

Abstract
Microviridins, tricyclic peptide natural products originally isolated from cyanobacteria, function as inhibitors of diverse serine-type proteases. Here we report the structure and biochemical characterization of AMdnB, a unique iterative macrocyclase involved in a microviridin biosynthetic pathway from Anabaena sp. PCC 7120. The ATP-dependent cyclase, along with the homologous AMdnC, introduce up to nine macrocyclizations on three distinct core regions of a precursor peptide, AMdnA. The results presented here provide structural and mechanistic insight into the iterative chemistry of AMdnB. In vitro AMdnB-catalyzed cyclization reactions demonstrate the synthesis of the two predicted tricyclic products from a multi-core precursor peptide substrate, consistent with a distributive mode of catalysis. The X-ray structure of AMdnB shows a structural motif common to ATP-grasp cyclases involved in RiPPs biosynthesis. Additionally, comparison with the non-iterative MdnB allows insight into the structural basis for the iterative chemistry. Overall, the presented results provide insight into the general mechanism of iterative enzymes in ribosomally synthesized and post-translationally modified peptide biosynthetic pathways.
Keywords: Peptide macrocyclase, ribosomally synthesized and post-translationally modified peptide, biosynthesis, ATP, microviridins
1. Introduction
The broad family of peptide-based natural products includes great structural and functional diversity that is commonly used as biological tools and therapeutics.1,2 Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a large subclass of peptide natural products with relatively simple biosynthetic pathways, providing unique opportunities for the engineered production of biologically-active molecules. The development of in silico genome mining methods, combined with molecular biology approaches, has allowed the characterization and prediction of numerous members of this superfamily.3 The RiPPs family currently encompasses over 40 subclasses, including lanthipeptides, linear azol(in)e-containing peptides, thiopeptides, lasso peptides, and cyanobactins.4 These biosynthetic pathways begin with a ribosome-derived precursor peptide, commonly consisting of leader (LP) and unmodified core peptide (UCP) regions. The core region constitutes the amino acids present in the final product. An N-terminal LP (or less commonly a C-terminal follower) is key for substrate recognition by post-translational modification (PTM) enzymes, producing a modified core peptide and subsequently the final natural product.4 Following various PTMs on the UCP, the leader (or follower) region is removed by proteolysis. Various enzyme chemistries are known to modify the core peptide, including macrocyclization, dehydration, heterocyclization, epimerization and prenylation.4,5 Peptide macrocyclization is a common modification in both non-ribosomal peptide and RiPPs biosynthesis, producing molecules with favorable in vivo stability and biological function.6,7
Microviridins are polycyclic peptides produced by several cyanobacterial strains,8,9,10 and are the first members of the recently named graspetide family.11 The structure of microviridin J bound to the bovine trypsin protease provides a structural basis for a substrate-mimic binding mode, with the analogous scissile peptide bond near the catalytic triad but not hydrolyzed.12 Due to the common and prevalent role of serine proteases in biology, microviridins and analogs are potentially useful small molecule tools and therapeutic lead compounds.13,14 The cage-like architecture of microviridins is biosynthesized by macrocyclases belonging to the ATP-grasp superfamily of enzymes.15 ATP-grasp ligases participate in various primary and secondary metabolic pathways commonly forming isopeptide or ester bonds.16,17 Several ATP-grasp superfamily members have been structurally characterized, revealing a conserved motif among the family despite relatively low sequence identity.18–20 An example is LysX, an essential enzyme in L-lysine biosynthesis, catalyzing the acylation of α-aminoadipate.15 The structure contains three α+β domains, each composed of a four or five-stranded β-sheet core flanked by α-helices. Additionally, a structurally conserved nucleotide binding site is common to the superfamily.21 Diverse ATP-grasp enzymes include d-Ala-d-Ala ligase, PurT glycinamide ribonucleotide transformylase, glutathione synthetase, and synapsin I, sharing a common scaffold despite varied chemistries.16 The crystal structures of both the isopeptide-forming MdnB and ester-forming MdnC have previously been determined from the microviridin J pathway of Microcystis aeruginosa UOWOCC MRC.22 The structures show that the cyclases have the canonical ATP-grasp fold. The structure of MdnC bound to the MdnA LP provides a molecular basis for substrate binding and activation of the enzyme for processing the UCP through macrocyclization. The LP binds in an α-helical conformation and binding results in a conformational change around the putative cyclization active site. Additionally, comparison of MdnC and MdnB to homologous ATP-grasp enzyme structures, along with molecular docking, allowed for identification of the putative ATP and modified core peptide (MCP) binding sites.22 Recently, the structure and characterization of the ester bond-forming CdnC, an ATP-grasp ligase in the chryseoviridin biosynthetic pathway, confirmed the conserved ATP-grasp fold for graspetide ligases along with a similar binding site and conformation of the leader peptide to that observed with MdnC.23 Structures of CdnC bound to substrate peptide illustrate an asymmetric binding mode and a basis for iterative, multicore cyclization chemistry.
Typically, RiPP gene clusters contain genes that encode for a single precursor peptide with one core region that multiple PTM enzymes elaborate to a mature final product.4 Our recent genome mining efforts identified 503 microviridin precursor peptides from four bacterial phyla, of which 24% carry 2 to 5 core peptides, indicating a prevalence of modular biosynthesis (Figure S1). A gene cluster (Figure 1a) from Anabaena sp. PCC 7120 contains a precursor peptide (AMdnA) with a LP region and three predicted UCP regions within the one ribosomal polypeptide. AMdnC catalyzes the formation of two esters between Ser/Glu and Thr/Asp residues,24 while AMdnB is expected to add a macrolactam, crosslinking Lys and Glu sidechains. After the formation of three rings, a peptidase-fused ATP-binding cassette transporter (AMdnE) putatively cleaves the modified AMdnA into three cyclized core peptides followed by capping of the N-termini by the N-acetyltransferase, AMdnD (Figure S2). The products of the cluster are predicted to be three structurally different microviridin tricycles (Figure 1b). This structural variability suggests the products could have unique targets for inhibition, in particular differences in the S1 site of a protease inhibitor. Multicore precursor peptides are known in several RiPPs pathways but are relatively rare in occurrence. Characterized examples of multicore RiPP precursor peptides include the cyanobactins, plesiocins, fungal dikaritins and cyclotides.25 A thorough biosynthetic understanding of the multicore RiPP processing will provide useful insights into natural product combinatorial biosynthesis, aiding genome-based discovery of new RiPP families and synthetic biology engineering.
FIGURE 1.
