

Abstract
Duchenne muscular dystrophy (DMD) is an inherited muscle wasting disease. Metabolic impairments and oxidative stress are major secondary mechanisms that severely worsen muscle function in DMD. Here, we sought to determine whether germline reduction or ablation of sarcolipin (SLN), an inhibitor of sarco/endoplasmic reticulum (SR) Ca2+ ATPase (SERCA), improves muscle metabolism and ameliorates muscle pathology in the mdx mouse model of DMD. Glucose and insulin tolerance tests show that glucose clearance rate and insulin sensitivity were improved in the SLN haploinsufficient mdx (mdx:sln+/−) and SLN-deficient mdx (mdx:sln−/−) mice. The histopathological analysis shows that fibrosis and necrosis were significantly reduced in muscles of mdx:sln+/− and mdx:sln−/− mice. SR Ca2+ uptake, mitochondrial complex protein levels, complex activities, mitochondrial Ca2+ uptake and release, and mitochondrial metabolism were significantly improved, and lipid peroxidation and protein carbonylation were reduced in the muscles of mdx:sln+/− and mdx:sln−/− mice. These data demonstrate that reduction or ablation of SLN expression can improve muscle metabolism, reduce oxidative stress, decrease muscle pathology, and protects the mdx mice from glucose intolerance.
Keywords: Duchenne muscular dystrophy, mdx, metabolism, mitochondria, sarcolipin
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder, epidemiologically reported to affect 1 in 5,000 male live births (1). DMD is caused by mutations in the dystrophin gene resulting in loss of functional dystrophin protein in skeletal and cardiac muscles (1). Dystrophin deficiency results in loss of fiber strength, stability, and structural fragility, which leads to defective membrane permeability (2, 3), metabolic crisis (4), and progressive muscle degeneration (5). Metabolic dysregulation with impaired ATP generation, mitochondrial structure and functional abnormalities, altered glycolytic flux, glycogen storage and utilization, fatty acid oxidation, and the purine nucleotide cycle has been reported in muscles of animal models, and DMD patients (6–14). Accumulating evidence suggests that the altered metabolic pathways occur both in the cytoplasmic compartments including glycolysis and purine nucleotide cycle and in the mitochondria such as citric acid cycle and electron transport chain and decrement of anaerobic metabolism impair the overall ATP synthesis in dystrophic muscles (7, 15–19). The low ATP synthesis is a major cause as well as an enhancer of contractile dysfunction, decreased intracellular Ca2+ (Ca2+i) homeostasis, diminished satellite cell function, and decreased protein synthesis. Therefore, improving the metabolic function in dystrophic muscles is anticipated to ameliorate muscle pathophysiology in DMD.
Ca2+i homeostasis plays a vital role in various biological processes including energy metabolism (20). An increase in cytosolic Ca2+ concentration is shown to promote hyperglycemia in patients susceptible to malignant hyperthermia (21). Chronic elevation of cytosolic Ca2+ concentration is the key pathological mechanism that promotes muscle wasting and cardiomyopathy in DMD (22–26). Mitochondria are suggested as potential targets of impaired Ca2+ homeostasis in muscular dystrophy (27). In dystrophic muscles, Ca2+ overloaded mitochondria undergo swelling, the opening of membrane permeability transition pore (mPTP), impaired ATP synthesis, and elevated reactive oxygen levels (ROS) and contribute to oxidative stress (28, 29). Studies have also shown that cytosolic Ca2+ overload is associated with mitochondrial dysfunction in dystrophic mice (26). It is now established that improving the sarco/endoplasmic reticulum (SR) Ca2+ ATPase (SERCA) function can reduce the cytosolic Ca2+ overload and ameliorate muscular dystrophy in animal models (26). A recent study shows that pharmacological activation of SERCA can reduce exercise-induced muscle damage and restore mitochondrial function in mdx mice (30). Thus, improving SERCA function could improve mitochondrial function and prevent metabolic dysfunction in DMD.
We have recently shown that sarcolipin (SLN), an inhibitor of SERCA, is upregulated in the heart and skeletal muscles of animal models and human DMD (31, 32). Using gene knockout mouse models and adeno-associated virus (AAV)-mediated SLN suppression, we have demonstrated that normalizing SLN expression is sufficient to improve the SERCA function and ameliorate muscle pathology and cardiomyopathy in mouse models of DMD (32, 33). In the DMD dog myoblasts (34) and mdx cardiac myocytes (33), SLN ablation normalizes the SR Ca2+ content and Ca2+ transients suggesting that SLN ablation can improve the Ca2+i cycling in dystrophic muscles. However, the relative contribution of SLN upregulation in metabolic dysfunction in the dystrophic muscle remains unknown. Transgenic overexpression of SLN in muscles has been shown to increase the rate of muscle glucose uptake, improve insulin sensitivity, activate mitochondrial biogenesis, and improve muscle performance by increasing oxidative capacity (35, 36). However, despite SLN upregulation, the rate of glucose clearance (4, 11), and mitochondrial content and function (10) are severely impaired in the dystrophic muscles, implying SLN upregulation may decrease mitochondrial function and cause energy demand in dystrophin-deficient muscles. Accordingly, we hypothesized that reducing or ablating SLN expression would improve SERCA function and Ca2+i cycling, resulting in reduced muscle damage, improved mitochondrial biogenesis, and oxidative capacity in dystrophic muscles. These changes would reduce energy demand and ameliorate muscular dystrophy in mdx mice. In this study, we have tested this hypothesis using SLN haploinsufficient mdx (mdx:sln+/−) and SLN-deficient mdx (mdx:sln−/−) mice. For the first time, our results demonstrate that reducing or abolishing SLN expression reduces muscle pathology, improves mitochondrial function, and reduces oxidative stress and promote glucose utilization, and prevents insulin resistance in mdx mice.
METHODS
Animal Studies
All animal experiments were conducted in accordance with policies of the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC), Rutgers, Newark, NJ. Mice were maintained in the animal facility at 22–24°C with 12-h light/dark cycles and 60%–70% of humidity. The animals were fed ad libitum under a normal chow diet and sterilized water. Mice were euthanized using pentobarbital (150 mg/kg, ip) according to the American Veterinary Medical Association guidelines approved by the IACUC. The mouse genotypes used in the study were wild type (WT), SLN knockout (sln−/−), mdx, mdx:sln+/−, and mdx:sln−/−, and all are in C57BL/10 background. The mdx:sln+/− and mdx:sln−/− mice were generated by crossing the mdx mice with sln−/− mice and genotyped as described earlier (33). Although DMD is an X-linked disorder, in mdx mice, the level of cardiac fibrosis was not significantly different between male and female mice. In addition, aged female mdx mice show severe cardiac dysfunction compared with males (33). Another study shows that both male and female mdx mice are equally susceptible to metabolic dysfunction (10). Therefore, for our studies, we have used both male and female mice. For all the experiments, tissues from 6-mo-old male and female mice in equal numbers were used.
Glucose and Insulin Tolerance Tests
Blood samples were collected by tail vein puncture and the glucose levels were measured using a biosensor-based glucometer (BG1000). The glucose clearance rate was determined by an intraperitoneal (ip) glucose tolerance test (ipGTT). Briefly, mice fasted overnight, and then fasting blood glucose concentration was measured through the tail-vein puncture. Mice were then administered intraperitoneally with 15% glucose solution (2 g/kg body wt) and blood glucose level was monitored at 15, 30, 60, and 120 min after glucose administration. Blood glucose versus time curve was plotted to determine the glycemic index after intraperitoneal administration. Glycemic response was expressed as area under the curve (AUC). Insulin sensitivity and counter regulatory responses were determined by an intraperitoneal insulin tolerance test (ITT) as previously described (37). After 4 h of fasting, mice were given intraperitoneal insulin injection (0.75 U/kg body weight). Blood samples were collected at 0, 15, 30, 45, and 60 min after insulin injection, and glucose level was estimated. AUC was calculated to evaluate insulin sensitivity.
Lipid Peroxidation Assay
The lipid peroxidation was measured in both diaphragm and pectorals by estimating malondialdehyde (MDA) levels using the QuantiChrom TBARS assay kit (DTBA-100, BioAssay Systems) following the manufacturer’s protocol. The assays were performed in triplicates. The absorbance was read at 532 nm and the amount of MDA was expressed as µmol/mg tissue.
