

Keywords: alcohol, chronic pancreatitis, deer mice, ER/oxidative stress, fatty acid ethyl esters
Abstract
Alcoholic chronic pancreatitis (ACP) is a fibroinflammatory disease of the pancreas. However, metabolic basis of ACP is not clearly understood. In this study, we evaluated differential pancreatic injury in hepatic alcohol dehydrogenase-deficient (ADH−) deer mice fed chronic ethanol (EtOH), chronic plus binge EtOH, and chronic plus binge EtOH and fatty acid ethyl esters (FAEEs, nonoxidative metabolites of EtOH) to understand the metabolic basis of ACP. Hepatic ADH− and ADH normal (ADH+) deer mice were fed Lieber-DeCarli liquid diet containing 3% (wt/vol) EtOH for 3 mo. One week before the euthanization, chronic EtOH-fed mice were further administered with an oral gavage of binge EtOH with/without FAEEs. Blood alcohol concentration (BAC), pancreatic injury, and inflammatory markers were measured. Pancreatic morphology, ultrastructural changes, and endoplasmic reticulum (ER)/oxidative stress were examined using H&E staining, electron microscopy, immunostaining, and/or Western blot, respectively. Overall, BAC was substantially increased in chronic EtOH-fed groups of ADH− versus ADH+ deer mice. A significant change in pancreatic acinar cell morphology, with mild to moderate fibrosis and ultrastructural changes evident by dilatations and disruption of ER cisternae, ER/oxidative stress along with increased levels of inflammatory markers were observed in the pancreas of chronic EtOH-fed groups of ADH− versus ADH+ deer mice. Furthermore, chronic plus binge EtOH and FAEEs exposure elevated BAC, enhanced ER/oxidative stress, and exacerbated chronic EtOH-induced pancreatic injury in ADH− deer mice suggesting a role of increased body burden of EtOH and its metabolism under reduced hepatic ADH in initiation and progression of ACP.
NEW & NOTEWORTHY We established a chronic EtOH feeding model of hepatic alcohol dehydrogenase-deficient (ADH−) deer mice, which mimics several fibroinflammatory features of human alcoholic chronic pancreatitis (ACP). The fibroinflammatory and morphological features exacerbated by chronic plus binge EtOH and FAEEs exposure provide a strong case for metabolic basis of ACP. Most importantly, several pathological and molecular targets identified in this study provide a much broader understanding of the mechanism and avenues to develop therapeutics for ACP.
INTRODUCTION
Chronic alcohol consumption is known to be the major cause of chronic pancreatitis (CP) and hospitalization for gastrointestinal disorders in the United States. The alcoholic chronic pancreatitis (ACP) characterized as a progressive inflammatory disease leads to irreversible destruction of the pancreatic gland in alcoholics (1). The most common pathologies reported in patients with a diagnosis of ACP are pancreatic necrotic cell death, inflammation, and fibrosis leading to progressive functional and structural loss of the parenchymal tissue. Several pathogenic and mechanistic hypotheses for the pathogenesis of ACP have been proposed (2–6), but the mechanism(s) linking chronic alcohol consumption to CP are complex and not well defined. The risk of developing pancreatic injury/pancreatitis appears to be directly proportional to the amount and duration of alcohol intake (5). Therefore, an increased body burden and metabolism of alcohol (ethanol, EtOH) itself can play a crucial role in the etiopathogenesis of ACP.
About 90% of ingested EtOH is predominately metabolized to acetaldehyde (a reactive and cytotoxic metabolite) in the liver primarily by two oxidative pathways involving cytoplasmic enzyme alcohol dehydrogenase 1 (ADH1) and microsomal cytochrome P450 2E1 (CYP2E1). Acetaldehyde is further metabolized to acetate by mitochondrial aldehyde dehydrogenase at the rate it is formed. The CYP2E1 catalyzed pathway of EtOH metabolism can generate reactive oxygen species (ROS) in addition to the formation of acetaldehyde. The chronic EtOH exposure in experimental animals and a long-term alcohol consumption in human subjects reduce hepatic ADH activity, with concomitant increase in hepatic CYP2E1 activity (7–12). Generally, oxidative stress has been suggested to be associated with the oxidative metabolism of EtOH (11).
A relatively lower expression of ADH as well as CYP2E1 in the pancreas than that in the liver suggests negligible capacity of the pancreas to metabolize EtOH via oxidative metabolism (13–15). Despite this fact, acetaldehyde can mediate some detrimental effects in pancreatic acinar cells (16, 17). However, nonoxidative metabolism of EtOH via esterification with endogenous fatty acids to fatty acid ethyl esters (FAEEs) catalyzed by FAEE synthase is a predominant pathway for EtOH disposition in the pancreas during chronic EtOH consumption (2, 15, 18). The expression of FAEE synthase is reported to be much higher in the pancreas than other vital organs and is remarkably induced upon EtOH exposure (18–21). Furthermore, we have reported a concentration-dependent formation of FAEEs with a significantly increased expression of carboxyl ester lipase (CEL, a key enzyme involved FAEE’s synthesis) in human pancreatic acinar cells treated with EtOH (22). Besides, a reduced expression of hepatic ADH substantially facilitates the increased formation of FAEEs in the pancreas (23). Similar findings were reported from our laboratory using the hepatic ADH− deficient (ADH−) deer mice, which mimic reduced hepatic ADH levels commonly found in chronic alcoholic subjects (21, 24, 25). Various toxic effects and inflammatory responses of FAEEs have been reported in the pancreas and pancreatic acinar cells (2, 17, 19, 23, 26).
Previously, we have demonstrated a dose and time-dependent metabolic effects of EtOH on pancreatic injury and associated changes in ER stress using 1-yr-old ADH− deer mice (21, 24, 25). In our continued efforts to develop an animal model that reproduce remarkable pancreatic fibroinflammatory responses reported in chronic alcoholic subjects with a diagnosis of ACP, we assessed chronic EtOH-induced pancreatic injury using 6-mo-old ADH− deer mice in this study. Furthermore, in addition to chronic EtOH feeding, a single oral dose (gavage) of binge EtOH with/without FAEEs was administered to evaluate differential pancreatic injury for a better understanding of metabolic basis of ACP and role of endogenously formed FAEEs (nonoxidative EtOH metabolites synthesized during chronic alcohol abuse) in etiopathogenesis of ACP.
MATERIALS AND METHODS
Antibodies and Reagents
The primary antibodies for glucose-regulated protein 78 (GRP78; 78 kDa; Cat. No. 3177), eukaryotic translation initiation factor 2 A (eIF2α; 38 kDa; Cat. No. 5324), phosphorylated (p)-eIF2α (Ser 51; 38 kDa; Cat. No. 3398), and β-actin (45 kDa; Cat. No. 4970) were purchased from Cell Signaling Technology (Danvers, MA).
Antibodies to spliced X-box binding protein (sXBP1; 40 kDa; Cat. No. 37152) and unspliced XBP1 (uXBP1; 29 kDa; Cat. No. 37152) were purchased from Abcam Inc. (Cambridge, MA). Antibodies to inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α; 110 kDa; Cat. No. NB100-2324), p-IRE1α (Ser 724) (110 kDa; Cat. No. NB100-232), activating transcription factor 6 (ATF6; 88 kDa; Cat. No. IMG273), collagen I (139 kDa; Cat. No. NB600-408), and collagen III (138 kDa; Cat. No. NB600-594) were from Novus Biologicals (Littleton, CO). Antibodies to cathepsin B (37 kDa procathepsin B, 25 kDa activated cathepsin-B; Cat. No. sc-365558), protein kinase RNA-like endoplasmic reticulum kinase (PERK; 150 kDa; Cat. No. 100–401-962), and CHOP (31 kDa; Cat. No. MA1-250) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX), Rockland (Limerick, PA) and Thermo Fisher Scientific (Houston, TX), respectively.
The chemicals, reagents, and HPLC and GC grade solvents used in this study were obtained from Sigma Aldrich (St. Louis, MO) and/or Thermo Fisher Scientific (Houston, TX).
Animal Experiments
Hepatic ADH1-negative phenotype (ADH−) deer mice, a natural genetic variant of Peromyscus maniculatus, and ADH1-positive (ADH+) deer mice (male, ∼6 mo old, ∼ 20 g body wt), obtained from Peromyscus Genetic Stock Center, University of South Carolina, Columbia, SC, were housed in the University of Texas Medical Branch (UTMB) Animal Resource Center. All the animal experiments were approved and conducted as per animal care protocols instituted by UTMB’s Institutional Animal Care and Use Committee.
The details of animal grouping and liquid diet (control and EtOH) feeding protocol are illustrated in Fig. 1. In brief, both the mice’s strains were randomly divided into control (n = 10) and experimental (n = 30) groups. The experimental groups were fed Lieber-DeCarli liquid diet (Cat. No. 710260, Dyets Inc., Bethlehem, PA) for a week, followed by a gradual increase in the EtOH concentration from 0.5% to 3.0% (wt/vol) in the liquid diet in 2-wk duration, followed by 3% EtOH in the liquid diet for additional 3 mo. In addition to chronic EtOH feeding, single binge EtOH [3 g/kg body weight (BW)]/combination of single binge EtOH (3 g/kg BW) and FAEEs mixture (100 mg/kg BW) was administered to chronic EtOH-fed groups of both strains of mice by oral gavage, a week before euthanization, sufficient time to develop chronic pancreatic pathology (27), (Fig. 1). Age-adjusted daily dose of 3% EtOH (wt/vol) in the liquid diet was found to be an optimal chronic tolerable dose for 6-mo-old ADH− deer mice in our preliminary studies. The diet was prepared fresh daily and provided in the late afternoon as mice consume most of their food during the dark phase (28). Each day, the amount of liquid diet consumed was noted. The calorie derived from these liquid diets was 1 kcal/mL. The mixture of FAEEs was prepared in the ratio 1:2.2:4.7:2 (wt/wt) of ethyl palmitate (Cat. No. P9009), ethyl oleate (Cat. No. 268011), ethyl linoleate (Cat. No. L1751), and ethyl arachidonate (Cat. No. A9135) respectively, purchased from Sigma-Aldrich, St. Louis, MO. The ratio of FAEEs used in this study was similar to that detected in the plasma of alcoholic subjects (29). Individual FAEEs were dissolved in EtOH (200 proof), used as a vehicle, and mixed in the ratio of 1:2.2:4.7:2 (wt/wt) to prepare a composite mixture of FAEEs. The contents of EtOH and FAEEs were adjusted to provide 20% EtOH (wt/vol, equivalent to EtOH-binge dose of 3 g/kg BW) and 100 mg/kg BW, respectively.
Figure 1.