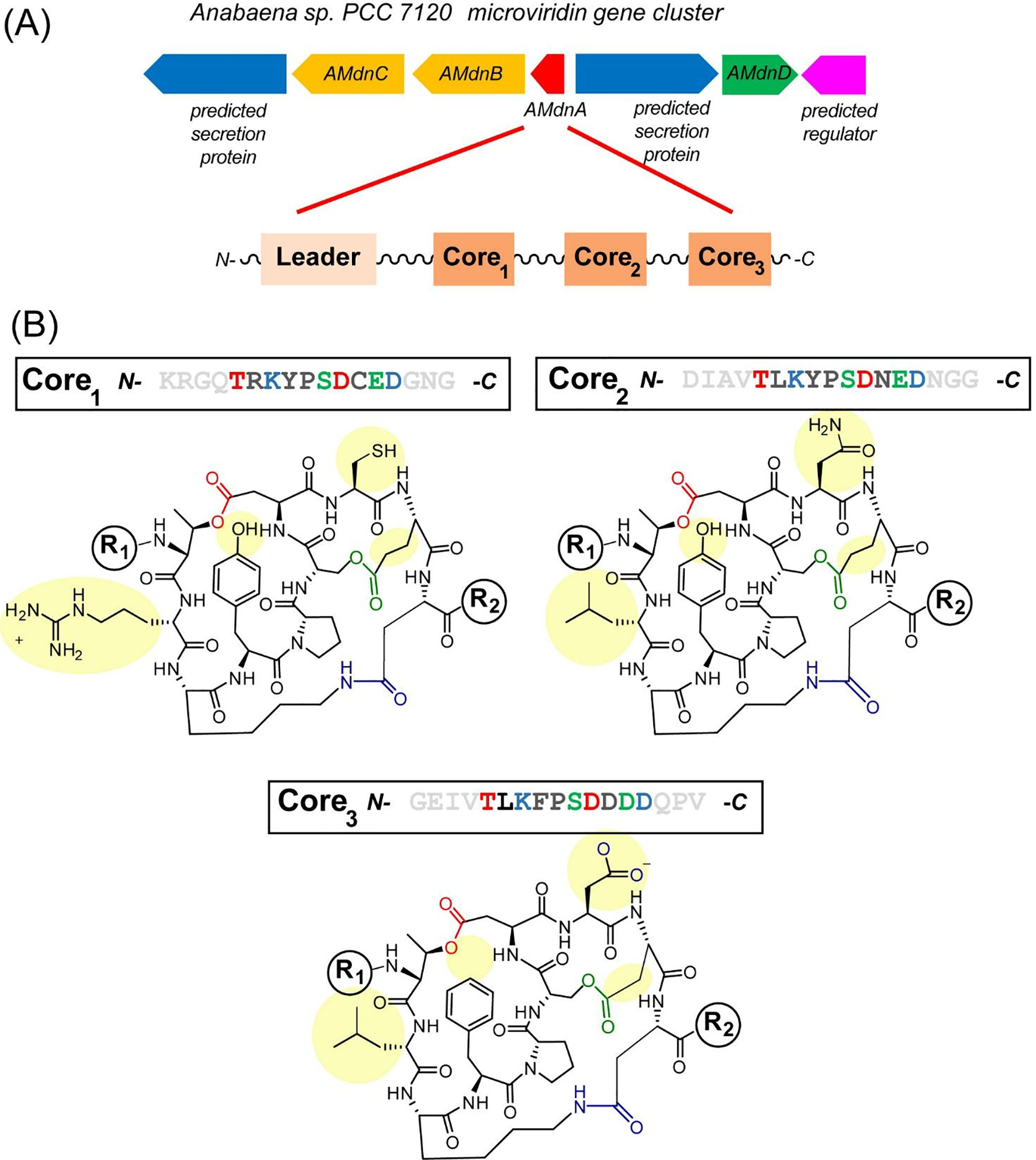
Iterative microviridin biosynthesis in the cyanobacterium, Anabaena sp. PCC 7120. (A) The gene cluster of the microviridin Anabaena sp. PCC 7120 pathway encodes: AMdnC and AMdnB, two macrocyclases; AMdnA, precursor peptide; AMdnD, acetyltransferase; and AMdnE, transporter/peptidase are illustrated along with a schematic representation of the multi-core peptide AMdnA. (B) Sequences of three unmodified core peptide (UCP) regions of AMdnA along with the predicted tricyclized peptide products. The regions of diversity among the three cyclized peptides are highlighted in yellow. Residues are colored based on the specific lactones/lactam formed. R1/R2 groups represent different potential peptidic structures at the N- and C-termini based on proteolytic processing.
Previously, we characterized modular catalysis by AMdnC from Anabaena sp. PCC 7120 responsible for the first PTM of the precursor peptide, AMdnA, forming two esters in the CP.22 The results demonstrated that AMdnC processes all three core regions within AMdnA in a distributive manner and a preferred N- to C- directionality. The catalysis was characterized as multiple cycles of substrate binding and release. In addition, AMdnC was shown to process non-natural peptide substrates containing different numbers of CPs. A distributive catalysis was also observed with enzymes processing cyanobactins, such as in the biosynthesis of patellamide from Prochloron didemni.26,27 These results suggest a common biosynthetic characteristic of multicore processing.
Here we report the structural and biochemical characterization of AMdnB, an iterative macrocyclase of the microviridin family acting downstream of AMdnC. The results provide a structural basis for iterative catalysis by a RiPP PTM macrocyclase. From comparison to the chemically homologous MdnB of the microviridin J pathway, unique structural features include a flexible active site and distinct differences in precursor peptide binding site. This comparative analysis highlights structural variation between iterative and non-iterative enzyme homologs. Additionally, biochemical characterization demonstrates that AMdnB generates two tricyclic CPs from an enzymatically synthesized macrolactone precursor substrate. Based on the results, a mechanism where precursor peptide binding and cyclization occurs on the homodimeric enzyme is proposed. Overall, this work provides molecular insight into iterative RiPP catalysis, with applications to other pathways and as tools for pathway engineering and catalysis.
2. RESULTS
2.1. Biochemical characterization of AMdnB
To biochemically characterize AMdnB, we first synthesized AMdnA-Δ5 (with Δ representing dehydration, loss of 18Da) from AMdnA/AMdnC. AMdnA-Δ5 consists of the monolactonized 1st CP (M1-Δ1) and bicyclic 2nd and 3rd CPs (M2-Δ2 and M3-Δ2). AMdnA-Δ5 is one of the potential UCP substrates of AMdnB.9 Purified AMdnA-Δ5 was incubated with recombinant AMdnB in the presence of Mg2+ and ATP. High resolution (HR)-MS analysis identified two species whose molecular weights were smaller than AMdnA-Δ5 by 1–2 repeats of 18 Da, along with those whose molecular weights were higher than AMdnA-Δ5 by repeats of 18 Da (Figure 2a). All these species were absent in controls with heat-inactivated AMdnB (Figure S3). We next treated the reaction mixture with the GluC endoproteinase and LC-HR-MS and MS/MS analysis of the digestion mixture revealed the presence of tricyclic M2 and M3 (Figures 2b–d, S4, Table S1), suggesting that AMdnB catalyzes the macrolactamization on the bicyclic 2nd and 3rd CPs of AMdnA-Δ5. The accumulation of AMdnA-Δ6 and AMdnA-Δ7 species in the AMdnB reaction suggests distributive catalysis, a potential common hallmark of the RiPP modular biosynthesis. Additionally, the data indicates that the first core peptide is not modified, while the second and third can be modified. Attempts to improve conversion to AMdnA-Δ7 using a higher enzyme concentration of AMdnB and/or longer reaction times were not successful.
FIGURE 2.
