

Abstract
Tissue clearing of gross anatomical samples was first described over a century ago and has only recently found widespread use in the field of microscopy. This renaissance has been driven by the application of modern knowledge of optical physics and chemical engineering to the development of robust and reproducible clearing techniques, the arrival of new microscopes that can image large samples at cellular resolution and computing infrastructure able to store and analyze large data volumes. Many biological relationships between structure and function require investigation in three dimensions and tissue clearing therefore has the potential to enable broad discoveries in the biological sciences. Unfortunately, the current literature is complex and could confuse researchers looking to begin a clearing project. The goal of this Primer is to outline a modular approach to tissue clearing that allows a novice researcher to develop a customized clearing pipeline tailored to their tissue of interest. Further, the Primer outlines the required imaging and computational infrastructure needed to perform tissue clearing at scale, gives an overview of current applications, discusses limitations and provides an outlook on future advances in the field.
INTRODUCTION
Tissue clearing refers to a collection of techniques that render biological samples transparent. These techniques enable the deep imaging of large tissue volumes using light microscopy approaches that are usually limited by the scattering of light by the tissue. Tissue clearing allows researchers to avoid time-consuming tissue sectioning approaches that can introduce artefacts and examine biological tissues in their native, three-dimensional state. Further, light microscopy enables the use of individual-molecule labelling techniques and imaging at sub-micrometre spatial resolutions, unlike traditional approaches for volume imaging such as magnetic resonance imaging (MRI), computed tomography (CT) and ultrasound imaging.
Tissue clearing protocols facilitate the passage of light through a biological sample by minimizing the refractive index (RI) differences between components of the tissue1. In general, this requires the removal of lipids (RI≈1.47) and replacement of intracellular and extracellular fluids (RI≈1.35) with a solution of equivalent RI to the remaining protein constituents (RI>1.50). Despite this simple underlying principle, the design of a tissue clearing project can easily become complex and requires optimization of multiple variables. Factors that should be considered include the need for retention of molecules of interest during processing (for example, proteins or mRNA), maintenance of the sample’s structural integrity, conformational protection of endogenously expressed fluorescent proteins, enhanced porosity to allow for the diffusion of small molecule or immunoglobulin labels, and preservation of the structure of relevant epitopes if immunolabelling.
Previous reviews have highlighted the basic physics1 and chemistry2 behind tissue clearing and highlighted the promise of tissue clearing for the field of neuroscience3. This Primer will instead focus on the methodology for clearing various tissue types, the technologies for imaging cleared tissue and how to store and analyse image data. Specifically, the Primer will enable a researcher to understand the purpose of each step in a clearing protocol and assemble a clearing pipeline specifically designed for their sample. It will discuss the advantages and disadvantages of several imaging techniques and describe several common image analysis and quantitation routines. This comprehensive description of clearing, imaging, and data analysis aims to provide the necessary information for individual researchers to design simple clearing experiment and also guide institutional core facilities and commercial entities on how to clear samples at scale.
EXPERIMENTATION
Historically, clearing techniques were assigned to one of two categories based on the composition of their final clearing solution: solvent-based (hydrophobic) or aqueous-based (hydrophilic). Originally, solvent-based techniques were faster, provided a closer RI match to the delipidated, proteinaceous sample and shrunk the sample in size. Conversely, aqueous techniques better preserved the emission of fluorescent proteins (if present), retained biomolecules to a higher degree and expanded samples in size. However, many of the original limitations of each category have been addressed and solvent and aqueous techniques have become nearly equivalent4–6. In fact, it is now recognized that combining aqueous-based and solvent-based delipidation steps can be advantageous and aqueous RI-matching solutions can be combined with solvent-based delipidation (and vice-versa). Therefore, it no longer seems relevant to label techniques aqueous-based or solvent-based but rather to view tissue clearing as a pipeline composed of several modules that can be mixed-and-matched. These modules are sample fixation, pre-treatment, delipidation, fluorescent labelling, RI matching and image acquisition and analysis (Fig. 1). Each clearing protocol must be customized to the sample type and the labelling method used, and the imaging modalities (such as microscopes) or IT infrastructure available to a researcher can influence the choice of clearing technique and inform the scale of the project. Below is an in-depth discussion of the various modules that can be combined to create a complete tissue clearing workflow, with details summarized in Table 1.
Figure 1: An overview of the components of a tissue clearing experiment.

Tissue clearing workflows are numerous, making them difficult to summarize. However, workflows can be defined as a series of modules (fixation, pre-treatment, delipidation, labelling and refractive index matching), where each module can be customized to suit the tissue that is to be cleared. Multiple modules must be combined to clear large organoids, organs and whole animals, whereas small samples (< 0.5 mm) may only require a refractive index matching step. Blue shading indicates that samples are immersed in polar, water-based, aqueous solutions whereas red shading indicates samples are in less/non-polar solvents. Expanded samples are hyperhydrated and grow in size, whereas dehydrated samples commonly shrink. ETC, electrophoretic tissue clearing; RI, refractive index.
Table 1:
Tissue Clearing Modules
| Modules and reagents | Indications for use | Required equipment | Additional considerations | Originating protocol |
|---|---|---|---|---|
| Fixation (1–12 hours) | ||||
| Paraformaldehyde (PFA) | • General fixation protocol | None | • Concentration and timing require optimization | Scale173, CLARITY33 |
| Glutaldehyde (GA) | • Used to retain low-abundance biomolecules • Used to stabilize fragile tissues |
None | • Concentration and timing require optimization • Often combined with PFA |
SWITCH20 |
| Polyglycerol-3-polyglycidyl ether (P3PE) | • Used to retain low-abundance biomolecules • Used to stabilize fragile tissues • Used to preserve fluorescent protein emission • Used to retain mRNA |
None | • High protein and mRNA fixation efficiency | SHIELD4 |
| 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) | • Used to retain mRNA | None | • High mRNA fixation efficiency • Poor protein fixation efficiency |
STARmap174 |
| Fixation with sodium dodecyl sulfate (SDS), urea, borate and heat | • Used for antigen retrieval | Oven | • May enhance antibody staining • Concentration of reagents and incubation length should be carefully determined |
CLARITY33, FLASH12 |
| Decolorization (5–24 hours) | ||||
| Amino alcohols (Quadrol, THEED) | • Used when tissues contain pigments such as kidney, liver, spleen and muscle (heme); skin (melanin); brain (lipofuscin) | None | • Used to remove heme from blood (hemoglobin) and muscle (myoglobin) | CUBIC24, FlyClear13 |
| N-alkylimidazole | None | • Effective at heme removal | CUBIC25 | |
| N-methyldiethanolamine (NMDEA) | None | • Effective at heme removal | SHANEL6 | |
| Bleaching by hydrogen peroxide | None | • Widely effective across many pigments • Can cause tissue damage if left too long or used at too high a concentration |
iDISCO47 | |
| Quenching/autofluorescence (12–24 hours) | ||||
| Photobleaching with high-intensity light | • Used when tissues have high autofluorescence (human brain) | LED array inside refrigeration unit | • Broadband white light is most effective • Will bleach genetically encoded fluorescent proteins |
ELAST31, Multispectral LED array30 |
| Use of red and far-red fluorophores | Antibody conjugated fluorescent dyes | • Use dyes with emission at wavelengths > 600 nm | iDISCO47 | |
| Hydrogel embedding (12–24 hours) | ||||
| Acrylamide, bis-acrylamide and PFA | • Used to retain low expression biomolecules • Used to stabilize fragile tissues • Used to retain mRNA • Required when performing tissue expansion |
Heat block under vacuum | • Best for retention of mRNA and low expressing proteins• Gel density can be adjusted to promote clearing and labelling (less dense) or biomolecule retention (more dense) | CLARITY33, PACT/ PARS34 |
| Acrylamide | Heat block under vacuum | • Acrylamide is polymerized in the absence of cross-linking molecules (bis-acrylamide), creating a stretchable gel | ELAST31 | |
| Acrylamide, acrylate, protein enzymes, detergents and imidazole | Incubator | • Hydrogels can expand many fold when placed in pure water (or an imidazole solution in the case of CUBIC-X) | ExM35, ProExM36, MAP37, CUBIC-X38, iExM39 | |
| Decalcification ( 12 hours–3 weeks) | ||||
| EDTA and imidazole | • Required when clearing tissue containing calcified bone | • Used to remove calcification in tissue | CUBIC-B25, PEGASOS42, PACT-deCAL43, bone CLARITY44, vDISCO45 | |
| Delipidation (1 hour–3 days) | ||||
| By solvent | • Required to clear any tissue > 0.2 mm thick | None | • Tissue must first be dehydrated with tetrahydrofuran (THF) or alcohol • If tissue contains fluorescent proteins, dehydrating solutions must be maintained at 4°C and > pH 9.0. Consider supplementing with: butylated hydroxytoluene or Quadrol and polyethlyene glycol (PEG) • Complete delipidation requires additional incubation in dichloromethane (DCM) |
fDISCO49, sDISCO50, uDISCO9, a-uDISCO52, FluoClearBABB51, PEGASOS42 |
| By detergent | Optional: electrophoretic tissue clearing (ETC) device | • Most common detergents: SDS, TritonX-100, CHAPS • Degree of clearing increased by alkaline pH, increased temperature and increased incubation time |
CLARITY33, PACT-PARS34, CUBIC23 | |
| By solvent and detergent | None | • Must be performed sequentially | Adipo-Clear57, SHANEL6 | |
| Refractive index matching (1 hour–2 days) | ||||
| In solvent | • Required for all tissue clearing experiments | None | • Commonly used solvents: BABB, dibenzyl ether (DBE) and ethyl cinnamate • If tissue contains fluorescent proteins, a pH > 9.0 should be maintained. Consider supplementing with triethylamine, Quadrol, DL-alpha-tocopherol, propyl gallate, DPE or PEG. • RI should be ~ 1.56 |
sDISCO50, uDISCO9, PEGASOS42 |
| In aqueous solution | None | • Commonly used reagents are antipyrine and nicotinamide; N-methylacetatamide and Histodenz; or TDE, DMSO and iohexol • RI should be ~ 1.52 |
CUBIC70, Ce3D71, ELAST31 | |
| With expansion | None | • Use pure water with RI = 1.33 (ExM, ProExM, iExM, MAP) or 5% imidazole/55% antipyrine with RI = 1.467 (CUBIC-X) solutions for RI matching | ExM35, ProExM36, MAP37, CUBIC-X38, iExM39 | |
BABB, benzyl alcohol benzyl benzoate; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; EDTA, ethylenediaminetetraacetic acid; RI, refractive index; THEED, 2,2′,2″,2″′-(Ethylenedinitrilo)tetraethanol;
Sample choice
Many commonly studied biological tissues from a host of organisms have been shown to be compatible with tissue clearing approaches. Entire rodents have been cleared and imaged while intact7–11, as have several other model organisms including drosophila12,13 and zebrafish14,15. Human tissues, including an entire human brain and kidney, have also been cleared successfully6. However, there are limitations to clearing and not all tissues clear equally well. For example, successful clearing of whole rodents usually requires the removal of the skin from the animal. Additionally, tissues with pigments (such as skin), calcification (bone), or extensive extracellular matrix and/or structural proteins (tumours and cardiac tissues) do not obtain the same level of clearing as tissues without these features.
Specific and bright fluorescent labelling is essential for imaging, regardless of the tissue. Small molecule organic dyes, dye-conjugated antibodies and genetically encoded fluorescent proteins are commonly used labels. Small-molecule dyes can rapidly penetrate and label thick tissue, but are limited in their recognizable targets. Antibodies have the widest utility but suffer from long labelling times and reproducibility issues owing to batch-to-batch variation. Genetically encoded fluorescent proteins may label a higher percentage of target molecules and allow labelling of specific cell types16. Viral expression of fluorescent proteins avoids the complex molecular biology of stably introducing a transgene to a tissue and can achieve high levels of fluorescence emission depending on virus titre, serotype and choice of promoter17,18. However, care must be taken as many fluorophores are susceptible to bleaching with hydrogen peroxide and fluorescent protein emission can be quenched by dehydration (see below).
Finally, it is important to match the final size of the sample to available imaging systems and data storage capacity. It is of note that cleared samples that are millimetres or centimetres in size can exceed the working distance of many microscope objectives and can produce TBs of data.
Fixation
Fixation of tissue is always required before clearing to avoid excessive loss of target biomolecules or tissue integrity. Fixing covalently crosslinks proteins and/or nucleic acids in the tissue to preserve the tissue’s mechanical integrity and biomolecular architecture. Fixation is a balancing act; if fixation is too weak, the tissue will not withstand chemical treatments in later clearing steps. Conversely, over-fixation can result in fixative molecules blocking access to the epitopes required for fluorescent labelling.
The degree of fixation is determined by the concentration of the fixative and amount of time the tissue spends immersed in it. The most commonly used tissue fixative is paraformaldehyde (PFA), which crosslinks the tissue’s biomolecules by reacting with amine groups on proteins and nucleic acids. PFA is usually used at a concentration of 3–4% w/v in phosphate-buffered saline (PBS). PFA diffuses rapidly into tissue, making it a good molecule for quickly and uniformly fixing samples greater than 500 µm in diameter. Glutaraldehyde (GA) can be used as an alternative to PFA, which — although chemically similar — reacts with biomolecular amine groups faster, resulting in more intermolecular covalent crosslinking and enhanced tissue fixation19. However, as GA is a larger molecule than PFA, diffusion into tissues is slower, leading to non-uniform tissue preservation. To address issues caused by diffusion, the SWITCH method can be used20. In this method, inactivated GA molecules diffuse through the sample in a low-pH “OFF” buffer and once the GA molecules are uniformly distributed, the tissue is transferred to a neutral pH “ON” buffer to initiate fixation. Using this protocol, GA was demonstrated to provide improvements in biomolecular preservation (3–5% protein loss) over PFA tissues (30–40% protein loss)20. GA-preserved tissues exhibit high autofluorescence [G] in the 500–600 nm range21 and retain mRNA molecules less well than PFA, and therefore GA is most suitable for strengthening delicate tissues or retaining poorly expressed proteins if imaging proteins in downstream imaging pipelines.
Chemical crosslinks formed by aldehyde fixatives can block antibody access to epitopes and reduce the effectiveness of downstream immunostaining (see below). Enzymatic digestion and/or a high heat and pressure treatment is routinely used to permeabilize tissue, reduce aldehyde crosslinks and restore antibody accessibility in thin histology sections ― a process referred to as antigen retrieval. However, applying these treatments to intact tissue is detrimental to its integrity. Interestingly, a combination of detergent, solvent and mild heat has been shown to have an antigen retrieval effect when included in a tissue clearing protocol (FLASH, see ref12).
Using polyepoxide molecules as fixatives, such as polyglycerol-3-polyglycidyl ether (P3PE), enables a high degree of intermolecular and intramolecular crosslinking for several biomolecules. Use of P3PE in the SHIELD protocol produces tissue that is more rigid and mechanically stable than tissue fixed with PFA or GA4. Additionally, this fixation method produces cleared tissue with significantly higher protein retention, mRNA retention and fluorescent protein emission than GA-fixed and PFA-fixed tissue. P3PE concentrations can be adjusted between 2–20% w/v, with epitope availability and chemical/label penetrability enhanced at low concentrations and protein preservation, mRNA preservation and tissue rigidity enhanced at high concentrations. In addition to the above fixation methods, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) has shown utility for fixing genetic material in place4.
It is best to evaluate several fixation protocols, including various concentrations and durations of each fixative, to determine which best retains your targets of interest while still allowing for sufficient and timely clearing. For example, thicker or denser tissues may require use of the SWITCH method coupled with relatively low fixative concentrations to enable uniform fixation within a reasonable amount of time.
