

Abstract
Background:
CD19-specific chimeric antigen receptor (CAR) T-cell therapies, including the FDA-approved tisagenlecleucel, induce high rates of remission in pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL). However, posttreatment relapse remains an issue. Optimal management of B-ALL after tisagenlecleucel treatment remains elusive and continued tracking of outcomes is necessary to establish a standard of care for this population.
Objective:
We sought to evaluate outcomes on the real-world use of tisagenlecleucel in a contemporary pediatric patient population, and to identify risk factors influencing event-free survival (EFS) and overall survival (OS). Additionally, we aimed to describe post tisagenlecleucel management strategies, including use of allogeneic hematopoietic cell transplant (AlloHCT) and/or repeat CAR T-cell infusions.
Study Design:
We report on 31 pediatric and adolescent and young adult patients (AYA) with B-ALL, treated with lymphodepleting chemotherapy followed by tisagenlecleucel. Patients were treated at Johns Hopkins Hospital and St. Jude Children’s Research Hospital between March 2018 and November 2020. Data on patient, disease and treatment characteristics were collected retrospectively from medical records and described. EFS and OS were estimated by the Kaplan-Meier method and compared by the log-rank test. Single-factor and multiple-factor analysis of EFS and OS were performed by fitting Cox regression models.
Results:
Of the 30 evaluable patients, 25 (83.3%) experienced a complete response, with 21 having negative minimal residual disease (MRD). Treatment was well tolerated, with expected rates of cytokine release syndrome (61.3%) and immune effector cell associated neurotoxicity (29%). After initial CR, 12 patients (48%) had subsequent disease recurrence, with CD19-negative relapse (n=6) occurring sooner than CD19-positive relapse (P = 0.0125). With a median follow-up time of 386 day s(range:11–1187 days), the EFS for the entire cohort (n = 31) at 6- and 12-months post infusion was 47% (95% confidence interval [CI]: 28.4–63.4%) and 35.2% (CI, 18.4–52.5%), respectively. In multivariate analysis, high pretreatment leukemic burden ≥5% bone marrow blasts) was an independent risk factor for inferior EFS (HR 5.98 [95% CI, 1.1–32.4], P =0.0380) and OS (HR 4.2 [95% CI, 1.33–13.39, P = 0.0148).
Conclusions:
Tisagenlecleucel induced high initial response rates in a contemporary cohort of pediatric and AYA patients with B-ALL. However, 48% of patients experienced subsequent disease relapse, including 6 with antigen-escape variants. This highlights a considerable limitation of single-agent autologous CD19-CAR T-cell therapy. Pretreatment leukemic disease burden of ≥5% blasts was significantly associated with worse outcomes in this study, including lower EFS and OS. Our findings suggest that reducing pre-infusion leukemic burden is a viable treatment strategy to improve outcomes of CAR T-cell therapy.
INTRODUCTION
Pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL) historically have poor survival outcomes due to inherent and/or acquired chemotherapy resistance (1). With the advent of CD19-specific chimeric antigen receptor (CD19-CAR) T-cell therapies, these patients are now experiencing remission at high rates (2–6). For pediatric patients without access to experimental T-cell products, the availability of the FDA-approved product tisagenlecleucel (Kymriah, Novartis) has changed the treatment paradigm (7, 8). Initial reports of real-world data from the Center for International Blood and Marrow Transplant Research (CIBMTR) registry showed that tisagenlecleucel has yielded efficacy and safety profiles similar to those from the registration trial (9).
Despite impressive initial response rates, relapse after CAR T-cell therapy is a significant concern, and questions remain regarding the optimal use of tisagenlecleucel (3, 4, 8, 10). These include how patient, disease, and treatment characteristics influence short-term and long-term outcomes, and how to employ post infusion management strategies and disease-monitoring techniques. Consequently, the standard of care of tisagenlecleucel treatment is not yet established. Studies of clinical and disease characteristics that predict outcomes are necessary to identify patients who can benefit from post-tisagenlecleucel treatment regimens, including allogeneic hematopoietic cell transplantation (AlloHCT).
