

Abstract
Background
Several SARS-CoV-2 variants of concern (VOC) have emerged through 2020 and 2021. There is need for tools to estimate the relative transmissibility of emerging variants of SARS-CoV-2 with respect to circulating strains.
Aim
We aimed to assess the prevalence of co-circulating VOC in Italy and estimate their relative transmissibility.
Methods
We conducted two genomic surveillance surveys on 18 February and 18 March 2021 across the whole Italian territory covering 3,243 clinical samples and developed a mathematical model that describes the dynamics of co-circulating strains.
Results
The Alpha variant was already dominant on 18 February in a majority of regions/autonomous provinces (national prevalence: 54%) and almost completely replaced historical lineages by 18 March (dominant across Italy, national prevalence: 86%). We found a substantial proportion of the Gamma variant on 18 February, almost exclusively in central Italy (prevalence: 19%), which remained similar on 18 March. Nationally, the mean relative transmissibility of Alpha ranged at 1.55–1.57 times the level of historical lineages (95% CrI: 1.45–1.66). The relative transmissibility of Gamma varied according to the assumed degree of cross-protection from infection with other lineages and ranged from 1.12 (95% CrI: 1.03–1.23) with complete immune evasion to 1.39 (95% CrI: 1.26–1.56) for complete cross-protection.
Conclusion
We assessed the relative advantage of competing viral strains, using a mathematical model assuming different degrees of cross-protection. We found substantial co-circulation of Alpha and Gamma in Italy. Gamma was not able to outcompete Alpha, probably because of its lower transmissibility.
Keywords: SARS-CoV-2 variant of concern, transmissibility, co-circulation, lineage
Introduction
Since the end of 2020, multiple severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOC) have emerged across the globe. Some of them are particularly concerning as their biological characteristics allowed them to outcompete and rapidly replace historical lineages in the countries where they probably emerged, and to spread rapidly to many other countries. The Alpha variant (Phylogenetic Assignment of Named Global Outbreak (Pango) lineage designation: B.1.1.7) was first detected in the United Kingdom (UK) in samples from September 2020 and became dominant throughout the country by early 2021 [1,2]. It has spread in most of Europe and has been reported in a majority countries worldwide [3-6]. The Gamma variant (P.1) was first reported in Japan among travellers returning from Brazil [7]. It was later found in almost half of the cases in December 2020 in Manaus, Brazil where, despite a very high estimated seroprevalence against historical lineages, a large upsurge of infections occurred throughout January 2021 [8-10]. The Beta variant (B.1.351) was first detected in South Africa, where it became dominant in late November 2020 [11].
The epidemiological success of these variants relies on evolutionary advantages such as increased transmissibility [1,2,12] and their ability (demonstrated for Gamma and Beta) to significantly reduce antibody neutralisation in convalescent and post-vaccination sera [13-17], probably resulting in reinfections through immune escape [18-21]. Besides their greater ability to spread, requiring more restrictive physical distancing measures to mitigate epidemics, these variants have caused additional concern regarding potentially increased morbidity and mortality [22,23] as well as their potential impact on vaccine effectiveness [24,25]. Because variants seem to have emerged in unconnected geographical areas, little is known as yet about their ecological interactions.
Here, we have developed a three-strain susceptible–infectious–recovered (SIR) model that simulates the co-circulation of historical SARS-CoV-2 lineages (wildtype) and the Alpha and Gamma VOC under different assumptions regarding the degree of cross-protection, to estimate the relative transmissibility of emerging variants vs historical lineages, using data from genomic surveillance in Italy. This model offers a simple and efficient tool to estimate the relative transmissibility of newly emerging variant of the SARS-CoV-2 virus such as the Delta variant (B.1.617.2) with respect to circulating variants.
Methods
Epidemiological information
Publicly available data on the number of newly diagnosed SARS-CoV-2 infections were taken from the Italian Civil Protection Department [26]. Data on the number of newly admitted patients in hospitals were obtained from the Italian integrated surveillance system curated by the Italian National Institute of Health (Istituto Superiore di Sanità, ISS) [27,28]. Data on physical distancing restrictions were collected from the Italian Ministry of Health [29]. Physical distancing restrictions were organised in tiers of different colours (white, yellow, orange and red, from most relaxed to most strict) [30] and updated for each Italian region on a weekly basis (with occasional exceptions). Restrictions corresponding to each tier are fully described in the Supplement. For purely descriptive reasons, we computed a tier index summarising the stringency of restrictions across the national territory: tiers were assigned an arbitrary value from 0 (white) to 3 (red) and their average weighted by the proportion of the population living under each tier was computed for each day. The resulting value was normalised to have values between 0 (representing 100.0% of the population under the white tier) and 1 (100.0% of the population under the red tier).
Survey methodology
Two cross-sectional surveys coordinated by the Italian National Institute of Health, in collaboration with the Ministry of Health and the laboratories of regions/autonomous provinces (AP) were conducted on 18 February and 18 March 2021, to estimate the prevalence of the Alpha, Gamma, and Beta variants [31,32].
The surveys involved all 19 regions and the two AP of Italy. Random samples of SARS-CoV-2-positive cases, diagnosed on 18 February and 18 March with a real-time reverse transcription PCR (RT-PCR) and a quantification cycle (Cq) < 28, were analysed in 101 and 129 laboratories, respectively, distributed across the national territory. Samples came from both passive surveillance and contact tracing and included symptomatic, pre-symptomatic and asymptomatic COVID-19 cases.
Samples were distributed across five macro-areas, defined according to the Eurostat NUTS1 classification: North-east, North-west, Centre, South and Islands. The sample size was calculated to have the statistical power to detect a prevalence of 1%, with 0.8% error within each macro-area, based on the number of cases notified on the day preceding each of the two surveys [33]. The collected samples were sequenced according to the local laboratory policy by either of the following techniques: (i) sequencing the entire S gene by Sanger technology, (ii) sequencing part of the S gene with identification of all mutations/deletions associated with SARS-CoV-2 variants or (iii) sequencing the whole genome by next generation sequencing. A small fraction of the sequenced samples could not be analysed because of insufficient sequencing coverage of the genome and were therefore discarded. In the second survey, one region (Marche) pre-screened 54 of its 65 RT-PCR-positive samples using an in-house test (see the Supplement for the methodology) that detects both H69-V70 and Y144 amino acid deletions which are specific for the Alpha variant; as a result, 46 cases positive in the in-house test were considered to be Alpha without sequencing. The eight cases negative in the test, plus the 11 cases that were not pre-screened were sequenced.
The point prevalence of the three lineages in each survey was computed as the fraction of infections confirmed for each lineage among sequenced samples, and corresponding multinomial 95% confidence intervals (CI) are provided. Multinomial CI were computed using the function MultinomlCI with Wilson method from package DescTools in R version 3.6.2. In the second survey, data from Marche were excluded from all analyses.