Real-Time Quantitative Reverse Transcription PCR Analysis
Total RNA from the pectorals and diaphragm was extracted using TRI reagent (Sigma-Aldrich Inc., USA). Complementary DNA (cDNA) was prepared using a High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific). The real-time quantitative reverse transcription-PCR (RT-qPCR) analysis was performed using the Superscript RT-PCR kit (Invitrogen). The cycle threshold (Ct) values obtained for each gene were normalized to Ct readings of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences used for qPCR were as follows: Fbp2 (forward, 5′-AGAAAGACCACGGAGGACGA-3′, reverse; 5′-CCCGCAGCCACGATGT-3′), Glut4 (forward, 5′- ATGAGAAACGGAAGTTGGAGAGA-3′; reverse, 5′-GTGGGTGCGGCTGCC-3′), Phkg1 (forward, 5′-CCTTAACCGAGAAGGAAACCA-3′; reverse, 5′- TGAGTTTGTGCAGGGTACAGA-3′), Pygm (forward, 5′-AGTGGAGGACGTGGAAAGG-3′; reverse, 5′- GCTCAGGAATTCGGTCGTAG-3′), Pgk1 (forward, 5′-TTGGACAAGCTGGACGTGAA-3′; reverse, 5′-TTTGGTTGTTTGTTATCTGGTTGTTC-3′), Pgc1α (forward, 5′- CATTTGATGCACTGACAGATGGA-3′; reverse, 5′- GTCAGGCATGGAGGAAGGAC-3′), Gpd1 (forward, 5′- AGACCTCATCACGACCTGCT-3′; reverse, 5′- CCAGCTGCTCAATGGACTTT-3′), Ppp1r3c (forward, 5′- CTGAGTGCGAAGTTGCTCAG-3′; reverse, 5′- TGGATCTAGCACATGGATCATT-3′), and GAPDH (forward, 5′- GTCGTGGATCTGACGTGCC-3′; reverse, 5′- ATGCCTGCTTCACCACCTTC-3′).
Mitochondrial Copy Number
Mitochondrial DNA copy number was calculated as the ratio of mitochondrial genome to nuclear genome following real-time quantitative PCR (qPCR) using total DNA isolated from muscle tissues as described by Malik et al. (38). The primers used were specific for mouse mitochondrial DNA (forward 5′- CTAGAAACCCCGAAACCAAA-3′; reverse, 5′- CCAGCTATCACCAAGCTCGT-3′) and nuclear DNA (β-2 microglobulin; forward, 5′- ATGGGAAGCCGAACATACTG-3′; reverse 5′- CAGTCTCAGTGGGGGTGAAT-3′).
Western Blot Analysis
Total protein extract preparations from pectorals and diaphragm, and Western blot analysis were carried out as described (32). Following primary antibodies were used in this study: antibodies specific for SLN (anti-rabbit, 1:3,000) (39), SERCA1 (anti-rabbit, 1:5,000) (39), SERCA2a (anti-rabbit, 1:5,000) (39), calsequestrin (CSQ; anti-rabbit, 1:5,000, Thermo Fisher Scientific, No. PA1-913), utrophin (anti-mouse, 1:100, 8A4; DSHB), pan-calcineurin A (anti-rabbit, 1:1,000, CST, No. 2614S), Fbp2 (anti-rabbit. 1:1,000, Invitrogen, PA5-89611), pyruvate dehydrogenase (PDH; anti-mouse, 1:1,000, Santa Cruz Biotechnology, sc377092), superoxide dismutase 2 (SOD2; anti-mouse, 1:1,000, Santa Cruz Biotechnology, sc137254), glutathione reductase (GR; anti-mouse, 1:1,000, Santa Cruz Biotechnology, sc133245), and GAPDH (anti-mouse, 1:10,000, Sigma, G8795). Western blot analysis for the subunits of oxidative phosphorylation protein complexes was carried out using OXPHOS Rodent WB Antibody Cocktail (anti-mouse, 1:1,000, Abcam, ab110413). Membranes were treated with appropriate secondary antibodies for 1 h at room temperature and visualized with SuperSignal West Dura Substrate kit (Thermo Fisher Scientific) using Bio-Rad ChemiDoc MP Imaging System. Quantitation of signals was performed using Image Lab v. 5.1 software and normalized to GAPDH levels. The validation of antibodies for SLN, SERCA1, SERCA2a, CSQ, and utrophin was performed using tissues from knockout or transgenic mouse models and cell lines expressing these proteins. The specificity of other protein-antibodies was validated using cell lines and/or blocking peptides according to the manufacturer’s instructions, and it was confirmed that the band with the reported molecular weight was specifically observed in each blotting.
To detect the oxidative modification of proteins, total protein extracts from the diaphragm or pectorals were derivatized using OxyBlot protein oxidation detection kit (Cat. No. S7150, Millipore), separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose membrane. The stable dinitrophenyl (DNP) proteins were detected by immunoblot using anti-DNP antibodies. Quantitation of signals was performed using Image Lab v. 5.1 software and normalized to GAPDH levels.
Mitochondrial Complex Assays
The mitochondrial complex I (NADH-dehydrogenase) and complex IV (Cytochrome c oxidase) activities were measured in freshly prepared tissue lysates using Abcam assay kits (Complex I, No. ab109721; Complex IV, No. ab109911) as per the manufacturer’s instructions. The optical densities were measured for complex I at 450 nm in kinetic mode at room temperature for 30 min and complex IV at 550 nm in kinetic mode at room temperature for 120 min.
Measurement of Mitochondrial Ca2+ Uptake and Release
Mitochondria were isolated from pectorals and diaphragm using differential centrifugation as previously described (40). Briefly, immediately after dissection, muscles were rinsed in ice-cold phosphate-buffered saline (PBS) containing 10 mM EDTA, minced, and incubated in 0.05% trypsin EDTA for 30 min. The digest was centrifuged for 5 min at 200 g, and the pellet was homogenized in the mitochondrial isolation buffer (in mmol, 67 sucrose, 50 KCl, 10 EDTA, 50 Tris. Cl, pH 7.4, and 0.2% BSA). The final suspension contained 1–5 mg of mitochondrial protein/mL.
Mitochondrial Ca2+ uptake and release were determined spectrophotometrically using arsenazo III [(2,2′-(1, 8-Dihydroxy-3,6-disulfonaphthylene-2,7-bisazo) bisbenzenearsonic acid, 2,7-Bis(2-arsonophenylazo) chromotropic acid] at 685 nm using a plate reader (Spectra max plus, Molecular devices) at 25°C as described (40). Briefly, skeletal muscle mitochondria (0.5 mg mitochondrial protein/mL) were suspended in an incubation medium (in mmol, 210 mannitol, 70 sucrose, 1 KH2PO4, and 10 HEPES-KOH, pH 7.4) containing arsenazo III (50 μmol) and EGTA (10 μmol) and energized with glutamate (2.5 mmol) and malate (2.5 mmol). About 50 μmol of CaCl2 was added to the mitochondrial suspension and the Ca2+ uptake was allowed to proceed for 5 min in the presence of 1 μmol cyclosporine (CsA). Mitochondrial Ca2+ release was determined after the addition of 0.5 μmol ruthenium red and 5 mmol Na+ to Ca2+-loaded mitochondrial suspension. At a neutral pH, Ca2+ forms a complex with arsenazo III, which produces a blue complex and the color intensity produced is proportional to the concentration of Ca2+ in the sample, which is expressed as nmol Ca2+/mg of mitochondrial protein. The concentration of the mitochondrial Ca2+ uptake and release were calculated based on free Ca2+ availability in the reaction system.
Histology
For histological examination, paraffin-embedded sections (10 microns) of pectorals and diaphragm were dehydrated and stained with hematoxylin and eosin (H&E) and Masson’s trichrome. The necrotic areas containing mononuclear cells from H&E staining and fibrosis indicated by the blue-stained collagen areas by trichrome were calculated using the NIH ImageJ 1.43 u program.
Seahorse Measurements
The freshly dissected diaphragm and pectorals tissues were rinsed in ice-cold 10 mM EDTA, minced and digested with 0.05% trypsin EDTA for 30 min, and centrifuged for 5 min at 200 g. The pellet was homogenized in the mitochondrial isolation buffer [in mmol, 210 D-Mannitol, 70 sucrose, 5 HEPES, 1 EGTA, and 0.5% (wt/vol) fatty acid-free BSA, pH 7.2], purified following differential centrifugation (41, 42) and suspended in mitochondrial assay buffer 1 [MAS1; in mmol, 220 D-Mannitol, 70 sucrose, 10 KH2PO4, 5 MgCl2, 2 HEPES, and 1 EGTA, and 0.2% (wt/vol) of fatty acid-free BSA, pH 7.2]. After protein estimation, mitochondria were pelleted again and resuspended in MAS2 (MAS1 containing 10 mM succinate and 2 µM rotenone). Succinate was used as a substrate to drive complex II respiration and rotenone was used simultaneously to inhibit complex I respiration. About 5 µg of mitochondria in 100 µL volume was added to each well of the Seahorse XFe 24 plate and centrifuged at 2,000 g for 20 min at 4°C. After ensuring the proper adherence, the flux assay was started by adding 400 μL of MAS2 with 5 mM ADP (prewarmed to 37°C) to each well. The state III respiration was measured first. Oxygen consumption rate (OCR) was measured after the sequential addition of1) oligomycin (3.2 µM final), a complex V ATP synthase inhibitor used to measure ATP-independent oxygen consumption; 2) FCCP (4 µM final), an uncoupler, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone used to measure maximum respiratory capacity; and 3) antimycin A (4 µM final), a complex III inhibitor used to stop mitochondrial respiration. The OCR, extracellular acidification rate (ECAR), various states of respiration (State III, State IV0, State IIIµ), and respiratory control ratio (RCR) were determined as described (41, 42). Briefly, the OCR values derived from ADP, oligomycin, and FCCP treatments were taken as state III, state IVo, and state IIIµ respiration, respectively. The ratio of highest reading after the ADP addition (state III) and lowest point after the oligomycin addition (state IVo) was designated as RCR value. Wave software (Agilent) was used to export OCR rates to GraphPad Prism 8.0.2. We used three technical replicates for each genotype and data from four mice per genotype were used for this experiment.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism v8.0.2 software. For the glucose and insulin tolerance tests, two-way ANOVA with Bonferroni post hoc analysis was used to compare between groups at different time points. One-way ANOVA with Tukey’s post hoc analysis or unpaired two-sided Student t test was used to compare multiple or between two groups. For all mouse experiments, cohort sizes match the common practice of the described experiments and are repeated three times. Experiments and data analysis were carried out in a blinded manner to ensure that the results are consistent and reproducible. No data points were excluded and the P < 0.05 was considered statistically significant in all analyses.