Herein, we report on the use of tisagenlecleucel to treat relapsed/refractory B-ALL in pediatric and adolescent and young adult (AYA) patients at two institutions. We highlight the impact of pre-infusion disease burden on the incidence of relapse and the risk of treatment resistance resulting from the emergence of CD19-negative antigen-loss variants.
SUBJECTS AND METHODS
Study design
This retrospective study reflects our real-world experience of treating pediatric and AYA patients with relapsed and/or refractory B-ALL with lymphodepletion and tisagenlecleucel, between March 2018 and November 2020 at Johns Hopkins Hospital (JHH) and St. Jude Children’s Research Hospital (St. Jude). A subset of these patients was included in a recent retrospective analysis (11). The study was approved by both institutional review boards, and informed consent/assent was obtained from patients/guardians as appropriate. Patient, disease, and treatment information was abstracted from prospectively collected clinical databases and supplemented by retrospective medical record review. High-risk leukemic genetics were categorized using published literature (12–14). Data were also collected for patients who received a second CAR T-cell infusion using previously manufactured product. The date of data cutoff is October 31, 2021.
Leukapheresis products were collected and cryopreserved in accordance with institutional standard operating procedures before shipment to Novartis’ centralized production site. All infused tisagenlecleucel products met the release specification. The use and type of bridging therapy before tisagenlecleucel infusion was determined by the treating physician. Pretreatment disease burden was evaluated after bridging therapy and before cellular infusion. Most patients underwent a bone marrow analysis to determine the levels of morphologic blasts and the disease burden using minimal residual disease (MRD) measurement by flow cytometry. MRD was used to categorize patients into disease burden. One patient had disease confirmed by peripheral blood only and was empirically categorized as having 26% marrow disease for our analysis. For another patient who did not have MRD measured, the morphologic blast percentage (52%) was used as a measurement of disease burden. Evaluation of extramedullary (EM) sites of disease included a cerebrospinal fluid (CSF) examination and, in a subset of patients, a positron emission tomography (PET) scan. CAR T-cell therapy included lymphodepletion followed by tisagenlecleucel infusion. Routine supportive care followed institutional standard operating procedures, including the use of seizure and/or infectious prophylaxis.
Toxicity and outcome evaluations
Cytokine release syndrome (CRS) was graded using the American Society of Transplantation and Cellular Therapy (ASTCT) consensus grading criteria (15). Before the ASTCT criteria were released, immune effector cell–associated neurotoxicity syndrome (ICANS) was graded using the Common Terminology Criteria for Adverse Events (CTCAE). Disease response was determined at approximately 4 weeks post infusion and categorized by comparing the leukemic burden in pretreatment and post treatment bone marrow specimens, using morphology and MRD detection (flow cytometry, RT-PCR, and/or next-generation sequencing [NGS] testing [Adaptive Biotechnologies], when available for a given patient). MRD-negative complete response (CR) was defined as <5% blasts by morphology AND <0.01% blasts by flow cytometry, <10−4 by PCR, and/or <10−5 by NGS in post therapy marrow. For patients with ≥5% morphologic disease before therapy, MRD-positive CR was defined as <5% blasts by morphology AND ≥0.01% blasts by flow cytometry, ≥10−4 by PCR, and/or ≥10−5 by NGS in the post therapy marrow. Patients with persistent disease after therapy were considered non-responsive if their disease burden remained at the same order of magnitude as before therapy (persistent MRD-positive or ≥5% blasts by morphology). Disease-response assessments of extramedullary sites were made independent of the marrow response.
Follow-up evaluations
Relapse was defined as the development of any detectable disease, including MRD-positive marrow (as defined above) or extramedullary disease, after initial CR post tisagenlecleucel treatment. Disease detected via flow-cytometry was determined to be CD19-positive or CD19-negative. Autologous B-cell recovery was defined as ≥1% CD19-positive cells (at two consecutive time-points ≥1 week apart) or an absolute CD19-positive cell count of ≥50/μL (at a single time-point) in the peripheral blood. Additional cell therapy (e.g., AlloHCT or CAR T-cell reinfusion) after CAR T-cell therapy was recorded.
