

Abstract
Persistent isolated microscopic hematuria is relatively common in pediatric practice, affecting around 0.25% of children. Isolated microscopic hematuria can be caused by a myriad of potentially benign or serious causes, including urologic issues; kidney stones; glomerular diseases, including disorders of the glomerular basement membrane; hematologic abnormalities; and others. The challenge for the pediatrician or pediatric nephrologist is to distinguish children with potentially progressive forms of kidney disease versus other causes while minimizing cost and inconvenience for the child and family. This manuscript will review the multiple potential causes of microscopic hematuria and provide a framework for the initial evaluation and monitoring of such patients.
Keywords: glomerular and tubulointerstitial diseases, Alport syndrome, hematuria, hypercalciuria, IgA nephropathy, IgA vasculitis, pediatric nephrology
Introduction
Microscopic hematuria is common in children, and it is present in 4.1% of school age children (1). Microscopic hematuria that persists on repeated evaluations is less common, with only 0.25% of children having blood in four consecutive urinalyses (UAs) (1). Of children with microscopic hematuria, only 16.2% of them also had proteinuria; thus, the majority of children identified with hematuria have isolated hematuria (1). A study of 1.2 million adolescents and young adults assessed at the time of Israeli army entrance found microscopic hematuria in 0.3% of patients (2). Importantly, the presence of microscopic hematuria was not a universally benign finding. The adjusted hazard ratio for ESKD in the patients with microscopic hematuria over 20 years of follow-up was 18.5 (95% confidence interval, 12.4 to 27.6), although absolute risk remained low.
Although routine screening UAs are not recommended for healthy, asymptomatic children in the United States, UAs are often obtained in pediatric practice due to symptoms of increased frequency of urination, dysuria, or fever with microscopic hematuria present as an incidental finding. Screening UAs are recommended for children who are at higher risk for CKD, such as those with a history of AKI, congenital anomalies of the kidney and urinary tract, acute nephritis, hypertension, active systemic disease, prematurity, intrauterine growth retardation, or a family history of kidney disease (3).
Isolated microscopic hematuria can be caused by a myriad of potentially benign or serious causes, including urologic issues; kidney stones; glomerular diseases including disorders of the glomerular basement membrane (GBM); hematologic abnormalities; and others. One biopsy series of children from the United States with persistent microscopic hematuria lasting longer than 6 months identified normal findings in 44%, thin GBMs in 22%, Alport syndrome in 12%, and IgA nephropathy (IgAN) in 11% (4). In contrast, a similar study in China demonstrated normal findings in 71%, thin GBMs in 3.2%, Alport syndrome in 1.4%, and IgAN in 16%, suggesting that practice and referral patterns or race/ethnicity may influence findings (5). The challenge for the pediatrician or pediatric nephrologist evaluating the child with persistent microscopic hematuria is to distinguish between children with potentially progressive forms of kidney disease versus other causes while minimizing cost and inconvenience for the child and family. This manuscript will review the multiple potential causes of isolated microscopic hematuria and provide a framework for the initial evaluation and monitoring of such patients. The presence of albuminuria along with microscopic hematuria is generally suggestive of kidney parenchymal disease that may require more urgent diagnosis or treatment. The evaluation of children with hematuria and albuminuria is not covered in this manuscript.
Urologic Causes
Hematuria with pyuria is often found in the setting of urinary tract infection in children. It can be seen with cystitis or acute pyelonephritis and can be microscopic or macroscopic in nature. When indicated clinically, UA should be obtained to evaluate for the presence of pyuria and/or bacteriuria, and infection should be confirmed with a urine culture before initiating antibiotics (6). Viral urinary tract infections may also be a cause of hematuria, including adenovirus and BK virus infections, particularly in immunosuppressed patients (7).
Microscopic hematuria can also be the result of a structural urinary tract lesion. The most serious lesions are malignant (such as Wilms tumor or renal cell carcinoma). However, malignancies are rare in children and more often present with other symptoms, such as abdominal pain or mass (8,9). Other etiologies may include bladder lesions, such as eosinophilic cystitis; hemangiomas; and malignancy or hydronephrosis (10).
Terminal hematuria is a pattern of typically painless gross hematuria at the end of the void. Microscopic hematuria may be present between occurrences. This is most often caused by idiopathic urethritis but can also be due to an infection, such as Schistosomiasis, in patients with a history of travel to endemic areas (11,12). Careful questioning about any history of gross hematuria and pattern of hematuria is important in children with microscopic hematuria.
Another potential structural cause for microscopic hematuria is nutcracker syndrome, in which the left renal vein is compressed by the aorta and/or superior mesenteric artery, causing kidney congestion and hematuria. Although previously thought to be a rare finding in children, recent studies suggest that it is a frequent cause of persistent microscopic hematuria in this population. A study of 216 children with hematuria (176 microscopic and 40 gross) showed that nutcracker syndrome was diagnosed in 33.3% (13). This study was performed at a single center by an experienced radiologist with expertise in the diagnosis of nutcracker syndrome. Referral bias, patient population, and expertise of the ultrasonographer may all have contributed to the high incidence of nutcracker syndrome in this study. Of 736 patients with nutcracker syndrome, 288 patients (39%) had microscopic hematuria, whereas the remaining 448 patients had gross hematuria (14). Nutcracker syndrome can be asymptomatic or associated with abdominal or flank pain (15). It can be diagnosed by careful kidney ultrasound with doppler (13,14), although more invasive testing (such as computed tomography angiography or magnetic resonance angiography or arteriography) might be required for diagnosis in some cases.