Optional pre-treatments
Preparing a cleared sample most often involves removing lipids from the sample and placing it in a solution matched to the RI of the remaining tissue components (usually protein, RI > 1.5). Refractive index matching alone may be adequate for clearing thin (< 0.2 mm), non-pigmented samples; however, most tissues require additional processing. The pre-treatment steps below are optional. Some are needed for specific tissue types, while others have a more general application. Experimentation on smaller samples is advised to identify which are necessary for a particular tissue of interest.
Decolorization
Many tissues contain natural endogenous pigments that can absorb light and prevent effective clearing and imaging, and in large tissue volumes even sparse pigments are obstacles to acquiring high-contrast images2,3,10,22. Heme is the most abundant chromophore found throughout tissues and is associated with hemoglobin and myoglobin pigments found in the blood and muscles, respectively. Blood can be removed after an animal is sacrificed by transcardial perfusion of large volumes of isotonic buffer; however, perfusion with PBS alone is insufficient to remove all heme from blood-rich and muscle-rich tissues such as the kidney, heart, muscle and liver. Extensive chemical screening has identified that amino alcohols effectively elute heme from both blood and muscle23; these chemicals (specifically Quadrol) also assist in lipid extraction, are compatible with fluorescent proteins and are a key component in the CUBIC tissue clearing protocols11,24. The amino alcohol N-methyldiethanolamine (NMDEA), in conjunction with 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) detergent, can effectively remove heme while permeabilizing and delipidating tissue6. N-alkylimidazole also has high utility for heme elution25. Sacrificed animals can be perfused with any of these heme solvating chemicals, or individual tissues ― including human tissue ― can be incubated with them after dissection.
Additional endogenous chromophores may be present depending on the tissue. Pigments of the compound eye such as pteridine and ommochrome and melanin in skin can be particularly difficult to remove. The amino alcohol 2,2′,2″,2″′-(Ethylenedinitrilo)tetraethanol (THEED) has been shown to be effective at removing pigments of the compound fly eye while preserving GFP and mCherry emission (FlyClear13).
A more generalized method for decolorization is to break the chemical bonds of pigments using peroxides, a process often referred to as bleaching15,26–29. Bleaching gives powerful decolorization performance, although it requires optimization of the peroxide concentration and treatment duration to avoid excessive damage to the tissue. Further, as peroxide treatment destroys the central chromophore of fluorescent proteins and bleaches some organic dye molecules, downstream immunostaining is required to visualize quenched fluorescent proteins and any fluorescent labelling must be carried out after the bleaching step.
Quenching autofluorescence
Autofluorescence can decrease the signal-to-noise ratio (SNR) and reduce contrast when imaging. To avoid this, samples can be photobleached to eliminate autofluorescence prior to staining. Photobleaching can be accomplished by exposing the tissue to broadband light for an extended period; for example, exposure using a custom device containing broadband visible LEDs in a cold room for up to three days can reduce autofluorescence in thick tissue samples30,31 (the cold room prevents tissue from overheating and degrading). A key disadvantage to this method is the need for immunostaining, as genetically encoded fluorescent proteins will be photobleached. A more common and efficient method to overcome autofluorescence is to select red and far-red dyes that fluoresce outside the 500–600 nm range where most autofluorescence occurs. If red and far-red dyes are selected, autofluorescence in the 500–550 nm range can be imaged to derive useful information, for example to identify anatomical structures or register the dataset against other samples or atlases (see Results section).
Hydrogel embedding
Hydrogel embedding techniques were originally developed to improve the mechanochemical properties of fixed tissue over standard PFA fixation. Hydrogel embedding technologies for tissue clearing employ the polymerization of monomer solutions containing varying compositions of acrylamide (AA), sodium acrylate (SA), and bis-acrylamide (Bis) molecules to generate polyacrylamide (pAAm) meshes that physically or chemically fix biomolecules in place. In recent years, researchers have harnessed the unique properties of hydrogels ― such as their expandability and elasticity ― to further enhance the applications of hydrogel embedding technologies. The fundamental principles and novel methods in tissue-hydrogel transformation technologies are covered in-depth in a recent review paper on the subject32.
CLARITY was the first widely-used hydrogel embedding technique33. CLARITY and its variants covalently anchor endogenous tissue biomolecules to a pAAm mesh that is generated from AA, PFA, and Bis (Fig. 2). Bis serves to chemically crosslink separate acrylamide chains to form the pAAm mesh, while PFA serves to crosslink tissue molecules to the mesh and decrease protein loss. Bis and PFA can be excluded from the monomer solution to increase pore size for enhanced clearing and immunostaining, with a reduction in mechanochemical preservation and stability34.
Figure 2: Concept of hydrogel embedding.

(a) Tissue biomolecules are fixed chemically or physically to a hydrogel mesh generated in situ, and then cell membranes are removed during delipidation to enable chemical transport and optical transparency. (b) In CLARITY hydrogel embedding, paraformaldehyde (PFA), bis-acrylamide (Bis) and acrylic acid (AA) are used to covalently link the primary amines of proteins and nucleic acids to a polyacrylamide (pAAm) hydrogel mesh generated through free radical polymerization. Free radical generation and subsequent polymerization is initiated by the chemical VA044 when it is warmed to a temperature of 37°C. (c) A tissue-hydrogel’s pore size, expandability, transparency, and biomechanical stability can be modulated based on the delipidation temperature and the concentrations of the sodium dodecyl sulfate (SDS) delipidation detergent, the PFA fixative and the Bis crosslinker.
More recently, the elastic properties of pAAm hydrogels have been harnessed to increase the toughness of hydrogel embedded tissue in a process called ELAST31. By significantly decreasing the Bis crosslinker concentration, more physical entanglement and less covalent crosslinking of long pAAm chains occurs. This decreases the rigidity and brittleness of the resulting tissue-gel, making it reversibly stretchable and more resistant to wear and tear when handled.
Certain hydrogels, such as the pAAm gels used in CLARITY, polymerize best when maintained in an oxygen-free environment and their polymerization is often activated by a temperature sensitive initiator. Custom (or commercial) equipment can be used to perform these steps; for example, a basic setup involves placing a conical tube or plate warmer inside a vacuum chamber. The samples are placed under vacuum prior to heating the sample to 37°C for approximately 3 hours.
Another interesting property of hydrogels is their ability to expand. These expansion properties have been harnessed to physically expand hydrogel-embedded tissue-gels and thus increase the achievable spatial resolution of conventional light microscopy35. Although expansion occurs during the final RI-matching step, the fixed tissue must first be embedded in a polyacrylamide-polyacrylate gel that will later facilitate expansion in each dimension. Additionally, protein digestion or denaturation is required to permit final tissue expansion. This has been achieved using Proteinase K (ExM35), LysC (Pro-ExM36), SDS (MAP37) or imidazole (CUBIC-X38). LysC, SDS and Triton X-100 are now the preferred strategies as these reagents are the least destructive to tissue and better preserve biomolecules for post-expansion labelling36–38. Recent protocols expand tissue up to 20x (iExM39) and allow for fine tuning of the degree of expansion (ZOOM40).
Decalcification
The clearing of limbs or whole vertebrates requires the decalcification of bone as calcified bone is a strong light scatterer2,3,10,22. 50–70% of bone consists of carbonated hydroxyapatite (HAp) crystals distributed throughout a collagen matrix. Elution of calcium ions from HAp reduces light scattering and renders bony material transparent. Decalcification can be performed by submerging the sample in an acidic solution (as illustrated by the classic classroom experiment in which immersing an egg in vinegar renders the shell transparent); however, this causes substantial soft tissue damage. Neutral buffered saline solutions that contain the calcium ion chelator EDTA are preferred41 and EDTA has been used in many tissue clearing methods, including CUBIC-B25, PEGASOS42, PACT-deCAL43, bone CLARITY44 and vDISCO45. Interestingly, imidazole may further enhance the decalcifying and clearing effects of EDTA on bone tissue25.
Delipidation
Removing lipids (delipidation) is critical for successful tissue clearing as lipids act as an optical barrier to light and a physical barrier to fluorescent probe delivery. Delipidation can be performed using solvents, detergents or a combination of both, and achieved passively or with the aid of an electric field (Fig.3). Delipidation of hydrogel embedded samples is primarily performed with detergents in aqueous buffer to prevent dehydration of the gel.
Figure 3: Protocols for delipidation.

A summary of methods for delipidating tissue using the most common detergents and solvents. Methodology example represents clearing of a murine brain. CHAPS, (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate; DCM, dichloromethane; SDS, sodium dodecyl sulfate; THF, tetrahydrofuran.
Solvent-based delipidation
Solvent-based delipidation involves submersing the tissue in a series of solutions (Fig.3). First, an initial dehydration step serves to permeabilize cell membranes, initiate some lipid solvation, and enhance tissue bleaching if the final dehydrating agent is combined with peroxides46,47. Dehydration is performed by moving the tissue through solutions with an increasing concentration of a water-miscible organic solvent. This process can cause tissue shrinkage, although increasing the number of steps in the gradient can minimize this effect and better preserve morphology46. Preservation of nominal sample size and morphology is often essential for registration to an anatomical atlas (see below).
Historically, tetrahydrofuran (THF), ethanol or methanol have been used for dehydration, although these reagents have notable disadvantages. Rapid dehydration using THF can cause unwanted morphological changes46. Further, tissues with a high lipid content do not clear well when dehydrated with ethanol as it does not have the same delipidating effect as methanol or THF (see below)48 Similarly, dehydrating tissues with THF, ethanol or methanol will quench fluorescent proteins, as removing water molecules from the chromophore of fluorescent proteins causes a conformational change that eliminates fluorescence emission. Methanol and ethanol quench fluorescent proteins within hours46–48. Fluorescence can be maintained in THF for a few days if the pH remains above 9.0 (as in fDISCO49), dehydration and clearing is performed at 4°C (fDISCO49) and/or by adding butylated hydroxytoluene (sDISCO50). Because of this quenching effect, THF, ethanol and methanol are rarely used to clear tissues expressing fluorescent proteins and these tissues are instead best dehydrated using 1-propanol or tert-butanol (FluoClearBABB51). If tert-butanol dehydration is performed step-wise (uDISCO9), in alkaline conditions (a-uDISCO52) or mixed with Quadrol and polyethylene glycol (PEGASOS42), GFP fluorescence can be preserved for over a month. Methanol, ethanol and THF are still used for dehydration of tissues that do not express fluorescent proteins as synthetic dyes are usually stable in organic solvents. Methanol, ethanol and THF provide the strongest dehydration and delipidation effects, which is important for achieving large tissue transparency. Tissue dehydration can affect the interactions of antibodies with their epitopes, either enhancing or preventing binding, and this effect must be independently tested for each novel antibody used47.
All of the dehydrating agents described above perform some degree of delipidation. However, this is not sufficient to produce a well-cleared final sample. Therefore, an additional incubation in dichloromethane (DCM) ― a water-immiscible solvent ― is performed to complete lipid removal from the tissue53,54. DCM induces the creation of inverse lipid micelles, which diffuse out of the tissue (Fig. 4). Owing to the high immiscibility of water in DCM, care must be taken to ensure that all water has been removed from the sample before submerging it in this solvent.
Figure 4: Mechanisms of delipidation.

(a) The long non-polar tails of ionic detergents such as sodium dodecyl sulfate (SDS) or Triton X100 intercalate with membrane lipids. If the detergent concentration is high enough, large micelles will form consisting of detergent and membrane lipids. (b) Intercalation of membrane lipids by ionic detergents can occur in the presence of a hydrogel mesh. (c) The hydrophobic moiety of zwitterionic detergents such as CHAPS embed into the top of the plasma membrane. If the detergent concertation is high enough, small micelles will form. (d) When tissues are transitioned from an aqueous liquid (polar) to a solvent (non-polar), membrane lipids form reverse micelles in which their non-polar tails are oriented toward the surrounding solvent environment. In all situations, micelles migrate out of the tissue and are washed away. CHAPS, (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate.
Detergent-based delipidation for aqueous-based clearing
As fixation is performed in aqueous buffers supplemented with fixative, these samples can be directly delipified using detergents (Fig.3). The most commonly used detergents are SDS, Triton X-100 and CHAPS. Detergents act by disrupting lipid bilayers and forming micelles that can be transported out of the tissue (Fig.4). Alkaline pH, higher temperature, and longer incubations can all enhance the degree of detergent-based delipidation and result in more chemically and optically permeable tissue. Delipidation buffers generally contain between 4%–10% detergent and are incubated at or above room temperature to prevent the solution from becoming supersaturated. Incubations can take from hours to months depending on the tissue type, size, method of micelle removal and the desired level of clearing5,6,11,24,25,33,55,56.
Detergents other than SDS and Triton X-100 have been used for delipidation, including sodium lauryl sulfate (SLS) in the CLARITY33 workflow, sodium dodecylbenzenesulfonate in CUBIC-HL25, Tween-20 in FocusClear1 and 1,2-Hexandiol25. CHAPS (used in the SHANEL workflow6) has also shown utility in very large samples as it generates smaller micelles than other detergents that can diffuse out of the tissue more quickly (Fig. 4).
Combined solvent and detergent delipidation
Physiological lipids have varying levels of polarity. Some lipids are better removed by detergents, others are better extracted through dehydration and delipidation by solvents. Lipid rich tissues can therefore benefit from the use of both detergents and solvents for delipidation (Fig.3). Adipose tissue, for example, can be delipified by first incubating with 0.1% (v/v) Triton X-100 followed by a methanol dehydration and further lipid extraction with DCM, as shown in the Adipo-Clear57 workflow. Likewise, insufficient delipidation by ethanol and DCM can be compensated for through an initial incubation with CHAPS, a strategy used in the SHANEL6 workflow.
Delipidation using electric fields
The speed of delipidation in large, thick tissue samples is limited by the rate that SDS and lipid micelles diffuse in and out of the sample, respectively. This rate can be increased using electric fields (Fig. 3). Unidirectional electrical fields were first used for this task33 and are now implemented in many commercial electrophoretic tissue clearing (ETC) devices. ETC devices can speed delipidation dramatically, accomplishing in a day what can take weeks by passive diffusion; however, unidirectional electric fields can produce unequal lipid extraction as lipid micelles nearest the anode are forced to traverse through the entire sample. This differential delipidation, along with the electrical force exerted on the tissue, can cause malformation of the tissue during the ETC process. A more efficient way to delipidate a sample is to use a rotating electric field or rotate the sample within a unidirectional field. This process, referred to as stochastic electrotransport, enhances the diffusion of highly electromobile species such as lipid micelles, while keeping species with low electromobility such as other tissue biomolecules relatively stationary58. Hydrogel embedding, and crosslinking of biomolecules of interest to the gel is a requirement for ETC in order to maintain tissue stability and to retain electromobile molecules of interest. The original publications contain design suggestions for ETC33 and stochastic electrotransport58 hardware and both can now be purchased commercially.
Labelling
Extracting biologically meaningful information from transparent tissues requires specific and uniform fluorescent staining of target molecules such as nucleic acids or proteins. Small molecule dyes such as the nuclear dyes DAPI, propidium iodide, RedDot2 and SYTO3,24,33,42 and membrane integrating dyes such as fixable DiI can diffuse or be perfused through biological tissue. Other fluorescent dyes can be targeted to molecules of interest using short peptide tags59 or antibodies. Passive diffusion of small molecules and antibodies is slow and the diffusion time scale correlates quadratically with the tissue thickness; when staining tissue at the organ scale, it can take days for the label to completely permeate to the tissue’s core60.
There are many well documented variables that need to be optimized for any immunostaining protocol including fixation conditions, antibody concentrations, temperature, and time61. These optimizations are best carried out on thin (~200 µm) tissue sections and imaged on a confocal microscope to ensure complete penetration through to the middle of the section. Canonical labelling with antibodies requires the use of two large molecules: a primary antibody that recognizes the molecular target and a secondary antibody that carries the fluorophore24,45,47. Labelling with multiple primary antibodies creates further complication as the optimal conditions for each antibody may be different. Once uniform staining of a thin section has been established, the antibody concentrations can likely stay the same for thicker tissue and the volume of antibody solution is increased. Each antibody also has a unique rate of denaturation that increases with temperature. Highly unstable antibodies must be incubated at lower temperatures and replenished if staining protocols last multiple days.
