

Abstract
Autoimmune polyglandular syndromes (APS) are rare disorders characterized by auto‐destruction of endocrine and non‐endocrine organs by organ‐specific antibody‐directed T‐lymphocytic infiltration. This case highlights a 29‐year‐old Caucasian man with vitiligo found to have significant neurological abnormalities in the setting of newly diagnosed pernicious anemia and thyroid autoimmune disease.
Keywords: autoimmune polyglandular syndrome, cobalamin deficiency, subacute combined degeneration, thyroid autoimmune disease, pernicious anemia, vitamin B12 deficiency
Autoimmune polyglandular syndromes can have widely variable presentations. If a patient is found to have newly diagnosed thyroid autoimmune disease, it would be valuable to screen for other autoimmune diseases such as pernicious anemia; as these associated endocrinopathies can lead to significant neurological consequences.
1. INTRODUCTION
Autoimmune polyglandular syndromes (APS) are rare disorders characterized by the destruction of endocrine and non‐endocrine organs by organ‐specific antibody‐directed T‐lymphocytic infiltration. There are understood patterns of polyendocrine deficiencies that are defined as monogenic and polygenic variances. Historically, these combinations of clinical features led to APS being categorized as Type 1, Type 2, Type 3, or Type 4. 1 , 2 This case highlights an unusual presentation of APS type 3.
2. CASE REPORT
A 29‐year‐old Caucasian man with only a past medical history of vitiligo presented to the emergency department with a constellation of symptoms including subacute mental status changes and progressive lower extremity weakness with gait abnormalities and upper extremity paresthesia, constipation, and a 15 pound weight loss over the past several months.
In the emergency department, he was unable to provide a detailed history due to altered mental status. His vitals were within normal limits. He was alert and oriented to person, place, and time but exhibited significantly delayed and brief answers to questions with word finding difficulty. A Montreal Cognitive Assessment was completed resulting in a score of 15/22 with deficits in short‐term memory, organization, word fluency, and executive functioning. Physical examination was notable for angular stomatitis, glossitis (Figure 1), patches of vitiligo throughout, cranial nerves II‐XII were intact, upper extremity and lower extremity strengths were 5/5 bilaterally with increased tone in the upper and lower extremities, sensation to touch, pinprick, and vibration were intact and symmetrical, reflexes were noted to be 2/4 in the upper extremities but 4/4 in the bilateral knees and bilateral ankles with >10 s of clonus in the right ankle and abnormal plantar reflexes in the bilateral feet. Coordination showed stiff, slow finger‐to‐nose testing without tremor or dysmetria, and his gait was noted to be abnormal with stiff straight leg movements with difficulty walking on toes and unable to walk on heels.