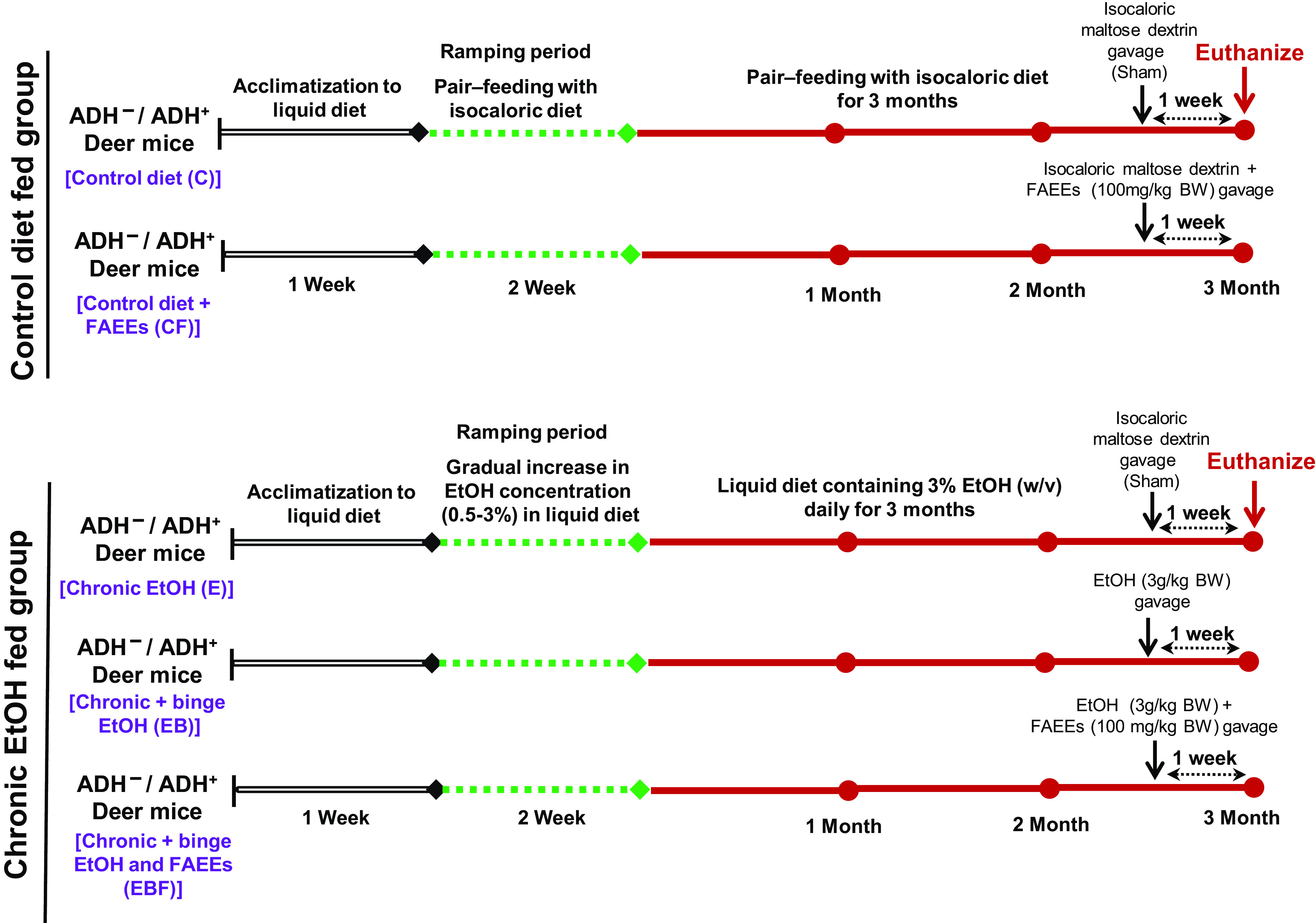
Experimental approach to determine chronic EtOH-induced pancreatic injury in ADH− and ADH+ deer mice. After initial acclimatization with liquid diet for a week, EtOH concentration was gradually increased from 0.5% to 3.0% (wt/vol) in 2 wk followed by 3.0% EtOH in the liquid diet daily for 3 mo (chronic EtOH). The chronic EtOH-fed group was divided into three groups: chronic EtOH, chronic plus binge EtOH (3.0 g/kg BW EtOH by gavage), and chronic plus binge EtOH and FAEEs (100 mg/kg BW by gavage), respectively, 1 wk before euthanizing the animals. Mice pair-fed with the liquid diet containing equivalent calories of EtOH substituted by maltose-dextrin with or without FAEEs (100 mg/kg BW by gavage) were used as controls. Binge EtOH or FAEEs dissolved in EtOH (equivalent to binge EtOH dose of 3.0 g/kg BW) were administered 1 wk before euthanizing the animals. ADH−, alcohol dehydrogenase-deficient; FAEEs, fatty acid ethyl esters.
Controls for both the strains were pair-fed with a liquid diet containing EtOH equivalent calories substituted by maltose-dextrin for 3 mo (Fig. 1). Similar to chronic EtOH-fed groups, the control groups of mice were either administered with isocaloric maltose-dextrin or a combination of isocaloric maltose-dextrin and FAEEs mix (100 mg/kg BW), by oral gavage, a week before euthanization, respectively (Fig. 1).
Animals were euthanized by carbon dioxide asphyxiation at the end of 3 mo of EtOH feeding, and blood was collected by cardiac puncture in a plasma separation tube (PST) with lithium heparin (Cat. No. 365985, Becton, Dickinson and Company, Franklin Lakes, NJ). Fifty microliters of whole blood were transferred to glass vials for analyzing alcohol and acetaldehyde concentrations, and the remaining blood was centrifuged at 10,000 g for ∼2 min, and the plasma was separated and stored at −80°C.
The pancreas was excised for gross examination, and a portion was fixed in 10% buffered formalin followed by dehydration in 70% EtOH and embedded in paraffin blocks for histology and immunohistochemistry. For electron microscopic examination, small (1–2 mm3) pieces of the pancreas were fixed in a buffered mixture of formaldehyde and glutaraldehyde and embedded in epoxy plastic (24). The remaining portions of the pancreas were snap-frozen in liquid nitrogen and stored at −80°C for biochemical and molecular studies.
Morphological and Immunohistochemical Studies
Standard sections of fixed pancreatic tissue embedded in paraffin blocks were cut (5 μm thick) and stained with hematoxylin and eosin (H&E) and Masson’s Trichrome for the light microscopic examination (24). The histological scoring was performed using semiquantitative scoring system to grade the extent of pancreatic parenchymal edema (0 – no edema, 1 – interlobular edema, 2 – interlobular and moderate intralobular edema, 3 – interlobular and severe intralobular edema), cell vacuolization (0 – none, 1 – <20% acini with vacuoles, 2 – <50% acini with vacuoles, 3 – >50% acini with vacuoles), inflammation (0 – no inflammation, 1 – inflammatory cells present at intralobular, 2 – inflammatory cells presents at interstitium fat and intralobular, 3 – inflammatory cells present at interacini or parenchyma), disorganized and degenerated acinar cells and fibrosis (0 – normal acini, 1 – < 25% of acini with disorganization, 2 – <50% of acini with disorganization and mild fibrosis, 3 – >50% acini with disorganization along with degenerated acini and fibrosis) (30, 31). The quantification of Masson’s Trichrome staining was performed using NIH Image J Software, v. 1.50i (Bethesda, MD, NIH) (32).
Immunohistochemical staining of pancreatic tissue sections was done as described previously (25), using antibodies against CD3 (Cat. No. ab16669; Abcam Inc., Cambridge, MA; 1:100 dilution, positive control – Human Tonsil) and F4/80 (Cat. No. 70076; Cell Signaling Technology, Danvers, MA, 1:200 dilution, positive control – mouse lung tissue) for evaluating the inflammatory response. The CD3 and F4/80 DAB (3, 3′-diaminobenzidine) positive-stained cells were counted using NIH Image J Software in a randomly selected 1/5 areas of whole pancreatic sections and the values represented as number of cells/mm2, respectively.
Similarly, ER and oxidative stress were evaluated by immunohistochemical staining using antibodies against GRP78 (Cat. No. ab21685; Abcam Inc., Cambridge, MA; 1:500 dilution, positive control – mouse pancreas) and 4-hydroxynonenal (4HNE, Cat. No. HNE11-S, Alpha diagnostics, San Antonio, TX, 1:300 dilution, positive control – Human lung fibrosis and mouse liver), respectively. The relative threshold for DAB staining intensity for GRP78 and 4HNE was quantified using NIH Image J Software and represented as an arbitrary unit.
For transmission electron microscopy (EM), ultrathin sections of the pancreas were cut as described earlier (25) and examined in a JEOL JEM-1400 electron microscope (JEOL, Inc., Peabody, MA) at 80 kV. Images were acquired with bottom-mounted CCD-camera Orius SC2001 (Gatan, Pleasanton, CA).
The ultrastructural findings of pancreatic acinar cell were further scored to assess overall cellular changes using Newcastle Pancreatic Acinar Stress Score (NPASS) (33) in various groups of control diet and chronic EtOH-fed ADH− and ADH+ deer mice, respectively. The nuclei, endoplasmic reticulum, mitochondria, and overall acinar cell structure were visualized and individually scored from a 0- to 3-point scale, with 0 indicating normal morphology and 3 being severe or abnormal pathology. The sum of individual scores was aggregated with the total score ranging from 0 to 12 to determine the degree/severity of pancreatic injury.
The pancreatic histology and EM scoring were done by an experienced pathologist who was blinded to the experimental groups.
Blood Alcohol and Acetaldehyde Levels and Pancreatic Injury Markers
Blood alcohol and acetaldehyde levels were analyzed using headspace gas chromatography (GC) as described previously (34).
Plasma amylase and lipase (key markers of pancreatic injury) were assayed using the kits from BioVision (Cat. No. K711, Milpitas, CA) and Cayman Chemical (Cat. No. 700640, Ann Arbor, MI), respectively, as per the manufacturer’s instructions. The absorbance/fluorescence was measured using Bio-Tek Epoch 2 microplate reader/Bio-Tek synergy/HTX multimode reader (Winooski, VT), respectively. The enzyme activity is expressed as units per liter.
The levels of trypsin (pancreatic injury marker) in the plasma and pancreatic homogenate were determined using a colorimetric assay kit from BioVision (Cat No. K771-100, Milpitas, CA) as per the manufacturer’s instructions. The absorbance was measured using Bio-Tek Epoch 2 microplate reader and the activity expressed as nmol/min/mg protein. Similarly, the relative expression of cathepsin-B (involved in pancreatic cell/necrotic pathways and pancreatitis) in the pancreatic homogenate was determined using Western blot analysis. In addition, the cathepsin B activity in the pancreatic homogenate was determined using fluorometric assay kit from BioVision (Cat. No. K140-100, Milpitas, CA) as per the manufacturer’s instructions. The fluorescence was measured with an excitation and emission wavelengths of 400 and 505 nm, respectively, using Bio-Tek synergy HTX multimode microplate reader. The fold increase in cathepsin B activity was determined by comparing relative fluorescence units (RFU) with level of control sample.
ER Stress Signaling Proteins
ER stress and related unfolded protein response (UPR) signaling in the pancreatic postnuclear fraction were analyzed by Western blot analysis. In brief, the frozen pancreatic tissue was thawed and homogenized using 1× RIPA buffer (Cat. No. 9806, Cell Signaling Technology, Danvers, MA) containing phosphatase and protease inhibitors (Cat. No. 78446, Thermo Fisher Scientific, Houston, TX). The protein content in the postnuclear fraction was measured by Bio-Rad protein assay reagent (Cat. No. 5000006, Bio-Rad Laboratories, Hercules, CA). About 30 μg protein from each fraction was electrophoresed using precasted 4%–12% NuPage mini-gels (Life Technologies, Carlsbad, CA). The resolved proteins were transferred onto the immune-blot PVDF membrane (Cat No. 1620177, Bio-Rad Laboratories, Hercules, CA) and blocked with 5% nonfat dry milk (Cat. No. 171–6404, Bio-Rad Laboratories, Hercules, CA) in Tris-buffered saline with Tween 20 (TBST) and probed with their respective primary antibodies (1:1,000 dilution). After extensive washes, the immunoreactivity was detected using specific horseradish peroxidase-conjugated secondary antibodies (1:5,000 dilution) followed by enhanced chemiluminescence, as described (25). The protein bands were quantified using NIH Image J Software, and the values were normalized to the loading control, β-actin. The ratio of phosphorylated protein to total protein was calculated.
Inflammatory Cytokines and Chemokines
Qualitative determination of proinflammatory cytokines and chemokines in plasma and the pancreatic homogenate was done by using a mouse cytokine membrane antibody array kit (Cat. No. 133993, Abcam, Cambridge, MA) following the manufacturer’s instructions.
Before the assays, the protein concentration of pancreatic lysate and plasma was determined by Bio-Rad protein assay. In brief, an aliquot of the pancreatic lysate (250 µg) or plasma (100 µg) protein was incubated on the array membrane, and the target cytokines were visualized using the chemiluminescence detection system. The intensity of the cytokine spot was measured using ImageJ. Data were normalized to the controls and presented as arbitrary units (a.u.).
Statistical Analysis
Data are expressed as means ± SEM (standard error of mean) of ≥4 animals per group unless indicated. The data sets were analyzed for statistical significance using Student’s t test and ANOVA, followed by Tukey’s multiple comparisons test. A P value ≤0.05 was considered significant.
RESULTS
Morphology and Ultrastructural Changes
At the microscopic level, the pancreatic tissue of control diet and control diet plus FAEEs-fed group of ADH− (Fig. 2, A1 and B1) and ADH+ (Fig. 2, A2 and B2) deer mice showed normal histology and tissue architecture.
Figure 2.
