. doi: 10.1016/j.cmet.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrosino J.M., Heiss V.J., Maurya S.K., Kalyanasundaram A., Periasamy M., LaFountain R.A., et al. Graded maximal exercise testing to assess mouse cardio-metabolic phenotypes. PLoS One. 2016;11(2) doi: 10.1371/journal.pone.0148010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Messer J.I., Jackman M.R., Willis W.T. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. American Journal of Physiology - Cell Physiology. 2004;286(3):C565–C572. doi: 10.1152/ajpcell.00146.2003. [DOI] [PubMed] [Google Scholar]
- 25.Fisher-Wellman K.H., Davidson M.T., Narowski T.M., Lin C.T., Koves T.R., Muoio D.M. Mitochondrial diagnostics: a multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Reports. 2018;24(13):3593–3606 e3510. doi: 10.1016/j.celrep.2018.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams A.S., Koves T.R., Davidson M.T., Crown S.B., Fisher-Wellman K.H., Torres M.J., et al. Disruption of acetyl-lysine turnover in muscle mitochondria promotes insulin resistance and redox stress without overt respiratory dysfunction. Cell Metabolism. 2020;31(1):131–147 e111. doi: 10.1016/j.cmet.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sale G.J., Randle P.J. Analysis of site occupancies in [32P]phosphorylated pyruvate dehydrogenase complexes by aspartyl-prolyl cleavage of tryptic phosphopeptides. European Journal of Biochemistry. 1981 doi: 10.1111/j.1432-1033.1981.tb05733.x. [DOI] [PubMed] [Google Scholar]
- 28.Yeaman S.J., Hutcheson E.T., Roche T.E., Pettit F.H., Brown J.R., Reed L.J., et al. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry. 1978 doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
- 29.Filipek A., Jastrzebska B., Nowotny M., Kuznicki J. CacyBP/SIP, a calcyclin and Siah-1-interacting protein, binds EF-hand proteins of the S100 family. Journal of Biological Chemistry. 2002;277(32):28848–28852. doi: 10.1074/jbc.M203602200. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee A., Rotwein P. Insulin-like growth factor-binding protein-5 inhibits osteoblast differentiation and skeletal growth by blocking insulin-like growth factor actions. Molecular Endocrinology. 2008;22(5):1238–1250. doi: 10.1210/me.2008-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salih D.A., Tripathi G., Holding C., Szestak T.A., Gonzalez M.I., Carter E.J., et al. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(12):4314–4319. doi: 10.1073/pnas.0400230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Witczak C.A., Jessen N., Warro D.M., Toyoda T., Fujii N., Anderson M.E., et al. CaMKII regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2010;298(6):E1150–E1160. doi: 10.1152/ajpendo.00659.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raney M.A., Turcotte L.P. Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. Journal of Applied Physiology (1985) 2008;104(5):1366–1373. doi: 10.1152/japplphysiol.01282.2007. [DOI] [PubMed] [Google Scholar]
- 34.Wu H., Kanatous S.B., Thurmond F.A., Gallardo T., Isotani E., Bassel-Duby R., et al. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296(5566):349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- 35.Eilers W., Jaspers R.T., de Haan A., Ferrie C., Valdivieso P., Fluck M. CaMKII content affects contractile, but not mitochondrial, characteristics in regenerating skeletal muscle. BMC Physiology. 2014;14:7. doi: 10.1186/s12899-014-0007-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y., Randall W.R., Schneider M.F. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. The Journal of Cell Biology. 2005;168(6):887–897. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fluck M., Booth F.W., Waxham M.N. Skeletal muscle CaMKII enriches in nuclei and phosphorylates myogenic factor SRF at multiple sites. Biochemical and Biophysical Research Communications. 2000;270(2):488–494. doi: 10.1006/bbrc.2000.2457. [DOI] [PubMed] [Google Scholar]
- 38.Fluck M., Waxham M.N., Hamilton M.T., Booth F.W. Skeletal muscle Ca(2+)-independent kinase activity increases during either hypertrophy or running. Journal of Applied Physiology (1985) 2000;88(1):352–358. doi: 10.1152/jappl.2000.88.1.352. [DOI] [PubMed] [Google Scholar]