Hypercalciuria and Kidney Stones
Hypercalciuria is one of the most common causes of persistent microscopic hematuria (16,17). It is a common finding in children, with an estimated prevalence of 3%–10% of the general population (18,19). It is most accurately diagnosed by a 24-hour urine collection showing urine calcium (Ca) >4 mg/kg per day, but this can be difficult to obtain in younger children where Ca-to-creatinine (Cr) ratio in random urine sample is often used. Hypercalciuria can be defined as a Ca-Cr ratio >0.86 mg/mg in children <7 months, >0.60 mg/mg in those age 7–18 months, >0.42 mg/mg in those age 19 months to 6 years, and >0.22 mg/mg in children older than 6 years (20). There is a good correlation between Ca-Cr ratio and total Ca in 24-hour urine collection (21,22).
Hypercalciuria is a common finding in patients with isolated microscopic hematuria, accounting for up to 30% of cases (16,23–25). A study of 325 children referred for evaluation of asymptomatic microscopic hematuria found hypercalciuria in 11% (26). In addition to hypercalciuria, hyperuricosuria and hypocitraturia have also been associated with microscopic hematuria in children (27).
Hypercalciuria is believed to cause hematuria by irritating the uroepithelial cells, and it can be associated with microscopic or gross hematuria and/or urinary tract symptoms, such as dysuria. It may also increase the risk of kidney stones (28,29). After it is diagnosed, serum Ca should be obtained to differentiate idiopathic hypercalciuria (where serum Ca is normal) from that secondary to hypercalcemia (such as hyperparathyroidism, vitamin D toxicity, etc.).
Similarly, nephrolithiasis can cause microscopic and/or macroscopic hematuria. Although not as common as in adults, the incidence of pediatric stone disease has increased in recent years (30). After it is suspected, kidney imaging should be obtained to confirm the presence of stones and evaluate for possible hydronephrosis. Kidney ultrasound is usually the initial modality of choice because it is effective to show kidney stones (>5–8 mm) while avoiding radiation. However, helical computed tomography continues to be the most sensitive modality to detect kidney or ureteral stones regardless of the size. Most children with kidney stones have identifiable risk factors (31), including hypercalciuria, hypocitraturia, hyperuricosuria, and/or urinary solute supersaturation.
Disorders of the GBM
The GBM imparts a significant barrier to the passage of red blood cells into the urine, and disorders of the GBM may lead to microscopic hematuria. Type IV collagen is the predominant collagen in the GBM, and it is composed of trimers of α3(IV), α4(IV), and α5(IV) encoded by genes COL4A3, COL4A4, and COL4A5, respectively. Mutations in COL4A5 on the X chromosome in boys cause classic Alport syndrome. Classic Alport syndrome manifests as microscopic hematuria from birth with progression to proteinuria and eventual loss of kidney function and ESKD, typically in the mid-20s but may be earlier or later depending on genotype and treatment (32). Affected individuals often have hearing loss and eye abnormalities as well due to presence of the α3α4α5(IV) trimer in the cochlea and lens of the eye. Girls with a heterozygous mutation in COL4A5 have a variable course ranging from only microscopic hematuria throughout life to ESKD in 10%–15% (33,34). Mutations in both copies of COL4A3 and COL4A4 cause autosomal recessive Alport syndrome with similar clinical features to men with X-linked Alport syndrome. Patients with a heterozygous mutation in COL4A3 or COL4A4 have autosomal dominant Alport syndrome. This disorder has wide variability in phenotype from only persistent microscopic hematuria to ESKD (35).
Alport syndrome can be diagnosed by a kidney biopsy or genetic testing. Initial biopsies may show only thinning of the GBM compared with age-matched controls. This may be the only finding in boys with XLAS or boys and girls with autosomal recessive Alport syndrome in the first decade of life or may be a consistent finding throughout life in women with XLAS. Thus, the presence of thin basement membranes on kidney biopsy may not be a benign finding and does not provide a prognosis or diagnosis for the patient. For these reasons, the Alport Syndrome Classification Working Group recently recommended that “thin basement membranes” be recognized as a pathologic finding and not a specific diagnosis (35). In patients with more advanced Alport syndrome, kidney pathology demonstrates thickening and splitting of the GBM with lamellation and basket weaving with interstitial fibrosis and podocyte foot process effacement.
Early diagnosis of Alport syndrome is beneficial for a number of reasons. First, treatment with ACE inhibitors or ARBs can be initiated, which can slow the progression of kidney disease (36). Early detection of hearing loss through screening can provide the opportunity for early assistive device use and improved school outcomes. Finally, Alport syndrome may be present in other family members who may benefit from diagnosis and early treatment as well.