FIGURE 1.
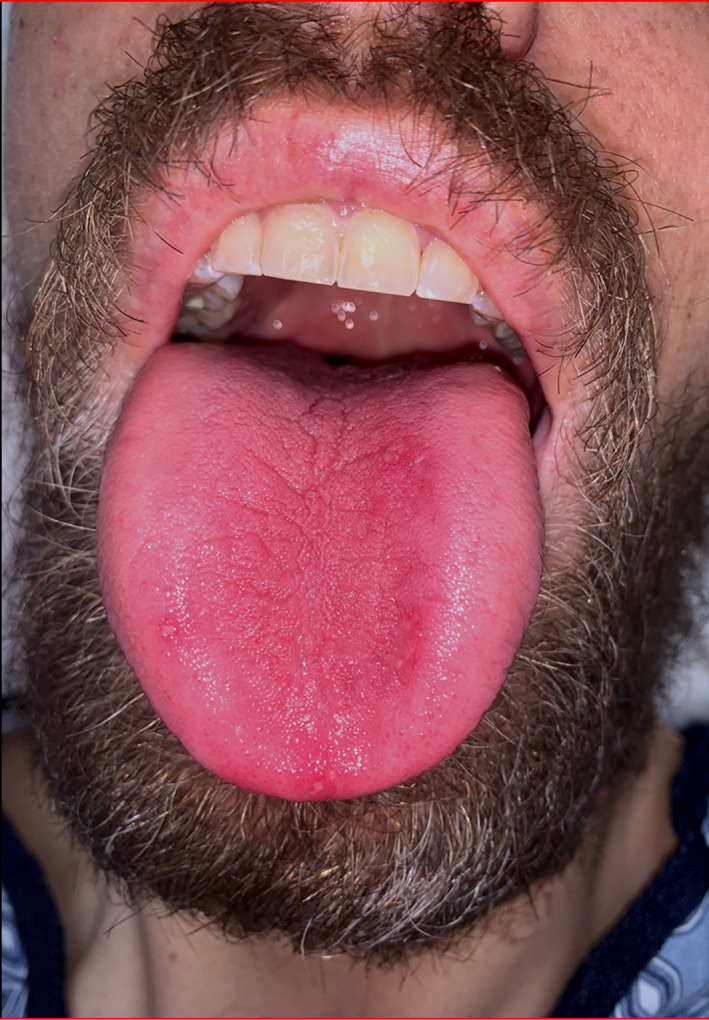
Image of patient’s mouth revealing glossitis
His laboratory tests revealed a complete blood cell count (CBC) remarkable for pancytopenia, including hemoglobin 7.6 g/dl [13.2–16.6], mean cell volume (MCV) 115.0 fl [78.2–97.9], white blood cell 1.8 × 109/L [3.4–9.6] with an absolute neutrophil count (ANC) of 1.00 × 109/L, absolute lymphocyte count 0.74 × 109/L, and a platelet count of 85 × 109/L. Serum chemistry analysis was all within normal limits. Endocrine studies revealed thyroid stimulating hormone (TSH) 20.6 milli‐international units/L [0.3–4.2] with a free T4 of 1.2 ng/dl [0.9–1.7], normal cosyntropin study, negative hydroxylase‐21 antibody, and normal fasting glucose. Iron studies showed iron 83 mg/dl [50–150], ferritin 163 mg/dl [31–409], total iron binding capacity 155 mg/dl [250–400], and percent saturation 54% [14%–50%]. An autoimmune panel was completed revealing antinuclear antibody 2.0 Units [≤1], extractable nuclear antigen antibody normal, C‐reactive protein <3, erythrocyte sedimentation rate 27 ml/hour, double‐stranded DNA normal, cyclic citrullinated peptide antibody <15.6 Units [<20], rheumatoid factor antibody <15 international units/milliliter [<15], thyroperoxidase (TPO) antibody >900 international units/milliliter [<9.0], intrinsic factor antibody positive, and tissue transglutaminase antibody negative with a normal IgA. Evaluation of vitamin deficiencies showed cobalamin (vitamin B12) <70 nanogram/L [180–914], thiamine (vitamin B1) 72 nanomole/L [70–180], folic acid 35 mg/L [> = 4.0], methylmalonic acid 39 nanomole/milliliter [≤0.40], homocysteine 205.9 nanomole/milliliter [6.4–11.8], vitamin D 9 ng/ml [20–50], pyridoxal 5‐phosphate 5 mg/L [5–50], pyridoxic acid 6 mg/L [3–30], and copper 0.91 mg/ml [0.75–1.45] with normal ceruloplasmin. A human immunodeficiency virus (HIV) screening was negative, and syphilis antibody was non‐reactive. A urine drug screen was negative for ethanol, amphetamine, barbiturates, benzodiazepines, cocaine, opiates, methadone, oxycodone, and tetrahydrocannabinol.
Our initial differential diagnosis for subacute encephalopathy associated with lower extremity weakness and ataxia remained broad to include nutritional and hematologic etiologies given his pancytopenia on initial laboratory evaluation. Vitamin B12 deficiency was suspected early on given evidence of glossitis on examination and macrocytic anemia with pancytopenia. Thiamine deficiency and Hashimoto encephalopathy were considered. Infectious encephalitis and malignancy, such as lymphoma and plasma cell dyscrasias, also remained on the differential. Neurosyphilis was ruled out. Concern for a neurologic disorder, such as autoimmune myelopathy, multiple sclerosis, and stiff person syndrome, prompted imaging of the brain and spine.