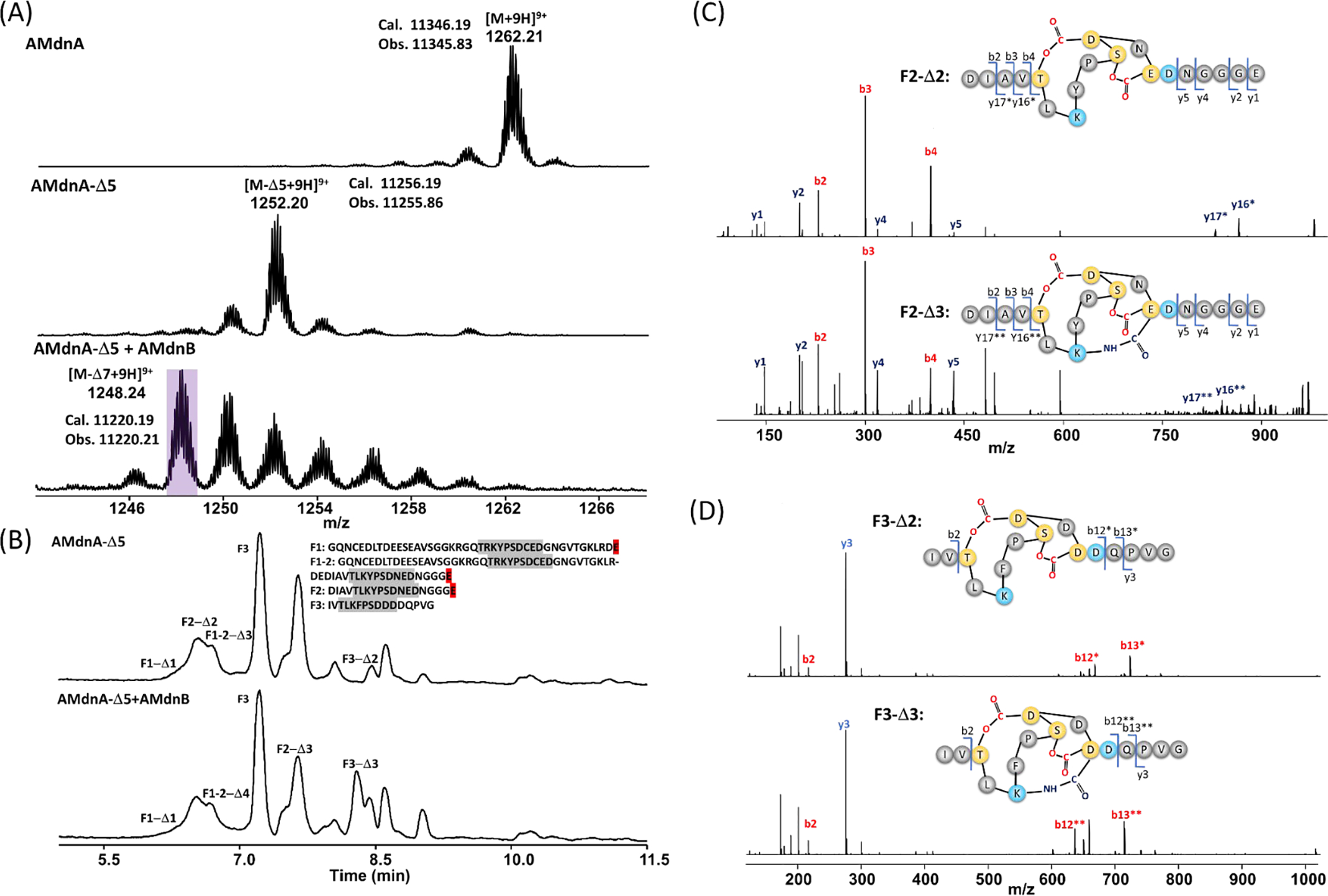
(A) HR-MS analysis detected new species with six to seven dehydrations (Δ) after incubating AMdnA-Δ5 with AMdnB at a molar ratio of 20:1 for 16 h. The most abundant species lost seven waters from AMdnA. (B) HPLC traces of AMdnA-Δ5 (top) and processed (bottom) AMdnA-Δ7 after GluC digestion. Key chromatographic peaks were labeled with the names of corresponding peptide fragments released by GluC (the F2-Δ3 peak coelutes with an unidentified peak). HR tandem MS analysis characterized the released F2-Δ2 (C) and F3-Δ2 (D) of AMdnA-Δ5 and the F2-Δ3 (C) and F3-Δ3 (D) of processed AMdnA. The samples were treated with GluC for 16 h.
2.2. X-ray structure of AMdnB
The structure of AMdnB (Figure 3a) confirms an ATP-grasp ligase fold with significant homology to the two MdnB and MdnC macrocyclases from the microviridin J pathway.22 As is common with ATP-grasp enzymes, AMdnB is a homodimer where each monomer consists of eight α-helices and 16 β-strands, comprising the three characteristic domains of the ATP-grasp superfamily: N-, C- and central. The central domain of AMdnB consists of a partially ordered helix (α7) at the precursor peptide binding site, consistent with the previous apo structure of MdnB.22 As expected, structure similarity searches28 reveal that AMdnB shares a similar structural layout with other ATP-grasp superfamily members despite a relatively low sequence identity (~<20%). After significant effort, we were not able to obtain co-complex structures with bound substrate peptides and/or ATP/ATP analogs. However, based on strong homology to MdnB and MdnC,21 the LP, CP and cofactor binding modes can be predicted (Figure 3a). A characteristic of ATP-grasp fold proteins is a functional homodimer. AMdnB likewise forms the dimer (Figure 3b) with extensive protein/protein interactions along the interface, specifically with α3 and the β3/β4. Structural alignment of the four monomers observed in the asymmetric unit of the crystal structure (Figure 3c) provides insight into the general flexibility of each protein monomer chain. Two protein regions predicted to respond to bound LP, a flexible loop region (residues ~172–186) and the α7 helix, are notably variable in comparison of the four structures. This is consistent with protein conformational changes observed with LP-bound MdnC to where the β9/β10 hairpin closes on the active site upon LP binding. The conformational flexibility on the LP binding regions is exemplified in the structure of the crystallographic tetramer of the asymmetric unit (Figure 3d), where a helical region of one dimer is bound into the LP binding site of another dimer.
FIGURE 3.