The largest barrier to scaling up an immunolabelling protocol is that the combination of the slow movement and high affinity of the antibody label results in the aggregation of the antibody at the sample’s surface5. Below, we outline several methods to prevent surface aggregation by reducing the distance the label must travel or temporarily manipulating its affinity for its target. Together, these methods permit more rapid and homogenous labelling of large, cleared tissues.
Reduce sample size
The simplest method to improve labelling speed is to reduce the size of the sample. Before initiating a clearing experiment, the tissue should be trimmed as small as possible. Dehydration can decrease sample volume up to 60% if used, with the rate that the tissue is dehydrated influencing the level of shrinking. Some researchers have embraced this shrinking effect to allow for more rapid labelling and imaging9. It should be noted that shrinking the sample will also decrease the achievable resolution when imaging as some components of the tissue will have moved closer than the diffraction limit of a light microscope62. Reversible shrinkage can be exploited to speed up the labelling process; if a tissue is infiltrated with a tough and elastic hydrogel, the sample can be reversibly compressed to one-sixth its original size31. Labelling can be performed in the compressed state, decreasing the distance that molecular labels must diffuse to their targets. Similarly, high-ionic-strength solutions can be used to temporarily shrink non-elastic tissue during the staining process5. If reversible shrinkage is used for labelling, the sample is returned to its original size prior to imaging to avoid compromising the final image resolution.
Reduce label size
Smaller labels can diffuse faster through a tissue and the use of small molecule fluorescent dyes with affinity for specific molecular targets is advantageous over antibodies when possible. Nanobodies (~15 kDa) are one-tenth the size of a conventional antibody (~150 kDa) and show high tissue penetration and high thermal/chemical stability; these labelled single-variable-domain antibodies from Camelidae or shark species63 have been shown to rapidly diffuse through fixed and cleared tissue, decreasing labelling time up to 5–fold45,60 over antibodies. Although labelling with nanobodies is fast, it should be noted it can also result in a relatively dim fluorescent signal. Standard immunolabelling protocols use polyclonal secondary antibodies, which can bind at multiple regions of a single primary antibody thereby amplifying the fluorescence signal. Nanobody labelling is a single step process were only one nanobody binds to each biomolecule of interest; therefore, no signal amplification occurs. Additionally, commercially available nanobodies are still limited to a small range of targets, although this range is rapidly increasing.
Increase porosity
Lipid membranes in biological tissue prevent the diffusion of imaging agents and immunolabelling is therefore often performed after delipidation. However, certain pre-treatment or delipidation steps may deplete or alter the target of interest, requiring immunolabelling to be performed first. In addition to delipidation by detergents6,23,33,34,64 or solvents6, weak fixation65, partial protein digestion5,65–67 or increasing the pore size of hydrogels can improve label diffusion34. Further, it is possible to harness the natural porosity of tissue for label delivery; for example, transcardial perfusion of fluorescent labels during pre-treatment is an excellent method for labelling whole animals11,34,45. Unfortunately, this perfusion technique is technically demanding and requires large amounts of label.
Manipulate label affinity
The binding affinity of fluorescent labels can be manipulated to allow them to diffuse freely throughout the tissue before their binding properties are reactivated in a way analogous to the SWITCH method described above. For ionic bonding stains such as nuclear stains, high salt concentrations can be used to inhibit binding until homogenous tissue distribution is achieved5,23. Alternatively, antibody binding can be temporarily inhibited (“turned off”) by introducing urea, Quadrol or SDS to a physiological buffer solution, or increasing the temperature or pH5,20,23,55. Once the antibody has distributed itself throughout the tissue, the tissue is returned to the standard buffer solution and binding will be activated (“turned on”). Careful optimization of solute concentration, temperature and pH is required if using this technique as antibodies can undergo irreversible denaturation and lose their binding capability when exposed to the above conditions. Changes in temperature and antibody concentration and/or the addition of Quadrol or urea appear to effect antibody penetration. Conversely, changes to salt and detergent concentrations tend to influence binding efficiency5,23,55. Finally, use of primary and secondary antibody complexes – pre-formed by co-incubating these antibodies prior to applying them to the sample – only requires a single diffusion event and results in a two-fold decrease in labelling time.
Apply an outside force
Certain fluorescent dyes and antibodies are electromobile and stochastic electrotransport can be used to increase the speed that these labels move through a cleared tissue sample20,58. When a SWITCH method is combined with stochastic electrotransport, multiple, rapid rounds of labelling can be performed. This methodology (eFLASH20) enables a highly multiplexed approach to investigating cleared tissues, where tens of target molecules can be investigated in a single sample rather than the standard 3–4. A custom (now commercially available) active immunolabelling device is needed to speed labelling; the device consists of a sample chamber across which an electric field can be applied and the sample or the electric field rotated. Active cooling is required to prevent heat damage to the sample. Another option to further increase the labelling speed is to focus the electrical field by adding magnets to the sides of the sample chamber. This technique, referred to as electromagnetic focused immunohistochemistry (EFIC) or magnetohydrodynamic force (MDH), drives the labels into the tissue and prevents them from flowing around the outside of the tissue68,69.
Refractive index matching
The last step of any clearing protocol involves immersing the sample in a final clearing solution. This solution must match the RI of the remaining biological components ― primarily protein ― and be miscible with the solutions used prior to it. The optimal RI for final clearing should be 1.50–1.60 to best match the remaining protein in the sample after delipidation (with an exception for expand samples, see below). Care must be taken to ensure fluorescent labels are compatible with the chosen solution. Post-fixation in 4% PFA may be required to better retain antibody labels, although this will prevent de-staining and is therefore incompatible with multi-round labelling. A wide variety of solutions have been used and are categorized as aqueous-based or solvent-based; formerly, aqueous solutions better preserved fluorescent protein emissions, whereas solvents better cleared tissue due to their higher RI. These differences are much less pronounced owing to recent advances.
Aqueous-based solutions
Many hydrophilic tissue-clearing reagents have been reported, from simple sugars such as sucrose and fructose to advanced chemistries such as the medical grade GI imaging reagent iohexol2,3,10,22. RI-matching solutions must achieve an RI of 1.50–1.60 in non-expanded samples; however, due to solubility limitations in water, many aqueous solutions of these reagents can only reach an RI of ~1.45. A new generation of aqueous solutions are now available that can match the clearing potential of solvents; these use high-RI chemicals that have high solubility in water, including antipyrine/nicotinamide (CUBIC-R70, RI = 1.52–1.54), N-methylacetamide/Histodenz (Ce3D71, RI = 1.50) and 2,2-thiodiethanol (TDE)/dimethylsulfoxide (DMSO)/iohexol (dPROTOS31, RI = 1.52).
It should be noted that the expansion or contraction of a sample will affect its RI. Aqueously cleared samples do not contract and therefore do not require as high an RI for their final clearing solution (RI~1.52) compared to dehydrated solvent cleared samples (RI~1.56). In the most extreme examples, where hydrogel embedded tissues are expanded 4–20 times, the sample is primarily composed of water and has an RI near 1.33; no specialized clearing solution is needed for these types of expansion methods.
Solvent-based solutions
If samples have been dehydrated and delipidated in solvents, the remaining tissue can be rendered optically transparent by impregnating it with an additional high RI solvent. Formerly, this has included benzyl alcohol benzyl benzoate (BABB) or dibenzyl ether (DBE)53,54. Like the dehydrating solutions described above, BABB and DBE can quench fluorophores contained in the sample. Several approaches can improve fluorescent protein preservation during BABB or DBE incubation, including maintaining a basic pH by supplementing the final clearing solution with triethylamine51 or Quadrol42 and preventing peroxide formation by adding DL-alpha-tocopherol9 or propyl gallate50. Additional fluorescent protein preservation has been achieved through the incorporation of diphenyl ether (DPE9) or polyethylene glycol (PEGMMA42). Supplementing BABB and DBE with the fluorescent protein protectants described above can preserve fluorescence emission for weeks to months in the cleared tissue8,42,50,51.
The toxicity of solvents, particularly BABB, remains a concern for some researchers and multi-user core facilities where cleared samples are often imaged. Ethyl cinnamate and 2,2-thiodiethanol (TDE) are less toxic alternatives that can be used to clear tissue. Ethyl cinnamate has shown equivalent clearing effects and prolonged fluorescent protein emission relative to BABB and DBE when clearing mouse organs72. A 97% TDE water solution results in a similar refractive index to BABB and DBE; however, many fluorescent dyes become unstable in TDE at this concentration73–75. Therefore, TDE is better suited to increase the RI of aqueous clearing solutions when used at concentrations below 70% (v/v) as described above.
Imaging
Cleared tissue samples require imaging techniques capable of optical sectioning [G] over scales of millimetres to centimetres. Some technologies, such as confocal microscopy and 2-photon microscopy, are widely accessible in individual labs and institutional core facilities whereas others, such as light sheet microscopy and optical projection tomography, are more limited. Additional components such as special objectives or sample holders may also be required (see ‘Results – Complications’ below). Table 2 displays a comparison of several microscope modalities commonly used for imaging cleared tissue samples.
Table 2:
Microscopy options for imaging cleared tissue
| Microscopy technique | Best for | Not for use with | Speed | Numerical aperture (NA) rangea | Approximate resolution (lateral, axial)b | Availability |
|---|---|---|---|---|---|---|
| Spinning disk confocal | • Samples < 100 µm thick • Fast imaging |
• Samples > 200 μm (when disk optimized for high NA) | Fast | Up to 1.4 (for < 200 µm working distance oil immersion objectives) | 250 nm, 400 nm | High |
| Point-scanning confocal (inverted) | • Samples < 2 mm thick • Subcellular resolution |
• Samples prone to bleaching | Slow | Up to 0.5 (for long-distance air objectives) | 700 nm, 4 µm | High |
| Point-scanning confocal (upright) | • Samples < 6 mm thick • Subcellular resolution |
• Samples prone to bleaching | Slow | Up to 1.1 with dipping objectives | 300 nm, 750, nm | High |
| 2-photon (upright) | • Samples < 6 mm thick • Subcellular resolution |
• Multicolour (>2) imaging | Slowest | Up to 1.1 with dipping objectives | 300 nm, 750, nm | High |
| Light sheet | • Samples < 6 cm thick • Cellular resolution |
• High resolution/ subcellular imaging | Fastest | Below 0.5 (for low magnification air objectives) | 700 nm, 5 µm | Lower |
| Optical projection tomography | • Samples < 5 mm thick • Isotropic imaging |
• High resolution/ subcellular imaging | Fast | Below 0.1 (for low magnification air objectives) | 3 µm, 3 µm | Lowest |
higher NAs give better resolution
assumes emission light at a wavelength of 550 nm; calculated at highest NA for each modality
Point-scanning and spinning disk confocal microscopes are the most widely available optical sectioning microscopes. Upright point-scanning confocal microscopes equipped with high numerical aperture (NA), long working distance dipping objectives [G] produce the highest quality images of cleared tissue. Inverted point-scanning confocal microscopes are limited in imaging depth and resolution (Table 2) owing to the need to use long working distance air objectives that cause sample-induced spherical aberrations when imaging thick, cleared tissues76. Spinning-disk microscopes allow fast acquisition of 3D volumes, but quickly experience a loss of confocality as they image deeper into tissue and are often optimized for use with high-NA objectives with working distances less than 200 µm. 2-photon excitation is advantageous for imaging cleared tissue as it can extend the imaging depth over standard confocal microscopy for less optimally cleared samples and eliminates out-of-plane photobleaching when imaging large axial volumes. Unfortunately, both point-scanning confocal and 2-photon microscopes are slow and can require tens of seconds to image a single field of view, and imaging samples greater than 1–3 mm3 in size requires a faster approach.
Light sheet microscopes have quickly become the preferred system for imaging cleared tissue. This is primarily owing to their speed, which arises from the perpendicular orientation of the excitation and detection objectives. Cleared tissues are well-suited to light sheet microscopy as the lack of scattering inside the tissue allows light sheets to be stably projected through the tissue and the emitted fluorescence can traverse large spans of tissue to the detection objective without scattering. Light sheet systems can be optimized for high resolution imaging using short, thin excitation sheets or lattice light sheets [G], although these configurations have steric hinderances [G] that prevent the imaging of cleared tissue greater than 100–200 µm in size. Systems can be optimized for larger samples using long, thick excitation sheets and low-resolution (NA < 0.5), large-field-of-view detection objectives (for example, the ultramicroscope48), where the light sheet (5–10 μm) determines the thickness of the optical section. Whenever we refer to light sheet microscopy in this Primer, we refer specifically to these high-capacity systems, which can image a cm3 volume in minutes-hours depending on the desired resolution and sample properties.
Optical projection tomography (OPT) can also be used for imaging cleared samples. By rotating the sample during imaging, OPT can achieve isotropic spatial resolution [G]. OPT achieves a lower lateral and higher axial resolution in comparison to light sheet or confocal microscopes using similar optics. Imaging can be accomplished in minutes to hours if the sample fits in a single field of view. A limitation of OPT is that the sample cannot be larger than a few millimetres in each dimension. OPT has primarily been used for imaging small vertebrate embryos and intact rodent organs that fit these dimensions and only require low-resolution imaging (cellular resolution at best)77,78.
Objectives
Two categories of detection objectives are used for imaging cleared tissue: clearing-specific, high-NA dipping objectives and low-NA air objectives. The decision of which objective to select is sample independent and solely based on the imaging system used and the level of resolution required. Dipping objectives are primarily used on upright point scanning confocal or multi-photon microscopes and are also be found in certain light sheet designs. Dipping objectives for cleared tissue imaging optimally have a high NA (> 0.9), long working distance (> 5 mm), low magnification (< 25x) and correction collars to allow imaging in solutions of varying refractive indexes. These objectives enable sub-cellular resolution imaging. Low-NA air objectives are more commonly used on light sheet systems and are not recommended for use with confocal or multi-photon microscopes as they have poor axial resolution and suffer from sample-induced spherical aberration. Preventing sample-induced spherical aberration requires methods that ensure the path length light travels through air is constant at all axial positions, for example by fixing objectives in place and translating the sample or placing a dipping cap over the objective76.
Data handling
Tissue clearing experiments, especially when performed at scale, produce large amounts of data. For simple experiments such as imaging single fields of view in tissues < 500 µm thick, the microscope’s acquisition computer or a standard desktop will suffice. However, imaging intact organs or animals is data intensive and can require extensive IT investments. Whereas confocal and multi-photon microscopes produce between 5–10 GB of data per hour per colour channel, light sheet microscopes can produce hundreds of GB of data per hour; indeed, a single sample labelled with 2–3 fluorophores and imaged using light sheet microscopy can produce over a TB of data. Researchers aiming to perform large-scale or high-throughput imaging of cleared tissue must carefully consider their IT infrastructure to account for these high rates of data production and it should be noted that the investment in IT infrastructure could equal or exceed the costs associated with purchasing the imaging system itself. The first consideration is whether to procure local or cloud storage. If a local data centre exists, expanding its storage capacity may be more cost effective than renting cloud space. Network upgrades may also be needed as the transfer of datasets 100 GBs to TBs in size is prohibitively slow over a standard 1 Gbit/s network. 10 Gbit/s or higher speeds are preferred. Installing microscopes, data storage and workstations in close physical proximity — preferable on the same network switch [G] — will increase transfer speeds and aid in network stability. If renting cloud storage, researchers may need to purchase dedicated bandwidth between their location and the cloud storage server to ensure uploads occur at practical speeds. It is essential to consider the data output of tissue clearing experiments from the early planning stages and ensure adequate IT infrastructure exists.
