

Abstract
In response to microbes and other danger signals, the NLRP3 inflammasome in immune cells triggers the activation of the protease caspase-1, which mediates the maturation of the inflammatory cytokine IL-1β. Here, we investigated how the NLRP3 inflammasome is regulated. We found that its activation in primary mouse macrophages induced the Src family kinase Lyn to phosphorylate NLRP3 at Tyr918, which correlated with a subsequent increase in its ubiquitination that facilitated its proteasome-mediated degradation. NLRP3 tyrosine phosphorylation and ubiquitination was abrogated in Lyn-deficient macrophages, which produced increased amounts of IL-1β. Furthermore, mice lacking Lyn were more susceptible to LPS-induced septic shock in an NLRP3-dependent manner. Our data demonstrate that Lyn-mediated tyrosine phosphorylation is a prerequisite for the ubiquitination that dampens NLRP3 inflammasome activity.
INTRODUCTION
Inflammasomes are multimeric protein complexes and their components are supposedly assembled upon recognition of microbes and endogenous danger signals (1). Activation of inflammasomes leads to the recruitment and autoactivation of caspase-1, which in turn mediates the cleavage of inactive pro-IL-1β and pro-IL-18 into bioactive forms and pyroptosis (2–5). The best characterized inflammasome is the NLRP3 inflammasome which plays a critical role in host defense responses to microbes such as E. coli and influenza virus (6, 7). In addition to microbes, host-derived stimulatory particles such as monosodium urate (MSU), cholesterol crystals, and islet amyloid-polypeptides damage the membrane of phagolysosomes, causing persistent activation of NOD-like receptors (NLR) family pyrin domain containing 3 (NLRP3) inflammasomes, leading to severe inflammatory diseases such as gout, atherosclerosis and type II diabetes (1, 8, 9). Moreover, NLRP3 mutations underlie conditions such as familial cold autoinflammatory syndrome (FCAS) (10, 11). To date, NLRP3 function has been characterized as mediating a rather “hard-wired” pro-inflammatory response. However, the current literature provides increasing evidence that pathogen recognition receptors (PRRs) must be regulated to enable site-specific host defense without causing excessive systemic inflammation.
Post-translational modifications such as phosphorylation and ubiquitination have emerged as the critical regulatory mechanisms for many physiological processes. It has been shown that phosphorylation of NLRP3 at Ser194 and Ser293 by JNK-1 and protein kinase D (PKD), respectively, is required for its activation (12, 13). However, phosphorylation of NLRP3 at Ser5, within the PYD domain, hinders the recruitment of ASC via the charge-charge interaction between NLRP3 PYD and ASC PYD, thus preventing the assembly of the NLRP3 inflammasome (14). Additionally, tyrosine phosphorylation of NLRP3 seems to down-regulate its activation (15). It has also been shown that NLRP3 undergoes ubiquitination which inhibits its activation (16–18). Here, we sought to uncover the protein tyrosine kinase (PTK) that phosphorylates NLRP3 and investigated the relationship between NLRP3 tyrosine phosphorylation and ubiquitination in cultured immune cells and in vivo.
RESULTS
Tyrosine phosphorylation of NLRP3 mediated by Src family PTK(s) is a prerequisite for its ubiquitination
We first explored how NLRP3 tyrosine phosphorylation inhibits its activation. Previously, we reported that C57BL/6 (B6) BMDMs undergo ubiquitination at 5 min after ATP stimulation, and IL-1β production at 30 min after ATP stimulation (18). Therefore, we measured NLRP3 tyrosine phosphorylation and ubiquitination at 5 min after ATP stimulation, and IL-1β production at 30 min after ATP stimulation. To determine whether and how tyrosine phosphorylation regulates NLRP3 inflammasome activation, BMDMs from B6 mice were primed with LPS, pretreated with the pan-Src PTK inhibitor Src-I1 or the selective Syk inhibitor Bay 61–3606, and then stimulated with the canonical NLRP3 inflammasome activators ATP, nigericin, MSU, or silica. Pretreatment of Src-I1 but not Bay 61–3606 significantly augmented IL-1β production in BMDMs stimulated with ATP and nigericin, but had no effect on IL-1β production induced by MSU and silica (Fig. 1A, left panel). These data suggest that inhibition of NLRP3 tyrosine phosphorylation induced by soluble canonical NLRP3 inflammasome stimuli activates the NLRP3 inflammasome. In support of this notion, we observed marked tyrosine phosphorylation of NLRP3 induced by ATP and nigericin, which was inhibited by Src-I1 (Fig. 1B). No tyrosine phosphorylation of NLRP3 was observed in BMDMs primed by LPS and stimulated by MSU and silica (fig. S1A). Src-I1 also potentiated disuccinimidyl suberate (DSS) crosslinking-induced ASC oligomerization (Fig. 1C). Note that Src-I1 treatment did not affect poly (dA:dT)- and flagellin-induced IL-1β production (Fig. 1A, right panel), suggesting that Src-I1 does not affect the activation of AIM-2 and NLRC4 inflammasomes. Similar results were obtained in peritoneal macrophages and THP-1 cells (fig. S1B). These data collectively suggest that tyrosine phosphorylation of NLRP3 negatively regulates NLRP3 inflammasome activity, and that one or more Src family PTKs is responsible for phosphorylating NLRP3 which facilitates its ubiquitination.
Fig. 1. NLRP3 tyrosine phosphorylation is required for its ubiquitination.
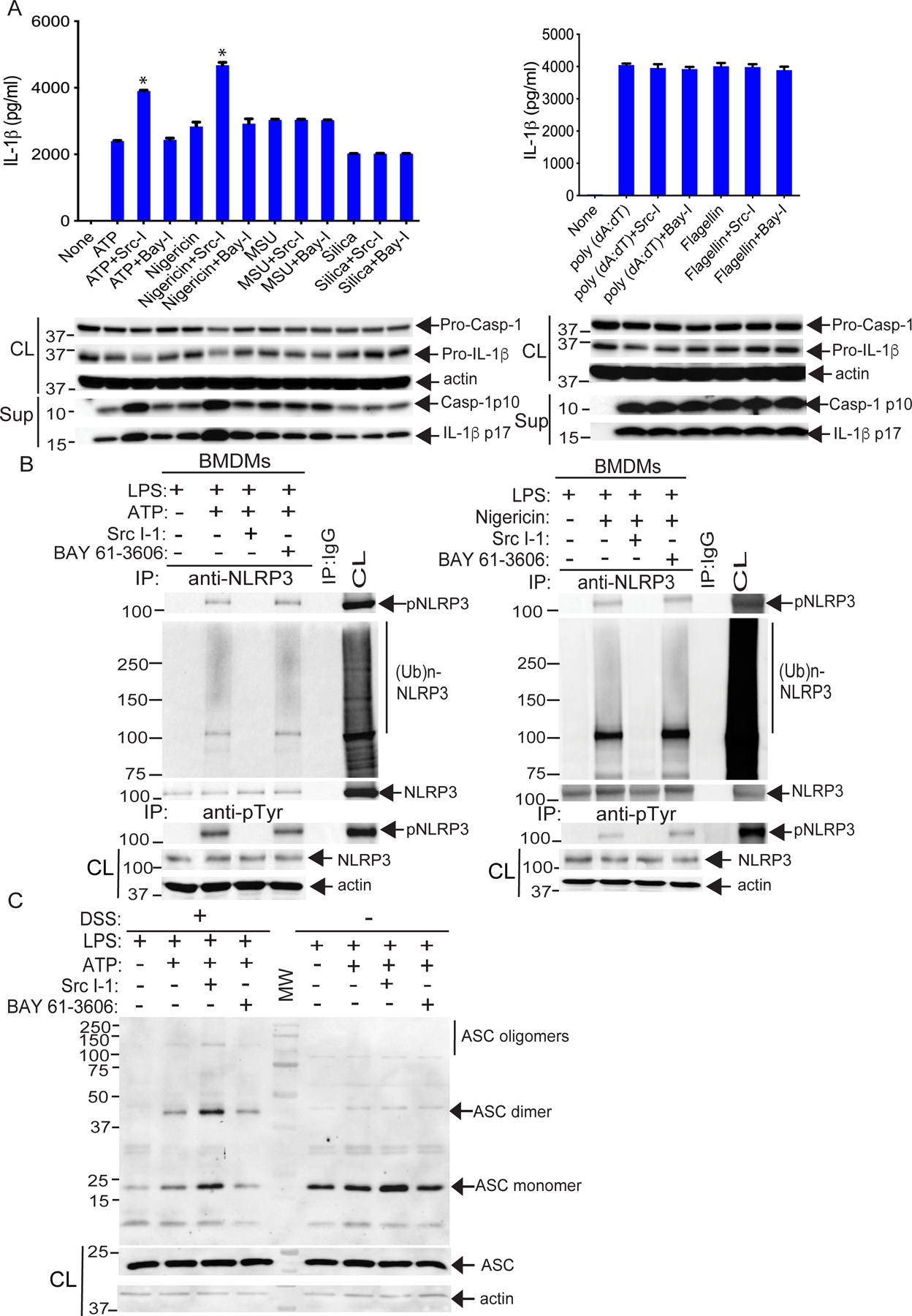
(A) ELISA detection of IL-1β in the supernatants from LPS-primed BMDMs treated as indicated with ATP(2.5 mM, 30 min), nigericin (20 μM, 3 hours), MSU (200 μg/ml, 4 hours), silica (500 μg/ml, 6 hours), poly(dA:dT) (1 μg/106 cells, 6 hours), or flagellin (6.25 μg/106 cells, 4 hours), each either alone or in the presence of Src-I1 (Src-I; 44 nM, 1 hour) or BAY 61–3606 (BAY-I; 10 nM, 1 hour). Below, Western blotting for caspase-1 (Casp-1) p10 and IL-1β p17 in the same supernatants (sup) and their pre-processed “pro” forms in whole cell extracts (CL). (B) WT BMDMs were primed with LPS and stimulated with ATP or nigericin in the presence of Src-I1 or BAY 61–3606, then lysed in RIPA buffer. The cell lysates were either immunoprecipitated with antibody to NLRP3 and blotted with antibodies to pTyr and ubiquitin, or they were immunoprecipitated with antibody to pTyr and blotted with antibody to NLRP3. (C) Western blotting for ASC in cell lysates (either cross-linked with DSS or not) from LPS-primed WT BMDMs pretreated with Src-I1 or BAY 61–3606 and stimulated with ATP. Input control (CL) is from pre-crosslinked aliquots. The data are from a representative one of three (A and B) or 4 (C) independent experiments. *P< 0.05, student t test.