RESULTS
Ablation of SLN Ameliorates Muscle Pathology in the Pectorals of Mdx Mice
The mdx:sln+/− and mdx:sln−/− mice were born normal and had a normal lifespan (33). For our studies, we chose the diaphragm, which has severe muscle degeneration, fibrosis, and functional deficit (43), and the pectorals, which are primarily composed of fast fibers and are prone to damage in the absence of dystrophin. We first examined the dystrophic phenotype of these tissues in 6-mo-old mdx:sln+/− and mdx:sln−/− mice by histopathological examinations. Age- and sex-matched mdx and WT littermates were used as controls. H&E and Masson’s trichrome staining showed increased mononuclear infiltration and fibrosis in diaphragm and pectorals of mdx mice. These pathological indices were significantly decreased in the pectorals of mdx:sln+/− and mdx:sln−/− mice (Fig. 1, A–C). Mononuclear infiltration was significantly reduced in the diaphragm of mdx:sln+/− mice but not in the diaphragm of mdx:sln−/− mice (Fig. 1, A and B). The percent fibrotic area in the diaphragm of mdx:sln+/− and mdx:sln−/− mice was similar to that of mdx mice (Fig. 1, A and C).
Figure 1.
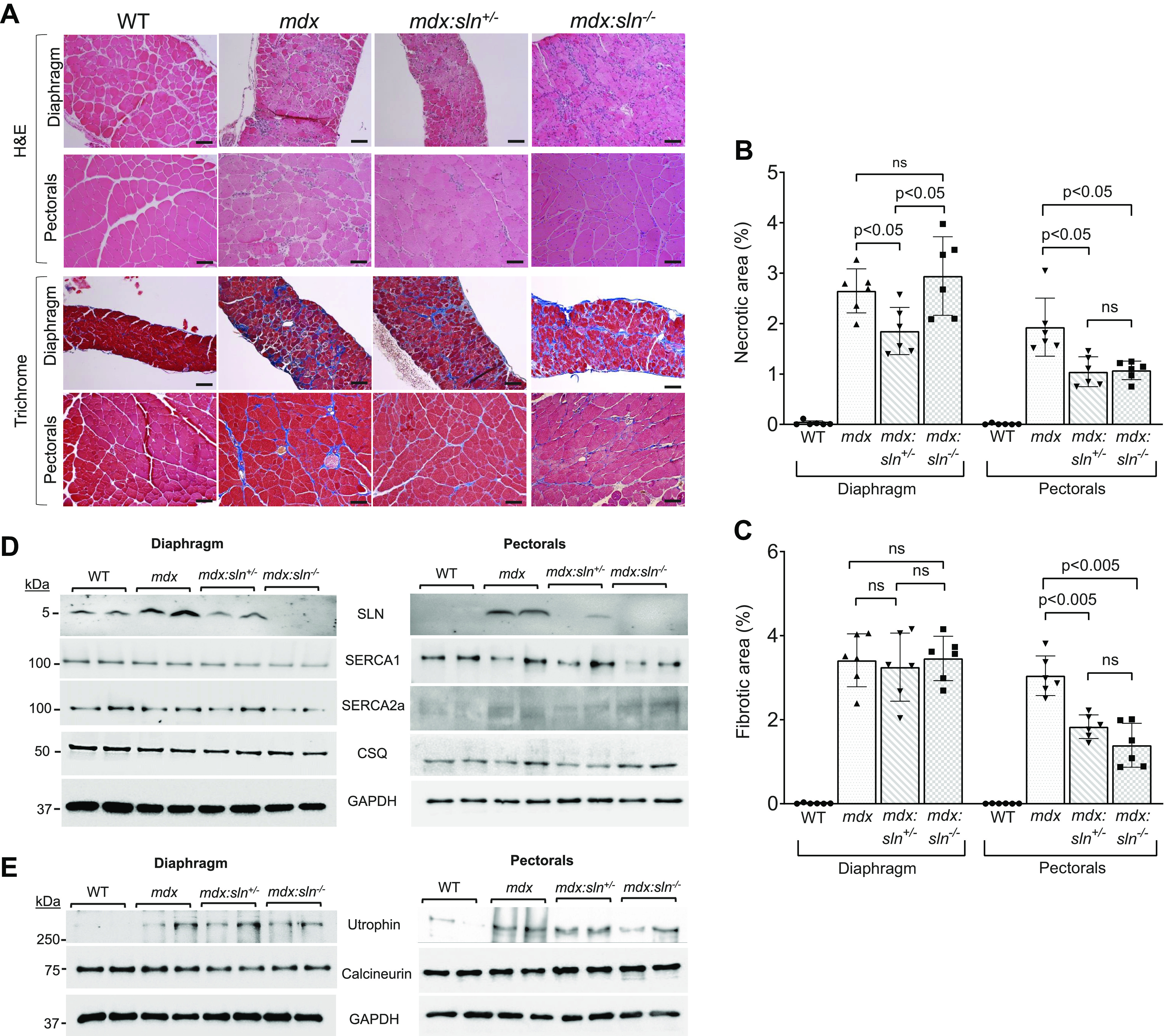

Sarcolipin ablation reduces muscle pathology in mdx mice. A: representative H&E and trichrome staining of sections of diaphragm and pectorals from WT, mdx, mdx:sln+/− and mdx:sln−/− mice. Original magnification is ×20 for the diaphragm and ×10 for pectorals. Scale bar, 100 µm. Quantitation showing necrotic (B) and fibrotic (C) areas in the diaphragm and pectorals of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Data represent means ± SD. n = 6 mice/group. P values are shown within the bar diagram. ns - not statistically significant. Representative Western blots showing the protein levels of SLN, SERCA1, SERCA2a, CSQ, and GAPDH (D) and utrophin, calcineurin, and GAPDH (E) in the diaphragm and pectorals of WT, mdx, mdx:sln+/− and mdx:sln−/− mice. n = 6 mice/group. Ca2+-dependent Ca2+ uptake and Vmax of Ca2+ in the diaphragm (F and G) and pectorals (H and I) of WT, mdx mdx:sln+/−, and mdx:sln−/− mice. Data represent means ± SD. n = 4 mice/group. Male and female mice were used in equal numbers in each experiment. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; H&E. hematoxylin and eosin; CSQ, calsequestrin; SLN, sarcolipin; WT, wild type.
We next examined whether SLN ablation has any effect on the expression levels of major SR Ca2+ handling proteins in the diaphragm and pectorals of mdx mice. Loss of one SLN allele (sln+/−) normalized SLN protein expression in both diaphragm and pectorals of mdx mice (Fig. 1D and Supplemental Fig. S1A; all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.14336660). The protein levels of SERCA1, SERCA2a, and CSQ were not statistically significant both in the diaphragm and pectorals of mdx, mdx:sln+/−, and mdx:sln−/− mice compared with that of WT controls (Fig. 1D and Supplemental Fig. S1, B and C).
It has been shown that the complete loss of SLN is associated with reduced expression of utrophin and calcineurin in the soleus and diaphragm of mdx mice (44). We, therefore, examined the protein levels of utrophin and calcineurin in the diaphragm and pectorals of mdx mice deficient for SLN. Western blot analysis shows that the utrophin protein levels were significantly high in both diaphragm and pectorals of mdx mice (Fig. 1E and Supplemental Fig. S1, D and E). The heterozygous and homozygous deletion of the SLN gene does not affect the utrophin upregulation in these dystrophic tissues. The calcineurin protein expression was unaltered in the diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice compared with that of both mdx and WT littermates (Fig. 1E and Supplemental Fig. S1, D and E).
We, next, examined the SERCA activity by measuring the SR Ca2+ uptake in protein extracts prepared from the diaphragm and pectorals. The rate, as well as the maximum velocity (Vmax) of Ca2+ dependent Ca2+ uptake, was decreased both in the diaphragm (Fig. 1, F and G) and pectorals (Fig. 1, H and I) of mdx mice, whereas the Vmax of Ca2+ uptake was significantly increased in the diaphragm and pectorals of both mdx:sln+/− and mdx:sln−/− mice.