RESULTS
At the completion of a tissue clearing experiment, a researcher can expect to obtain a sample through which a fluorescence microscope can image beyond the standard limit of 50–100 µm imposed by light scatter. How much deeper will be a function of the tissue composition and the clearing protocol. It is common for large cleared tissues to retain a yellow tint owing to the continued absorption of blue and near-UV light by the remaining protein in the sample (Fig. 5) and a lack of this yellow colour in large samples may even indicate a loss of protein during the clearing process. The most common qualitative method for determining the effectiveness of clearing is to place the tissue over printed text and attempt to read the words below. A transilluminator with text or a grid printed on a transparent film can allow for better viewing. A more quantitative approach is to remove a > 1 mm section of cleared tissue and view it under a dark-field microscope; when compared to uncleared tissue, the cleared tissue should not be visible under dark-field illumination owing to a lack of light scatter. The most quantitative method is to analyse proton concentration across a sample using MRI; again, the less contrast that is visible, the better cleared the tissue is79.
Figure 5: Examples of the clearing process.

(a) Four pieces of mouse skeletal muscle at various stages of the clearing process. From left to right: muscle extracted from the mouse and fixed in paraformaldehyde (PFA); muscle tissue extracted from a mouse that was transcardially perfused with phosphate buffered saline (PBS) and hydrogel embedded; muscle tissue passively delipidated for 4 days in SDS/boric acid; muscle tissue that has been refractive index matched in an RI = 1.47 aqueous solution. (b) Mouse brains cleared using the CLARITY process. Left: hydrogel embedded sample. Centre: sample that has been delipidated using active electrophoretic tissue clearing (ETC) for 24 hours. Right: Sample after refractive index matching in a 1.47 RI solution for 48 hours. (c) 2mm-thick coronal human brain hemisphere slabs after formalin banking. Scale bars, 1cm. Left: slab before clearing. Right: the same sample after ELAST tissue transformation and RI matching. (d) Mouse whole-organ clearing using the CUBIC process and nuclear staining with propidium iodide. RI matching was performed using the CUBIC R +(N) protocol (RI= 1.522). Scale bars, 5 mm.
The integration of tissue clearing and high-resolution light-sheet microscopy has enabled the volumetric visualization of large biological specimens while simultaneously creating challenges for the subsequent assessment of cellular content. Analysis of cleared tissue imaging data can be intensive and time consuming by eye and requires image analysis routines, algorithms and software. Many open-source and commercial tools now exist in this space. Some general software tools capable of handling large data files include the BigDataBrowser for Fiji/ImageJ80, Napari, Arivis Vision4D, Imaris, and Aivia. Data analysis routines for cleared tissue follow five basic steps: correction of imaging induced artefacts; filtering to improve contrast; segmentation of objects and assessment of segmentation accuracy; registration to anatomical atlases (optional); and quantitative measurements of segmented objects. Quantitative measurements can take several forms, including object counting, volume measurements, distance measurements, determination of colocalization and tracing of filamentous objects such as neurons and blood vessels. We discuss these steps in more detail below, along with details of specialized software tools for each.
Image stitching
Cleared samples will exceed the field of view of most imaging systems. Therefore, imaging of the entire tissue requires the acquisition of multiple Z-stack “tiles” that must be fused together by image stitching. During stitching, neighbouring tiles are translated horizontally and vertically until a best fit if found within overlapping regions of the tiles; it is therefore important to acquire each tile with an overlap of 5–10% with all neighbouring views. This approach serves to correct two common artefacts: the misalignment of objects that cross tile boundaries (caused by sample movement or non-perfect stage translation) and the inhomogeneity of signal intensity across the field of view81,82. Several software tools are available for stitching including the BigStitcher for Fiji/ImageJ83, Arivis Vision4D and Imaris.
Correction of sample-induced artefacts
The two most common imaging artefacts associated with imaging cleared tissue are spherical and chromatic aberration. Spherical aberration occurs when light rays striking the outer portion of a spherical lens do not focus to the same point on the optical axis as the rays that enter at the centre of the lens. Spherical aberration in fluorescence microscopes manifest as a dim, blurry, and axially compressed image. Chromatic aberration occurs if different wavelengths of light fail to focus at the same point of the optical axis. This is the result of light passing through a material with high dispersion [G] and in a fluorescent image manifests as a shift of each colour channel ―most obvious axially ― relative to one another. Microscope objectives contain several lens elements that together reduce spherical and chromatic aberration; however, these lens elements are selected assuming that only immersion and mounting media with specified RI and dispersion values will be used (usually those of water, 80% glycerol or oil). If a clearing solution does not exactly match the RI and dispersion values specified in the objective’s design, sample-induced spherical and chromatic aberrations are produced76. Spherical aberration can be avoided by using an objective specifically calibrated to the RI and dispersion of the clearing solution (see Objectives, above), fixing the objectives in place and translating the sample through the clearing solution, installing a dipping cap over the front lens of the detection objective or using software (available from certain microscope manufacturers or a recent tutorial for correcting spherical abberation76) to correct the axial distortion after acquisition. Chromatic aberration in the axial dimension can be corrected by identifying a fiducial marker (such as a broadband autofluorescent structure or a position where all fluorophores colocalize) and obtaining a short Z-stack from which the axial shift of this singular object can be calculated for each colour channel. Z-stacks for each colour channel can then be offset (confocal) or the beam of a lightsheet microscope can be shifted axially for each colour channel according to this calculation. Alternatively, channel registration software can be used to correct chromatic aberration post-acquisition using a similar approach. If several fiducial markers can be identified, the program can automatically calculate the required axial shift and realign the dataset. Post-processing has the advantage of correcting for lateral chromatic aberrations as well. If the dataset does not contain easily identifiable objects, a tissue phantom comprised of agarose and a sparse concentration (1–30 per field of view) of 200 nm broad-band fluorescent or gold beads submerged in the final RI-matching medium of choice can be used. The required channel offset is identified by imaging the phantom and then applied to the dataset set through the hardware or software methods described above. Fluorescent beads (such as TetraSpeck from ThermoFisher) are preferred; however, they are unstable in most solvents, necessitating the use of gold beads.
Filtering to improve contrast
Microscopy data is inherently noisy and this noise determines how successfully objects of interest can be segmented and analysed in automated routines. Therefore, some degree of filtering is often required prior to segmentation. Because cleared tissue datasets are large, it is important to select filtering techniques with the least computational burden. A simple median filter with a diameter (in pixels) set to the same size as the objects of interest in the data set is often sufficient. Where a median filter is insufficient or the data was acquired with low-NA objectives via confocal microscopy or thick light sheets, deconvolution may be required to eliminate out-of-focus light and improve optical sectioning. Nearly all open source and commercial image analysis software packages offer these methods.
Segmentation of objects
Segmentation refers to the grouping of pixels or voxels from a data set into objects. Once defined, these objects can be 3D rendered free of background noise or measured in numerous ways to obtain quantitative data. The simplest method for segmentation is to choose a single intensity threshold and count all pixels above that value as being part of one or more objects. Any pixels below this threshold are considered background. When applied to an entire image, this method is referred to as global thresholding and can be performed using the open source9,46 and commercial software45,84 packages described above to segment objects in cleared tissue volumes. The most common object segmented in cleared tissue is nuclei. In most tissues, nuclei are spaced far enough apart to permit highly accurate segmentation even when general nuclear stains that label all cells are used7,22. Fig.6 displays an example of single-cell counting in several different mouse organs, which were cleared with the CUBIC-R+ protocol and imaged by light-sheet microscopy; here, over 75 million cells are identified in each organ by using a 3D Hessian-based difference of Gaussian algorithm24. The accuracy of nuclear segmentation can be further improved by only labelling a subset of cells, which can also produce biologically relevant insights when linked to a physiological readout such as identification of active neurons (via c-fos staining) after providing a stimulus to an animal46. Segmentation of individual cell bodies labelled with cytoplasmic or membrane expressed fluorescent proteins is also possible if the fluorophore is sparsely expressed17.
Figure 6: Whole-organ cell profiling using the latest CUBIC-L/R+ protocol.
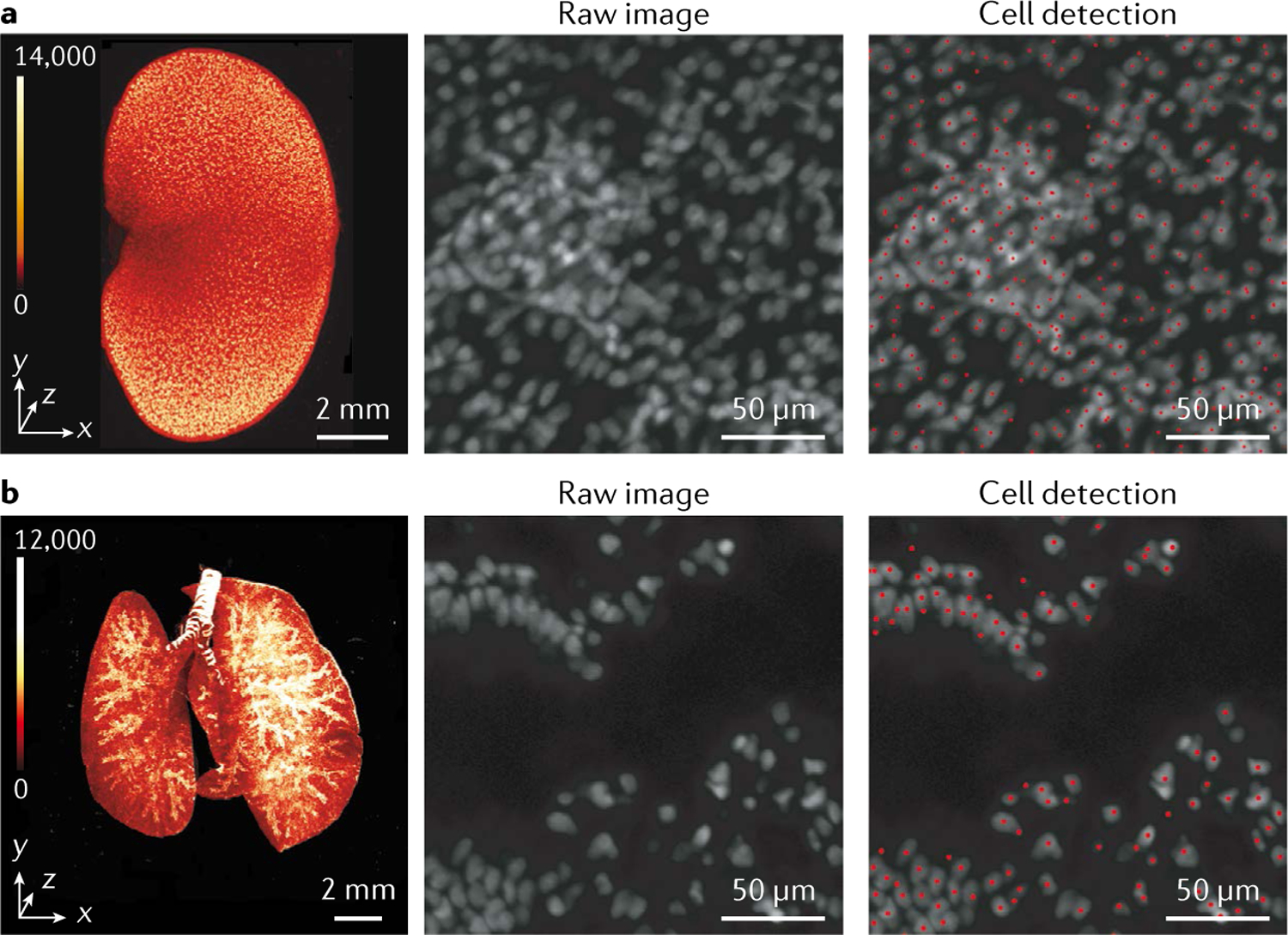
Volume-rendered and single-plane images of mouse organs of 8-week-old C57BL/6N male mice that were cleared using CUBIC-L/R+. (a,b) The organs were stained with propidium iodide (PI) and individual cells were detected using a custom, machine-learning, GPU-based cell detection algorithm with over 90 % accuracy (see ref. 24). (a) Imaging of the mouse kidney identified 79–83 million cells. (b) Imaging of the mouse lung identified 99.3 million cells.
Global thresholding is difficult to perform in large cleared tissue as the signal-to-noise ratio can vary dramatically from region to region within the sample, making selecting a single intensity volume that can accurately separate signal from noise impossible. In recent years, the application of machine learning-based algorithms to segment objects of interest in microscopy images has provided robust methodologies for cellular profiling85. At least two studies have shown that machine learning algorithms are superior to global thresholding when used to detect, segment and quantify cells in large cleared tissues6,86.
Quality control for automated segmentation
It is impossible for a human to assess the accuracy of segmentation by eye throughout a whole dataset. However, it is possible to perform quality control on portions of a dataset to infer the overall accuracy. For example, a researcher could count sparse objects by eye within multiple, small, representative regions throughout the sample. The automated segmentation algorithm can then be run on the same regions and the results compared. Counting by eye quickly becomes impossible as objects become denser and “by hand” segmentation can be used, where a researcher makes annotations on each object on sequential 2D planes by circling or painting with a mouse or pen that are then combined into 3D objects. Again, a series of smaller regions are annotated by hand, counted, and the results are compared to those obtained by automated segmentation. All open source and commercial software programs identified above have annotation functionality. Automated segmentation should be > 90% accurate and preferably > 95% accurate when compared to by-eye or by-hand methods.
When approaching machine learning for the first time, a researcher should determine if pre-trained algorithms already exist for their structure of interest (for example, StarDist87 for detecting nuclei). Due to the unique labelling patterns of individual experiments, it is likely that such resources will not be available and in these cases, multiple user-friendly options exist for training machine learning algorithms using the annotate-by-hand approach described above, including Trainable Weka Segmentation (ImageJ/Fiji), ilastik, Intellisis (Zeiss), Vision4D (Arivis), and Aivia. When using these types of painting approaches for training, it is important to paint the edges of objects so that the algorithm is trained using pixels at object boundaries. At least two objects, or classes, must be trained: “Object1” and background. It should be noted that many of these software packages are not compatible with the large data sets obtained by imaging cleared tissue and custom coding is often required. First, training data must be established by manual annotation, as described above,86 or by creating synthetic training data that mimics the structural features to be imaged88. Enough training data must be produced so that a portion can be reserved to test the algorithm’s ability to achieve satisfactory results with data it has not seen previously. Secondly, a machine learning approach must be chosen. Random forests [G] are the simplest approach and require the least computing power, although they are less flexible than complicated neural networks89. In this approach, the only parameter to set is the number of decision trees, with more decision trees improving the performance of the model but requiring additional computing power and processing time. Neural networks require in-depth understanding to correctly design their architecture including the number of layers, number of neurons in each layer, activation functions, and initialization rates; therefore, an equal or greater number of hyperparameters must also be set for the training algorithm85,90. Therefore, customized neural networks should only be attempted by those with expertise in deep learning and proper testing is essential. A good algorithm can correctly identify objects chosen at random from the training data with few false positives and few false negatives; that is, its precision (correct annotation rate among all the annotations it made) and its recall (correct annotation rate among all the elements in the dataset that should have discovered) are high. Although the manual annotation step can be time-consuming, it is a worthwhile investment. A well-established algorithm facilitates the 3D analysis of large specimens, leading to a better understanding of various biological and biomedical questions86,88.
Quantitative measurements of segmented objects
Quantitative measurements performed in cleared tissue are similar to those performed in volumetric images of non-cleared tissue, although all measurements must be performed in three dimensions and the size of the dataset can cause an analysis routine to exceed the maximum capacity of many current image analysis software programs.