SARS-CoV-2 three-strain transmission model
We adopted a three-strain SIR mathematical model to simulate co-circulation of historical lineages of SARS-CoV-2 (wildtype) and the VOC Alpha and Gamma. We did not consider the Beta variant because the surveys found little or no circulation of this lineage (see Results). We assumed that a previous infection with the wildtype provides complete protection against the Alpha variant [34] and that infection with either the wildtype or Alpha confers the same degree of cross-protection against the Gamma variant. In addition, we assumed that the transmissibility of variants Alpha and Gamma are scaled with respect to the wildtype transmissibility by a lineage-specific factor representing their relative transmissibility. We assumed that the three strains had identical generation time (set at 6.6 days to reflect the serial interval estimated for SARS-CoV-2 in Italy [35]). The model was initialised on 15 January 2021, assuming that an unknown fraction of all infections on that date belonged to the Alpha and Gamma variant (see the Supplement for full model details). Unknown model parameters (namely, the transmissibility of the wildtype strain, the relative transmissibility parameters of Alpha and Gamma and their respective initial prevalence) were estimated by calibrating the model against prevalence data from the two surveys, using a Markov chain Monte Carlo (MCMC) approach based on Metropolis–Hastings sampling and uninformative priors. Different scenarios for cross-protection from wildtype and Alpha against Gamma were explored. The likelihood was defined as:
Where M(·) is the multinomial probability density distribution, t is the date of the survey, Sl t is the number of cases observed at date t for lineage l, Qt = ∑lSl t is the total number of analysed samples at date t, and pl t is the model-estimated fraction of infections of lineage l over the total at date t. In order to reproduce the observed epidemiological temporal trends, we assigned L = 0 to simulations for which the model’s mean squared error on the observed daily hospital admissions between 18 January and 18 March 2021 exceeded 1.5 times the variance of observations. We ran the MCMC algorithm for 50,000 iterations and convergence was assessed by checking that, after a burn-in period of 10,000 iterations, the trace plots associated with different chains, i.e. the sequence of accepted parameter values, were characterised by an approximately constant standard deviation and average, therefore proving good mixing of the parameters.
The model was run on four different geographical aggregations of regional data, i.e. the national level and the Centre, North-east and South macro-areas. Macro-areas North-west and Islands did not have samples positive for the Gamma variant. Data from Marche were excluded because of heterogeneity in data collection, but we performed a sensitivity analysis to evaluate the impact of this choice. We also ran additional sensitivity analyses to evaluate the robustness of results (see the Supplement for the results of these sensitivity analyses). Specifically, we estimated the relative transmissibility of Alpha alone in Lombardy and Veneto, where Gamma was not co-circulating and for which a previous data point on the prevalence of Alpha was available from a survey conducted with the same methodology on cases confirmed on 3–4 February (the relative transmissibility of Alpha in this scenario is provided in the Supplement). To evaluate the potential impact of geographical aggregation on estimates of the relative transmissibility of Alpha and Gamma, we re-ran the analysis in two individual regions with high co-circulation (Tuscany and Lazio). Finally, we ran an analysis where, instead of fixing the degree of cross-protection between historical, Alpha and Gamma lineages, we left it as a free model parameter.
Estimated posterior distributions of free parameters are summarised by posterior means and 95% credible intervals (CrI).
Ethical statement
Ethical approval for sequencing SARS-CoV-2 genomes from clinical samples sent to ISS was obtained from the Ethical Committee of ISS (ref. PRE BIO CE n.26259, 29 July 2020).
Results
SARS-CoV-2 epidemiology in Italy, January–March 2021
The daily incidence of newly diagnosed SARS-CoV-2 cases and COVID-19 patients admitted to hospital remained stable in Italy between mid-January and mid-February 2021, then increased in the second half of February and first half of March (Figure 1A) [26]. Strict measures in the second half of January (ca 80% of the population in orange/red tiers, Figure 1B) were relaxed in February and remained similar until March (ca 70–80% of citizens in yellow tier, none in red tier). Starting on 14 March, restrictions were rapidly tightened to control the rise in infections and hospitalisations, and more than 70% of the national population ended up in the strictest tier.
Figure 1.
Epidemiological COVID-19 situation in Italy between 15 January and 18 March 2021
COVID-19: coronavirus disease.
Data sources for panel A: new diagnoses [26]; hospital admissions [27]. Data source for panel B: population in white, yellow, orange and red tiers [29]. A value of 0 for the tier index implies that the entire population is in the most relaxed tier (white) and a value of 1 implies that the entire population is in the most strict tier (red).

Prevalence of variants of concern in Italy, 18 February and 18 March
During the genomic survey conducted on 18 February, 1,296 samples were sequenced, of which 57 (4%) were discarded for the analysis because of insufficient sequencing coverage of the genome. Among the remaining 1,239 analysed samples, the Alpha variant was confirmed in 658, Gamma in 62 and Beta in six, for a national prevalence of 53.1% (95% CI: 50.2–56.1), 5.0% (95% CI: 2.1–7.9) and 0.5% (95% CI: 0.0–3.4), respectively. The Alpha variant was found in 20 of 21 regions/AP, Gamma in six and Beta in three (Figure 2 and Table 1). The prevalence of Alpha was highest in the North-west macro-area (60.4%; 95% CI: 55.1–66.1) and lowest in the Centre (44.7%; 95% CI: 38.8–50.8), while Gamma was almost exclusively concentrated in the Centre (mean prevalence: 18.8%; 95% CI: 12.9–24.9, as opposed to 1% or less elsewhere; see Table 1 and Figure 2). The Beta variant was identified only in Lombardy (three cases), in the AP Bolzano (two cases) and in Sicily (one case).
Figure 2.
Geographical distribution of SARS-CoV-2 variants of concern, Italy, point prevalence on 18 February 2021 (n = 1,296) and 18 March 2021 (n = 1,938)
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
Abbreviations of region names are reported in panel C and listed in the Supplement. Borders of mainland macro-areas are highlighted in blue, and the names of macro-areas are reported in panel D.
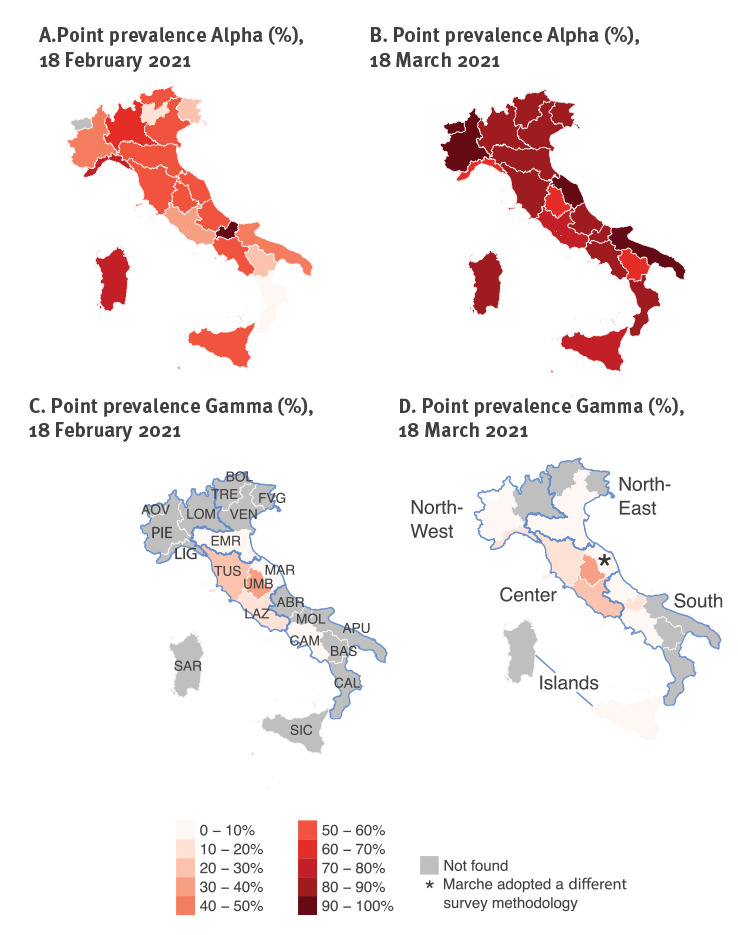
Table 1. Results of the first SARS-CoV-2 point prevalence survey across the 21 participating regions/AP, Italy, 18 February 2021 (n = 1,296).