Glucose Homeostasis is Improved in Mdx Mice Deficient for SLN
Metabolic alterations and impaired glucose clearance have been reported in mdx mice (4, 11) and DMD patients (45). We, therefore, first determined the glucose homeostasis and its metabolic rate in mdx and SLN-deficient mdx mice. The fasting blood glucose level was not significantly different between WT, mdx, mdx:sln+/−, and mdx:sln−/− mice (Fig. 2A). The ipGTT showed a decreased rate of glucose clearance in mdx mice. Also, the glycemic index represented as the area under the curve (AUC) was significantly high in mdx mice. On the other hand, the rate of glucose clearance and glycemic index in the mdx:sln+/− and mdx:sln−/− mice was near to that of WT mice (Fig. 2B). After insulin bolus, blood glucose levels remained high in mdx mice throughout the experimental time points. The glycemic index in response to insulin injection was also significantly high in mdx mice compared with that of WT mice. Insulin-mediated glucose clearance and glycemic index were comparable in mdx:sln+/− and mdx:sln−/− mice to WT controls (Fig. 2C).
Figure 2.

Sarcolipin ablation ameliorates glucose homeostasis in mdx mice. A: fasting blood glucose levels in WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Glucose clearance rate measured by intraperitoneal glucose tolerance test (ipGTT) and area under the curve (AUC; B) and insulin tolerance test (ITT) and AUC (C) in WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Data represent means ± SD. n = 8 mice/group. *P < 0.05 vs. other groups. Male and female mice were used in equal numbers in each experiment. WT, wild type.
Expressions of Genes Involved in the Cytosolic Energy Metabolism Are Unaltered in the Muscles of Mdx:sln+/− and Mdx:sln−/− Mice
Intracellular glucose homeostasis is mainly regulated by glucose anabolic (gluconeogenesis and glycogenolysis), catabolic, and storage (glycolysis and glycogenesis) pathways. We, therefore, next studied the expression levels of genes involved in glucose metabolism in dystrophic muscles. The RT-qPCR analyses showed significant downregulation of major glucoregulatory genes such as Glut4 and Fbp2 in the diaphragm of mdx mice. The expression levels of these genes were not restored in the diaphragm of mdx:sln+/− and mdx:sln−/− mice (Fig. 3A). The mRNA levels of Ppp1r3c were unaltered in the diaphragm of mdx mice, whereas its levels significantly decreased in the diaphragm of mdx:sln+/− and mdx:sln−/− mice compared with that of WT and mdx mice (Fig. 3A). In the pectorals, the mRNA levels of Glut4 were unaltered, whereas the mRNA levels of Fbp2 and Ppp1r3c were significantly downregulated in the pectorals of mdx, mdx:sln+/−, and mdx:sln−/− mice compared with that of WT controls. (Fig. 3B). There was no significant change in the expression levels of other muscle-specific glucoregulatory genes such as Pygm, Phkg1, Pgk1, Pgc1α, and Gpd1 in the diaphragm (Fig. 3C) and pectorals (Fig. 3D) of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Consistent with mRNA expression, the protein levels of Fbp2 were significantly low in both pectorals and diaphragm of mdx, mdx:sln+/−, and mdx:sln−/− mice (Fig. 3E). PDH, which catalyzes the conversion of pyruvate to acetyl-CoA, was not significantly altered in the diaphragm and pectorals of all four groups of mice studied (Fig. 3E and Supplemental Fig. S1, D and E). Taken together, these findings suggest that the cytosolic energy metabolism is not improved at least at the gene expression levels in both pectorals and diaphragm of mdx:sln+/− and mdx:sln−/− mice.
Figure 3.

Effect of SLN ablation on the expression of genes/proteins involved in glucose transport, gluconeogenesis, and glycogen metabolism. RT-qPCR data showing the mRNA levels of Glut4, Fbp2, and Ppp1r3c in the diaphragm (A) and pectorals (B). RT-qPCR data showing mRNA levels of Pygm, Phkg1, Pgk1, Pgc1α, and Gpd1 in the diaphragm (C) and pectorals (D). E: representative Western blots and quantitation showing the protein levels of Fbp2 and PDH in the diaphragm and pectorals of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Data represent means ± SD. n = 6 mice/group. **P < 0.001 vs. other groups, *P < 0.05 vs. WT and mdx mice. ns - not statistically significant. Male and female mice were used in equal numbers in each experiment. PDH, pyruvate dehydrogenase; RT-qPCR, real-time quantitative reverse transcription-PCR; SLN, sarcolipin; WT, wild type.
SLN Ablation Restores Mitochondrial Energy Metabolism in Both Diaphragm and Pectorals of Mdx Mice
Mitochondria play a vital role in energy metabolism, and therefore we next examined mitochondrial metabolism. Mitochondrial total OXPHOS Western blot shows that the complex I (CI) and complex IV (CIV) proteins were significantly decreased in both diaphragm (Fig. 4A) and pectorals (Fig. 4B) of mdx mice. The expression levels of these proteins were significantly increased in the diaphragm of mdx:sln+/− and mdx:sln−/− mice compared with that of mdx mice (Fig. 4A). The expression levels of CI and CIV proteins were also increased but not to significant levels in the pectorals of mdx:sln+/− and mdx:sln−/− mice (Fig. 4B). The expression level of complex II (CII) protein was decreased in the diaphragm of mdx, mdx:sln+/−, and mdx:sln−/− mice (Fig. 4A). The expression level of CII protein was decreased in the pectorals of mdx mice, whereas its level increased in the pectorals of mdx:sln+/− and mdx:sln−/− mice (Fig. 4B). Complex III (CIII) protein expression was unaltered in both diaphragm (Fig. 4A) and pectorals (Fig. 4B) of all four groups of mice. The complex V (CV) protein level was significantly increased in the diaphragm (Fig. 4A) but not in the pectorals (Fig. 4B) of mdx:sln+/− and mdx:sln−/− mice compared with that of mdx mice.
Figure 4.
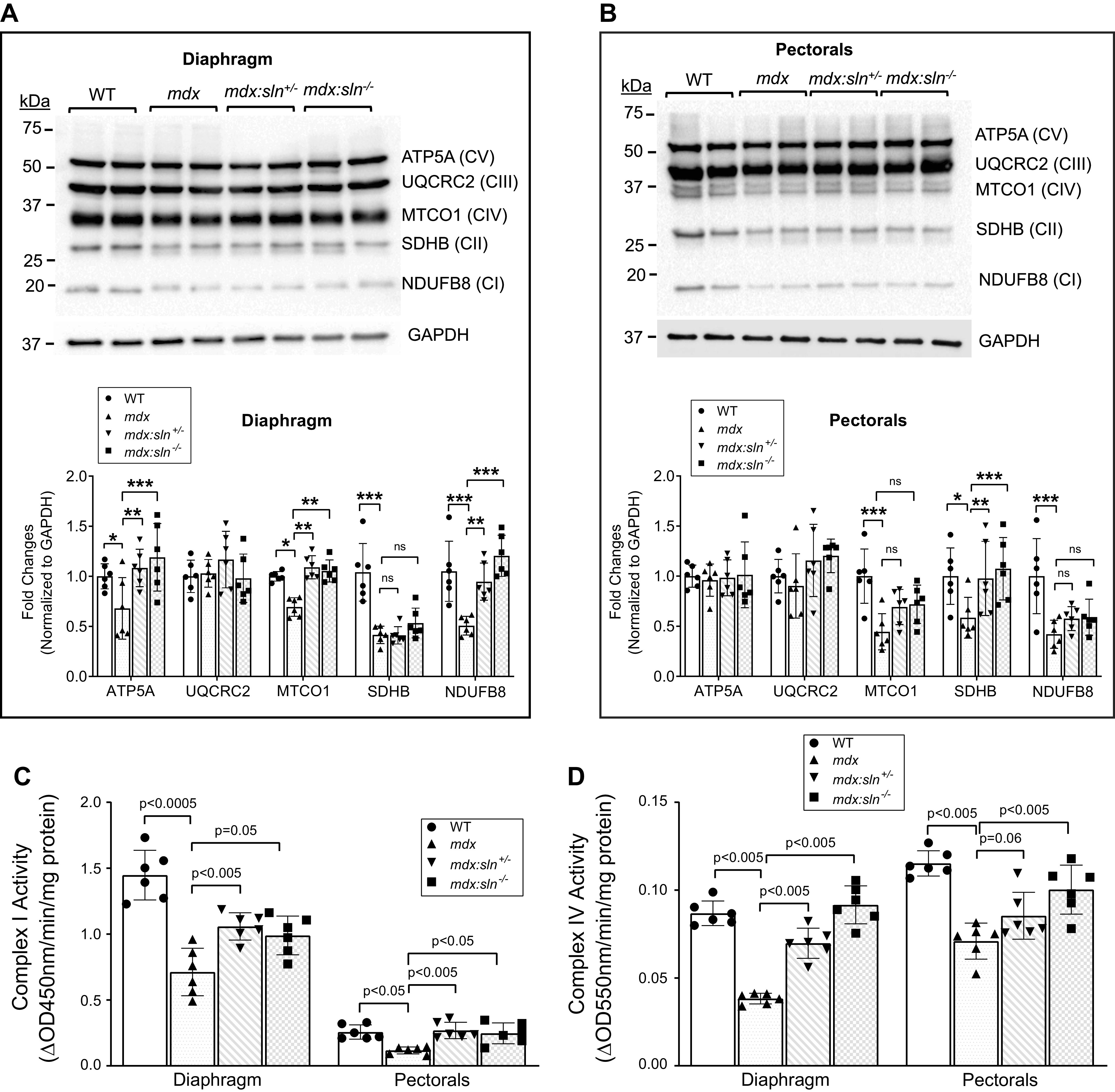

SLN ablation improved mitochondrial metabolism in mdx muscles. Representative OXPHOS Western blots and quantitation showing mitochondrial complex proteins in the diaphragm (A) and pectorals (B) of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Data represent means ± SD. n = 6 mice/group. *P < 0.05; **P < 0.005; ***P < 0.0001. ns - not statistically significant. Bar graphs representing mitochondrial complex I (C) and complex IV (D) activities in the diaphragm and pectorals of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. n = 6 mice/group. qPCR analysis showing mitochondrial copy number in the diaphragm (E) and pectorals (F) of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. P values are shown within the bar graphs. Data represent means ± SD. n = 6 mice/group. Graph showing the cumulative OCR in the isolated mitochondria from the diaphragm (G) and pectorals (H) of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice with Seahorse XFe 24. n = 4 mice/group. Data represent means ± SD. n = 4 mice/group. *P < 0.05. Male and female mice were used in equal numbers in each experiment. OCR, oxygen consumption rate; SLN, sarcolipin; WT, wild type.