The least computationally complex and most-performed quantitation of 3D imaging data from cleared tissue is object counting. Once objects have been segmented (although this is not a requirement), they can easily be counted by scanning the dataset for areas of intensity maxima. Objects that are easily counted include nuclei23,46 and sparsely labelled cell bodies17. Volume measurements are easily calculated if the X, Y and Z dimensions of a voxel are known and this information is often contained in the metadata of the image file and automatically read by most image analysis software; the volume of an object can be calculated as the number of voxels in the object multiplied by the volume of the voxel. Several distance measurements can also be performed; for example, the centre of mass for each object can be identified and a region-growing technique ― in which a virtual sphere is extended pixel-by-pixel until the sphere contacts the next nearest centre of mass ― can be used to measure the distance between objects in a sample91. If two or more fluorescent labels are used, the region-growing technique can be specified to only detect objects in another channel, providing a readout of the distance between different molecules of interest. In cleared tissue, co-localization measurements usually require segmentation of objects of interest in two or more channels, locating their centres of mass and setting a colocalization limit – the maximum distance two centres of mass can be from one another and still be considered colocalized. This is usually set to 0.5–1x the diameter of the object92. Finally, filament tracing of blood vessels and neurons are popular quantitative measures. Filament tracing often remains semi-automated and requires substantial user input to direct the propagation of these structures through poorly labelled or noisy regions of the image93. Once filaments have been segmented, several measurements can be performed, including length, volume, orientation, tortuosity [G] and number of branch points. See the Applications section below for more detailed descriptions of common applications for each of these quantitative measures.
Registration to tissue atlases
Atlases are three dimensional maps that are used to delineate different anatomical or functional regions of an organ. In complex organs, the boundaries between these regions require hundreds of labels to be visualized94. As incorporating these labels into an experiment is currently not achievable, determining the exact region of an organ in which a fluorescent signal resides requires registering the 3D dataset to an existing atlas24,38,95–104. Atlases are used extensively in the analysis of cleared tissue 3D datasets; for example, several studies have mapped data from multiple cleared brains onto the Franklin and Paxinos Atlas17, the Waxholm Space Atlas23,100 and the Allen Brain Atlas46. Open-source software for performing these types of registration are available including ClearMap46 and large deformation diffeomorphic metric mapping (LDDMM)105–107. When mapping data to an atlas there are two key considerations. First, the dataset must contain a fluorescence channel with a similar fluorescence intensity across the entire organ, most often a channel containing tissue autofluorescence or a general nuclear dye. Second, the resolution of the sample image must be similar to the resolution of the images in the atlas. A limitation of many established atlases is that their regional-level resolution, often developed using MRI or CT108,109, is not sufficient for registering the cellular resolution of microscopy data. To address this, a single-cell resolution atlas was developed for the mouse brain (CUBIC-Atlas) along with a cloud-based framework to perform 3D analysis at single-cell resolution (CUBIC-Cloud)104. High-resolution reference atlases of entire organisms are needed to assist whole-body clearing methods. Currently, manual annotation86 of individual organs is being replaced with deep-learning-based organ segmentation110; this approach has potential to transform the analysis of whole-body scans, eventually leading to the generation of more accurate mouse body atlases.
Required computing specifications
High-end workstations are needed to perform the above analyses. For optimal performance, data must be rapidly accessible, ideally in a solid state drive (SSD); if SSDs are cost prohibitive, multiple hard disk drives (HDDs) in a RAID array can suffice. Large amounts of RAM (> 200 GB) can reduce the need for disc access, which is usually an image analysis bottleneck. Finally, the amount of GPU memory and number of GPU processing cores are also important. For image analysis routines that can be GPU accelerated such as CLIJ111, DeepMACT86, TubeMap112 and others, these specifications will determine how rapidly the processing can be completed. Additionally, GPU memory will determine the resolution limits when displaying 3D data. For example, when running a deep learning-based algorithm such as DeepMACT, a workstation with 64 GB RAM and a 12 GB Nvidia Titan XP GPU is capable of processing a series of whole-mouse images in 45 min86. Workstations equipped with 512 GB RAM and a 24 GB NVIDIA Quadro P6000 GPU have been used to process the TubeMap algorithm112.
APPLICATIONS
Tissue clearing now provides a viable alternative to serial sectioning of large biological samples. In comparison to sectioning, tissue clearing is often a lengthier process owing to the requirement for incubations in various solutions and the additional time required for diffusion of labels; however, these incubations require very little hands-on time and no specialized skills. Clearing protocols that do not require specialized equipment allow for a high degree of parallelization that cannot be achieved with a single vibratome or cryostat. Further, tissue clearing avoids mechanical damage that can occur within the tissue during sectioning and the heavy computational burden of aligning, de-warping and assembling hundreds or even thousands of 2D images into a 3D rendering. Analysis of intact, cleared samples shows improved accuracy compared to quantitative stereology113 and traditional 2D histopathology114. Finally, clearing allows for studies across entire organs (such as long-distance nerve tracing17) or entire organisms (such as identification of cancer metastasis7,86) that cannot be accomplished through serial sectioning.
Biologically, the applications of tissue clearing methods are wide-ranging. Tissue clearing has primarily shown use in the fields of neuroscience, oncology and cardiology, although nearly every organ and organ system in rodents has now been cleared, including the brain, intestine, spleen, lymph node, heart, kidney, lung, eye, and bone7,10,11,27,34,43–45,70,71,110. Other model and non-model systems such as fish and salamanders have also been used15,115,116. The ever-increasing size of in vitro models such as spheroids and organoids ― now many millimetres in size117 ― necessitates the use of tissue clearing to image them in their entirety. Finally, methods to clear living samples and enhance imaging depth for in vivo studies now show promise.
Applications in neuroscience
In many organisms including mice and humans, neurons are the largest cells in the body with processes up to several centimetres long. Their large size requires that they be investigated in complete volumes spanning a whole organ or even organism, and for most animals tissue clearing or whole animal sectioning118 is required (aside from in the most translucent aquatic creatures, as demonstrated in a recent pre-print article119). Nervous system tissues are abundant in lipids and relatively lacking in pigment and extracellular matrix proteins such as collagen and elastin, making them ideally suited to clearing as once the lipid is removed, only a sparse protein matrix is left behind. Solvent-based and aqueous-based techniques have both6,57 proven successful in clearing tissues of the nervous system; however, techniques that combine both may be best suited for regions of high lipid density such as the spinal cord and myelinated axon bundles in the brain.
A major goal in neuroscience is to dissect the complete wiring diagram of the brain, a field known as connectomics. Even after tissue clearing, the density of neurons remains beyond the resolution limit of fluorescence microscopes120 and current practice is to create maps of inter-areal projections by sparsely labelling collections of neurons121,122 (Fig.7). Several techniques exist to sparsely label subsets of neurons throughout the brain, including genetically encoded fluorescent proteins under the control of a cell-type specific promoter and immunolabelling with known markers of specific cell types. Expression of fluorescent proteins may involve a single fluorescent protein per cell type or use combinatorial expression of multiple fluorescent proteins to enable the identification of hundreds of individual cells (for example Brainbow123,124 and other similar approaches). Transgenic animals can be produced expressing fluorescent proteins or they can be expressed by infecting cells with a virus carrying DNA encoding a fluorescent protein. Rabies and AAV viruses are commonly used for transducing cells of the nervous system; for example, a combination of AAV viruses were directly injected into the brains of mice in order to determine patterns of dopamine neuron innervation in the midbrain17. A less invasive approach is to use AAV viruses that can be injected into the blood stream and infect specific cell types of the brain18. To aid in the analysis of sparsely labelled neurons in cleared tissue volumetric, neuronal tracing algorithms have been developed that can perform manual or semi-automated segmentation93. Most promising are new, fully automated algorithms such as NeuroGPS-Tree, which has traced over 1000 neurons in thick, cleared tissue sections125, and a software package called BIRDS that uses deep learning to perform long-distance neuronal projection tracing and parallel counting of cell populations in different brain regions126
Figure 7: Example of neuronal staining in the mouse brain.

(a) 3D rendering of an entire mouse expressing GFP downstream of the thy1 promoter122 in a subset of cells. The brain was cleared using a CLARITY protocol. Major fibre tracks are clearly visible. (b) Zoomed cortical region of the brain shown in panel a. Individual neuronal processes can be visualized. (c) Brainbow-expressing neuromuscular junctions in mouse skeletal muscle. Muscle was cleared using a passive hydrogel-embedding method. Each neuron expresses a unique ratio of blue, green and red fluorophore, allowing for the observation of individual neurons.
Tissue clearing has also been used to map areas of brain activity after an organism performs a behaviour or is administered a stimulus. Brain activity can be inferred by immunostaining for the protein products of immediate-early genes such as c-fos. If c-fos is present in a neuron, this indicates it was recently activated. Using this method, brain region activity mapping was accomplished in cleared mouse brain, harvested after sensory whisker stimulation or observed parenting behaviours in mice46.
Finally, tissue clearing shows potential to aid in the investigation of neuronal pathologies. The SWITCH clearing and immunolabelling protocol was recently used to show that amyloid-beta peptides (Aβ) that contribute to the onset and progression of Alzheimer’s dementia can be labelled and imaged in a brain-wide manner127. This methodology allowed for microscopic mapping of Aβ plaques and identified several novel sub-cortical hubs where Aβ accumulates.
To date, tissue clearing of brains has primarily focused on rodent tissue as human tissue presents several barriers to tissue clearing, including enhanced autofluoresence, over-fixation from long-term preservation and large physical size. Recently, the SHANEL and ELAST tissue clearing protocols have been adapted to overcome many of these barriers and permit investigation of large brain slabs or even whole brain6,31. Numerous exciting applications exist in this domain and we refer readers to a recent review for further applications3.
Applications in oncology
Whole animal clearing using aqueous-based (CUBIC) and solvent-based (vDISCO) techniques has enabled the visualization of micro-metastases throughout an entire rodent at single-cell resolution7,86. Additionally, by fluorescently labelling a therapeutic antibody, it is possible to detect the percentage of metastatic sites receiving treatment across the entire animal86 (Fig.8).
Figure 8: Representative results of vDISCO panoptic imaging.

a, 3D rendering of a Thy1-GFPM mouse after vDISCO whole-body immunolabelling/clearing and imaging by light-sheet microscopy. Neuronal fibres expressing GFP are enhanced by anti-GFP nanobodies conjugated with Atto647 dye and shown in green, bones and internal organs highlighted by propidium iodide are in cyan, and the background signal — mainly from autofluorescence of the muscle tissue — is in white. b and c, Zoomed-in images of the boxed regions in panel a, showing detailed neuronal structures with sub-cellular level resolution. d, 3D reconstruction of a vDISCO-processed NSG mouse implanted with MDA-231 human breast cancer cells in the mammary fat pad for approximately 2 months, followed by intravenous injection of an anti-human CA12 therapeutic antibody named 6A10 which is conjugated with Alexa-568 dye. The cancer cells were previously transduced by mCherry viral vectors and further enhanced by anti-mCherry nanobodies conjugated with Atto647 dye. The 6A10-targeted primary tumour (pointed by the white arrowhead) and metastases are shown in yellow, untargeted metastases are in red, and selected organs including the brain, lungs, liver, kidneys are manually segmented and shown in cyan. e and f, enlarged view of the boxed regions in d showing greater details of the distribution of micrometastases and targeting efficacy of the 6A10 therapeutic antibody.
In the field of cancer pathology, there is interest in determining whether three-dimensional samples can provide a more accurate diagnosis or prognosis than traditional two-dimensional surgical sampling techniques. A form of expansion microscopy, termed ExPATH, suggested that imaging of cleared biopsies from breast cancer patients was able to discriminate between early breast neoplastic lesions better than surgical sampling114. Large (30 × 20 × 5 mm) surgically resected breast tumours were cleared in less than 48 hours using the PathoDISCO solvent-based clearing technique128; in PathoDISCO the key step is using 2,2-dimethoxypropane to react with water to create acetone and methanol, which rapidly dehydrates the tissue.
The microenvironment that surrounds a tissue is highly heterogeneous and influences its growth and development129. Hydrogel embedding protocols have been shown to preserve important elements of the microenvironment surrounding developing breast tissue and allow for its investigation in 3D130. In this manner, the CUBIC tissue clearing method revealed the role of TGF-β in the tumour microenvironment by profiling multicellular interactions in a mouse model of experimental lung metastasis.
Interestingly, studies investigating the administration of novel, non-lipid-based nanoparticles ― which are being investigated as potential delivery vehicles for therapeutics ― showed both solvent-based131 and aqueous-based132,133 techniques allow for the retention of these nanoparticles in the tissue. These studies demonstrated that tissue clearing can be used for measuring the biodistribution of novel cancer therapeutics.
Applications in cardiovascular research
Blood vessels are easily labelled prior to clearing through the perfusion of labelling reagents. Multiple labels have been used, including antibodies targeting molecules that line the vasculature134, wheat germ agglutinin and Evans blue88, or lipophilic dyes135. It is important that any label contain a moiety that can react with fixatives such as PFA or else it will be washed away during subsequent clearing steps. Fig.9 displays several cleared tissues in which blood vessels have been stained. Tissue clearing allows for the study of vessel repair and remodelling during development or after injury in any organ throughout the body; of particular interest are studies of blood vessel rupture in the brain in the case of aneurysm and stroke, and infarctions of the heart136. Cardiovascular studies investigating blood vessel structure and function are now aided by several semi-automated vasculature segmentation, and quantification methods have been developed in commercial137 and custom138 software to analyse small regions of interest (ROI) in cleared mouse brains. These methods are not sufficient to reliably detect capillaries with variable signal to noise ratio (SNR) in different brain regions or to systematically analyse the vascular alterations at the whole brain or organ scale88,112. Machine learning-based strategies have overcome these hurdles and currently two machine learning-based methods (VesSAP88 and TubeMap112) are available to reliably detect, segment and register images of whole-brain vasculature to the Allen brain atlas. Recently, tissue clearing has been used to map the parasympathetic and sympathetic circuits that control heart rate in mice139, produce a single-neuron-scale map of the intrinsic cardiac nervous system of the rat140, and quantify sympathetic hyperinnervation of murine hearts after infarction141.
Figure 9: Example of vascular staining in the mouse brain.

(a) 3D render of an entire mouse brain that was perfused with CM-DiI lipophilic dye prior to fixation. After fixation, the brain was cleared using a modified iDISCO protocol and imaged via light sheet microscopy. (b) Zoomed region of brain on left showing the level of detail in vasculature staining.
Non-murine applications
Aquatic organisms
Genome modification technologies such as CRISPR have enabled biological investigation in species beyond traditional model systems. Clearing protocols must be adapted to each species as many express unique pigments throughout their tissues that aid in protecting them from dangerous UV radiation (such as protection of the haematopoietic niche by melanin in fish)142. Many pigments such as melanin, ommochromes and pterins are resistant to hydrogen peroxide bleaching and do not elute with heme in amino alcohols. The DEEP-Clear method has shown that combining hydrogen peroxide, THEED and acetone can bleach or remove most of the pigments from annelids, molluscs, bony fish and tetrapods15. Others have shown that the addition of decalcification and bleaching steps render crustaceans clear and may enable the creation of developmental atlases with single-cell resolution116. Finally, an enzymatic treatment with collagenase was added to a customized tissue clearing protocol for flatfish to show that thyroid hormone is involved in their eye migration during metamorphosis115.
Plants
The plant wall imposes an additional RI mismatch that is not present in mammalian tissue and must be addressed. Additionally, chloroplasts acts as a pigment absorbing blue and red light and generates fluorescence during imaging, which may or may not be desired depending on the experiment. Plant tissue has historically been cleared using a single solution of sodium hydroxide and chloral hydrate that breaks down cell walls and equilibrates the RI throughout the sample143. Urea144, xylitol powder and sodium deoxycholate145 and NaClO146 have been used as less-toxic alternatives to chloral hydrate. Interestingly, the general principle of RI matching has even been used to create transparent wood with the potential to replace glass as a building material147.
