

Abstract
Selective autophagy of mitochondria, known as mitophagy, is a major quality control pathway in the heart that is involved in removing unwanted or dysfunctional mitochondria from the cell. Baseline mitophagy is critical for maintaining fitness of the mitochondrial network by continuous turnover of aged and less-functional mitochondria. Mitophagy is also critical in adapting to stress associated with mitochondrial damage or dysfunction. The removal of damaged mitochondria prevents reactive oxygen species-mediated damage to proteins and DNA and suppresses activation of inflammation and cell death. Impairments in mitophagy are associated with the pathogenesis of many diseases, including cancers, inflammatory diseases, neurodegeneration, and cardiovascular disease. Mitophagy is a highly regulated and complex process that requires the coordination of labeling dysfunctional mitochondria for degradation while simultaneously promoting de novo autophagosome biogenesis adjacent to the cargo. In this review, we provide an update on our current understanding of these steps in mitophagy induction and discuss the physiological and pathophysiological consequences of altered mitophagy in the heart.
Keywords: autophagy, heart, mitochondria, mitophagy, COVID-19
INTRODUCTION
Mitochondria are essential for many metabolic processes, including oxidative phosphorylation, fatty acid oxidation, and calcium homeostasis (1). Mitochondria are also key participants in other processes, including apoptosis, necrosis, and initiation of inflammation (1). They are a major source of reactive oxygen species (ROS), a byproduct of oxidative phosphorylation (2). Although ROS are not inherently detrimental, excessive ROS production can cause oxidative damage to mitochondrial lipids, proteins, and DNA, as well as cytosolic macromolecules and other organelles (2, 3). Excessive ROS can also lead to opening of the mitochondrial permeability transition pore, an initiating step of outer membrane rupture and necrotic cell death (4). In addition, release of mitochondrial DNA (mtDNA) in response to stress or injury can activate proinflammatory responses that can persist as chronic inflammation and subsequently increase susceptibility to disease (5, 6).
Maintaining a healthy functional mitochondrial network is critical for cellular homeostasis, especially in terminally differentiated cells such as neurons and cardiac myocytes that are enriched in mitochondria. Mitochondrial dysfunction is associated with both neurodegenerative and cardiovascular diseases (4). Several mitochondrial quality control mechanisms are in place to facilitate repair or degradation of damaged mitochondria to ensure that they do not pose a hazard to the rest of the network. Mitochondrial chaperones ensure proper folding of proteins, whereas resident proteases are responsible for protein degradation (7). When these mechanisms are overwhelmed, the cell can activate a mitochondrial unfolded protein response that initiates transcription of mitochondrial chaperones and proteases. If the local repair mechanism is unable to restore mitochondrial protein homeostasis, then the cell activates autophagy as a mechanism to remove the damaged organelle from the cellular environment.
In general, autophagy is a catabolic degradation pathway through which cargos such as damaged organelles and protein aggregates are broken down and recycled. In this process, the cargo is engulfed by a double-membraned vesicle called the autophagosome and degraded upon fusion with the lysosome (Fig. 1). Mitochondrial autophagy (mitophagy) is a selective form of autophagy and the main pathway responsible for degrading mitochondria in cells (8). Mitophagy is essential in ensuring mitochondrial quality and quantity at baseline and in response to stress, and impairments in mitophagy are linked to aging as well as many diseases (9). Mitophagy involves both labeling of the mitochondrion for degradation and de novo biogenesis of the autophagosome membrane near the cargo. Although decreased mitophagy will lead to accumulation of harmful mitochondria that can activate cell death, excessive degradation of mitochondria can lead to energy deficiency and death. Cardiac myocytes are terminally differentiated cells that rely on mitochondria to sustain contraction. They are therefore highly enriched in mitochondria and rely on mitophagy for both baseline turnover and adaptation to stress. Thus, the pathways that regulate mitophagy are tightly controlled to safeguard against unnecessary removal of this key organelle. In this review, we discuss the current understanding of the proteins and pathways involved in regulating mitophagy. We also discuss the importance of functional mitophagy in cardiovascular physiology and its role in suppressing disease development and aging.
Figure 1.
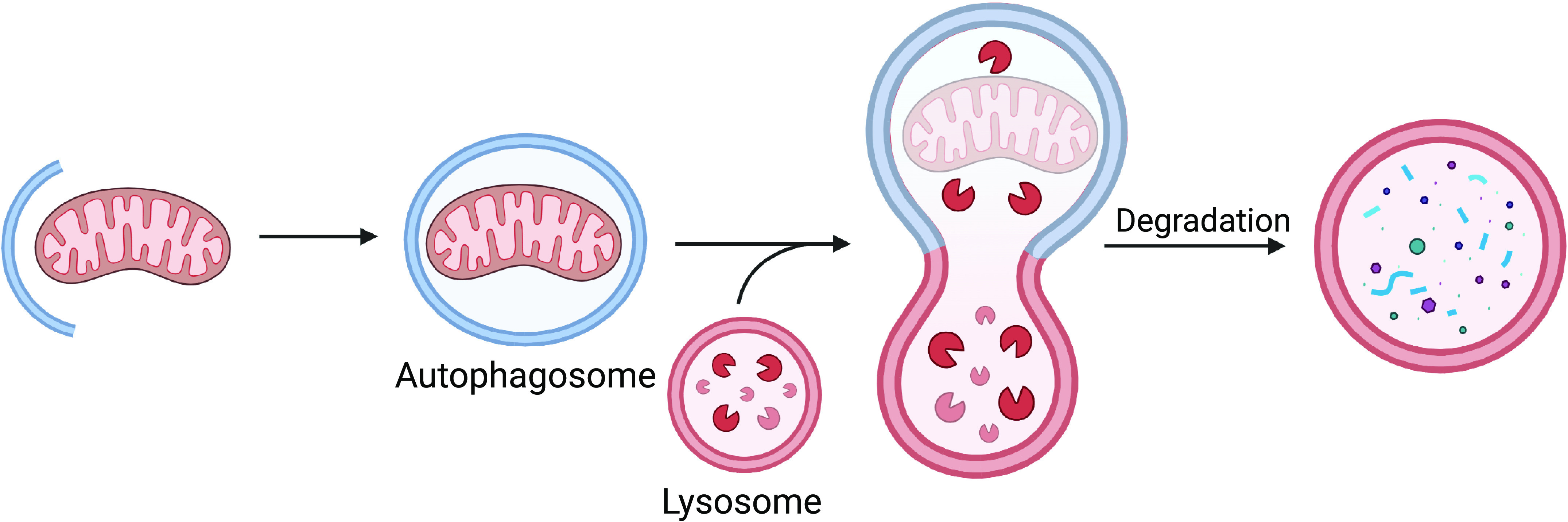
Overview of mitophagy. When damaged mitochondria are labeled for degradation, de novo autophagosome formation is initiated at the mitochondrion. The phagophore, a precursor of the autophagosome, forms and elongates around its cargo. Once fully enclosed around the mitochondrion, the mature autophagosome fuses with the lysosome and the contents are degraded. (Created with Biorender.com)
MECHANISMS OF MITOPHAGY
PINK1/Parkin-Mediated Mitophagy
The regulation of mitophagy by PINK1 and Parkin is one of the most well-studied pathways in the field. There is a strong interest in these proteins because loss-of-function mutations in the genes encoding PINK1 or Parkin (PARK2) are associated with development of juvenile recessive Parkinson’s disease (PD) (10, 11). Activation of PINK1/Parkin-mediated mitophagy is linked to a loss of mitochondrial membrane potential, a common consequence of stress and damage. In the absence of mitochondrial damage, the serine/threonine kinase PINK1 is partially imported into mitochondria, where it is cleaved by mitochondrial proteases and then released into cytosol for proteasomal degradation (12, 13) (Fig. 2A). When mitochondria become depolarized, import of PINK1 is abrogated and it accumulates on the outer mitochondrial membrane (14, 15). PINK1 then phosphorylates ubiquitin, which leads to recruitment and activation of the E3 ubiquitin ligase Parkin (16, 17) (Fig. 2A). Parkin is also phosphorylated by PINK1 on Ser65 in its ubiquitin-like domain (8, 18, 19). Parkin then proceeds to ubiquitinate different proteins on the outer mitochondrial membrane, which functions as a degradation signal for the autophagic machinery. The polyubiquitin chains are recognized by different autophagy adaptor proteins that are recruited to mitochondria by PINK1 (20). These adaptors tether mitochondria to autophagosomes via motifs that bind to ubiquitin on mitochondria and to microtubule-associated protein 1A/1B-light chain 3 (LC3) on the autophagosome membranes (21).
