

Introduction
IgG4-related disease (IgG4-RD) is a rare immune-mediated fibroinflammatory condition mostly presenting as tumor-like lesions with distinct histopathology. All ethnic groups can be affected with a slight male predominance.1 The initial description of IgG4-RD involved the pancreas, presenting as autoimmune pancreatitis.2 However, IgG4-RD can affect nearly any organ system. To date, there are few reports of genital organ involvement in IgG4-RD. Here we present a case of extensive infiltrative mass of the vagina as a rare manifestation of IgG4-RD.
Case report
A 39-year–old woman with a diagnosis of diffuse large B-cell lymphoma in remission and an 8-year history of HIV infection presented with a slowly enlarging infiltrative mass with central necrotic debris on the right labium majus (Fig 1, A). One year prior to presentation, she had acute urinary retention, and a cystoscopy revealed a mass at the bladder neck. The tissue biopsy showed nonspecific, acute and chronic inflammation with no evidence of malignancy or infection. Gynecologic examination revealed an 8-cm mass with a firm-to-hard consistency at the anterior aspect of the vagina extending to the right labium majus. Abdominal magnetic resonance imaging showed an irregular heterogeneous mass, involving the anterior aspect of the vagina with urethral and urinary bladder invasion and extending to the parametrium (Fig 1, B and C). Repeat biopsies at the right labium majus and vagina were performed and revealed only numerous plasma cells, lymphocytes, and eosinophils infiltrating the tissue without evidence of malignancy or infection. Cultures, as well as polymerase chain reaction for bacteria, mycobacteria, and fungi, were all negative. Multiple courses of oral antibacterials were ineffective. The prominence of plasma-cell infiltration on histopathology could lead to many differential diagnoses, including infection, such as syphilis; neoplasm, especially hematologic malignancy, such as multiple myeloma; plasmablastic lymphoma; and inflammatory disorders, such as cutaneous plasmacytosis, IgG4-RD, as well as Castleman disease. Previous biopsies were reviewed and revealed numerous IgG+ mature plasma cells, positive IgG4 with IgG4 count >100 cells/high-power field, and an IgG4/IgG ratio of >40% (Fig 2). Whole abdominal computed tomography revealed an enlarged right external iliac lymph node. A core needle biopsy of the lymph node revealed predominant plasma-cell proliferation, which stained positive for CD138, IgG4, IgG, and CD19, without kappa/lambda light chain restriction; and negative for human herpesvirus-8, CD56, anaplastic lymphoma kinase, CD3, CD5, CD20, CD117, cylinD1, and Epstein-Barr virus-encoded small RNAs. Her serum protein electrophoresis demonstrated a polyclonal pattern, and the serum IgG4 level was >5320 mg/dL (normal range, 5-125 mg/dL). Autoimmune profile including tests for antinuclear antibodies and antineutrophilic cytoplasmic antibodies were negative. IgG4-RD was diagnosed. The patient was initially treated with prednisolone 1 mg/kg/day but later switched to melphalan and dexamethasone due to partial clinical response (Fig 3). The follow-up serum IgG4 level was reduced to 2050 mg/dL. Unfortunately, after the first cycle of chemotherapy, the patient’s general condition deteriorated, with multiple organ involvement, so further cycles of chemotherapy could not be administered. The patient expired while receiving supportive care.
Fig 1.

A, Infiltrative mass causing erythematous, edematous, and enlarged labia with central necrosis and yellowish exudate. B, C, Magnetic resonance imaging. Coronal (B) and sagittal (C) T1-weight images exhibiting an irregular heterogeneous mass involving the lateral and anterior walls of the vagina, which shows upward invasion to the urinary bladder base. The yellow arrowheads outlined the tumor from imaging studies.
Fig 2.

Vaginal tissue demonstrating prominent lymphoplasmacytic infiltration with some eosinophils. (Hematoxylin-eosin stain; original magnification: ×200.) Immunohistochemical studies revealed that mature plasma cells were positive for CD138, negative for CD20, and positive for IgG4 with an IgG4/IgG ratio of more than 40%.
Fig 3.