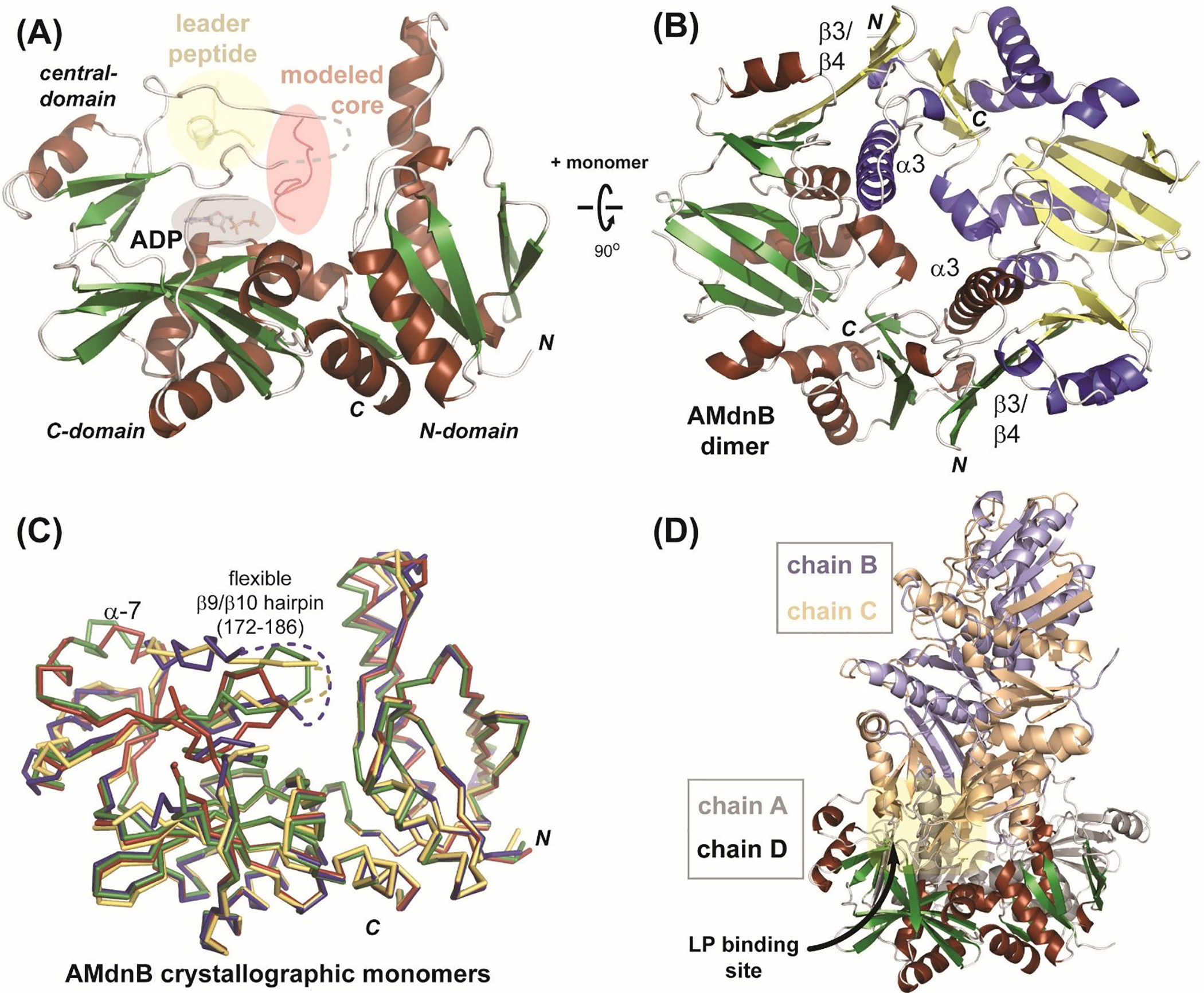
X-ray structure of AMdnB. (A) Monomeric AMdnB illustrating the domains of the ATP-grasp fold with modeled binding regions for LP region of the precursor peptide (yellow), UCP (red), and ADP (grey). Residues 172–186 are disordered (dashed line) (B) Dimeric structure of AMdnB, with the two monomers colored red/green and blue/yellow. (C) Four crystallographic AMdnB monomers aligned with each other; the flexible active site loop (β9/β10 hairpin) is highlighted. (D) Tetrameric structure in the crystal asymmetric unit. The orientation of chain D is the same as panels A and C.
2.3. Structural comparison to non-iterative MdnB
In order to gain structural insight into the iterative-acting AMdnB, the structure was compared with MdnB22 from the microviridin J pathway (Figure 4). As mentioned, AMdnB shares many of the common structural features of the superfamily and in particular with MdnB. Some differences are noteworthy, including variation of the active site pocket and the protein surface. In both structures, the α7 helix is partially ordered but differs slightly in location relative to the substrate binding site (Figure 4a). Additionally, the β9/β10 hairpin motif, adjacent to the active site, is disordered in AMdnB but ordered into distinct secondary structure elements in MdnB. As a comparison, in the structure of MdnC, along with CdnC, with LP bound,23 both the α7 helix and the β9/β10 hairpin are well ordered. Considering the rigidity of the α7 helix in LP-bound MdnC and CdnC (Figure S6), it is reasonable to assume that the disordered α7 helix is due to the absence of the substrate. The observed disordered β9/β10 hairpin region, along with a likewise shift of the β6/α3 loop, suggests additional flexibility in the AMdnB active site. This plasticity could function to accommodate the three UCP regions of a large cyclized AMdnA precursor peptide in a distributive manner. Of note, the three UCP regions further demonstrate sequence variations (Figure 1b). Structural differences of MdnB/AMdnB were examined by sequence alignment (Figure S7) along with analysis of the structures focusing on the surface of the precursor peptide binding area. A comparison of the electrostatic surfaces (Figure 4b) reveals a distinct charge localization in AMdnB at the dimer interface. Additionally, sequence variations (discounting surface loops) between AMdnB and MdnB are also localized to the dimer interface (orange residues, Figure 4c). The differences suggest variations that favor a multi-CP iterative mode of catalysis are at the homodimer interface where the precursor peptide linkers are predicted to interact.
FIGURE 4.