We next determined the complex activities in freshly prepared tissue lysates. The mitochondrial CI (Fig. 4C) and CIV activities (Fig. 4D) were significantly decreased in both diaphragm and pectorals of mdx mice. The activities of CI and CIV were significantly increased in the diaphragm of both mdx:sln+/− and mdx:sln−/− mice (Fig. 4, C and D). CI activity was increased in the pectorals of both mdx:sln+/− and mdx:sln−/− mice (Fig. 4C). On the other hand, CIV activity was significantly increased only in the pectorals of mdx:sln−/− mice but not in the mdx:sln+/− mice (Fig. 4D).
We next determined the mitochondrial copy number by qPCR. The results show that there was a significant reduction in mitochondrial DNA copy number in both the diaphragm (Fig. 4E) and pectorals (Fig. 4F) of mdx mice when compared with that of WT mice. On the other hand, there was a significant increase in mitochondrial copy number in both diaphragm (Fig. 4E) and pectorals (Fig. 4F) of mdx:sln+/− and mdx:sln−/− mice when compared with that of mdx mice.
Mitochondrial function in intact mitochondria isolated from diaphragm and pectorals tissues was determined using a Seahorse XF24 flux analyzer. Mitochondrial OCR was significantly reduced in the mitochondria isolated from both diaphragm (Fig. 4G) and pectorals (Fig. 4H) of mdx mice compared with that of WT controls. On the other hand, the OCR was significantly increased in both diaphragm (Fig. 4G) and pectorals (Fig. 4H) of both mdx:sln+/− and mdx:sln−/− mice. The ECAR was significantly low in both diaphragm and pectorals of mdx muscles, whereas ECAR was increased in both diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice (Supplemental Fig. S2, A and B) The state III and state IIIµ respiration of mitochondria from the diaphragm (Supplemental Fig. S2C) and pectorals (Supplemental Fig. S2D) of mdx mice were significantly reduced compared with that of WT, mdx:sln+/− and mdx:sln−/− mice. The state IV respiration was similar in both diaphragm (Supplemental Fig. S2C) and pectorals (Supplemental Fig. S2D) of all four groups of mice. The RCR an indicator of mitochondrial health was low in both diaphragm (Supplemental Fig. S2E) and pectorals (Supplemental Fig. S2F) of mdx mice, whereas the RCR was increased, although not statistically significant, in both diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice.
SLN Ablation Improves Mitochondrial Ca2+ Cycling in the Mdx Pectorals
Ca2+ is the key modulator of mitochondrial function. We, therefore, studied whether SLN ablation has any effect on mitochondrial Ca2+ (Ca2+m) cycling in dystrophic muscles. Ca2+m uptake (Fig. 5A) and release (Fig. 5B) were unaffected in the diaphragm of mdx, mice compared with that of WT mice. In addition, reduction, or ablation of SLN expression did not affect the Ca2+m uptake (Fig. 5A) and release (Fig. 5B) in the dystrophic diaphragm. Ca2+m uptake was significantly increased (Fig. 5C) and Ca2+m release was significantly decreased (Fig. 5D) in the mdx pectorals, indicating impairment in Ca2+m cycling. In the pectorals of mdx:sln+/− and mdx:sln−/− mice, Ca2+m uptake and release were near to the levels of WT pectorals (Fig. 5, C and D).
Figure 5.

SLN ablation improves mitochondrial Ca2+ (Ca2+m) cycling in the pectorals of mdx mice. Ca2+m uptake (A) and Ca2+m release (B) are unaltered in the diaphragm of all four groups of mice. Ca2+m uptake (C) and release (D) are decreased in mdx pectorals, whereas Ca2+m uptake and release are restored in the pectorals of mdx:sln+/− and mdx:sln−/− mice. Data represent means ± SD. n = 6 mice/group. P values are shown within the bar graphs. ns - not statistically significant. Male and female mice were used in equal numbers in each experiment. SLN, sarcolipin.
SLN Ablation Prevents Oxidative Stress in Dystrophic Muscles
To determine the effect of SLN ablation on oxidative stress, we measured the oxidative modification of proteins as an indication of protein damage. Results showed that the protein carbonylation was significantly higher in the diaphragm of mdx mice, whereas it was significantly reduced in the diaphragm of mdx:sln+/− and mdx:sln−/− mice (Fig. 6A). The protein carbonylation in the pectorals of mdx, mdx:sln+/−, and mdx:sln−/− mice was significantly low compared with that of WT mice (Fig. 6B).
Figure 6.


Oxidative stress is reduced in muscles of mdx:sln+/− and mdx:sln−/− mice. Representative oxyblot and quantitation showing protein carbonyl content in the diaphragm (A) and pectorals (B) of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. Data represent means ± SD. n = 6 mice/group. Quantitation showing malondialdehyde (MDA) levels in the diaphragm (C) and pectorals (D) of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice. n = 6 mice/group. Data represent means ± SD. P values are shown within the bar graphs. E: representative Western blot and quantitation showing the protein levels of superoxide dismutase 2 (SOD2) and glutathione reductase (GR) in the diaphragm and pectorals of WT, mdx, mdx:sln+/−, and mdx:sln−/− mice (F and G). Data represent means ± SD. n = 6 mice/group. *P < 0.05 vs. other groups; **P < 0.005 vs. other groups. Male and female mice were used in equal numbers in each experiment. WT, wild type.
We next determined lipid peroxidation by measuring the MDA levels. The MDA levels were significantly high in the diaphragm (Fig. 6C) and pectorals (Fig. 6D) of mdx mice compared with that of WT mice, whereas its level was significantly reduced both in diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice.
The protein levels of major antioxidants, SOD2 (Fig. 6, E and F) and GR (Fig. 6, E and G), were significantly high in both diaphragm and pectorals of mdx, mdx:sln+/−, and mdx:sln−/− mice compared with that of WT mice.
DISCUSSION
The major findings of this study indicate that reduction or ablation of SLN expression can improve the rate of glucose utilization and prevent insulin resistance in mdx mice. Furthermore, reduction or ablation of SLN expression can improve the SR Ca2+ uptake, mitochondrial copy number, and metabolism, and reduce oxidative stress in dystrophic muscles of mdx mice. These changes could protect the dystrophic muscle from energy demand and ameliorate DMD.
Our findings demonstrate that fibrosis and necrosis were significantly reduced in the pectorals of mdx:sln+/− and mdx:sln−/− mice. Similarly, necrosis was significantly reduced in the diaphragm of mdx:sln+/− mice. In contrast, another study suggests that total loss of SLN may worsen the dystrophic phenotype of the diaphragm and soleus muscles of mdx mice possibly via downregulation of calcineurin and utrophin (44). However, we found the protein levels of calcineurin and utrophin were unaltered in the diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice compared with that of mdx controls. Furthermore, SLN ablation did not exacerbate necrosis or fibrosis in the diaphragm of mdx mice. We did not find reasons for these discrepancies in findings. However, in support of our data, another recent study found improved cytosolic Ca2+ homeostasis and amelioration of dystrophic muscle phenotype in mdx mice lacking SLN (46).
