

Abstract
The field of cardio-oncology has emerged in response to the increased risk of cardiovascular disease (CVD) in patients with cancer. However, recent studies suggest a more complicated CVD-cancer relationship, wherein development of CVD, either prior to or following a cancer diagnosis, can also lead to increased risk of cancer and worse outcomes for patients. In this review, we describe the current evidence base, across epidemiological as well as preclinical studies, which supports the emerging concept of ‘reverse-cardio oncology’, or CVD-induced acceleration of cancer pathogenesis.
Introduction
The field of cardio-oncology has evolved from observations of increased risk of cardiovascular disease (CVD) following a cancer diagnosis [1]. The increased CVD risk is linked to both direct (e.g. cardiotoxic) and indirect (e.g. sedentary lifestyle) complications of cancer treatments [2], and the cardio-oncology field continues to grow with the introduction of new immunotherapies, with various cardiotoxic sequelae, and expansion of their clinical use [3]. This expansion, alongside the evolving management and treatment of CVD in patients with cancer, has also led to an adjacent line of investigation: can the presence of CVD reciprocally influence cancer pathogenesis? Indeed, recent studies suggest that the CVD-cancer relationship may be more complex than previously appreciated, leading to a new concept of CVD-induced cancer risk and progression that has been termed ‘reverse cardio-oncology’ [4].
Cardiovascular disease (CVD) is now well established as a systemic disease [5–8]. CVD-induced dysregulation of systemic inflammation, immunity, and metabolism have been shown to have direct effects on both CVD (e.g. pre-existing atherosclerotic plaques) [8] and non-CVD tissues (e.g. adipose tissue) [6], leading to increased morbidity and mortality (e.g. recurrent myocardial infarction (MI), insulin resistance and diabetes). It is therefore plausible that the systemic effects of CVD can also drive other disease entities, including cancer. Cancer and CVD, the two leading causes of death in developed countries, share numerous modifiable and non-modifiable risk factors, including smoking, obesity, physical inactivity, hypertension, dyslipidaemia, aging, and genetic predisposition [9, 10]. Over the last decade, observational data have shown a positive relationship between CVD and pan-cancer incidence [11–13], with emerging support from preclinical studies [14, 15]. Whether this relationship is causal or due to shared risk factors remains in debate [16], but evidence continues to mount that the systemic changes associated with CVD can have pro-tumorigenic effects [17]. Further, recent work that suggests incident CVD following a primary cancer diagnosis may drive cancer progression [18] has spurred further interest in understanding the impact of this bi-directional relationship on disease progression and clinical practice [19].
In this review, we outline the emerging data exploring how the development of CVD, either prior to or following a cancer diagnosis, relates to cancer initiation and progression. First, we briefly overview a number of selected CVD risk factors that have cancer-promoting effects. We next describe the extant observational data outlining the role of established CVD on cancer incidence and progression, as well as post-cancer diagnosis CVD on cancer outcomes. We then provide a detailed overview of recent mechanistic studies that draw causal connections in preclinical models between CVD and cancer pathogenesis. Finally, we propose future directions, across basic, translational, and clinical levels for the field.
1. Common Risk Factors in CVD and Cancer
The growing recognition of the interplay between CVD and cancer is placed on the background of the increased prevalence of CVD in patients with cancer, and vice versa [9, 20]. Today, it is acknowledged that these two diseases have various similarities, including risk factors that explain, at least in part, their co-occurrence. This section aims to summarize select CVD modifiable and non-modifiable shared risk factors [21] and the potential biological pathways by which such risk factors contribute to cancer incidence and progression. A more comprehensive overview of these and other risk factors are provided in recent reviews [9, 10].
1.1. The Link Between Modifiable CVD Risk Factors and Cancer
Smoking
Smoking, like CVD [22], is an indisputable risk factor for cancer [23]. Beyond the elevated risk in lung cancer, where 80–90% of cancer deaths are due to smoking, chronic smoke exposure also increases cancer risk in up to 17 other cancer subtypes, and current estimates suggest that ~30% of all cancer deaths are due to smoking [23–25]. A multitude of pro-tumorigenic mechanisms of smoking have been identified, centring on the direct carcinogenic effects of smoke exposure on mutagenesis, epigenetic modifications and inflammation [23].
Obesity
Obesity is also a CVD risk factor that is associated with cancer risk and progression [26]. A recent analysis of ~1000 observational studies identified that high body mass index (BMI) is associated with increased risk of 13 different cancers [27]. Further, a prospective study of ~1 million adults identified that high BMI is associated with increased risk of cancer-specific mortality across 10 different cancers in men and 12 different cancers in women [28]. Mechanistically, obesity is associated with increased levels of various circulating factors including leptin, glucose, insulin, and insulin-like growth factor 1, all of which activate numerous growth factor signalling pathways resulting in tissue microenvironments primed for cell growth, proliferation, and survival [23]. Obesity also promotes the production of chronic inflammatory cytokines, increases oxidative stress through production of mutagenic reaction oxygen species, and induces immune suppression [29–31]. Collectively, these alterations can reduce the barrier to oncogenic transformation [29, 32], as well as promote disease progression [29–31].
Physical Inactivity
Mounting evidence suggests that physical inactivity, a CVD risk factor [33], also increases risk of cancer incidence and progression. Pooled data from 12 prospective cohort studies demonstrated that high levels of self-reported physical activity are associated with reduced risk of cancer incidence across 13 cancer subtypes compared to those reporting low levels of physical activity [34]. Further, pooled estimates across 26 prospective studies of breast, colorectal, and prostate cancer show that high levels of post cancer diagnosis self-reported physical activity are also associated with reduced risk of recurrence and cancer-specific mortality compared to those reporting low levels [35]. The mechanisms by which physical inactivity drives cancer incidence and progression are multifactorial and several emerging mechanisms by which its inverse, physical activity, may protect from cancer and its progression have been identified, including modulation of immunity, metabolism, and angiogenesis [36–40].
Hypertension
The causal role of hypertension (i.e., chronically elevated blood pressure) in cancer remains ambiguous. A prospective study of seven population-based cohorts totalling more than half a million adults identified a small increased risk of cancer incidence in patients with elevated blood pressure across several cancer types in men, but not women, yet increased risk for cancer-specific mortality across both men and women [41]. Hypertension also independently predicts cancer-specific mortality in women with early-stage breast cancer [42]. While speculative, several mechanistic links between CVD and carcinogenesis have been proposed, including hypertension-induced increases in vascular endothelial growth factor and angiotensin II, as well as oxidative stress [9].