| Region | Laboratories | RT-PCR-positive | Sequenced samples | Analysed samples | Confirmed cases | Point prevalence (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Alpha | Gamma | Beta | Alpha | Gamma | Beta | |||||
| Islands | ||||||||||
| Sardinia | 6 | 38 | 25 | 12 | 9 | 0 | 0 | 75% (46.8–91.1) | 0% (0–24.2) | 0% (0–24.2) |
| Sicily | 5 | 268 | 63 | 58 | 32 | 0 | 1 | 55.2% (42.5–67.3) | 0% (0–6.2) | 1.7% (0.3–9.1) |
| Total islands | 11 | 306 | 88 | 70 | 41 | 0 | 1 | 58.6% (46.9–69.4) | 0% (0–5.2) | 1.4% (0.3–7.7) |
| South | ||||||||||
| Abruzzo | 2 | 374 | 61 | 61 | 31 | 0 | 0 | 50.8% (38.6–62.9) | 0% (0–5.9) | 0% (0–5.9) |
| Apulia | 7 | 59 | 59 | 59 | 28 | 0 | 0 | 47.5% (35.3–60) | 0% (0–6.1) | 0% (0–6.1) |
| Basilicata | 5 | 7 | 7 | 5 | 1 | 0 | 0 | 20% (3.6–62.4) | 0% (0–43.4) | 0% (0–43.4) |
| Calabria | 3 | 166 | 11 | 11 | 1 | 0 | 0 | 9.1% (1.6–37.7) | 0% (0–25.9) | 0% (0–25.9) |
| Campania | 2 | 366 | 86 | 86 | 51 | 2 | 0 | 59.3% (48.7–69.1) | 2.3% (0.6–8.1) | 0% (0–4.3) |
| Molise | 1 | 114 | 15 | 15 | 14 | 0 | 0 | 93.3% (70.2–98.8) | 0% (0–20.4) | 0% (0–20.4) |
| Total South | 20 | 1,086 | 239 | 237 | 126 | 2 | 0 | 53.2% (46.8–59.4) | 0.8% (0.2–3) | 0% (0–1.6) |
| Centre | ||||||||||
| Lazio | 5 | 169 | 169 | 144 | 49 | 19 | 0 | 34% (26.8–42.1) | 13.2% (8.6–19.7) | 0% (0–2.6) |
| Marche | 8 | 38 | 38 | 38 | 22 | 3 | 0 | 57.9% (42.2–72.1) | 7.9% (2.7–20.8) | 0% (0–9.2) |
| Tuscany | 3 | 88 | 80 | 80 | 43 | 19 | 0 | 53.8% (42.9–64.3) | 23.8% (15.8–34.1) | 0% (0–4.6) |
| Umbria | 4 | 247 | 48 | 47 | 24 | 17 | 0 | 51.1% (37.2–64.7) | 36.2% (24–50.5) | 0% (0–7.6) |
| Total Centre | 20 | 542 | 335 | 309 | 138 | 58 | 0 | 44.7% (39.2–50.2) | 18.8% (14.8–23.5) | 0% (0–1.2) |
| North-east | ||||||||||
| AP Bolzano | 1 | 320 | 70 | 70 | 40 | 0 | 2 | 57.1% (45.5–68.1) | 0% (0–5.2) | 2.9% (0.8–9.8) |
| AP Trento | 1 | 20 | 20 | 14 | 2 | 0 | 0 | 14.3% (4–39.9) | 0% (0–21.5) | 0% (0–21.5) |
| Emilia-Romagna | 2 | 99 | 99 | 99 | 57 | 2 | 0 | 57.6% (47.7–66.8) | 2% (0.6–7.1) | 0% (0–3.7) |
| Friuli Venezia Giulia | 4 | 133 | 28 | 27 | 8 | 0 | 0 | 29.6% (15.9–48.5) | 0% (0–12.5) | 0% (0–12.5) |
| Veneto | 12 | 92 | 92 | 92 | 52 | 0 | 0 | 56.5% (46.3–66.2) | 0% (0–4) | 0% (0–4) |
| Total North-east | 20 | 664 | 309 | 302 | 159 | 2 | 2 | 52.6% (47–58.2) | 0.7% (0.2–2.4) | 0.7% (0.2–2.4) |
| North-west | ||||||||||
| Aosta Valley | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0% (0–79.3) | 0% (0–79.3) | 0% (0–79.3) |
| Liguria | 6 | 227 | 22 | 22 | 16 | 0 | 0 | 72.7% (51.8–86.8) | 0% (0–14.9) | 0% (0–14.9) |
| Lombardy | 9 | 213 | 213 | 213 | 137 | 0 | 3 | 64.3% (57.7–70.4) | 0% (0–1.8) | 1.4% (0.5–4.1) |
| Piedmont | 14 | 93 | 89 | 85 | 41 | 0 | 0 | 48.2% (37.9–58.7) | 0% (0–4.3) | 0% (0–4.3) |
| Total North-west | 30 | 534 | 325 | 321 | 194 | 0 | 3 | 60.4% (55–65.6) | 0% (0–1.2) | 0.9% (0.3–2.7) |
| Total Italy | 101 | 3,132 | 1,296 | 1,239 | 658 | 62 | 6 | 53.1% (50.3–55.9) | 5% (3.9–6.4) | 0.5% (0.2–1.1) |
CI: confidence interval; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
All percentages in the table are approximated to the first decimal, the value zero may therefore stand for < 0.05%. ‘RT-PCR-positive’ represent only cases tested in the laboratories involved and not all positive cases in an area.
During the genomic survey conducted on 18 March, 1,938 samples were sequenced (not including data from Marche), of which 24 (1%) were discarded for the analysis because of insufficient coverage of the genome sequencing (Table 2). Among the remaining 1,914 analysed samples, Alpha was confirmed in 1,641 infections, Gamma in 92 and Beta in three, for a national prevalence of 85.7% (95% CI: 84.3–87.3), 4.8% (95% CI: 3.3–6.3) and 0.2% (95% CI: 0.0–1.7), respectively. Alpha was found in all 21 regions/AP, Gamma in 12, and Beta in three (Figure 2 and Table 2). According to the survey conducted on 18 March, the Alpha variant had become dominant in all Italian regions, with regional prevalence estimates ranging from 63.6% to 100.0% (Figure 2 and Table 2). Regional prevalence estimates for the Gamma variant ranged from 0.0% to 32%; the highest prevalence estimates for this VOC were still obtained for central regions (Figure 2), however, it was detected in six additional regions compared with 18 February. The Beta variant was identified only in Lombardy, Sardinia and Veneto (one case each).