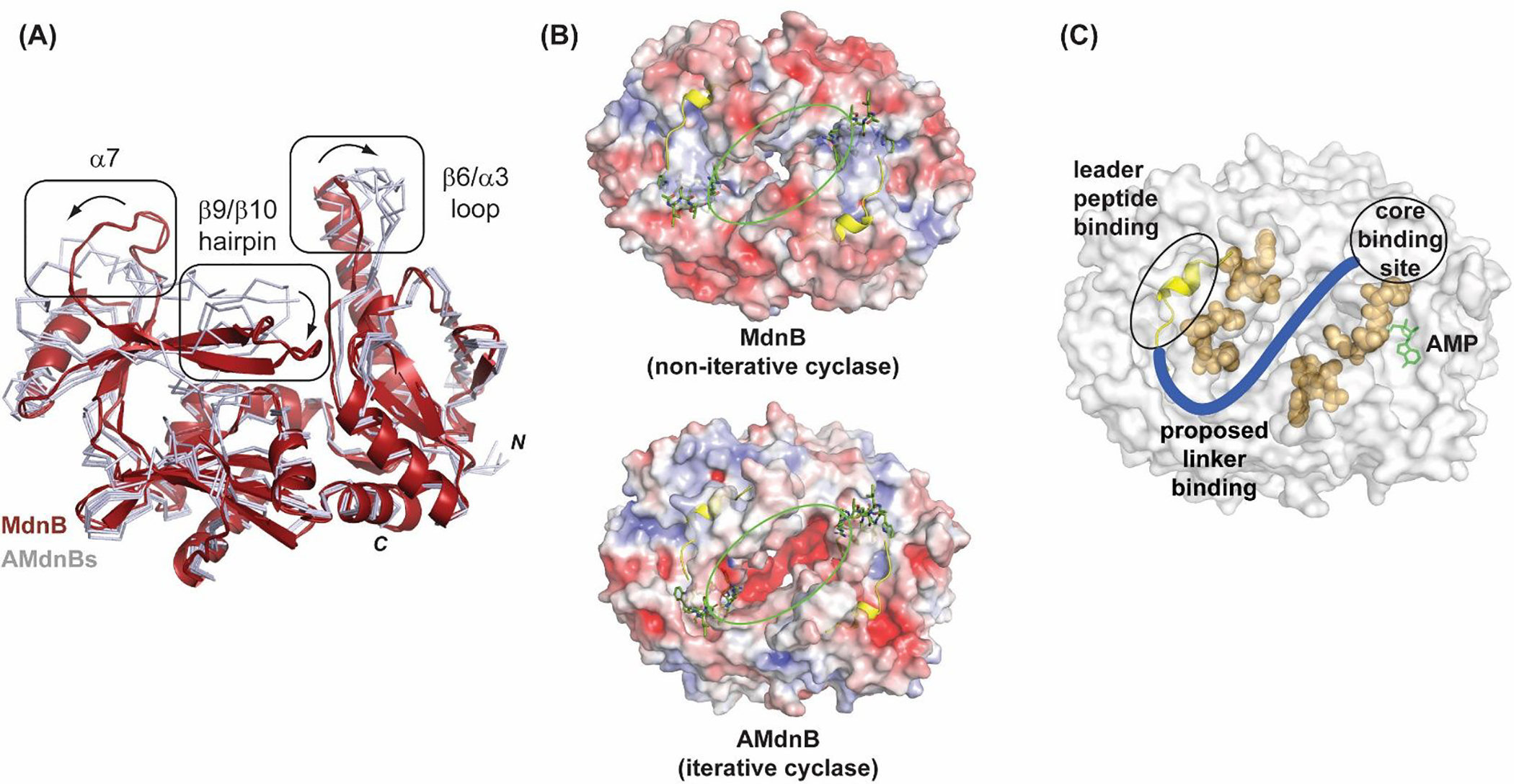
Structural differences between AMdnB (iterative-acting) and MdnB (non-iterative). (A) Comparison of AMdnB with MdnB from the microviridin J pathway. Crystallographic AMdnB monomers (grey ribbons) are aligned with MdnB (red cartoon representation). Major observed differences between the two structures are boxed. (B) Comparison of solvent accessible protein surfaces of AMdnB and MdnB. LP (yellow cartoon representation) and modeled UCP (green stick representation) are shown. Surfaces are colored by electrostatic charge, red(−)/ blue(+). (C) Surface representation of AMdnB homodimer with LP and cyclization active site indicated. Proposed linker binding channel (blue) with divergent residues (AMdnB vs. MdnB) in orange spheres. Modeled22 AMP (green stick representation) is shown near core binding site.
2.4. Precursor peptide interactions and model for catalysis
The common mode for precursor peptide binding and catalysis for RiPP enzymes is discrete binding sites for recognition (LP) and catalysis (UCP).29,30 X-ray structures for full-length precursor peptide/PTM enzyme co-complex structures have remained a challenge to obtain. This is not unanticipated for RiPP PTM enzymes, as substrate binding is generally localized at the LP/enzyme interface. In order to provide insight into AMdnA interactions with AMdnB, we examined general structural features of the apo- protein. The distance between the LP and UCPs of AMdnA can be estimated from the number of residues and the distance between the LP and UCP M1 is significant (> 50 Å). The distance between the LP and UCP binding sites of the AMdnB monomer (Figure 3A) is <10 Å. In comparison, the distance across the dimer interface is ~25 Å, a distance that is more feasible to accommodate lengthy linkers and multiple UCPs. This is particularly relevant to large multicore precursor peptides such as AMdnA. Additionally, as described above, notable differences between AMdnB and MdnB are present along this channel between the homomonomers. This includes prominent sequence variation at residues lining this channel (Figure 4c) and variation of surface charge, with AMdnB containing a strongly acidic charged patch at the channel. The linker regions of AMdnA (and homologs) are also largely acidic and a charge repulsion interaction can allow iterative AMdnB to bind and release multiple UCPs. Based on this model, an inter-CP linker will pass through a channel at the dimer interface from the LP binding site of one monomer to the active site of the second monomer.
2.5. Binding Affinity of AMdnB/AMdnA LP
To probe the interaction between AMdnA LP and AMdnB, substrate binding was assayed using isothermal titration calorimetry. The LP of AMdnA contains the canonical PFFARFL motif that forms an α-helical LP structural motif. Based on previous results, LP binding affinity is largely localized to the PFFARFL motif and is not dependent on metal ion or cofactor ATP.21 The results show that AMdnA1–24 peptide (truncated AMdnA LP) binds to AMdnB with a measured binding constant (Kd) of 12.3 ± 4.0 μM (Figure S8). The binding is lower than that measured for MdnC (84-fold lower) and MdnB (4.4-fold lower) from the microviridin J biosynthetic pathway.22 The relatively weaker binding could indicate greater mobility of the substrate during the macrocyclization reactions where the precursor peptide would bind in a relatively loose conformation.
2.6. Bioinformatic mining of multi-core microviridin gene clusters
To evaluate the general prevalence of precursor peptides containing multiple core peptides in microviridin biosynthesis, we performed a bioinformatic survey of the microviridin precursor peptides on the basis of previously reported data.8 Using homology-based method, we were able to identify 503 microviridin precursor peptides with various length and composition from 312 putative microviridin and marinostatin biosynthetic gene clusters (BGCs) (Figure S1). To identify microviridin gene clusters, BlastP searches (E < 1e−50) were performed using the AMdnA (WP_010999573) peptide sequence and other microviridin precursor peptide sequences as the queries in the nonredundant database at NCBI. The individual adjacent open reading frames (ORFs) were manually inspected with the Conserved Domain Database analysis to identify putative ATP-grasp ligases and other enzymes involved in microviridin biosynthesis (e.g.: MdnD analogs for N-acetylation, MdnE analogs for cross‐membrane transportation). This allowed the identification of candidate microviridin gene clusters with sequence identities of 40−100%. Multiple sequence alignment of the precursor peptides was conducted using a web server T-Coffee31 and visualized using Jalview.32 Intriguingly, three microviridin precursor peptides from two microviridin BGCs were identified from the phylum of acidobacteria. We believe this is the first time microviridin BGCs have been identified outside the bacterial phyla of cyanobacteria, bacteroidetes and proteobacteria. A sequence similarity network of the 503 microviridin precursor sequences was generated using the EFI-Enzyme Similarity Tool (EFI-EST).33 The sequence similarity network contained 503 nodes with 47633 edges constructed by pairwise BLAST alignments with an E value better than 10−5. Cytoscape 3.2.1 was used to visualize the sequence similarity network using the organic layout. In line with previous findings, this network revealed clear clustering of three clades (Figure S1), i.e. Class I: cyanobacterial precursor peptides with single core peptide; Class II: mostly precursor peptides containing multiple core peptides (2–5) from all aforementioned bacterial phyla; Class III: mostly precursor peptides from bacteriodetes with single core peptide featured with longer linker regions after the conserved leader peptide PFFAXF/YL/I compared to Class I. Although the precursor peptides containing multiple core peptides account for approximately 24% of the whole collection and span over all four phyla, the popularity of the phenomenon differs significantly among the different phylum (35% in cyanobacteria, 8% in bacteroidetes, 76% in proteobacteria and 100% in acidobacteria; the ratio in acidobacteria may be biased due to small sample volume).