Lyn is the PTK that phosphorylates NLRP3
Several PTKs including Lyn, Src, Hck, Yes, and c-Fgr are expressed in macrophages (19). To determine which PTK(s) specifically phosphorylates NLRP3, we primed BMDMs with LPS, and stimulated them with ATP for 5 min. The BMDM lysates were incubated with GST-NLRP3 and the GST-NLRP3-binding proteins were subjected to mass spectrometric analysis. We found that Lyn was the only PTK associated with NLRP3 on the top list over 95% probability legend in both LPS priming and LPS priming/ATP-stimulated BMDM samples (Fig. 2A and data file S1). To confirm this result, we performed a co-immunoprecipitation assay. We stimulated LPS-primed BMDMs with ATP for 5 and 15 min and lysed them in 0.5% NP40 lysis buffer. The cell lysates were immunoprecipitated with anti-NLRP3 and blotted with antibodies against Lyn. We found that Lyn constitutively associated with NLRP3, and this association was increased at 15 min after ATP stimulation (Fig. 2B). These data suggest that the Lyn SH3 domain may bind to the proline-rich region of NLRP3, and once NLRP3 is tyrosine phosphorylated, it binds to Lyn via the Lyn SH2 domain. To determine whether Lyn associates with NLRP3 in the absence of LPS stimulation, we transfected HEK293T cells with Flag-tagged NLRP3 and Lyn and lysed the cells in 0.5% NP40 lysis buffer. The cell lysates were immunoprecipitated with antibody to Lyn and blotted with antibody to Flag. In support of this notion, we found that Lyn bound to NLRP3 in the absence of LPS signal 1 (Fig. 2C)
Fig. 2. Lyn directly phosphorylates NLRP3.
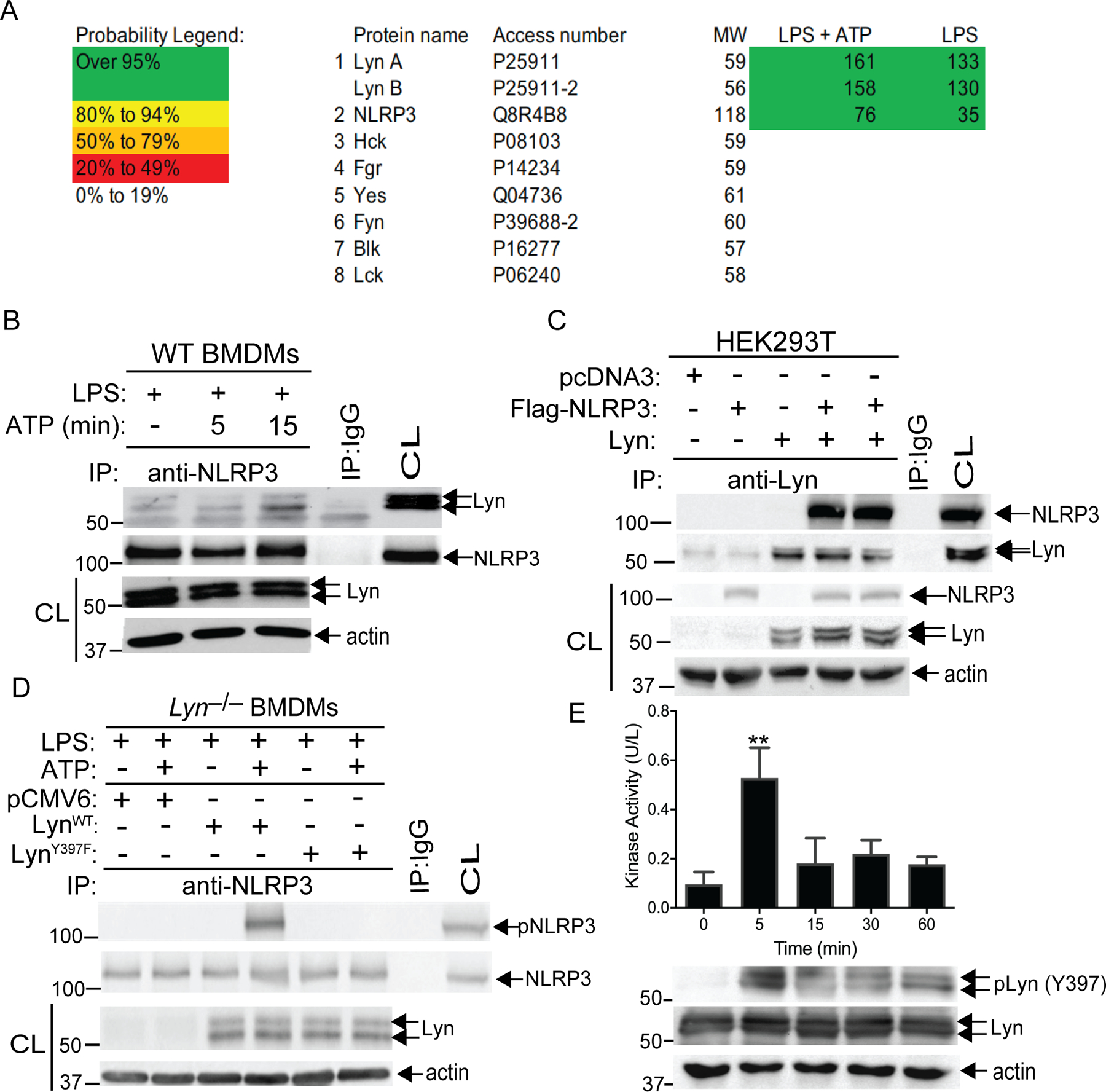
(A) LS-MS analysis of GST-NLRP3 pull-down proteins. Spectral counts of high confidence proteins are listed at the right. Thirty-four peptides with confidence >95% are assigned to two Lyn isoforms. Coverage of Lyn increased from 44% to 66% with ATP in addition to LPS. (B) Lysates from LPS-primed BMDMs treated with ATP at indicated time-points were immunoprecipitated with antibody to NLRP3 and blotted with antibodies to Lyn and NLRP3. (C) Lysates from HEK293T cells transfected with Flag-tagged NLRP3 and Lyn, were immunoprecipitated with antibody to Lyn and blotted with antibody to Flag. (D) Lysates from Lyn–/– BMDMs were transfected with the constructs expressing WT Lyn or a kinase-dead Lyn (LynY397F) upon LPS priming and ATP stimulation were immunoprecipitated with antibody to NLRP3 and blotted with antibody to pTyr. Whole cell lysates were immunoblotted with antibodies to Lyn and actin. (E) Lysates from LPS-primed BMDMs treated with ATP at indicated time-points were immunoprecipitated with antibody to Lyn, incubated with GST-NLRP3 in kinase buffer in the presence of ATP, and then detected using an EnzyChrom™ kinase assay kit. An aliquot of kinase reaction mixture was blotted with antibodies to phospho-Lyn (Tyr397) and Lyn. Data are from a representative one of two (A and D) or three (C, B and E) independent experiments. **P< 0.01, student t test
To verify this finding, we transfected Lyn–/– BMDMs with plasmids encoding LynWT or LynY397F, a Lyn kinase dead mutant. As predicted, Lyn deficiency abrogated NLRP3 tyrosine phosphorylation, and reconstituting Lyn–/– BMDMs with LynWT, but not LynY397F, rescued NLRP3 tyrosine phosphorylation (Fig. 2D). A recent report showed that LPS priming induces NLRP3 tyrosine phosphorylation, while ATP stimulation results in the dephosphorylation of NLRP3 (15). These data are contradicted by our observations. To determine whether this discrepancy is due to different priming times or cell types used, we primed WT and Lyn–/– BMDMs and BMDCs with LPS for 12 hours, as described (15), or 4 hours, and then stimulated the cells with or without ATP. Indeed, priming BMDMs and BMDCs for 12 hours induced NLRP3 tyrosine phosphorylation, whereas this phosphorylation disappeared upon ATP stimulation (fig. S2A) as reported (15). This data is in sharp contrast with our data (fig. S2B). The failure to detect the basal level of NLRP3 tyrosine phosphorylation in our study is likely due to the different priming time used. It is possible that prolonged priming with LPS activates the non-canonical NLRP3 inflammasome, and additional triggering of the NLRP3 inflammasome with ATP may facilitate the activation of PTPN22 which dephosphorylates NLRP3. This notion is further supported by the fact that FITC-conjugated LPS was markedly co-localized with caspase-11 when priming WT BMDMs with FITC-conjugated LPS for 12 hours (fig. S2C). It has been shown that LPS undergoes spontaneous internalization without transfection (20) and that LPS priming induces the expression of caspase-11 protein, which is dependent on TRIF (6).
To further confirm these data, we performed an in vitro Lyn kinase assay using GST- NLRP3 as a substrate. To this end, B6 BMDMs were primed with LPS, and stimulated with ATP for 5, 15, 30, and 60 min, and lysed in 1% Triton-X100 lysis buffer as previously described (21). The cell lysates were immunoprecipitated with anti-Lyn, incubated with GST-NLRP3 in kinase buffer in the presence of ATP, and then detected using an EnzyChrom™ kinase assay kit. Indeed, Lyn phosphorylated NLRP3 at 5 min after ATP stimulation (Fig. 2E) which correlated with Lyn autophosphorylation at Tyr397 (Fig. 2E). Therefore, our data strongly indicate that Lyn is the PTK responsible for phosphorylating NLRP3.