- 39.Casado P., Rodriguez-Prados J.C., Cosulich S.C., Guichard S., Vanhaesebroeck B., Joel S., et al. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Science Signaling. 2013;6(268):rs6. doi: 10.1126/scisignal.2003573. [DOI] [PubMed] [Google Scholar]
- 40.Wiredja D.D., Koyuturk M., Chance M.R. The KSEA App: a web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics. 2017;33(21):3489–3491. doi: 10.1093/bioinformatics/btx415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Viard P., Butcher A.J., Halet G., Davies A., Nurnberg B., Heblich F., et al. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nature Neuroscience. 2004;7(9):939–946. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- 42.Camors E., Valdivia H.H. CaMKII regulation of cardiac ryanodine receptors and inositol triphosphate receptors. Frontiers in Pharmacology. 2014;5:101. doi: 10.3389/fphar.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan J., Jiang X., Yin G., He L., Liu J., Long Z., et al. Anacardic acid induces cell apoptosis of prostatic cancer through autophagy by ER stress/DAPK3/Akt signaling pathway. Oncology Reports. 2017;38(3):1373–1382. doi: 10.3892/or.2017.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujiwara N., Usui T., Ohama T., Sato K. Regulation of beclin 1 protein phosphorylation and autophagy by protein phosphatase 2A (PP2A) and death-associated protein kinase 3 (DAPK3) Journal of Biological Chemistry. 2016;291(20):10858–10866. doi: 10.1074/jbc.M115.704908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirzoev T.M., Sharlo K.A., Shenkman B.S. The role of GSK-3beta in the regulation of protein turnover, myosin phenotype, and oxidative capacity in skeletal muscle under disuse conditions. International Journal of Molecular Sciences. 2021;22(10) doi: 10.3390/ijms22105081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei Y., An Z., Zou Z., Sumpter R., Su M., Zang X., et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife. 2015;4 doi: 10.7554/eLife.05289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schiaffino S., Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skeletal Muscle. 2011;1(1):4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verhees K.J., Pansters N.A., Schols A.M., Langen R.C. Regulation of skeletal muscle plasticity by glycogen synthase kinase-3beta: a potential target for the treatment of muscle wasting. Current Pharmaceutical Design. 2013;19(18):3276–3298. doi: 10.2174/1381612811319180011. [DOI] [PubMed] [Google Scholar]
- 49.Egerman M.A., Glass D.J. Signaling pathways controlling skeletal muscle mass. Critical Reviews in Biochemistry and Molecular Biology. 2014;49(1):59–68. doi: 10.3109/10409238.2013.857291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richter E.A., Nielsen N.B. Protein kinase C activity in rat skeletal muscle. Apparent relation to body weight and muscle growth. FEBS Letters. 1991;289(1):83–85. doi: 10.1016/0014-5793(91)80913-n. [DOI] [PubMed] [Google Scholar]
- 51.Kolobova E., Tuganova A., Boulatnikov I., Popov K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochemical Journal. 2001;358(Pt 1):69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeaman S.J., Hutcheson E.T., Roche T.E., Pettit F.H., Brown J.R., Reed L.J., et al. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry. 1978;17(12):2364–2370. doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
- 53.Strumilo S. Short-term regulation of the mammalian pyruvate dehydrogenase complex. Acta Biochimica Polonica. 2005;52(4):759–764. [PubMed] [Google Scholar]
- 54.Turkan A., Hiromasa Y., Roche T.E. Formation of a complex of the catalytic subunit of pyruvate dehydrogenase phosphatase isoform 1 (PDP1c) and the L2 domain forms a Ca2+ binding site and captures PDP1c as a monomer. Biochemistry. 2004;43(47):15073–15085. doi: 10.1021/bi048901y. [DOI] [PubMed] [Google Scholar]
- 55.Denton R.M., Richards D.A., Chin J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochemical Journal. 1978;176(3):899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacDonald M.J., Brown L.J. Calcium activation of mitochondrial glycerol phosphate dehydrogenase restudied. Archives of Biochemistry and Biophysics. 1996;326(1):79–84. doi: 10.1006/abbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- 57.Lawlis V.B., Roche T.E. Regulation of bovine kidney alpha-ketoglutarate dehydrogenase complex by calcium ion and adenine nucleotides. Effects on S0.5 for alpha-ketoglutarate. Biochemistry. 1981;20(9):2512–2518. doi: 10.1021/bi00512a023. [DOI] [PubMed] [Google Scholar]