3. DISCUSSION
In this study, we describe the structural and biochemical characterization of an iterative ribosomal peptide macrocyclase, AMdnB, involved in microviridin biosynthesis, providing insight into multicore RiPPs biosynthetic pathways. AMdnB and MdnB are useful model systems to understand differences between iterative and non-iterative PTM enzymes as the two enzymes are highly homologous and perform the same chemistry. AMdnB was assayed against a precursor peptide, after pre-esterification by AMdnC, and was shown to catalyze the synthesis of two predicted macrolactams. However, as the natural product(s) microviridins from Anabaena sp. PCC 7120 are as of yet unknown, it is not clear how this in vitro result corresponds to that of the in vivo pathway.
The presented AMdnB X-ray structure illustrates a conserved structural organization of the ATP-grasp superfamily. Comparison of the AMdnB and MdnB structures reveals several structural differences, including a more open, flexible active site for the iterative AMdnB and differences in a channel connecting the monomers of the dimeric structure. This larger active site of AMdnB may be attributed to multiple UCPs within a larger multicore precursor peptide substrate and the differences in the interfacial channel can play a role in interacting with the multiple UCPs and linker regions. Based on these results, a mode of catalysis where LP binds to one monomer and catalysis occurs on the second monomer is presented. A similar binding mode was observed for the recently determined CdnC crystal structure, a dual-function ATP-grasp ligase from a Chryseobacterium species that catalyzes the formation of macrolactone cycles on a multicore precursor peptide. CdnC was cocrystallized with ADP, a truncated conserved leader peptide and a core peptide. As mentioned with other microviridin ATP-grasp ligases, the CdnC structure is also a homodimer.23 However, it is present as an asymmetric homodimer where one monomer contains ADP, leader peptide, and core peptide while the other monomer is lacking the core peptide, further supporting our model.
4. CONCLUSIONS
Presented is the structural and biochemical characterization of a multi-core processing peptide macrocyclase from a graspetide RiPPs pathway. The results provide mechanistic insight into the iterative chemistry of AMdnB and into the broad family of ATP-grasp macrocyclases. AMdnB-catalyzed cyclization was shown to produce multi-core tricyclic products, employing a distributive mode of catalysis. Structural comparison with other ATP-grasp enzymes provides a basis for substrate binding modes and the iterative chemistry. Overall, the work provides details that can guide peptide engineering efforts and expanded cyclic peptide libraries.
4. MATERIALS AND METHODS
5.1. In vitro reconstruction of AMdnB activity
AMdnA-Δ5 was prepared using AMdnA and AMdnC following a previous protocol.24 The resultant product was further purified by HPLC and lyophilized to remove AMdnC. A Shimadzu Prominence UHPLC system (Kyoto, Japan) fitted with a Vydac 218tp54 column (4.6 × 250 mm 300 Å), coupled with a PDA detector, was used for preparative HPLC. Solvent A was H2O containing 0.1% formic acid and solvent B was CH3CN containing 0.1% formic acid. Solvent B was applied with the following gradient: 0–5 min 10% B; 5–23 min, a linear gradient to 90% B; 27–27.5 min, a linear gradient to 10% B at a flow rate of 1.0 mL/min. The detection wavelengths were set at 210, 254 and 365 nm. AMdnA-Δ5 eluted at a retention time of 15.5 min.
The biochemical characterization of AMdnB using AMdnA-Δ5 as substrate was carried out in the mixture (100 μL) containing 20 μM AMdnA-Δ5, 50 mM HEPES pH 8.0, 5% glycerol, 2 mM ATP, 10 mM MgCl2 and 50 mM KCl. The reactions were incubated at 37 °C for 16 h and terminated by an equal volume of methanol. The resulting mixtures were centrifuged (14,000 g, 20 min, 4°C) and 50 μL of supernatants were used for HPLC and LC-MS analysis, whose parameters are detailed below. Enzyme dose elevation of AMdnB (1.6 μM, 3.2 μM, and 6.4 μM) was performed but failed to push the reaction to completion. The optimal reaction for the one-pot macrocyclization of AMdnA was as follows: 50 mM HEPES, pH 8.0, 5% glycerol, 2 mM ATP, 50 mM KCl, 20 μM AMdnA, 1.6 μM AMdnC and 1.6 μM AMdnB. The reactions were incubated at 37 °C for 16 h and terminated by an equal volume of methanol. The resulting mixtures were centrifuged (14,000 g, 20 min, 4°C) and 50 μL of supernatants were used for HPLC and LC-MS analysis, whose parameters are detailed below.
5.2. HPLC, LC-ESI-MS and LC-ESI-MS/MS analysis
A Shimadzu Prominence UHPLC system (Kyoto, Japan) fitted with an Agilent Poroshell 120 EC-C18 column (2.7 μm, 3.0 × 50 mm), coupled with a PDA detector, was used for HPLC analysis. Solvent A was H2O containing 0.1% TFA and solvent B was CH3CN containing 0.1% TFA. Solvent B was applied with the following gradient: 0–1 min 10% B; 1–15 min, a linear gradient to 90% B; 18–20 min, a linear gradient to 10% B at a flow rate of 0.3 mL/min. The detection wavelengths were set at 210, 254 and 365 nm. Data from LC-HR-MS and MS/MS were obtained using a Thermo Fisher Q Exactive Focus mass spectrometer equipped with electrospray probe on Universal Ion Max API source. Acetonitrile (B)/water (A) containing 0.1% formic acid were used as mobile phases with a linear gradient program (10‐90% solvent B over 15 min) to separate chemicals by the above reverse phase HPLC column at a flow rate of 0.3 mL/min. A pre-wash phase of 10 min with 10% solvent B was added at the beginning of each run, in which the eluate was diverted to the waste by a diverting valve. MS1 were acquired under Full Scan mode of the Orbitrap, in which a mass range of m/z 150‐2000 was covered and data were collected in the positive ion mode. Fragmentation was introduced by HCD technique with optimized collision energy ranging from 20 to 30. For each selected peptide, the ion with the highest intensity was selected as the precursor ion for MS/MS analysis. Other settings for the Orbitrap scan included resolution at 15000 and AGC target at 5×105. Full scan mass spectra and targeted MS/MS spectra for each of the pre-selected peptides were extracted from the raw files of the HPLC-MS/MS Experiment II using Xcalibur™ 2.1 (Thermo Scientific).
5.3. Proteomic analysis to determine the ring topology of processed AMdnA
The reactions were set up as described above and stopped at 0.5 h, 2 h, and 16 h by an equal volume of methanol. The mixtures were reduced to 10 to 20 uL using speedvac (Savant DNA120 Speed Vac Concentrator DNA RNA Sample Dryer with 36 Well Rotor). 50 μg Endoproteinase GluC was reconstituted in 500 μL autoclaved Milli-Q water. 35 μl H2O,10 μl protein substrate (10 μg), 50 μL 2X GluC reaction buffer, and 5 μL 100 ng/μl GluC (mass ratio between peptide substrate and GluC was kept at 1:20) were combined. The digestion mixtures were incubated at 37°C for 16 hours before being subjected to LC-MS analysis.