Lyn inhibits NLRP3 inflammasome activation by facilitating NLRP3 degradation in the proteasome
Both Lyn and Syk have been shown to facilitate malarial hemozoin-induced NLRP3 inflammasome activation via an unknown mechanism (22). To further define the physiological effect of Lyn on the NLRP3 inflammasome activation, we first silenced the Lyn gene in B6 BMDMs using Lyn-specific siRNA. Lyn gene silencing abrogated ATP-induced NLRP3 tyrosine phosphorylation, ubiquitination, and degradation (Fig. 3, A and B), which correlated with heightened IL-1β production in the culture supernatants (Fig. 3C). Lyn deficiency did not alter the production of TNF-α and IL-6, or the expression of NLRP3, during LPS priming (fig. S3, A and B). To further confirm these data, we generated BMDMs from Lyn–/– mice. We primed WT and Lyn–/– BMDMs with LPS and stimulated them with ATP. As expected, Lyn deficiency completely abrogated NLRP3 tyrosine phosphorylation, ubiquitination, and subsequent degradation (Fig. 3D and E), and Lyn–/– BMDMs produced significantly higher IL-1β than WT BMDMs (Fig. 3F). Lyn deficiency did not affect AIM-2 and NLRC4 inflammasomes as revealed by comparable IL-1β production by BMDMs from mice deficient or sufficient for Lyn stimulated with flagellin and poly(dA:dT) (Fig. 3G), consistent with the data described above (Fig. 1). Previously, it has been shown that autophagasome induction can degrade tyrosine-phosphorylated NLRP3 within the NLRP3/ASC complex (23). To determine whether NLRP3 undergoes proteasomal degradation in the soluble NLRP3 fraction versus the insoluble NLRP3 inflammasome complex, we primed BMDMs with LPS in the presence of caspase-1 inhibitor VX-765, which prevents cell death triggered by caspase-1 (pyroptosis) and retains all of the NLRP3 within the cells (24) (Fig. 3, H and I). The primed BMDMs were then stimulated with ATP and soluble and insoluble cellular fractions were isolated. NLRP3 was degraded in the absence of MG-132 in the soluble fraction, whereas MG-132 treatment facilitated the translocation of NLRP3 from the soluble fraction to insoluble fraction (Fig. 3I). This suggests that NLRP3 mainly undergoes proteasome-mediated degradation before it assembles the complex with ASC and caspase-1. Therefore, our data collectively indicate that Lyn keeps NLRP3 inflammasome in check by phosphorylating NLRP3 which facilitates its ubiquitination and proteasome-mediated degradation. The discrepancy between our findings and those reported by others (22) may be due to different stimuli used.
Fig. 3. Lyn suppresses the activation of NLRP3 inflammasome.
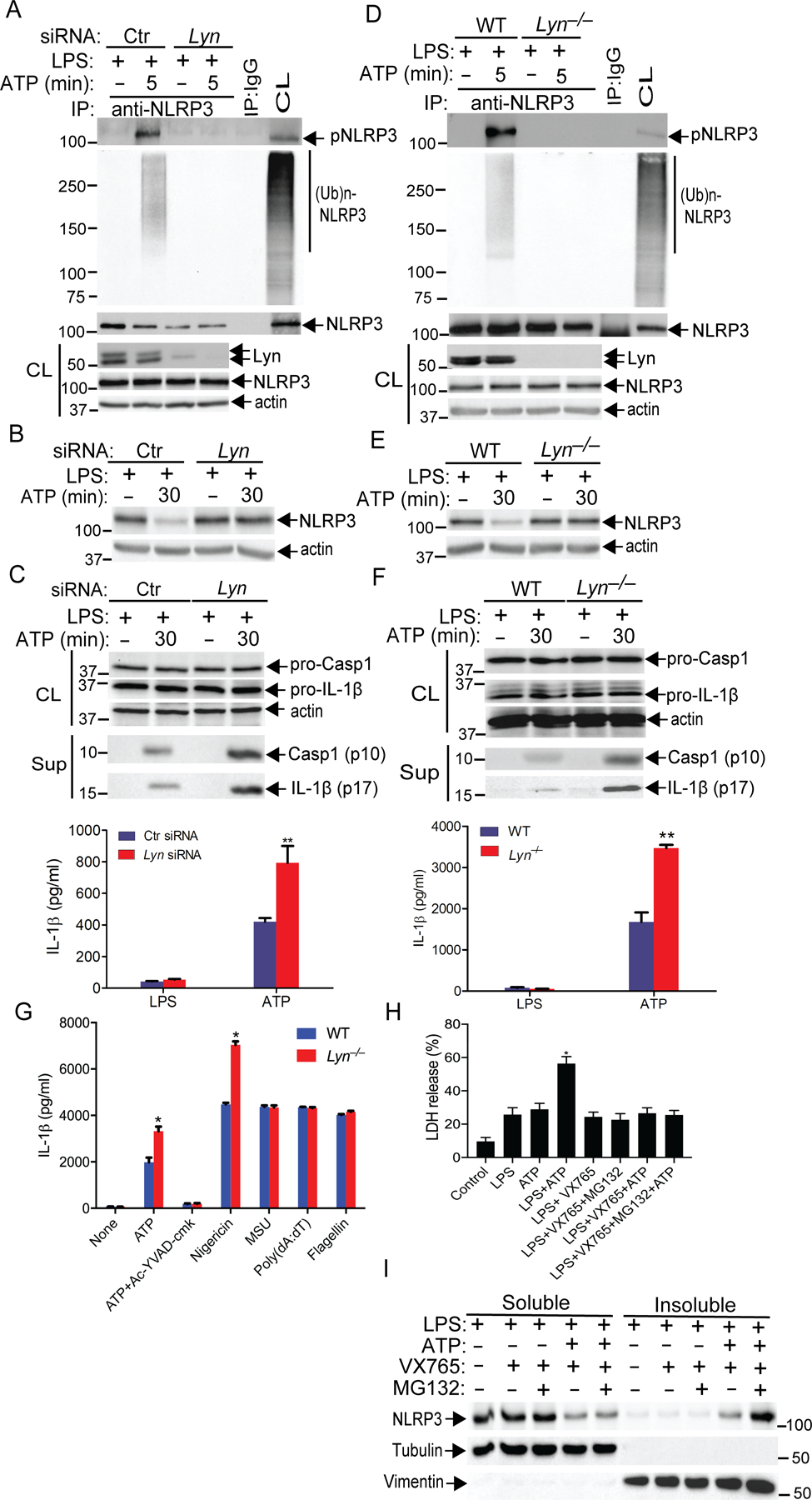
(A) WT BMDMs were transfected with Ctr siRNA or Lyn siRNA by lipofectamine 2000, primed with LPS for 4 hours, then stimulated with ATP for 5 min, and lysed in RIPA buffer. The cell lysates were immunoprecipitated with anti-NLRP3 and blotted with antibodies to pTyr and ubiquitin. (B) WT BMDMs treated with siRNA and primed as in A, and then stimulated with ATP for 30 min. The expression of NLRP3 was determined by immunoblot analysis. (C) Immunoblot analysis for caspase-1 (Casp-1) p10 and IL-1β p17 pre-processed “pro” forms in whole cell extracts (CL) from (B) and their cleaved form in the same supernatants (sup). IL-1β production in supernatants under the same conditions was measured by ELISA. (D) BMDMs from WT and Lyn–/– mice, primed with LPS, and stimulated with ATP. The cell lysates were immunoprecipitated with antibody to NLRP3 and blotted with antibodies to pTyr and ubiquitin. (E) Immunoblot analysis for NLRP3 in cell lysates from LPS-primed WT and Lyn–/– BMDMs stimulated with ATP. (F) Immunoblot analysis for caspase-1 (Casp-1) p10 and IL-1β p17 pre-processed “pro” forms in whole cell extracts (CL) from (E) and their cleaved form in the same supernatants (sup). IL-1β production in supernatants under the same conditions was measured by ELISA. (G) ELISA detection of IL-1β in the supernatants from LPS-primed BMDMs from WT and Lyn–/– mice treated as indicated with ATP or ATP with Ac-YVAD-cmk, nigericin, MSU, poly(dA:dT), and flagellin. (H) LDH assay of LPS-primed BMDMs and stimulated with ATP in the presence of VX765 or VX765 plus MG132. (I) Immunoblot analysis for NLRP3 in cell lysates (both solube and insoluble fractions) from LPS-primed WT BMDMs treated as in (H). The purity of soluble and insoluble fractions was determined by immunoblotting with tubulin and vimentin, respectively. The data are from a representative one of three (A to F), two (G and H), or four (I) independent experiments. Data are mean ± SD for IL-1β in (C, G, and H). *P< 0.05; **P< 0.01, student t test.
We previously demonstrated that K63- and K48-linked ubiquitination of NLRP3 by RNF125 and Cbl-b restrains LPS-induced endotoxemia (18). We then wanted to address whether tyrosine phosphorylation of NLRP3 results in a specific K63- or K48-linked ubiquitination. To this end, we primed WT and Lyn–/– BMDMs with LPS and stimulated them with ATP. We found that both K63- and K48-linked ubiquitination of NLRP3 was abrogated in BMDMs lacking Lyn (fig. S4). These data suggest that tyrosine phosphorylation of NLRP3 induced by Lyn facilitates RNF125 and Cbl-b to associate with NLRP3, thus targeting NLRP3 for ubiquitination.
