

Abstract
TCF7L2 encodes transcription factor 7-like 2 (OMIM 602228), a key mediator of the evolutionary conserved canonical Wnt signaling pathway. Although several large-scale sequencing studies have implicated TCF7L2 in intellectual disability and autism, both the genetic mechanism and clinical phenotype have remained incompletely characterized. We present here a comprehensive genetic and phenotypic description of 11 individuals who have been identified to carry de novo variants in TCF7L2, both truncating and missense. Missense variation is clustered in or near a high mobility group box domain, involving this region in these variants’ pathogenicity. All affected individuals present with developmental delays in childhood, but most ultimately achieved normal intelligence or had only mild intellectual disability. Myopia was present in approximately half of the individuals, and some individuals also possessed dysmorphic craniofacial features, orthopedic abnormalities, or neuropsychiatric comorbidities including autism and attention-deficit/hyperactivity disorder (ADHD). We thus present an initial clinical and genotypic spectrum associated with variation in TCF7L2, which will be important in informing both medical management and future research.
Keywords: autism, intellectual disability, myopia, neurodevelopmental disorder, TCF7L2
Introduction:
TCF7L2 encodes a high mobility group (HMG) box-containing transcription factor and is located on chromosome 10q25.2-q25.3. Although it was initially identified and referred to as TCF4 (Castrop et al., 1992; Clevers, 2006), it should not be confused with the currently designated TCF4 (ITF2/SEF2-1B/SEF2/E2-2, MIM 602272), which is located on Chromosome 18 and associated with Pitt–Hopkins syndrome. TCF7L2 mediates canonical Wnt signaling. Signaling by secreted Wnt proteins through this pathway leads to release of the protein beta-catenin (CTNNB1) from a repressive degradation complex in the cytoplasm, allowing it to accumulate and translocate to the nucleus, where it acts with DNA-binding factors including TCF7L2 to turn on Wnt-responsive target genes. TCF7L2 thus acts with beta-catenin as an on/off switch for transcriptional regulation. Through mostly genome-wide association studies, TCF7L2 has been involved in a variety of human disease, including Type 2 diabetes mellitus, colon cancer, and schizophrenia (Alkelai et al., 2012; Folsom et al., 2008; Grant et al., 2006). TCF7L2 is also known to be critical in central nervous system development (Chodelkova et al., 2018; Lee et al., 2017; Nagalski et al., 2013). It has been directly involved in processes as diverse as neurogenesis and thalamic development to mediating the effects of neuropsychiatric pharmacological agents including lithium and nicotine (Chodelkova et al., 2018; Duncan et al., 2019; Lee et al., 2017; Misztal et al., 2017; Nagalski et al., 2013). Large-scale sequencing studies have also identified a handful of isolated patients with de novo variants in TCF7L2 in association with neurodevelopmental disorders, but clinical details are lacking (Iossifov et al., 2014; De Rubeis et al., 2014; Lelieveld et al., 2016; Jeremy F McRae et al., 2017 (Deciphering Developmental Disorders [DDD] Study), 2017; Guo et al., 2018; Liu et al., 2018; Satterstrom et al., 2020; Wang et al., 2020). TCF7L2 encodes multiple alternatively spliced transcripts and alternative splicing has been demonstrated to play an important role in the function and specificity of the transcriptional repertoire of TCF7L2 in a variety of tissues and contexts, including the brain (Nagalski et al., 2013; Prokunina-Olsson et al., 2009; Weise et al., 2009). TCF7L2 is significantly intolerant to loss-of-function (LOF) variation, with significantly fewer observed LOF variants as compared to predicted, as indicated in the probability of being loss-of-function intolerant (pLI) score of 0.99–1 reported in the gnomAD and ExAC databases. There is also a region of missense constraint encompassing the HMG box domain indicating additional intolerance to missense variation (Samocha et al., 2017). We describe here the genotypic and clinical phenotypic spectrum of 11 individuals with de novo, heterozygous variants in TCF7L2 presenting with a neurodevelopmental disorder.