Table 2. Results from the second SARS-CoV-2 point prevalence survey across the 21 participating regions/AP, Italy, 18 March 2021 (n = 1,938).
| Region | Laboratories | RT-PCR-positive | Sequenced samples | Analysed samples | Confirmed cases | Point prevalence (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Alpha | Gamma | Beta | Alpha | Gamma | Beta | |||||
| Islands | ||||||||||
| Sardinia | 6 | 85 | 21 | 21 | 18 | 0 | 1 | 85.7% (65.4–95) | 0% (0–15.5) | 4.8% (0.8–22.7) |
| Sicily | 5 | 632 | 132 | 129 | 97 | 3 | 0 | 75.2% (67.1–81.8) | 2.3% (0.8–6.6) | 0% (0–2.9) |
| Total Islands | 11 | 717 | 153 | 150 | 115 | 3 | 1 | 76.7% (69.3–82.7) | 2% (0.7–5.7) | 0.7% (0.1–3.7) |
| South | ||||||||||
| Abruzzo | 2 | 293 | 87 | 80 | 66 | 4 | 0 | 82.5% (72.7–89.3) | 5% (2–12.2) | 0% (0–4.6) |
| Apulia | 11 | 126 | 126 | 126 | 117 | 0 | 0 | 92.9% (87–96.2) | 0% (0–3) | 0% (0–3) |
| Basilicata | 6 | 62 | 27 | 20 | 13 | 0 | 0 | 65% (43.3–81.9) | 0% (0–16.1) | 0% (0–16.1) |
| Calabria | 4 | 404 | 26 | 26 | 22 | 0 | 0 | 84.6% (66.5–93.8) | 0% (0–12.9) | 0% (0–12.9) |
| Campania | 3 | 1400 | 261 | 261 | 232 | 4 | 0 | 88.9% (84.5–92.2) | 1.5% (0.6–3.9) | 0% (0–1.5) |
| Molise | 1 | 63 | 16 | 16 | 13 | 2 | 0 | 81.2% (57–93.4) | 12.5% (3.5–36) | 0% (0–19.4) |
| Total South | 27 | 2,348 | 543 | 529 | 463 | 10 | 0 | 87.5% (84.4–90.1) | 1.9% (1–3.4) | 0% (0–0.7) |
| Centre | ||||||||||
| Lazio | 11 | 214 | 205 | 205 | 161 | 42 | 0 | 78.5% (72.4–83.6) | 20.5% (15.5–26.5) | 0% (0–1.8) |
| Marchea | 11 | 65 | Excluded from analysisa | 83.1% (72.2–90.3) | 3.1% (0.8–10.5) | 0% (0–5.6) | ||||
| Tuscany | 3 | 144 | 103 | 99 | 85 | 10 | 0 | 85.9% (77.7–91.4) | 10.1% (5.6–17.6) | 0% (0–3.7) |
| Umbria | 5 | 80 | 26 | 25 | 16 | 8 | 0 | 64% (44.5–79.8) | 32% (17.2–51.6) | 0% (0–13.3) |
| Total Centreb | 30 | 503 | 334 | 329 | 262 | 60 | 0 | 79.6% (75–83.6) | 18.2% (14.4–22.8) | 0% (0–1.2) |
| North-east | ||||||||||
| Ap Bolzano | 1 | 69 | 15 | 15 | 12 | 0 | 0 | 80% (54.8–93) | 0% (0–20.4) | 0% (0–20.4) |
| Ap Trento | 1 | 16 | 16 | 16 | 16 | 0 | 0 | 100% (80.6–100) | 0% (0–19.4) | 0% (0–19.4) |
| Emilia-Romagna | 2 | 175 | 175 | 175 | 154 | 13 | 0 | 88% (82.4–92) | 7.4% (4.4–12.3) | 0% (0–2.1) |
| Friuli Venezia Giulia | 7 | 126 | 55 | 55 | 49 | 0 | 0 | 89.1% (78.2–94.9) | 0% (0–6.5) | 0% (0–6.5) |
| Veneto | 13 | 156 | 156 | 156 | 138 | 2 | 1 | 88.5% (82.5–92.6) | 1.3% (0.4–4.6) | 0.6% (0.1–3.5) |
| Total North-east | 24 | 542 | 417 | 417 | 369 | 15 | 1 | 88.5% (85.1–91.2) | 3.6% (2.2–5.8) | 0.2% (0–1.3) |
| North-west | ||||||||||
| Aosta Valley | 1 | 32 | 2 | 2 | 2 | 0 | 0 | 100% (34.2–100) | 0% (0–65.8) | 0% (0–65.8) |
| Liguria | 8 | 179 | 22 | 22 | 14 | 3 | 0 | 63.6% (43–80.3) | 13.6% (4.7–33.3) | 0% (0–14.9) |
| Lombardy | 12 | 314 | 314 | 312 | 278 | 0 | 1 | 89.1% (85.2–92.1) | 0% (0–1.2) | 0.3% (0.1–1.8) |
| Piedmont | 16 | 155 | 153 | 153 | 138 | 1 | 0 | 90.2% (84.5–94) | 0.7% (0.1–3.6) | 0% (0–2.4) |
| Total North-west | 37 | 680 | 491 | 489 | 432 | 4 | 1 | 88.3% (85.2–90.9) | 0.8% (0.3–2.1) | 0.2% (0–1.1) |
| Total Italyb | 129 | 4,790 | 1,938 | 1,914 | 1,641 | 92 | 3 | 85.7% (84.1–87.2) | 4.8% (3.9–5.9) | 0.2% (0.1–0.5) |
CI: confidence interval; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
All percentages in the table are approximated to the first decimal, the value zero may therefore stand for < 0.05%. ‘RT-PCR-positive’ represent only cases tested in the laboratories involved and not all positive cases in an area.
a Marche followed a different experimental design (see Methods).
b The prevalence and totals for Italy and macro-area Centre do not include results from Marche.
Relative transmissibility of SARS-CoV-2 variants Alpha and Gamma
The three-strain transmission model was able to fit the epidemiological trends on hospital admissions and the estimated prevalence of the Alpha and Gamma VOC in all geographical aggregations and independently of the assumed degree of cross-protection (Figure 3 shows results for Italy when assuming no cross-protection or complete cross-protection).
Figure 3.