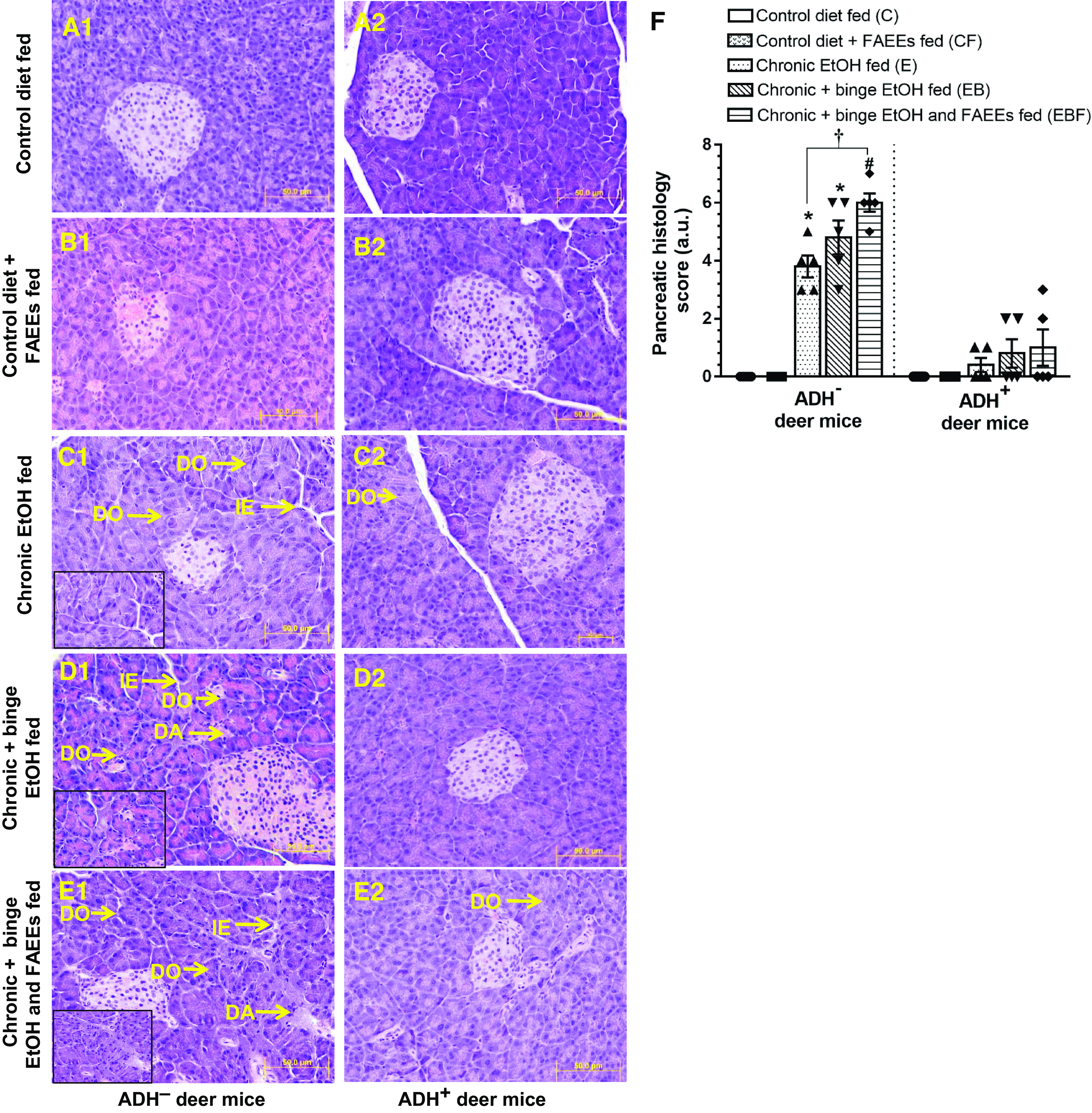
Hematoxylin and eosin (H&E)-stained representative pancreatic tissue sections of ADH− (A1–E1; left) and ADH+ deer mice (A2–E2; right), respectively. Pancreas of ADH− and ADH+ deer mice pair-fed control diet (A1 and A2) or with FAEEs (B1 and B2) showing normal histology. Chronic EtOH-fed ADH− deer mice (C1) show disorganization (DO) of acinar cells and interstitial edema (IE). A mild disorganization (DO) of acinar cells was also observed in chronic EtOH-fed ADH+ deer mice (C2). Pancreas of chronic plus binge EtOH-fed ADH− deer mice shows a significant acinar disorganization (DO) along with degenerative changes (DA), and interstitial edema (IE) (D1), as compared with normal histology in chronic plus binge EtOH-fed ADH+ deer mice (D2). Pancreas of chronic plus binge EtOH and FAEEs-fed ADH− deer mice shows a remarkable disorganization (DO) and degenerative changes (DA) in acinar structure and interstitial edema (IE) (E1) as compared with the pancreas of chronic plus binge EtOH and FAEEs-fed ADH+ deer mice (E2). Inset shows area of higher magnification (X40). (Original magnification ×20, bar = 50 μm, n = 5 mice/group). Cumulative histology score (F) in the pancreas of ADH− and ADH+ deer mice. Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 5 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group. †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group. ADH−, alcohol dehydrogenase-deficient; FAEEs, fatty acid ethyl esters.
However, the pancreatic tissue from EtOH-fed groups of ADH− deer mice (chronic EtOH, chronic plus binge EtOH, and chronic plus binge EtOH and FAEEs) exhibited marked interstitial edema and disorganization of acinar cells with decreased intra glandular contents/secretion (Fig. 2, C1, D1, and E1). Furthermore, a significant degenerative change in the acinar structure was observed in the chronic plus binge EtOH and FAEEs-fed group of ADH− deer mice (Fig. 2E1). Of note, mild changes in the acinar structure were observed in various chronic EtOH-fed groups of ADH+ deer mice (Fig. 2, C2, D2, and E2).
Histological analysis showed relatively well-preserved and intact islets of Langerhans in both the chronic EtOH-fed and control groups of ADH− (Fig. 2, A1–E1) and ADH+ (Fig. 2, A2–E2) deer mice, respectively.
As shown in Fig. 2F, the pancreatic histology score was significantly greater (∼4-fold) for chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively. Furthermore, the score was significantly greater (∼ 1.5-fold) for chronic plus binge EtOH- and FAEEs-fed ADH− deer mice compared with chronic EtOH-fed group. Overall, marked changes in pancreatic morphology were observed in chronic EtOH-fed ADH− versus ADH+ deer mice, and the degree of pancreatic injury was relatively greater for chronic plus binge EtOH and FAEEs-fed ADH− deer mice than that for chronic plus binge EtOH and chronic EtOH-fed ADH− deer mice, respectively. In general, the extent of pancreatic injury followed this order: chronic plus binge EtOH and FAEEs > chronic plus binge EtOH > chronic EtOH.
The Masson’s trichrome staining in the pancreas of control diet and control diet plus FAEEs-fed ADH− (Fig. 3, A1 and B1) and ADH+ (Fig. 3, A2 and B2) deer mice did not reveal any positive-stained areas. However, significant staining of collagen fibers was observed in the pancreas of various chronic EtOH-fed groups of ADH− deer mice (Fig. 3, C1, D1, and E1) indicating fibrosis. On the other hand, a minimal staining for fibrosis was found in the chronic EtOH-fed groups of ADH+ deer mice (Fig. 3, C2, D2, and E2), respectively.
Figure 3.
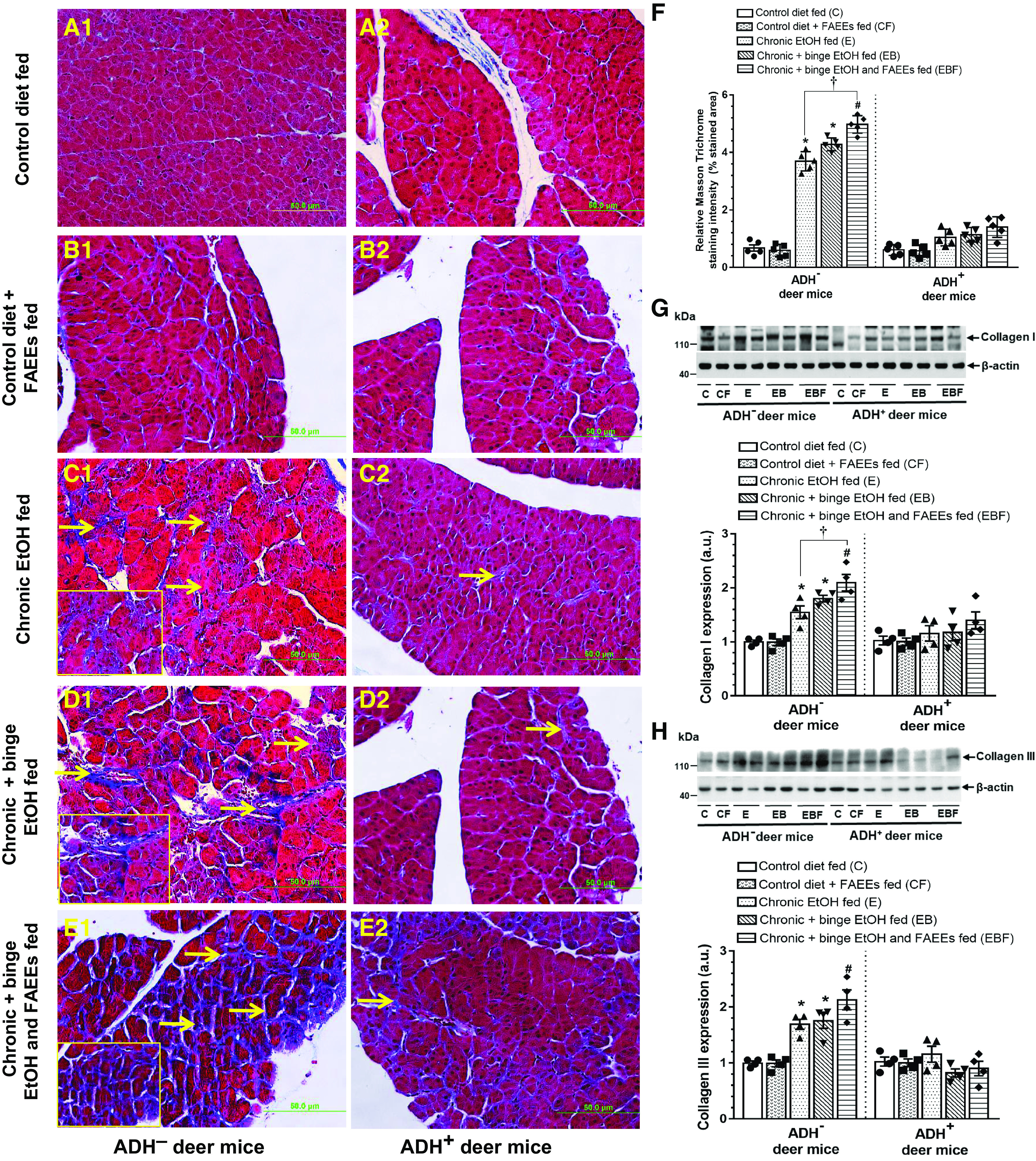
Histological evaluation of fibrosis by Masson’s trichrome staining in pancreatic tissue sections of ADH− (A1–E1; left) and ADH+ (A2–E2; right) deer mice (Original magnification ×20, bar = 50 μm, n = 5 mice/group). Arrows represent deposition of collagen fibers implicating fibrosis. Pancreas of pair-fed control diet and control diet plus FAEEs-fed ADH− (A1 and B1) and ADH+ (A2 and B2) deer mice, show normal histology. Pancreas of chronic EtOH-fed (C1), chronic plus binge EtOH-fed (D1), and chronic plus binge EtOH and FAEEs-fed (E1) ADH− deer mice shows positive staining for Trichrome with mild to moderate degree of fibrosis, whereas chronic EtOH-fed (C2), chronic plus binge EtOH-fed (D2), and chronic plus binge EtOH and FAEEs-fed (E2) ADH+ deer mice show relatively normal histology. Inset shows area of higher magnification (X40). Quantification of the Masson Trichrome staining (F) in the pancreas of ADH− and ADH+ deer mice. Data are presented as means ± SE (n = 5 replicates). Representative immunoblots along with respective bar diagram show relative protein expression for collagen 1 (G) and collagen III (H) in pancreatic homogenate of various chronic EtOH-fed groups of ADH− and ADH+ deer mice. Intensities were normalized to β-actin (loading control). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 4 replicates). #P ≤ 0.05 chronic plus single binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group and †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group, respectively. ADH−, alcohol dehydrogenase-deficient; C, pair-fed control diet group; CF, control diet plus FAEEs group; E, chronic EtOH-fed group; EB, chronic plus binge EtOH-fed group; EBF, chronic plus binge EtOH and FAEEs-fed group; FAEEs, fatty acid ethyl esters.
As shown in Fig. 3F, an overall intensity of Masson trichrome staining was relatively greater (∼3.6-fold) for chronic EtOH-fed ADH− versus ADH+ deer mice, respectively. Of note, the pancreatic fibrotic response was significantly increased (∼ 1.5-fold) in chronic plus binge EtOH and FAEEs-fed ADH− deer mice than that in chronic EtOH-fed group.
A significantly increased expression of pancreatic collagen type I (Fig. 3G) and type III (Fig. 3H) was found in chronic EtOH-fed groups of ADH− versus ADH+ deer mice. Relatively, increased levels of collagen I and collagen III were found in the chronic plus binge EtOH and FAEEs-fed ADH− versus chronic plus binge EtOH- and chronic EtOH-fed ADH− deer mice. The levels of collagens I and III were not significantly altered in chronic EtOH-fed groups of ADH+ deer mice as compared with respective pair-fed controls.
The electron microscopy (EM) of pancreatic sections of control diet and control diet plus FAEEs-fed ADH− (Fig. 4, A1 and B1) and ADH+ (Fig. 4, A2 and B2) deer mice showed normal endoplasmic reticulum (ER) morphology and well-preserved nucleus and mitochondria.
Figure 4.