Dyslipidaemia
Dyslipidaemia, a well-established CVD risk factor [43], has also been implicated as a risk factor for cancer, although evidence is mixed [44]. In prostate cancer, low levels of total cholesterol are associated with decreased risk of high-grade prostate cancer [45, 46], and high levels are associated with increased recurrence risk [47]. In breast cancer, while conflicting reports have yielded it unclear whether total, low density lipoprotein (LDL), or high density lipoprotein (HDL) cholesterol impact risk of cancer incidence [48], a prospective study of 520 women with early-stage breast cancer showed that high circulating total cholesterol and low density lipoprotein cholesterol levels were correlated with recurrence risk [49]. Preclinical studies have identified that high cholesterol levels, and particularly the primary cholesterol metabolite 27-hydroxycholesterol, which acts as a selective estrogen receptor modulator, drives estrogen receptor positive breast cancer [50–52] through pro-metastatic shifts in both innate and adaptive immunity [50, 51]. Further, given that intracellular cholesterol homeostasis and dysregulation is implicated in cancer development and progression across a variety of cancers [44], future studies that continue to resolve equivocal epidemiologic data alongside mechanistic studies of systemic and/or intracellular cholesterol dysregulation are warranted.
1.2. Shared Non-modifiable Risk Factors in Cancer and CVD
Non-modifiable CVD risk factors including genetics, age, and sex also influence the incidence and progression of cancer. For example, genetic mutations related to the Wnt/b-catenin pathway play a role in both the development of CVD by mediating hypertrophy, fibrosis, and ischemia [53], as well as malignant transformation and cancer cell proliferation in many cancer types [54]. Further, mutations in the protein kinase dual specificity tyrosine phosphorylation‐regulated kinase 1B (DYRK1B) gene are associated with individual CVD risk factors, namely obesity, coronary artery disease (CAD), hypertension, and diabetes [55], while in cancer, DYRK1B regulates cellular quiescence and survival [56]. Age-associated mutations in hematopoietic stem cells also contribute to a condition known as clonal haematopoiesis of indeterminate potential (CHIP), which has been linked to both CVD and cancer [57–59]. In CVD, clinical and preclinical data show CHIP carriers of specific mutations have increased coronary-artery calcification and overexpression of several chemokines and cytokine genes that are known to induce atherosclerosis [58]. In cancer, CHIP is a major risk factor for haematologic malignancy [57].
2. Increased cancer incidence and worse cancer-specific outcomes in patients with prevalent CVD
Beyond common risk factors between CVD and cancer that may drive their co-occurrence, a growing body of clinical evidence also demonstrates that prevalent CVD is itself associated with higher cancer incidence. While these studies are of substantial hypothesis-generating value, they should also be critically assessed for their limitations and validity.
One of the central outstanding issues is if prevalent CVD can initiate new cancer formation (tumorigenesis), or that rather the internal milieu in CVD patients is such that it accelerates early existing tumors to grow or metastasize. Preclinical models mostly have focused on tumor acceleration and growth. So, in the literature, when incident cancer is described, it may be that cancer already existed, but remained occult and only started to manifest after CVD ensued.
Having said that, an abundance of data has hinted at an association between heart failure (HF) and cancer incidence. A cohort study showed that patients who develop HF within one month after MI were more prone to develop cancer in comparison to participants with no HF [11]. Four Danish registries (The Danish Civil Registration System registry, the NPR (Danish National Patient Registry), the National Causes of Death Registry, and the Danish National Prescription Registry) evaluated cancer risk and cancer death in patients with MI. All age groups of patients demonstrated higher incidence rates of cancer after 1 year from the diagnosis of MI [12]. A long-term prospective study evaluated the clinical features and prevalence of malignant neoplasm in patients with acute coronary syndrome (ACS) during a 17-year follow-up. This group reported a higher malignancy risk in ACS patients, and those who developed malignancies after the ACS diagnosis demonstrated a worse prognosis [13].
Utilizing the PREVEND study, a community-based cohort study of middle-aged participants, Meijers et al identified that NT-proBNP, which is the gold standard biomarker for HF detection, was also associated with incident cancer [14]. Specifically, with a median follow-up of 11.5 years (n=8319), where 13.2% of participants developed cancer (n=1132), higher levels of NT-proBNP were associated with new-onset cancer (HR: 1.06, 95%CI 1.00–1.12) after adjustment for age, smoking and body mass index (BMI). Similar adjusted analyses showed comparable effect sizes (although not significant possibly due to limited power) for incident colorectal cancer, but interestingly, high levels of NT-proBNP were associated with female reproductive cancer incidence (adjusted HR: 1.30, 95%CI 1.08–1.56). In addition to natriuretic peptides, high-sensitivity troponin, as well as the pro-inflammatory cytokines pro-adrenomedullin, pro-endothelin, and C-reactive Protein (CRP) were also associated with incident cancer, the latter with the strongest association (HR 1.08; 95%CI 1.04–1.13).
Cancer risk in other CVD, including stroke and cardiac arrythmias, has also been assessed. The Swedish Inpatient Register found that 4% of patients with venous thromboembolism were diagnosed with cancer within the first year after enrolment [60]. In the Vitamin Intervention for Stroke Prevention study, ischemic stroke survivors demonstrated a higher annual rate of age-adjusted cancer risk compared to the general population [61]. Moreover, it appears that atrial fibrillation can also predict cancer. In the Women’s Health Study, 10% of patients who had new-onset AF developed subsequent cancer [62]. Similarly, the investigators of the Danish population-based cohort study found that 11.1% of women and 15% of men who presented with new-onset AF were diagnosed with cancer later [63]. In a study that analysed echocardiographic data from more than 80,000 patients, of which nearly 5,000 patients had aortic stenosis and over 8,000 patients developed non-haematological cancers during a median follow-up of 5.4 years, Avraham et al [15] showed that the crude incidence rate and death of non-haematological cancer were higher in patients with moderate to severe aortic stenosis. However, when adjusted for covariates, including age, ethnicity, alcohol abuse, smoking, obesity, diabetes, history of cancer, and aspirin and statin use, the association between aortic stenosis and cancer only held in patients between 40–60 years of age.
It should be noted that CVD patients are more exposed to medical surveillance in comparison to the general population. Consequently, the increased cancer risk in these patients can be due to detection bias, and regular lab tests, chest X-rays, CT scans, PET scans, and MRI scans may unmask occult malignancies [64]. In the Swedish Inpatient Register and Women’s Health Study, the increased short-term cancer prevalence (within one year after venous thromboembolism diagnosis) confirms this assumption [60, 62]. Nevertheless, the longer-term increase in the relative risk of cancer in patients with atrial fibrillation and venous thromboembolism cannot be explained by surveillance bias exclusively. In addition, CVD management, like anticoagulants administration to treat atrial fibrillation, may contribute to the earlier detection of cancers due to bleeding.