5.4. Protein cloning, overexpression and purification
Codon-optimized, full-length AMdnB was cloned into pET-28(a) vector with BamHI and NotI as the restriction enzyme sites. The plasmid was transformed into E. coli BL21(DE3) for overexpression. Overnight cultures were made a day before protein overexpression in 5 mL LB media and incubated at 37 °C overnight. Then cell cultures (1L) were inoculated with 2 mL of the overnight culture. The cell cultures were grown at 37 °C for about 4 hours until OD600 reached 0.6, after which the overexpression was induced with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Overexpression of the target protein was continued at 16 °C for 18 hours. Cells were harvested by centrifugation at 3,500 rpm for 25 minutes. After resuspension of the cell pellet in 45 mL lysis buffer (0.3 M NaCl, 50 mM Tris-HCl, pH 8.5 and 5% glycerol), cells were lysed at 14,000 psi through a nitrogen-pressure microfluidizer cell (M-110L Pneumatic). The cell lysate was then centrifuged at 13,500 rpm at 4°C to clarify the protein solution. The resultant solution was then subjected to Ni-affinity chromatography with a 1-hour long incubation. AMdnB was washed three times with 3×15 mL wash buffer (0.3 M NaCl, 50 mM Tris-HCl, pH 8.5, 5% glycerol and 20 mM imidazole) and eluted three times with 5 mL elution buffer (0.3 M NaCl, 50 mM Tris-HCl, pH 8.5, 5% glycerol and 250 mM imidazole). Each elution was incubated for 5 minutes on ice before collecting the fraction. After this, the purified protein sample was dialyzed against FPLC buffer (100 mM NaCl, 50 mM Tris-HCl, pH 8.5, 5% glycerol and 3 mM 2-mercaptoethanol).
The AMdnB sample was then subjected to anion-exchange chromatography (HiTrap Q HP, AKTA FPLC System, GE Healthcare) with a NaCl gradient from 0 M to 1 M for further purification. Then size-exclusion chromatography was performed in FPLC buffer (HiLoad 16/60 Superdex 200 column, AKTA FPLC System, GE Healthcare) at a flow rate of 1 mL/min. Protein fractions eluted around 40 minutes and were collected and concentrated for crystallization.
5.5. Crystallization, data collection and structural determination of AMdnB
Purified AMdnB was concentrated to 2.7 mg/mL and incubated with truncated MdnA (residues 1–24) at a molar ratio of 1:1.15. The sample was equilibrated at 4°C overnight before setting up crystallization trays. Initial crystallization screening was performed through the sitting drop vapor diffusion method against commercial sparse matrix screens (Hampton Index HT, Hampton PEG/Ion, Hampton Crystal Screen, Rigaku Wizard Classic 1 and 2, Qiagen NeXtal Tubes JCSG Core Suite I and II). An initial hit was found in Core Suite I G1, which contained 0.18 M Tri-ammonium citrate and 20% (w/v) PEG 3350. Optimization of the salt concentration and percentage of the precipitant as well as additive screening was performed, and the final crystals were obtained in 0.18 M Tri-ammonium citrate, 20% (w/v) PEG 3350 and 0.012 M praseodymium (III) acetate hydrate.
Diffraction data for AMdnB crystals were collected on beamline 23-ID-D at the Advanced Photon Source-Argonne National Laboratory (APS-ANL) at a wavelength of 1.033 Å and a detector distance at 400 mm. The data were collected at 100 K, integrated, merged and scaled with the XDS package.35 AMdnB was indexed into space group P21 with four molecules in the asymmetric unit. The previously solved MdnB structure (PDB code: 5IG8) shares 60.38% sequence identity with AMdnB and thus was used for molecular replacement. Amino acid residues were mutated and manually built into the electron density map in COOT.36 The resultant model was refined in PHENIX.37 Water molecules were added during the last few rounds of refinement and manually checked and further added or removed based on the electron density map. The final AMdnB model had an Rwork value of 19.9% and Rfree value of 26.5%. Data collection and refinement statistics are shown in Table S1. Structure images were generated with PyMOL. Additionally, an apo-AMdnB crystal structure, without pre-equilibration with LP, was determined and gave an identical structure.
5.6. Leader peptide binding affinity determination
Isothermal titration calorimetry (ITC) was used to determine the substrate binding affinity of AMdnB to AMdnA1–24. AMdnB was purified in 50 mM KCl and 100 mM HEPES-NaOH, pH 8.0 as the final buffer. The AMdnB sample was concentrated to 0.1 mM and placed in the sample cell. Commercially synthesized AMdnA1–24 (Bon Opus Biosciences, 90% purity, residues 1–24: MPENRQEDLNAQAVPFFARFLEGQ) was dissolved in an identical buffer and the final concentration was adjusted to 1.0 mM. The AMdnA leader peptide was titrated into AMdnB at 25°C. The titration was continued for 75 minutes with 33 titrations made in total, each titration containing 1 μL of AMdnA1–24 solution. The initial titration contained 0.2 μL AMdnA1–24 sample solution. The spacing time between each titration was 100 seconds. Titration of AMdnA1–24 to the buffer was used as a blank reaction. The binding parameters were calculated by the Origin software package, during which the background was removed by subtracting the heat change generated from the blank reaction.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded in part by the University of Florida and NIH R35GM128742 (YD). We acknowledge the Advanced Photon Source, particularly the staff and resources at GM/CA CAT. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
X-ray crystallographic data is deposited in the RCSB Protein Data Bank: accession code 7M4S.