Materials and Methods:
Patients were ascertained from GeneMatcher through the Match-maker Exchange Network between May 2019 and December 2020 (Philippakis et al., 2015; Sobreira et al., 2015). TCF7L2 variants were detected on exome sequencing in 10 individuals, and on a trio autism/ intellectual disability gene panel at a commercial lab in one individual. No additional plausible candidate gene variants were identified (Supplementary Table 1). One additional patient was excluded from the cohort because the phenotype was confounded by perinatal hypoxic–ischemic injury; the data for this individual (S1) are included in Supplementary Table 1. Institutional review board approval was obtained. The reported variants in TCF7L2 in our cohort are annotated on the coding sequence and protein structure in Figure 1. We found a marked pattern of clustering of the variants, with all missense variants located in or immediately adjacent to the PFAM predicted HMG box domain. Two residues, Tyr423 and Asn381, are each affected by two different missense variants (Figure 1). All of the missense variants occur at highly conserved locations, and none are found in the gnomAD database v2.1.1. All truncating variants occurred greater than 55 nucleotides upstream of the last exon–exon junction and are predicted to be subject to nonsense-mediated decay. The two splice variants we report are predicted by splice prediction tools (MaxEnt, NNSPLICE,SSF) with high likelihood to affect splicing.
Figure 1:
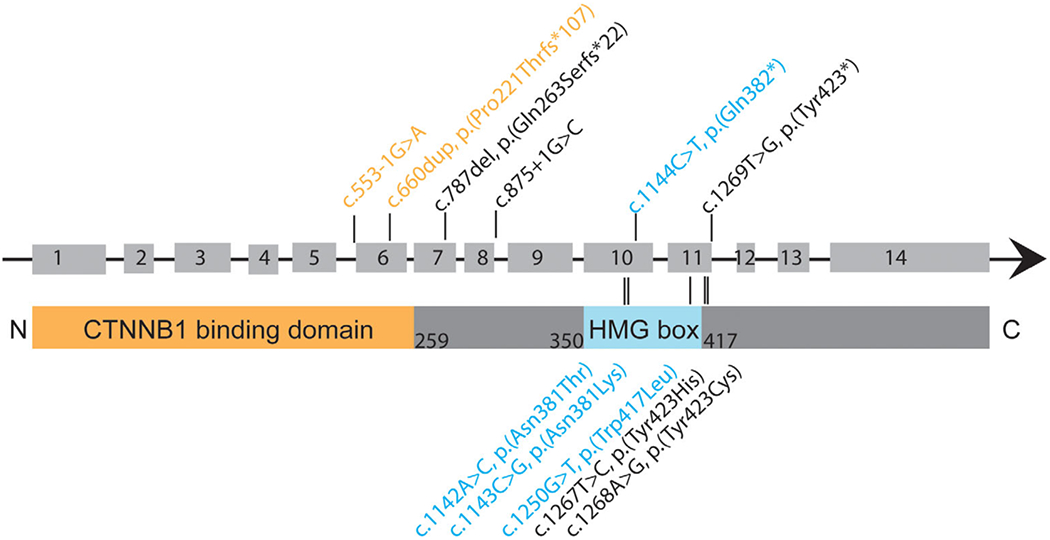
Annotation of loss of function and missense variation in TCF7L2. Top bar indicates exon structure of NM_001146274.1. Second bar represents protein structure with PFAM amino acid ranges overlaid, that is, the CTNNB1-binding domain (orange) spanning amino acids 1– 259, and the HMG box domain (light blue) spanning 350–417. Predicted splicing and loss-of-function variants are annotated above the figure and missense variants are annotated below
Individuals with truncating variants and missense variants in our cohort present with largely indistinguishable phenotypes, although sample size is too small to make definitive conclusions regarding this (see Table 1 and Supplementary Table 1). All individuals present with developmental delays, including delayed speech and motor milestones. Intellectual abilities range from average cognitive functioning to mild/moderate intellectual disability. Variability in speech language abilities is notable regardless of intellectual functioning; abilities range from individuals who are completely non-verbal to individuals with hypophonia, dysphasia, and dysarthria. Autism and/or social communication deficits are frequently observed, and comorbid attention-deficit/hyperactivity disorder (ADHD) and executive functioning challenges are also seen. One individual has a history of glioma status post resection and focal motor seizures; this individual was also found to have a heterozygous TP53 variant of uncertain significance, and is being managed as Li–Fraumeni syndrome. Myopia is seen in 6 of 11 individuals, and is very severe in two We also reviewed previous reports of variation in TCF7L2 to evaluate whether phenotypes were consistent with this cohort (Supplemental Table 2); however, our assessment of previous reports was limited due to lack of validation, absent clinical descriptions, and varied methodological approaches. Thus, these limitations preclude definitive interpretation of previously reported variants.