Lyn phosphorylates NLRP3 at Tyr918 and controls NLRP3 ubiquitination
It has been shown that the NLRP3 leucine-rich repeat (LRR) domain is a major domain harboring the ubiquitin chain (16). To further define which tyrosine residue(s) is phosphorylated by Lyn, we used the NetPhos 3.1 server to predict the potential tyrosine phosphorylation sites of NLRP3. Two potential phosphorylation sites were found to have high confidence within NLRP3 LRR domain (Tyr861 and Tyr918). We mutated them individually or in combination to phenylalanine to generate NLRP3Y861F, NLRP3Y918F, and NLRP3Y861F/918F mutants. To determine whether these tyrosine residues are the phosphorylation sites of NLRP3, we utilized Nlrp3–/– mice which avoid any effect of endogenous NLRP3. We reconstituted Nlrp3–/– BMDMs with Flag-tagged NLRP3WT, NLRP3Y861F, NLRP3Y918F, or NLRP3Y861F/Y918F, primed them with LPS, and stimulated them with ATP. Reconstitution of Nlrp3–/– BMDMs with NLRP3WT or NLRP3Y861F, but not NLRP3Y918F or NLRP3Y861F/Y918F restored NLRP3 tyrosine phosphorylation and ubiquitination (Fig. 4A), further confirming that Tyr918 of NLRP3 is the phosphorylation site. In support of these data, increased IL-1β production was observed in Nlrp3–/– BMDMs reconstituted with NLRP3Y918F mutant (Fig. 4B) which abrogated NLRP3 degradation (Fig. 4C). Thus, our data indicate that Tyr918 is the tyrosine residue phosphorylated by Lyn. It has been reported that Tyr861 is the phosphorylation site of NLRP3 when PTP22N is inhibited (15); however, the reason for this discrepancy is currently unknown.
Fig. 4. Src kinase Lyn phosphorylates NLRP3 at Tyr 918, which facilitates NLRP3 ubiquitination and subsequent degradation.
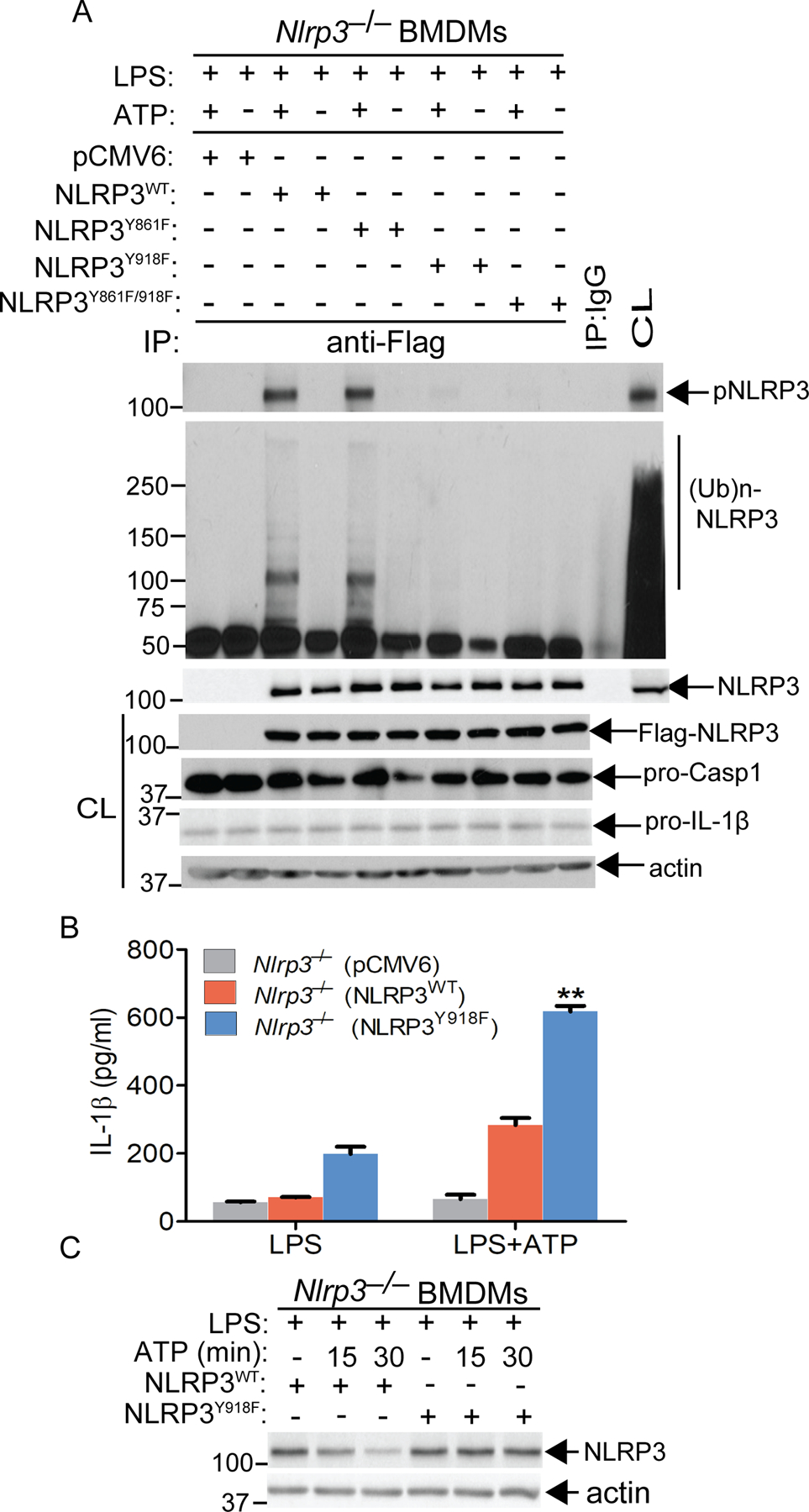
(A) Nlrp3–/– BMDMs were reconstituted with the constructs expressing pCMV6, NLRP3WT, NLRP3Y861F, NLRP3Y918F and NLRP3Y861F/918F, primed with LPS, stimulated with ATP. The cell lysates were immunoprecipitated with antibody to NLRP3, and blotted with antibodies to pTyr and ubiquitin, respectively. The cell lysates were blotted with antibodies to Flag and actin, respectively. (B) Nlrp3–/– BMDMs were reconstituted with the constructs expressing NLRP3WT and NLRP3Y918F, primed with LPS, and stimulated with ATP. The IL-1β in the supernatants was determined by ELISA. (C) Nlrp3–/– BMDMs were reconstituted with the constructs expressing NLRP3WT and NLRP3Y918F, primed with LPS and stimulated with ATP. The cell lysates were blotted with antibodies to NLRP3 and actin. Data are representative of three independent experiments (A to C). **P< 0.01, student t test.
Lyn–/– mice are highly susceptible to LPS-induced septic shock via a NLRP3-dependent manner
To determine whether Lyn is indeed a negative regulator of the NLRP3 inflammasome, we generated Lyn–/– mice on a C57BL/6 background using CRISPR-Cas technology (Fig. 5A and B). We utilized LPS-induced septic shock as a model since we and others have shown that LPS-induced septic shock is NLRP3-dependent (18, 25). To test whether Lyn regulates NLRP3 inflammasomes in vivo, we challenged WT and Lyn–/– mice with a sub-lethal dose of LPS (5 mg/kg) (E. coli 0111:B4; Sigma) by intraperitoneal injection. Although WT mice all survived 40 hours after LPS injection, all Lyn–/– mice died within 24 hours after LPS injection (Fig. 5C). Serum IL-1β was increased at 2–6 hours post-injection in the Lyn–/– mice compared to WT mice, and TNF-α was significantly higher in the Lyn–/– mice than WT mice at 2 hours (Fig. 5D and E). To determine whether heightened activation of the NLRP3 inflammasome is the major cause of the LPS-induced lethality observed in Lyn–/– mice, we injected Lyn–/– mice with MCC950, a specific NLRP3 inhibitor, prior to LPS challenge. Inhibiting NLRP3 prevented the death of Lyn–/– mice after LPS injection (Fig. 5C). This prevention was associated with attenuated serum levels of IL-1β and TNF-α (Fig. 5D and E). Serum IL-6 levels also peaked at 2 hours after LPS injection in WT and Lyn–/– mice, but no difference observed between WT and Lyn–/– mice upon LPS injection, and MCC950 treatment (Fig. 5F). Therefore, our data collectively indicate that Lyn inhibits the NLRP3 inflammasome in vivo.
Fig. 5. Loss of Lyn increases the susceptibility of mice to sublethal LPS-induced septic shock via a NLRP3-dependent manner.