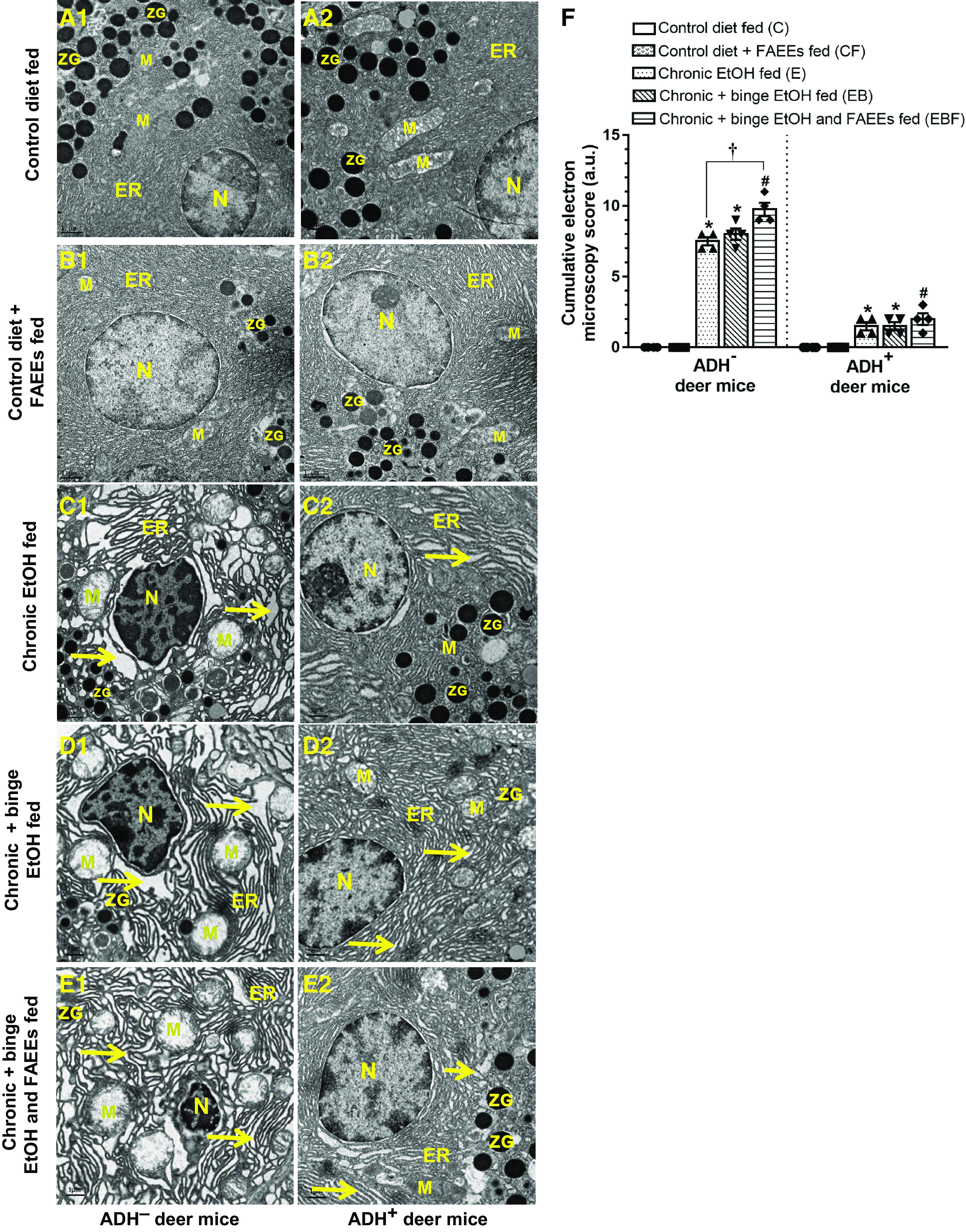
Electron micrographs of pancreatic sections of ADH− (A1–E1; left) and ADH+ deer mice (A2–E2; right). (Scale bars = 1 µm). Pancreas of pair-fed control and control diet and FAEEs-fed ADH− (A1 and B1) and ADH+ (A2 and B2) deer mice show normal ultrastructure with intact nucleus mitochondria and ER cisteranes. Pancreas of chronic EtOH-fed (C1), chronic plus binge EtOH-fed (D1), and chronic plus binge EtOH and FAEEs-fed (E1) ADH− deer mice shows extensive dilatations of ER cisternae along with swollen mitochondria with short and broken cristae and shrunken nucleus. Pancreas of chronic EtOH-fed (C2), chronic plus binge EtOH-fed (D2), and chronic plus binge EtOH and FAEEs-fed (E2) ADH+ deer mice show mild to moderate dilatations in ER cisteranes with normal and intact mitochondria and nucleus. Cumulative electron microscopy score in the pancreatic ultrastructure of ADH− and ADH+ deer mice (F). Arrows indicate the dilatation of ER cisternae. Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 4 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus single binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group and †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group, respectively. ADH−, alcohol dehydrogenase-deficient; ER, granular endoplasmic reticulum; FAEEs, fatty acid ethyl esters; M, mitochondria; N, nucleus; ZG, zymogen granules.
However, the EM of pancreatic sections of chronic EtOH-fed groups of ADH− deer mice showed significant disorganization of granular ER with extensive dilatations of ER cisternae, pyknosis of the nucleus with chromatin condensation/clumping, and swollen mitochondria with short and broken cristae and degranulated zymogen granules (Fig. 4, C1, D1, and E1), respectively.
On the other hand, the pancreatic ultrastructure of various chronic EtOH-fed groups of ADH+ deer mice exhibited mild dilatations of ER cisternae (Fig. 4, C2, D2, and E2) with a well-preserved nucleus and mitochondria, and intact cristae.
Of note, the cumulative EM scoring for pancreatic injury was increased (∼6-fold) in chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively (Fig. 4F). The cumulative EM score was significantly increased (∼1.5-fold) for chronic plus binge EtOH and FAEEs-fed ADH− deer mice as compared with the chronic EtOH-fed group. Overall, significant pancreatic ultrastructural changes were observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively, and an exposure to binge EtOH and FAEEs resulted in significant ultrastructural changes in chronic EtOH-fed ADH− deer mice.
Inflammation and Proinflammatory Cytokines and Chemokines
The pancreatic tissue sections of control diet and control diet plus FAEEs-fed ADH− (Fig. 5, A1 and B1) and ADH+ (Fig. 5, A2 and B2) deer mice did not show any positive staining for the CD3 cells. However, the pancreatic tissue sections of chronic EtOH-fed groups of ADH− deer mice showed positive immunostaining for CD3, which confirms the infiltration of inflammatory cells in the exocrine parenchyma (Fig. 5, C1, D1, and E1).
Figure 5.
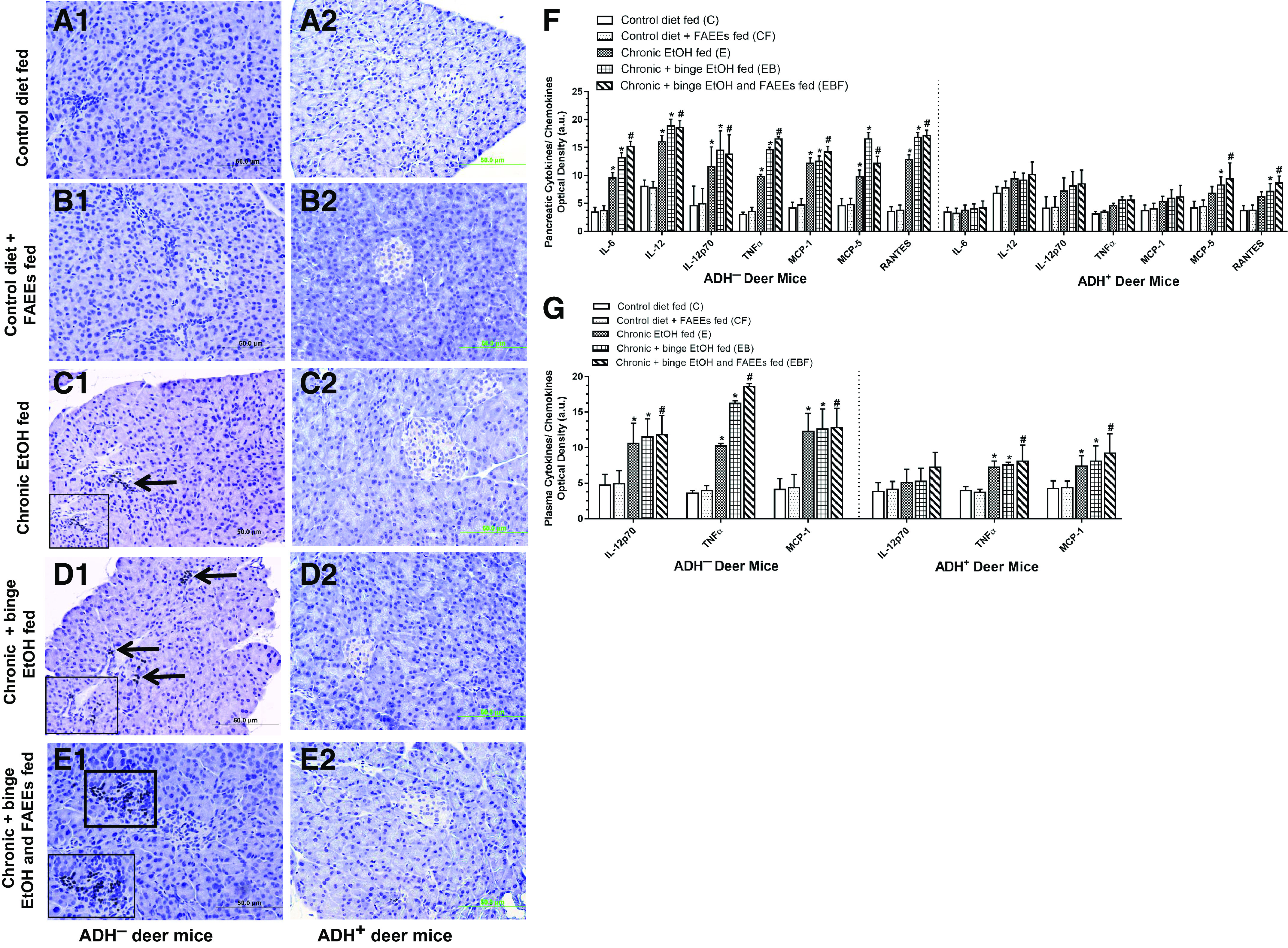
Immunohistochemical staining using antibodies against CD3 (marker for inflammation) in representative pancreatic sections of ADH− (A1–E1; left) and ADH+ (A2–E2; right) deer mice. Presence of inflammatory cells was found in the exocrine parenchyma of various chronic EtOH-fed groups of ADH− deer mice (C1–E1) as compared with ADH+ deer mice (C2–E2), respectively. No positive staining for CD3 was observed in the pancreas of pair-fed control diet and control diet plus FAEEs-fed ADH− (A1 and B1) and ADH+ (A2 and B2) deer mice, respectively. Arrows and square box represent positive staining for CD3. Inset shows area of higher magnification (×40). Levels of cytokines/chemokines in pancreatic homogenate and plasma of ADH− and ADH+ deer mice are shown in F and G, respectively. Substantially increased levels of cytokines/chemokines were detected in pancreatic homogenate (7 of 22) (F) and plasma (3 of 22) (G) of various chronic EtOH-fed ADH− vs. ADH+ deer mice, respectively. Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n =4 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group, respectively. Following cytokines/chemokines were tested but not detected in the pancreatic homogenate and plasma: IL-2; IL-3, IL-4; IL-5; IL-9; IL-10; IL-12 p40/p70; IL-13; IL-17; IFN-γ; SCF; sTNFRI; GCSF; GM-CSF; VEGF; and thrombopoietin. ADH−, alcohol dehydrogenase-deficient; FAEEs, fatty acid ethyl esters; IL, interleukin; MCP, monocyte chemoattractant protein; TNF, tumor necrosis factor.
Furthermore, DAB staining showed relatively greater number of CD3-positive cells in the pancreatic sections of ADH− deer mice fed chronic plus binge EtOH and FAEEs (11 cells/mm2) as compared with chronic plus binge EtOH (8 cells/mm2) and chronic EtOH (5 cells/mm2)-fed group, respectively.
Of note, a similar positive immunostaining for CD3 was not observed in the pancreas of various chronic EtOH-fed groups of ADH+ deer mice (Fig. 5, C2, D2, and E2).
Besides, an increase of 1.5- to 2-fold for the pancreatic levels of inflammatory cytokines (IL-6, Il-12, IL-12p70, and TNFα) and chemokines (MCP-1, MCP-5, and RANTES) were observed in chronic EtOH-fed groups of ADH− deer mice as compared with its respective controls (Fig. 5F). Of note, the levels of only two chemokines MCP-5 and RANTES were significantly greater in chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed groups of ADH+ deer mice compared with chronic EtOH-fed ADH+ deer mice and respective pair-fed controls (Fig. 5F). Otherwise, similar levels of inflammatory cytokines (IL-6, Il-12, IL-12p70, and TNFα) and chemokine (MCP-1) were observed for various chronic EtOH-fed groups of ADH+ deer mice and respective controls (Fig. 5F). Overall, pancreatic inflammatory cytokines and chemokines were two- to fourfold greater in chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively, (Fig. 5F).