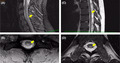
Magnetic resonance imaging (MRI) of the brain was completed that showed T2 hyperintensity around the right and left frontal horn and right occipital horn without any evidence of acute ischemic change. MRI of the cervical, thoracic, and lumbar spine was notable for extensive long segment T2 cord signal abnormality along the posterior columns throughout the thoracic spine (Figure 2). These findings of the spinal cord are consistent with radiological findings of subacute combined degeneration, which occurs in the setting of significant vitamin B12 deficiency.
FIGURE 2.
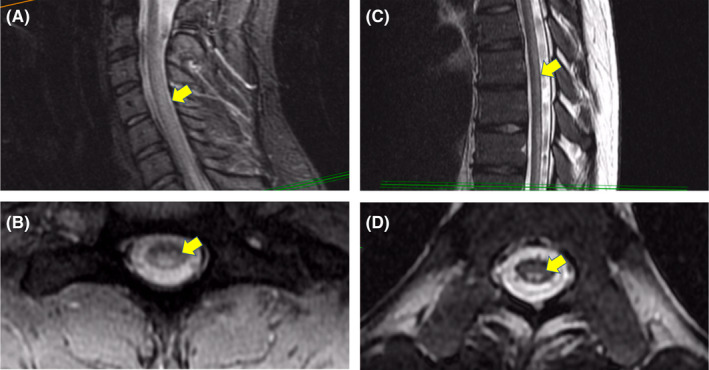
T2‐weighted magnetic resonance (MR) imaging. Sagittal slice of the cervical spine with diffuse hyperintensity of the dorsal column and mid cord (A). Axial slice of the cervical spine with diffuse hyperintensity of dorsal column and mid cord (B). Sagittal slice of the thoracic spine with diffuse hyperintensity of the dorsal column (C). Axial slice of the thoracic spine with symmetric hyperintensity of the dorsal column (D)
He was diagnosed with autoimmune polyglandular syndrome given the discovery of newly diagnosed pernicious anemia (PA) with profound vitamin B12 deficiency, newly diagnosed Hashimoto thyroiditis with hypothyroidism and known vitiligo.
3. DISCUSSION
This case report of APS is remarkable for the severity of neurologic impairment due to pernicious anemia resulting in profound vitamin B12 deficiency. Sequelae of his vitamin B12 deficiency included metabolic encephalopathy and subacute combined degeneration of the spine leading to progressive ataxia with upper motor neuron signs. Additional manifestations of his vitamin B12 deficiency included glossitis, pancytopenia, and megaloblastic anemia. His hypothyroidism due to Hashimoto’s thyroiditis potentially contributed to his encephalopathy, pancytopenia, and glossitis. However, hypothyroidism was unlikely to explain his subacute combined degeneration of the spine or megaloblastic anemia. In consultation with our endocrinology colleagues, our patient was diagnosed with APS type 3 given his constellation of pernicious anemia, thyroid autoimmune disease, and vitiligo.