IgAN
IgAN is the most common chronic GN in the world and the most common primary glomerulopathy in young adult whites (37). Its prevalence is higher in patients with European and Asian ancestry. It is diagnosed by kidney biopsy showing a dominant staining of IgA in the mesangium of the glomeruli.
Although IgAN has a wide spectrum of clinical presentations, most children will have recurrent episodes of gross hematuria with persistent microscopic hematuria in between the episodes (38). Gross hematuria commonly presents within 1–2 days of an upper respiratory infection. However, IgAN can also present with persistent microscopic hematuria with or without proteinuria, heavy proteinuria with or without nephrotic syndrome, and even rapid progression GN. A systemic review of 1092 children identified 857 with isolated microscopic hematuria and 235 with combined microscopic hematuria and proteinuria (17). In this series, IgAN was the cause in 10.4% of patients with isolated microscopic hematuria and 44.3% of patients with hematuria and proteinuria. Similar studies have shown that IgAN is one of the most encountered biopsy diagnoses in children with persistent microscopic hematuria (5,39).
The clinical course of IgAN is quite variable, and progression to ESKD can be as high as 40% of cases (40,41). However, this can vary on the basis of patient’s ethnicity and course of treatment used, among other factors. IgAN in children most often has a favorable outcome (38,42). The presence of uncontrolled hypertension, significant proteinuria (>0.5–1 g/d), and/or decreased kidney function at presentation are risk factors for CKD progression (43,44). Kidney biopsy findings also can provide further prognostic factors, and the Oxford classification of kidney pathology in IgAN has a good correlation to kidney outcomes (45), although findings in pediatric cohorts have been inconsistent (46–48).
Other GN
Persistent microscopic hematuria is also seen in the setting of other GN. One of the most common GNs encountered in children is acute postinfectious GN (APIGN), with an incidence of 0.3 cases per 100,000 person-years in developed countries (49). It is most common between 4 and 14 years of age, and although it is classically induced by β-hemolytic streptococcal infection, it can also be seen following infections with different pathogens, including those with bacterial, viral, and fungal causes (50).
APIGN has a variable clinical presentation. It classically presents after a latent period (7–10 days following a throat infection or 3–5 weeks after skin infections) with cola-colored urine, proteinuria, hypertension with edema, and decreased kidney function. A more severe presentation occurs in some children with rapidly progressive GN or hypertensive encephalopathy (51,52). APIGN can present with microscopic hematuria with or without proteinuria and no significant hypertension or decreased kidney function, especially at times of streptococcal outbreaks in the community. It is also common to see persistent microscopic hematuria after resolution of APIGN, and this may last for a few months to over a year (53).
Microscopic hematuria can also be seen with other types of glomerular disease, such as lupus nephritis, membranoproliferative GN, or FSGSs; however, these are unlikely without the presence of proteinuria, hypertension, and/or extrarenal manifestation. Vasculitis can also be associated with microscopic hematuria among other clinical manifestations, such as pulmonary and sinus disease, skin rash, and general constitutional symptoms (such as fatigue or fever). The most common vasculitis in children is IgA vasculitis (previously known as Henoch–Schönlein purpura). This leukocytoclastic vasculitis usually presents with a typical purpuric rash over the lower extremities, arthritis, and abdominal pain. Kidney involvement (IgAVN) occurs in 30%–50% of the cases, more often in the first 8 weeks of presentation (54,55). Kidney manifestations may vary significantly between children, but most cases present with isolated microscopic hematuria, occasionally associated with non-nephrotic–range proteinuria. Less frequently, IgAVN presents with nephrotic syndrome or severe GN associated with significant hypertension and/or kidney dysfunction. Children presenting with proteinuria or significant crescentic GN often have a worse outcome (56). However, patients may have prolonged kidney sequelae even in absence of crescents (57).
Hematologic Causes
Hemophilia is a rare, inherited bleeding disorder caused by deficiency in factor VIII (hemophilia A, X linked), factor IX (hemophilia B, X linked), or factor XI (hemophilia C, autosomal recessive). Patients may have signs and symptoms of easy bruising with minor trauma, hemarthrosis, gingival bleeding, or bleeding after injury or procedures, such as neonatal circumcision. Hematuria is relatively common in children with hemophilia, with an incidence of 19%–45% in various series (58,59). Episodes of gross hematuria are also common and may require hospitalization and treatment with recombinant factors. Diagnosis can be made by careful bleeding history along with targeted laboratory examinations showing prolonged aPTT with correction with mixing study. Factor levels can also be measured for confirmation.
Sickle cell anemia, sickle cell trait, or thalassemias are other potential causes of microscopic hematuria in children. Sickle cell anemia is an autosomal recessive hemoglobinopathy caused by mutations in HBB, the β-globin gene. It leads to hemolytic anemia and vaso-occlusive crises in affected patients. Thalassemias are a collection of hemoglobinopathies that lead to imbalance in the normal ratio of α- and β-chains of hemoglobin leading to hemolysis, ineffective erythropoiesis, and severe anemia. These disorders have higher prevalence in patients of African or Asian origin. Microscopic hematuria is present in 4.1%–23.2% of children with sickle cell disease (60) because of sickling predominantly in the renal medulla due to the hypoxic, hyperoxmotic, and acidic milieu (61). In severe cases, this may progress to renal infarction and papillary necrosis. Renal medullary carcinoma is a rare but serious cause of hematuria in patients with sickle cell disease or trait. It is most common in early adulthood but can also manifest in childhood (62). Microscopic hematuria is present in 9.8% of children with β-thalassemia (63). Potential mechanisms of hematuria include side effects of iron chelators, such as deferoxamine; iron deposition in the kidney; or vascular thrombosis and renal infarction.