Table 1:
Clinical features of affected patients.
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 | Totals | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant Information | ||||||||||||
| Coding variant (c. ) ( NM_001146274.1) | c.553-1G>A | c.1269T>G | c.787del | c.1144C>T | c.660dup | c.875+1G>C | c.1143C>G | c.1142A>C | c.1250G>T | c.1267T>C | c.1268A>G | |
| Amino acid variant (p. ) | p.(Tyr423*) | p.(Gln263 Serfs*22) | p.(Gln382*) | p.(Pro221 Thrfs*107) | p.(Asn381Lys) | p.(Asn381Thr) | p.(Trp417Leu) | p.(Tyr423His) | p.(Tyr423Cys) | |||
| Inheritance | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | |
| Demographics | ||||||||||||
| Sex | male | male | female | female | female | male | male | male | male | male | male | |
| Age at evaluation | 12 y | 11 y | 18 y | 8 y 7 m | 5 y 7 m | 11 y | 4 y | 17 y 10 m | 3 y 9 m | 5 y 7 m | 7 y 6 m | |
| Development | ||||||||||||
| Motor delay? | yes | yes | yes | yes | yes | yes | no | yes | no | no | yes | n=8/11 |
| Age at walking | 16 m | 24 m | 18 m | 12-13 m | 15 m | 14 m | 14 m | 15 m | 18 m | 14 m | 24 m | |
| Speech delay? | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | n=11/11 |
| Age at first words | 24 m | delayed | 7 years | 4 y | unknown | unknown | 2.5 y | unknown | 18 m | unknown | not verbal | |
| Verbal at evaluation? | yes | minimally | yes | yes | yes | yes | yes | yes | yes | no | no | n=8/11 |
| Intellectual disability? | no | yes | yes | no | no | no | no | yes | no | yes | yes | |
| Behavioral Features | ||||||||||||
| Autism? | no | yes | yes | yes | no | no | no | no | no | no | yes | n=4/11 |
| ADHD? | yes | yes | no | yes | no | yes | no | no | no | no | no | n=4/11 |
| Sleep disturbances ? | no | yes | yes | no | yes | no | yes | no | no | no | no | n=4/11 |
| Neurologic features | ||||||||||||
| Tone abnormalities? | no | yes | no | no | no | no | no | yes | no | no | yes | n=3/11 |
| Other clinical features | ||||||||||||
| Dysmorphic features? | no | yes | no | yes | yes | yes | yes | yes | no | yes | yes | n=8/11 |
| Ophthalmology findings? | yes | no | no | yes | yes | yes | no | yes | no | yes | yes | n=7/11 |
| Dermatology findings? | no | yes | no | yes | yes | no | yes | no | no | no | yes | n=5/11 |
| Orthopedic findings? | no | no | no | yes | yes | yes | yes | yes | no | no | yes | n=6/11 |
We also reviewed previous reports of variation in TCF7L2 to evaluate whether phenotypes were consistent with this cohort (Supplemental Table 2); however, our assessment of previous reports was limited due to lack of validation, absent clinical descriptions, and varied methodological approaches. Thus, these limitations preclude definitive interpretation of previously reported variants.
Discussion:
We present a series of 11 patients with de novo, heterozygous variants in TCF7L2 manifesting with neurodevelopmental abnormalities. All individuals had initial developmental delays, and intellectual and verbal abilities ultimately demonstrate significant heterogeneity. Some individuals have average cognitive functioning and fluent speech, while others are nonverbal. Other phenotypic features variably included autism spectrum disorder, social communication disorder, ADHD, speech–language impairment, dysmorphic features, myopia, hypertrichosis, and orthopedic abnormalities.