Similarly, two- to threefold increases for the plasma levels of inflammatory cytokines (IL-12p70 and TNFα) and chemokine (MCP-1) were observed in EtOH-fed groups of ADH− deer mice as compared with control diet-fed ADH− deer mice (Fig. 5G). As shown in Fig. 5G, twofold increases for the plasma levels of TNFα and MCP-1 were observed in various chronic EtOH-fed groups of ADH+ deer mice as compared with its respective controls. However, two- to threefold increases for the plasma levels of IL-12p70, TNFα, and MCP-1 were observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice (Fig. 5G). The markers of inflammation were significantly increased in the plasma and pancreas of chronic EtOH-fed ADH− versus ADH+ deer mice, respectively. Furthermore, their levels were relatively greater in chronic plus binge EtOH and FAEEs-fed ADH− deer mice followed by chronic plus binge EtOH and chronic EtOH-fed ADH− deer mice.
Of importance, immunostaining of pancreatic tissue sections for F4/80 (macrophage marker) in control diet and control diet plus FAEEs-fed ADH− (Fig. 6, A1 and B1) and ADH+ deer mice (Fig. 6, A2 and B2) did not show any positive immunostaining for F4/80. However, the pancreatic tissue sections in chronic EtOH-fed groups of ADH− deer mice showed positive immunostaining, which confirms the presence of macrophages in the exocrine parenchyma (Fig. 6, C1, D1, and E1).
Figure 6.

Immunohistochemical staining using antibodies against F4/80 (macrophage marker) in representative pancreatic sections of ADH− (A1–E1; left) and ADH+ (A2–E2; right) deer mice. Macrophages or positive staining for F4/80 were found in the exocrine parenchyma of various chronic EtOH-fed groups of ADH− deer mice (C1–E1) as compared with ADH+ deer mice (C2–E2), respectively. No positive staining for F4/80 was observed in the pancreas of pair-fed control diet and control diet plus FAEEs-fed ADH− (A1 and B1) and ADH+ (A2 and B2) deer mice, respectively. Arrows represents positive staining for F4/80. Inset shows area of higher magnification (×40). ADH−, alcohol dehydrogenase-deficient; FAEEs, fatty acid ethyl esters.
Furthermore, the DAB staining showed relatively greater positive immunostained cells for F4/80 antibodies in pancreatic sections of ADH− deer mice fed chronic plus binge EtOH and FAEEs (10 cells/mm2) as compared with chronic plus binge EtOH (7 cells/mm2) and chronic EtOH (4 cells/mm2)-fed group, respectively.
Of note, a similar positive immunostaining for F4/80 was not observed in EtOH-fed groups of ADH+ deer mice (Fig. 6, C2, D2, and E2), respectively.
Blood Alcohol and Acetaldehyde Concentration and Pancreatic Injury Markers
Significantly increased blood alcohol concentration (BAC) was observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice (Fig. 7A). BAC was exponentially increased for both the strains of chronic EtOH-fed deer mice. However, the average BAC was ∼14-fold greater in the chronic EtOH-fed ADH− (∼178 mg%) versus ADH+ deer mice (∼12 mg%). Similarly, the average BAC was ∼10-fold (∼215 vs. ∼21 mg%) and ∼20-fold (∼234 vs. 12 mg%) greater in chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed groups of ADH− versus ADH+ deer mice, respectively. Overall, the BACs were relatively higher in chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed groups of ADH− deer mice than the chronic EtOH-fed group.
Figure 7.

Blood alcohol concentration (A) and blood acetaldehyde concentration (B) in various EtOH-fed groups of ADH− and ADH+ deer mice. Values are means ± SE (n = 5 replicates). Enzymatic activities of plasma amylase (C), plasma lipase (D), pancreatic trypsin (E), and cathepsin B (F) in various EtOH-fed groups of ADH− and ADH+ deer, respectively. Values are means ± SE (n = 4 replicates). Representative immunoblot along with respective bar diagram shows relative protein expression for Cathepsin B (G) in pancreatic homogenates of various chronic EtOH-fed ADH− and ADH+ deer mice. Intensities were normalized to β-actin (loading control). Data are analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 4 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus single binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group and †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group, respectively. ADH−, alcohol dehydrogenase-deficient; C, pair-fed control diet group; CF, control diet plus FAEEs-fed group; E, chronic EtOH-fed group; EB, chronic plus binge EtOH-fed group; EBF, chronic plus single binge EtOH and FAEEs-fed group; FAEEs, fatty acid ethyl esters.
Blood acetaldehyde levels were increased by two- to threefold in chronic EtOH-fed groups of ADH− and ADH+ deer mice compared with their respective controls (Fig. 7B). Average blood acetaldehyde levels were ∼1.5-foldgreater in the chronic EtOH-fed groups of ADH− (∼0.336 mg%) versus ADH+ deer mice (∼0.224 mg%). Furthermore, the blood acetaldehyde concentration was relatively higher in chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed groups of ADH− deer mice than those fed chronic EtOH.
As shown in Fig. 7C, a relative increase in plasma amylase activity was observed in chronic EtOH-fed groups of ADH− deer mice. However, a significant increase was observed in chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed groups of ADH− deer mice (∼1.6-fold) compared with the respective controls. As shown in Fig. 7D, a relative increase for plasma lipase activity was observed in chronic EtOH-fed groups of ADH− deer mice as compared with respective controls. However, a significant increase for plasma lipase activity was observed only in chronic plus binge EtOH and FAEEs-fed groups of ADH− deer mice (∼1.4-fold) as compared with its respective control. Of note, no changes were observed for the plasma amylase and lipase activity in any chronic EtOH-fed groups of ADH+ deer mice.
Of importance, a significant increase for pancreatic trypsin (Fig. 7E) and cathepsin B (Fig. 7F) activities were observed in EtOH-fed groups of ADH− deer mice (∼2-fold) compared with its respective controls. The pancreatic trypsin and cathepsin B activities were relatively higher in chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed versus chronic EtOH-fed ADH− deer mice. Furthermore, a significant increase in pancreatic trypsin and cathepsin B activities were observed in chronic plus binge EtOH and FAEEs-fed (∼1.5-fold) versus chronic EtOH-fed ADH− deer mice. However, the changes were not significant for the pancreatic trypsin and cathepsin B activities in various groups of chronic EtOH-fed ADH+ deer mice, as well as for the plasma trypsin activity in various chronic EtOH-fed ADH− and ADH+ deer mice (data not shown).
Similarly, a significant increase for activated cathepsin-B expression was observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice (Fig. 7G). A relative expression of activated cathepsin-B expression in the pancreas was increased for chronic plus binge EtOH and chronic plus binge EtOH and FAEEs-fed ADH− deer mice than that for the chronic EtOH-fed ADH− deer mice. Besides, a significant increase for activated cathepsin-B expression was observed in chronic plus binge EtOH and FAEEs-fed ADH− deer mice versus chronic EtOH-fed ADH− deer mice. However, activated cathepsin-B expression was not altered in chronic EtOH-fed groups of ADH+ deer mice. Overall, relative increases for pancreatic injury markers were observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively, and the levels of pancreatic injury markers were relatively increased in chronic plus binge EtOH and FAEEs-fed versus chronic plus binge EtOH and chronic EtOH-fed ADH− deer mice.
ER/Oxidative Stress and UPR Signaling
Significant pancreatic oxidative stress observed in various chronic EtOH-fed groups of ADH− deer mice as compared with its respective controls is supported by immunohistochemical findings using antibodies against 4HNE, a lipid peroxidation product and a marker for oxidative stress (Fig. 8, A1–E1). Of note, a significantly higher intensity of 4HNE immunostaining (∼1.8-fold) was observed in chronic plus binge EtOH and FAEEs-fed versus chronic EtOH-fed ADH− deer mice (Fig. 8F). However, the staining was not appreciably changed between various groups of ADH+ deer mice fed chronic EtOH and their respective pair-fed controls (Fig. 8, A2–E2). Overall, the intensity for 4HNE staining was three- to fourfold greater in chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively (Fig. 8F).
Figure 8.
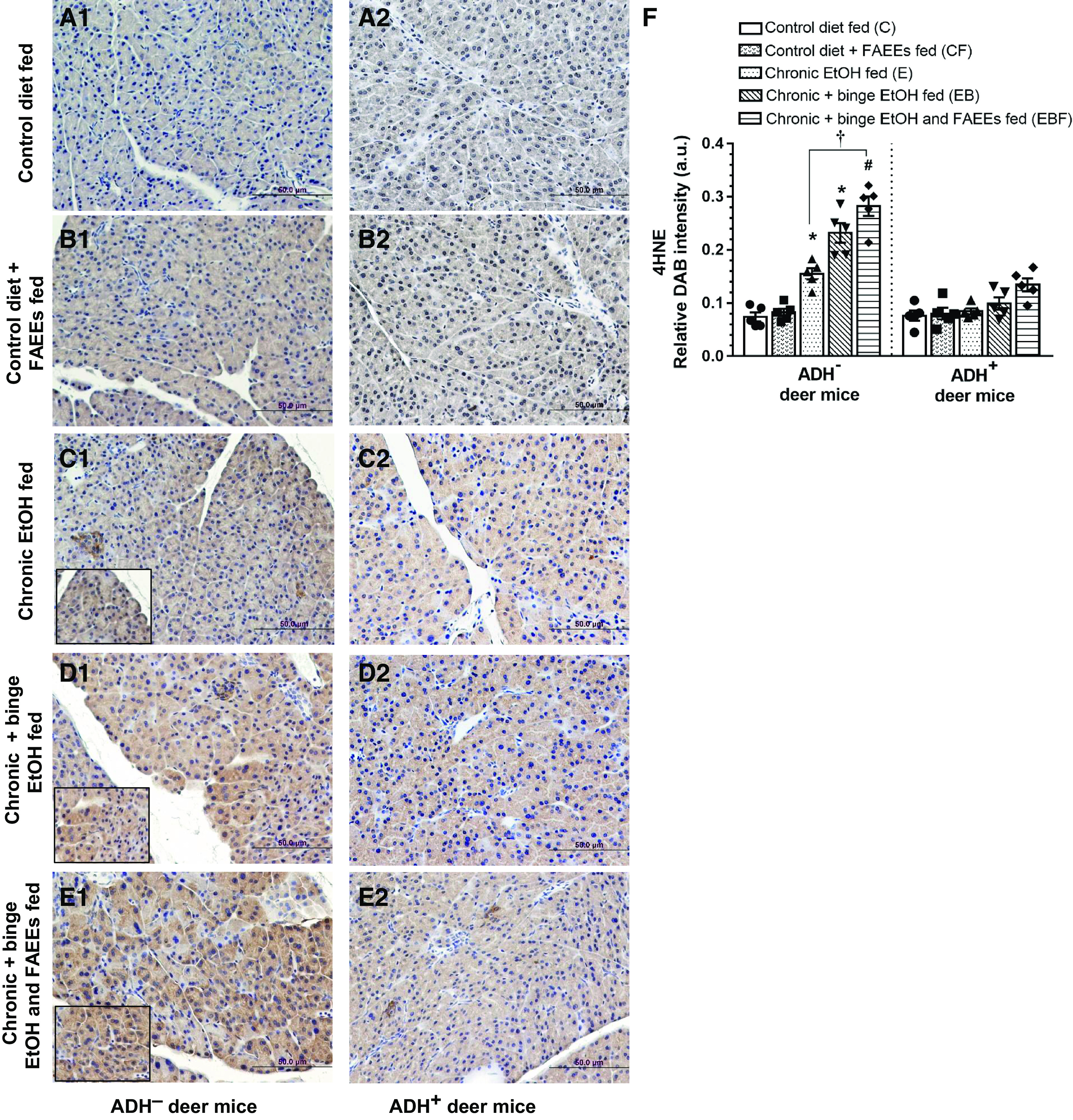
Immunohistochemical staining using 4HNE (marker for oxidative stress) antibodies in representative pancreatic sections of ADH− (A1–E1; left) and ADH+ (A2–E2; right) deer mice, respectively. No positive staining for 4HNE was observed in the pancreas of pair-fed control diet and control diet plus FAEEs-fed ADH− (A1 and B1) and ADH+ (A2 and B2) deer mice, respectively. A significant positive staining for 4HNE was observed in various chronic EtOH-fed groups of ADH− vs. ADH+ deer mice, respectively. Pancreas of chronic EtOH-fed (C1), chronic plus binge EtOH-fed (D1), and chronic plus binge EtOH and FAEEs-fed (E1) ADH− deer mice shows significant positive staining for 4HNE. However, the positive staining for 4HNE was not observed in the pancreas of various chronic EtOH-fed groups of ADH+ deer mice (C2–E2). Quantification of the intensity of immunohistochemical staining for 4HNE (F) in the pancreas of ADH− and ADH+ deer mice. Inset shows area of higher magnification (×40). Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 5 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus single binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group and †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group, respectively. ADH−, alcohol dehydrogenase-deficient; FAEEs, fatty acid ethyl esters; 4HNE, 4-hydroxynonenal.