In sum, while these provocative studies suggest a relationship between prevalent CVD, incident cancer, and worse cancer outcomes, it is important to acknowledge their limitations. Primarily, many of these associations are identified in retrospectives analyses, in which causality is not guaranteed. Also, these studies are hampered by their design not being powered toward specific cancer outcomes in CVD patients. Thus, targeted and independent analyses are needed to reach clinically relevant conclusions.
3. Increased risk of recurrence and cancer-specific mortality in patients with incident (post-cancer diagnosis) CVD
A more recent line of investigation has been understanding the relationship between the onset of CVD following a cancer diagnosis and progression of underlying malignancy. Koelwyn et al [18] performed a retrospective analysis of two prospective case cohort studies in early-stage breast cancer, the LACE and Pathways studies, interrogating the relationship between a post diagnosis CVD event (i.e., MI, CAD, stroke, HF, and arrythmia), and cancer outcomes (i.e. recurrence and breast cancer-specific mortality) (n=1724, median follow-up 11.7 years). Patients were excluded if they had established CVD, or CVD risk factors (i.e., dyslipidaemia, hypertension, and diabetes). After adjustment for multiple covariates, including age, race, smoking status, body mass index at diagnosis date, tumor stage and adjuvant therapy (chemotherapy, radiation, endocrine therapy), patients who experienced a CV event had an adjusted 59% increased risk of cancer recurrence (95% CI: 1.23–2.06) and 60% increased risk cancer-specific mortality (95% CI: 1.16–2.22) compared to patients who did not experience a CV event. These data suggest that CV events drive progression of breast cancer. Mechanistic studies in pre-clinical models of breast cancer suggest that MI may reprogram subsequent immune responses leading to a pro-tumorigenic environment (described further below). However, validation of this relationship in independent and larger trials, as well as in other cancer populations at high risk of CVD post-cancer diagnosis are warranted.
4. Mechanisms of CVD-induced cancer pathogenesis in preclinical models
Given the growing body of observational data describing the effects of CVD on cancer incidence and outcomes, a new field has emerged exploring the causal mechanistic links that may enable cross-disease communication between CVD and cancer. These studies have combined observational findings in patients (discussed above) with relevant preclinical models of CVD, including surgical models of MI and subsequent HF, as well as aortic stenosis/constriction, identifying a number of candidate systemic factors that drive CVD-induced acceleration of colon, breast, and lung cancer.
MI-induced heart failure and colon cancer pathogenesis
In the first study to assess the role of CVD in cancer pathogenesis, Meijers and colleagues [14] discerned the effects of MI-induced HF on intestinal polyp formation in the APCmin model of colon cancer. This model forms spontaneous intestinal adenomas, developing ~30 adenomas throughout the intestinal tract, which lead to colon obstruction and mortality starting at ~17 weeks of age. The researchers performed surgical MI by permanent ligation of the left anterior descending coronary artery at 6 weeks of age. HF was confirmed at 12 weeks of age by MRI or echo imaging, and post-mortem by increased LV fibrosis and atrial, spleen and liver weight, as well as elevated cardiac and plasma levels of fibrosis and inflammatory-associated gene and protein products. Intestinal tissue taken at the same time point (6 weeks following MI) showed that MI-induced HF increased polyp number, size, and cumulative tumor volume. Interestingly, cumulative tumor volume positively and negatively correlated with LV fibrosis and LVEF, respectively, suggesting a dose response effect. Further, measures of proliferation by immunostaining for Ki67 in the gut showed greater proliferation in HF mice compared to sham control.
The authors subsequently investigated if the cancer-promoting effects of HF were driven by the presence of a failing heart, independent of the hemodynamic changes induced by HF (i.e., ‘forward failure’ due to reduced systolic blood pressure or ‘backward failure’ due to congestion from increased filling pressures). To experimentally test this question, hearts were excised from donor APCmin mice one week following surgical MI or sham surgery and transplanted into the cervical region of recipient APCmin mice at 7 weeks of age, and connected to the circulation via the external jugular vein and carotid artery. This enabled recipient mice to maintain hemodynamic function via their native (endogenous) heart but be exposed to the secretome of a failing heart (or sham control). In support of the hypothesis that the systemic effects of HF drove cancer outgrowth, the presence of a failing heart increased polyp number, size and tumor volume, as well as spleen weight, compared to sham transplant. Proliferation as measured by Ki67 was also increased, and similar positive and negative correlations of LV fibrosis and LVEF with tumor volume were observed. Such evidence showed that systemic factors released from the failing heart were a central driver of colon cancer outgrowth, independent of HF-associated hemodynamic changes.
To discern relevant candidate factors released from the failing heart that promote colon polyp formation and outgrowth, the authors next performed a literature search of HF-associated circulating factors (ligands) with corresponding intestine-specific receptors, identifying 5 potential circulating candidates: SerpinA1, SerpinA3, Fibronectin, Ceruloplasmin and Paraoxonase 1. The authors identified elevated levels of all 5 proteins in the plasma of 101 patients with chronic HF compared to 180 age and sex matched controls, and validated increased cardiac-specific gene expression of these factors in the failing hearts of mice compared to sham control, as well as three genes (SerpinA3, Fibronectin, Paraoxonase1) in transplanted hearts. In vitro, only SerpinA3 exerted consistent proliferative effects on HT29 cells, a human colorectal cancer cell line, which was shown to occur via activation of the AKT pathway.
In sum, this study provided the first causal evidence in a preclinical model that the systemic effects of HF directly regulate colon cancer pathogenesis. These effects were independent of hemodynamic changes, implicating the HF-induced cardiac secretome as a driver of colon cancer, of which a number of candidate factors in mice were identified and shown to also be upregulated in HF patients, most notably SerpinA3.