In this cohort, all missense variants occurred in or directly adjacent to the HMG box domain. The HMG box domain is an evolutionary conserved region that mediates interactions with DNA. This region is highly missense constrained. Interestingly, we also identify one splice variant that occurs at the start of an exon immediately following an alternatively spliced exon that is absent in both the canonical and the highest brain expressed isoforms (c.553-1G>A, Gnomad v2.1.1). Although speculative, it is thus possible that in addition to LoF as a possible mechanism of pathogenicity, this variant may lead to inappropriate exon retention.
No clear phenotypic differences were observed between individuals in this cohort with truncating and missense variants. For example, the five individuals with missense variants demonstrated a wide range of cognitive functioning similar to the individuals with truncating variants. Across the different forms of variation, individuals also shared findings like myopia and hypertrichosis. We hypothesize that the missense variation clustering at the HMG domain may interfere with appropriate DNA binding and interaction, contributing to a similar LoF effect as the truncating variants.
Given the genomic wide association study findings of intronic variants associated with diabetes risk, it is also interesting that there are no reported endocrine abnormalities, including diabetes mellitus, in any of the patients presented here, although it is important to note that this cohort reflects a predominately pediatric population and thus may not yet manifest certain findings.
TCF7L2 has been implicated in oligodendrocyte development and it has recently been posited that expression in this cellular subtype may represent an underappreciated mechanism of pathogenicity in neurodevelopmental disorders (Polioudakis et al., 2019; Ye et al., 2009; Zhao et al., 2016). Interestingly, recent work on Pitt– Hopkins syndrome, caused by mutations in TCF4, as well as idiopathic autism, has recently implicated oligodendrocyte pathology in autism (Phan et al., 2020). Further work will be needed to identify more definitively the cellular subtypes and neuronal circuitry responsible for mediating the effects of variation in TCF7L2, as well as functional interrogation of the described variation.
In conclusion, we present 11 patients with de novo, heterozygous variants in TCF7L2 presenting with a distinct neurodevelopmental disorder associated with initial developmental delay, speech–language difficulties, and variable risk for intellectual disability, autism, ADHD, myopia, and orthopedic abnormalities. All reported missense variants occurred in or adjacent to the HMG box domain. The phenotypes of individuals with missense and truncating variants were indistinguishable. Based on this, we hypothesize that the molecular mechanism for this disorder is haploinsufficiency, although further work to confirm this is required on a research basis.
Supplementary Material
Acknowledgements:
The authors thank the families for their invaluable participation in this study. For Family 11, we thank Muna Al-Saffar (research coordination), Ganeshwaran H. Mochida (neurological evaluation), Ramzi H. Nasir (developmental evaluation), and Lariza Rento and Samantha Kirkham (Sanger sequencing). We thank Abbe Lai for assistance with computational analysis.
Some of this work was supported by PROGETTO GENE (GENE— Genomic analysis Evaluation NEtwork) founded by PROGETTI DI INNOVAZIONE IN AMBITO SANITARIO E SOCIO SANITARIO (BANDO EX DECRETO N. 2713 DEL 28/02/2018). Caroline Dias is supported in part by the National Institute of Mental Health (T32MH112510, Translational Post-doctoral Training in Neurodevelopment). For family 11, this project was supported by the Qatar National Research Fund (NPRP 5-175-3-051) (Tawfeg Ben-Omran, Laila Mahmoud, Christopher A. Walsh), as well as by NINDS 1R01 NS035129 to Christopher A. Walsh. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) funded by the National Human Genome Research Institute; the National Eye Institute; and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. Christopher A. Walsh is an Investigator of the Howard Hughes Medical institute. One individual (P4) was ascertained in the Duke Genome Sequencing Clinic (Duke Protocol number 00032301). Funding for the Duke Genome Sequencing Clinic is supported by the Duke University Health System. Francois Lecoquierre and Anne-Marie Guerrot received funding from European Union and Région Normandie in the context of Recherche Innovation Normandie (RIN 2018). Europe is involved in Normandie with the European Regional Development Fund (ERDF).