Figure 2.
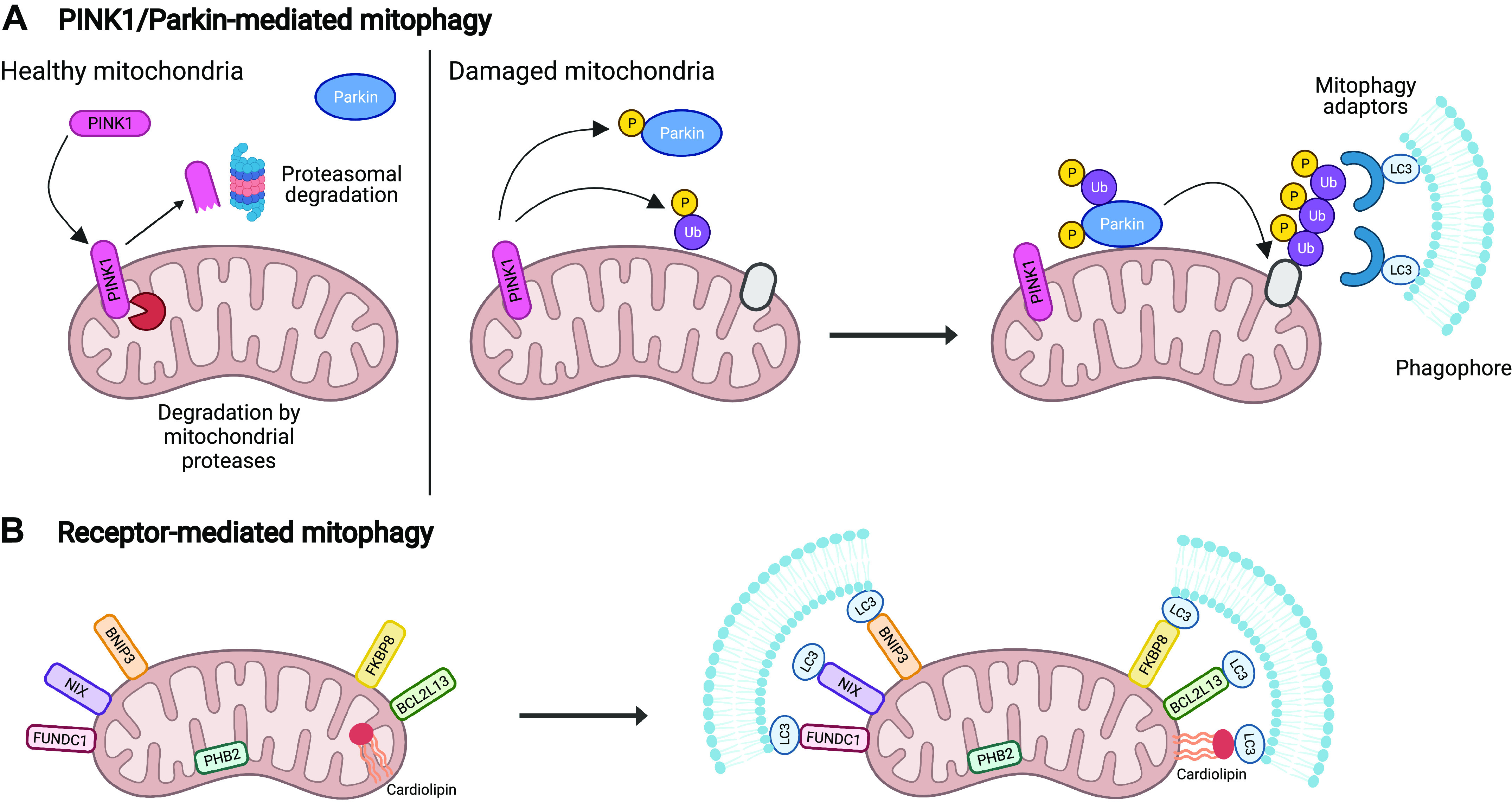
Mechanisms of mitophagy. A: under normal conditions, PINK1 is imported into mitochondria where it is cleaved by proteases. Upon loss of mitochondrial membrane potential, PINK1 is stabilized on the outer mitochondrial membrane due to inhibition of import. Activated PINK1 phosphorylates ubiquitin molecules and the E3 ubiquitin ligase Parkin. Once activated, Parkin ubiquitinates specific substrates in the outer mitochondrial membrane. Autophagy adaptor proteins subsequently bind to the poly-ubiquitin chains and to LC3 on the growing phagophore adjacent to mitochondria. B: mitophagy receptors BNIP3, NIX, FUNDC1, BCL2L13, and FKBP8 are anchored in the outer mitochondrial membrane via their transmembrane domains and contain an LC3-interacting region (LIR), a motif that binds LC3. Upon induction of mitophagy, these proteins directly bind to LC3 in an ubiquitin-independent manner to link the mitochondrion to the autophagosome membrane. Cardiolipin, a lipid that relocalizes to the outer mitochondrial membrane during stress, can also function as a mitophagy receptor by interacting with LC3. PHB2 is a mitophagy receptor that localizes to the inner mitochondrial membrane and mainly functions in the event of mitochondrial rupture. LC3, 1A/1B-light chain 3; PHB2, prohibitin 2. (Created with Biorender.com)
Because aberrant mitophagy can have severe consequences for the cell, there are several mechanisms in place to fine-tune mitophagy and prevent excessive removal of mitochondria. Deubiquitinating enzymes (DUBs) at the mitochondria are responsible for counteracting Parkin-mediated ubiquitination and diminishing mitophagic activity. Usp15 and Usp30 have been identified as DUBs that specifically antagonize mitophagy by opposing Parkin-mediated ubiquitination of proteins in the outer mitochondrial membrane (22, 23). Parkin activation is also associated with auto-ubiquitination (24, 25). Niu and colleagues (26) recently identified Usp33 as a DUB that specifically targets Parkin to antagonize its activity. Mitophagy is also limited by the mitochondrial ubiquitin ligase (MITOL; also known as MARCH5). Specifically, MITOL mediates ubiquitination of Parkin, which promotes its proteasomal degradation (27). Thus, limiting protein ubiquitination and Parkin levels are key steps in suppressing mitophagy. Regulating mitophagy at the level of Parkin is clearly important, as overexpression of Parkin in cells can lead to degradation of the majority of the mitochondrial population upon activation of mitophagy (8, 28). This also suggests that Parkin might represent a rate-limiting step in this pathway. PINK1 can also limit the level of mitophagy by activating a noncanonical function of the mitochondrial Tu translocation elongation factor (TUFm) (29). Lin and colleagues discovered that PINK1 phosphorylates TUFm at Ser222, restricting its localization to the cytosol where it then blocks the formation of the Atg5-12 lipid conjugation complex and subsequent elongation of the autophagosome membrane.
Mitophagy Receptors
Mitophagy receptors are anchored in the outer mitochondrial membrane and facilitate mitophagy by directly interacting with LC3 on the autophagosome membrane via an LC3-interacting region (LIR) motif (Fig. 2B) (21). Thus, unlike PINK1/Parkin-mediated mitophagy, these receptors facilitate removal of mitochondria in a ubiquitin-independent manner. Many proteins, including Nix, Bnip3, Fundc1, BCL2L13, FKBP8, and PHB2, and even lipids (cardiolipin) can function as mitophagy receptors depending on the context (30–38). For instance, Nix is critical for programmed mitophagy and plays an important role in eliminating mitochondria during maturation of erythroid cells (39, 40), whereas Bnip3 and Fundc1 induce mitophagy in response to oxygen deprivation and ischemia/reperfusion injury (36, 37, 41). Although some of these receptors are activated by similar conditions, it is currently unclear why such redundancies are in place. Prohibitin 2 (PHB2) is the only mitophagy receptor that is localized in the inner membrane and is responsible for removal of mitochondria after outer membrane rupture (33). PHB2 can also promote PINK1/Parkin-mediated mitophagy by suppressing proteolytic cleavage of PINK1 by mitochondrial protease PARL, which leads to increased recruitment of Parkin to mitochondria (42). Many of these mitophagy receptors have alternative functions in other pathways, which has made it challenging to dissect how they regulate mitophagy in cells. For instance, Bnip3 and Nix were initially identified as proapoptotic BH3-only proteins (43–45), whereas PHB2 is a mitochondrial scaffolding protein that is important for mitochondrial stabilization and function (46). Altering expression of mitophagy receptors that are critical effectors of other mitochondrial signaling pathways could result in off-target effects that are not a direct result of these proteins’ role in mitophagy.