Model fits for prevalence of SARS-CoV-2 variants of concern and hospital admissions, Italy, 18 January–18 March 2021
CrI: credible intervals; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
Panels A–D: Model estimates, solid black lines: mean values; shaded areas: 95% CrI. Observed data, red points: mean values: red lines: 95% CrI. At each time, the model-estimated prevalence for a given lineage is computed as the ratio of simulated number of infectious individuals with that lineage and the simulated total number of infectious individuals. Panels E and F: Model-estimated and observed hospital admissions over time under the assumption of no cross-protection between wildtype or Alpha and Gamma. Black lines: mean values of model-estimated overall daily hospitalisations; shaded areas: 95%CrI. Coloured lines: mean values of model-estimated daily hospitalisations; black points: observed daily hospitalisations
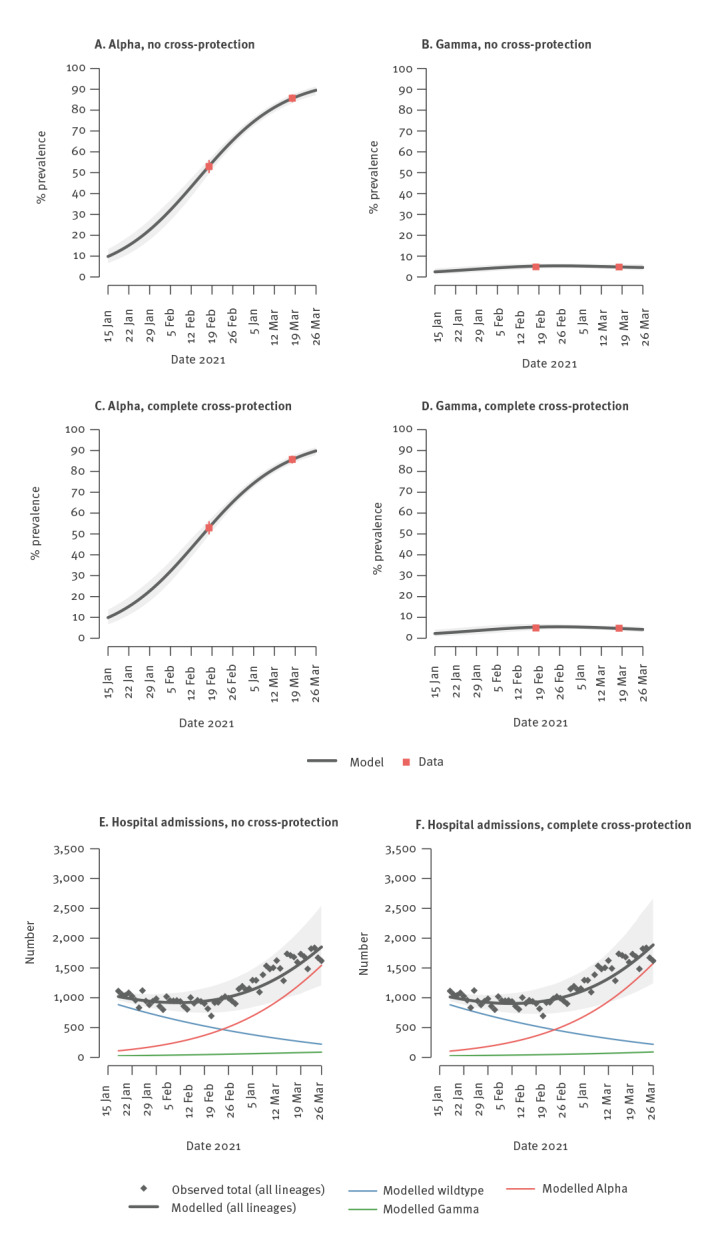
Independently of the geographical aggregation and of the assumed degree of cross-protection, we found a robust mean estimate for the relative transmissibility of Alpha, ranging between 1.48 and 1.73, with CrI ranging between 1.31 and 1.97 (Figure 4). Considering the national aggregation, estimates varied between 1.55 and 1.57 with CrI ranging between 1.45 and 1.66. When estimating the relative transmissibility of Alpha in the absence of significant co-circulation of Gamma, using data from Veneto and Lombardy, we found consistent values of 1.49 (95% CrI: 1.36–1.66) and 1.72 (95% CrI: 1.52–1.98) respectively (see the Supplement for results in this scenario).
Figure 4.
Estimates of the relative transmissibility of SARS-CoV-2 variants Alpha and Gamma, Italy, 18 February and 18 March 2021
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
Estimates are provided for different assumed values on the degree of cross protection (0: no cross-protection; 1: complete cross-protection) conferred by previous infection with the wildtype or Alpha against reinfection with Gamma. Points indicate the mean value of the estimated relative transmissibility; lines indicate 95% credible intervals. The panels represent four geographical aggregations for which the analysis was possible given the presence of the Gamma. Data from Marche were excluded from the national estimate and the macro-area Centre because of heterogeneity in data collection.

The estimated relative transmissibility of the Gamma variant was systematically lower than the one estimated for Alpha. For the national aggregation and for the three regions of the macro-area Centre (Lazio, Tuscany and Umbria) where the observed prevalence of Gamma was higher, we estimate a relative transmissibility of 1.12–1.24 under the assumption that there is no cross-protection (95% CrI: 1.03–1.42), growing linearly for increasing values of cross-protection, up to 1.39–1.46 (95% CrI: 1.26–1.63) under the assumption of complete cross-protection. Results for the macro-areas North-east and South reproduced a similar pattern, although these estimates were more variable because of the limited number of reported Gamma. The results remained robust when considering individual regions with high co-circulation, and when the degree of cross-protection was not assumed a priori but considered as a free model parameter (see the Supplement for detailed results of this sensitivity analysis).
Discussion
We provide a mathematical framework to estimate the relative transmissibility of competing SARS-CoV-2 variants of concern, taking as case study the co-circulation of Alpha and Gamma in Italy at the beginning of 2021. Based on two genomic surveillance surveys conducted across the whole Italian territory on 18 February and 18 March 2021, we showed that the Alpha variant was already dominant on 18 February in a majority of regions/AP (national prevalence: 54%) and had almost completely replaced historical lineages by 18 March (dominant in all regions/AP, national prevalence: 86%). At the same time, we found a substantial proportion of cases of the Gamma variant on 18 February, almost exclusively in Regions of central Italy (Lazio, Tuscany, Umbria and Marche, with an overall prevalence of 19%). Gamma was also identified in samples from Campania and Emilia Romagna, both with prevalences below 3%. The prevalence of Gamma remained similar on 18 March, suggesting that this VOC was not able to outcompete Alpha. However, on 18 March, lineage Gamma was identified in cases from six additional regions in northern (Piedmont, Veneto, Liguria) and southern Italy (Abruzzo, Molise, Sicily). We found only six cases of the Beta variant among the 1,239 analysed samples on 18 February, and only three of 1,908 on 18 March. Using data from these two surveys, we made use of a mathematical transmission model to estimate the relative advantage of these two VOC over the wildtype virus. Compared with historical lineages, we estimated a mean relative transmissibility of Alpha ranging between 1.55 and 1.57 (with 95% CrI between 1.45 and 1.66) in Italy. These values are consistent with available estimates from the UK [1,2,36,37] and France [12].
The estimated relative transmissibility of Gamma (compared with historical lineages) varied according to different assumptions on the degree of cross-protection granted by previous infection with historical lineages or Alpha: the estimate at the national level ranged from 1.12 (95% CrI: 1.03–1.23) in the case of complete immune evasion by Gamma to 1.39 (95% CrI: 1.26–1.56) in the case of complete cross-protection. This transmissibility advantage estimated for Gamma would have been sufficient for replacement of historical lineages in the absence of Alpha, independently of the degree of cross-protection (see the Supplement for detailed additional results). Previous estimates on the relative transmissibility of Gamma provided from a study in Manaus, Brazil, where the variant rapidly replaced historical lineages, were very broad (between 1.03 and 2.87), with an estimate of cross-protection between 12% and 90.0%.
As suggested by our work, the true degree of cross-protection between variants of concern is likely to be critical for their coexistence, and a key role will be played by the effectiveness of licensed vaccines against emerging strains. The slight decrease in Gamma prevalence over 1 month occurred under a condition of strict mitigation measures; if Gamma could have at least partially escaped immunity from infection with Alpha and from existing vaccines, this may have posed challenges to the roll-out of vaccination programmes. Furthermore, if some degree of cross-protection exists, a higher proportion of asymptomatic infections may have been observed, posing challenges to surveillance and control. It is crucial to keep continuously monitoring the relative advantage of newly emerging VOC to efficiently contain or mitigate outbreaks and evaluate the impact of the adopted interventions.