Humans
Three-dimensional imaging of clinical samples could have additional diagnostic potential over two-dimensional histopathology investigations148; however, imaging these samples is difficult owing to over fixation, high levels of autofluorescence, the need for exogenous fluorescent labels and the overall high density of tissue from older patients. For very dense tissue, images of routine, thin, pathology tissue sections can be improved via expansion microscopy114. For larger samples ― including intact human organs ― the use of relatively small detergent (CHAPS) and solvent molecules for delipidation are needed overcome the density of human tissue6. Elasticizing human tissue has been shown to improve probe delivery when the tissue is stretched, while simultaneously providing better preservation over time to allow for multiple rounds of labelling31. Conversely, human embryonic and foetal tissue is less dense and far easier to clear. Solvent-based clearing methods have been used to clear human embryos and foetuses from gestational weeks 6–14149.
In vitro applications
Tissue clearing protocols have become essential for imaging spheroids and organoids in their entirety as traditional light microscopy approaches cannot image more than 100–200 μm beyond their surface. Even the smallest organoids (on the scale of 50–100 µm) can benefit from simple RI matching. Human airway, colon, kidney, liver and breast tumour, and mouse mammary gland organoids of this size have been cleared with simple RI matching in high-concentration sugar solutions150. More complex tissue clearing methods are needed for larger and denser organoids such as 3D neuronal spheres (200–300 µm diameter)151 and retinal organoids152. Finally, for organoids many mm in diameter, the SCOUT protocol has been used to clear, label, image and correlate hundreds of phenotypes between control and experimentally treated organoids117. SCOUT was able to identify several effects of Zika infection on cerebral organoids approximately two millimetres in diameter at the single-cell level117.
In vivo and label-free applications
The modular tissue clearing methodologies described in this Primer are targeted toward fixed, non-living tissues stained with fluorescent labels. Although not the focus of this article, researchers have demonstrated clearing strategies that increase the imaging depth for several optical technologies in in vivo samples, including single-photon and multi-photon fluorescence, second harmonic generation (SHG) and optical coherence tomography (OCT) (reviewed in 153). To date, in vivo methods have primarily focused on the topical application of biocompatible high-RI solutions to the skin154–158, skull159 and superficial tumours160,161 to provide increased imaging depth in both animal and human systems. As fluorescent labelling of live human tissue can be difficult owing to the inability to use genetic modification, human samples are often imaged by label-free techniques such as SHG and OCT. The ability to perform deep, label-free imaging in vivo is intriguing as it may hold clinical potential for the diagnosis and understanding of certain pathologies such as tumour biology.
Applications for systems biology
Systems biology generally entails analysing hundreds to thousands of targets within a tissue. Often, labelling is the limiting step as it requires substantial time and. although studies exist where tens of organs17 have been imaged, it is more practical to perform systems-level investigations on smaller samples. Tissue clearing can be used on this smaller scale; for example, spatial transcriptomic techniques like MER-FISH and seqFISH use imaging to detect thousands of mRNA species in a sample162,163 and tissue clearing has been adopted with these techniques to enable three-dimensional spatial transcriptomics. Incorporation of tissue clearing with the MER-FISH technique allowed a read out of the positions of thousands of mRNA molecules in each cell of a tissue sample in 3D164.
REPRODUCIBILITY AND DATA DEPOSITION
The reliability and reproducibility of tissue clearing protocols has dramatically improved in recent years. Solvent-based clearing methods now generally use chemical components that are affordable and effective. Aqueous-based techniques have been aided by commercially available equipment for hydrogel polymerization and ETC. Further, the availability of commercial pre-formulated clearing solutions has allowed for standardization of clearing protocols between research groups.
Fluorescent labelling in tissue clearing experiments still primarily relies on antibodies. Commercially available antibodies are prone to reproducibility issues caused by batch-to-batch variation, which are more apparent (specifically background caused by off-target binding) when labelling large tissues. Antibodies should be carefully selected based on past results and validated on thin, uncleared tissue sections. An online repository for tissue clearing-validated antibodies can also be used.
Many journals and funding bodies encourage the deposition of raw data in publicly accessible searchable databases. Recently, imaging data repositories such as the Image Data Resource and the Cell Image Library have been made available165 and authors are encouraged to deposit their raw data in a publicly accessible database if possible. In addition to raw images, essential metadata should be reported to ensure reproducibility and set expectations for a researcher who would like to replicate a tissue clearing experiment (Box 1).
Box 1: Suggested metadata to report when preparing and imaging cleared tissue.
Approximate dimensions of the sample being cleared.
Composition of all clearing solutions used, duration of incubation in each solution and the purpose/function of each solution.
Degree of expansion or contraction of the sample.
Antibody manufacturer, lot number, concentration, incubation temperature and incubation duration.
Imaging modality used.
Numerical Aperture (NA) of imaging objective.
Light sheet thickness (if used).
Voxel size of raw images.
Total raw data size.
A summary of all data processing steps (SA correction, chromatic correction, filtering).
Description of data quantitation methods.
Deposition of any custom software in an open source depository.
A subset of manually annotated ground-truth data to assess accuracy of automated data quantitation.
Studies that involve clearing tens or hundreds of samples may produce hundreds of TBs or even PBs of data, and public image repositories are limited in the amount of data they can store. Additionally, available bandwidth and a researcher’s proximity to the repository’s server can prevent uploads from occurring in a reasonable amount of time. Powerful lossy and non-lossy compression algorithms such as JPEG XS, AVIF, and WebP can reduce data size with little reduction in image quality; these provide better quality and compression than using .jpeg or.png files.
LIMITATIONS AND OPTIMIZATIONS
There are some outstanding limitations of clearing techniques. First, the methods described in this Primer are most often applied to fixed tissue as in vivo applications of tissue clearing are still limited153. Second, several factors restrict the throughput of tissue clearing experiments such as the limited sample capacity in tissue clearing equipment, high cost of antibodies and clearing solutions, the long time required for immunolabelling, lack of common conditions for antibody binding, sample capacity limitations in imaging devices, and limitations in the IT infrastructure for data storage and analysis. Although one could imagine developing an automated pipeline for clearing, labelling and imaging, this would require an extensive investment in parallel equipment for sample preparation and imaging. Third, imaging of large samples requires microscope objectives with equally large working distances. Most imaging systems designed for cleared samples use moderate-NA, low-magnification, long-working-distance objectives from zoom microscopes. In general, increasing the working distance of an objective decreases its NA and therefore its achievable resolution81. The goal of tissue clearing experiments is primarily to survey a large tissue or organ; however, if subcellular resolution is also required, secondary imaging of select areas may be better suited to a second, higher resolution imaging device. Finally, the maximum size of samples that can be cleared now exceeds the maximum sample size able to fit under a microscope6,113. Before investigation of human organs can be fully realized, imaging devices able to accommodate samples of this size must be developed. It is important to note that every tissue is different and researchers should not automatically follow a previously published method; for example, protocols that clear high lipid content tissue such as brain are not optimal for pigmented, protein-rich kidney tissue. By viewing tissue clearing protocols as modular, a researcher can select and assemble an optimal pipeline for clearing their tissue of interest.
OUTLOOK
The past decade has seen dramatic innovations in tissue clearing methods. Clearing solutions are no longer discovered serendipitously; researchers with extensive knowledge of optical physics, chemistry and tissue engineering are designing tailored solutions that improve on past methods. A decade ago, tissue clearing techniques were complex, unreliable and each had obvious limitations. Now, through incremental improvements, both aqueous and solvent-based techniques are able to preserve emission from fluorescent proteins and dyes and their final clearing solutions have coalesced around an ideal RI of 1.52–1.56. Commercially manufactured systems for steps such as hydrogel polymerization and electrophoretic tissue clearing and labelling are now available from multiple manufacturers, as are microscopes and objectives that have been designed specifically for cleared tissue.
Barriers do remain despite the positive outlook. The use of genetically encoded fluorescent proteins is not always possible and immunolabelling techniques must still be used in many systems. No matter how quickly antibodies are delivered to their target, if the antibody is not specific or stable, the result will be a failed experiment. Further, the current variability in ideal labelling conditions between individual antibodies makes it unlikely that a researcher’s unique panel of probes will all function under identical conditions. We hope that nanobodies will be able to solve the many variability and stability issues of antibodies, although they are not without limitations. First, the random attachment of a fluorophore to a nanobody can block its binding site, necessitating directed attachment of the fluorophore with genetically encoded click chemistry166,167. Second, fewer fluorophores can be attached to a nanobody than an antibody owing to its small size, providing a weaker signal. Third, nanobodies are primarily monoclonal and can be directly conjugated to a fluorescent dye. Therefore, polyclonal secondary antibodies that normally produce substantial fluorescent signal amplification are not used. Amplification strategies that are widely used in the single-molecule-RNA FISH field such as SABER168 and HCR169 could be adopted for nanobodies (as they have been for antibodies170) to alleviate the above limitations. Single chain variable fragments (scFvs) - small immunolabels known to penetrate faster through biological tissue than nanobodies - have seen little application in tissue clearing to date but may be a promising technology for labelling large samples171.
As tissue clearing experiments become commonplace, deep biological insight will follow. It is difficult to imagine a field of biology that will not benefit from the ability to investigate the 3D relationships between all components of a tissue. The recent clearing of entire, rhesus monkey172 and human brains6 has shown that clearing methods are outpacing imaging technology. Labs looking to image on this scale will need to partner with industry to design and build objectives and optical systems with working distances of tens of centimetres that can still achieve reasonable lateral and axial resolution. There can be no doubt that the democratization of tissue clearing has begun and exciting biological insights are sure to follow.
Acknowledgements
We thank K. Matsumoto and S.Y. Yoshida for help constructing Figures 5 and 6, respectively and Erin Diel and Ian Boothby for preparing samples in Figures 5, 7 and 9 This work was supported by a Japan Science and Technology Corporation (JST) Exploratory Research for Advanced Technology (ERATO) Grant (JPMJER2001). H.R.U. was supported by the Science and Technology Platform Program for Advanced Biological Medicine (AMED/MEXT), a Japan Society of the Promotion of Science (JSPS) KAKENHI grant-in-aid for scientific research (JP18H05270), a Grant-in-Aid from the Human Frontier Science Program and a MEXT Quantum Leap Flagship Program (MEXT QLEAP) Grant (JPMXS0120330644). K.M. was supported by a JSPS KAKENHI grant-in-aid for scientific research (20K06885) and a JST Moonshot R&D Grant (JPMJMS2023). A.E. was supported by the European Research Council (ERC) Calvaria project, the Vascular Dementia Research Foundation and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy, ID 390857198). K.C. was supported by a Burroughs Wellcome Fund Career Awards at the Scientific Interface, the Searle Scholars Program, the Packard award in Science and Engineering, the NARSAD Young Investigator Award, the McKnight Foundation Technology Award, the JPB Foundation (PIIF and PNDRF), the Institute for Basic Science (IBS-R026-D1) and the National Institutes of Health (NIH) grants (1-DP2-ES027992) and (U01MH117072). J.W.L. is supported by the NIH grants U19NS104653 and P50MH094271. Resources that may help enable general users to establish the methodology are freely available online at http://www.chunglabresources.org.
Glossary
- Autofluorescence
Fluorescence arising from endogenous fluorescent molecules contained within a biological specimen. Can also be introduced exogenously (i.e. some hydrogels autofluorescence).
- Dipping objectives
Objective lenses that are designed to be submerged into a liquid. Dipping objectives are found on upright microscopes and samples are mounted without a coverslip.
- Optical section
An image of a 2D plane within a 3D object that is derived by optical, rather than mechanical, means.
- Lattice light sheet
A light sheet that is formed using a specialized interference pattern that results in the projection of thin beams of excitation light into a sample, which are rapidly dithered to form the light sheet.
- Steric hindrances
Refers to the inability to mount a sample on a microscope when the objective’s working distance is shorter than the thinnest dimension of the sample.
- Isotropic spatial resolution
Refers to specialized light microscopy techniques that produce an identical lateral and axial resolution
- Network switch
A computer network hardware device that allows multiple computers to communicate.
- Dispersion
A measure of the change in refractive index relative to the wavelength of light passing through a substance. If a substance has high dispersion, it means blue light and red light will refract differently when passing through it.
- Random forest
A machine learning algorithm that is comprised of many “estimators” that each make a prediction as to which segmentation group a pixel should belong. When many estimators are combined into a “forest” the final prediction is highly accurate.
- Tortuosity
Describes the degree of curvature/twist in a blood vessel.
Footnotes
Competing interests
H.R.U. is co-founder of CUBICStars Inc and a co-inventor on the following patent applications covering the CUBIC reagents: PCT/JP2014/070618 (pending, patent applicant is RIKEN, other co-inventors are E. A. Susaki and K. Tainaka); PCT/JP2017/016410 (pending, patent applicant is RIKEN, other co-inventors are K. Tainaka and T. Murakami). K.C. is an inventor for patent applications covering some technologies described in this paper and co-founder of LifeCanvas Technologies. A.E. and C.P. have filed a patent on whole-body clearing and imaging related technologies.
Peer review information
Nature Reviews Methods Primers thanks Alan King Lun Liu, Woong Sun, Valery Tuchin and the other, anonymous, reviewers for their contribution to the peer review of this work.