Cystic Kidney Disease
Hematuria may be present in up to 40% of patients with autosomal dominant polycystic kidney disease and may be the presenting sign (64). Hematuria in these patients may be caused by hemorrhage into a cyst, kidney stone, or malignancy.
Recommended Evaluation of Microscopic Hematuria
The first step in evaluation of microscopic hematuria is to determine the source of hematuria via urine microscopy. Urine erythrocytes from patients with a nonglomerular (urologic) cause of hematuria appear homogenous and normal in shape (65). The finding of deformed red blood cells or red blood cell casts under microscopy suggests a glomerular cause of the hematuria, and this can be seen on routine light microscopy (Figure 1) (66). There are no uniform criteria for determination of glomerular versus nonglomerular hematuria; however, one commonly used classification scheme considers “glomerular” hematuria when ≥40% of at least 100 red blood cells are dysmorphic (67). Other potential findings on urine microscopy include pyuria and/or bacteriuria (infection), presence of ova and parasites (schistosomiasis), or crystaluria (hypercalciuria).
Figure 1.

Urine microscopy of hematuria from a glomerular source demonstrates red blood cell casts and dysmorphic red blood cells. (Left panel) Image shows a red blood cell cast. (Right panel) Image shows deformed red blood cells (arrows). Both findings are consistent with a glomerular source for hematuria.
When isolated microscopic hematuria is identified on a UA performed for any cause, this should be noted, and a urine culture should be performed when clinically indicated. UA should be repeated two times over the next 1–2 months to exclude transient hematuria for which reassurance alone is appropriate. Although most cases of isolated persistent microscopic hematuria are idiopathic, the most common causes are hypercalciuria (with or without kidney stones), urinary tract infections and urinary tract malformations (including nutcracker syndrome), disorders of the GBM (Alport syndrome), and IgAN. Thus, the initial approach should focus on evaluating for those diagnoses by getting a thorough medical history, family history and physical examination, urine protein-Cr ratio, urine Ca-Cr ratio, BUN/Cr and electrolytes, and a kidney ultrasound (Figure 2). Screening for sickle cell disease or coagulopathies should also be considered in high-risk populations.
Figure 2.
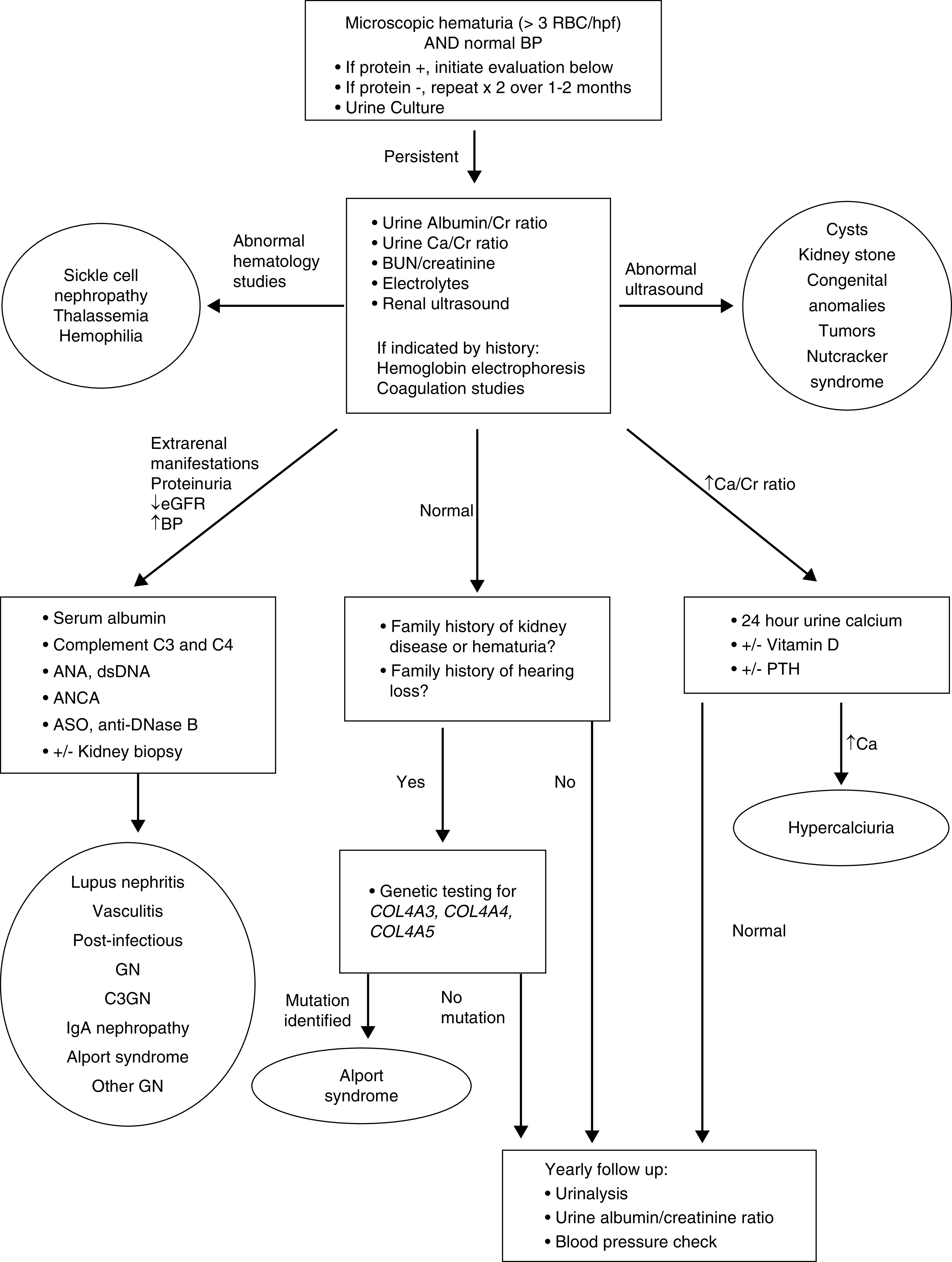
Algorithm for evaluation of a child with microscopic hematuria. ANA, antinuclear antibody; ASO, antistreptolysin O antibody; Ca, calcium; Cr, creatinine; C3GN, complement factor 3 glomerulopathy; dsDNA, antidouble-stranded DNA antibody; hpf, high-powered field; PTH, parathyroid hormone; RBC, red blood cell.
For children with extrarenal manifestations, albuminuria, decreased kidney function, or hypertension, further evaluation should be performed to exclude GN, including C3, C4, antinuclear antibody, antidouble-stranded DNA antibody, ANCA, serum albumin, antistreptolysin O antibody titers, and anti-DNase B titers. Kidney biopsy may be required in these situations for diagnosis.
For children with a family history of early-onset kidney failure, hearing loss, or recurrent gross hematuria, the chance of finding abnormalities on kidney biopsy is higher (4). Genetic testing for Alport syndrome via screening for mutations in COL4A3, COL4A4, and COL4A5 is recommended to allow for early diagnosis and initiation of treatment if indicated.
Children with no albuminuria, normal kidney function and BP, and no hypercalciuria should receive reassurance that the prognosis of isolated microscopic hematuria is often good (1,68,69). However, providing long-term follow-up is recommended for early detection of signs, such as proteinuria or hypertension, suggestive of Alport syndrome or IgAN that may require further evaluation or treatment.
Disclosures