Glucose intolerance and insulin resistance are characteristics of DMD patients (47, 48). The mdx mice show glucose intolerance as reported before (11). The decreased glucose clearance and increased glycemic index after insulin injection in mdx mice suggest insulin resistance. In addition, blood glucose levels remained high throughout the experimental time points after glucose or insulin injection in mdx mice suggesting a possible defect in insulin receptor function and/or impairment in the glucose catabolic pathways. On the other hand, reduction, or ablation of SLN improved the efficacy of glucose clearance after ipGTT and insulin bolus and helps in maintaining the blood glucose levels. In mdx:sln+/− mice, the glucose clearance rate was improved near to that of WT mice. In mdx:sln−/− mice, glucose milieu was not significantly increased following an ipGTT and was maintained in a normal range throughout the experiment which could probably be due to compensatory insulin action. These findings also contradict the studies which show that in normal or high-fat administered mice, SLN overexpression increased ATP utilization, thereby attenuating the diet-induced obesity and glucose intolerance (49). It is also important to note that the rate of glucose clearance after glucose or insulin bolus was similar between WT and sln−/− mice (Supplemental Fig. S3) suggesting that SLN ablation in WT mice does not affect glucose homeostasis. Together our studies suggest that the mechanisms associated with SLN overexpression or ablation in healthy and dystrophic muscles are different. Furthermore, our studies suggest that the reduction/ablation of SLN expression can prevent glucose intolerance and insulin resistance in mdx mice.
In dystrophic muscles, impaired metabolism is associated with decreased expression of metabolic genes (50–52). Downregulation of genes involved in glycogen synthesis, glycolysis, and fatty acid β-oxidation has been reported in the mdx diaphragm (53). Consistent with these reports, in the mdx diaphragm, the expression levels of Glut4 and Fbp2 genes were significantly downregulated. However, reduction or total loss of SLN does not affect the expression levels of these genes in the mdx diaphragm. The mRNA levels of PPP1r3c, a protein phosphatase involved in glycogen metabolism was significantly decreased in the diaphragm of mdx:sln+/− and mdx:sln−/− mice. PPP1r3c activates glycogen synthase, reduces glycogen phosphorylase activity, and limits glycogen breakdown (54). Therefore, decreased expression of PPP1r3c could be a beneficial effect of SLN ablation in the dystrophic diaphragm and help to enhance glycogen breakdown and release glucose to compensate for the energy demand. Like the diaphragm, the expression levels of Fbp2 and Ppp1r3c were significantly decreased in the pectorals of mdx mice. Furthermore, reduction or total loss of SLN did not improve the expression of these genes in the pectorals of mdx mice. Taken together, our findings suggest that SLN ablation may not improve the cytosolic energy metabolism in dystrophic muscles.
An abnormal elevation of cytoplasmic Ca2+ is accepted as a major disease-causing mechanism in DMD (23, 26). To reduce cytoplasmic Ca2+ overload, mitochondria buffer some of this Ca2+, which results in mitochondrial Ca2+ overload and, subsequently, mitochondrial dysfunction and myofiber necrosis (40, 55). The findings presented here and in previous studies (32–34) have shown that reducing or eliminating SLN can improve SERCA function in dystrophic muscles. The enhanced SERCA activity is anticipated to reduce the cytosolic Ca2+ load and thereby normalize Ca2+m cycling. In support of this notion, the maximal Ca2+m uptake and release, which were significantly impaired in the pectorals of mdx mice, are restored in the pectorals of mdx:sln+/− and mdx:sln−/− mice. On the other hand, the maximal Ca2+m uptake and release are unaffected in the mdx diaphragm. Furthermore, both reduction and ablation of SLN expression do not affect the Ca2+m cycling in the dystrophic diaphragm. At present we do not have any possible explanation for the difference in the Ca2+m uptake and release properties between the dystrophic diaphragm and pectorals of mdx mice. Our future studies will focus on studying the mechanisms associated with Ca2+m uptake and release in these tissues.
It has been shown that transgenic overexpression of SLN in skeletal muscle induces mitochondrial biogenesis and energy metabolism (35). On the other hand, in dystrophin-deficient muscles, SLN expression is high (31), whereas mitochondrial function is defective (10). A recent study shows that skeletal muscle wasting in asthma is associated with decreased SERCA function, increased SLN expression, mitochondrial dysfunction, and oxidative stress (56). We, therefore, speculate that in dystrophic muscle, the increased levels of SLN can uncouple the SERCA pump and increase ATP utilization. This can further increase the energy demand and contributes to mitochondrial dysfunction and oxidative stress. Thus, in dystrophic muscles, reduction or ablation of SLN expression is anticipated to improve mitochondrial function. In support of this notion, reduction and/or ablation of SLN restored the complex proteins as well as their activities in both diaphragm and pectorals of mdx mice. It is also important to note that SLN ablation in normal muscle does not affect the expression levels of mitochondrial complex proteins (Supplemental Fig. S4A) as well as complex activities (Supplemental Fig. S4, B and C). In addition to the increase in mitochondrial copy number, Seahorse measurements in isolated mitochondria indicate that both reduction and ablation of SLN expression improved the mitochondrial metabolism in the dystrophic diaphragm and pectorals. Taken together, these findings imply that the mechanisms underlying SLN function in healthy and dystrophic muscles, which have yet to be identified, may differ. Our findings also suggest that reducing or ablating SLN expression in dystrophin-deficient muscles is beneficial in preventing abnormal Ca2+ cycling and promoting mitochondrial health and oxidative metabolism to meet their higher energy demand.
Mitochondrial Ca2+ overload can cause mitochondrial swelling, mitochondrial reactive oxygen species (ROS) production, and mPTP opening and contributes to oxidative stress in the dystrophin-deficient muscle (22, 29). ROS-induced oxidative modification to the protein leads to carbonylation, which is a reliable indicator of oxidative damage in response to the activation of stress pathways (57, 58). Myofibers with dystrophin deficiency were proven to be more susceptible to ROS-induced muscle injury (59, 60). Accordingly, increased lipid peroxidation, protein thiol oxidation, and protein carbonyl contents were reported in dystrophic muscles (61–64). Consistent with these reports, we found increased protein carbonyl content and lipid peroxidation in the mdx diaphragm. Lipid peroxidation was high in mdx pectorals, whereas protein carbonylation was significantly low in the pectorals of mdx mice. On the other hand, both in the diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice, protein carbonyl content and lipid peroxidation were significantly reduced indicating reduced oxidative stress. Increased expression of SOD2, catalase, and GR has been reported in the diaphragm and limb muscles of mdx mice (65, 66). Consistent with these reports, the protein levels of SOD2 and GR were significantly high in the diaphragm and pectorals of mdx mice. These levels remained high in the diaphragm and pectorals of mdx:sln+/− and mdx:sln−/− mice. It has been shown that overexpression of SOD2 can partially protect mitochondrial function in the rat diaphragm (67). Thus, the increased expression of SOD2 and GR could partly protect the mitochondrial function in dystrophic muscles. Together these findings indicate that reduction or ablation of SLN expression can reduce the oxidative stress in dystrophic muscles. Recently using RNA-Seq analysis, we have shown that reduction in SLN expression is associated with the activation of several cardioprotective pathways in mdx mice (33). Therefore, further studies are warranted to identify similar beneficial signaling pathways that could be activated in skeletal muscles of mdx:sln+/− and mdx:sln−/− mice.
Diaphragm, severely affected muscle tissue in mdx mice (43) has relatively high SLN expression compared with other muscle tissues. Based on the current findings and our previous studies on mdx:utr−/− mice (32), the total loss of SLN does not ameliorate diaphragm muscle pathology in mouse models of DMD. On the other hand, SLN ablation in the mdx diaphragm is associated with improved SERCA function, mitochondrial complex activities, and mitochondrial metabolism. The diaphragm contains a mixed composition of fast- and slow-fibers and fibrous tissues. We have previously shown that reducing SLN expression can prevent the fiber-type switch in fast-twitch muscles like pectorals and quadriceps of mdx:utr−/− mice (32). However, we do not know the relative proportions of different fiber types in the dystrophic diaphragm or whether SLN ablation affects the fiber types. Further study into these aspects may explain the lack of improvements in the diaphragm pathology in mdx:sln−/− mice. Although SLN ablation did not reduce muscle pathology in tissues like the diaphragm, the total loss of SLN is not detrimental to the life of mdx mice. In support of this notion, our recent studies demonstrate that SLN ablation can prevent the development of cardiomyopathy and improve cardiac function in aged mdx mice (33).
In conclusion, our findings suggest that reduction or ablation of SLN expression can improve the SR Ca2+ uptake, mitochondrial energy metabolism in dystrophic muscles and help to maintain glucose homeostasis in mdx mice. These changes could reduce oxidative stress and contribute to the amelioration of muscular dystrophy in mdx mice.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.14336660.
GRANTS
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, US National Institutes of Health (NIH) Grant (AR069107 to G.J.B.) and the American Heart Association (Founders Affiliates) Grant in aid (16GRNT30960034 to G.J.B.).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.B. and G.J.B. conceived and designed research; R.B., S.M., and G.J.B. performed experiments; R.B., S.M., and G.J.B. analyzed data; R.B., S.M., and G.J.B. interpreted results of experiments; R.B., S.M., and G.J.B. prepared figures; R.B., S.M., and G.J.B. drafted manuscript; R.B., S.M., and G.J.B. edited and revised manuscript; R.B., S.M., and G.J.B. approved final version of manuscript.