Funding information:
Duke University Health System; European Union and Région Normandie; National Human Genome Research Institute grant, Grant/ Award Number: HG009141; National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute, Grant/Award Number: UM1 HG008900; National Institute of Mental Health (Translational Post-doctoral Training in Neurodevelopment), Grant/Award Number: T32MH112510; NINDS, Grant/Award Number: NS035129; PROGETTO GENE, (GENE - Genomic analysis Evaluation NEtwork) founded by PROGETTI DI INNOVAZIONE IN AMBITO SANITARIO E SOCIO SANITARIO (BANDO EX DECRETO N. 2713 DEL 28/02/2018); Qatar National Research Fund, Grant/Award Number: NPRP 5-175-3-051
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References:
- Alkelai A, Greenbaum L, Lupoli S, Kohn Y, & Sarner-Kanyas K (2012). Association of the Type 2 diabetes mellitus susceptibility gene, TCF7L2, with schizophrenia in an Arab-Israeli family sample. PLoS One, 7(1), 29228. 10.1371/journal.pone.0029228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrop J, van Norren K, & Clevers H (1992). A gene family of HMG- box transcription factors with homology to TCF-1. Nucleic Acids Research, 20(3), 611–611. 10.1093/nar/20.3.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodelkova O, Masek J, Korinek V, Kozmik Z, & Machon O (2018). Tcf7L2 is essential for neurogenesis in the developing mouse neocortex. Neural Development, 13(1), 8. 10.1186/s13064-018-0107-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/β-catenin signaling in development and disease. Cell, 127, 469–480. 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfield JM, Cai J, … Buxbaum JD (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515(7526), 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A, Heyer MP, Ishikawa M, Caligiuri SPB, Liu X-A, Chen Z, Micioni di Bonaventura MV, Elayouby KS, Ables JL, Howe WM, Bali P, Fillinger C, Williams M, O’Connor RM, Wang Z, Lu Q, Kamenecka TM, Ma’ayan A, O’Neill HC, … Kenny PJ (2019). Habenular TCF7L2 links nicotine addiction to diabetes. Nature, 574(7778), 372–377. 10.1038/s41586-019-1653-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsom AR, Pankow JS, Peacock JM, Bielinski SJ, Heiss G, & Boerwinkle E (2008). Variation in TCF7L2 and increased risk of colon cancer: The atherosclerosis risk in communities (ARIC) study. Diabetes Care, 31, 905–909. 10.2337/dc07-2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters G, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason, Saemundsdottir J, Wilensky RL, … Stefansson K (2006). Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics, 38(3), 320–323. 10.1038/ng1732 [DOI] [PubMed] [Google Scholar]
- Guo H, Wang T, Wu H, Long M, Coe BP, Li H, Xun J, Ou J, Chen B, Duan G, Bai T, Zhao N, Shen Y, Li Y, Wang Y, Zhang Y, Baker C, Liu Y, Pang N, … Xia K (2018). Inherited and multiple De novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Molecular Autism, 9, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, … Wigler M (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515(7526), 216–221. 10.1038/nature13908, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Yoon J, Song H, Lee B, Lam DT, Yoon J, Baek K, Clevers H, & Jeong Y (2017). Tcf7l2 plays crucial roles in forebrain development through regulation of thalamic and habenular neuron identity and connectivity. Developmental Biology, 424(1), 62–76. 10.1016/j.ydbio.2017.02.010 [DOI] [PubMed] [Google Scholar]
- Lelieveld SH, Reijnders MRF, Pfundt R, Yntema HG, Kamsteeg E-J, de Vries P, de Vries BBA, Willemsen MH, Kleefstra T, Löhner K, Vreeburg M, Stevens SJC, Van der Burgt I, Bongers EMHF, Stegmann APA, Rump P, Rinne T, Nelen MR, Veltman JA, … Gilissen C (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nature Neuroscience, 19(9), 1194–1196. 10.1038/nn.4352 [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhang N, Zhang U, Du Y, Zhang T, Li Z, Wu J, & Wang X (2018). Prioritized high-confidence risk genes for intellectual disability reveal molecular convergence. Frontiers in Genetics, 9(349), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, Alvi M, Ambridge K, Barrett DM, Bayzetinova T, Jones P, Jones WD, King D, Krishnappa N, Mason LE, Singh T, … Deciphering Developmental Disorders Study. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542(7642), 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misztal K, Brozko N, Nagalski A, Szewczyk LM, Krolak M, Brzozowska K, Kuznicki J, Wisniewska MB (2017). TCF7L2 mediates the cellular and behavioral response to chronic lithium treatment in animal models. Neuropharmacology, 113, 490–501. 10.1016/j.neuropharm.2016.10.027 [DOI] [PubMed] [Google Scholar]