Transverse Aortic Constriction and breast cancer and lung cancer progression
Avraham and colleagues [15] investigated the effect of transverse aortic constriction (TAC), a model of pressure overload-induced cardiac hypertrophy and HF, on tumor growth and metastasis in mouse models of breast and lung cancer. First, the researchers performed TAC 10 days prior to orthotopic injection of tumor cells isolated from the genetically engineered MMTV-PyMT mouse model of breast cancer. Characterization of heart function 9 days post TAC showed that fractional shortening was decreased, heart to body weight ratio was elevated, and cardiac expression of hypertrophic genes including Anp, Bnp, bMHC and Acta1 were increased in mice exposed to TAC compared to sham or control. No changes in LV fibrosis were noted, suggesting mild cardiac remodelling and hypertrophy with reduced contractile function, without overt signs of HF. In this model, TAC accelerated breast cancer tumor growth over 25 days compared to sham and control mice. Using a second cancer model, the Lewis Lung Carcinoma (LLC) model, the authors similarly found that TAC accelerated subcutaneous tumor growth in the flank over 20 days compared to sham and control. Cell proliferation, as assessed by Ki67 immunostaining, was greater in tumors from mice with TAC compared to sham in both the PyMT and LLC models; however, no differences were noted for tumor angiogenesis. Further, delaying PyMT tumor injection to 30 days post TAC compared to 10 days post, where cardiac remodelling was more pronounced (e.g., further reductions in fractional shortening, greater heart weight/body weight ratio), lead to greater acceleration of tumor growth, suggesting that more advanced cardiac remodelling conferred a larger primary tumor growth advantage. Next, the authors sought to investigate the effects of TAC on metastasis using an experimental metastasis model in which TAC was performed (or no surgery control) on mice 45 days prior to tail vein injection of PyMT or LLC cells. TAC resulted in a greater number of lung metastatic lesions, as well a greater average lesion area, in both the PyMT and LLC models after 10 days. Together, these models show that TAC, resulting in varying levels of early cardiac remodeling, has tumor- and metastasis-promoting effects in models of breast and lung cancer.
To explore the factors and/or processes that may be responsible for TAC-accelerated tumor growth, the authors investigated the requirement of an intact immune system using NOD/SCID mice, which lack T and B lymphocytes, and have reduced natural killer and myeloid cell function. In these experiments, TAC similarly accelerated PyMT primary tumor growth compared to non-surgery control mice, suggesting the effects were independent of effects on a fully functioning immune system. The authors also performed TAC in the maladaptive-cardiac remodeling-resistant (MCRR) mouse model, which failed to result in significant differences in cardiac function and remodeling, or TAC-accelerated tumor growth. To identify potential candidate factors of TAC-induced tumor growth, the authors next investigated whether the systemic (circulating) milieu associated with TAC altered tumor cell behaviour. Both PyMT and LLC cells cultured in vitro with serum from mice exposed to TAC showed increased proliferation compared to serum from either sham or non-surgery control mice. Using bulk RNA sequencing of the TAC-hearts 55 days following surgery the authors identified 520 differentially expressed genes, of which 33 were upregulated and encoded for secreted proteins. Two of those, CTgF and Periostin, which are known regulators of cancer progression [65], were upregulated in TAC-operated hearts of both tumor (PyMT and LLC) and non-tumor bearing mice, as were protein levels in the serum. Finally, periostin increased proliferation of PyMT and LLC cells in vitro, and periostin-depleted serum from TAC-operated mice failed to increase cell proliferation in vitro. Together, these data suggest that periostin may be a candidate driver of tumor growth following TAC via its effects on cancer cell proliferation. In sum, this study supports the concept that early cardiac remodelling in response to aortic constriction, similar to models of HF, has cancer promoting effects through altering the systemic host milieu.
Myocardial infarction accelerates breast cancer
While the aforementioned studies discerned the role of pre-existing CVD (early and late-stage HF) on cancer pathogenesis, Koelwyn et al [18] interrogated whether incident CVD events, such as MI, following primary cancer could alter cancer progression. To address this question in preclinical models, the authors first implanted syngeneic E0771 cancer cells into the mammary fat pad of C57BL/6J mice, then subjected to surgical MI or sham surgery 3 days following implantation. Over 17 days, MI accelerated tumor growth compared to sham, resulting in increased tumor volume and tumor weight. MI also increased intratumoral cell proliferation at the tumor border, as assessed by Ki67 immunostaining, which occurred in both the non-immune (CD45-) and immune (CD45+) cell fractions. The authors validated that MI-accelerated tumor growth in MMTV-PyMT mice – a transgenic mouse model of spontaneous breast cancer on the C57BL/6 background. Surgical MI, performed upon palpable tumor formation, accelerated tumor growth and metastasis to the lung over a period of 18 days, compared to sham surgery. Together, these experiments identified in mouse models of breast cancer that MI following primary breast cancer accelerates disease progression.
To discern how MI accelerates cancer outgrowth, the authors performed intratumoral immune profiling by flow cytometry. In the E0771 model, MI increased the proportion of CD45+ immune cells in tumors compared to sham, which was driven by an increased accumulation of CD11b+Ly6Chi monocytes. Monocytes, as well as monocyte-derived tumor associated macrophages, have numerous cancer-promoting functions, including immune suppression [66]. MI also decreased tumoral CD3+ T cells as a percentage of CD45+ cells, but induced a proportional increase in immunosuppressive CD3+FoxP3+ regulatory T cells in the tumors. These MI-induced alterations in the tumor immune landscape were also noted in the MMTV-PyMT model, wherein MI increased levels of monocyte-derived CD11bloMHCIIhi tumor associated macrophages and CD3+FoxP3+ regulatory T cells in primary tumors, as well as increasing Ly6Chi monocytes in metastasis-bearing lungs.
High circulating monocyte levels are known to correlate with worse cancer outcomes across multiple cancers [67, 68]. Following MI, E0771 tumor-bearing mice had a sustained monocytosis within the circulation compared to sham mice. This may be due to increased haematopoiesis, as the proportion of common myeloid progenitors within the bone marrow, a precursor of Ly6Chi monocytes, was increased after MI. Using adoptive transfer experiments, the authors showed that MI also increased recruitment of monocytes to tumors during early tumor growth. These increases in the systemic availability and recruitment of Ly6Chi monocytes to tumors were required for MI-accelerated tumor growth, as depletion of monocytes 10 days following E0771 tumor injections using the CCR2-diptheria toxin receptor mouse model abrogated the MI-induced tumor growth advantage. Intriguingly, removal of intratumoral Ly6Chi monocytes reversed the MI-induced immunosuppressive microenvironment in the tumor, by reducing the proportion of regulatory T cells and increasing the proportion of activated (Granzyme B+) CD8+ T cells. To explore how MI altered monocyte phenotypes in the tumor, the authors isolated tumor Ly6Chi monocytes and tested their ability to alter CD8+ T cell activation and proliferation. While no differences were noted for CD8+ T cell proliferation, monocytes from MI mice more potently suppressed CD8+ T cell activation (as measured by GrB+, iFNg+, and TNFa+) compared to tumoral monocytes from sham controls. Consistent with this, RNA sequencing of tumoral Ly6Chi monocytes isolated 17 days following MI identified pathways associated with immunosuppression, including inhibition of lymphocyte activation, adaptive immune responses and IFNg signalling. In support of the central role of CD8+ T cell-induced immunosuppression in tumor growth, depletion of these cells using anti-CD8 accelerated tumor growth in sham mice, while no tumor growth differences were noted in mice with MI, consistent with the established dysfunctional phenotype of CD8+ cells following MI.