- 58.Rutter G.A., Pralong W.F., Wollheim C.B. Regulation of mitochondrial glycerol-phosphate dehydrogenase by Ca2+ within electropermeabilized insulin-secreting cells (INS-1) Biochimica et Biophysica Acta. 1992;1175(1):107–113. doi: 10.1016/0167-4889(92)90016-5. [DOI] [PubMed] [Google Scholar]
- 59.Kwong J.Q., Huo J., Bround M.J., Boyer J.G., Schwanekamp J.A., Ghazal N., et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight. 2018;3(22) doi: 10.1172/jci.insight.121689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maus M., Cuk M., Patel B., Lian J., Ouimet M., Kaufmann U., et al. Store-operated Ca(2+) entry controls induction of lipolysis and the transcriptional reprogramming to lipid metabolism. Cell Metabolism. 2017;25(3):698–712. doi: 10.1016/j.cmet.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Collins H.E., Pat B.M., Zou L., Litovsky S.H., Wende A.R., Young M.E., et al. Novel role of the ER/SR Ca(2+) sensor STIM1 in the regulation of cardiac metabolism. American Journal of Physiology - Heart and Circulatory Physiology. 2019;316(5):H1014–H1026. doi: 10.1152/ajpheart.00544.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashby M.C., Tepikin A.V. ER calcium and the functions of intracellular organelles. Seminars in Cell & Developmental Biology. 2001;12(1):11–17. doi: 10.1006/scdb.2000.0212. [DOI] [PubMed] [Google Scholar]
- 63.Haystead T.A. ZIP kinase, a key regulator of myosin protein phosphatase 1. Cellular Signalling. 2005;17(11):1313–1322. doi: 10.1016/j.cellsig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 64.Wright D.C., Geiger P.C., Han D.H., Jones T.E., Holloszy J.O. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. Journal of Biological Chemistry. 2007;282(26):18793–18799. doi: 10.1074/jbc.M611252200. [DOI] [PubMed] [Google Scholar]
- 65.Roux P.P., Shahbazian D., Vu H., Holz M.K., Cohen M.S., Taunton J., et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. Journal of Biological Chemistry. 2007;282(19):14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salmond R.J., Emery J., Okkenhaug K., Zamoyska R. MAPK, phosphatidylinositol 3-kinase, and mammalian target of rapamycin pathways converge at the level of ribosomal protein S6 phosphorylation to control metabolic signaling in CD8 T cells. The Journal of Immunology. 2009;183(11):7388–7397. doi: 10.4049/jimmunol.0902294. [DOI] [PubMed] [Google Scholar]
- 67.Behera S., Kapadia B., Kain V., Alamuru-Yellapragada N.P., Murunikkara V., Kumar S.T., et al. ERK1/2 activated PHLPP1 induces skeletal muscle ER stress through the inhibition of a novel substrate AMPK. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2018;1864(5 Pt A):1702–1716. doi: 10.1016/j.bbadis.2018.02.019. [DOI] [PubMed] [Google Scholar]
- 68.Hwang S.L., Chang H.W., Lee I.K., Yang B.K., Magae J., Chang Y.C. Ascofuranone prevents ER stress-induced insulin resistance via activation of AMP-activated protein kinase in L6 myotube cells. Biochemical and Biophysical Research Communications. 2010;396(4):967–972. doi: 10.1016/j.bbrc.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 69.Ozcan L., Cristina de Souza J., Harari A.A., Backs J., Olson E.N., Tabas I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metabolism. 2013;18(6):803–815. doi: 10.1016/j.cmet.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bracken C., Beauverger P., Duclos O., Russo R.J., Rogers K.A., Husson H., et al. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. American Journal of Physiology - Renal Physiology. 2016;310(11):F1414–F1422. doi: 10.1152/ajprenal.00426.2015. [DOI] [PubMed] [Google Scholar]
- 71.Michelucci A., Garcia-Castaneda M., Boncompagni S., Dirksen R.T. Role of STIM1/ORAI1-mediated store-operated Ca(2+) entry in skeletal muscle physiology and disease. Cell Calcium. 2018;76:101–115. doi: 10.1016/j.ceca.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schuler M., Ali F., Metzger E., Chambon P., Metzger D. Temporally controlled targeted somatic mutagenesis in skeletal muscles of the mouse. Genesis. 2005;41(4):165–170. doi: 10.1002/gene.20107. [DOI] [PubMed] [Google Scholar]