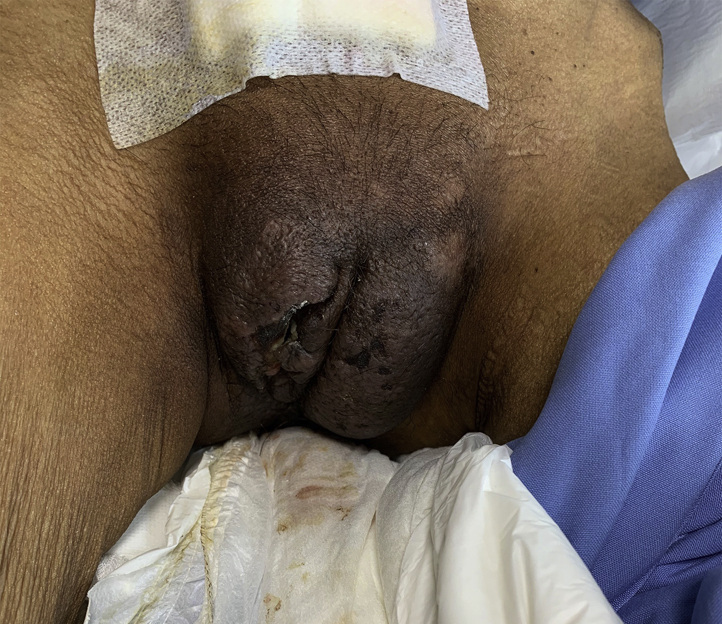
Follow-up image after treatment with prednisolone and the first cycle of the melphalan and dexamethasone regimen. A marked reduction in size and degree of edema and erythema of the labia was observed.
Discussion
IgG4-RD is an immune-mediated fibroinflammatory condition, which was recognized as a new distinct disease entity in the early 2000s.1 The typical clinical presentation is a slowly progressive lesion, which may mimic other diseases such as malignancy or chronic infections. Frequently affected organs include the lacrimal glands, salivary glands, pancreas, lymph nodes, and the retroperitoneum.3,4 The proposed pathogenesis of the disease is a biphasic process, including an inflammatory phase and a fibrotic phase.1 The onset involves multiple potential triggers, including genetic susceptibility (either human leukocyte antigen genes or nonhuman leukocyte antigen risk genes), infectious agents, self-antigens, as well as occupational or environmental substances, such as asbestos.1,3,4 These could initiate antigen presentation, immune activation, as well as an anti-inflammatory reaction. Studies revealed that follicular T helper lymphocytes could generate the Th2 cytokines as well as activation of IgG4-producing plasmablasts and CD4+ cytotoxic T lymphocytes, which release fibrotic mediators, leading to fibrosis formation.1,4 However, the complete immunopathogenesis of the disease remains to be elucidated.
Currently, there are few reports of female genital organ involvement in IgG4-RD.5, 6, 7 We believe that this is the first case of IgG4-RD with vaginal involvement. To define the new organ involvement in IgG4-RD, the 2020 revised comprehensive diagnostic criteria for IgG4-RD consisting of clinical symptoms, histopathologic features, and serum IgG4 level, must be fulfilled.8 The markedly enlarged and infiltrative mass with subsequent tissue necrosis of the vagina and labia along with our patient’s underlying diseases could lead to a myriad of differential diagnoses. Exclusion of chronic infections or malignancy is necessary. Meticulous clinical examination and repeated follow-up imaging, as well as selection of the most representative biopsy site are critical. Histopathologic examination, together with appropriate immunohistochemistry studies, is of utmost importance for an accurate diagnosis. Predominant IgG4+ plasma-cell infiltration along with storiform fibrosis and obliterative phlebitis are pathognomonic.9 However, these characteristic features are not commonly observed in some organs, such as lymph nodes, lungs, and kidneys.9 Our case emphasized a point that clinicians need to have a very high index of suspicion and that repeat biopsy should be considered, if necessary.