- Nagalski A, Irimia M, Szewczyk L, Ferran JL, Misztal K, Kuznicki J, & Wisniewska MB (2013). Postnatal isoform switch and protein localization of LEF1 and TCF7L2 transcription factors in cortical, thalamic, and mesencephalic regions of the adult mouse brain. Brain Structure and Function, 218(6), 1531–1549. 10.1007/s00429-012-0474-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan BDN, Bohlen JF, Davis BA, Ye Z, Chen HY, Mayfield B, Sripathy SR, Cerceo Page S, Campbell MN, Smith HL, Gallop D, Kim H, Thaxton CL, Simon JM, Burke EE, Shin JH, Kennedy AJ, Sweatt JD, Philpot BD, … Maher BJ (2020). A myelin-related transcriptomic profile is shared by Pitt– Hopkins syndrome models and human autism spectrum disorder. Nature Neuroscience, 23, 375–385. 10.1038/s41593-019-0578-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, Brunner HG, Buske OJ, Carey K, Doll C, Dumitriu S, Dyke SOM, den Dunnen JT, Firth HV, Gibbs RA, Girdea M, Gonzalez M, Haendel MA, Hamosh A, … Rehm HL (2015). The matchmaker exchange: A platform for rare disease gene discovery. Human Mutation, 36(10), 915–921. 10.1002/humu.22858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, Vuong CK, Nichterwitz S, Gevorgian M, Opland CK, Lu D, Connell W, Ruzzo EK, Lowe JK, Hadzic T, Hinz FI, Sabri S, Lowry WE, Gerstein MB, … Geschwind DH (2019). A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron, 103, 785–801.e8. 10.1016/j.neuron.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokunina-Olsson L, Welch C, Hansson O, Adhikari N, Scott LJ, Usher N, Tong M, Sprau A, Swift A, Bonnycastle LL, Erdos MR, He Z, Saxena R, Harmon B, Kotova O, Hoffman EP, Altshuler D, Groop L, Boehnke M, … Hall JL (2009). Tissue- specific alternative splicing of TCF7L2. Human Molecular Genetics., 18, 3795–3804. 10.1093/hmg/ddp321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, Neale BM, & Daly MJ (2017). Regional missense constraint improves variant deleteriousness prediction. BioRxiv. 10.1101/148353 [DOI] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, Stevens C, Reichert J, Mulhern MS, Artomov M, Gerges S, Sheppard B, Xu X, Bhaduri A, Norman U, & Buxbaum JD (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell, 180(3), 568–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, & Hamosh A (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36, 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Hoekzema K, Vecchio D, Wu H, Sulovari A, Coe BP, Gillentine MA, Wilfert AB, Perez-Jurado LA, Kvarnung M, Sleyp Y, Earl RK, Rosenfeld JA, Geisheker MR, Han L, Du B, Barnett C, Thompson E, Shaw M, … Eicher EE (2020). Large-scale targeted sequencing identifies risk genes for Neurodevelopental disorders. Nature Communications, 11(1), 4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise A, Bruser K, Elfert S, Wallmen B, Wittel Y, Wöhrle S, & Hecht A (2009). Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/β-catenin targets. Nucleic Acids Research, 38, 1964–1981. 10.1093/nar/gkp1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao X, Bu H, Hu T, Taketo MM, Van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, & Lu QR (2009). HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nature Neuroscience, 12(7), 829–838. 10.1038/nn.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng Y, Liu L, Yu K, Zhang L, Wang H, He X, Wang J, Lu C, Wu LN, Weng Q, Mao M, Li J, Van Es JH, Xin M, Parry L, Goldman SA, Clevers H, & Lu QR (2016). Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nature Communications, 7(1), 10883. 10.1038/ncomms10883 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.