Given that MI dysregulated systemic immune processes, the authors next investigated whether the immunosuppressive transcriptional signature noted in tumor Ly6Chi monocytes was also observed in monocytes in the circulation and bone marrow reservoir, prior to tumor recruitment. Indeed, geneset analysis of the top 1000 differentially expressed genes in tumor monocytes showed that these changes correlated with those observed in circulating and bone marrow monocytes, suggesting that MI reprograms systemic monocytes and these changes are maintained upon tumor entry. Further, transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) analysis performed on bone marrow monocytes identified that MI reduced chromatin accessibility at loci associated with immune and inflammatory responses, lymphocyte activation and cytokine production. Analysis of transcription factor binding motifs in these regions of less accessible chromatin after MI identified the pioneer factors PU.1 and CEBP, as well as the interferon regulatory factor (IRF)-8, which is known to regulate myeloid cell differentiation. Interestingly, repression of IRF-8 has previously been shown to induce a myeloid-derived suppressor cell phenotype [69, 70], which is consistent with the immunosuppressive phenotype of monocytes observed after MI. Subsequent integration of ATAC- and RNA-seq monocyte datasets found numerous genes regulated by PU.1, CEBP and IRF-8 that showed both less accessible chromatin in the bone marrow and reduced gene expression in the tumor, including genes involved in T cell activation (e.g. Cd40, Cd86), Il12, and Irf8 itself. To confirm that changes in the bone marrow were driving MI-accelerated tumor growth, the authors performed a bone marrow transplant from tumor-bearing mice exposed to MI or sham surgery to wildtype donor mice, and assessed monocyte levels and tumor growth 14 weeks later. Strikingly, mice with MI-donor bone marrow exhibited a circulating monocytosis compared to mice with sham-donor bone marrow, and accelerated growth of E0771 tumors upon implantation. These data suggest long-term alterations to the chromatin (e.g., epigenetic) status of monocyte precursors following MI, which drives sustained haematopoiesis and an immunosuppressive phenotype that accelerates tumor growth. Together, this study highlights that post-cancer CVD events such as MI, similar to prevalent HF prior to cancer, can promote a pro-tumorigenic systemic host milieu.
In sum, this emerging collection of mechanistic studies highlights numerous candidate mechanisms by which CVD (e.g. models of MI, MI-induced heart failure, and aortic stenosis/constriction) accelerates cancer, including cardiac-specific circulating factors (e.g. SerpinA3, Periostin) and innate immune-specific changes (Figure 1). However, given the pleotropic effects that CVD exerts on the systemic host milieu, these changes likely explain only part of this complicated cross-disease interaction. It is plausible that a combination of changes across systemic regulatory networks (e.g. autonomic function, inflammatory and immune responses, metabolism) as well tissue-specific alterations (e.g. bone marrow, spleen, heart, lung, muscle, adipose, liver, kidney) that occur in both CVD and cancer are leading to deleterious interactions that potentiate risk for cancer cell transformation, proliferation and cancer progression. Such interactions, however, will likely be dependent both on CVD and cancer type. In a recent study, Shi and colleagues found that MI-induced HF did not accelerate renal cancer progression in the RENCA mouse model, and tumour weights were comparable between the MI and sham groups [71]. These outcomes suggest that the effects of HF on tumor growth are not generic, and likely the underlying mechanisms might be specific for HF etiologies, cancer types, and animal models. Further, as basic and translational scientists continue to explore the systemic, tissue, and cell-specific factors that enable CVD-induced cancer pathogenesis, it will be essential to consider the appropriate development and utilization of CVD and cancer model systems that are designed to recapitulate clinical observational findings and subsequent translation to patients.
Figure 1: Graphical summary of preclinical studies [14, 15, 18, 71] interrogating the effects of cardiovascular disease on cancer pathogenesis.
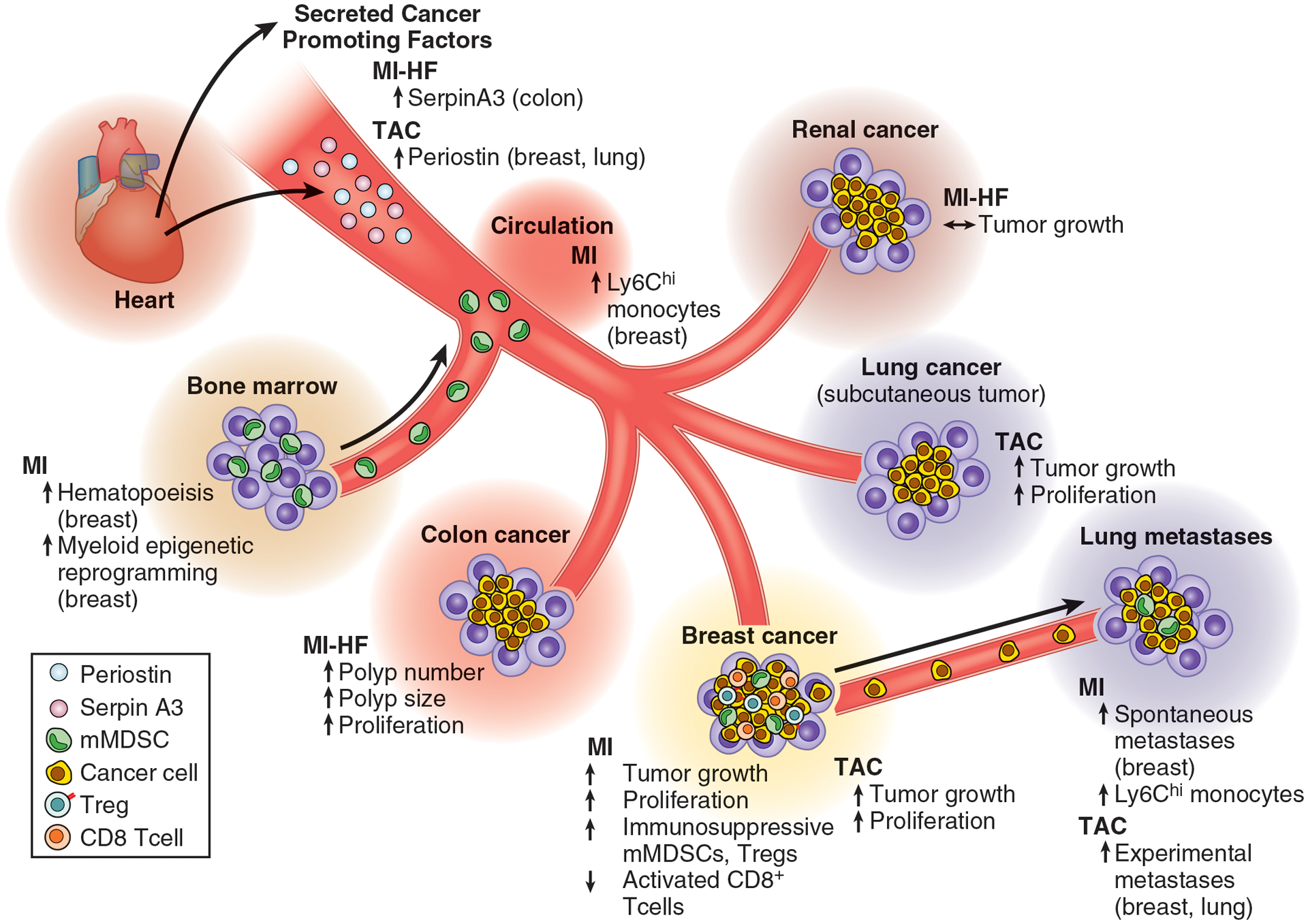
MI: myocardial infarction; HF: heart failure; TAC: transverse aortic constriction, a model of pressure overload-induced cardiac hypertrophy and heart failure; mMDSC: monocytic myeloid-derived suppressor cell; Treg: regulatory T cell.