REFERENCES
- 1.Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers 7: 13, 2021. doi: 10.1038/s41572-021-00248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90: 3710–3714, 1993. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emery AE. The muscular dystrophies. Lancet 359: 687–695, 2002. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 4.Strakova J, Kamdar F, Kulhanek D, Razzoli M, Garry DJ, Ervasti JM, Bartolomucci A, Townsend D. Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci Rep 8: 13624, 2018. doi: 10.1038/s41598-018-31753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deconinck N, Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol 36: 1–7, 2007. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 6.Rybalka E, Timpani CA, Cooke MB, Williams AD, Hayes A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS One 9: e115763, 2014. doi: 10.1371/journal.pone.0115763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuznetsov AV, Winkler K, Wiedemann FR, von Bossanyi P, Dietzmann K, Kunz WS. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol Cell Biochem 183: 87–96, 1998. doi: 10.1023/a:1006868130002. [DOI] [PubMed] [Google Scholar]
- 8.Timpani CA, Hayes A, Rybalka E. Revisiting the dystrophin-ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med Hypotheses 85: 1021–1033, 2015. doi: 10.1016/j.mehy.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Pant M, Sopariwala DH, Bal NC, Lowe J, Delfin DA, Rafael-Fortney J, Periasamy M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of Duchenne muscular dystrophy. PLoS One 10: e0123875, 2015. doi: 10.1371/journal.pone.0123875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore TM, Lin AJ, Strumwasser AR, Cory K, Whitney K, Ho T, Ho T, Lee JL, Rucker DH, Nguyen CQ, Yackly A, Mahata SK, Wanagat J, Stiles L, Turcotte LP, Crosbie RH, Zhou Z. Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front Physiol 11: 690, 2020. doi: 10.3389/fphys.2020.00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stapleton DI, Lau X, Flores M, Trieu J, Gehrig SM, Chee A, Naim T, Lynch GS, Koopman R. Dysfunctional muscle and liver glycogen metabolism in mdx dystrophic mice. PLoS One 9: e91514, 2014. doi: 10.1371/journal.pone.0091514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saini-Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: thematic review series: genetics of human lipid diseases. J Lipid Res 53: 4–27, 2012. doi: 10.1194/jlr.R012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camiña F, Novo-Rodriguez MI, Rodriguez-Segade S, Castro-Gago M. Purine and carnitine metabolism in muscle of patients with Duchenne muscular dystrophy. Clin Chim Acta 243: 151–164, 1995. [Erratum in Clin Chim Acta 252: 105, 1996]. doi: 10.1016/0009-8981(95)06164-9. [DOI] [PubMed] [Google Scholar]
- 14.van Bennekom CA, Oerlemans FT, Kulakowski S, De Bruyn CH. Enzymes of purine metabolism in muscle specimens from patients with Duchenne-type muscular dystrophy. Adv Exp Med Biol 165: 447–450, 1984. doi: 10.1007/978-1-4757-0390-0_85. [DOI] [PubMed] [Google Scholar]
- 15.Chinet AE, Even PC, Decrouy A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 50: 602–605, 1994. doi: 10.1007/BF01921731. [DOI] [PubMed] [Google Scholar]
- 16.Glesby MJ, Rosenmann E, Nylen EG, Wrogemann K. Serum CK, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle Nerve 11: 852–856, 1988. doi: 10.1002/mus.880110809. [DOI] [PubMed] [Google Scholar]
- 17.Gardan-Salmon D, Dixon JM, Lonergan SM, Selsby JT. Proteomic assessment of the acute phase of dystrophin deficiency in mdx mice. Eur J Appl Physiol 111: 2763–2773, 2011. doi: 10.1007/s00421-011-1906-3. [DOI] [PubMed] [Google Scholar]
- 18.Timpani CA, Goodman CA, Stathis CG, White JD, Mamchaoui K, Butler-Browne G, Gueven N, Hayes A, Rybalka E. Adenylosuccinic acid therapy ameliorates murine Duchenne muscular dystrophy. Sci Rep 10: 1125, 2020. doi: 10.1038/s41598-020-57610-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capitanio D, Moriggi M, Torretta E, Barbacini P, De Palma S, Viganò A, Lochmuller H, Muntoni F, Ferlini A, Mora M, Gelfi C. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: changes contributing to preserve muscle function in Becker muscular dystrophy patients. J Cachexia Sarcopenia Muscle 11: 547–563, 2020. doi: 10.1002/jcsm.12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagur R, Hajnóczky G. Intracellular Ca2+ sensing: its role in calcium homeostasis and signaling. Mol Cell 66: 780–788, 2017. doi: 10.1016/j.molcel.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tammineni ER, Kraeva N, Figueroa L, Manno C, Ibarra CA, Klip A, Riazi S, Rios E. Intracellular calcium leak lowers glucose storage in human muscle, promoting hyperglycemia and diabetes. Elife 9: e53999, 2020. doi: 10.7554/eLife.53999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burr AR, Molkentin JD. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ 22: 1402–1412, 2015. doi: 10.1038/cdd.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law ML, Cohen H, Martin AA, Angulski ABB, Metzger JM. Dysregulation of calcium handling in Duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J Clin Med 9: 520, 2020. doi: 10.3390/jcm9020520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Westering TL, Betts CA, Wood MJ. Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne muscular dystrophy. Molecules 20: 8823–8855, 2015. doi: 10.3390/molecules20058823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin J, Tajrishi MM, Ogura Y, Kumar A. Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol 45: 2266–2279, 2013. doi: 10.1016/j.biocel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mareedu S, Million ED, Duan D, Babu GJ. Abnormal calcium handling in Duchenne muscular dystrophy: mechanisms and potential therapies. Front Physiol 12: 647010, 2021. doi: 10.3389/fphys.2021.647010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robert V, Massimino ML, Tosello V, Marsault R, Cantini M, Sorrentino V, Pozzan T. Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol Chem 276: 4647–4651, 2001. doi: 10.1074/jbc.M006337200. [DOI] [PubMed] [Google Scholar]
- 28.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch 458: 915–928, 2009. doi: 10.1007/s00424-009-0670-2. [DOI] [PubMed] [Google Scholar]
- 29.Kyrychenko V, Poláková E, Janíček R, Shirokova N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 58: 186–195, 2015. doi: 10.1016/j.ceca.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nogami K, Maruyama Y, Sakai-Takemura F, Motohashi N, Elhussieny A, Imamura M, Miyashita S, Ogawa M, Noguchi S, Tamura Y, Kira JI, Aoki Y, Takeda S, Miyagoe-Suzuki Y. Pharmacological activation of SERCA ameliorates dystrophic phenotypes in dystrophin-deficient mdx mice. Hum Mol Genet 30: 1006–1019, 2021. doi: 10.1093/hmg/ddab100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider JS, Shanmugam M, Gonzalez JP, Lopez H, Gordan R, Fraidenraich D, Babu GJ. Increased sarcolipin expression and decreased sarco(endo)plasmic reticulum Ca2+ uptake in skeletal muscles of mouse models of Duchenne muscular dystrophy. J Muscle Res Cell Motil 34: 349–356, 2013. doi: 10.1007/s10974-013-9350-0. [DOI] [PubMed] [Google Scholar]
- 32.Voit A, Patel V, Pachon R, Shah V, Bakhutma M, Kohlbrenner E, McArdle JJ, Dell'Italia LJ, Mendell JR, Xie LH, Hajjar RJ, Duan D, Fraidenraich D, Babu GJ. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat Commun 8: 1068, 2017. doi: 10.1038/s41467-017-01146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mareedu S, Pachon R, Thilagavathi J, Fefelova N, Balakrishnan R, Niranjan N, Xie LH, Babu GJ. Sarcolipin haploinsufficiency prevents dystrophic cardiomyopathy in mdx mice. Am J Physiol Heart Circ Physiol 320: H200–H210, 2021. doi: 10.1152/ajpheart.00601.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niranjan N, Mareedu S, Tian Y, Kodippili K, Fefelova N, Voit A, Xie LH, Duan D, Babu GJ. Sarcolipin overexpression impairs myogenic differentiation in Duchenne muscular dystrophy. Am J Physiol Cell Physiol 317: C813–C824, 2019. doi: 10.1152/ajpcell.00146.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maurya SK, Herrera JL, Sahoo SK, Reis FCG, Vega RB, Kelly DP, Periasamy M. Sarcolipin signaling promotes mitochondrial biogenesis and oxidative metabolism in skeletal muscle. Cell Rep 24: 2919–2931, 2018. doi: 10.1016/j.celrep.2018.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sopariwala DH, Pant M, Shaikh SA, Goonasekera SA, Molkentin JD, Weisleder N, Ma J, Pan Z, Periasamy M. Sarcolipin overexpression improves muscle energetics and reduces fatigue. J Appl Physiol (1985) 118: 1050–1058, 2015. doi: 10.1152/japplphysiol.01066.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rekha B, Velmurugan G, Freddy AJ, Anusha S, Ramprasath T, Karthik KV, Suresh S, Kulshrestha P, Mithieux G, Lyon AR, Selvam GS, Ramasamy S. Chronic intake of 4-methylimidazole induces hyperinsulinemia and hypoglycaemia via pancreatic β cell hyperplasia and glucose dyshomeostasis. Sci Rep 8: 17037, 2018. doi: 10.1038/s41598-018-35071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malik AN, Czajka A, Cunningham P. Accurate quantification of mouse mitochondrial DNA without co-amplification of nuclear mitochondrial insertion sequences. Mitochondrion 29: 59–64, 2016. doi: 10.1016/j.mito.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 39.Babu GJ, Bhupathy P, Carnes CA, Billman GE, Periasamy M. Differential expression of sarcolipin protein during muscle development and cardiac pathophysiology. J Mol Cell Cardiol 43: 215–222, 2007. doi: 10.1016/j.yjmcc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dubinin MV, Talanov EY, Tenkov KS, Starinets VS, Mikheeva IB, Sharapov MG, Belosludtsev KN. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim Biophys Acta Mol Basis Dis 1866: 165674, 2020. doi: 10.1016/j.bbadis.2020.165674. [DOI] [PubMed] [Google Scholar]