All authors have nothing to disclose.
Funding
None.
Acknowledgments
Dr. Michelle N. Rheault receives research funding from Advicenne, Genentech, Reata, Retrophin, and Sanofi, outside the submitted work.
Author Contributions
M. Rheault conceptualized the study; M. Kallash and M. Rheault wrote the original draft; and M. Kallash and M. Rheault reviewed and edited the manuscript.
References
- 1.Vehaskari VM, Rapola J, Koskimies O, Savilahti E, Vilska J, Hallman N: Microscopic hematuria in school children: Epidemiology and clinicopathologic evaluation. J Pediatr 95: 676–684, 1979 [DOI] [PubMed] [Google Scholar]
- 2.Vivante A, Afek A, Frenkel-Nir Y, Tzur D, Farfel A, Golan E, Chaiter Y, Shohat T, Skorecki K, Calderon-Margalit R: Persistent asymptomatic isolated microscopic hematuria in Israeli adolescents and young adults and risk for end-stage renal disease. JAMA 306: 729–736, 2011 [DOI] [PubMed] [Google Scholar]
- 3.American Academy of Pediatrics—Section on Nephrology and the American Society of Pediatric Nephrology: Choosing wisely: Five things physicians and patients should question, 2018. Available at: https://www.choosingwisely.org/societies/american-academy-of-pediatrics-section-on-nephrology-and-the-american-society-of-pediatric-nephrology/. Accessed June 10, 2020
- 4.Trachtman H, Weiss RA, Bennett B, Greifer I: Isolated hematuria in children: Indications for a renal biopsy. Kidney Int 25: 94–99, 1984 [DOI] [PubMed] [Google Scholar]
- 5.Feng CY, Xia YH, Wang WJ, Xia J, Fu HD, Wang X, Shen HJ, Qian GL, Liu AM, Mao JH: Persistent asymptomatic isolated hematuria in children: Clinical and histopathological features and prognosis. World J Pediatr 9: 163–168, 2013 [DOI] [PubMed] [Google Scholar]
- 6.Roberts KB; Subcommittee on Urinary Tract Infection, Steering Committee on Quality Improvement and Management : Urinary tract infection: Clinical practice guideline for the diagnosis and management of the initial UTI in febrile infants and children 2 to 24 months. Pediatrics 128: 595–610, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Paduch DA: Viral lower urinary tract infections. Curr Urol Rep 8: 324–335, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raney RB Jr., Palmer N, Sutow WW, Baum E, Ayala A: Renal cell carcinoma in children. Med Pediatr Oncol 11: 91–98, 1983 [DOI] [PubMed] [Google Scholar]
- 9.Howlader NNA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA: SEER Cancer Statistics Review, 1975-2017, Bethesda, MD, National Cancer Institute, 2020 [Google Scholar]
- 10.Malkan AD, Loh A, Bahrami A, Navid F, Coleman J, Green DM, Davidoff AM, Sandoval JA: An approach to renal masses in pediatrics. Pediatrics 135: 142–158, 2015 [DOI] [PubMed] [Google Scholar]
- 11.Prakash V, Caplivski D, Vassalotti JA: Terminal hematuria. Kidney Int 88: 204, 2015 [DOI] [PubMed] [Google Scholar]
- 12.Degheili JA, Dickson AP: Childhood and adolescent idiopathic urethritis: What does the current literature say? J Pediatr Urol 16: 276–283, 2020 [DOI] [PubMed] [Google Scholar]
- 13.Shin JI, Park JM, Lee JS, Kim MJ: Effect of renal Doppler ultrasound on the detection of nutcracker syndrome in children with hematuria. Eur J Pediatr 166: 399–404, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vianello FA, Mazzoni MB, Peeters GG, Fossali EF, Camozzi P, Bianchetti MG, Milani GP: Micro- and macroscopic hematuria caused by renal vein entrapment: Systematic review of the literature. Pediatr Nephrol 31: 175–184, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Alaygut D, Bayram M, Soylu A, Cakmakcı H, Türkmen M, Kavukcu S: Clinical course of children with nutcracker syndrome. Urology 82: 686–690, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Parekh DJ, Pope JC 4th, Adams MC, Brock JW 3rd: The association of an increased urinary calcium-to-creatinine ratio, and asymptomatic gross and microscopic hematuria in children. J Urol 167: 272–274, 2002 [PubMed] [Google Scholar]