Autoimmune polyglandular syndromes has been categorized as monogenetic and polygenetic types, which exhibit particular clinical patterns. APS type 1 is a rare monogenetic defect in the autoimmune regulator gene found in chromosome 21. Clinical significance typically arises during childhood with chronic mucocutaneous candida infections. This is then followed by endocrine dysfunction hallmarked by hypoparathyroidism and primary adrenal insufficiency (Addison’s disease). Other associated dysfunctions include but are not limited to gonadal dysfunction, enamel hypoplasia, and enteropathies. 1 , 2 , 3
Autoimmune polyglandular syndromes type 2 is a more common polygenetic syndrome that is typically early adult onset and predominantly seen in females. Endocrine dysfunction classically seen in APS type 2 includes primary adrenal insufficiency with type 1 diabetes mellitus and/or thyroid autoimmune disease (TAD). Other associated dysfunctions include but are not limited to celiac disease and pernicious anemia (PA). 1 , 2 , 4 , 5
Autoimmune polyglandular syndromes type 3 is the most common APS type, hallmarked by TAD, and typically observed in middle aged females. 1 , 5 There are several subtypes which are categorized based on other organ‐specific autoimmune involvement; type 3a exhibits TAD with type 1 diabetes, type 3b exhibits TAD with pernicious anemia (PA), and 3c exhibits TAD with vitiligo, alopecia, or other organ‐specific autoimmune disease. 1 , 5 , 6 APS type 4 is characterized as findings of two or more autoimmune endocrine organ destruction, which does not fit the other APS types. 1
There are no universally established guidelines for APS type 3 diagnosis and management. However, it may be prudent to keep this diagnosis in mind in patients with TAD as other studies have suggested that 52% of patients with TAD could fit an APS type 3 diagnosis. 5 , 6 Since there are no established guidelines for APS, we have highlighted important diagnosis and management information for the commonly involved organ‐specific autoimmune findings of TAD and pernicious anemia in APS type 3.
Evaluation for thyroid dysfunction by TSH measurement is recommended in the presence of symptoms. If TSH is elevated, subsequent measurement of free serum T4 is indicated and diagnosis of primary hypothyroidism may be made if free serum T4 is low. 7 Detection of elevated IgG4 or presence of anti‐thyroperoxidase antibodies is inherently suggestive of thyroid autoimmune disease, even in a euthyroid state. 8 Additionally, the presence of vitiligo alone may have a positive predictive value for diagnosing autoimmune thyroid disease. Meta‐analysis estimates the prevalence of thyroid dysfunction and thyroid autoantibodies in patients with vitiligo at 15.1% and 20.8%, with corresponding relative risks of 1.9 and 5.2, respectively. 9 Regarding management, the American Thyroid Association recommends the initiation of levothyroxine monotherapy in patients with elevated TSH, low T4, and symptoms of hypothyroidism with serial re‐evaluation for clinical improvement and TSH normalization. The use of glucocorticoids is generally not recommended in the context of autoimmune thyroid disease. 7 Thyroid hormone replacement therapy increases hepatic corticosteroid metabolism, and evaluation for underlying adrenal insufficiency is warranted before initiation of thyroid supplementation to prevent frank adrenal failure. 10
Due to a lack of guideline recommendations for the diagnosis of PA, a combination of studies must be undertaken and analyzed in conjunction with overall clinical features. Serum cobalamin measurement remains integral to diagnosing deficiency, and plasma methylmalonic acid and homocysteine measurement are useful in delineating B12 deficiency from other underlying primary biochemical derangements. 11 Anti‐parietal cell antibody (APCA) and anti‐intrinsic factor (anti‐IF) antibody have been utilized to screen for pernicious anemia. APCA targets the H+/K+ ATP‐ase found in parietal cells that plays a major role in acidifying the gastric content for optimal vitamin B12 absorption. APCA can be detected in 85%–90% of patients with pernicious anemia. However, APCA is seen in 7.8%–19.5% in the general adult population (incidence increases with age) and more prevalent in patients with known organ‐specific autoimmune disease such as those seen in APS with TAD, type 1 diabetes mellitus, or vitiligo. Ultimately, the specificity for APCA in the setting of PA is only 50%. 12 , 13 Anti‐IF antibody targets intrinsic factor secreted from parietal cells which complexes with cobalamin to allow for improved absorption. In contrast to APCA, anti‐IF antibody is more specific at 98% but less sensitive at 50%–70% as incidence vary by gender and race. 13 , 14 , 15 Given the high sensitivity of APCA and the specificity of anti‐IF, both auto‐antibody tests should be obtained when evaluating for pernicious anemia.