AMPK-Ulk1 Axis in Mitophagy
A key step in mitophagy is signal emission from damaged mitochondria to the autophagic machinery to initiate autophagosome formation. One well-studied signaling pathway that is activated by defective mitochondria involves the cellular energy sensor AMP-activated protein kinase (AMPK) (47). Damage to mitochondria often leads to decreased oxidative phosphorylation and diminished ATP generation. The resulting increase in cellular AMP and ADP levels leads to activation of AMPK and phosphorylation of its downstream targets function to shift metabolism toward decreased anabolism and increased catabolism (47). Several studies have also linked AMPK activation to mitophagy (48–51). This function was initially reported by Egan et al. (48), who discovered that AMPK-deficient cells have increased numbers of mitochondria due to impaired mitophagy. Subsequent studies have identified that AMPK can selectively regulate the induction of mitophagy through at least three different mechanisms, including 1) activation of the serine/threonine kinase unc-51-like kinase 1 (Ulk1) (48, 49), 2) interaction with the Atg16L complex (50), and 3) induction of mitochondrial fission factor (Mff)-mediated mitochondrial fission (51).
Ulk1
The primary pathway through which AMPK regulates mitophagy appears to be via Ulk1. This specific function of Ulk1 in mitophagy was discovered when Kundu and colleagues (52) noted that Ulk1 deficiency is associated with abrogation of mitochondrial clearance in red blood cells during maturation. Additional studies using tissue-specific Ulk1-deficient mice have confirmed that Ulk1 is required for functional mitophagy (48, 53, 54). Mechanistically, AMPK initiates mitophagy by phosphorylating Ulk1 on Ser555 (48), which leads to translocation of Ulk1 to damaged mitochondria (55). Ulk1 then recruits the Beclin1-Vps34-Vps15 complex, a downstream effector involved in nucleation of the phagophore membrane, ensuring that autophagosome formation is initiated at the cargo for more efficient removal of mitochondria (56). Although Ulk1 is activated by AMPK during nutrient starvation (48), activation of mitophagy is not strictly dependent on AMPK. A recent study reported that while Parkin-mediated mitophagy of depolarized mitochondria is impaired in Ulk1/2-deficient cells, this process remains intact in AMPK α 1/2 double knockout cells (57).
Moreover, autophagy adaptor proteins facilitate autophagy by linking ubiquitinated cargo to LC3 at the growing autophagosome membrane. Recently, these proteins have also been linked to the recruitment and retention of Ulk1 at the mitochondria during mitophagy (57). To date, at least five adaptor proteins have been reported to facilitate mitophagy by tethering mitochondria and the autophagosome membrane together, and HeLa cells lacking all five adaptor proteins exhibit impairments in mitophagy (20). The authors also discovered that these cells have a defect in Ulk1 recruitment to damaged mitochondria, suggesting the adaptor proteins also play a role in recruiting Ulk1 to mitochondria during mitophagy. In a subsequent study, this group found that the presence of the adaptor protein NDP52 on mitochondria is sufficient to recruit and activate Ulk1 independently of AMPK (57).
In addition to initiating autophagosome formation at the mitochondria, Ulk1 also phosphorylates the mitophagy receptor FUNDC1 (FUN14 domain-containing protein 1), facilitating the interaction between FUNDC1 and LC3 on the autophagosome (58). More recently, Hung et al. (59) reported that Ulk1 is also involved in promoting translocation of Parkin to mitochondria, where phosphorylation of Parkin at Ser108 by Ulk1 in the cytosol represents one of the initial events in mitophagy induction. Inhibition of Ulk1 or mutation of this phosphorylation site leads to delayed activation of Parkin and defects in mitophagy. These findings suggest that Ulk1 functions as a central regulator of several distinct steps of mitophagy: 1) initiating autophagosome formation by activating the Beclin1-Vps34-Vps15 complex, 2) enhancing the tethering between mitochondria and the autophagosome membrane via FUNDC1, and 3) promoting Parkin translocation to mitochondria.
AMPK and Atg16
The second mechanism by which AMPK can regulate the induction of mitophagy is via Atg16 (Atg16L1 in mammals), a core autophagy protein that forms a complex with Atg12 and Atg5 during autophagosome biogenesis (60, 61). Atg12-Atg5 exhibits the E3-like ligase activity responsible for conjugating phosphatidylethanolamine (PE) to LC3. The main function of Atg16 is to bring the conjugation complex to the phagophore (62, 63), and Atg16-deficiency leads to abrogation of LC3 lipidation due to a lack of Atg12-Atg5 complex at the phagophore (64). When AMPK associates with damaged mitochondria during mitophagy, it also recruits the Atg16-Atg5-Atg12 complex to the site of phagophore formation to initiate mitophagy (50). The same study reported that treating cells with the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) leads to increased association between AMPK and the Atg16-Atg12-Atg5 complex at depolarized mitochondria. Interestingly, this study also discovered that AMPK is responsible for retaining the conjugation complex at the mitochondria. Overall, the recruitment and retention of the Atg16-Atg12-Atg5 complex at the mitochondria by AMPK ensure the localized formation of autophagosome near the cargo.
AMPK and Mff-Induced Mitochondrial Fission
Finally, AMPK can facilitate mitophagy by promoting mitochondrial fission via phosphorylation of Mff (51, 65, 66). Mitochondria can undergo symmetric or asymmetric division depending on the physiological context. Symmetric division of mitochondria occurs during cell division to facilitate mitochondrial biogenesis, whereas asymmetric division is associated with clearance of damaged mitochondria through mitophagy (67, 68). Thus, once separated from the healthy mitochondrial network, these dysfunctional mitochondrial fragments can be degraded via mitophagy (67, 69). AMPK-mediated phosphorylation of Mff leads to recruitment of the large GTPase Drp1 from the cytosol to the outer mitochondrial membrane. Upon recruitment, Drp1 promotes the constriction and subsequent fission of mitochondria (51). Abrogation of Mff phosphorylation leads to inhibition of both mitochondrial fission and mitophagy, confirming the importance of the AMPK-Mff/Drp1 pathway for functional mitophagy (51).
Although the mechanisms regulating asymmetric and symmetric division of mitochondria are mostly unclear, a recent study reported that mitochondria have distinct fission signatures that dictate their fate (68). This study discovered that division at the periphery of the mitochondria (asymmetrical fission) generates fragments that are destined for mitophagy, whereas division at the midzone (symmetrical fission) is associated with mitochondrial biogenesis. Although both peripheral and midzone divisions require Drp1, the authors found that Mff is primarily associated with midzone division while Fis1 is enriched in peripheral sites (68). Thus, the findings in this study contradict the report that the AMPK-Mff axis is involved in regulating mitophagy. Moreover, Mff-deficient mice develop lethal heart failure within 13 wk of birth, and these hearts have reduced mitochondrial respiration along with decreased mitochondrial content and enhanced mitophagy (70). Clearly, the exact functions of Mff in regulating mitochondrial fission and mitophagy need to be investigated further to determine if these contrasting reports are due to differences in experimental systems. Although these studies indicate that Mff-mediated fission might not be required for mitophagy, it is well accepted that functional mitochondrial fission is important for mitochondrial and cardiac health (71–73).
Mitochondria-Associated Membranes and Mitophagy
Autophagosome formation requires a source of lipids, and various cellular compartments have been reported to supply lipids to the growing autophagosome. It has been reported that the lipids can come from the plasma membrane, Golgi apparatus, or nuclear membrane, but the endoplasmic reticulum (ER) seems to be the major contributor of membrane lipids to the autophagosome (74). The phagophore originates from a subcompartment of the ER called the omegasome that is enriched in phosphatidylinositol 3-phosphate (PI3P) (75). In addition, the ER forms contact sites with mitochondria called mitochondria-associated ER membranes (MAMs), and these MAMs have been shown to be instrumental in autophagosome formation (76). MAMs mark the site of phagophore nucleation, which is where the autophagic machinery assemble to initiate formation and expansion of the autophagosome (Fig. 3) (76). The Atg2-Atg18 complex accumulates at MAMs and is responsible for tethering the phagophore membrane to the ER to facilitate membrane elongation (77). Atg2 is also crucial for autophagosome growth by promoting lipid transfer from donor membranes to the growing phagophore (78–80). Abrogating Atg2 recruitment to MAMs leads to impaired phagophore expansion and autophagic flux, which suggests that Atg2 may source lipids from the ER and mitochondria for autophagosome biogenesis at these contact sites (81). In mitophagy, the localized formation of the autophagosome at MAMs on dysfunctional mitochondria ensures their efficient removal. However, the mechanism through which autophagic machinery is selectively recruited to MAMs on damaged mitochondria is unclear and needs to be investigated further.