- 73.Weir J.B. New methods for calculating metabolic rate with special reference to protein metabolism. Journal of Physiology. 1949;109(1–2):1–9. doi: 10.1113/jphysiol.1949.sp004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Riachi M., Himms-Hagen J., Harper M.E. Percent relative cumulative frequency analysis in indirect calorimetry: application to studies of transgenic mice. Canadian Journal of Physiology and Pharmacology. 2004;82(12):1075–1083. doi: 10.1139/y04-117. [DOI] [PubMed] [Google Scholar]
- 75.Koves T.R., Sparks L.M., Kovalik J.P., Mosedale M., Arumugam R., DeBalsi K.L., et al. PPARgamma coactivator-1alpha contributes to exercise-induced regulation of intramuscular lipid droplet programming in mice and humans. The Journal of Lipid Research. 2013;54(2):522–534. doi: 10.1194/jlr.P028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lorentz K. Improved determination of serum calcium with 2-cresolphthalein complexone. Clinica Chimica Acta. 1982;126(3):327–334. doi: 10.1016/0009-8981(82)90308-4. [DOI] [PubMed] [Google Scholar]
- 77.Shefer G., Yablonka-Reuveni Z. Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods in Molecular Biology. 2005;290:281–304. doi: 10.1385/1-59259-838-2:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jensen M.V., Joseph J.W., Ilkayeva O., Burgess S., Lu D., Ronnebaum S.M., et al. Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. Journal of Biological Chemistry. 2006;281(31):22342–22351. doi: 10.1074/jbc.M604350200. [DOI] [PubMed] [Google Scholar]
- 79.Muoio D.M., Noland R.C., Kovalik J.P., Seiler S.E., Davies M.N., DeBalsi K.L., et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metabolism. 2012;15(5):764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crown S.B., Marze N., Antoniewicz M.R. Catabolism of branched chain amino acids contributes significantly to synthesis of odd-chain and even-chain fatty acids in 3T3-L1 adipocytes. PLoS One. 2015;10(12) doi: 10.1371/journal.pone.0145850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernandez C.A., Des Rosiers C., Previs S.F., David F., Brunengraber H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. Journal of Mass Spectrometry. 1996;31(3):255–262. doi: 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 82.Grimsrud P.A., Carson J.J., Hebert A.S., Hubler S.L., Niemi N.M., Bailey D.J., et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metabolism. 2012;16(5):672–683. doi: 10.1016/j.cmet.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Walejko J.M., Christopher B.A., Crown S.B., Zhang G.F., Pickar-Oliver A., Yoneshiro T., et al. Branched-chain alpha-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nature Communications. 2021;12(1):1680. doi: 10.1038/s41467-021-21962-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kall L., Canterbury J.D., Weston J., Noble W.S., MacCoss M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature Methods. 2007;4(11):923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 85.Taus T., Kocher T., Pichler P., Paschke C., Schmidt A., Henrich C., et al. Universal and confident phosphorylation site localization using phosphoRS. Journal of Proteome Research. 2011;10(12):5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 86.Davidson M.T., Grimsrud P.A., Lai L., Draper J.A., Fisher-Wellman K.H., Narowski T.M., et al. Extreme acetylation of the cardiac mitochondrial proteome does not promote heart failure. Circulation Research. 2020;127(8):1094–1108. doi: 10.1161/CIRCRESAHA.120.317293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.R Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2018. R: a language and environment for statistical computing.https://www.R-project.org/ Available online at: [Google Scholar]
- 88.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ulgen E., Ozisik O., Sezerman O.U. pathfindR: an R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Frontiers in Genetics. 2019;10:858. doi: 10.3389/fgene.2019.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data from proteomics were submitted to ProteomeXchange.
No unpublished custom code, software, or algorithm was used in this study. All software packages and methods used in this study have been properly detailed and referenced under the Software and algorithms listed in reporting summary. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.