Vitamin B12 deficiency from pernicious anemia can manifest with neurologic and hematologic abnormalities. Vitamin B12 is an important cofactor in DNA synthesis and odd chain fatty acid metabolism. Vitamin B12 deficiency therefore impairs synthesis and methylation of myelin basic protein and lipids, accumulation of abnormal fatty acid metabolites, and ultimately damage of the myelin sheath. 16 This is postulated as the primary mechanism of demyelination in subacute combined degeneration of the spinal cord, which is only evident with advanced deficiency such as in this case. 16 , 17 Subacute combined degeneration can be appreciated with T2‐weighted MRI, with classical findings of increased signal intensity of the bilateral dorsal columns. 18 Homocysteine methyltransferase is another vitamin B12‐dependent enzyme responsible for the conversion of 5‐methyl‐tetrahydrofolate (5‐MTF) to tetrahydrofolate, which is pivotal in DNA synthesis. In bone marrow, buildup of 5‐MTF and impaired DNA synthesis leads to the disruption of hematopoiesis and development of megaloblastic anemia. 16
Standard treatment of PA includes regular, lifelong injections of supplemental cyanocobalamin. Recommendations include doses of 1000 mg multiple days weekly for 1–2 weeks, then weekly for approximately 4 weeks until clinical improvement is noted, then monthly for life. 19 , 20 Limited data suggests that daily high‐dose oral supplementation of cobalamin may be an effective alternative treatment for PA, with criticisms arising from poorly defined dosing recommendations and possibility of poor compliance or follow‐up. 11 , 14 , 20 Hematologic abnormalities should be monitored and are expected to improve within one week, often resolving within 6 weeks of supplementation. Neurologic abnormalities typically improve more gradually but may never fully resolve depending on initial severity. Factors associated with improved short‐term neurological outcomes include brevity of disease course, absence of Babinski’s and Romberg’s signs, age less than 50 years, absence of sensory deficits, and specific spinal MRI findings including involvement of fewer segments, presence of contrast enhancement and absence of cord atrophy. 21 , 22 Higher levels of serum cobalamin at the time of presentation are also associated with decreased clinical severity and improved neurological outcomes. 23 Systematic review of global data suggests a pooled incidence rate of gastric cancer in PA patients to be 0.27% per person‐years, and meta‐analysis demonstrates an overall relative risk of 6.8 across ethnicities. 24 The American Society for Gastrointestinal Endoscopy recommends a one‐time endoscopic evaluation after initial diagnosis with subsequent repeat endoscopy based on the emergence or evolution of gastrointestinal symptoms and increased risk of malignancy. 25
While our patient’s neurological abnormalities and encephalopathy can be explained by his vitamin B12 deficiency and hypothyroidism, it is pertinent to remember that autoimmune polyglandular syndromes can also cause neurologic complications due to other endocrinopathies. These complications can manifest with Addison disease leading to adrenal insufficiency, diabetes mellitus leading to hyperosmolar hyperglycemic state or diabetic ketoacidosis, and hypoparathyroidism leading to hypocalcemia. Other described associated malabsorption, vitamin and mineral deficiencies that can lead to ataxic gait are celiac sprue, vitamin E deficiency, and hypomagnesemia. 26
There have also been case reports of patients with APS type 3 and primary neurological diseases. Yokote et al. 28 described a case of a 73‐year‐old female who was hospitalized for diabetic ketoacidosis then found to have APS type 3 with type 1 diabetes mellitus and chronic thyroiditis, and also diagnosed with late‐onset conventional multiple sclerosis based. There are also several reports of patients with APS type 3 who develop myasthenia gravis. When including APS type 1 and type 2, other primary neurological diseases have been concomitantly reported including Guillain–Barré, Miller Fisher syndrome, autoimmune central nervous system demyelination, and autoimmune cerebellar degeneration. It is hypothesized that there is a genetic component as APS and these neurological diseases are associated with human leukocyte antigen (HLA). 26 , 27 , 28 Because our patient’s neurologic complications were secondary to metabolic and endocrine disorders, HLA testing was not complete.