Figure 3.
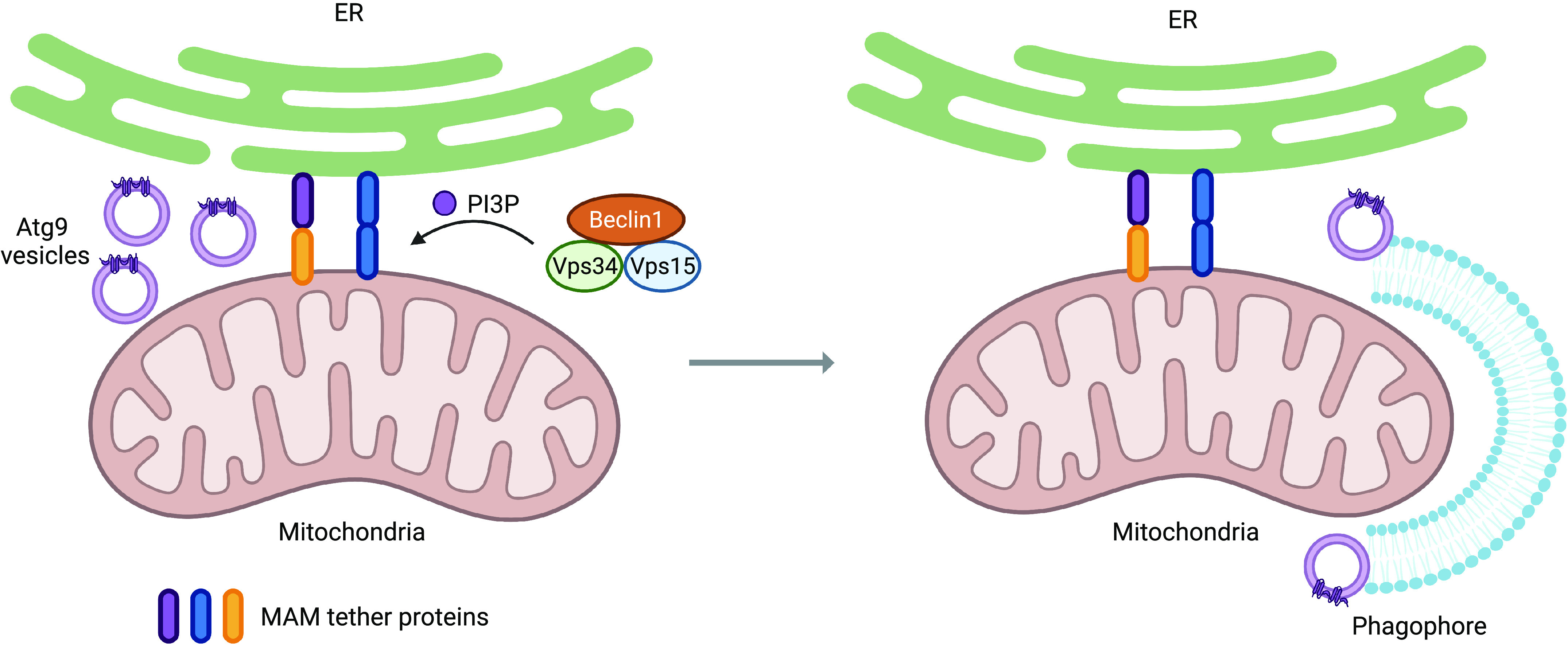
Mitophagy initiation at MAMs. MAMs are contact sites between the ER and mitochondria that are maintained by a variety of tethering proteins. When mitophagy is initiated, the initiation machinery, which includes the Beclin1-Vps34-Vps15 complex and Atg9-containing vesicles, translocates to MAMs to promote autophagosome biogenesis. The Beclin1-Vps34-Vps15 complex generates PI3P, which promotes the recruitment of downstream autophagy regulators. Atg9-positive vesicles also localize to MAMs, where they serve as platforms for autophagosome nucleation and subsequent membrane elongation. ER, endoplasmic reticulum; MAMs, mitochondria-associated ER membranes; PI3P, phosphatidylinositol 3-phosphate. (Created with Biorender.com)
Moreover, fission of mitochondria has been reported to occur at MAMs (82). The fission protein Drp1 localizes to MAMs to induce mitochondrial fission, which allows for more efficient degradation of these fragments (82, 83). Interestingly, the mitophagy receptor FUNDC1 also localizes to MAMs (84), where it may regulate fission (85, 86). Wu et al. (86) reported that FUNDC1 accumulates at MAMs and facilitates fission by directly interacting with DRP1 during hypoxia in HeLa cells. Additional key proteins involved in mitophagy have also been shown to localize to MAMs. For instance, both Beclin1 and PINK1 relocalize to MAMs during mitophagy to facilitate enhancement of ER-mitochondria contacts and initiation of autophagy (87). Parkin is also recruited to the MAMs by PINK1 upon induction of mitophagy and may play a role in regulating MAM formation and stability during mitophagy (87, 88). Specifically, Parkin enhances the ER-mitochondria connection by ubiquitinating the mitochondria-ER tether protein Mfn2 (89). Overall, these findings demonstrate the important role of MAMs as sites of both autophagosome formation and mitochondrial fission for more efficient removal of dysfunctional mitochondria.
Beclin1 and Mitophagy
Beclin1 is the mammalian ortholog of Atg6 in yeast and a positive regulator of autophagy (87, 90). Early studies identified Beclin1 as a Bcl-2-interacting protein and tumor suppressor in cancer cells (90, 91). Subsequent studies revealed that Beclin1 is a scaffold protein that interacts with the lipid kinase Vps34 and its regulatory subunit Vps15 to form the Class III phosphatidylinositol 3-kinase (PI3K) complex. This core complex binds to additional proteins to activate its proautophagic functions, including initiation of autophagosome formation and autophagosome maturation. Binding of Atg14L to the Beclin1-Vps34-Vps15 complex facilitates proper localization of the complex to the site of autophagosome biogenesis and local generation of PI3P (56, 75, 76, 92). This pool of PI3P promotes the recruitment of downstream autophagic regulators containing motifs that directly bind to PI3P (56, 75, 92). The Beclin1-Vps34-Vps15 complex also functions at later stages of autophagy through binding with UV radiation resistance-associated gene protein (UVRAG) to facilitate autophagosome maturation (93).
Studies have also reported that Beclin1 plays a selective role in mitophagy. For instance, Beclin1 interacts with Parkin and facilitates its translocation to mitochondria upon induction of mitophagy (94, 95). However, a different study found that although silencing of Becn1 leads to reduced mitophagy, it does not alter Parkin recruitment to mitochondria (87). Beclin1 also interacts with PINK1, and this interaction is essential for both MAM and autophagosome formation during general autophagy and mitophagy (87, 96). The importance of Beclin1 in MAM formation or stability in the heart has also been confirmed in vivo, where Becn1+/− mice exhibit decreased MAM levels and Becn1 transgenic mice exhibit enhanced MAM formation in hearts (97). Altogether, these studies implicate Beclin1 in autophagosome formation at MAMs during mitophagy.
Despite the findings implicating Beclin1 in mitophagy, it is important to note that other groups have reported that loss of Beclin1 does not significantly impair mitochondrial clearance or LC3 lipidation during mitophagy (98, 99). These findings suggest the existence of Beclin1-independent mechanisms of autophagy. However, considering the importance of mitophagy for cellular survival, it is not surprising that redundancies in these pathways would exist.