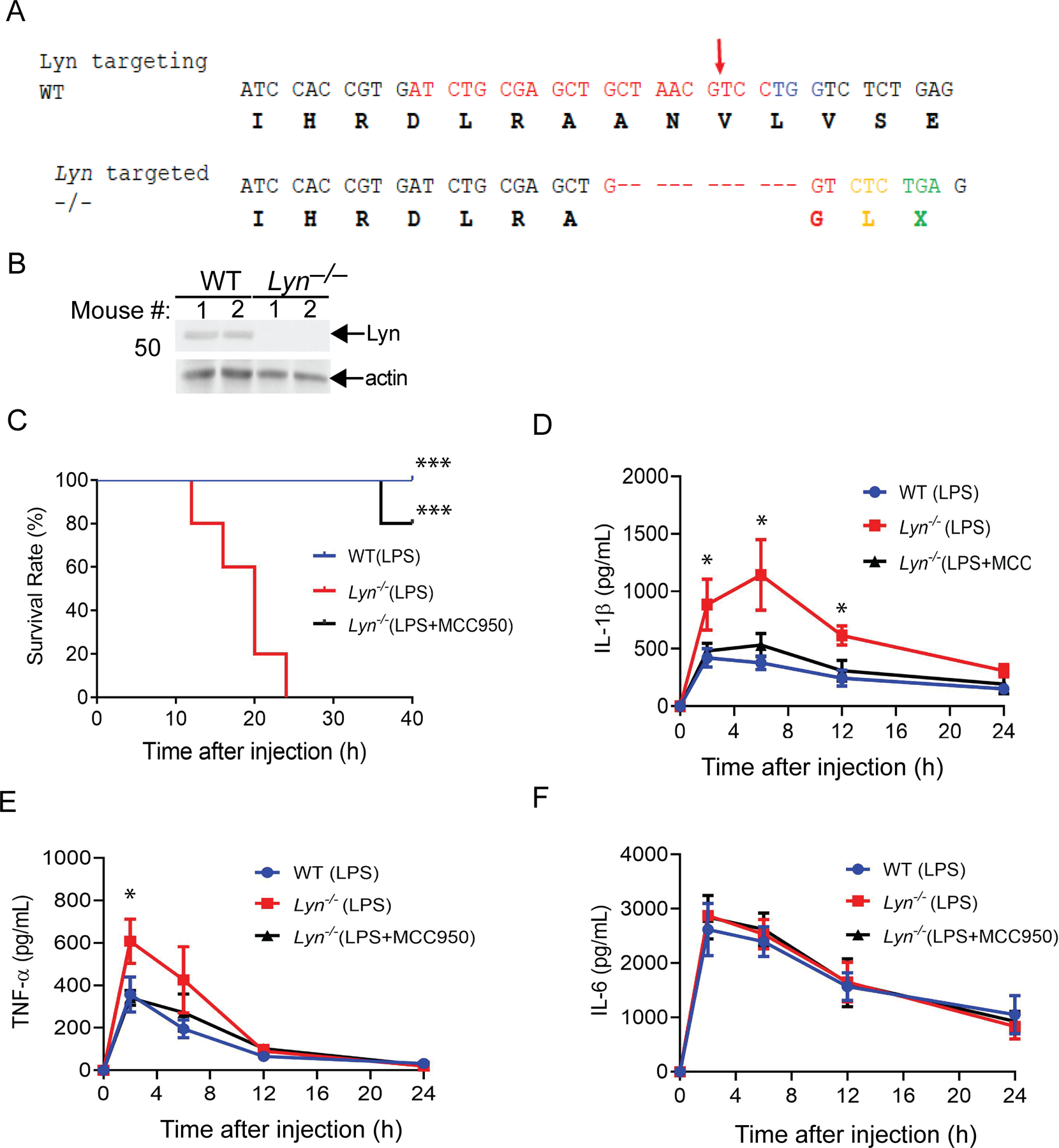
(A) Generation of Lyn–/– mice by CRISPR Cas9 technology. Lyn–/– mice were generated by CRISPR-Cas9-mediated targeting of exon11 of mouse Lyn. The DNA sequence for specific gRNA is shown in red, and the PAM sequence in blue (top panel). Arrow indicates the Cas9 cleavage site. Amino acids are shown below the respective codes. The sequence of a targeted allele with 11-nucleotide deletion is presented (lower panel). The frameshift and an immediate premature stop codon TGA [X] are shown. The Lyn homozygous knockout mice with the 11bp deletion (delCTAACGTCCTG) were used in this study. (B) Splenocytes from two WT and two Lyn–/– mice were lysed and blotted with antibodies to Lyn and actin. (C) Kaplan-Meier Survival curve of WT and Lyn–/– mice injected with a sub-lethal dose of LPS (5 mg/kg) in the presence or absence of MCC950 (50 mg/kg) pretreatment. N = 5 each. ***P< 0.001 (Lyn–/– + LPS vs WT + LPS or Lyn–/– + LPS/MCC950), by log rank test. (D to F) ELISA of serum IL-1β, TNF-α, and IL-6 levels from WT and Lyn–/– mice injected with LPS (1 mg/kg) with or without MCC950 pretreatment. N = 5. *P< 0.05(WT + LPS vs Lyn–/– + LPS; Lyn–/– + LPS vs Lyn–/– + LPS/MC950, by student’s t-test. In (B to F), data are representative of three independent experiments.
DISCUSSION
It has been shown that the NLRP3 inflammasome is essential for host defense against certain pathogen infections, but aberrant NLRP3 inflammasome activation is associated with development of neurodegenerative, metabolic, and autoimmune/autoinflammatory diseases as well as cancers (1, 3, 26, 27). Although the positive regulation of the NLRP3 inflammasome has been extensively studied during the last several years, the negative control of NLRP3 inflammasome activity is less so. In this study, we unveiled a new role of tyrosine phosphorylation of NLRP3 by Lyn, which is a prerequisite for its ubiquitination, thus controlling NLRP3 inflammasome activity.
Phosphorylation of NLRP3 has been shown to positively or negatively regulate NLRP3 inflammasome activation. Recent studies suggested that tyrosine phosphorylation of NLRP3, when PTPN22 is inhibited, down-regulates NLRP3 inflammasome activation possibly via an autophagasome-dependent mechanism (15, 23). Furthermore, the PTK(s) responsible for phosphorylating NLRP3, and the relationship between NLRP3 tyrosine phosphorylation and ubiquitination, are unknown. Using GST-NLRP3 pull-downs coupled with mass spectrometric analysis we found that Lyn is the PTK with the highest probability. Indeed, we confirmed that NLRP3 associates with Lyn in macrophages using a co-immunoprecipitation assay, and that this association is enhanced upon NLRP3 inflammasome stimuli such as ATP. Because Lyn contains an SH3 and an SH2 domain that mediate protein-protein interactions (28), it is possible that Lyn may associate with NLRP3 via Lyn’s SH3 domain and the proline-rich region within NLRP3. Once NLRP3 is tyrosine phosphorylated, the phospho-tyrosine residues of NLRP3 may interact with Lyn’s SH2 domain. We further confirmed that Lyn is the PTK that phosphorylates NLRP3 by showing (i) that reconstituting Lyn–/– BMDMs with LynWT, but not LynY397F, restores NLRP3 tyrosine phosphorylation upon LPS priming and ATP stimulation and (ii) that Lyn directly phosphorylates NLRP3 by an in vitro Lyn kinase assay. Notably, we identified NLRP3 Tyr918 as the site that is tyrosine phosphorylated by Lyn by showing that mutation of Tyr918 abrogates NLRP3 tyrosine phosphorylation. In support of a role for Lyn in phosphorylating NLRP3, Lyn deficiency results in heightened IL-1β production by macrophages upon NLRP3 inflammasome activation. These data are further supported by the fact that the NLRP3 Y918F mutation, which abrogates its tyrosine phosphorylation, elicits aberrant IL-1β production. Our data are contradicted by those by Spalinger et al. (15) in which they found that NLRP3 is tyrosine phosphorylated in BMDCs during LPS priming, and that ATP stimulation induces PTPN22-mediated NLRP3 dephosphorylation (15). One possibility is that these authors primed BMDCs with LPS for 12 hours. Under this condition, LPS not only primes BMDCs to induce NLRP3 expression, but also undergoes endocytosis which triggers caspase-11, a cytosolic sensor for LPS, and activates caspase-11-dependent non-canonical NLRP3 inflammasome. Additional stimulation with ATP may activate PTPN22 under this specific condition which dephosphorylates NLRP3. In support of this notion, we found that priming of BMDMs and BMDCs with LPS for 12, but not 4, hours induces the tyrosine phosphorylation of NLRP3.
It has been shown that ASC also undergoes tyrosine phosphorylation by Syk (29). However, we do not know whether Lyn phosphorylates ASC. ASC is not among the target proteins identified in our mass spectrometry data. It is possible that Lyn may not directly act on ASC. In our ASC oligomerization assays, 5 min was determined to be the optimal time point at which to observe clear ASC oligomerization. We also believe that ASC oligomerization, measured by either microscopy or biochemical methodology, is sufficient to support the conclusion. Our data indicate that 30 to 60 min of ATP stimulation results in the degradation of a significant amount of NLRP3 (18). Therefore, these time-points are not ideal for ASC oligomerization. Gasdermin D (GSDMD) is cleaved by both caspase-11 and caspase-1. It is expected that GSDMD cleavage is increased in Lyn–/– BMDMs upon ATP stimulation because canonical NLRP3 inflammasome activation is heightened in Lyn–/– BMDMs upon LPS priming and ATP stimulation. GSDMD cleavage is beyond the scope of our current study.
It has been evident for many years that there are multiple connections between phosphorylation and ubiquitination, and that they can act either positively or negatively in both directions to regulate these processes (30). Using a Src kinase inhibitor, we found that inhibition of Src kinases results in the abrogation of NLRP3 ubiquitination in macrophages triggered by ATP. Indeed, Lyn deficiency, which completely diminishes NLRP3 tyrosine phosphorylation, also abrogates its ubiquitination. In keeping with this datum, NLRP3 Y918F mutation abrogates its ubiquitination. Notably, mice lacking Lyn were highly susceptible to LPS-induced septic shock in an NLRP3-dependent manner. Therefore, our data collectively indicate that NLRP3 tyrosine phosphorylation is required for its ubiquitination, which provides the molecular mechanism by which NLRP3 tyrosine phosphorylation negatively regulates NLRP3 inflammasome activation. It is possible that tyrosine phosphorylation at Tyr918 may recruit RNF125 and Cbl-b to NLRP3, resulting in its ubiquitination and proteasome-mediated degradation. This notion is supported by the fact that both K63- and K48-linked ubiquitination of NLRP3 is compromised in BMDMs lacking Lyn upon LPS priming and ATP stimulation. The reason for the discrepancy relating to the tyrosine residue that we identified is currently unknown (15). It is possible that deficiency of PTPN22 may lead to aberrant activation of other PTK(s), which phosphorylate NLRP3 at Tyr861, whereas under normal conditions this tyrosine residue is not phosphorylated.
In summary, we have unveiled a previously uncharacterized function of Lyn which phosphorylates NLRP3 at Tyr918. This phosphorylation event is critical for NLRP3 ubiquitination, thus dampening the NLRP3 inflammasome activity (Fig. 6). Therefore, Lyn is essential for keeping the NLRP3 inflammasome in check both in vitro and in vivo.
Fig. 6. A schematic model for Lyn in NLRP3 inflammasome activation.