4. CONCLUSION
This patient’s constellation of symptoms can be explained by his findings of profound vitamin B12 deficiency secondary to pernicious anemia with subacute combined degeneration of the spinal cord and hypothyroidism in the setting of newly diagnosed Hashimoto’s thyroiditis. Given his history of vitiligo and newly diagnosed pernicious anemia and Hashimoto’s thyroiditis, he meets criteria for autoimmune polyglandular syndrome type 3. He was treated with supplemental support with cyanocobalamin, levothyroxine, and thiamine. After the initiation of vitamin and thyroid supplementation, he quickly experienced the improvement of his symptoms over several days. Though his mentation had cleared, he continued to have significant weakness and required discharge to a skilled nursing facility for further skilled needs. Follow‐up was planned with an endocrinologist and gastroenterologist for gastric cancer screening; however, the patient was lost to follow‐up.
CONFLICT OF INTEREST
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
AUTHOR CONTRIBUTIONS
Michael Apolinario, Mayo Clinic Arizona, involved in design and conceptualization, literature review, conceptualized and drafted figures, and revised the manuscript for intellectual content. Aaron Brussels and Curtiss B. Cook and Shaun Yang, Mayo Clinic Arizona, involved in design and conceptualization, literature review, and revised the manuscript for intellectual content.
CONSENT
The written informed consent was obtained from the patient for the publication of this case report. A copy of the written consent is available for review by the Editor‐in‐Chief of this journal.
ACKNOWLEDGEMENTS
None.
Apolinario M, Brussels A, Cook CB, Yang S. Autoimmune polyglandular syndrome type 3: A case report of an unusual presentation and literature review. Clin Case Rep. 2022;10:e05391. doi: 10.1002/ccr3.5391
Funding information
The author(s) received no financial support for the research, authorship, and/or publication of this article
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Neufeld M, Blizzard RM, Pinchera A, Doniach D, Fenzi GF, Baschieri L Polyglandular Autoimmune Disease. Symposium on Autoimmune Aspects of Endocrine Disorders. Academic Press Inc.; 1980:357‐365. [Google Scholar]
- 2. Husebye ES, Anderson MS, Kämpe O. Autoimmune polyendocrine syndromes. N Engl J Med. 2018;378:2543–2544. [DOI] [PubMed] [Google Scholar]
- 3. Nwosu I, Oladiran O, Ogbonna‐Nwosu C, Anyata A. Autoimmune polyglandular syndrome type 1: a case report and brief review. J Community Hosp Intern Med Perspect. 2019;9(3):252–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sonthalia N, Ray S, Maiti A, Maitra S. Autoimmune polyglandular syndrome type II presenting as an endocrine emergency: a case report. Oman Med J. 2013;28(3):e048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tincani A, Ceribelli A, Cavazzana I, Franceschini F, Sulli A, Cutolo M. Autoimmune polyendocrine syndromes. In: Shoenfeld Y, Cervera R, Gershwin ME, eds. Diagnostic Criteria in Autoimmune Diseases. Humana Press; 2008:265–269. [Google Scholar]
- 6. Tian S, Xu B, Liu Z, Liu R. Autoimmune polyglandular syndrome type III associated with antineutrophil cytoplasmic autoantibody‐mediated crescentic glomerulonephritis: a case report and literature review. Medicine (Baltimore). 2020;99(7):e19179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jonklaas J, Bianco AC, Bauer AJ, et al. Guidelines for the treatment of hypothyroidism: prepared by the American thyroid association task force on thyroid hormone replacement. Thyroid. 2014;24(12):1670–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rotondi M, Carbone A, Coperchini F, Fonte R, Chiovato L. Diagnosis of endocrine disease: IgG4‐related thyroid autoimmune disease. Eur J Endocrinol. 2019;180(5):R175–R183. [DOI] [PubMed] [Google Scholar]