- 17.Clark M, Aronoff S, Del Vecchio M: Etiologies of asymptomatic microscopic hematuria in children—systematic review of 1092 subjects. Diagnosis (Berl) 2: 211–216, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Moore ES, Coe FL, McMann BJ, Favus MJ: Idiopathic hypercalciuria in children: Prevalence and metabolic characteristics. J Pediatr 92: 906–910, 1978 [DOI] [PubMed] [Google Scholar]
- 19.Escribano J, Balaguer A, Martin R, Feliu A, Espax R: Childhood idiopathic hypercalciuria--clinical significance of renal calyceal microlithiasis and risk of calcium nephrolithiasis. Scand J Urol Nephrol 38: 422–426, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Sargent JD, Stukel TA, Kresel J, Klein RZ: Normal values for random urinary calcium to creatinine ratios in infancy. J Pediatr 123: 393–397, 1993 [DOI] [PubMed] [Google Scholar]
- 21.Mir S, Serdaroglu E: Quantification of hypercalciuria with the urine calcium osmolality ratio in children. Pediatr Nephrol 20: 1562–1565, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Choi IS, Jung ES, Choi YE, Cho YK, Yang EM, Kim CJ: Random urinary calcium/creatinine ratio for screening hypercalciuria in children with hematuria. Ann Lab Med 33: 401–405, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stapleton FB, Roy S 3rd, Noe HN, Jerkins G: Hypercalciuria in children with hematuria. N Engl J Med 310: 1345–1348, 1984 [DOI] [PubMed] [Google Scholar]
- 24.Stapleton FB: Idiopathic hypercalciuria: Association with isolated hematuria and risk for urolithiasis in children. The Southwest Pediatric Nephrology Study Group. Kidney Int 37: 807–811, 1990 [DOI] [PubMed] [Google Scholar]
- 25.Valavi E, Nickavar A, Aeene A: Urinary metabolic abnormalities in children with idiopathic hematuria. J Pediatr Urol 15: 165.e1-165.e4, 2019 [DOI] [PubMed] [Google Scholar]
- 26.Feld LG, Meyers KE, Kaplan BS, Stapleton FB: Limited evaluation of microscopic hematuria in pediatrics. Pediatrics 102: E42, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Spivacow FR, Del Valle EE, Rey PG: Metabolic risk factors in children with asymptomatic hematuria. Pediatr Nephrol 31: 1101–1106, 2016 [DOI] [PubMed] [Google Scholar]
- 28.Spivacow FR, Negri AL, del Valle EE, Calviño I, Fradinger E, Zanchetta JR: Metabolic risk factors in children with kidney stone disease. Pediatr Nephrol 23: 1129–1133, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Polito C, La Manna A, Cioce F, Villani J, Nappi B, Di Toro R: Clinical presentation and natural course of idiopathic hypercalciuria in children. Pediatr Nephrol 15: 211–214, 2000 [DOI] [PubMed] [Google Scholar]
- 30.Ward JB, Feinstein L, Pierce C, Lim J, Abbott KC, Bavendam T, Kirkali Z, Matlaga BR; NIDDK Urologic Diseases in America Project : Pediatric urinary stone disease in the United States: The urologic diseases in America Project. Urology 129: 180–187, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gearhart JP, Herzberg GZ, Jeffs RD: Childhood urolithiasis: Experiences and advances. Pediatrics 87: 445–450, 1991 [PubMed] [Google Scholar]
- 32.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC: X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J Am Soc Nephrol 11: 649–657, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Dahan K, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC: X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport syndrome Concerted action” study. J Am Soc Nephrol 14: 2603–2610, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, Finlay M, Flinter F: Alport syndrome in women and girls. Clin J Am Soc Nephrol 11: 1713–1720, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, Nozu K, Renieri A, Rheault M, Wang F, Gross O: Alport syndrome: A unified classification of genetic disorders of collagen IV α345: A position paper of the Alport syndrome classification working group. Kidney Int 93: 1045–1051, 2018 [DOI] [PubMed] [Google Scholar]