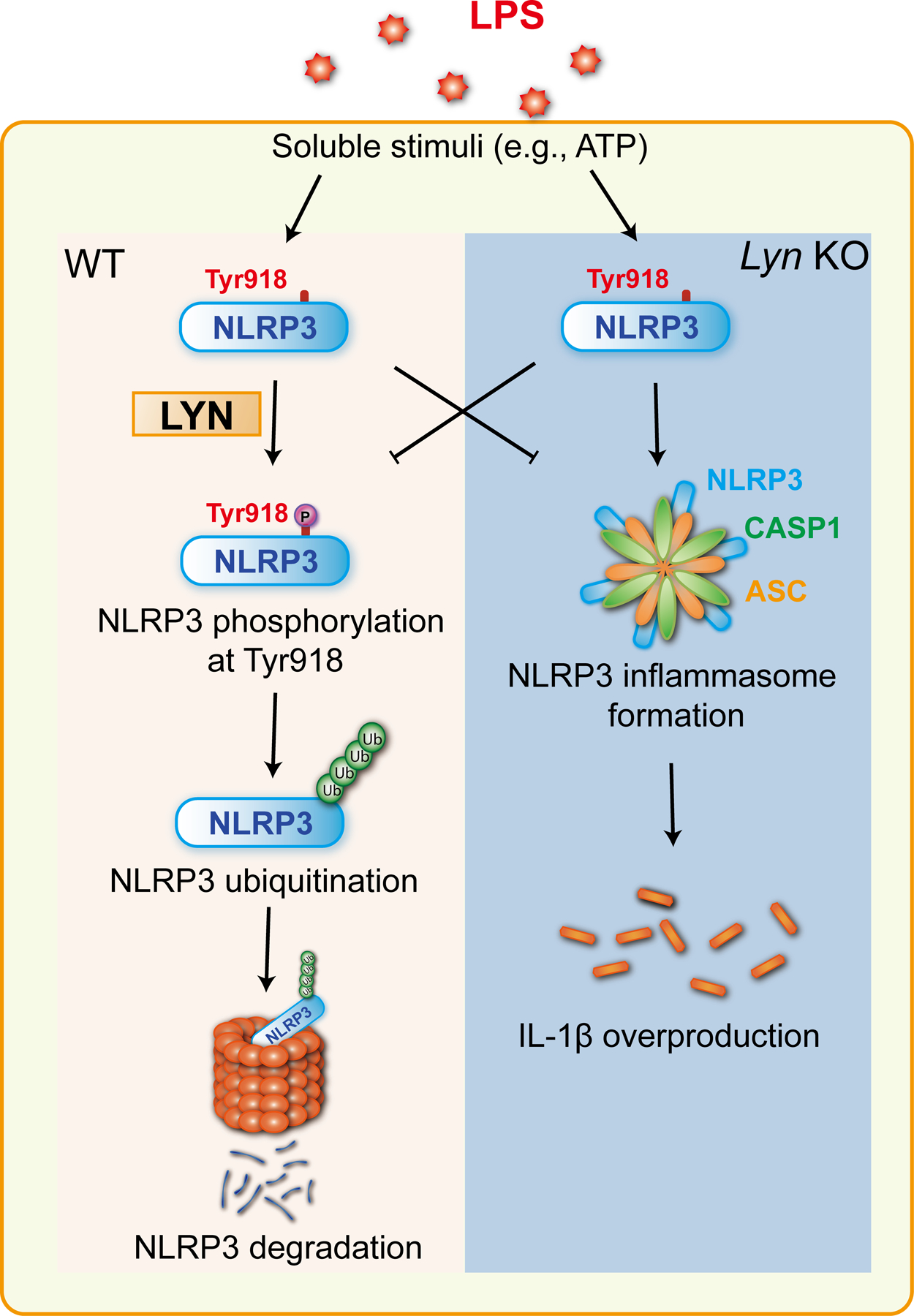
When macrophages from WT mice are primed with LPS, and stimulated with soluble stimuli such as ATP, Lyn is activated, and then phosphorylates NLRP3 at Tyr 918 which is a prerequisite for NLRP3 ubiquitination and subsequent degradation in the proteasome. In the absence of Lyn, NLRP3 tyrosine phosphorylation and subsequent ubiquitination is abrogated, which results in heightened NLRP3 protein expression, and activation.
Materials and Methods
Mice
C57BL/6J (B6) mice and Nlrp3−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Lyn−/− mice (31) were kindly provided by Dr. Anthony L. DeFranco (University of California at San Francisco, San Francisco, CA). We also generated additional Lyn−/− mice using CRISPR-Cas9 technology. The mouse Lyn gene has 13 exons, which appear to generate different mRNA and protein isoforms. To efficiently knockout all the isoforms, we chose to target exon 11, so that all the isoforms would be targeted, and any frameshift insertions/deletions are expected to result in nonsense-mediated mRNA decay (NMD). We selected a 21-nucleotide sequence in exon 11 of mouse Lyn as the target sequence [5’-atctgcgagctgctaacgtcc-3’ (Pam:tgg)]. The target sequence is highly specific, as shown by lacking any off-targets with mismatches of <4 nucleotides in the mouse genome. Microinjection of the sgRNA (50 ng/μl) and Cas9 mRNAs (from 100 ng/μl) into the cytoplasm of fertilized C57BL/6 eggs and embryo transplantation was performed in the Northwestern University Transgenic and Targeted Mutagenesis Facility. Founder mice were genotyped by a direct PCR-sequencing approach. Two intronic primers flanking the exon 11 (mLyn-E11F: 5’-catgacagcctatctagaatac-3’; mLyn-E11R: 5’-cacaagcctgcagtgagatgg-3’) were used for PCR amplification of the mouse tail DNA samples. The 246 bp PCR fragment was directly analyzed by Sanger sequencing. In 12 pups (24 alleles), we identified 20 targeted alleles, which involved insertions/deletions of 1–15 bp. The founder mice with out-of-frame insertion/deletion mutations were maintained for the generation of the homozygous knockout mice. The Lyn homozygous knockout mice with an 11bp deletion (delCTAACGTCCTG) were used in this study. All mice were 8 to12 weeks of age when used, and both male and female mice were used in this study. All animal experimentation involving LPS injection and in vivo delivery of MCC950 was approved by the Institutional Animal Care and Use Committees (IACUCs) of the University of Iowa.
Antibodies and reagents
The following antibodies (Abs) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA): rabbit anti-caspase-1 Ab (P10) (M20; sc-514), mouse anti-ubiquitin mAb (P4D1; sc-8017), rabbit anti-Lyn Ab (sc-15), Mouse anti-IL-1β mAb (3A6; #12242) Mouse anti-phospho-tyrosine mAb (P-Tyr-100) (#9411) was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Rabbit anti-Flag (M2) mAb (F2555), Src-I1 inhibitor (S2075), and BAY 61–3606 (B9685), and FLAG® M Purification Kit (CELLMM2) were obtained from Sigma-Aldrich (St. Louis, MO). Mouse anti-actin was purchased from Millipore (1A4; 113200) (Massachusetts, MA). Rabbit anti-caspase 11 mAb (ab180673), Rat monoclonal anti-mouse kappa light chain (HRP) (ab99632), and mouse monoclonal anti-rabbit IgG light chain (HRP) (ab99697) were obtained from Abcam (Cambridge, MA). HRP-conjugated goat anti-rabbit IgG or rabbit anti-mouse IgG were purchased from Kirkegaard & Perry Laboratories (Gaithersburg, MD). Protein G-Sepharose and Gutathione Sepharose beads were purchased from GE Healthcare (Piscataway, NJ). ELISA kits for mouse IL1β, TNF-α, and IL-6, and recombinant mouse GM-CSF were purchased from BioLegend (San Diego, CA). Lyn siRNA was purchased from Dharmacon RNA Technologies. Lipofectamine 2000 kit (#18324012) and Alex Fluor Plus 594-conjugated goat anti-rabbit IgG (H+L) (A32740) were purchased from Invitrogen (Carlsbad, CA). MCC950 was purchased from InvivoGen (San Diego, CA). Cytotoxicity Detection KitPlus (LDH) was purchased from Roche (#04744926001; Mannheim, Germany). EnzyChromTM Kinase Assay Kit was obtained from BioAssay Systems (EKIN-400; Hayward, CA)
Generation of bone marrow-derived macrophages (BMDMs) and bone marrow-derived dendritic cells (BMDCs)
Bone-marrow (BM) cells from WT and various knockout mouse strains were harvested from the femurs and tibias of mice. Cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM) containing 10% FBS and 50% conditioned medium from L929 cells expressing M-CSF as described (32). After one week of culture, non-adherent cells were removed, and adherent cells were 80–90% F4/80+CD11b+ as determined by flow cytometric analysis.
BMDCs were generated as previously described (33). In brief, BM cells from WT and various knockout mouse strains were harvested from the femurs and tibias of mice. Cells were cultured in RPMI1640 containing 10% FBS and 20 ng/ml GM-CSF and added an additional 20 ng/mL GM-CSF at day 3. Then half of the media were removed and same volume of fresh RPMI1640 containing 10% FBS and 20 ng/ml GM-CSF were added on day 6. The cells were harvested on day 10, were 80% CD11c+ as determined by flow cytometric analysis.
Plasmids and transfection
Flag-tagged NLRP3 plasmids were kindly provided by Dr. Fabio Martinon (University of Lausanne, Switzerland). NLRP3Y861F, NLRP3Y918F, and NLRP3Y861F/918F mutants were generated at the Mutagenex Inc. (Suwanee, GA). The murine Lyn expression plasmids pCA-Lyn and kinase-inactive mutant pCA-LynY397F were provided by Dr. M. Hibbs (Monash University, Melbourne, Australia). Nlrp3−/− or Lyn–/– BMDMs were transfected with constructs expressing Flag-tagged NLRP3, NLRP3Y861F, NLRP3Y918F, or NLRP3Y861F/918F , or WT Lyn or LynY397F (1 μg/106 cells) by 2 μl Lipofectamine 2000 reagent according to the manufacturer’s instructions for 24 hours. For determination of Lyn and NLRP3 association in the absence of LPS signaling, HEK293T cells were transfected with Flag-tagged NLRP3 by calcium precipitation as described (18, 34).
Differentiation of THP-1 cells into macrophages by PMA
THP-1 cells were cultured in 24-well plates for 3 hours with 300 ng/ml PMA at a density of 5 or 6 × 105 cells per well, then cells were washed in PBS and cultured for 3 days in complete RPMI 1640 containing 10% FBS and 1% L-glutamine.