5. Future Directions and Clinical Implications
Cardio-oncology for a long time has been exclusively focused on CVD development during or after cancer and cancer treatment. Just recently, it has been appreciated that CVD may also be accompanied or complicated by incident cancer. As described, epidemiological studies show that the presence of CVD is associated with higher incidence and worse outcomes for cancer patients, which has been backed by compelling experimental studies, which identify various CVD-specific secreted factors and alterations to host immunity, which in turn induce a pro-tumorigenic milieu that is favourable to cancer outgrowth. Such interrogation provides a window into the mechanistic underpinnings of such clinical observations.
We are now at the stage where we need scientific expansion of this field to allow clinical translation. Clearly, the first studies, that we have contributed to and have reviewed in this article, are a simplification of the complex human (patho-) physiology, yet at the same time, have explored several very attractive mechanistic pathways. Circulating factors may be employed for detection of cancer risk, that may be CVD specific, or generic. And as has recently been discussed by several groups [72–74], biomarkers may be further developed into biotargets. The observed changes in the immune system obviously are also very feasible and attractive targets for treatment, in the era that immune therapy is becoming the mainstay of cancer treatment.
What needs to be done? In Table 1, we provide three overarching areas of future investigation. First, we will need cardiologists and oncologists who are dedicated to move outside their comfort zone, and systematically and precisely map the scope of the problem. Cancer trials need meticulous CV phenotyping, and CV trials need meticulous cancer phenotyping. In reality, this rarely happens. Second, databases, biobanks and repositories coming from such concerted actions will prove invaluable in deep phenotyping of the intimate relationship, and generate insights into potential pathways. Third, translational and basic researchers should become involved to test the pathways. Ultimately, this should improve the understanding of the complex interplays between cancer and CVD and improve outcomes for patients.
Table 1 –
Future areas of research in the field of ‘reverse cardio-oncology’
| Future Area of Research | Lines of Investigation |
|---|---|
| 1. Epidemiological Discovery | |
| Prevalent CVD and Cancer Incidence, Recurrence, Cancer-specific Mortality | |
|
|
| Post Cancer Diagnosis Incident CVD and Cancer Recurrence, Cancer-specific Mortality | |
|
|
| 2. Molecular Epidemiology | |
|
|
| 3. Preclinical Studies | |
|
Funding:
Drs. Aboumsallem and de Boer are supported by a grant from the European Research Council (ERC CoG 818715, SECRETE-HF). This work was further supported by grants from the Netherlands Heart Foundation (CVON SHE-PREDICTS-HF, grant 2017-21; CVON RED-CVD, grant 2017-11; CVON PREDICT2, grant 2018-30; and CVON DOUBLE DOSE, grant 2020B005), and by a grant from the leDucq Foundation (Cure PhosphoLambaN induced Cardiomyopathy (Cure-PLaN). Dr. Moore is supported by the National Institutes of Health (R35HL135799 and P01HL131481).
Potential conflicts of interest:
The UMCG, which employs Drs. Aboumsallem and De Boer, has received research grants and/or fees from AstraZeneca, Abbott, Boehringer Ingelheim, Cardior Pharmaceuticals Gmbh, Ionis Pharmaceuticals, Inc., Novo Nordisk, and Roche. Dr. de Boer received speaker fees from Abbott, AstraZeneca, Bayer, Novartis, and Roche. Drs. Koelwyn and Moore have no conflicts of interest.