- 36.Gross O, Tonshoff B, Weber LT, Pape L, Latta K, Fehrenbach H, Lange-Sperandio B, Zappel H, Hoyer P, Staude H, Konig S, John U, Gellermann J, Hoppe B, Galiano M, Hoecker B, Ehren R, Lerch C, Kashtan CE, Harden M, Boeckhaus J, Friede T; German Pediatric Nephrology Study Group, EARLY PRO-TECT Alport Investigators : A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int 97: 1275–1286, 2020 [DOI] [PubMed] [Google Scholar]
- 37.Nair R, Walker PD: Is IgA nephropathy the commonest primary glomerulopathy among young adults in the USA? Kidney Int 69: 1455–1458, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Wyatt RJ, Kritchevsky SB, Woodford SY, Miller PM, Roy S 3rd, Holland NH, Jackson E, Bishof NA: IgA nephropathy: Long-term prognosis for pediatric patients. J Pediatr 127: 913–919, 1995 [DOI] [PubMed] [Google Scholar]
- 39.Bergstein J, Leiser J, Andreoli S: The clinical significance of asymptomatic gross and microscopic hematuria in children. Arch Pediatr Adolesc Med 159: 353–355, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Shen P, He L, Li Y, Wang Y, Chan M: Natural history and prognostic factors of IgA nephropathy presented with isolated microscopic hematuria in Chinese patients. Nephron Clin Pract 106: c157–c161, 2007 [DOI] [PubMed] [Google Scholar]
- 41.D’Amico G: Natural history of idiopathic IgA nephropathy: Role of clinical and histological prognostic factors. Am J Kidney Dis 36: 227–237, 2000 [DOI] [PubMed] [Google Scholar]
- 42.Ronkainen J, Ala-Houhala M, Autio-Harmainen H, Jahnukainen T, Koskimies O, Merenmies J, Mustonen J, Ormälä T, Turtinen J, Nuutinen M: Long-term outcome 19 years after childhood IgA nephritis: A retrospective cohort study. Pediatr Nephrol 21: 1266–1273 [DOI] [PubMed] [Google Scholar]
- 43.Reich HN, Troyanov S, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry : Remission of proteinuria improves prognosis in IgA nephropathy. J Am Soc Nephrol 18: 3177–3183, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L: Predicting the risk for dialysis or death in IgA nephropathy. J Am Soc Nephrol 22: 752–761, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, Liu ZH, Roberts IS, Yuzawa Y, Zhang H, Feehally J; IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology SocietyConference Participants : Oxford classification of IgA nephropathy 2016: An update from the IgA nephropathy classification working group. Kidney Int 91: 1014–1021, 2017 [DOI] [PubMed] [Google Scholar]
- 46.Coppo R, Lofaro D, Camilla RR, Bellur S, Cattran D, Cook HT, Roberts IS, Peruzzi L, Amore A, Emma F, Fuiano L, Berg U, Topaloglu R, Bilginer Y, Gesualdo L, Polci R, Mizerska-Wasiak M, Caliskan Y, Lundberg S, Cancarini G, Geddes C, Wetzels J, Wiecek A, Durlik M, Cusinato S, Rollino C, Maggio M, Praga M, K Smerud H, Tesar V, Maixnerova D, Barratt J, Papalia T, Bonofiglio R, Mazzucco G, Giannakakis C, Soderberg M, Orhan D, Di Palma AM, Maldyk J, Ozluk Y, Sudelin B, Tardanico R, Kipgen D, Steenbergen E, Karkoszka H, Perkowska-Ptasinska A, Ferrario F, Gutierrez E, Honsova E: Risk factors for progression in children and young adults with IgA nephropathy: An analysis of 261 cases from the VALIGA European cohort [published correction appears in Pediatr Nephrol 32: 193–194, 2017]. Pediatr Nephrol 32: 139–150, 2017 [DOI] [PubMed] [Google Scholar]
- 47.Fabiano RCG, Araújo SA, Bambirra EA, Oliveira EA, Simões E Silva AC, Pinheiro SVB: The Oxford Classification predictors of chronic kidney disease in pediatric patients with IgA nephropathy. J Pediatr (Rio J) 93: 389–397, 2017 [DOI] [PubMed] [Google Scholar]
- 48.Shima Y, Nakanishi K, Hama T, Mukaiyama H, Togawa H, Hashimura Y, Kaito H, Sako M, Iijima K, Yoshikawa N: Validity of the Oxford classification of IgA nephropathy in children. Pediatr Nephrol 27: 783–792, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Carapetis JR, Steer AC, Mulholland EK, Weber M: The global burden of group A streptococcal diseases. Lancet Infect Dis 5: 685–694, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Balasubramanian R, Marks SD: Post-infectious glomerulonephritis. Paediatr Int Child Health 37: 240–247, 2017 [DOI] [PubMed] [Google Scholar]