We acknowledge a number of limitations for this study. The sample size was calculated to have the statistical power to detect different lineages at the macro-area level. As such, regional estimates of prevalence should be taken with caution because the number of sequenced samples was small. Samples were randomly selected for sequencing among cases diagnosed by the laboratories, but some degree of correlation between them (e.g. cases belonging to an over-represented cluster on that day) cannot be completely excluded, especially in regions with smaller sample sizes. One region used a different laboratory methodology in the second survey and those data were excluded from the computation of the national prevalence of the variants. Possible biases in the estimate are expected to be minimal, since cases from this region represented only ca 1.5% of the total. The model was calibrated using available data on two genomic surveys only, and this represents a limitation of our study. However, for biological reasons, we assumed a simple exponential growth model for the estimation of the prevalence of Alpha and Gamma variants, according to which the prevalence over time is a logistic function, and this function is univocally defined by two points in time. As future measurements of the prevalence of co-circulating variants become available, a validation of the estimated prevalence over time is warranted.
Regarding model estimates on transmissibility, we could not take into account major determinants of SARS-CoV-2 transmission dynamics such as possible differences across strains in the generation time and duration of viral shedding [38], age-specific susceptibility or transmissibility [39-41], the age-specific proportion of asymptomatic individuals and their relative transmissibility [39], the severity of symptoms [42,43], because this kind of data on the variants was not available. For example, we show that if the duration of infectiousness, rather than the transmissibility, were responsible for the competitive advantage of Alpha over the historical lineages, we would expect a much higher increase in the duration of infection (80–150%) compared with the estimated increase in transmissibility (see the Supplement for detailed results of this sensitivity analysis). This would have implications for the preventive effectiveness of timely isolation of individuals and for quarantine times. Owing to these uncertainties, it is virtually impossible at this stage to model the potential lineage-specific impact of existing mitigation measures, nor to factor in the potential effect of vaccines administered until now. These factors are likely to shape the future outcome of the epidemiological competition across strains. For these reasons, we acknowledge that this work represents only a first step towards the understanding of the relationship among multiple SARS-CoV-2 variants and that our findings need to be corroborated by further evidence taking into account more details on the characteristics of emerging lineages as they become consolidated. Finally, our model did not consider possible differences in the interventions in place at regional level, and this may have resulted in heterogeneous estimates of the relative transmissibility of the variants in different areas of interest.
Despite these limitations, we provide a simple and useful modelling framework to assess the relative advantage of competing strains, by assuming different degrees of cross-protection among them. We found that the Gamma variant was not able to outcompete Alpha in Italy under existing mitigation measures, with Alpha becoming largely dominant and Gamma remaining stable over a month of co-circulation. We suggest that this may be due to a lower transmissibility of Gamma compared with Alpha and independent of its ability to re-infect individuals with previous infections with historical strains or the Alpha variant.
Conclusion
We believe that our work provides an important tool to evaluate the relative transmissibility advantage of the SARS-CoV-2 Delta variant and of other newly emerging strains in the foreseeable future.
Acknowledgements
The authors thank Francesco Maraglino, Ministry of Health, for supporting coordination activities, and the laboratory staff throughout the country who participated to the surveys.
Funding: G.G. and S.M. acknowledge funding from EU grant 874850 MOOD (catalogued as MOOD 019).
Supplementary Data
COVID-19 National Microbiology Surveillance Study Group
Angela Di Martino, Luigina Ambrosio, Alessandra Lo Presti, Stefano Fiore, Concetta Fabiani, Eleonora Benedetti, Giuseppina Di Mario, Marzia Facchini, Simona Puzelli, Laura Calzoletti, Stefano Fontana, Giulietta Venturi, Claudia Fortuna, Giulia Marsili, Antonello Amendola, Department of Infectious Diseases, Istituto Superiore di Sanità, Rome; Liborio Stuppia, Laboratorio di Genetica Molecolare, Center for Advanced Studies and Technology (CAST), Università degli Studi “G. d'Annunzio”, Chieti; Giovanni Savini, Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise “Giuseppe Caporale”, Teramo; Antonio Picerno, Teresa Lopizzo, UOC Analisi Chimico Cliniche e Microbiologiche, AOR “San Carlo”, Potenza; Domenico Dell’Edera, UOSD Laboratorio di Genetica Medica, P.O. “Madonna delle Grazie”, Matera; Pasquale Minchella, SOC Microbiologia e Virologia, AO “Pugliese-Ciaccio”, Catanzaro; Francesca Greco, UOC Microbiologia e Virologia, PO “Annunziata”, Cosenza; Giuseppe Viglietto, Laboratorio di Genomica Funzionale e Patologia Molecolare, Università degli Studi “Magna Graecia”, Catanzaro; Luigi Atripaldi, AORN “Azienda Sanitaria dei Colli”, Napoli; Antonio Limone, Istituto Zooprofilattico Sperimentale del Mezzogiorno, Napoli; Pierlanfranco D’Agaro, SC UCO Igiene e Sanità Pubblica, Azienda Sanitaria Universitaria Giuliano-Isontina (ASUGI), Trieste; Danilo Licastro, Laboratorio Genomica ed Epigenomica, Area Science Park, Basovizza, Trieste; Stefano Pongolini, Unità di Analisi del Rischio ed Epidemiologia Genomica, Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia-Romagna, Parma; Vittorio Sambri, Dipartimento di Medicina Specialistica Diagnostica e Sperimentale (DIMES), Università di Bologna, Bologna, & UOC Microbiologia, AUSL della Romagna, Cesena; Giorgio Dirani, Silvia Zannoli, UOC Microbiologia, AUSL della Romagna, Cesena; Paola Affanni, Maria Eugenia Colucci, Laboratorio di Igiene e Sanità Pubblica, Dipartimento di Medicina e Chirurgia, Università degli Studi di Parma, Parma; Maria Rosaria Capobianchi, Laboratorio di Virologia, Istituto Nazionale Malattie Infettive IRCCS “L. Spallanzani”, Roma; Giancarlo Icardi, Bianca Bruzzone, Flavia Lillo, Andrea Orsi, Laboratorio di Riferimento Regionale per le Emergenze di Sanità Pubblica (LaRESP), Liguria; Elena Pariani, Dipartimento di Scienze Biomediche per la Salute, Università degli Studi di Milano, Milano; Fausto Baldanti, Unità Virologia Molecolare, Fondazione IRCCS Policlinico “San Matteo”, Università di Pavia, Pavia; Maria Rita Gismondo, UOC Microbiologia Clinica, Virologia e diagnostica delle Bioemergenze, ASST “Fatebenefratelli-Sacco”, Milano; Fabrizio Maggi, SC Laboratorio Microbiologia, ASST “Sette Laghi”, Varese; Arnaldo Caruso, Laboratorio di Microbiologia e Virologia, ASST “Spedali Civili di