The pancreatic ER stress, as evaluated by immunostaining using antibodies against GRP78, was significantly increased in various chronic EtOH-fed groups of ADH− deer mice as compared with their respective controls (Fig. 9, A1–E1). Besides, the intensity of GRP78 immunostaining was significantly higher (∼1.7-fold) in chronic plus binge EtOH and FAEEs-fed versus chronic EtOH-fed ADH− deer mice (Fig. 9F). However, GRP78 staining was not changed significantly between chronic EtOH (Fig. 9C2) and chronic plus binge EtOH (Fig. 9D2) fed groups of ADH+ deer mice versus respective controls (Fig. 9A2). Of note, the intensity of GRP78 staining was significantly greater in chronic plus binge EtOH and FAEEs-fed ADH+ deer mice only as compared with respective control (Fig. 9F). Overall, the intensity for GRP78 staining was approximately two- to fivefold greater in chronic EtOH-fed groups of ADH− versus ADH+ deer mice, respectively (Fig. 9F).
Figure 9.
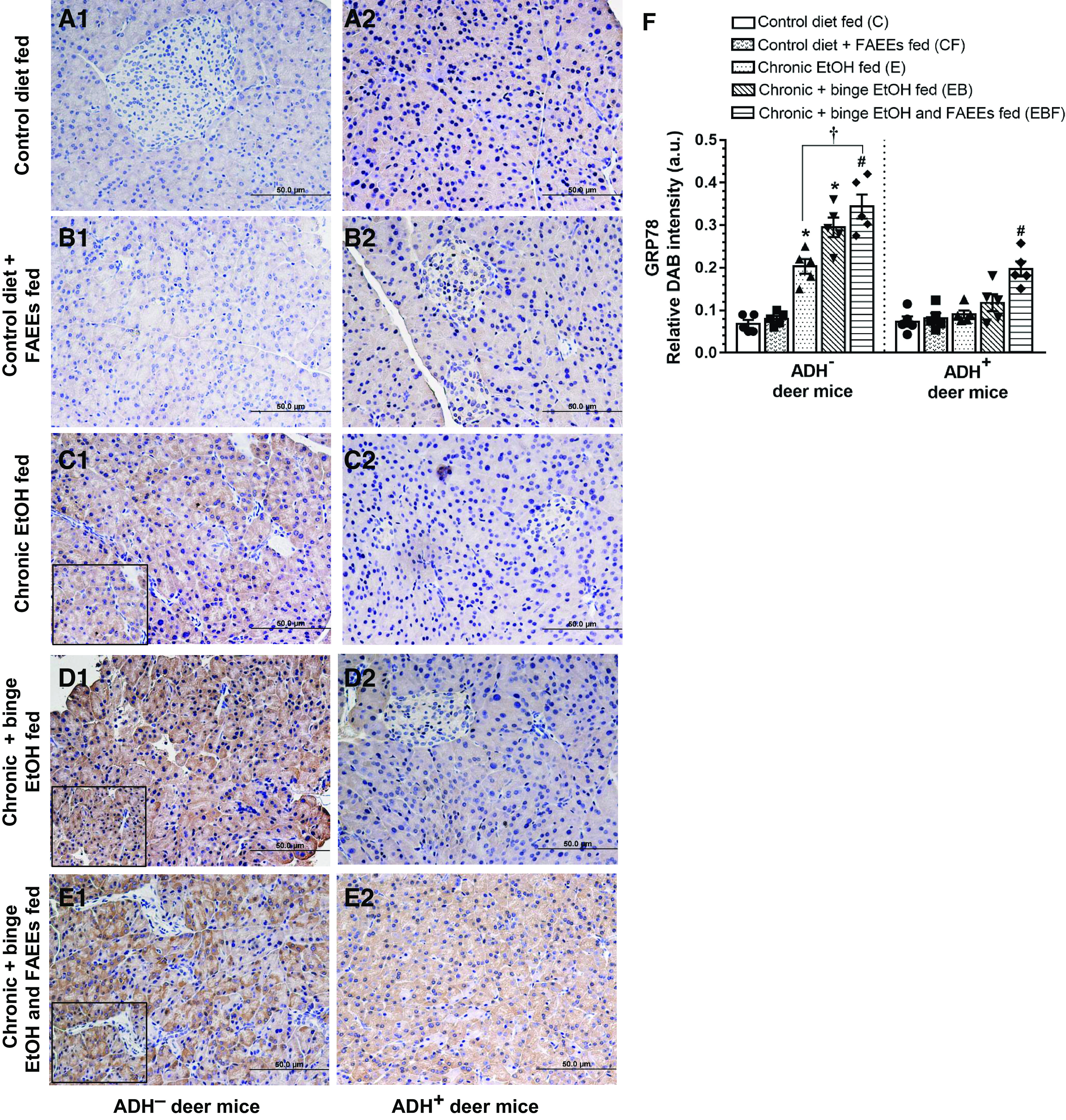
Immunohistochemical staining for GRP78 (marker for ER Stress) in representative pancreatic sections of ADH− (A1–E1; left) and ADH+ deer mice (A2–E2; right). No positive staining for GRP78 was observed in the pancreas of pair-fed control and control plus FAEEs-fed ADH− (A1 and B1) and ADH+ (A2 and B2) deer mice. A significant positive staining for GRP78 was observed in various chronic EtOH-fed groups of ADH− vs. ADH+ deer mice, respectively. Pancreas of chronic EtOH-fed (C1), chronic plus binge EtOH-fed (D1), and chronic plus binge EtOH and FAEEs-fed (E1) ADH− deer mice shows significant positive staining for GRP78. The positive staining for GRP 78 was not observed in the pancreas of chronic EtOH-fed (C2) and chronic plus binge EtOH-fed (D2) ADH+ deer mice, respectively. However, a significant staining was observed only in chronic plus binge EtOH and FAEEs-fed ADH+ deer mice (E2) as compared with its respective pair-fed control. Quantification of relative intensity of immunohistochemical staining for GRP78 is shown in the pancreas of ADH− and ADH+ deer mice (F). Inset shows area of higher magnification (×40). Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 5 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus single binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group and †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group, respectively. ADH−, alcohol dehydrogenase-deficient; ER, endoplasmic reticulum; FAEEs, fatty acid ethyl esters; UPR, unfolded protein response.
Expression of pancreatic ER stress and UPR regulators in ADH− and ADH+ deer mice fed chronic EtOH are shown in Fig. 10, A–F. A significantly increased expression of GRP78 (Fig. 10A), unspliced XBP1 (uXBP1) (Fig. 10C), p-PERK (Fig. 10D), p-EIF2α (Fig. 10E), and CHOP (Fig. 10F) were observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice. Expression of ER stress and UPR proteins were relatively higher in chronic plus binge EtOH and FAEEs-fed ADH− deer mice as compared with chronic plus binge EtOH and chronic EtOH-fed ADH− deer mice, respectively. Furthermore, the expression for GRP78 (Fig. 10A) and p-PERK (Fig. 10D) was significantly increased in chronic plus binge EtOH and FAEEs-fed versus chronic EtOH-fed ADH− deer mice, respectively.
Figure 10.

ER stress and UPR signaling in the pancreas of ADH− and ADH+ deer mice fed chronic EtOH, chronic plus binge EtOH, and chronic plus single binge EtOH and FAEEs. Representative immunoblots along with respective bar diagram show expression levels of GRP78 (A), p-IRE1α/IRE1α (B), sXBP1/uXBP1 (C), p-PERK (D), p-EIF2α/EIF2α (E), and CHOP (F). For the A, C, D, and F, the same β-actin blot was reused as the target proteins expressed in a single blot were normalized to the corresponding β-actin. Similarly, for B and E, same β-actin blot was reused as loading control. Data were analyzed using ANOVA, followed by Tukey’s multiple comparisons test, and presented as means ± SE (n = 4 replicates). *P ≤ 0.05 chronic EtOH-fed group and chronic plus binge EtOH-fed group vs. pair-fed control diet group. #P ≤ 0.05 chronic plus single binge EtOH and FAEEs-fed group vs. control diet plus FAEEs-fed group and †P ≤ 0.05 chronic plus binge EtOH and FAEEs-fed group vs. chronic EtOH-fed group, respectively. ADH−, alcohol dehydrogenase deficient; C, pair-fed control diet group; CF, control diet plus FAEEs-fed group; E, chronic EtOH-fed group; EB, chronic plus binge EtOH-fed group; EBF, chronic plus binge EtOH with FAEEs-fed group; ER, endoplasmic reticulum; FAEEs, fatty acid ethyl esters; UPR, unfolded protein response.
In contrast, a significant increase for the protein levels of p-IRE1α (Fig. 10B) and spliced XBP1 (sXBP1) (Fig. 10C) were observed in chronic EtOH-fed ADH+ versus ADH− deer mice. However, expression of ATF-6 was not altered in both the strains (data not shown).
Overall, the degree of pancreatic ER/oxidative stress was relatively higher in chronic plus binge EtOH and FAEEs-fed ADH− deer mice followed by those fed with chronic plus binge EtOH and chronic EtOH, respectively. Of note, an increased expression of p-IRE1α/sXBP1 arm of UPR could be a key factor for the homeostasis of ER in the pancreas of ADH+ deer mice fed chronic EtOH.
DISCUSSION
In this study, we evaluated differential pancreatic injury using ADH− deer mice to determine the metabolic basis of ACP and the role of endogenously formed FAEEs (nonoxidative EtOH metabolites commonly synthesized during chronic alcohol abuse) in etiopathogenesis of ACP.
A reduced level of hepatic ADH expression as observed in chronic alcoholics and after the chronic EtOH feeding to experimental animals (7–10) could largely enhance the nonoxidative metabolism of EtOH, particularly in the pancreas having a minimal capacity to metabolize alcohol via oxidative pathway. Of importance, pharmacological inhibition of hepatic ADH1 in human subjects’ increases plasma concentration of FAEEs (35) and results in two- to threefold increased production of FAEEs in the pancreas of rats fed EtOH than those in controls (23). Substantially elevated BAC in chronic alcoholic subjects is shown to be associated with increased biosynthesis and bioaccumulation of FAEEs (18, 29, 36). Of importance, previously we have demonstrated a substantial formation of FAEEs in the pancreas and plasma of chronic EtOH-fed hepatic ADH− versus ADH+ deer mice (21, 24, 25). Thus, we hypothesized that increased biosynthesis and accumulation of FAEEs in the pancreas of chronic EtOH-fed ADH− versus ADH+ deer mice are associated with increased BAC, which is supported, in part, by the studies in alcoholic subjects (29, 37, 38). Furthermore, comparable blood acetaldehyde levels in chronic EtOH-fed ADH− and ADH+ deer mice suggest that CYP2E1 may not be significantly contributing to EtOH metabolism and associated pancreatic injury in our animal model, despite the increased expression of hepatic CYP2E1 by chronic EtOH exposure (12). Being a reactive aldehyde, much of the acetaldehyde formed could bind to the proteins at the locus of its production in the liver or elsewhere with little in circulation. Thus, hepatic ADH deficiency appears to be key determinant for the increased body burden of EtOH and associated toxicity in the pancreas of ADH− versus ADH+ deer mice fed chronic EtOH.
The exocrine pancreas (primarily composed of acinar cells) has a vast network of endoplasmic reticulum (ER) membranes, a major site for the synthesis of pancreatic digestive proteins required for the digestion of the food (39). EtOH-induced perturbations in ER homeostasis have evolved as one of the mechanisms in experimental pancreatitis (40). ER stress triggers unfolded protein response (UPR) by activating three main ER transducers (IRE1α, ATF-6, and PERK). In general, activation of UPR results in reduced synthesis of nascent proteins, recruitment of chaperones, and degradation of misfolded proteins, leading to the restoration of ER homeostasis. However, prolonged or severe ER stress promotes cell death and inflammation, a basis for many chronic diseases (41).
In pancreatitis, ER stress is manifested by morphological changes in the ER membranes including dilatations of ER cisternae and loss of granulation as well as increased phosphorylation of PERK, splicing of XBP1 (sXBP1), and CHOP expression (39, 42–44). The activation of IRE1α/sXBP1 arm of UPR may protect the pancreas against injury by restoring ER homeostasis in contrast to activation of the PERK/CHOP arm of UPR resulting from prolonged/severe ER stress, which could also be associated with acinar cell injury/death with an elevated inflammatory response (22, 45). An increased expression of GRP78 along with PERK/CHOP arm of UPR in addition to extensive dilatations and disorganization of ER/ER cisternae in the pancreas of chronic EtOH-fed groups of ADH− deer mice as found in this study suggests the lack of adaptive UPR to maintain ER homeostasis. On the other hand, the activation of IRE1α/XBP1 arm of UPR, with mild dilatations of ER/ER cisternae in chronic EtOH-fed groups of ADH+ deer mice, explains the restoration of ER homeostasis to a greater extent and protection against EtOH-induced pancreatic injury in our animal model. Furthermore, increased fibrosis as confirmed by Trichrome staining together with increased collagen I and collagen III expression in chronic EtOH-fed groups of ADH− versus ADH+ deer mice could be linked to prolonged ER stress and inflammation (46).