Related links
Napari: https://napari.org/
Arivis Vision4D: https://imaging.arivis.com/en/imaging-science/arivis-vision4d
Imaris: https://imaris.oxinst.com/
Aivia: https://www.aivia-software.com/
Trainable Weka Segmentation: https://imagej.net/plugins/tws/
ilastik: https://www.ilastik.org/
Online repository for tissue clearing-validated antibodies: https://idisco.info/validated-antibodies/
Image Data Resource: http://idr.openmicroscopy.org/about/
Cell Image Library: http://www.cellimagelibrary.org/home
REFERENCES
- 1.Richardson DS & Lichtman JW Clarifying Tissue Clearing. Cell 162, 246–257, doi: 10.1016/j.cell.2015.06.067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tainaka K, Kuno A, Kubota SI, Murakami T & Ueda HR Chemical Principles in Tissue Clearing and Staining Protocols for Whole-Body Cell Profiling. Annual review of cell and developmental biology 32, 713–741, doi: 10.1146/annurev-cellbio-111315-125001 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Ueda HR et al. Tissue clearing and its applications in neuroscience. Nature Reviews Neuroscience 21, 61–79, doi: 10.1038/s41583-019-0250-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park YG et al. Protection of tissue physicochemical properties using polyfunctional crosslinkers. Nature biotechnology, doi: 10.1038/nbt.4281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Susaki EA et al. Versatile whole-organ/body staining and imaging based on electrolyte-gel properties of biological tissues. Nature Communications 11, 1982, doi: 10.1038/s41467-020-15906-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao S et al. Cellular and Molecular Probing of Intact Human Organs. Cell 180, 796–812.e719, doi: 10.1016/j.cell.2020.01.030 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubota SI et al. Whole-Body Profiling of Cancer Metastasis with Single-Cell Resolution. Cell Rep 20, 236–250, doi: 10.1016/j.celrep.2017.06.010 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Lee E et al. ACT-PRESTO: Rapid and consistent tissue clearing and labeling method for 3-dimensional (3D) imaging. Scientific reports 6, 18631, doi: 10.1038/srep18631 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan C et al. Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nature methods 13, 859–867, doi: 10.1038/nmeth.3964 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Susaki EA & Ueda HR Whole-body and Whole-Organ Clearing and Imaging Techniques with Single-Cell Resolution: Toward Organism-Level Systems Biology in Mammals. Cell Chem Biol 23, 137–157, doi: 10.1016/j.chembiol.2015.11.009 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Tainaka K et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell 159, 911–924, doi: 10.1016/j.cell.2014.10.034 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Messal HA et al. Antigen retrieval and clearing for whole-organ immunofluorescence by FLASH. Nature protocols 16, 239–262, doi: 10.1038/s41596-020-00414-z (2021). [DOI] [PubMed] [Google Scholar]
- 13.Pende M et al. High-resolution ultramicroscopy of the developing and adult nervous system in optically cleared Drosophila melanogaster. Nat Commun 9, 4731, doi: 10.1038/s41467-018-07192-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindsey BW, Douek AM, Loosli F & Kaslin J A Whole Brain Staining, Embedding, and Clearing Pipeline for Adult Zebrafish to Visualize Cell Proliferation and Morphology in 3-Dimensions. Front Neurosci 11, 750, doi: 10.3389/fnins.2017.00750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pende M et al. A versatile depigmentation, clearing, and labeling method for exploring nervous system diversity. Science advances 6, eaba0365–eaba0365, doi: 10.1126/sciadv.aba0365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deverman BE et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nature biotechnology 34, 204–209, doi: 10.1038/nbt.3440 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menegas W et al. Dopamine neurons projecting to the posterior striatum form an anatomically distinct subclass. Elife 4, e10032, doi: 10.7554/eLife.10032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravindra Kumar S et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nature methods 17, 541–550, doi: 10.1038/s41592-020-0799-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hopwood D The reactions between formaldehyde, glutaraldehyde and osmium tetroxide, and their fixation effects o bovine serum albumin and on tissue blocks. Histochemie 24, 50–64, doi: 10.1007/BF00310003 (1970). [DOI] [PubMed] [Google Scholar]
- 20.Murray E et al. Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems. Cell 163, 1500–1514, doi: 10.1016/j.cell.2015.11.025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins JS & Goldsmith TH Spectral properties of fluorescence induced by glutaraldehyde fixation. J Histochem Cytochem 29, 411–414, doi: 10.1177/29.3.6787116 (1981). [DOI] [PubMed] [Google Scholar]
- 22.Ueda HR et al. Whole-Brain Profiling of Cells and Circuits in Mammals by Tissue Clearing and Light-Sheet Microscopy. Neuron 106, 369–387, doi: 10.1016/j.neuron.2020.03.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Susaki EA et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739, doi: 10.1016/j.cell.2014.03.042 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto K et al. Advanced CUBIC tissue clearing for whole-organ cell profiling. Nature protocols 14, 3506–3537, doi: 10.1038/s41596-019-0240-9 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Tainaka K et al. Chemical Landscape for Tissue Clearing Based on Hydrophilic Reagents. Cell Reports 24, 2196–2210.e2199, doi: 10.1016/j.celrep.2018.07.056 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Kim S-Y & Assawachananont J A New Method to Visualize the Intact Subretina From Retinal Pigment Epithelium to Retinal Tissue in Whole Mount of Pigmented Mouse Eyes. Translational vision science & technology 5, 6–6, doi: 10.1167/tvst.5.1.6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Futami K, Furukawa O, Maita M & Katagiri T Application of Hydrogen Peroxide-Melanin Bleaching and Fluorescent Nuclear Staining for Whole-Body Clearing and Imaging in Fish. Fish Pathology 54, 101–103, doi: 10.3147/jsfp.54.101 (2020). [DOI] [Google Scholar]
- 28.Kuroda M & Kuroda S Whole-body clearing of beetles by successive treatment with hydrogen peroxide and CUBIC reagents. Entomological Science 23, 311–315, doi: 10.1111/ens.12427 (2020). [DOI] [Google Scholar]
- 29.Henning Y, Osadnik C & Malkemper EP EyeCi: Optical clearing and imaging of immunolabeled mouse eyes using light-sheet fluorescence microscopy. Exp Eye Res 180, 137–145, doi: 10.1016/j.exer.2018.12.001 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Duong H & Han M A multispectral LED array for the reduction of background autofluorescence in brain tissue. Journal of neuroscience methods 220, 46–54, doi: 10.1016/j.jneumeth.2013.08.018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ku T et al. Elasticizing tissues for reversible shape transformation and accelerated molecular labeling. Nature methods 17, 609–613, doi: 10.1038/s41592-020-0823-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi SW, Guan W & Chung K Basic principles of hydrogel-based tissue transformation technologies and their applications. Cell 184, 4115–4136, doi: 10.1016/j.cell.2021.07.009 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chung K et al. Structural and molecular interrogation of intact biological systems. Nature 497, 332–337, doi: 10.1038/nature12107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang B et al. Single-Cell Phenotyping within Transparent Intact Tissue through Whole-Body Clearing. Cell 158, 945–958, doi: 10.1016/j.cell.2014.07.017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen F, Tillberg PW & Boyden ES Optical imaging. Expansion microscopy. Science 347, 543–548, doi: 10.1126/science.1260088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tillberg PW et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nature biotechnology 34, 987–992, doi: 10.1038/nbt.3625 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ku T et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nature biotechnology 34, 973–981, doi: 10.1038/nbt.3641 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami TC et al. A three-dimensional single-cell-resolution whole-brain atlas using CUBIC-X expansion microscopy and tissue clearing. Nature neuroscience 21, 625–637, doi: 10.1038/s41593-018-0109-1 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Chang JB et al. Iterative expansion microscopy. Nature methods 14, 593–599, doi: 10.1038/nmeth.4261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park HE et al. Scalable and Isotropic Expansion of Tissues with Simply Tunable Expansion Ratio. Adv Sci (Weinh) 6, 1901673, doi: 10.1002/advs.201901673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiviranta I, Tammi M, Lappalainen R, Kuusela T & Helminen HJ The rate of calcium extraction during EDTA decalcification from thin bone slices as assessed with atomic absorption spectrophotometry. Histochemistry 68, 119–127, doi: 10.1007/BF00489507 (1980). [DOI] [PubMed] [Google Scholar]
- 42.Jing D et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell research 28, 803–818, doi: 10.1038/s41422-018-0049-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Treweek JB et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature protocols 10, 1860–1896, doi: 10.1038/nprot.2015.122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenbaum A et al. Bone CLARITY: Clearing, imaging, and computational analysis of osteoprogenitors within intact bone marrow. Sci Transl Med 9, doi: 10.1126/scitranslmed.aah6518 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Cai R et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull–meninges connections. Nature neuroscience 22, 317–327, doi: 10.1038/s41593-018-0301-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renier N et al. Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes. Cell 165, 1789–1802, doi: 10.1016/j.cell.2016.05.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Renier N et al. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910, doi: 10.1016/j.cell.2014.10.010 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Dodt HU et al. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nature methods 4, 331–336, doi: 10.1038/nmeth1036 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Qi Y et al. FDISCO: Advanced solvent-based clearing method for imaging whole organs. Science advances 5, eaau8355, doi: 10.1126/sciadv.aau8355 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hahn C et al. High-resolution imaging of fluorescent whole mouse brains using stabilised organic media (sDISCO). Journal of biophotonics 12, e201800368, doi: 10.1002/jbio.201800368 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Schwarz MK et al. Fluorescent-protein stabilization and high-resolution imaging of cleared, intact mouse brains. PloS one 10, e0124650, doi: 10.1371/journal.pone.0124650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Xu J, Wan P, Yu T & Zhu D Optimization of GFP Fluorescence Preservation by a Modified uDISCO Clearing Protocol. Frontiers in neuroanatomy 12, 67, doi: 10.3389/fnana.2018.00067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Becker K, Jahrling N, Saghafi S, Weiler R & Dodt HU Chemical clearing and dehydration of GFP expressing mouse brains. PloS one 7, e33916, doi: 10.1371/journal.pone.0033916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Erturk A et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nature protocols 7, 1983–1995, doi: 10.1038/nprot.2012.119 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Hama H et al. ScaleS: an optical clearing palette for biological imaging. Nature neuroscience 18, 1518–1529, doi: 10.1038/nn.4107 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Chen L et al. UbasM: An effective balanced optical clearing method for intact biomedical imaging. Scientific reports 7, 12218, doi: 10.1038/s41598-017-12484-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chi J et al. Three-Dimensional Adipose Tissue Imaging Reveals Regional Variation in Beige Fat Biogenesis and PRDM16-Dependent Sympathetic Neurite Density. Cell Metab 27, 226–236 e223, doi: 10.1016/j.cmet.2017.12.011 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Kim SY et al. Stochastic electrotransport selectively enhances the transport of highly electromobile molecules. Proceedings of the National Academy of Sciences of the United States of America 112, E6274–6283, doi: 10.1073/pnas.1510133112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoelzel CA & Zhang X Visualizing and Manipulating Biological Processes by Using HaloTag and SNAP-Tag Technologies. ChemBioChem 21, 1935–1946, doi: 10.1002/cbic.202000037 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fang T et al. Nanobody immunostaining for correlated light and electron microscopy with preservation of ultrastructure. Nature methods 15, 1029–1032, doi: 10.1038/s41592-018-0177-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burry RW Immunocytochemistry: A Practical Guide for Biomedical Research. Immunocytochemistry: A Practical Guide for Biomedical Research, 1–223, doi: 10.1007/978-1-4419-1304-3 (2010). [DOI] [Google Scholar]
- 62.Sahl SJ, Hell SW & Jakobs S Fluorescence nanoscopy in cell biology. Nat Rev Mol Cell Biol 18, 685–701, doi: 10.1038/nrm.2017.71 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Muyldermans S Nanobodies: natural single-domain antibodies. Annual review of biochemistry 82, 775–797, doi: 10.1146/annurev-biochem-063011-092449 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Lai HM et al. Next generation histology methods for three-dimensional imaging of fresh and archival human brain tissues. Nature Communications 9, 1066, doi: 10.1038/s41467-018-03359-w (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gleave JA, Lerch JP, Henkelman RM & Nieman BJ A Method for 3D Immunostaining and Optical Imaging of the Mouse Brain Demonstrated in Neural Progenitor Cells. PloS one 8, e72039, doi: 10.1371/journal.pone.0072039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar V et al. Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell–lymphotoxin-dependent pathway. Blood 115, 4725–4733, doi: 10.1182/blood-2009-10-250118 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Sillitoe RV & Hawkes R Whole-mount Immunohistochemistry: A High-throughput Screen for Patterning Defects in the Mouse Cerebellum. Journal of Histochemistry & Cytochemistry 50, 235–244, doi: 10.1177/002215540205000211 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Na M, Kim K, Lim HR, Ha CM & Chang S Rapid immunostaining method for three-dimensional volume imaging of biological tissues by magnetic force-induced focusing of the electric field. Brain Struct Funct 226, 297–309, doi: 10.1007/s00429-020-02160-0 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Dwyer J, Ramirez MD, Katz PS, Karlstrom RO & Bergan J Accelerated clearing and molecular labeling of biological tissues using magnetohydrodynamic force. Scientific reports 11, 16462, doi: 10.1038/s41598-021-95692-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi K, Kubota SI, Ehata S, Ueda HR & Miyazono K Protocol for Imaging and Analysis of Mouse Tumor Models with CUBIC Tissue Clearing. STAR Protoc 1, 100191, doi: 10.1016/j.xpro.2020.100191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li W, Germain RN & Gerner MY Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proceedings of the National Academy of Sciences 114, E7321–E7330, doi: 10.1073/pnas.1708981114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klingberg A et al. Fully Automated Evaluation of Total Glomerular Number and Capillary Tuft Size in Nephritic Kidneys Using Lightsheet Microscopy. Journal of the American Society of Nephrology : JASN 28, 452–459, doi: 10.1681/ASN.2016020232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aoyagi Y, Kawakami R, Osanai H, Hibi T & Nemoto T A Rapid Optical Clearing Protocol Using 2,2′-Thiodiethanol for Microscopic Observation of Fixed Mouse Brain. PloS one 10, e0116280, doi: 10.1371/journal.pone.0116280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costantini I et al. A versatile clearing agent for multi-modal brain imaging. Scientific reports 5, 9808, doi: 10.1038/srep09808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Staudt T, Lang MC, Medda R, Engelhardt J & Hell SW 2,2′-Thiodiethanol: A new water soluble mounting medium for high resolution optical microscopy. Microscopy research and technique 70, 1–9, doi: 10.1002/jemt.20396 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Diel EE, Lichtman JW & Richardson DS Tutorial: avoiding and correcting sample-induced spherical aberration artifacts in 3D fluorescence microscopy. Nature protocols 15, 2773–2784, doi: 10.1038/s41596-020-0360-2 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Vallejo Ramirez PP et al. OptiJ: Open-source optical projection tomography of large organ samples. Scientific reports 9, 15693, doi: 10.1038/s41598-019-52065-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mayer J et al. OPTiSPIM: integrating optical projection tomography in light sheet microscopy extends specimen characterization to nonfluorescent contrasts. Optics letters 39, 1053–1056, doi: 10.1364/OL.39.001053 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Baek K et al. Quantitative assessment of regional variation in tissue clearing efficiency using optical coherence tomography (OCT) and magnetic resonance imaging (MRI): A feasibility study. Scientific reports 9, 2923, doi: 10.1038/s41598-019-39634-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pietzsch T, Saalfeld S, Preibisch S & Tomancak P BigDataViewer: visualization and processing for large image data sets. Nature methods 12, 481–483, doi: 10.1038/nmeth.3392 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Jonkman J, Brown CM, Wright GD, Anderson KI & North AJ Tutorial: guidance for quantitative confocal microscopy. Nature protocols 15, 1585–1611, doi: 10.1038/s41596-020-0313-9 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Jost AP & Waters JC Designing a rigorous microscopy experiment: Validating methods and avoiding bias. J Cell Biol 218, 1452–1466, doi: 10.1083/jcb.201812109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horl D et al. BigStitcher: reconstructing high-resolution image datasets of cleared and expanded samples. Nature methods 16, 870–874, doi: 10.1038/s41592-019-0501-0 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Prahst C et al. Mouse retinal cell behaviour in space and time using light sheet fluorescence microscopy. Elife 9, doi: 10.7554/eLife.49779 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moen E et al. Deep learning for cellular image analysis. Nature methods 16, 1233–1246, doi: 10.1038/s41592-019-0403-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pan C et al. Deep Learning Reveals Cancer Metastasis and Therapeutic Antibody Targeting in the Entire Body. Cell 179, 1661–1676 e1619, doi: 10.1016/j.cell.2019.11.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmidt U, Weigert M, Broaddus C & Myers G Cell Detection with Star-convex Polygons 265–273 (Springer International Publishing; ). [Google Scholar]