REFERENCES
- 1.Cragg GM, Newman DJ Natural products: A continuing source of novel drug leads. Biochim Biophys Acta. 2013, 1830(6): 3670–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harvey AL, Edrada-Ebel AL, Quinn R, R.J.The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14 pp. 111–129 [DOI] [PubMed] [Google Scholar]
- 3.Medema MH & Fischbach MA Computational approaches to natural product discovery. Nat. Chem. Biol. 2015,11(9):639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnison PG et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30(1):108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Truman AW Cyclisation Mechanisms in the Biosynthesis of Ribosomally Synthesised and Post-Translationally Modified Peptides. Beilstein J. Org. Chem. 2016, 12 (1):1250–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baeriswyl V, Heinis C Polycyclic Peptide Therapeutics. Chem. Med. Chem. 2013, 8, 377–384. [DOI] [PubMed] [Google Scholar]
- 7.McIntosh JA, Donia MS & Schmidt EW Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Natural Product Reports 26, 537 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed MN, Reyna-González E, Schmid B, Wiebach V, Süssmuth RD, Dittmann E, Fewer DP Phylogenomic analysis of the microviridin biosynthetic pathway coupled with targeted chemo-enzymatic synthesis yields potent protease inhibitors. ACS Chem. Biol. 2017. 12(6):1538–1546. [DOI] [PubMed] [Google Scholar]
- 9.Lange J, Demir F, Huesgen PF, Baumann U, von Elert E, Pichlo C Heterologous expression and characterization of a novel serine protease from Daphnia magna: A possible role in susceptibility to toxic cyanobacteria. Aquat Toxicol. 2018, 205:140–147. [DOI] [PubMed] [Google Scholar]
- 10.Agrawa l M.K., Zitt A, Bagchi D, Weckesser J, Bagchi SN, von Eler t E. Characterization of proteases in guts of Daphnia magna and their inhibition by Microcystis aeruginosa PCC 7806. Environ Toxicol. 2005, 20(3):3143–22. [DOI] [PubMed] [Google Scholar]
- 11.Montalbán-López M, Scott TA, Ramesh S, Rahman IR, van Heel AJ, Vie l J.H., Bandarian V, Dittmann E, Genilloud O, Goto Y, Grande Burgos MJ, Hill C, Kim S, Koehnke J, Latham JA, Link AJ, Martínez B, Nair SK, Nicolet Y, Rebuffat S, Sahl HG, Sareen D, Schmidt EW, Schmitt L, Severinov K, Süssmuth RD, Truman AW, Wang H, Weng JK, van Wezel GP, Zhang Q, Zhong J, Piel J, Mitchell DA, Kuipers OP, van der Donk WA. New developments in RiPP discovery, enzymology and engineering. Nat Prod Rep. 2021. Jan 1;38(1):130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiz AR, Ishida K, Quitterer F, Meyer S, Kehr JC, Müller KM, Groll M, Hertweck C, Dittmann E Harnessing the evolvability of tricyclic microviridins to dissect protease-inhibitor interactions. Angew Chem Int Ed Engl, 53(14):3735–3738. [DOI] [PubMed] [Google Scholar]
- 13.Al-Awadhi FH, Luesch H Targeting eukaryotic proteases for natural products-based drug development. Nat. Prod. Rep. 2020, 37, 827–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.do Amaral SC, Monteiro PR, Neto JDSP, Serra GM, Gonçalves EC, Xavier LP, Santos AV current knowledge on microviridin from cyanobacteria. Mar. Drugs. 2021, 19(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiz AR, Ishida K, Makower K, Ziemert N, Hertweck C, Dittmann E Leader peptide and a membrane protein scaffold guide the biosynthesis of the tricyclic peptide microviridin. Chem Biol. 2011, 18(11):1413–1421. [DOI] [PubMed] [Google Scholar]
- 16.Fawaz MV, Topper ME, Firestine SM The ATP-grasp enzymes. Bioorg. Chem. 2011, 39, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ziemert N, Ishida K, Liaimer A, Hertweck C, Dittmann E Ribosomal synthesis of tricyclic depsipeptides in bloom-forming cyanobacteria. Angew Chem Int Ed Engl. 2008, 47, 7756–7759. [DOI] [PubMed] [Google Scholar]
- 18.Zhao G, Jin Z, Wang Y, Allewell NM, Tuchman M, Shi D Structure and function of Escherichia coli RimK, an ATP-grasp fold, L-glutamyl ligase enzyme. Proteins. 2013, 81(10):1847–1854. [DOI] [PubMed] [Google Scholar]
- 19.Wang W, Kappock TJ, Stubbe J, Ealick SE X-ray crystal structure of glycinamide ribonucleotide synthetase from Escherichia coli. Biochemistry. 1998. 37(45):15647–15662. [DOI] [PubMed] [Google Scholar]
- 20.Thoden JB, Holden HM, Firestine SM Structural analysis of the active site geometry of N5-carboxyaminoimidazole ribonucleotide synthetase from Escherichia coli. Biochemistry. 200, 47(50):13346–13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakai H, Vassylyeva MN, Matsuura T, Sekine S, Gotoh K, Nishiyama M, Terada T, Shirouzu M, Kuramitsu S, Vassylyev DG, Yokoyama S Crystal structure of a lysine biosynthesis enzyme, LysX, from Thermus thermophilus HB8. J. Mol. Biol. 2003, 332(3):729–740. [DOI] [PubMed] [Google Scholar]
- 22.Li K, Condurso HL, Li G, Ding Y, Bruner SD Structural basis for precursor protein-directed ribosomal peptide macrocyclization. Nat. Chem. Biol. 2016, 12(11):973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao G, Kosek D, Liu HB, Ohlemacher SI, Blackburne B, Nikolskaya A, Makarova KS, Sun J, Barry CE, Koonin EV, Dyda F, Bewley CA Structural Basis for a Dual Function ATP Grasp Ligase That Installs Single and Bicyclic ω-Ester Macrocycles in a New Multicore RiPP Natural Product. J. Am. Chem. Soc. 143(21):8056–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Li K, Yang G, McBride JL, Bruner SD, Ding Y A distributive peptide cyclase processes multiple microviridin core peptides within a single polypeptide substrate. Nat Commun. 2018, 9(1):1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin GM, Ding Y Recent advances in the biosynthesis of RiPPs from multicore-containing precursor peptides. J. Ind. Microbiol. Biotechnol. 2020, 47(9–10):659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koehnke J, Bent AF, Zollman D, Smith K, Houssen WE, Zhu X, Mann G, Lebl T, Scharff R, Shirran S, Botting CH, Jaspars M, Schwarz-Linek U, Naismith JH The cyanobactin heterocyclase enzyme: a processive adenylase that operates with a defined order of reaction. Angew Chem Int Ed Engl. 2013, 52, 13991–13996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sardar D, Lin Z, Schmidt EW Modularity of RiPP enzymes enables designed synthesis of decorated Peptides. Chemistry & Biology, 2015, 22, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holm L Benchmarking fold detection by DaliLite v.5. Bioinformatics. 2019, 35(24):5326–5327 [DOI] [PubMed] [Google Scholar]
- 29.Oman TJ & van der Donk WA Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol, 2010, 6, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montalbán-López M et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep., 2021, 38(1):130–239.30. Zhao, G.; Kosek, D.; Liu, H.-B.; Ohlemacher, S. I.; Blackburne, B.; Nikolskaya, A.; Makarova, K. S.; Sun, J.; Barry III, C. E.; Koonin, E. V.; Dyda, F.; Bewley, C. A. Structural Basis for a Dual Function ATP Grasp Ligase That Installs Single and Bicyclic ω-Ester Macrocycles in a New Multicore RiPP Natural Product. J. Am. Chem. Soc. 2021, 143 (21), 8056–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000,302(1):205–217. [DOI] [PubMed] [Google Scholar]
- 32.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ Jalview Version 2—a multiple sequence alignment editor and analysis workbench. 2009, Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerlt JA et al. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochimica et Biophysica Acta - Proteins and Proteomics, 2015, 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao G, Kosek D, Liu HB, Ohlemacher SI, Blackburne B, Nikolskaya A, Makarova KS, Sun J, Barry Iii CE, Koonin EV, Dyda F, Bewley CA. Structural Basis for a Dual Function ATP Grasp Ligase That Installs Single and Bicyclic ω-Ester Macrocycles in a New Multicore RiPP Natural Product. J Am Chem Soc. 2021, 143, 8056–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabsch W XDS. Acta Crystallogr D Biol Crystallogr. 2010, 66(Pt 2):125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004, 60(Pt 12):2126–2132. [DOI] [PubMed] [Google Scholar]
- 37.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012, 68(Pt 4):352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
X-ray crystallographic data is deposited in the RCSB Protein Data Bank: accession code 7M4S.