Brescia”, Brescia; Ferruccio Ceriotti, Fondazione IRCCS “Ca' Granda” Ospedale Maggiore Policlinico di Milano, Milano; Maria Beatrice Boniotti, Ilaria Barbieri, Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna, Brescia; Patrizia Bagnarelli, Stefano Menzo, AOU “Ospedali Riuniti”, Ancona; Silvio Garofalo, Massimiliano Scutellà, Ospedale “Cardarelli”, Campobasso; Elisabetta Pagani, Laboratorio Aziendale di Microbiologia e Virologia, Azienda Sanitaria dell’Alto Adige, Bolzano; Lucia Collini, Microbiologia e Virologia, Presidio Ospedaliero “Santa Chiara”, Trento; Valeria Ghisetti, Laboratorio di Microbiologia e Virologia, ASL Città di Torino, Torino; Silvia Brossa, IRCCS Fondazione del Piemonte per l'Oncologia, Candiolo, Torino; Giuseppe Ru, Elena Bozzetta, Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d’Aosta, Torino; Maria Chironna, Laboratorio di Epidemiologia Molecolare e Sanità Pubblica, AOUC Policlinico di Bari, Bari; Antonio Parisi, Istituto Zooprofilattico Sperimentale della Puglia e della Basilicata, Putignano, Bari; Salvatore Rubino, Caterina Serra, Department of Biomedical Science, University of Sassari, SC Microbiologia e Virologia, AOU Sassari, Sassari; Giovanna Piras, UOC Ematologia, PO “San Francesco,” Azienda Tutela Salute, ASSL Nuoro, Nuoro; Ferdinando Coghe, Laboratorio Generale (HUB) di analisi chimico cliniche e microbiologia, PO “Duilio Casula”, AOU di Cagliari, Cagliari; Francesco Vitale, Fabio Tramuto, Laboratorio di Riferimento Regionale per la Sorveglianza Epidemiologica e Virologica del PROMISE - AOUP “Giaccone”, Palermo; Guido Scalia, Concetta Ilenia Palermo, Laboratorio di Virologia Clinica, AOUP “V. Emanuele”, PO “Gaspare Rodolico”, Catania; Giuseppe Mancuso, Laboratorio di Virologia, UOC Microbologia, AOU “G. Martino ”, Messina; Teresa Pollicino, Laboratorio di Diagnostica Molecolare, UOSD Gestione Centralizzata dei Laboratori, AOU “G. Martino ”, Messina; Francesca Di Gaudio, Centro Regionale per la Qualità (CRQ), Palermo; Stefano Vullo, Stefano Reale, Istituto Zooprofilattico Sperimentale della Sicilia, Palermo; Maria Grazia Cusi, UOC Microbiologia e Virologia, Azienda Ospedaliera Universitaria Senese, Dipartimento di Biotecnologie Mediche, Università degli Studi di Siena, Siena; Gian Maria Rossolini, SOD Microbiologia e Virologia, AOU “Careggi”, Firenze; Mauro Pistello, UOC Virologia, AOU Pisana, Pisa; Antonella Mencacci, Barbara Camilloni, Microbiology and Clinical Microbiology, Department of Medicine and Surgery, University of Perugia, Perugia; Silvano Severini, Istituto Zooprofilattico Sperimentale dell’Umbria e delle Marche, Perugia; Massimo Di Benedetto, Laboratorio Analisi Cliniche, Ospedale “Parini”, Aosta; Calogero Terregino, Isabella Monne, Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Padova; Valeria Biscaro, UOC Microbiologia-Virologia, AULSS2 La Marca, PO Treviso, Treviso.
Conflict of interest: M.A. reports receiving funding for research not related to COVID-19 from Seqirus S.r.l. (San Martino, Italy). None of the other authors reported any potential conflicts of interest.
Authors’ contributions: PS, FT, GG, SM and GR conceived the study. PS,FT,GG and SM, wrote the manuscript. FT, VM, AM, MSS, PP, CMG, MM and MA, elaborated the data for mathematical model. MDM and XA collected the epidemiological data. SB helped in the discussion on the results. SM revised critically the study and the results. The members of the COVID-19 National Microbiology Surveillance Study Group collected the samples and sequenced the RNA for the molecular typing.
References
- 1. Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372(6538):eabg3055. 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature. 2021;593(7858):266-9. 10.1038/s41586-021-03470-x [DOI] [PubMed] [Google Scholar]
- 3.European Centre for Disease Prevention and Control (ECDC). SARS-CoV-2 - increased circulation of variants of concern and vaccine rollout in the EU/EEA, 14th update. Risk assessment. Stockholm: ECDC; 2021. Available from: https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-variants-vaccine-fourteenth-update-february-2021
- 4. Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, et al. Emergence of SARS-CoV-2 B.1.1.7 lineage - United States, December 29, 2020-January 12, 2021. MMWR Morb Mortal Wkly Rep. 2021;70(3):95-9. 10.15585/mmwr.mm7003e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Claro IM, da Silva Sales FC, Ramundo MS, Candido DS, Silva CAM, de Jesus JG, et al. Local transmission of SARS-CoV-2 lineage B.1.1.7, Brazil, December 2020. Emerg Infect Dis. 2021;27(3):970-2. 10.3201/eid2703.210038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pango Lineages. Summary of B.1.1.7 data. Oxford: Pango network. [Accessed: 17 Jan 2022]. Available from: https://cov-lineages.org/global_report_B.1.1.7.html
- 7.National Institute of Infectious Diseases (NIID). Brief report: New variant strain of SARS-CoV-2 Identified in travelers from Brazil. Tokyo; NIID;2021. Available from: https://www.niid.go.jp/niid/en/2019-ncov-e/10108-covid19-33-en.html
- 8.Faria NR, Claro IM, Candido D, Moyses Franco LA, Andrade PS, Coletti TM, et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings. Virological. 2021. Available from: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586
- 9. Buss LF, Prete CA, Jr, Abrahim CMM, Mendrone A, Jr, Salomon T, de Almeida-Neto C, et al. Three-quarters attack rate of SARS-CoV-2 in the Brazilian Amazon during a largely unmitigated epidemic. Science. 2021;371(6526):288-92. 10.1126/science.abe9728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sabino EC, Buss LF, Carvalho MPS, Prete CA, Jr, Crispim MAE, Fraiji NA, et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet. 2021;397(10273):452-5. 10.1016/S0140-6736(21)00183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021;592(7854):438-43. 10.1038/s41586-021-03402-9 [DOI] [PubMed] [Google Scholar]
- 12. Gaymard A, Bosetti P, Feri A, Destras G, Enouf V, Andronico A, et al. Early assessment of diffusion and possible expansion of SARS-CoV-2 lineage 20I/501Y.V1 (B.1.1.7, variant of concern 202012/01) in France, January to March 2021. Euro Surveill. 2021;26(9):2100133. 10.2807/1560-7917.ES.2021.26.9.2100133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dejnirattisai W, Zhou D, Supasa P, Liu C, Mentzer AJ, Ginn HM, et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell. 2021;184(11):2939-2954.e9. 10.1016/j.cell.2021.03.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen RE, Zhang X, Case JB, Winkler ES, Liu Y, VanBlargan LA, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. 2021;27(4):717-26. 10.1038/s41591-021-01294-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593(7857):130-5. 10.1038/s41586-021-03398-2 [DOI] [PubMed] [Google Scholar]