Atg9-Positive Lipid Vesicles in Mitophagy
Atg9 is the only membrane-spanning core autophagy protein and is embedded in single-membrane vesicles 30–60 nm in diameter derived from the Golgi apparatus (100). The Atg9-positive vesicles cycle between the trans-Golgi network, endosomal compartments, and plasma membrane under basal conditions (101–103). Upon induction of autophagy, Atg9-positive vesicles redistribute to sites of autophagosome formation (104). Interestingly, these vesicles associate only transiently with the growing autophagosome and are not integrated into the autophagosome membrane (104). Although it was initially reported that Atg9-positive vesicles are responsible for promoting autophagosome membrane expansion by delivering lipids, it is now clear that these small vesicles are not able to transport enough lipids to form mature autophagosomes. Instead, the ER is likely the main source of lipids for the autophagosome. It has been reported that the Atg9-positive vesicles can supply proteins that are involved in autophagosome biogenesis. For instance, these vesicles contain proteins involved in lipid synthesis, such as acetyl-coA synthetases Faa1 and Faa4 (105), and have been shown to deliver phosphatidylinositol 4 kinases PI4KIIIβ and PI4KIIα to the forming autophagosome (106). More recently, a study demonstrated that Atg9-positive vesicles serve as platforms for the recruitment and assembly of downstream autophagic machinery to initiate autophagosome formation and elongation (105). Atg9 can also serve as an acceptor of Atg2-mediated lipid transfer from ER to the phagophore (105). Indeed, Tang et al. (81) found that interactions between Atg9a and MAM-localized Atg2 are required for proper phagophore expansion.
For proper maturation of the autophagosome, there is also a need for lipid scramblase activity to facilitate the transfer of lipids from the cytoplasmic leaflet of the autophagosome membrane to its luminal leaflet (78). Recent structural studies have discovered that ATG9A exhibits lipid scramblase activity (107, 108). Proteomic analysis also found that flippases Drs2 and Neo1 are associated with Atg9-positive vesicles isolated from yeast (105). Thus, it is possible that these flippases are transferred to the growing phagophore. Because the interaction between Atg9-positive vesicles and the growing autophagosome is transient (104), this redundancy could ensure that there is machinery in place to continue performing the functions of Atg9 at the autophagosomes once the vesicles are recycled elsewhere.
Specific roles for Atg9a in Parkin-mediated mitophagy have also been identified. First, Atg9a-positive vesicles have been observed to localize to depolarized mitochondria in a Ulk1-independent manner (109). Atg9a deficiency also suppresses autophagosome formation during mitophagy, which subsequently leads to accumulation of depolarized mitochondria (109). The fact that autophagosome formation is not completely abrogated suggests that the paralog Atg9b can potentially compensate for the loss of Atg9a in mammalian cells. Atg9a and Atg9b are often expressed in the same cells (110), but whether they have distinct functions in regulating autophagy and mitophagy are unknown. A recent study also found that the autophagy adaptor optineurin (OPTN) forms a complex with ATG9A and LC3 at ubiquitinated mitochondria during mitophagy. Disrupting this complex results in loss of Parkin-mediated mitophagy (111). Thus, it is likely that the OPTN-ATG9A complex serves as a platform for the recruitment of downstream autophagic machinery to damaged mitochondria for localized autophagosome formation.
MITOCHONDRIAL QUALITY CONTROL IN THE HEART
The heart is enriched in mitochondria due to its high energy demand, and mitophagy plays an important function in maintaining cardiac homeostasis. Cardiac myocytes rely on mitophagy to sustain the quality and quantity of mitochondria at baseline. They also need mitophagy to adapt to stressors that are associated with mitochondrial damage to suppress activation of inflammation and protect against cell death. Studies in mice have demonstrated the impact of defective mitophagy on baseline cardiac homeostasis. For example, both Parkin-deficient and Bnip3/Nix double knockout mice accumulate dysfunctional mitochondria in the heart at an accelerated rate with aging (112, 113). Similarly, PINK1-deficient mice display abnormal mitochondrial function and develop cardiac hypertrophy and contractile dysfunction at an early age (114). PINK1 protein levels are also reduced in human heart failure (114), but whether this is part of the underlying cause or a consequence of heart failure in humans is still unknown. In contrast, overexpression of Parkin in the heart ameliorates the decline in mitochondrial and cardiac function observed in aging mouse hearts (115, 116).
Decreases in autophagy and mitophagy levels in the myocardium are observed with aging (116, 117), and interventions that enhance these processes are associated with improved cardiac function and extended longevity (115, 116, 118, 119). For instance, the compromised myocardial function and mitochondrial morphology observed in aged (>24 mo old) mice is preventable in mice with transgenic overexpression of Parkin (116). Although this study found that Parkin overexpression had no effect on cardiac function in young mice (116), another study reported increased cardiac fibrosis with aging in transgenic mice with cardiac-specific overexpression of Parkin (120). It is likely that differences in the level of Parkin overexpression are responsible for the different findings in these two studies. Although Parkin is likely a good therapeutic target in the heart, its levels must be carefully regulated to prevent any detrimental outcomes and effects.
Interestingly, mitochondrial activity and turnover in tissues closely follow circadian rhythms, and disruptions to the circadian clock lead to altered mitochondrial morphology and impaired function (121–123). Fis1, Pink1, and Bnip3, key regulators of mitochondrial dynamics and mitophagy, are transcriptional targets of regulators of the circadian clock (124). Therefore, it is not surprising that disruptions to these cycles lead to accumulation of enlarged and dysfunctional mitochondria. Rabinovich-Nikitin et al. (125) recently identified a link between transcription factor circadian locomotor output cycles kaput (CLOCK) activity and susceptibility to cardiac injury. They demonstrated that loss of CLOCK activity in myocytes leads to decreased expression of various autophagy and mitophagy regulators, including Atg7, Rab7a, Tfeb, and Sqstm1. The authors also found that disruption of CLOCK is associated with impaired activation of mitophagy and increased susceptibility to cardiac stress (125). Because it is well established that circadian disruption is associated with increased risk of developing heart disease and increased damage after cardiac injury in humans, it is possible that dysregulation of mitophagy is a major underlying factor.
Moreover, there is strong evidence that mitophagy is important for adapting to various cardiac stressors. For instance, Parkin-mediated mitophagy protects against myocardial infarction (126) and hemodynamic overload (127). Mitochondrial dysfunction is also often observed in diabetic cardiomyopathy, but how mitophagy is altered and whether this contributes to or is a consequence of the pathology are still unclear. Depending on the model being studied, autophagy and mitophagy have been reported to be either downregulated (128–130) or upregulated (131). However, a more recent study performed by Junichi Sadoshima’s group using new tools and mouse models demonstrated that a high-fat diet (HFD) in mice is associated with activation of autophagy and mitophagy in the heart. They also reported that Parkin-deficient mice with impaired mitophagy are more susceptible to HFD-induced cardiac impairment and exhibit increased mitochondrial dysfunction and lipid accumulation. In contrast, activating mitophagy in wild-type mice by administering the Tat-Beclin1 peptide (TB1), a potent autophagy activator, provided protection against diabetic cardiomyopathy induced by HFD (132). Overall, the findings in this study suggest that mitophagy is activated as an adaptive and cardioprotective response to high fat intake.
Myocardial damage induced by inflammation is a key feature of various cardiovascular diseases, and mitophagy has been reported to play an important role in suppressing inflammation. Excessive generation of mitochondrial ROS and release of mtDNA can stimulate the assembly and activation of the NLRP3 inflammasome (5, 133). Thus, mitophagy plays a critical role in suppressing inflammation by eliminating ROS-producing and leaky mitochondria. The importance of mitophagy in suppressing inflammation in the heart was demonstrated by Sun and colleagues (134), who reported that cardiac-specific overexpression of Beclin1 protects mice against LPS exposure by reducing inflammation and fibrosis. Specifically, Beclin1 overexpression or activation with the TB1 peptide leads to enhanced activation of PINK1/Parkin-mediated mitophagy and reduced release of mitochondrial danger-associated molecular patterns (mtDAMPs). Similarly, another group discovered that activation of FUNDC1-mediated mitophagy protects the heart against LPS-induced sepsis by preserving mitochondrial function and attenuating inflammation (6). Thus, both PINK1/Parkin- and mitophagy receptor-mediated mitophagy can suppress inflammation in the heart during LPS challenge, suggesting that there is redundancy in these pathways’ function. Finally, Kawasaki disease (KD) is associated with inflammation in blood vessels and is the most common cause of acquired heart disease among children (135). Studies in both mouse models and human patients indicate that activation of the NLRP-IL-1β pathway is the main factor underlying the pathology of KD (136). A recent study reported that autophagy and mitophagy are both impaired in a mouse model of KD vasculitis and that activating autophagy and mitophagy attenuates NLRP3 activation and cardiovascular inflammation (137). This study also found that Parkin-deficient mice are more susceptible to KD development and exhibit increased heart inflammation relative to wild-type controls. Overall, these studies clearly demonstrate the importance of mitophagy in suppressing NLRP3 activation and inflammation.