Of note, chronic EtOH exposure has been demonstrated to delay the regeneration of the damaged pancreas (47). Moreover, FAEEs have been shown to have a fibrogenic effect in the exocrine pancreas with increased levels of extracellular matrix proteins (48). Thus, an increased body burden of EtOH and formation of FAEEs together with extensive ER stress could be contributing factors for the pancreatic injury observed in chronic EtOH-fed ADH− versus ADH+ deer mice instituting a basis for the pathogenesis of ACP.
The first pathologic response in alcoholic pancreatitis is manifested by inflammation as indicated by significant neutrophil and macrophage infiltration in pancreatic parenchyma of ADH− deer mice. Besides, the elevated levels of inflammatory cytokines and chemokines in the pancreas and plasma of ADH− deer mice fed chronic EtOH are parallel to those reported in patients/other experimental models of pancreatitis (49–54). Together, a lack of activation of adaptive UPR and increased levels of proinflammatory cytokines observed in the pancreas of chronic EtOH-fed ADH− deer mice are like those commonly observed in subjects with ACP (44).
Of importance, chronic EtOH administration has been shown to cause oxidative stress in the pancreas, one of the putative mechanisms involved in the development of EtOH-induced pancreatic injury (55, 56). Besides, the presence of such reactive aldehydes as 4-hydroxy-2-nonenal (4HNE), a biomarker for oxidative stress, has been found in acinar cells in the resected pancreatic specimens of patients with ACP (57). Metabolism of EtOH has been demonstrated to induce oxidative stress, which may inhibit mitochondrial functions and various pathways of cellular energetics (11, 58, 59). A positive staining for 4HNE, as found only in the pancreas of chronic EtOH-fed ADH− deer mice, demonstrates a metabolic basis and role of alternative mechanisms related to the metabolism of lipids and/or their oxidation in chronic EtOH-induced pancreatic injury in our ADH− deer mouse model. Besides, our ultrastructural findings of swollen mitochondria with small and irregular cristae clearly show mitochondrial damage in chronic EtOH-fed groups of ADH− deer mice. Therefore, it is likely that the interplay of ER stress and oxidative stress in coordination to impaired cellular energetics underlies EtOH-induced pancreatic injury.
Irrespective of various experimental models of alcoholic pancreatitis, we have demonstrated that chronic EtOH feeding alone can cause a spectrum of morphological and inflammatory changes in the pancreas of ADH− versus ADH+ deer mice. An acute injury is generally associated with a marked proinflammatory cytokine and chemokine response, which may diminish within days. Although, marked morphological and fibrotic changes, ER/oxidative stress and immunological (CD3 and F4/89 positive) changes were observed between chronic EtOH versus chronic EtOH plus binge EtOH and FAEEs-fed ADH− deer mice, cytokine response after 7 days of binge EtOH with/without FAEEs could be at reparative stage (60, 61), which could also explain limited effects of single binge EtOH in aggravating chronic EtOH-induced pancreatic injury in our model.
Of note, oral administration of FAEEs alone in control diet-fed ADH− and ADH+ deer mice and chronic EtOH-fed ADH+ deer mice could not induce pancreatic injury most likely due to lack of formation and/accumulation of requisite amount of FAEEs or their rapid degradation in the presence of low blood alcohol concentration (62). Thus, significant formation and accumulation of FAEEs in the pancreas during chronic alcohol consumption under reduced hepatic ADH levels suggest their role in pathophysiology of ACP (21, 23–25, 37). Recently, we have demonstrated a differential cytotoxicity of EtOH, FAEEs, and acetaldehyde in human primary pancreatic acinar cells (17). The expression of ADH1 and CYP2E1 in pancreatic acinar cells is very low and is not inducible by EtOH exposure (17), and a similar observation was found in the pancreas of chronic EtOH-fed ADH− deer mice fed chronic EtOH (unpublished data). Thus, an exacerbated pancreatic injury observed in ADH− deer mice with oral administration of chronic plus binge EtOH and FAEEs could be linked to an increased body burden of FAEEs in the pancreas and suggest its role in mediating pancreatic injury during chronic alcohol use/abuse.
Although plasma and pancreatic FAEEs levels are not included in this study, we have previously demonstrated a dose-dependent substantial formation of FAEEs in the pancreas and plasma of chronic EtOH-fed hepatic ADH− versus ADH+ deer mice (21, 24, 25). Of importance, evidence show that FFAs (free fatty acids) are more pancreatotoxic than FAEEs, and the levels of FAEEs are parallel to levels of FFAs released during the fat breakdown in alcoholic pancreatitis (38, 63). However, FAEEs alone could cause pancreatic injury (2, 17, 26). Thus, further studies to characterize the lipidomics of the pancreas of chronic EtOH-fed deer mouse model are warranted to identify differentially altered lipids or their conjugation with EtOH and its metabolites for a better understanding of pathogenesis of ACP. In addition of direct toxic effects of alcohol on the pancreas, the pancreatic injury might also be mediated by inflammatory mediators or metabolites secreted from other organs, as alcohol is known to cause systemic toxicity (64, 65). Thus, exploring interorgan cross talk that contributes to the development and progression of alcoholic pancreatitis would be important in understating the mechanism of ACP.
Conclusions
In summary, a greater extent of pancreatic injury as observed in chronic EtOH-fed groups of ADH− versus ADH+ deer mice could be associated with increased body burden of EtOH and its nonoxidative metabolism to FAEEs in the pancreatic gland. A significant degenerative change in ER cisternae and mitochondria observed in the pancreas of chronic EtOH-fed groups of ADH− versus ADH+ deer mice, suggests an interplay among increased body burden of EtOH, and its metabolism under reduced hepatic ADH activity and ER and oxidative stress, key factors involved in the etiopathogenesis of ACP. Our findings on exacerbation of pancreatic injury by chronic binge EtOH and FAEEs-fed ADH− deer mice further support our hypothesis that chronic EtOH-induced pancreatitis has a strong metabolic basis.
GRANTS
This work is supported by funds from Grants AA24699 and AA25850 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA), National Institutes of Health (NIH).
DISCLAIMERS
Its contents are solely the authors’ responsibility and do not necessarily represent the official views of the NIAAA/NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.P.S., L.K., and B.S.K. conceived and designed research; M.P.S., K.K.B., and A.A.C. performed experiments; M.P.S. and A.A.C. analyzed data; M.P.S., K.K.B., B.G., V.L.P., and P.J.B. interpreted results of experiments; M.P.S. prepared figures; M.P.S. drafted manuscript; M.P.S., K.K.B., A.A.C., L.K., B.G., V.L.P., P.J.B., G.A.S.A., and B.S.K. revised and approved the final version of manuscript.
ACKNOWLEDGMENTS
We thank Lori Weaver, UTMB veterinarian staff for the assistance in blood collection from mice and oral gavage feeding. The graphical abstract presented in this study was created with BioRender.com
REFERENCES
- 1.Cote GA, Yadav D, Slivka A, Hawes RH, Anderson MA, Burton FR, Brand RE, Banks PA, Lewis MD, Disario JA, Gardner TB, Gelrud A, Amann ST, Baillie J, Money ME, O'Connell M, Whitcomb DC, Sherman S, North A, Pancreatitis Study G. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol 9: 266–273, 2011. doi: 10.1016/j.cgh.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaphalia BS, Ansari GA. Fatty acid ethyl esters and ethanol-induced pancreatitis. Cell Mol Biol (Noisy-le-grand) 47: OL173–OL179, 2001. [PubMed] [Google Scholar]
- 3.Pandol SJ, Lugea A, Mareninova OA, Smoot D, Gorelick FS, Gukovskaya AS, Gukovsky I. Investigating the pathobiology of alcoholic pancreatitis. Alcohol Clin Exp Res 35: 830–837, 2011. doi: 10.1111/j.1530-0277.2010.01408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clemens DL, Schneider KJ, Arkfeld CK, Grode JR, Wells MA, Singh S. Alcoholic pancreatitis: new insights into the pathogenesis and treatment. World J Gastrointest Pathophysiol 7: 48–58, 2016. doi: 10.4291/wjgp.v7.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasineni K, Srinivasan MP, Balamurugan AN, Kaphalia BS, Wang S, Ding WX, Pandol SJ, Lugea A, Simon L, Molina PE, Gao P, Casey CA, Osna NA, Kharbanda KK. Recent advances in understanding the complexity of alcohol-induced pancreatic dysfunction and pancreatitis development. Biomolecules 10: 669, 2020. doi: 10.3390/biom10050669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Miao Y, Kang F, Dolai S, Gaisano HY. Susceptibility factors and cellular mechanisms underlying alcoholic pancreatitis. Alcohol Clin Exp Res 44: 777–789, 2020. doi: 10.1111/acer.14304. [DOI] [PubMed] [Google Scholar]
- 7.Nuutinen H, Lindros KO, Salaspuro M. Determinants of blood acetaldehyde level during ethanol oxidation in chronic alcoholics. Alcohol Clin Exp Res 7: 163–168, 1983. doi: 10.1111/j.1530-0277.1983.tb05432.x. [DOI] [PubMed] [Google Scholar]
- 8.Sharkawi M. In vivo inhibition of liver alcohol dehydrogenase by ethanol administration. Life Sci 35: 2353–2357, 1984. doi: 10.1016/0024-3205(84)90527-7. [DOI] [PubMed] [Google Scholar]
- 9.Panes J, Caballeria J, Guitart R, Pares A, Soler X, Rodamilans M, Navasa M, Pares X, Bosch J, Rodes J. Determinants of ethanol and acetaldehyde metabolism in chronic alcoholics. Alcohol Clin Exp Res 17: 48–53, 1993. doi: 10.1111/j.1530-0277.1993.tb00725.x. [DOI] [PubMed] [Google Scholar]
- 10.Kaphalia BS, Khan MF, Caroll RM, Aronson J, Ansari GA. Subchronic toxicity of 2-chloroethanol and 2-bromoethanol in rats. Res Commun Pharmacol Toxicol 1: 173–186, 1996. [Google Scholar]
- 11.Cederbaum AI. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig Dis 28: 802–811, 2010. doi: 10.1159/000324289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasan MP, Bhopale KK, Amer SM, Wan J, Kaphalia L, Ansari GS, Kaphalia BS. Linking dysregulated AMPK signaling and ER stress in ethanol-induced liver injury in hepatic alcohol dehydrogenase deficient deer mice. Biomolecules 9: 560, 2019. doi: 10.3390/biom9100560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haber PS, Apte MV, Applegate TL, Norton ID, Korsten MA, Pirola RC, Wilson JS. Metabolism of ethanol by rat pancreatic acinar cells. J Lab Clin Med 132: 294–302, 1998. doi: 10.1016/S0022-2143(98)90042-7. [DOI] [PubMed] [Google Scholar]
- 14.Norton ID, Apte MV, Haber PS, McCaughan GW, Pirola RC, Wilson JS. Cytochrome P4502E1 is present in rat pancreas and is induced by chronic ethanol administration. Gut 42: 426–430, 1998. doi: 10.1136/gut.42.3.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Werner J, Saghir M, Fernandez-del Castillo C, Warshaw AL, Laposata M. Linkage of oxidative and nonoxidative ethanol metabolism in the pancreas and toxicity of nonoxidative ethanol metabolites for pancreatic acinar cells. Surgery 129: 736–744, 2001. doi: 10.1067/msy.2001.113891. [DOI] [PubMed] [Google Scholar]
- 16.Shalbueva N, Mareninova OA, Gerloff A, Yuan J, Waldron RT, Pandol SJ, Gukovskaya AS. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 144: 437–446.e6, 2013. doi: 10.1053/j.gastro.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srinivasan MP, Bhopale KK, Caracheo AA, Kaphalia L, Loganathan G, Balamurugan AN, Rastellini C, Kaphalia BS. Differential cytotoxicity, ER/oxidative stress, dysregulated AMPKα signaling, and mitochondrial stress by ethanol and its metabolites in human pancreatic acinar cells. Alcohol Clin Exp Res 45: 961–978, 2021. doi: 10.1111/acer.14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laposata EA, Lange LG. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 231: 497–499, 1986. doi: 10.1126/science.3941913. [DOI] [PubMed] [Google Scholar]