Isolation of murine peritoneal macrophages
Peritoneal macrophages were isolated according to the protocol previously described (35). The peritoneal cells (4 × 105) in DMEM/F12–10 medium were added to 24-well plates, and cultured for 2 hours at 30°C. Nonadherent cells were removed by gently washing three times with warm PBS. The adherent macrophages were used for the experiments.
Activation of NLRP3 inflammasomes
LPS-primed BMDMs were stimulated with ATP (2.5 mM) for 30 min, nigericin (20 μM) for 3 hours, MSU (200 μg/ml) for 4 hours, and silica (500 μg/ml) for 6 hours. Poly(dA:dT) (1 μg/106 cells, for 6 hours) and flagellin (6.25 μg/106 cells, for 4 hours) were transfected by 2 μl Lipofectamine 2000 reagent according to the manufacturer’s instructions.
ELISA and cell death assay
Cell culture supernatants and serum were assayed by ELISA for IL-1β, IL-6 and TNF-α using kits from BioLegend, and cell death was measured by a lactate dehydrogenase (LDH) assay kit from Roche, each according to kit protocols.
Immunoprecipitation and Western blot analysis
BMDMs from various groups of mice were primed with LPS (100 ng/ml) for 4 hours and pretreated with Src-I1 (44 nM), Bay 61–3606 (10 nM) for 1 hour, and then stimulated with ATP (2.5 mM) for 5 min. Cells were lysed in RIPA buffer containing 2% SDS, sonicated, heated to 95°C, and then diluted to 0.2% of SDS. The cell lysates were immunopreciptated with antibody to NLRP3 and blotted with antibodies to phospho-tyrosine (pTyr) and ubiquitin. To detect the active forms of caspase-1 and mature IL-1β in the supernatants, cell supernatants were chloroform-methanol precipitated as described (36). Samples were analyzed by SDS-PAGE. Immunoblot analysis were done with Abs to Caspase-1 p10 (1:1000), IL-1β p17 (1:1000). The cell lysates treated with LPS (100 ng/ml, 4 hours) and ATP (2.5 mM, 5 min) were determined by immunoblot with Abs against NLRP3 (1:1000), Lyn (1:1000) and actin (1:3000).
For the co-immunoprecipitation assay, B6 BMDMs were primed with LPS (100 ng/ml) for 4 hours and stimulated with ATP (2.5 mM) for various times. Cells were lysed in 0.5% NP40 lysis buffer in order to better keep protein-protein interaction. The cell lysates were immunoprecipitated with antibody to NLRP3 and blotted with antibody to Lyn (1:500). In some experiments, lysates of HEK293T cells transfected with Flag-tagged NLRP3 were immunoprecipitated with antibody to Lyn and blotted with antibody to Flag (1:1000).
Confocal microscopy
WT BMDMs were primed with FITC-conjugated LPS (100 μg/ml) for 12 or 4 hours. The cells were fixed in 1% paraformaldehyde, permeabilized in 0.05% saponin, and intracellularly stained with antibody to caspase-11 (1:250), followed by Alex Fluor Plus 594-conjugated goat antibody to rabbit IgG (1: 300) and then DAPI. Imaging was performed on a Leica TCS SPE confocal microscope (Buffalo Grove, IL).
Preparation of GST-NLRP3 fusion protein
GST-NLRP3 protein was purified from the GST Fusion Protein Purification kit as described previously (34). Briefly, T7 Express Competent E. Coli cells were transformed with 100 ng GST-NLRP3 plasmid. When the OD reached 0.6, 0.1 mM IPTG was added to induce the expression of GST-NLRP3. Then the bacteria were shaken at 25˚C overnight. The bacteria pellets were lysed in lysis buffer and sonicated. Soluble proteins were purified with fast flow glutathione sepharose beads (GE Healthcare).
In vitro kinase assay
In vitro Lyn kinase assays were performed using EnzyChromTM Kinase Assay Kits (EKIN-400; BioAssay Systems, Hayward, CA). WT BMDMs were primed with 100 ng LPS for 4 hours, and stimulated with 2.5 mM ATP at 5, 15, 30, and 60 min, and lysed in ice-cold 1% Triton X-100 lysis buffer containing 10 mM Tris (pH 7.5), 150 mM NaCl, 2 mM EGTA, 50 mM β-glycerophosphate, 2 mM Na3VO4, 10 mM NaF, 1 mM DTT, 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml apoptinin (21). The cell lysates were incubated for 1 hour at 4°C with antibody to Lyn and further reacted with protein G agarose for an additional 1 hour at 4°C. Lyn immunoprecipitates were washed three times with lysis buffer, and twice with kinase buffer [20 mM HEPES (pH 7.5), 20 mM MgCl2, 20 mM MnCl2, 2 mM DTT, 25 mM β-glycerophosphate, and 100 nM Na3VO4], then resuspended in kinase assay buffer. Immunoprecipitates of Lyn were incubated with 2 μg GST-NLRP3 and 100 μM ATP in kinase buffer at room temperature for 30 min in the dark. The working reagents to detect ADP provided in the kinase assay kit were added and fluorescence intensity at lexc = 530 nm and lem = 590 nm were measured 10 minutes later. The kinase activity was calculated accordingly to fluorescence intensity. An aliquot of the kinase reaction mixtures was resolved by SDS-PAGE. The membranes were blotted with antibodies to phospho-Lyn (Tyr397) and Lyn, respectively.
Detection of ASC oligomerization
For detection of ASC oligomerization, 2 × 106 cells were primed with LPS (100 ng/ml), pretreated with Srcl-1 (44 nM) and BAY61–3606 (10 nM) for 1 hour, and stimulated with ATP (2.5 mM) for 5 min. The cells were washed twice with PBS, scraped from the plate and sheared 30 times through a 21-gauge needle. The cell lysates were washed twice with PBS and centrifuged at 5,000 x g to pellet ASC oligomers. The cell lysates were resuspended in 50 μl CHAPS buffer (20 mM HEPES-KOH, pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, 0.1 mM PMSF, 0.1% CHAPS). 4 mM disuccinimidyl suberate (DSS; AA39267, Thermo Fisher Scientific; Waltham, MA) was added, and incubated the reaction mixture for 30 min at room temperature. The reactions were quenched by adding 2x protein loading buffer, then heated for 5 min at 99 °C. The samples were separated by SDS-PAGE and immunoblotted with antibody to ASC (37, 38).
Isolation of soluble and insoluble fractions
The soluble and insoluble proteins were extracted by using the FOCUS™ Soluble & Insoluble Protein Extraction kit (Cat. #786–247; G-Biosciences, St. Louis, MO). In brief, BMDMs were washed twice with PBS, scraped from the plate and centrifuged. SPE Buffer was added and the cells were incubated 30 min on ice, followed by sonication. The lysates were centrifuged, and the supernatant was regarded as soluble fraction. The pellets were suspended in FPS Buffer and vortexed to solubilize the insoluble proteins. After centrifugation, the clear supernatants were collected for analysis as the insoluble fraction. The soluble fraction was concentrated before analysis. The purities of soluble and insoluble fractions were determined by tubulin and vimentin immunoblotting, respectively (24).
Identification of PTKs that binds to NLRP3
To identify NLRP3-binding PTKs, we incubated GST and GST-NLRP3 proteins with lysates from WT BMDMs primed by LPS and stimulated by ATP for 5 min, followed by incubation with glutathione-sepharose beads as described (18).
Suspension trapping filters for cleanup and digestion
Home-made Suspension Trapping filters were fashioned from 8 × 3mm punches of QM-A Whatman quartz discs, stacked above two layers of C18 resin (Empore C18, 3M) in a 200 μl pipet tip (39). This was filled with 100 mM Tris-HCl (pH 7.4.) in methanol. The sample was solubilized with SDS (4% wt/vol), 20 mM DTT and incubated at 95°C for 5 min then alkylated with chloroacetamide in the dark for 30 min. Phosphoric acid was added to 3% and the sample transferred to the Trapping pipet. Proteins were captured on quartz fibers as a fine dispersion while MS-incompatible materials pass through the tip. The stack was then rinsed and refilled with cold trypsin in 50 mM AmBiC and set to digest at 47°C for 2 hours. Peptides were eluted with 300 μl of 50% ACN, 0.1% TFA, lyophilized and stored at −20°C.
LC-MS/MS
Full MS1 profile data were acquired on a Q-Exactive hf (Thermo Fisher) from 380 to 1700 m/z at a resolution of 60,000 (40). The 10 most abundant precursors were selected with a mass window of 2 Th and subjected to higher-energy collisional dissociation (HCD) at 37% activation efficiency. A 30 second dynamic exclusion improved selection of lower abundant ions. Fragmentation data was acquired in centroid at 30,000 resolution.
Initial spectral searches were performed with both Mascot version 2.6.2 (MatrixScience) and Byonic search engines (Protein Metrics ver. 2.8.2) against the 2/06/2016 UniprotKB and reverted entry database for mouse. With 5 and 10 ppm tolerance for precursor and fragments respectively, searches assume fixed Cys modifications of 57 Da, as well as variable mods of 16 (M), 80 (S, T) and 114 (K) Th. Final discriminant scores were determined by Scaffold Q + S ver. 4.7 (Proteome Software) at 1.2% False Discovery Rate (FDR).
In vitro knock down of Lyn
WT BMDMs were plated at 10 cm dish and transiently transfected with 12 μg Lyn Accell siRNA or nonsense siRNA (Dharmacon) plus 30 μl Lipofectamine 2000 according to manufacturer’s protocol. Cells were harvested 36 hours later and lysed for Western blotting analysis.
LPS-induced septic shock
WT and Lyn–/– mice (n = 5) at 8–12 weeks of age were injected i.p. with a sub-lethal dose of LPS (5 mg/kg) (E. coli 0111:B4), and monitored for the survival rate for 40 hours. One group of Lyn–/– mice were injected by i.p. with MCC950 (50 mg/kg) 1 hour prior to LPS injection. For serum pro-inflammatory cytokines, 1 mg/kg of LPS was injected i.p., and serum was collected at 0, 2, 6, 12, and 24 hours.