MITOCHONDRIAL QUALITY CONTROL IN OTHER DISEASES
Neurodegeneration
Accumulation of dysfunctional mitochondria is a hallmark of many neurodegenerative disorders, and studies have demonstrated that this is, at least in part, due to decreased mitophagy (138). Loss-of-function mutations in PINK1 and PARK2, two key mitophagy regulators, lead to development of juvenile recessive PD (10, 11). Many neurodegenerative diseases are also age-dependent, and mitochondrial quality control mechanisms, including mitophagy, are known to be impaired with aging, leaving cells with diminished ability to efficiently remove dysfunctional mitochondria (9, 110). For example, Alzheimer’s disease (AD), which is typically characterized by the presence of amyloid-β plaques and hyperphosphorylation of tau protein, has also been associated with accumulation of dysfunctional mitochondria and impaired mitophagy (138). A recent study reported that restoration of mitophagy reduces the size of amyloid-β plaques, inhibits tau hyperphosphorylation, and improves cognition in Caenorhabditis elegans and in mouse models of AD (139). The pathophysiology of many neurodegenerative diseases is poorly understood, which has made the development of treatments extremely challenging (138). However, therapeutic interventions that restore or enhance mitochondrial quality control mechanisms represent a promising path forward in the search for treatments.
Cancer
Mitochondria play an important role in the commitment of cancer cells to either proliferation or apoptosis. During the earlier stages of tumorigenesis, accumulation of dysfunctional mitochondria due to impaired mitophagy can lead to a metabolic switch to glycolysis, hence promoting tumor growth and survival (140, 141). In more established tumors, mitophagy is vital for survival, potentially by preventing activation of apoptosis in the hypoxic conditions of the tumor microenvironment (142). Loss of function or downregulation of Parkin has been identified as a contributor to tumorigenesis in several different cancers (143). The tumor-suppressing functions of Parkin include its positive regulation of mitophagy and its suppression of metabolic reprogramming, which shifts energy production toward glycolysis (144). Bnip3-mediated mitophagy is also responsible for suppressing tumor progression by preventing accumulation of dysfunctional mitochondria and metabolic reprogramming (140). However, the roles that many other core mitophagy effector proteins play in cancer are still unclear and seem to be either stage- or cell type-dependent. Downregulation of many mitophagy regulators has been observed in different cancers, but they have also been identified as biomarkers of poor prognosis because of their tumor-protective functions (145). Clearly, the role of mitophagy in cancer is highly complex, and further studies are required to understand the importance of mitophagy during different stages and types of cancer.
COVID-19 Infection
Although mitochondria are directly involved in cellular responses to infection by stimulating inflammation and cytokine production, many pathogens can directly target mitochondria to evade host immunity and use them as a base for expansion (146). Interestingly, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA contains mitochondrial localization sequences, and computational modeling predicts that the virus is likely trafficked to the mitochondria (147). Other coronaviruses have been shown to interfere with mitochondrial function, “hijacking” the mitochondria to optimize conditions for replication and suppress viral immunity (148, 149). For example, SARS-CoV-1, the virus responsible for the 2002 SARS outbreak, encodes a protein that promotes proteasomal degradation of Drp1 (150), thereby inhibiting mitochondrial fission and potentially limiting mitophagy (68). Analysis of the SARS-CoV-2-host interactome also revealed that proteins encoded by SARS-CoV-2 can interact with host mitochondrial proteins, suggesting that SARS-CoV-2 may use a similar strategy (151). In addition, there are predictions that SARS-CoV-2 may indirectly impair mitochondrial function through its binding to the angiotensin-converting enzyme carboxypeptidase 2 (ACE2) receptor, a regulator of calcium signaling, mitochondrial respiration, and ATP production (148). Because aging is associated with accumulation of dysfunctional mitochondria and inefficient mitophagy (9), studies on mitochondria and SARS-CoV-2 might provide insights into why aged individuals are more susceptible to COVID-19 infection and at increased risk for hospitalization and death. Also, if the virus is indeed hijacking mitochondria for proliferation, enhancing mitophagy might then represent a therapeutic intervention to limit the infection.
CONCLUSIONS
It is clear that functional mitophagy in cardiac myocytes is necessary to preserve a healthy mitochondrial network. There is overwhelming evidence in the literature that functional and properly regulated mitophagy is essential for cellular homeostasis and survival and that impaired mitophagy contributes to both aging and disease development, raising the exciting possibility that this pathway represents a desirable future therapeutic target. However, much still needs to be learned before proteins in this pathway can be safely manipulated pharmacologically. For instance, it is unknown how this process can be safely targeted without causing excessive mitochondrial clearance or undesirable off-target effects due to the involvement of many key mitophagy proteins in other critical cellular processes. In the heart, most efforts to date have focused on investigating autophagy and mitophagy in cardiac myocytes, but there are still very few studies on mitophagy in the other cell populations in the heart, such as fibroblasts, smooth muscle cells, and endothelial cells. Therapies aimed at targeting mitophagy will also affect this process in these cells. Therefore, it will be important to determine how targeting mitophagy will impact their function.
GRANTS
Å. B. Gustafsson is supported by NIH Grants R01HL138560, R01HL132300, R01HL155281, and HL157265. R. Y. Diao is supported by the University of California, San Diego Graduate Training Program in Cellular and Molecular Pharmacology Grant T32GM007752.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.Y.D. prepared figures; R.Y.D. and A.B.G. drafted manuscript; R.Y.D. and A.B.G. edited and revised manuscript; R.Y.D. and A.B.G. approved final version of manuscript.
REFERENCES
- 1.Friedman JR, Nunnari J. Mitochondrial form and function. Nature 505: 335–343, 2014. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950, 2014. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 1863: 2977–2992, 2016. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21: 85–100, 2020. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 5.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191: 5230–5238, 2013. doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol 45: 102049, 2021. doi: 10.1016/j.redox.2021.102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quiles JM, Gustafsson AB. Mitochondrial quality control and cellular proteostasis: two sides of the same coin. Front Physiol 11: 515, 2020. doi: 10.3389/fphys.2020.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G, Kroemer G, Kepp O. Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol 8: 200, 2020. doi: 10.3389/fcell.2020.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608, 1998. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 11.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: 1158–1160, 2004. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 12.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13: 378–385, 2012. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy 9: 1758–1769, 2013. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298, 2010. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221, 2010. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153, 2014. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166, 2014. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 18.Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2: 120080, 2012. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2: 1002, 2012. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314, 2015. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gustafsson ÅB, Dorn GW 2nd.. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol Rev 99: 853–892, 2019. doi: 10.1152/physrev.00005.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510: 370–375, 2014. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 23.Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, Vandenberghe W. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23: 5227–5242, 2014. doi: 10.1093/hmg/ddu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimura H, Hattori N, Kubo S-i, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302–305, 2000. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA 97: 13354–13359, 2000. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu K, Fang H, Chen Z, Zhu Y, Tan Q, Wei D, Li Y, Balajee AS, Zhao Y. USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 16: 724–734, 2020. doi: 10.1080/15548627.2019.1656957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiiba I, Takeda K, Nagashima S, Ito N, Tokuyama T, Yamashita SI, Kanki T, Komatsu T, Urano Y, Fujikawa Y, Inatome R, Yanagi S. MITOL promotes cell survival by degrading parkin during mitophagy. EMBO Rep 22: e49097, 2021. doi: 10.15252/embr.201949097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, Li H, Kitsis RN, Dorn GW II, Sadoshima J, Ellisman MH, Gustafsson AB. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 8: 14050, 2017. doi: 10.1038/ncomms14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin J, Chen K, Chen W, Yao Y, Ni S, Ye M, Zhuang G, Hu M, Gao J, Gao C, Liu Y, Yang M, Zhang Z, Zhang X, Huang J, Chen F, Sun L, Zhang X, Yu S, Chen Y, Jiang Y, Wang S, Yang X, Liu K, Zhou HM, Ji Z, Deng H, Haque ME, Li J, Mi LZ, Li Y, Yang Y. Paradoxical mitophagy regulation by PINK1 and TUFm. Mol Cell 80: 607–620.e12, 2020.doi: 10.1016/j.molcel.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15: 1197–1205, 2013. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antón Z, Landajuela A, Hervas JH, Montes LR, Hernandez-Tiedra S, Velasco G, Goni FM, Alonso A. Human Atg8-cardiolipin interactions in mitophagy: specific properties of LC3B, GABARAPL2 and GABARAP. Autophagy 12: 2386–2403, 2016. doi: 10.1080/15548627.2016.1240856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, Kim T, Sussman MA, Gustafsson ÅB. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15: 1182–1198, 2019. doi: 10.1080/15548627.2019.1580095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168: 224–238.e10, 2017. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marinković M, Šprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 17: 1232–1243, 2021. doi: 10.1080/15548627.2020.1755120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhujabal Z, Birgisdottir ÅB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S, Kirkin V, Lamark T, Johansen T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 18: 947–961, 2017. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 14: 146–157, 2007. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 37.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185, 2012. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 38.Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12: 689–702, 2016. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104: 19500–19505, 2007. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235, 2008. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117: 2825–2833, 2007. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan C, Gong L, Chen L, Xu M, Abou-Hamdan H, Tang M, Désaubry L, Song Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 16: 419–434, 2020. doi: 10.1080/15548627.2019.1628520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem 274: 7–10, 1999. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- 44.Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW 2nd.. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 117: 396–404, 2008. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91: 226–231, 2002. doi: 10.1161/01.RES.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 46.Signorile A, Sgaramella G, Bellomo F, De Rasmo D. Prohibitins: a critical role in mitochondrial functions and implication in diseases. Cells 8: 71, 2019. doi: 10.3390/cells8010071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salt IP, Hardie DG. AMP-activated protein kinase: an ubiquitous signaling pathway with key roles in the cardiovascular system. Circ Res 120: 1825–1841, 2017. doi: 10.1161/CIRCRESAHA.117.309633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461, 2011. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang J, Xu ZX, Ding Z, Lu Y, Yu Q, Werle KD, Zhou G, Park YY, Peng G, Gambello MJ, Mills GB. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun 6: 7926, 2015. doi: 10.1038/ncomms8926. [DOI] [PubMed] [Google Scholar]
- 51.Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Lóson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351: 275–281, 2016. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112: 1493–1502, 2008. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saito T, Nah J, Oka SI, Mukai R, Monden Y, Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li H, Baljinnyam E, Fraidenraich D, Fritzky L, Zhai P, Ichinose S, Isobe M, Hsu CP, Kundu M, Sadoshima J. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest 129: 802–819, 2019. doi: 10.1172/JCI122035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8: 548, 2017. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, Zhong W, Chen Y, Wu W, Lin C, Chen H, Hou X, Zhang L, Sui S, Zhao B, Hu Z, Li L, Feng D. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett 589: 1847–1854, 2015. doi: 10.1016/j.febslet.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 56.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750, 2013. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74: 347–362.e6, 2019. doi: 10.1016/j.molcel.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhao B, Feng D. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15: 566–575, 2014. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hung CM, Lombardo PS, Malik N, Brun SN, Hellberg K, Van Nostrand JL, Garcia D, Baumgart J, Diffenderfer K, Asara JM, Shaw RJ. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci Adv 7: eabg4544, 2021. doi: 10.1126/sciadv.abg4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. Embo J 18: 3888–3896, 1999. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277: 18619–18625, 2002. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 62.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282: 37298–37302, 2007. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 63.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19: 2092–2100, 2008. doi: 10.1091/mbc.e07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268, 2008. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 65.Ducommun S, Deak M, Sumpton D, Ford RJ, Núñez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, Morrice NA, Sakamoto K. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal 27: 978–988, 2015. doi: 10.1016/j.cellsig.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 66.Seabright AP, Fine NHF, Barlow JP, Lord SO, Musa I, Gray A, Bryant JA, Banzhaf M, Lavery GG, Hardie DG, Hodson DJ, Philp A, Lai YC. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J 34: 6284–6301, 2020. doi: 10.1096/fj.201903051R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J 27: 433–446, 2008. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, Ruberto FP, Nemir M, Wai T, Pedrazzini T, Manley S. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593: 435–439, 2021. doi: 10.1038/s41586-021-03510-6. [DOI] [PubMed] [Google Scholar]
- 69.Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, Youle RJ. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 216: 3231–3247, 2017. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen H, Ren S, Clish C, Jain M, Mootha V, McCaffery JM, Chan DC. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol 211: 795–805, 2015. doi: 10.1083/jcb.201507035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd.. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab 26: 872–883.e5, 2017.doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116: 264–278, 2015. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 73.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33: 2798–2813, 2014. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nishimura T, Tooze SA. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov 6: 32, 2020. doi: 10.1038/s41421-020-0161-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701, 2008. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature 495: 389–393, 2013. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 77.Kotani T, Kirisako H, Koizumi M, Ohsumi Y, Nakatogawa H. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc Natl Acad Sci USA 115: 10363–10368, 2018. doi: 10.1073/pnas.1806727115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, Ohsumi Y, Noda NN. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol 26: 281–288, 2019. doi: 10.1038/s41594-019-0203-4. [DOI] [PubMed] [Google Scholar]
- 79.Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. eLife 8: e45777, 2019. doi: 10.7554/eLife.45777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valverde DP, Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, Reinisch KM, Melia TJ. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol 218: 1787–1798, 2019. doi: 10.1083/jcb.201811139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang Z, Takahashi Y, He H, Hattori T, Chen C, Liang X, Chen H, Young MM, Wang HG. TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep 28: 1744–1757.e5, 2019. doi: 10.1016/j.celrep.2019.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Friedman JR, Lackner LL, West M, Dibenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science 334: 358–362, 2011. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang J-Y, Yang WY. Bit-by-bit autophagic removal of Parkin-labelled mitochondria. Nat Commun 4: 2428, 2013. doi: 10.1038/ncomms3428. [DOI] [PubMed] [Google Scholar]
- 84.Wang X, Wen Y, Dong J, Cao C, Yuan S. Systematic in-depth proteomic analysis of mitochondria-associated endoplasmic reticulum membranes in mouse and human testes. Proteomics 18: 1700478, 2018. doi: 10.1002/pmic.201700478. [DOI] [PubMed] [Google Scholar]
- 85.Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou MH. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 136: 2248–2266, 2017. doi: 10.1161/CIRCULATIONAHA.117.030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, Zhuang H, Zhang X, Chen H, Li S, Yang Y, Lu Y, Wang J, Zhu R, Zhang L, Sui S, Tan N, Zhao B, Zhang J, Li L, Feng D. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J 35: 1368–1384, 2016. doi: 10.15252/embj.201593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM, Valente EM. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13: 654–669, 2017. doi: 10.1080/15548627.2016.1277309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gautier CA, Erpapazoglou Z, Mouton-Liger F, Muriel MP, Cormier F, Bigou S, Duffaure S, Girard M, Foret B, Iannielli A, Broccoli V, Dalle C, Bohl D, Michel PP, Corvol J-C, Brice A, Corti O. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet 5: 2972–2984, 2016. doi: 10.1093/hmg/ddw148. [DOI] [PubMed] [Google Scholar]