- 88.Todorov MI et al. Machine learning analysis of whole mouse brain vasculature. Nature methods 17, 442–449, doi: 10.1038/s41592-020-0792-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qi Y in Ensemble Machine Learning: Methods and Applications (eds Zhang Cha & Ma Yunqian) 307–323 (Springer US, 2012). [Google Scholar]
- 90.Dhillon A & Verma GK Convolutional neural network: a review of models, methodologies and applications to object detection. Progress in Artificial Intelligence 9, 85–112, doi: 10.1007/s13748-019-00203-0 (2020). [DOI] [Google Scholar]
- 91.Callara AL, Magliaro C, Ahluwalia A & Vanello N A Smart Region-Growing Algorithm for Single-Neuron Segmentation From Confocal and 2-Photon Datasets. Frontiers in neuroinformatics 14, doi: 10.3389/fninf.2020.00009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Richardson DS et al. SRpHi ratiometric pH biosensors for super-resolution microscopy. Nat Commun 8, 577, doi: 10.1038/s41467-017-00606-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao R et al. Cortical column and whole-brain imaging with molecular contrast and nanoscale resolution. Science 363, doi: 10.1126/science.aau8302 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Randlett O et al. Whole-brain activity mapping onto a zebrafish brain atlas. Nature methods 12, 1039–1046, doi: 10.1038/nmeth.3581 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hawrylycz MJ et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399, doi: 10.1038/nature11405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mikula S, Trotts I, Stone JM & Jones EG Internet-enabled high-resolution brain mapping and virtual microscopy. NeuroImage 35, 9–15, doi: 10.1016/j.neuroimage.2006.11.053 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amunts K et al. BigBrain: An Ultrahigh-Resolution 3D Human Brain Model. Science 340, 1472–1475, doi: 10.1126/science.1235381 (2013). [DOI] [PubMed] [Google Scholar]
- 98.Calabrese E et al. A diffusion tensor MRI atlas of the postmortem rhesus macaque brain. NeuroImage 117, 408–416, doi: 10.1016/j.neuroimage.2015.05.072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rohlfing T et al. The INIA19 Template and NeuroMaps Atlas for Primate Brain Image Parcellation and Spatial Normalization. Frontiers in neuroinformatics 6, 27, doi: 10.3389/fninf.2012.00027 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Johnson GA et al. Waxholm space: an image-based reference for coordinating mouse brain research. NeuroImage 53, 365–372, doi: 10.1016/j.neuroimage.2010.06.067 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Papp EA, Leergaard TB, Calabrese E, Johnson GA & Bjaalie JG Waxholm Space atlas of the Sprague Dawley rat brain. NeuroImage 97, 374–386, doi: 10.1016/j.neuroimage.2014.04.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dong HW The Allen reference atlas: A digital color brain atlas of the C57Bl/6J male mouse (John Wiley & Sons Inc, 2008). [Google Scholar]
- 103.Kuan L et al. Neuroinformatics of the Allen Mouse Brain Connectivity Atlas. Methods 73, 4–17, doi: 10.1016/j.ymeth.2014.12.013 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Mano T et al. CUBIC-Cloud provides an integrative computational framework toward community-driven whole-mouse-brain mapping. Cell Reports Methods 1, 100038, doi: 10.1016/j.crmeth.2021.100038 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ye L et al. Wiring and Molecular Features of Prefrontal Ensembles Representing Distinct Experiences. Cell 165, 1776–1788, doi: 10.1016/j.cell.2016.05.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kutten K. S. a. V., Joshua T and Charon Nicolas and Ye Li and Deisseroth Karl and Miller Michael I. Deformably registering and annotating whole CLARITY brains to an atlas via masked LDDMM. Optics, Photonics and Digital Technologies for Imaging Applications IV, doi: 10.1117/12.2227444 (2016). [DOI] [Google Scholar]
- 107.Gradinaru V, Treweek J, Overton K & Deisseroth K Hydrogel-tissue chemistry: Principles and applications. Annual review of biophysics 47, 355–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baiker M et al. Atlas-based whole-body segmentation of mice from low-contrast Micro-CT data. Medical image analysis 14, 723–737, doi: 10.1016/j.media.2010.04.008 (2010). [DOI] [PubMed] [Google Scholar]
- 109.Dogdas B, Stout D, Chatziioannou AF & Leahy RM Digimouse: a 3D whole body mouse atlas from CT and cryosection data. Physics in medicine and biology 52, 577–587, doi: 10.1088/0031-9155/52/3/003 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schoppe O et al. Deep learning-enabled multi-organ segmentation in whole-body mouse scans. Nature Communications 11, 5626, doi: 10.1038/s41467-020-19449-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haase R et al. CLIJ: GPU-accelerated image processing for everyone. Nature methods 17, 5–6, doi: 10.1038/s41592-019-0650-1 (2020). [DOI] [PubMed] [Google Scholar]
- 112.Kirst C et al. Mapping the Fine-Scale Organization and Plasticity of the Brain Vasculature. Cell 180, 780–795 e725, doi: 10.1016/j.cell.2020.01.028 (2020). [DOI] [PubMed] [Google Scholar]
- 113.Hahn M et al. 3D imaging of human organs with micrometer resolution - applied to the endocrine pancreas. Commun Biol 4, 1063, doi: 10.1038/s42003-021-02589-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao Y et al. Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nature biotechnology 35, 757–764, doi: 10.1038/nbt.3892 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Campinho MA et al. A thyroid hormone regulated asymmetric responsive centre is correlated with eye migration during flatfish metamorphosis. Scientific reports 8, 12267, doi: 10.1038/s41598-018-29957-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Konno A & Okazaki S Aqueous-based tissue clearing in crustaceans. Zoological Lett 4, 13, doi: 10.1186/s40851-018-0099-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Albanese A et al. Multiscale 3D phenotyping of human cerebral organoids. Scientific reports 10, 21487, doi: 10.1038/s41598-020-78130-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Winnubst J et al. Reconstruction of 1,000 Projection Neurons Reveals New Cell Types and Organization of Long-Range Connectivity in the Mouse Brain. Cell 179, 268–281 e213, doi: 10.1016/j.cell.2019.07.042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weissbourd B et al. Functional modules within a distributed neural network control feeding in a model medusa. bioRxiv, 2021.2002.2022.432372, doi: 10.1101/2021.02.22.432372 (2021). [DOI] [Google Scholar]
- 120.Morgan JL & Lichtman JW Digital tissue and what it may reveal about the brain. BMC Biol 15, 101, doi: 10.1186/s12915-017-0436-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Economo MN, Winnubst J, Bas E, Ferreira TA & Chandrashekar J Single-neuron axonal reconstruction: The search for a wiring diagram of the brain. The Journal of comparative neurology 527, 2190–2199, doi: 10.1002/cne.24674 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Feng G et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51, doi: 10.1016/s0896-6273(00)00084-2 (2000). [DOI] [PubMed] [Google Scholar]
- 123.Cai D, Cohen KB, Luo T, Lichtman JW & Sanes JR Improved tools for the Brainbow toolbox. Nature methods 10, 540–547 (2013). [PubMed] [Google Scholar]
- 124.Livet J et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62, doi: 10.1038/nature06293 (2007). [DOI] [PubMed] [Google Scholar]
- 125.Quan T et al. NeuroGPS-Tree: automatic reconstruction of large-scale neuronal populations with dense neurites. Nature methods 13, 51–54, doi: 10.1038/nmeth.3662 (2016). [DOI] [PubMed] [Google Scholar]
- 126.Wang X et al. Bi-channel image registration and deep-learning segmentation (BIRDS) for efficient, versatile 3D mapping of mouse brain. Elife 10, doi: 10.7554/eLife.63455 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gail Canter R et al. 3D mapping reveals network-specific amyloid progression and subcortical susceptibility in mice. Commun Biol 2, 360, doi: 10.1038/s42003-019-0599-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sabdyusheva Litschauer I et al. 3D histopathology of human tumours by fast clearing and ultramicroscopy. Scientific reports 10, 17619, doi: 10.1038/s41598-020-71737-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ma Y et al. 3D Spatiotemporal Mechanical Microenvironment: A Hydrogel-Based Platform for Guiding Stem Cell Fate. Advanced Materials 30, 1705911, doi: 10.1002/adma.201705911 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Lloyd-Lewis B Multidimensional Imaging of Mammary Gland Development: A Window Into Breast Form and Function. Front Cell Dev Biol 8, 203, doi: 10.3389/fcell.2020.00203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang L et al. Three-Dimensional Quantitative Co-Mapping of Pulmonary Morphology and Nanoparticle Distribution with Cellular Resolution in Nondissected Murine Lungs. ACS Nano 13, 1029–1041, doi: 10.1021/acsnano.8b07524 (2019). [DOI] [PubMed] [Google Scholar]
- 132.Cuccarese MF et al. Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D organ imaging. Nat Commun 8, 14293, doi: 10.1038/ncomms14293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sindhwani S, Syed AM, Wilhelm S & Chan WC Exploring Passive Clearing for 3D Optical Imaging of Nanoparticles in Intact Tissues. Bioconjug Chem 28, 253–259, doi: 10.1021/acs.bioconjchem.6b00500 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Schimmenti LA, Yan HC, Madri JA & Albelda SM Platelet endothelial cell adhesion molecule, PECAM-1, modulates cell migration. J Cell Physiol 153, 417–428, doi: 10.1002/jcp.1041530222 (1992). [DOI] [PubMed] [Google Scholar]
- 135.Konno A, Matsumoto N & Okazaki S Improved vessel painting with carbocyanine dye-liposome solution for visualisation of vasculature. Scientific reports 7, 10089, doi: 10.1038/s41598-017-09496-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nehrhoff I, Ripoll J, Samaniego R, Desco M & Gomez-Gaviro MV Looking inside the heart: a see-through view of the vascular tree. Biomed Opt Express 8, 3110–3118, doi: 10.1364/BOE.8.003110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lugo-Hernandez E et al. 3D visualization and quantification of microvessels in the whole ischemic mouse brain using solvent-based clearing and light sheet microscopy. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 37, 3355–3367, doi: 10.1177/0271678X17698970 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Di Giovanna AP et al. Whole-Brain Vasculature Reconstruction at the Single Capillary Level. Scientific reports 8, 12573, doi: 10.1038/s41598-018-30533-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rajendran PS et al. Identification of peripheral neural circuits that regulate heart rate using optogenetic and viral vector strategies. Nat Commun 10, 1944, doi: 10.1038/s41467-019-09770-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Achanta S et al. A Comprehensive Integrated Anatomical and Molecular Atlas of Rat Intrinsic Cardiac Nervous System. iScience 23, 101140, doi: 10.1016/j.isci.2020.101140 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yokoyama T et al. Quantification of sympathetic hyperinnervation and denervation after myocardial infarction by three-dimensional assessment of the cardiac sympathetic network in cleared transparent murine hearts. PloS one 12, e0182072, doi: 10.1371/journal.pone.0182072 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kapp FG et al. Protection from UV light is an evolutionarily conserved feature of the haematopoietic niche. Nature 558, 445–448, doi: 10.1038/s41586-018-0213-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lersten NR Modified clearing method to show sieve tubes in minor veins of leaves. Stain Technol 61, 231–234, doi: 10.3109/10520298609109942 (1986). [DOI] [PubMed] [Google Scholar]
- 144.Warner CA et al. An optical clearing technique for plant tissues allowing deep imaging and compatible with fluorescence microscopy. Plant Physiol 166, 1684–1687, doi: 10.1104/pp.114.244673 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kurihara D, Mizuta Y, Sato Y & Higashiyama T ClearSee: a rapid optical clearing reagent for whole-plant fluorescence imaging. Development 142, 4168–4179, doi: 10.1242/dev.127613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lu L et al. A rapid and effective optical-clearing technique for deep tissue fluorescence imaging in trees. Trees-Struct Funct 34, 783–790, doi: 10.1007/s00468-020-01957-0 (2020). [DOI] [Google Scholar]
- 147.Xia Q et al. Solar-assisted fabrication of large-scale, patternable transparent wood. Science advances 7, doi: 10.1126/sciadv.abd7342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nojima S et al. CUBIC pathology: three-dimensional imaging for pathological diagnosis. Scientific reports 7, 9269, doi: 10.1038/s41598-017-09117-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Belle M et al. Tridimensional Visualization and Analysis of Early Human Development. Cell 169, 161–173 e112, doi: 10.1016/j.cell.2017.03.008 (2017). [DOI] [PubMed] [Google Scholar]
- 150.Dekkers JF et al. High-resolution 3D imaging of fixed and cleared organoids. Nature protocols 14, 1756–1771, doi: 10.1038/s41596-019-0160-8 (2019). [DOI] [PubMed] [Google Scholar]
- 151.Rigamonti A et al. Large-Scale Production of Mature Neurons from Human Pluripotent Stem Cells in a Three-Dimensional Suspension Culture System. Stem Cell Reports 6, 993–1008, doi: 10.1016/j.stemcr.2016.05.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cora V et al. A Cleared View on Retinal Organoids. Cells 8, doi: 10.3390/cells8050391 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Costantini I, Cicchi R, Silvestri L, Vanzi F & Pavone FS In-vivo and ex-vivo optical clearing methods for biological tissues: review. Biomed Opt Express 10, 5251–5267, doi: 10.1364/BOE.10.005251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Deng ZJ et al. Viscous optical clearing agent for in vivo optical imaging. Journal of Biomedical Optics 19, doi:Artn 076019 10.1117/1.Jbo.19.7.076019 (2014). [DOI] [PubMed] [Google Scholar]
- 155.Millon SR, Roldan-Perez KM, Riching KM, Palmer GM & Ramanujam N Effect of optical clearing agents on the in vivo optical properties of squamous epithelial tissue. Lasers in Surgery and Medicine 38, 920–927, doi: 10.1002/lsm.20451 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Tuchin VV, Bashkatov AN, Genina EA, Sinichkin YP & Lakodina NA In vivo investigation of the immersion-liquid-induced human skin clearing dynamics. Tech Phys Lett+ 27, 489–490, doi:Doi 10.1134/1.1383834 (2001). [DOI] [Google Scholar]
- 157.Wen X, Mao ZZ, Han ZZ, Tuchin VV & Zhu D In vivo skin optical clearing by glycerol solutions: mechanism (vol 3, pg 44, 2010). Journal of biophotonics 3, 252–252, doi:DOI 10.1002/jbio.201090004 (2010). [DOI] [PubMed] [Google Scholar]
- 158.Zhu D, Wang J, Zhi ZW, Wen X & Luo QM Imaging dermal blood flow through the intact rat skin with an optical clearing method. Journal of Biomedical Optics 15, doi:Artn 026008 10.1117/1.3369739 (2010). [DOI] [PubMed] [Google Scholar]
- 159.Zhao YJ et al. Skull optical clearing window for in vivo imaging of the mouse cortex at synaptic resolution (vol 7, pg 17153 2018). Light-Sci Appl 7, doi:ARTN 6 10.1038/s41377-018-0021-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pires L et al. Optical clearing of melanoma in vivo: characterization by diffuse reflectance spectroscopy and optical coherence tomography. J Biomed Opt 21, 081210, doi: 10.1117/1.JBO.21.8.081210 (2016). [DOI] [PubMed] [Google Scholar]
- 161.Zhao H et al. A versatile strategy for improving phototherapeutic efficacy on deep-sited tumor by tissue optical clearing technique. Nano Today 36, doi:ARTN 101058 10.1016/j.nantod.2020.101058 (2021). [DOI] [Google Scholar]
- 162.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090, doi: 10.1126/science.aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Eng CL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239, doi: 10.1038/s41586-019-1049-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Moffitt JR et al. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proceedings of the National Academy of Sciences of the United States of America 113, 14456–14461, doi: 10.1073/pnas.1617699113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dance A Find a home for every imaging data set. Nature 579, 162–163, doi: 10.1038/d41586-020-00594-4 (2020). [DOI] [PubMed] [Google Scholar]
- 166.Pleiner T et al. Nanobodies: site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation. Elife 4, e11349, doi: 10.7554/eLife.11349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schumacher D, Helma J, Schneider AFL, Leonhardt H & Hackenberger CPR Nanobodies: Chemical Functionalization Strategies and Intracellular Applications. Angewandte Chemie 57, 2314–2333, doi: 10.1002/anie.201708459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kishi JY et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nature methods 16, 533–544, doi: 10.1038/s41592-019-0404-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shah S et al. Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development 143, 2862–2867, doi: 10.1242/dev.138560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lin R et al. A hybridization-chain-reaction-based method for amplifying immunosignals. Nature methods 15, 275–278, doi: 10.1038/nmeth.4611 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Saritas T, Puelles VG, Su XT, Ellison DH & Kramann R Optical Clearing and Imaging of Immunolabeled Kidney Tissue. J Vis Exp, doi: 10.3791/60002 (2019). [DOI] [PubMed] [Google Scholar]
- 172.Xu F et al. High-throughput mapping of a whole rhesus monkey brain at micrometer resolution. Nature biotechnology, doi: 10.1038/s41587-021-00986-5 (2021). [DOI] [PubMed] [Google Scholar]
- 173.Hama H et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nature neuroscience 14, 1481–1488, doi: 10.1038/nn.2928 (2011). [DOI] [PubMed] [Google Scholar]
- 174.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, doi: 10.1126/science.aat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]