Another interesting point regarding our patient is whether the preceding lymphoma (especially considering that it was a B-cell lymphoma) and the HIV infection, a condition in which overproduction of immunoglobulins is rather frequent, could lead to activation of the immune system or exposure to unknown potential autoantigens, resulting in development of the disease. A previous study revealed that there may be an association between previous malignancy and IgG4-RD, with prostate cancer and lymphoma being the conditions most commonly involved.10 The average duration after the diagnosis of primary malignancy is 8 years.10 Further studies are required to further elucidate the underlying mechanism by which previous malignancy or chronic infection increases the risk of IgG4-RD.
The treatment goal for IgG4-RD is induction and maintenance of the remission.1 Treatment in the early inflammatory and proliferative stage can lead to good clinical response. However, the fibrotic stage is less responsive and can progress to irreversible damage. To date, systemic administration of corticosteroids is considered the first-line treatment.1,3 Other steroid-sparing immunosuppressive agents as well as anti-CD20 biologics, such as rituximab, are also used.1,3 Currently, many novel targeted therapies for IgG4-RD are being studied. These agents directly target different levels of immunopathogenesis of the disease, such as XmAb5871 for B-cell inhibition, or abatacept, which inhibits co-stimulatory molecules involved in T-cell activation.1 Other cytokine blockers, such as dupilumab and tumor necrosis factor alpha inhibitors, have also been proposed for the treatment of IgG4-RD.1
In summary, we present an extremely rare case of IgG4-RD involving the vagina that extended to the vulva, urethra, bladder, and lymph nodes. The progressive tumor-like lesion, which can mimic other diseases, often leads to misdiagnosis. Maintaining a high index of suspicion is necessary for reaching an accurate diagnosis, initiating proper treatment, and preventing irreversible damage.
Conflicts of interest
None disclosed.
Footnotes
Funding sources: None.
IRB approval status: Not applicable.
References
- 1.Lanzillotta M., Mancuso G., Della-Torre E. Advances in the diagnosis and management of IgG4 related disease. BMJ. 2020;369:m1067. doi: 10.1136/bmj.m1067. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T., Funata N., Hayashi Y., et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982–984. doi: 10.1007/s00535-003-1175-y. [DOI] [PubMed] [Google Scholar]
- 3.Chen L.Y.C., Mattman A., Seidman M.A., Carruthers M.N. IgG4-related disease: what a hematologist needs to know. Haematologica. 2019 Mar;104(3):444–455. doi: 10.3324/haematol.2018.205526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perugino C.A., Stone J.H. IgG4-related disease: an update on pathophysiology and implications for clinical care. Nat Rev Rheumatol. 2020;16(12):702–714. doi: 10.1038/s41584-020-0500-7. [DOI] [PubMed] [Google Scholar]
- 5.Maruyama S., Sato Y., Taga A., Emoto I., Shirase T., Haga H. Immunoglobulin G4-related disease presenting as bilateral ovarian masses and mimicking advanced ovarian cancer. J Obstet Gynaecol Res. 2016;42(1):103–108. doi: 10.1111/jog.12835. [DOI] [PubMed] [Google Scholar]
- 6.Mizuno R., Yamanishi Y., Uda S., Terashima T., Higashi T., Higuchi T. Invasive cervical cancer accompanied by IgG4-related disease. J Obstet Gynaecol Res. 2016;42(9):1198–1202. doi: 10.1111/jog.13030. [DOI] [PubMed] [Google Scholar]
- 7.Ohkubo H., Miyazaki M., Oguri T., Arakawa A., Kobashi Y., Niimi A. A rare case of IgG4-related disease involving the uterus. Rheumatology (Oxford) 2015;54(6):1124–1125. doi: 10.1093/rheumatology/kev024. [DOI] [PubMed] [Google Scholar]
- 8.Umehara H., Okazaki K., Kawa S., et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021;31(3):529–533. doi: 10.1080/14397595.2020.1859710. [DOI] [PubMed] [Google Scholar]
- 9.Deshpande V., Zen Y., Chan J.K., et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–1192. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 10.Wallace Z.S., Wallace C.J., Lu N., Choi H.K., Stone J.H. Association of IgG4-related disease with history of malignancy. Arthritis Rheumatol. 2016;68(9):2283–2289. doi: 10.1002/art.39773. [DOI] [PMC free article] [PubMed] [Google Scholar]