- 16. Supasa P, Zhou D, Dejnirattisai W, Liu C, Mentzer AJ, Ginn HM, et al. Reduced neutralization of SARS-CoV-2 B.1.1.7 variant by convalescent and vaccine sera. Cell. 2021;184(8):2201-2211.e7. 10.1016/j.cell.2021.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoffmann M, Arora P, Groß R, Seidel A, Hörnich BF, Hahn AS, et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell. 2021;184(9):2384-2393.e12. 10.1016/j.cell.2021.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nonaka CKV, Franco MM, Gräf T, de Lorenzo Barcia CA, de Ávila Mendonça RN, de Sousa KAF, et al. Genomic evidence of SARS-CoV-2 reinfection involving E484K spike mutation, Brazil. Emerg Infect Dis. 2021;27(5):1522-4. 10.3201/eid2705.210191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naveca F, da Costa C, Nascimento V, Souza A, Corado A, Nascimento F, et al. SARS-CoV-2 reinfection by the new variant of concern (VOC) P.1 in Amazonas, Brazil. Virological. 2021. Available from: https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596
- 20. Faria NR, Mellan TA, Whittaker C, Claro IM, Candido DDS, Mishra S, et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science. 2021;372(6544):815-21. 10.1126/science.abh2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zucman N, Uhel F, Descamps D, Roux D, Ricard JD. Severe reinfection with South African African severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant 501Y.V2. Clin Infect Dis. 2021;73(10):1945-6. 10.1093/cid/ciab129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horby P, Bell I, Breuer J, Cevik M, Challen R, Davies N, et al. Update note on B.1.1.7 severity. London: New and Emerging Respiratory Virus Threats Advisory Group; 2021. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/961042/S1095_NERVTAG_update_note_on_B.1.1.7_severity_20210211.pdf
- 23. Challen R, Brooks-Pollock E, Read JM, Dyson L, Tsaneva-Atanasova K, Danon L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ. 2021;372:n579. 10.1136/bmj.n579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Madhi SA, Baillie V, Cutland CL, Voysey M, Koen AL, Fairlie L, et al. Efficacy of the ChAdOx1 nCoV-19 covid-19 vaccine against the B. 1.351 variant. N Engl J Med. 2021;384(20):1885-98. 10.1056/NEJMoa2102214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou D, Dejnirattisai W, Supasa P, Liu C, Mentzer AJ, Ginn HM, et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell. 2021;184(9):2348-2361.e6. 10.1016/j.cell.2021.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dipartimento di Protezione Civile. COVID-19 Italia. [COVID-19 data Italy]. Data repository. Rome; Protezione Civile; 24 Feb 2020. Italian. Available from: https://github.com/pcm-dpc/COVID-19
- 27.Istituto Superiore di Sanità (ISS). COVID-19 ISS OpenData. Rome: ISS; 24 Feb 2020. Italian. Available from: https://www.epicentro.iss.it/coronavirus/open-data/covid_19-iss.xlsx
- 28. Riccardo F, Ajelli M, Andrianou XD, Bella A, Del Manso M, Fabiani M, et al. Epidemiological characteristics of COVID-19 cases and estimates of the reproductive numbers 1 month into the epidemic, Italy, 28 January to 31 March 2020. Euro Surveill. 2020;25(49):2000790. 10.2807/1560-7917.ES.2020.25.49.2000790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ministero della Salute. Covid-19, il nuovo Dpcm del Governo. [Covid-19, the new Decree of the President of the Council of Ministers]. Rome: Ministero della Salute; 3 Mar 2021. Italian. Available from: https://www.salute.gov.it/portale/nuovocoronavirus/dettaglioNotizieNuovoCoronavirus.jsp?lingua=italiano&menu=notizie&p=dalministero&id=5354
- 30. Manica M, Guzzetta G, Riccardo F, Valenti A, Poletti P, Marziano V, et al. Impact of tiered restrictions on human activities and the epidemiology of the second wave of COVID-19 in Italy. Nat Commun. 2021;12(1):4570. 10.1038/s41467-021-24832-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Istituto Superiore di Sanità. Prevalenza delle varianti VOC 202012/01 (lineage B.1.1.7), P.1, e 501.V2 (lineage B.1.351) in Italia. Indagine del 18 febbraio 2021. [Prevalence of VOC variants 202012/01 (lineage B.1.1.7), P1 and 501.V2 (lineage B.1.351) in Italy]. Rome: ISS; 2021. Italian. Available from: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-indagini-rapide-18-febbraio-2021.pdf
- 32.Istituto Superiore di Sanità. Prevalenza delle varianti VOC (Variant Of Concern) in Italia: lineage B.1.1.7, P.1, P.2, lineage B.1.351, lineage B.1.525 (Indagine del 18/3/2021). [Prevalence of VOC (variant of concern) variants in Italy: lineage B.1.1.7, P.1, P.2, lineage B.1.351, lineage B.1.525 (survey from 18/3/2021)]. Rome: ISS; 2021. Italian. Available from: https://www.epicentro.iss.it/coronavirus/pdf/sars-cov-2-monitoraggio-varianti-indagini-rapide-18-marzo-2021.pdf
- 33.Ministero della Salute. Indagine rapida per la valutazione della prevalenza delle varianti VOC 202012/01 (ovvero lineage B.1.1.7-Regno Unito), P1 (ovvero Brasiliana), e 501.V2 (ovvero lineage B.135- Sud Africana) in Italia. [Rapid survey for the assessment of the prevalence of VOC variants 202012/01 (i.e. lineage B.1.1.7-United Kingdom), P1 (i.e. Brazilian), and 501.V2 (i.e. lineage B.135- South African) in Italy]. Rome: Ministero della Salute; 17 Feb 2021. Italian. Available from: https://www.trovanorme.salute.gov.it/norme/renderNormsanPdf?anno=2021&codLeg=78864&parte=1%20&serie=null
- 34.Centers for Disease Control (CDC). SARS-CoV-2 variant classifications and definitions. Atlanta: CDC. [Accessed: 17 Jan 2022]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html
- 35. Cereda D, Manica M, Tirani M, Rovida F, Demicheli V, Ajelli M, et al. The early phase of the COVID-19 epidemic in Lombardy, Italy. Epidemics. 2021;37:100528. 10.1016/j.epidem.2021.100528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao S, Lou J, Cao L, Zheng H, Chong MKC, Chen Z, et al. Quantifying the transmission advantage associated with N501Y substitution of SARS-CoV-2 in the UK: an early data-driven analysis. J Travel Med. 2021;28(2):taab011. 10.1093/jtm/taab011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leung K, Shum MH, Leung GM, Lam TT, Wu JT. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill. 2021;26(1):2002106. 10.2807/1560-7917.ES.2020.26.1.2002106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kissler SM, Fauver JR, Mack C, Tai CG, Breban MI, Watkins AE, et al. Viral dynamics of SARS-CoV-2 variants in vaccinated and unvaccinated persons. N Engl J Med. 2021;385(26):2489-91. 10.1056/NEJMc2102507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu S, Wang W, Wang Y, Litvinova M, Luo K, Ren L, et al. Infectivity, susceptibility, and risk factors associated with SARS-CoV-2 transmission under intensive contact tracing in Hunan, China. Nat Commun. 2021;12(1):1533. 10.1038/s41467-021-21710-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brookman S, Cook J, Zucherman M, Broughton S, Harman K, Gupta A. Effect of the new SARS-CoV-2 variant B.1.1.7 on children and young people. Lancet Child Adolesc Health. 2021;5(4):e9-10. 10.1016/S2352-4642(21)00030-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walker AS, Vihta KD, Gethings O, Pritchard E, Jones J, House T, et al. Increased infections, but not viral burden, with a new SARS-CoV-2 variant. medRxiv 2021.01.13.21249721. Preprint. 10.1101/2021.01.13.21249721 [DOI]
- 42. Davies NG, Jarvis CI, Edmunds WJ, Jewell NP, Diaz-Ordaz K, Keogh RH. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. 2021;593(7858):270-4. 10.1038/s41586-021-03426-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Challen R, Brooks-Pollock E, Read JM, Dyson L, Tsaneva-Atanasova K, Danon L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ. 2021;372(579):n579. 10.1136/bmj.n579 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.