- 51.Kanjanabuch T, Kittikowit W, Eiam-Ong S: An update on acute postinfectious glomerulonephritis worldwide. Nat Rev Nephrol 5: 259–269, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Gümüş H, Per H, Kumandaş S, Yikilmaz A: Reversible posterior leukoencephalopathy syndrome in childhood: Report of nine cases and review of the literature. Neurol Sci 31: 125–131, 2010 [DOI] [PubMed] [Google Scholar]
- 53.White AV, Hoy WE, McCredie DA: Childhood post-streptococcal glomerulonephritis as a risk factor for chronic renal disease in later life. Med J Aust 174: 492–496, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Chen JY, Mao JH: Henoch-Schönlein purpura nephritis in children: Incidence, pathogenesis and management. World J Pediatr 11: 29–34, 2015 [DOI] [PubMed] [Google Scholar]
- 55.Davin JC, Coppo R: Henoch-Schönlein purpura nephritis in children. Nat Rev Nephrol 10: 563–573, 2014 [DOI] [PubMed] [Google Scholar]
- 56.Narchi H: Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: A systematic review. Arch Dis Child 90: 916–920, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goldstein AR, White RH, Akuse R, Chantler C: Long-term follow-up of childhood Henoch-Schönlein nephritis. Lancet 339: 280–282, 1992 [DOI] [PubMed] [Google Scholar]
- 58.Davis KA, Stanek JR, Dunn AL: Screening urinalysis demonstrates that haematuria is a frequent finding in persons with haemophilia treated at a paediatric haemophilia treatment centre. Haemophilia 25: 782–788, 2019 [DOI] [PubMed] [Google Scholar]
- 59.Hamed AA, Shalaby MH, El-Kinawy NS, Elamawy AA, Abd El-Ghany SM: Renal abnormalities among Egyptian children with hemophilia A using renal Scintigraphy: Relation to risk factors and disease Severity. Clin Appl Thromb Hemost 23: 478–486, 2017 [DOI] [PubMed] [Google Scholar]
- 60.Akubuilo UC, Ayuk A, Ezenwosu OU, Okafor UH, Emodi IJ: Persistent hematuria among children with sickle cell anemia in steady state [published online ahead of print October 19, 2019]. Hematol Transfus Cell Ther doi: 10.1016/j.htct.2019.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nath KA, Hebbel RP: Sickle cell disease: Renal manifestations and mechanisms. Nat Rev Nephrol 11: 161–171, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alvarez O, Rodriguez MM, Jordan L, Sarnaik S: Renal medullary carcinoma and sickle cell trait: A systematic review. Pediatr Blood Cancer 62: 1694–1699, 2015 [DOI] [PubMed] [Google Scholar]
- 63.Fallahzadeh MH, Fallahzadeh MK, Shahriari M, Rastegar S, Derakhshan A, Fallahzadeh MA: Hematuria in patients with Beta-thalassemia major. Iran J Kidney Dis 4: 133–136, 2010 [PubMed] [Google Scholar]
- 64.Dell KM: The spectrum of polycystic kidney disease in children. Adv Chronic Kidney Dis 18: 339–347, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Becker GJ, Garigali G, Fogazzi GB: Advances in urine microscopy. Am J Kidney Dis 67: 954–964, 2016 [DOI] [PubMed] [Google Scholar]
- 66.Ohisa N, Yoshida K, Kaku M, Sato H: [Comparison between optical microscopic examination and phase contrast microscopic examination for diagnosing the origin of urinary bleeding]. Nippon Jinzo Gakkai Shi 48: 401–406, 2006 [PubMed] [Google Scholar]
- 67.Fogazzi GB, Edefonti A, Garigali G, Giani M, Zolin A, Raimondi S, Mihatsch MJ, Messa P: Urine erythrocyte morphology in patients with microscopic haematuria caused by a glomerulopathy. Pediatr Nephrol 23: 1093–1100, 2008 [DOI] [PubMed] [Google Scholar]
- 68.Wyatt RJ, McRoberts JW, Holland NH: Hematuria in childhood: Significance and management. J Urol 117: 366–368, 1977 [DOI] [PubMed] [Google Scholar]
- 69.Ruberto U, D’Eufemia P, Giardini O: [Long-term prognosis in monosymptomatic microscopic hematuria]. Pediatr Med Chir 10: 475–479, 1988 [PubMed] [Google Scholar]