- 41.Iuso A, Repp B, Biagosch C, Terrile C, Prokisch H. Assessing mitochondrial bioenergetics in isolated mitochondria from various mouse tissues using seahorse XF96 analyzer. Methods Mol Biol 1567: 217–230, 2017. doi: 10.1007/978-1-4939-6824-4_13. [DOI] [PubMed] [Google Scholar]
- 42.Sakamuri SSVP, Sperling JA, Sure VN, Dholakia MH, Peterson NR, Rutkai I, Mahalingam PS, Satou R, Katakam PVG. Measurement of respiratory function in isolated cardiac mitochondria using seahorse XFe24 analyzer: applications for aging research. Geroscience 40: 347–356, 2018. doi: 10.1007/s11357-018-0021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352: 536–539, 1991. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 44.Fajardo VA, Chambers PJ, Juracic ES, Rietze BA, Gamu D, Bellissimo C, Kwon F, Quadrilatero J, Russell Tupling A. Sarcolipin deletion in mdx mice impairs calcineurin signalling and worsens dystrophic pathology. Hum Mol Genet 27: 4094–4102, 2018. doi: 10.1093/hmg/ddy302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saure C, Caminiti C, Weglinski J, de Castro Perez F, Monges S. Energy expenditure, body composition, and prevalence of metabolic disorders in patients with Duchenne muscular dystrophy. Diabetes Metab Syndr 12: 81–85, 2018. doi: 10.1016/j.dsx.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Tanihata J, Nagata T, Ito N, Saito T, Nakamura A, Minamisawa S, Aoki Y, Ruegg UT, Takeda S. Truncated dystrophin ameliorates the dystrophic phenotype of mdx mice by reducing sarcolipin-mediated SERCA inhibition. Biochem Biophys Res Commun 505: 51–59, 2018. doi: 10.1016/j.bbrc.2018.09.039. [DOI] [PubMed] [Google Scholar]
- 47.Rodríguez-Cruz M, Sanchez R, Escobar RE, Cruz-Guzmán Odel R, López-Alarcón M, Bernabe García M, Coral-Vázquez R, Matute G, Velázquez Wong AC. Evidence of insulin resistance and other metabolic alterations in boys with Duchenne or Becker muscular dystrophy. Int J Endocrinol 2015: 867273, 2015. doi: 10.1155/2015/867273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bostock EL, Edwards BT, Jacques MF, Pogson JTS, Reeves ND, Onambele-Pearson GL, Morse CI. Impaired glucose tolerance in adults with Duchenne and Becker muscular dystrophy. Nutrients 10: 1947, 2018. doi: 10.3390/nu10121947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pant M, Bal NC, Periasamy M. Sarcolipin: a key thermogenic and metabolic regulator in skeletal muscle. Trends Endocrinol Metab 27: 881–892, 2016. doi: 10.1016/j.tem.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nghiem PP, Bello L, Stoughton WB, López SM, Vidal AH, Hernandez BV, Hulbert KN, Gourley TR, Bettis AK, Balog-Alvarez CJ, Heath-Barnett H, Kornegay JN. Changes in muscle metabolism are associated with phenotypic variability in golden retriever muscular dystrophy. Yale J Biol Med 90: 351–360, 2017. [PMC free article] [PubMed] [Google Scholar]
- 51.Santacatterina F, Chamorro M, de Arenas CN, Navarro C, Martín MA, Cuezva JM, Sánchez-Aragó M. Quantitative analysis of proteins of metabolism by reverse phase protein microarrays identifies potential biomarkers of rare neuromuscular diseases. J Transl Med 13: 65, 2015. doi: 10.1186/s12967-015-0424-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chi MM, Hintz CS, McKee D, Felder S, Grant N, Kaiser KK, Lowry OH. Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 36: 761–767, 1987. doi: 10.1016/0026-0495(87)90113-2. [DOI] [PubMed] [Google Scholar]
- 53.Porter JD, Merriam AP, Leahy P, Gong B, Feuerman J, Cheng G, Khanna S. Temporal gene expression profiling of dystrophin-deficient (mdx) mouse diaphragm identifies conserved and muscle group-specific mechanisms in the pathogenesis of muscular dystrophy. Hum Mol Genet 13: 257–269, 2004. doi: 10.1093/hmg/ddh033. [DOI] [PubMed] [Google Scholar]
- 54.Agius L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med 46: 34–45, 2015. doi: 10.1016/j.mam.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 55.Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50: 222–233, 2011. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 56.Qaisar R, Qayum M, Muhammad T. Reduced sarcoplasmic reticulum Ca2+ ATPase activity underlies skeletal muscle wasting in asthma. Life Sci 273: 119296, 2021. doi: 10.1016/j.lfs.2021.119296. [DOI] [PubMed] [Google Scholar]
- 57.Petrillo S, Pelosi L, Piemonte F, Travaglini L, Forcina L, Catteruccia M, Petrini S, Verardo M, D'Amico A, Musarò A, Bertini E. Oxidative stress in Duchenne muscular dystrophy: focus on the NRF2 redox pathway. Hum Mol Genet 26: 2781–2790, 2017. doi: 10.1093/hmg/ddx173. [DOI] [PubMed] [Google Scholar]
- 58.Grounds MD, Terrill JR, Al-Mshhdani BA, Duong MN, Radley-Crabb HG, Arthur PG. Biomarkers for Duchenne muscular dystrophy: myonecrosis, inflammation and oxidative stress. Dis Model Mech 13: dmm043638, 2020. doi: 10.1242/dmm.043638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Disatnik MH, Chamberlain JS, Rando TA. Dystrophin mutations predict cellular susceptibility to oxidative stress. Muscle Nerve 23: 784–792, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 60.Dudley RW, Danialou G, Govindaraju K, Lands L, Eidelman DE, Petrof BJ. Sarcolemmal damage in dystrophin deficiency is modulated by synergistic interactions between mechanical and oxidative/nitrosative stresses. Am J Pathol 168: 1276–1287, 2006. doi: 10.2353/ajpath.2006.050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.El-Shafey AF, Armstrong AE, Terrill JR, Grounds MD, Arthur PG. Screening for increased protein thiol oxidation in oxidatively stressed muscle tissue. Free Radic Res 45: 991–999, 2011. doi: 10.3109/10715762.2011.590136. [DOI] [PubMed] [Google Scholar]
- 62.Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, Reggiani C, Di Lisa F, Bonaldo P, Canton M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum Mol Genet 19: 4207–4215, 2010. doi: 10.1093/hmg/ddq339. [DOI] [PubMed] [Google Scholar]
- 63.Messina S, Altavilla D, Aguennouz M, Seminara P, Minutoli L, Monici MC, Bitto A, Mazzeo A, Marini H, Squadrito F, Vita G. Lipid peroxidation inhibition blunts nuclear factor-κB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol 168: 918–926, 2006. doi: 10.2353/ajpath.2006.050673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spassov A, Gredes T, Gedrange T, Pavlovic D, Lupp A, Kunert-Keil C. Increased oxidative stress in dystrophin deficient (mdx) mice masticatory muscles. Exp Toxicol Pathol 63: 549–552, 2011. doi: 10.1016/j.etp.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 65.Kaczor JJ, Hall JE, Payne E, Tarnopolsky MA. Low intensity training decreases markers of oxidative stress in skeletal muscle of mdx mice. Free Radic Biol Med 43: 145–154, 2007. doi: 10.1016/j.freeradbiomed.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 66.Hermes TA, Mizobuti DS, da Rocha GL, da Silva HNM, Covatti C, Pereira ECL, Ferretti R, Minatel E. Tempol improves redox status in mdx dystrophic diaphragm muscle. Int J Exp Pathol 101: 289–297, 2020. doi: 10.1111/iep.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morton AB, Smuder AJ, Wiggs MP, Hall SE, Ahn B, Hinkley JM, Ichinoseki-Sekine N, Huertas AM, Ozdemir M, Yoshihara T, Wawrzyniak NR, Powers SK. Increased SOD2 in the diaphragm contributes to exercise-induced protection against ventilator-induced diaphragm dysfunction. Redox Biol 20: 402–413, 2019. doi: 10.1016/j.redox.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.14336660.