- 9. Vrijman C, Kroon MW, Limpens J, et al. The prevalence of thyroid disease in patients with vitiligo: a systematic review. Br J Dermatol. 2012;167(6):1224–1235. [DOI] [PubMed] [Google Scholar]
- 10. Schatz DA, Winter WE. Autoimmune polyglandular syndrome. II: clinical syndrome and treatment. Endocrinol Metab Clin North Am. 2002;31(2):339–352. [DOI] [PubMed] [Google Scholar]
- 11. Devalia V, Hamilton MS, Molloy AM. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496–513. [DOI] [PubMed] [Google Scholar]
- 12. Rusak E, Chobot A, Krzywicka A, Wenzlau J. Anti‐parietal cell antibodies ‐ Diagnostic significance. Adv Med Sci. 2016;61(2):175–179. [DOI] [PubMed] [Google Scholar]
- 13. Wong CW. Chapter 16 ‐ Vitamin B12 deficiency in the elderly. In: Watson RR, ed. Nutrition and Functional Foods for Healthy Aging. Academic Press; 2017:159–166. [Google Scholar]
- 14. Carmel R. How I treat cobalamin (vitamin B12) deficiency. Blood. 2008;112(6):2214–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carmel R. Reassessment of the relative prevalences of antibodies to gastric parietal cell and to intrinsic factor in patients with pernicious anaemia: influence of patient age and race. Clin Exp Immunol. 1992;89(1):74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scott JM, Dinn JJ, Wilson P, Weir DG. Pathogenesis of subacute combined degeneration: a result of methyl group deficiency. Lancet. 1981;2(8242):334–337. [DOI] [PubMed] [Google Scholar]
- 17. Scalabrino G. Cobalamin (vitamin B(12)) in subacute combined degeneration and beyond: traditional interpretations and novel theories. Exp Neurol. 2005;192(2):463–479. [DOI] [PubMed] [Google Scholar]
- 18. Kumar A, Singh AK. Teaching NeuroImage: Inverted V sign in subacute combined degeneration of spinal cord. Neurology. 2009;72(1):e4. [DOI] [PubMed] [Google Scholar]
- 19. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441–1448. [DOI] [PubMed] [Google Scholar]
- 20. Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(21):2041–2042. [DOI] [PubMed] [Google Scholar]
- 21. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao J, Su ZY, Xu SB, Liu CC. Subacute combined degeneration: a retrospective study of 68 cases with short‐term follow‐up. Eur Neurol. 2018;79(5–6):247–255. [DOI] [PubMed] [Google Scholar]
- 23. Jain KK, Malhotra HS, Garg RK, Gupta PK, Roy B, Gupta RK. Prevalence of MR imaging abnormalities in vitamin B12 deficiency patients presenting with clinical features of subacute combined degeneration of the spinal cord. J Neurol Sci. 2014;342(1–2):162–166. [DOI] [PubMed] [Google Scholar]
- 24. Vannella L, Lahner E, Osborn J, Annibale B. Systematic review: gastric cancer incidence in pernicious anaemia. Aliment Pharmacol Ther. 2013;37(4):375–382. [DOI] [PubMed] [Google Scholar]
- 25. Evans J, Chandrasekhara V, Chathadi K, et al. The role of endoscopy in the management of premalignant and malignant conditions of the stomach. Gastrointest Endosc. 2015;82(1):1–8. [DOI] [PubMed] [Google Scholar]
- 26. Berger JR, Weaver A, Greenlee J, Waglen GE. Neurologic consequences of autoimmune polyglandular syndrome type 1. Neurology. 2008;70(23):2248–2251. doi: 10.1212/01.wnl.0000313837.45525.b6 [DOI] [PubMed] [Google Scholar]
- 27. Yokote H, Nagasawa M, Ichijo M, Amino T, Fujigasaki H. Autoimmune polyendocrine syndrome‐3 in a patient with late‐onset multiple sclerosis. Neurologist. 2012;18(2):83–84. doi: 10.1097/NRL.0b013e318248ea2a. PMID: 22367836. [DOI] [PubMed] [Google Scholar]
- 28. Inoue H, Yamada K, Fujii A, et al. A Patient with fulminant myasthenia gravis is seropositive for both AChR and LRP4 antibodies, complicated by autoimmune polyglandular syndrome type 3. Intern Med. 2020;59(17):2177–2181. doi: 10.2169/internalmedicine.4708-20. Epub 2020 May 26. PMID: 32461531; PMCID: PMC7516320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.