Data analysis and statistical analysis
ELISA data were analyzed by using the Student’s t-test. Survival data were analyzed by using the Kaplan–Meier log-rank test. Differences were considered significant at p < 0.05. No animals were excluded from the analysis. Mice were allocated to experimental groups based on their genotypes and were randomized within their sex- and age-matched groups. No statistical method was used to predetermine sample size. It was assumed that normal variance occurs between the experimental groups.
Supplementary Material
ACKNOWLEDGEMENTS:
We thank M.L. Hibbs and F. Martinon for providing Lyn and NLRP3 constructs, and A. L. DeFranco for providing the Lyn−/− mice.
FUNDING:
This work was supported by the US National Institutes of Health (R01 AI090901, R01 AI121196, and R01 AI123253 to J.Z.; and R01 AR067705 to N.G.). The University of Iowa Proteomic facility is supported by an endowment from the Carver Foundation and by a Howard Hughes Medical Institute (HHMI) grant to Dr. K. Campbell. J.Z. is a University of Iowa Health Care Distinguished Scholar.
Footnotes
COMPETING INTERESTS: The authors declare that they have no competing interests.
Data and materials availability: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium through the PRIDE partner repository with the data identifier PXD028582. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Martinon F, Mayor A, Tschopp J, The inflammasomes: guardians of the body. Annu Rev Immunol 27, 229–265 (2009) 10.1146/annurev.immunol.021908.132715). [DOI] [PubMed] [Google Scholar]
- 2.Schroder K, Tschopp J, The inflammasomes. Cell 140, 821–832 (2010); published online EpubMar 19 ( 10.1016/j.cell.2010.01.040). [DOI] [PubMed] [Google Scholar]
- 3.Davis BK, Wen H, Ting JP, The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29, 707–735 (2011) 10.1146/annurev-immunol-031210-101405). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broz P, Dixit VM, Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420 (2016); published online EpubJul ( 10.1038/nri.2016.58). [DOI] [PubMed] [Google Scholar]
- 5.Schroder K, Zhou R, Tschopp J, The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300 (2010); published online EpubJan 15 ( 10.1126/science.1184003). [DOI] [PubMed] [Google Scholar]
- 6.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA, TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619 (2012); published online EpubAug 3 ( 10.1016/j.cell.2012.07.007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP, The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565 (2009); published online EpubApr 17 ( 10.1016/j.immuni.2009.02.005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamkanfi M, Dixit VM, Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28, 137–161 (2012) 10.1146/annurev-cellbio-101011-155745). [DOI] [PubMed] [Google Scholar]
- 9.Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I, Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 179, 903–913 (2009); published online EpubMay 15 ( 10.1164/rccm.200808-1274OC). [DOI] [PubMed] [Google Scholar]
- 10.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD, Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29, 301–305 (2001); published online EpubNov ( 10.1038/ng756). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J, NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004); published online EpubMar ( [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Meszaros G, He WT, Xu Y, de Fatima Magliarelli H, Mailly L, Mihlan M, Liu Y, Puig Gamez M, Goginashvili A, Pasquier A, Bielska O, Neven B, Quartier P, Aebersold R, Baumert TF, Georgel P, Han J, Ricci R, Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med 214, 2671–2693 (2017); published online EpubSep 04 ( 10.1084/jem.20162040). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, Liu X, Huang YJ, Cai H, Zhan XY, Han QY, Wang H, Chen Y, Li HY, Li AL, Zhang XM, Zhou T, Li T, NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell 68, 185–197 e186 (2017); published online EpubOct 5 ( 10.1016/j.molcel.2017.08.017). [DOI] [PubMed] [Google Scholar]
- 14.Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, Brinkschulte R, Geyer M, Meissner F, Latz E, NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med 214, 1725–1736 (2017); published online EpubJun 5 ( 10.1084/jem.20160933). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, Scharl S, Gutte PM, Grutter MG, Beer HD, Contassot E, Chan AC, Dai X, Rawlings DJ, Mair F, Becher B, Falk W, Fried M, Rogler G, Scharl M, NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest 126, 1783–1800 (2016); published online EpubMay 02 ( 10.1172/JCI83669). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J, Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49, 331–338 (2013); published online EpubJan 24 ( 10.1016/j.molcel.2012.11.009). [DOI] [PubMed] [Google Scholar]
- 17.Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X, Zhang L, Gao C, The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol 18, 214–224 (2017); published online EpubFeb ( 10.1038/ni.3641). [DOI] [PubMed] [Google Scholar]
- 18.Tang J, Tu S, Lin G, Guo H, Yan C, Liu Q, Huang L, Tang N, Xiao Y, Pope RM, Rajaram MVS, Amer AO, Ahmer BM, Gunn JS, Wozniak DJ, Tao L, Coppola V, Zhang L, Langdon WY, Torrelles JB, Lipkowitz S, Zhang J, Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J Exp Med 217, (2020); published online EpubApr 6 ( 10.1084/jem.20182091). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byeon SE, Yi YS, Oh J, Yoo BC, Hong S, Cho JY, The role of Src kinase in macrophage-mediated inflammatory responses. Mediators Inflamm 2012, 512926 (2012) 10.1155/2012/512926). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM, Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013); published online EpubSep 13 ( 10.1126/science.1240248). [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Salojin KV, Gao JX, Cameron MJ, Bergerot I, Delovitch TL, p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J Immunol 162, 3819–3829 (1999); published online EpubApr 1 ( [PubMed] [Google Scholar]
- 22.Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, Sutterwala FS, Bohle DS, Descoteaux A, Flavell RA, Olivier M, Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog 5, e1000559 (2009); published online EpubAug ( 10.1371/journal.ppat.1000559). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spalinger MR, Lang S, Gottier C, Dai X, Rawlings DJ, Chan AC, Rogler G, Scharl M, PTPN22 regulates NLRP3-mediated IL1B secretion in an autophagy-dependent manner. Autophagy 13, 1590–1601 (2017); published online EpubSep 2 ( 10.1080/15548627.2017.1341453). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider KS, Gross CJ, Dreier RF, Saller BS, Mishra R, Gorka O, Heilig R, Meunier E, Dick MS, Cikovic T, Sodenkamp J, Medard G, Naumann R, Ruland J, Kuster B, Broz P, Gross O, The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep 21, 3846–3859 (2017); published online EpubDec 26 ( 10.1016/j.celrep.2017.12.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, He Z, Zeng Y, Hu Y, Sun S, Li J, Wu X, Wang X, Strober W, Chen C, Meng G, Sun B, Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 23, 201–212 (2013); published online EpubFeb ( 10.1038/cr.2013.6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo H, Callaway JB, Ting JP, Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21, 677–687 (2015); published online EpubJul ( 10.1038/nm.3893). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broz P, Inflammasomes in Host Defense and Autoimmunity. Chimia (Aarau) 70, 853–855 (2016); published online EpubDec 21 ( 10.2533/chimia.2016.853). [DOI] [PubMed] [Google Scholar]
- 28.Ingley E, Functions of the Lyn tyrosine kinase in health and disease. Cell Commun Signal 10, 21 (2012); published online EpubJul 17 ( 10.1186/1478-811X-10-21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, Mitsuyama M, Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol 14, 1247–1255 (2013); published online EpubDec ( 10.1038/ni.2749). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunter T, The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell 28, 730–738 (2007); published online EpubDec 14 ( 10.1016/j.molcel.2007.11.019). [DOI] [PubMed] [Google Scholar]
- 31.Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA, Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity 7, 69–81 (1997); published online EpubJul ( [DOI] [PubMed] [Google Scholar]
- 32.Xiao Y, Tang J, Guo H, Zhao Y, Tang R, Ouyang S, Zeng Q, Rappleye CA, Rajaram MV, Schlesinger LS, Tao L, Brown GD, Langdon WY, Li BT, Zhang J, Targeting CBLB as a potential therapeutic approach for disseminated candidiasis. Nat Med 22, 906–914 (2016); published online EpubAug ( 10.1038/nm.4141). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, Brinster C, Isolation of dendritic cells. Curr Protoc Immunol Chapter 3, Unit 3 7 (2009); published online EpubAug ( 10.1002/0471142735.im0307s86). [DOI] [PubMed] [Google Scholar]
- 34.Qiao G, Ying H, Zhao Y, Liang Y, Guo H, Shen H, Li Z, Solway J, Tao E, Chiang YJ, Lipkowitz S, Penninger JM, Langdon WY, Zhang J, E3 Ubiquitin Ligase Cbl-b suppresses proallergic T cell development and allergic airway inflammation. Cell Rep 6, 709–723 (2014); published online EpubFeb 27 ( 10.1016/j.celrep.2014.01.012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Goncalves R, Mosser DM, The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14, Unit 14 11 (2008); published online EpubNov ( 10.1002/0471142735.im1401s83). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM, Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011); published online EpubNov 3 ( 10.1038/nature10558). [DOI] [PubMed] [Google Scholar]
- 37.Lugrin J, Martinon F, Detection of ASC Oligomerization by Western Blotting. Bio Protoc 7, (2017); published online EpubMay 20 ( 10.21769/BioProtoc.2292). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Micco A, Frera G, Lugrin J, Jamilloux Y, Hsu ET, Tardivel A, De Gassart A, Zaffalon L, Bujisic B, Siegert S, Quadroni M, Broz P, Henry T, Hrycyna CA, Martinon F, AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc Natl Acad Sci U S A 113, E4671–4680 (2016); published online EpubAug 9 ( 10.1073/pnas.1602419113). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zougman A, Selby PJ, Banks RE, Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 14, 1006–1000 (2014); published online EpubMay ( 10.1002/pmic.201300553). [DOI] [PubMed] [Google Scholar]
- 40.Yu CL, Summers RM, Li Y, Mohanty SK, Subramanian M, Pope RM, Rapid identification and quantitative validation of a caffeine-degrading pathway in Pseudomonas sp. CES. J Proteome Res 14, 95–106 (2015); published online EpubJan 2 ( 10.1021/pr500751w). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.