References
- [1].Moslehi JJ, Cardiovascular Toxic Effects of Targeted Cancer Therapies, N Engl J Med 375(15) (2016) 1457–1467. [DOI] [PubMed] [Google Scholar]
- [2].Jones LW, Haykowsky MJ, Swartz JJ, Douglas PS, Mackey JR, Early breast cancer therapy and cardiovascular injury, J Am Coll Cardiol 50(15) (2007) 1435–41. [DOI] [PubMed] [Google Scholar]
- [3].Johnson DB, Reynolds KL, Sullivan RJ, Balko JM, Patrinely JR, Cappelli LC, et al. , Immune checkpoint inhibitor toxicities: systems-based approaches to improve patient care and research, Lancet Oncol 21(8) (2020) e398–e404. [DOI] [PubMed] [Google Scholar]
- [4].Aboumsallem JP, Moslehi J, de Boer RA, Reverse Cardio-Oncology: Cancer Development in Patients With Cardiovascular Disease, J Am Heart Assoc 9(2) (2020) e013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nahrendorf M, Swirski FK, Innate immune cells in ischaemic heart disease: does myocardial infarction beget myocardial infarction?, Eur Heart J 37(11) (2016) 868–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vasamsetti SB, Coppin E, Zhang X, Florentin J, Koul S, Gotberg M, et al. , Apoptosis of hematopoietic progenitor-derived adipose tissue-resident macrophages contributes to insulin resistance after myocardial infarction, Sci Transl Med 12(553) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Libby P, Nahrendorf M, Swirski FK, Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”, J Am Coll Cardiol 67(9) (2016) 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. , Myocardial infarction accelerates atherosclerosis, Nature 487(7407) (2012) 325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Koene RJ, Prizment AE, Blaes A, Konety SH, Shared Risk Factors in Cardiovascular Disease and Cancer, Circulation 133(11) (2016) 1104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Meijers WC, de Boer RA, Common risk factors for heart failure and cancer, Cardiovasc Res 115(5) (2019) 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hasin T, Gerber Y, Weston SA, Jiang R, Killian JM, Manemann SM, et al. , Heart Failure After Myocardial Infarction Is Associated With Increased Risk of Cancer, J Am Coll Cardiol 68(3) (2016) 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Malmborg M, Christiansen CB, Schmiegelow MD, Torp-Pedersen C, Gislason G, Schou M, Incidence of new onset cancer in patients with a myocardial infarction - a nationwide cohort study, BMC Cardiovasc Disord 18(1) (2018) 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Berton G, Cordiano R, Cavuto F, Bagato F, Segafredo B, Pasquinucci M, Neoplastic disease after acute coronary syndrome: incidence, duration, and features: the ABC-4* Study on Heart Disease, J Cardiovasc Med (Hagerstown) 19(10) (2018) 546–553. [DOI] [PubMed] [Google Scholar]
- [14].Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, de Jong S, et al. , Heart Failure Stimulates Tumor Growth by Circulating Factors, Circulation 138(7) (2018) 678–691. [DOI] [PubMed] [Google Scholar]
- [15].Avraham S, Abu-Sharki S, Shofti R, Haas T, Korin B, Kalfon R, et al. , Early Cardiac Remodeling Promotes Tumor Growth and Metastasis, Circulation 142(7) (2020) 670–683. [DOI] [PubMed] [Google Scholar]
- [16].Boffetta P, Malhotra J, Impact of Heart Failure on Cancer Incidence: A Complicated Question, J Am Coll Cardiol 71(14) (2018) 1511–1512. [DOI] [PubMed] [Google Scholar]
- [17].de Boer RA, Hulot JS, Tocchetti CG, Aboumsallem JP, Ameri P, Anker SD, et al. , Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC), Eur J Heart Fail 22(12) (2020) 2272–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koelwyn GJ, Newman AAC, Afonso MS, van Solingen C, Corr EM, Brown EJ, et al. , Myocardial infarction accelerates breast cancer via innate immune reprogramming, Nat Med 26(9) (2020) 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].de Boer RA, Meijers WC, van der Meer P, van Veldhuisen DJ, Cancer and heart disease: associations and relations, Eur J Heart Fail 21(12) (2019) 1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bertero E, Canepa M, Maack C, Ameri P, Linking Heart Failure to Cancer, Circulation 138(7) (2018) 735–742. [DOI] [PubMed] [Google Scholar]
- [21].Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. , 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines, Circulation 140(11) (2019) e596–e646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Banks E, Joshy G, Korda RJ, Stavreski B, Soga K, Egger S, et al. , Tobacco smoking and risk of 36 cardiovascular disease subtypes: fatal and non-fatal outcomes in a large prospective Australian study, BMC Med 17(1) (2019) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Golemis EA, Scheet P, Beck TN, Scolnick EM, Hunter DJ, Hawk E, et al. , Molecular mechanisms of the preventable causes of cancer in the United States, Genes Dev 32(13–14) (2018) 868–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. , Mutational signatures associated with tobacco smoking in human cancer, Science 354(6312) (2016) 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lortet-Tieulent J, Goding Sauer A, Siegel RL, Miller KD, Islami F, Fedewa SA, et al. , State-Level Cancer Mortality Attributable to Cigarette Smoking in the United States, JAMA Intern Med 176(12) (2016) 1792–1798. [DOI] [PubMed] [Google Scholar]
- [26].Ligibel JA, Alfano CM, Courneya KS, Demark-Wahnefried W, Burger RA, Chlebowski RT, et al. , American Society of Clinical Oncology position statement on obesity and cancer, J Clin Oncol 32(31) (2014) 3568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, et al. , Body Fatness and Cancer--Viewpoint of the IARC Working Group, N Engl J Med 375(8) (2016) 794–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ, Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults, N Engl J Med 348(17) (2003) 1625–38. [DOI] [PubMed] [Google Scholar]
- [29].Hopkins BD, Goncalves MD, Cantley LC, Obesity and Cancer Mechanisms: Cancer Metabolism, J Clin Oncol 34(35) (2016) 4277–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA, Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation, J Clin Oncol 34(35) (2016) 4270–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Quail DF, Dannenberg AJ, The obese adipose tissue microenvironment in cancer development and progression, Nat Rev Endocrinol 15(3) (2019) 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song M, Vogelstein B, Giovannucci EL, Willett WC, Tomasetti C, Cancer prevention: Molecular and epidemiologic consensus, Science 361(6409) (2018) 1317–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lavie CJ, Ozemek C, Carbone S, Katzmarzyk PT, Blair SN, Sedentary Behavior, Exercise, and Cardiovascular Health, Circ Res 124(5) (2019) 799–815. [DOI] [PubMed] [Google Scholar]
- [34].Moore SC, Lee IM, Weiderpass E, Campbell PT, Sampson JN, Kitahara CM, et al. , Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults, JAMA Intern Med 176(6) (2016) 816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Friedenreich CM, Neilson HK, Farris MS, Courneya KS, Physical Activity and Cancer Outcomes: A Precision Medicine Approach, Clin Cancer Res 22(19) (2016) 4766–4775. [DOI] [PubMed] [Google Scholar]
- [36].Betof AS, Lascola CD, Weitzel D, Landon C, Scarbrough PM, Devi GR, et al. , Modulation of murine breast tumor vascularity, hypoxia and chemotherapeutic response by exercise, J Natl Cancer Inst 107(5) (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Koelwyn GJ, Quail DF, Zhang X, White RM, Jones LW, Exercise-dependent regulation of the tumour microenvironment, Nat Rev Cancer 17(10) (2017) 620–632. [DOI] [PubMed] [Google Scholar]