- 19.Gukovskaya AS, Mouria M, Gukovsky I, Reyes CN, Kasho VN, Faller LD, Pandol SJ. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology 122: 106–118, 2002. doi: 10.1053/gast.2002.30302. [DOI] [PubMed] [Google Scholar]
- 20.Pfutzer RH, Tadic SD, Li HS, Thompson BS, Zhang JY, Ford ME, Eagon PK, Whitcomb DC. Pancreatic cholesterol esterase, ES-10, and fatty acid ethyl ester synthase III gene expression are increased in the pancreas and liver but not in the brain or heart with long-term ethanol feeding in rats. Pancreas 25: 101–106, 2002. doi: 10.1097/00006676-200207000-00021. [DOI] [PubMed] [Google Scholar]
- 21.Amer SM, Bhopale KK, Kakumanu RD, Popov VL, Rampy BA, El-Mehallawi IH, Ashmawy MM, Shakeel Ansari GA, Kaphalia BS. Hepatic alcohol dehydrogenase deficiency induces pancreatic injury in chronic ethanol feeding model of deer mice. Exp Mol Pathol 104: 89–97, 2018. doi: 10.1016/j.yexmp.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivasan MP, Bhopale KK, Caracheo AA, Amer SM, Khan S, Kaphalia L, Loganathan G, Balamurugan AN, Kaphalia BS. Activation of AMP-activated protein kinase attenuates ethanol-induced ER/oxidative stress and lipid phenotype in human pancreatic acinar cells. Biochem Pharmacol 180: 114174, 2020. doi: 10.1016/j.bcp.2020.114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werner J, Saghir M, Warshaw AL, Lewandrowski KB, Laposata M, Iozzo RV, Carter EA, Schatz RJ, Fernandez-Del Castillo C. Alcoholic pancreatitis in rats: injury from nonoxidative metabolites of ethanol. Am J Physiol Gastrointest Liver Physiol 283: G65–G73, 2002. doi: 10.1152/ajpgi.00419.2001. [DOI] [PubMed] [Google Scholar]
- 24.Bhopale KK, Wu H, Boor PJ, Popov VL, Ansari GA, Kaphalia BS. Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol 39: 179–188, 2006. doi: 10.1016/j.alcohol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 25.Kaphalia BS, Bhopale KK, Kondraganti S, Wu H, Boor PJ, Ansari GA. Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol Appl Pharmacol 246: 154–162, 2010. doi: 10.1016/j.taap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci U S A 101: 10738–10743, 2004. doi: 10.1073/pnas.0403431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ansari GA, Kaphalia BS, Boor PJ. Selective pancreatic toxicity of palmitoylpentachlorophenol. Toxicology 46: 57–63, 1987. doi: 10.1016/0300-483x(87)90137-5. [DOI] [PubMed] [Google Scholar]
- 28.Guo F, Zheng K, Benede-Ubieto R, Cubero FJ, Nevzorova YA. The Lieber-DeCarli Diet-A flagship model for experimental alcoholic liver disease. Alcohol Clin Exp Res 42: 1828–1840, 2018. doi: 10.1111/acer.13840. [DOI] [PubMed] [Google Scholar]
- 29.Kaphalia BS, Cai P, Khan MF, Okorodudu AO, Ansari GA. Fatty acid ethyl esters: markers of alcohol abuse and alcoholism. Alcohol 34: 151–158, 2004. doi: 10.1016/j.alcohol.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 30.Moreno C, Nicaise C, Gustot T, Quertinmont E, Nagy N, Parmentier M, Louis H, Deviere J. Chemokine receptor CCR5 deficiency exacerbates cerulein-induced acute pancreatitis in mice. Am J Physiol Gastrointest Liver Physiol 291: G1089–G1099, 2006. doi: 10.1152/ajpgi.00571.2005. [DOI] [PubMed] [Google Scholar]
- 31.Bettaieb A, Chahed S, Tabet G, Yang J, Morisseau C, Griffey S, Hammock BD, Haj FG. Effects of soluble epoxide hydrolase deficiency on acute pancreatitis in mice. PLoS One 9: e113019, 2014. doi: 10.1371/journal.pone.0113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Yu Q, Xu C-B. A convenient method for quantifying collagen fibers in atherosclerotic lesions by ImageJ software. Int J Clin Exp Med 10: 14904–14910, 2017. [Google Scholar]
- 33.Kattner N, Dyson N, Bury Y, Tiniakos D, White K, Davey T, Eliasson L, Tindale L, Wagner BE, Honkanen-Scott M, Doyle J, Ploeg RJ, Shaw JA, Scott WE. Development and validation of a quantitative electron microscopy score to assess acute cellular stress in the human exocrine pancreas. J Pathol Clin Res 7: 173–187, 2021. [PMC]doi: 10.1002/cjp2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H, Cai P, Clemens DL, Jerrells TR, Ansari GA, Kaphalia BS. Metabolic basis of ethanol-induced cytotoxicity in recombinant HepG2 cells: role of nonoxidative metabolism. Toxicol Appl Pharmacol 216: 238–247, 2006. [Erratum in Toxicol Appl Pharmacol 220: 111–112, 2007]. doi: 10.1016/j.taap.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Best CA, Sarkola T, Eriksson CJ, Cluette-Brown JE, Laposata M. Increased plasma fatty acid ethyl ester levels following inhibition of oxidative metabolism of ethanol by 4-methylpyrazole treatment in human subjects. Alcohol Clin Exp Res 30: 1126–1131, 2006. doi: 10.1111/j.1530-0277.2006.00138.x. [DOI] [PubMed] [Google Scholar]
- 36.Doyle KM, Bird DA, Al-Salihi S, Hallaq Y, Cluette-Brown JE, Goss KA, Laposata M. Fatty acid ethyl esters are present in human serum after ethanol ingestion. J Lipid Res 35: 428–437, 1994. [PubMed] [Google Scholar]
- 37.Doyle KM, Cluette-Brown JE, Dube DM, Bernhardt TG, Morse CR, Laposata M. Fatty acid ethyl esters in the blood as markers for ethanol intake. JAMA 276: 1152–1156, 1996. [Erratum in JAMA 227: 792, 1997]. doi: 10.1309/6F39-EAR2-L4GY-X5G6. [DOI] [PubMed] [Google Scholar]
- 38.Vela S, Guerra A, Farrell G, Trivedi S, Chaffin H, Rood C, Singh R, Kostenko S, Chang YH, Snozek C, Patel K, Khatua B, Singh VP. Pathophysiology and biomarker potential of fatty acid ethyl ester elevation during alcoholic pancreatitis. Gastroenterology 161: 1513–1525, 2021. doi: 10.1053/j.gastro.2021.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrera K, Stanek A, Okochi K, Niewiadomska Z, Mueller C, Ou P, John D, Alfonso AE, Tenner S, Huan C. Acinar cell injury induced by inadequate unfolded protein response in acute pancreatitis. World J Gastrointest Pathophysiol 9: 37–46, 2018. doi: 10.4291/wjgp.v9.i2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 140: 987–997, 2011. doi: 10.1053/j.gastro.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 197: 857–867, 2012. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helin H, Mero M, Markkula H, Helin M. Pancreatic acinar ultrastructure in human acute pancreatitis. Virchows Arch A Pathol Anat Histol 387: 259–270, 1980. doi: 10.1007/BF00454829. [DOI] [PubMed] [Google Scholar]
- 43.Ghadially FN. Ultrastructural Pathology of the Cell and Matrix. Boston: Butterworth-Heinemann, 1997. [Google Scholar]
- 44.Sah RP, Garg SK, Dixit AK, Dudeja V, Dawra RK, Saluja AK. Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J Biol Chem 289: 27551–27561, 2014. doi: 10.1074/jbc.M113.528174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lugea A, Gerloff A, Su HY, Xu Z, Go A, Hu C, French SW, Wilson JS, Apte MV, Waldron RT, Pandol SJ. The combination of alcohol and cigarette smoke induces endoplasmic reticulum stress and cell death in pancreatic acinar cells. Gastroenterology 153: 1674–1686, 2017. doi: 10.1053/j.gastro.2017.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest 128: 64–73, 2018. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clemens DL, Jerrells TR. Ethanol consumption potentiates viral pancreatitis and may inhibit pancreas regeneration: preliminary findings. Alcohol 33: 183–189, 2004. doi: 10.1016/j.alcohol.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 48.Lugea A, Gukovsky I, Gukovskaya AS, Pandol SJ. Nonoxidative ethanol metabolites alter extracellular matrix protein content in rat pancreas. Gastroenterology 125: 1845–1859, 2003. doi: 10.1053/j.gastro.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 49.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-α. Role in regulating cell death and pancreatitis. J Clin Invest 100: 1853–1862, 1997. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brivet FG, Emilie D, Galanaud P. Pro- and anti-inflammatory cytokines during acute severe pancreatitis: an early and sustained response, although unpredictable of death. Parisian Study Group on Acute Pancreatitis. Crit Care Med 27: 749–755, 1999. doi: 10.1097/00003246-199904000-00029. [DOI] [PubMed] [Google Scholar]
- 51.Hirota M, Nozawa F, Okabe A, Shibata M, Beppu T, Shimada S, Egami H, Yamaguchi Y, Ikei S, Okajima T, Okamoto K, Ogawa M. Relationship between plasma cytokine concentration and multiple organ failure in patients with acute pancreatitis. Pancreas 21: 141–146, 2000. doi: 10.1097/00006676-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 52.Yang BM, Demaine AG, Kingsnorth A. Chemokines MCP-1 and RANTES in isolated rat pancreatic acinar cells treated with CCK and ethanol in vitro. Pancreas 21: 22–31, 2000. doi: 10.1097/00006676-200007000-00048. [DOI] [PubMed] [Google Scholar]
- 53.Regner S, Appelros S, Hjalmarsson C, Manjer J, Sadic J, Borgstrom A. Monocyte chemoattractant protein 1, active carboxypeptidase B and CAPAP at hospital admission are predictive markers for severe acute pancreatitis. Pancreatology 8: 42–49, 2008. doi: 10.1159/000114866. [DOI] [PubMed] [Google Scholar]
- 54.Aoun E, Chen J, Reighard D, Gleeson FC, Whitcomb DC, Papachristou GI. Diagnostic accuracy of interleukin-6 and interleukin-8 in predicting severe acute pancreatitis: a meta-analysis. Pancreatology 9: 777–785, 2009. doi: 10.1159/000214191. [DOI] [PubMed] [Google Scholar]
- 55.Norton ID, Apte MV, Lux O, Haber PS, Pirola RC, Wilson JS. Chronic ethanol administration causes oxidative stress in the rat pancreas. J Lab Clin Med 131: 442–446, 1998. doi: 10.1016/S0022-2143(98)90145-7. [DOI] [PubMed] [Google Scholar]
- 56.Ren Z, Wang X, Xu M, Yang F, Frank JA, Ke ZJ, Luo J. Binge ethanol exposure causes endoplasmic reticulum stress, oxidative stress and tissue injury in the pancreas. Oncotarget 7: 54303–54316, 2016. doi: 10.18632/oncotarget.11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Casini A, Galli A, Pignalosa P, Frulloni L, Grappone C, Milani S, Pederzoli P, Cavallini G, Surrenti C. Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J Pathol 192: 81–89, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 58.Lange LG, Sobel BE. Mitochondrial dysfunction induced by fatty acid ethyl esters, myocardial metabolites of ethanol. J Clin Invest 72: 724–731, 1983. doi: 10.1172/JCI111022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130: 781–793, 2006. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 60.Jakkampudi A, Jangala R, Reddy R, Mitnala S, Rao GV, Pradeep R, Reddy DN, Talukdar R. Acinar injury and early cytokine response in human acute biliary pancreatitis. Sci Rep 7: 15276, 2017. doi: 10.1038/s41598-017-15479-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manohar M, Jones EK, Rubin SJS, Subrahmanyam PB, Swaminathan G, Mikhail D, Bai L, Singh G, Wei Y, Sharma V, Siebert JC, Maecker HT, Husain SZ, Park WG, Pandol SJ, Habtezion A, Pandol SJ, Habtezion A. Novel circulating and tissue monocytes as well as macrophages in pancreatitis and recovery. Gastroenterology 161: 2014–2029.e14, 2021. doi: 10.1053/j.gastro.2021.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saghir M, Werner J, Laposata M. Rapid in vivo hydrolysis of fatty acid ethyl esters, toxic nonoxidative ethanol metabolites. Am J Physiol 273: G184–G190, 1997. doi: 10.1152/ajpgi.1997.273.1.G184. [DOI] [PubMed] [Google Scholar]
- 63.Patel K, Durgampudi C, Noel P, Trivedi RN, de Oliveira C, Singh VP. Fatty acid ethyl esters are less toxic than their parent fatty acids generated during acute pancreatitis. Am J Pathol 186: 874–884, 2016. doi: 10.1016/j.ajpath.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Osna NA, Kharbanda KK. Multi-organ alcohol-related damage: mechanisms and treatment. Biomolecules 6: 20, 2016. doi: 10.3390/biom6020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Armutcu F. Organ crosstalk: the potent roles of inflammation and fibrotic changes in the course of organ interactions. Inflamm Res 68: 825–839, 2019. doi: 10.1007/s00011-019-01271-7. [DOI] [PubMed] [Google Scholar]