- 89.Basso V, Marchesan E, Peggion C, Chakraborty J, von Stockum S, Giacomello M, Ottolini D, Debattisti V, Caicci F, Tasca E, Pegoraro V, Angelini C, Antonini A, Bertoli A, Brini M, Ziviani E. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol Res 138: 43–56, 2018. doi: 10.1016/j.phrs.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 90.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature 402: 672–676, 1999. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 91.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100: 15077–15082, 2003. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 55: 238–252, 2014. doi: 10.1016/j.molcel.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liang C, Lee J-S, Inn K-S, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10: 776–787, 2008. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M, Liiv M, Anwar T, Eskelinen E-L, Kaasik A. BECN1 is involved in the initiation of mitophagy. Autophagy 10: 1105–1119, 2014. doi: 10.4161/auto.28615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar A, Shaha C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci Rep 8: 615, 2018. doi: 10.1038/s41598-017-19102-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, Torosantucci L, Cassina L, Russo MA, Dallapiccola B, Valente EM, Casari G. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 17: 962–974, 2010. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- 97.Sun Y, Cai Y, Qian S, Chiou H, Zang QS. Beclin‐1 improves mitochondria‐associated membranes in the heart during endotoxemia. FASEB Bioadv 3: 123–135, 2021. doi: 10.1096/fba.2020-00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating parkin/PINK1-dependent mitophagy. Mol Cell 55: 451–466, 2014. doi: 10.1016/j.molcel.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 99.Chen D, Chen X, Li M, Zhang H, Ding W-X, Yin X-M. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem Biophys Res Commun 432: 226–230, 2013. doi: 10.1016/j.bbrc.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198: 219–233, 2012. doi: 10.1083/jcb.201202061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Young AR, Chan EY, Hu XW, Köchl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 119: 3888–3900, 2006. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 102.Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, Luo Q, Feng D, Zhu Y, Chen Q. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res 27: 184–201, 2017. doi: 10.1038/cr.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Imai K, Hao F, Fujita N, Tsuji Y, Oe Y, Araki Y, Hamasaki M, Noda T, Yoshimori T. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J Cell Sci 129: 3781–3791, 2016. doi: 10.1242/jcs.196196. [DOI] [PubMed] [Google Scholar]
- 104.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23: 1860–1873, 2012. doi: 10.1091/mbc.E11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sawa-Makarska J, Baumann V, Coudevylle N, von Bülow S, Nogellova V, Abert C, Schuschnig M, Graef M, Hummer G, Martens S. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 369: eaaz7714, 2020. doi: 10.1126/science.aaz7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Judith D, Jefferies HBJ, Boeing S, Frith D, Snijders AP, Tooze SA. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J Cell Biol 218: 1634–1652, 2019. doi: 10.1083/jcb.201901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, Sugita Y, Nomura N, Iwata S, Ohsumi Y, Fujimoto T, Nakatogawa H, Kikkawa M, Noda NN. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol 27: 1185–1193, 2020. [Erratum in Nat Struct Mol Biol 27:1209, 2020]. doi: 10.1038/s41594-020-00518-w. [DOI] [PubMed] [Google Scholar]
- 108.Maeda S, Yamamoto H, Kinch LN, Garza CM, Takahashi S, Otomo C, Grishin NV, Forli S, Mizushima N, Otomo T. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat Struct Mol Biol 27: 1194–1201, 2020. doi: 10.1038/s41594-020-00520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 125: 1488–1499, 2012. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 110.Liang W, Moyzis AG, Lampert MA, Diao RY, Najor RH, Gustafsson ÅB. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell 19: e13187, 2020.doi: 10.1111/acel.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, Shoda T, Demizu Y, Naito M, Tanaka K, Matsuda N. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol 219: e201912144, 2020.doi: 10.1083/jcb.201912144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dorn GW 2nd. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3: 374–383, 2010. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol 6: e24511, 2013. doi: 10.4161/cib.24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 108: 9572–9577, 2011. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun 4: 2308, 2013. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 116.Gao B, Yu W, Lv P, Liang X, Sun S, Zhang Y. Parkin overexpression alleviates cardiac aging through facilitating K63-polyubiquitination of TBK1 to facilitate mitophagy. Biochim Biophys Acta Mol Basis Dis 1867: 165997, 2021. doi: 10.1016/j.bbadis.2020.165997. [DOI] [PubMed] [Google Scholar]
- 117.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Takuji Shirasawa TS, Mizushima N, Otsu K. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6: 600–606, 2010. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 118.Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 22: 1428–1438, 2016. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ, Rabinovitch PS. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13: 529–539, 2014. doi: 10.1111/acel.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Woodall BP, Orogo AM, Najor RH, Cortez MQ, Moreno ER, Wang H, Divakaruni AS, Murphy AN, Gustafsson AB. Parkin does not prevent accelerated cardiac aging in mitochondrial DNA mutator mice. JCI Insight 4: e127713, 2019. doi: 10.1172/jci.insight.127713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sardon Puig L, Valera-Alberni M, Cantó C, Pillon NJ. Circadian Rhythms and Mitochondria: connecting the dots. Front Genet 9: 452, 2018. doi: 10.3389/fgene.2018.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmitt K, Grimm A, Dallmann R, Oettinghaus B, Restelli LM, Witzig M, Ishihara N, Mihara K, Ripperger JA, Albrecht U, Frank S, Brown SA, Eckert A. Circadian control of DRP1 activity regulates mitochondrial dynamics and bioenergetics. Cell Metab 27: 657–666.e5, 2018. doi: 10.1016/j.cmet.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 123.Neufeld-Cohen A, Robles MS, Aviram R, Manella G, Adamovich Y, Ladeuix B, Nir D, Rousso-Noori L, Kuperman Y, Golik M, Mann M, Asher G. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proc Natl Acad Sci USA 113: E1673–E1682, 2016. doi: 10.1073/pnas.1519650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jacobi D, Liu S, Burkewitz K, Kory N, Knudsen NH, Alexander RK, Unluturk U, Li X, Kong X, Hyde AL, Gangl MR, Mair WB, Lee C-H. Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab 22: 709–720, 2015. doi: 10.1016/j.cmet.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rabinovich-Nikitin I, Rasouli M, Reitz CJ, Posen I, Margulets V, Dhingra R, Khatua TN, Thliveris JA, Martino TA, Kirshenbaum LA. Mitochondrial autophagy and cell survival is regulated by the circadian Clock gene in cardiac myocytes during ischemic stress. Autophagy 17: 3794–3719, 2021. doi: 10.1080/15548627.2021.1938913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926, 2013. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133: 1249–1263, 2016. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60: 1770–1778, 2011. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M, Seishima M, Minatoguchi S. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 11: 1146–1160, 2015. doi: 10.1080/15548627.2015.1051295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 288: 18077–18092, 2013. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol 50: 1035–1043, 2011. doi: 10.1016/j.yjmcc.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 132.Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 124: 1360–1371, 2019. doi: 10.1161/CIRCRESAHA.118.314607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, Xie Y, Carlson D, Rothermel BA, Sun Y, Levine B, Hill JA, Wolf SE, Minei JP, Zang QS. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 138: 2247–2262, 2018. doi: 10.1161/CIRCULATIONAHA.117.032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Soni PR, Noval Rivas M, Arditi M. A comprehensive update on Kawasaki disease vasculitis and myocarditis. Curr Rheumatol Rep 22: 6, 2020. doi: 10.1007/s11926-020-0882-1. [DOI] [PubMed] [Google Scholar]
- 136.Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, Fishbein MC, Lehman TJ, Arditi M. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation 125: 1542–1550, 2012. doi: 10.1161/CIRCULATIONAHA.111.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Marek-Iannucci S, Ozdemir AB, Moreira D, Gomez AC, Lane M, Porritt RA, Lee Y, Shimada K, Abe M, Stotland A, Zemmour D, Parker S, Sanchez-Lopez E, Van Eyk J, Gottlieb RA, Fishbein MC, Karin M, Crother TR, Rivas MN, Arditi M. Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight 6: e151981, 2021. doi: 10.1172/jci.insight.151981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang Y, Liu N, Lu B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci Ther 25: 859–875, 2019. doi: 10.1111/cns.13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22: 401–412, 2019. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163, 2015. doi: 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chang HW, Kim MR, Lee HJ, Lee HM, Kim GC, Lee YS, Nam HY, Lee M, Jang HJ, Lee KE, Lee JC, Byun Y, Kim SW, Kim SY. p53/BNIP3-dependent mitophagy limits glycolytic shift in radioresistant cancer. Oncogene 38: 3729–3742, 2019. doi: 10.1038/s41388-019-0697-6. [DOI] [PubMed] [Google Scholar]
- 142.Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol 6: 104, 2018. doi: 10.3389/fcell.2018.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu L, Lin DC, Yin D, Koeffler HP. An emerging role of PARK2 in cancer. J Mol Med (Berl) 92: 31–42, 2014. doi: 10.1007/s00109-013-1107-0. [DOI] [PubMed] [Google Scholar]
- 144.Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA 108: 16259–16264, 2011. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang Y, Liu H-H, Cao Y-T, Zhang L-L, Huang F, Yi C. The role of mitochondrial dynamics and mitophagy in carcinogenesis, metastasis and therapy. Front Cell Dev Biol 8: 413, 2020. doi: 10.3389/fcell.2020.00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tiku V, Tan MW, Dikic I. Mitochondrial functions in infection and immunity. Trends Cell Biol 30: 263–275, 2020. [Erratum in Trends Cell Biol 30: 748, 2020]. doi: 10.1016/j.tcb.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu KE, Fazal FM, Parker KR, Zou J, Chang HY. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst 11: 102–108.e3, 2020. doi: 10.1016/j.cels.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singh KK, Chaubey G, Chen JY, Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol 319: C258–C267, 2020. doi: 10.1152/ajpcell.00224.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nunn AVW, Guy GW, Brysch W, Botchway SW, Frasch W, Calabrese EJ, Bell JD. SARS-CoV-2 and mitochondrial health: implications of lifestyle and ageing. Immun Ageing 17: 33, 2020. doi: 10.1186/s12979-020-00204-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shi C-S, Qi H-Y, Boularan C, Huang N-N, Abu-Asab M, Shelhamer JH, Kehrl JH. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 193: 3080–3089, 2014. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468, 2020. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]