- [38].Koelwyn GJ, Zhuang X, Tammela T, Schietinger A, Jones LW, Exercise and immunometabolic regulation in cancer, Nat Metab 2(9) (2020) 849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pedersen L, Idorn M, Olofsson GH, Lauenborg B, Nookaew I, Hansen RH, et al. , Voluntary Running Suppresses Tumor Growth through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution, Cell Metab 23(3) (2016) 554–62. [DOI] [PubMed] [Google Scholar]
- [40].Wennerberg E, Lhuillier C, Rybstein MD, Dannenberg K, Rudqvist NP, Koelwyn GJ, et al. , Exercise reduces immune suppression and breast cancer progression in a preclinical model, Oncotarget 11(4) (2020) 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stocks T, Van Hemelrijck M, Manjer J, Bjorge T, Ulmer H, Hallmans G, et al. , Blood pressure and risk of cancer incidence and mortality in the Metabolic Syndrome and Cancer Project, Hypertension 59(4) (2012) 802–10. [DOI] [PubMed] [Google Scholar]
- [42].Braithwaite D, Moore DH, Satariano WA, Kwan ML, Hiatt RA, Kroenke C, et al. , Prognostic impact of comorbidity among long-term breast cancer survivors: results from the LACE study, Cancer Epidemiol Biomarkers Prev 21(7) (2012) 1115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nelson RH, Hyperlipidemia as a risk factor for cardiovascular disease, Prim Care 40(1) (2013) 195–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kuzu OF, Noory MA, Robertson GP, The Role of Cholesterol in Cancer, Cancer Res 76(8) (2016) 2063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Platz EA, Till C, Goodman PJ, Parnes HL, Figg WD, Albanes D, et al. , Men with low serum cholesterol have a lower risk of high-grade prostate cancer in the placebo arm of the prostate cancer prevention trial, Cancer Epidemiol Biomarkers Prev 18(11) (2009) 2807–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Platz EA, Clinton SK, Giovannucci E, Association between plasma cholesterol and prostate cancer in the PSA era, Int J Cancer 123(7) (2008) 1693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Allott EH, Howard LE, Cooperberg MR, Kane CJ, Aronson WJ, Terris MK, et al. , Serum lipid profile and risk of prostate cancer recurrence: Results from the SEARCH database, Cancer Epidemiol Biomarkers Prev 23(11) (2014) 2349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nelson ER, The significance of cholesterol and its metabolite, 27-hydroxycholesterol in breast cancer, Mol Cell Endocrinol 466 (2018) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bahl M, Ennis M, Tannock IF, Hux JE, Pritchard KI, Koo J, et al. , Serum lipids and outcome of early-stage breast cancer: results of a prospective cohort study, Breast Cancer Res Treat 94(2) (2005) 135–44. [DOI] [PubMed] [Google Scholar]
- [50].Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. , The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells, Nat Commun 8(1) (2017) 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ma L, Wang L, Nelson AT, Han C, He S, Henn MA, et al. , 27-Hydroxycholesterol acts on myeloid immune cells to induce T cell dysfunction, promoting breast cancer progression, Cancer Lett 493 (2020) 266–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. , 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology, Science 342(6162) (2013) 1094–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Haybar H, Khodadi E, Shahrabi S, Wnt/beta-catenin in ischemic myocardium: interactions and signaling pathways as a therapeutic target, Heart Fail Rev 24(3) (2019) 411–419. [DOI] [PubMed] [Google Scholar]
- [54].Polakis P, Wnt signaling in cancer, Cold Spring Harb Perspect Biol 4(5) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Keramati AR, Fathzadeh M, Go GW, Singh R, Choi M, Faramarzi S, et al. , A form of the metabolic syndrome associated with mutations in DYRK1B, N Engl J Med 370(20) (2014) 1909–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Becker W, A wake-up call to quiescent cancer cells - potential use of DYRK1B inhibitors in cancer therapy, FEBS J 285(7) (2018) 1203–1211. [DOI] [PubMed] [Google Scholar]
- [57].Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. , Age-related clonal hematopoiesis associated with adverse outcomes, N Engl J Med 371(26) (2014) 2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. , Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease, N Engl J Med 377(2) (2017) 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Libby P, Sidlow R, Lin AE, Gupta D, Jones LW, Moslehi J, et al. , Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week, J Am Coll Cardiol 74(4) (2019) 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Baron JA, Gridley G, Weiderpass E, Nyren O, Linet M, Venous thromboembolism and cancer, Lancet 351(9109) (1998) 1077–80. [DOI] [PubMed] [Google Scholar]
- [61].Qureshi AI, Malik AA, Saeed O, Adil MM, Rodriguez GJ, Suri MF, Incident cancer in a cohort of 3,247 cancer diagnosis free ischemic stroke patients, Cerebrovasc Dis 39(5–6) (2015) 262–8. [DOI] [PubMed] [Google Scholar]
- [62].Conen D, Wong JA, Sandhu RK, Cook NR, Lee IM, Buring JE, et al. , Risk of Malignant Cancer Among Women With New-Onset Atrial Fibrillation, JAMA Cardiol 1(4) (2016) 389–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vinter N, Christesen AMS, Fenger-Gron M, Tjonneland A, Frost L, Atrial Fibrillation and Risk of Cancer: A Danish Population-Based Cohort Study, J Am Heart Assoc 7(17) (2018) e009543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].de Boer RA, Aboumsallem JP, Bracun V, Leedy D, Cheng R, Patel S, et al. , A new classification of cardio-oncology syndromes, Cardiooncology 7(1) (2021) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. , Pre-metastatic niches: organ-specific homes for metastases, Nat Rev Cancer 17(5) (2017) 302–317. [DOI] [PubMed] [Google Scholar]
- [66].Ugel S, De Sanctis F, Mandruzzato S, Bronte V, Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages, J Clin Invest 125(9) (2015) 3365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shipp C, Speigl L, Janssen N, Martens A, Pawelec G, A clinical and biological perspective of human myeloid-derived suppressor cells in cancer, Cell Mol Life Sci 73(21) (2016) 4043–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Engblom C, Pfirschke C, Pittet MJ, The role of myeloid cells in cancer therapies, Nat Rev Cancer 16(7) (2016) 447–62. [DOI] [PubMed] [Google Scholar]
- [69].Kurotaki D, Nakabayashi J, Nishiyama A, Sasaki H, Kawase W, Kaneko N, et al. , Transcription Factor IRF8 Governs Enhancer Landscape Dynamics in Mononuclear Phagocyte Progenitors, Cell Rep 22(10) (2018) 2628–2641. [DOI] [PubMed] [Google Scholar]
- [70].Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. , Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis, J Clin Invest 123(10) (2013) 4464–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shi C, Aboumsallem JP, de Wit S, Schouten EM, Bracun V, Meijers WC, et al. , Evaluation of renal cancer progression in a mouse model of heart failure, Cancer Commun (Lond) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Suthahar N, Meijers WC, Sillje HHW, Ho JE, Liu FT, de Boer RA, Galectin-3 Activation and Inhibition in Heart Failure and Cardiovascular Disease: An Update, Theranostics 8(3) (2018) 593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shi C, van der Wal HH, Sillje HHW, Dokter MM, van den Berg F, Huizinga L, et al. , Tumour biomarkers: association with heart failure outcomes, J Intern Med 288(2) (2020) 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bracun V, Aboumsallem JP, van der Meer P, de Boer RA, Cardiac Biomarkers in Patients with Cancer: Considerations, Clinical Implications, and Future Avenues, Curr Oncol Rep 22(7) (2020) 67. [DOI] [PMC free article] [PubMed] [Google Scholar]