Statistical methods
Descriptive statistics were compared using Fisher’s exact test, the Wilcoxon rank-sum test, or the Kruskal–Wallis test as appropriate. Event-free survival (EFS) and overall survival (OS) functions were estimated by the Kaplan–Meier method and compared by the log-rank test. EFS was defined as the time from CAR T-cell infusion to being non-responsive at week 4 evaluation, relapse, or death; patients who underwent HCT before one of these events were censored at the time of HCT. OS was defined as the time from CAR T-cell infusion to death. Single-factor and multiple-factor analysis of EFS and OS were performed by fitting Cox regression models. The cumulative incidence function of relapse was compared by Gray's test accounting for competing risks. Relapse analysis included censoring of patients at the time of death or HCT. Risk factors with univariate P values of 0.25 or less were included in the multivariable models for EFS and OS. Statistical analyses were performed with SAS version 9.4.
RESULTS
Patient and disease characteristics
Thirty-three pediatric and AYA patients with relapsed/refractory B-ALL were considered for tisagenlecleucel treatment; two patients were not treated because of poor clinical status or manufacturing failure. Baseline patient and disease characteristics of the 31 treated patients are shown in Table 1, and Figure 1A gives an overview of treatment outcomes. Four patients received repeat CAR T-cell infusions subsequent to the first infusion without interval therapy, using previously manufactured product; the resulting data are reported separately in a subsequent section.
Table 1:
Patient and Disease Characteristics (n = 31)
| Treating institution | |
| JHH | 18 (58.1) |
| St. Jude | 13 (41.9) |
|
| |
| Sex | |
| Male | 18 (58.1) |
| Female | 13 (41.9) |
|
| |
| Age at diagnosis | 6.5 [0.3–21.0] |
|
| |
| Age at time of CAR T-cell infusion | 7.9 [0.8–23.6] |
|
| |
| Indication for tisagenlecleucel | |
| Primary refractory disease | 11 (35.5) |
| Relapse 1 | 14 (45.2) |
| Relapse 2 | 5 (16.1) |
| Relapse 3+ | 1 (3.2) |
|
| |
| High-risk genetics | |
| BCR-ABL1 | 1 (3.2) |
| Ph-like | 6 (19.4) |
| KMT2A rearranged | 5 (19.4) |
| p53 alteration (without associated hypodiploidy) | 2 (6.5) |
| TCF3-PBX1 | 1 (3.2) |
| Myc translocation | 1 (3.2) |
| Low hypodiploidy | 3 (9.7) |
| Monosomy 7 | 1 (3.2) |
|
| |
| Prior allogeneic HCT | 4 (12.9) |
|
| |
| Prior CD19- or CD22-directed therapy | |
| Any agent | 8 (25.8) |
| Blinatumomab | 6 (19.3) |
| Inotuzumab | 5 (16.1) |
| CD19-CAR T-cell therapy | 1 (3.2) |
|
| |
| Pre-infusion disease evaluation * | |
| Marrow burden (median disease % [range]) | |
| MRD-negative (n = 3) | — |
| MRD-positive >0 to <5% (n = 15) | 0.6 [0.003–2.7] |
| ≥5% (n = 13**) | 52 [7.6–100] |
| CNS3 | 1 (3.2) |
| Non-CNS extramedullary disease^ | 3 (9.7) |
JHH: Johns Hopkins Hospital; St. Jude: St. Jude Children’s Research Hospital; HCT: hematopoietic cell transplantation; MRD: minimal residual disease (by flow cytometry). Numerical data are presented as the n (%) or median [range]
After bridging chemotherapy and before CAR T-cell therapy
One patient without available disease % by flow cytometry was classified using morphologic blasts (52%), and one patient had disease confirmed by peripheral blood only and was empirically categorized as having 26% disease for this analysis
Increased metabolic uptake on PET-CT [evaluated in a subset of patients (n = 7)]
Figure 1. Disease response and toxicities of pediatric and AYA patients treated with lymphodepletion and tisagenlecleucel.
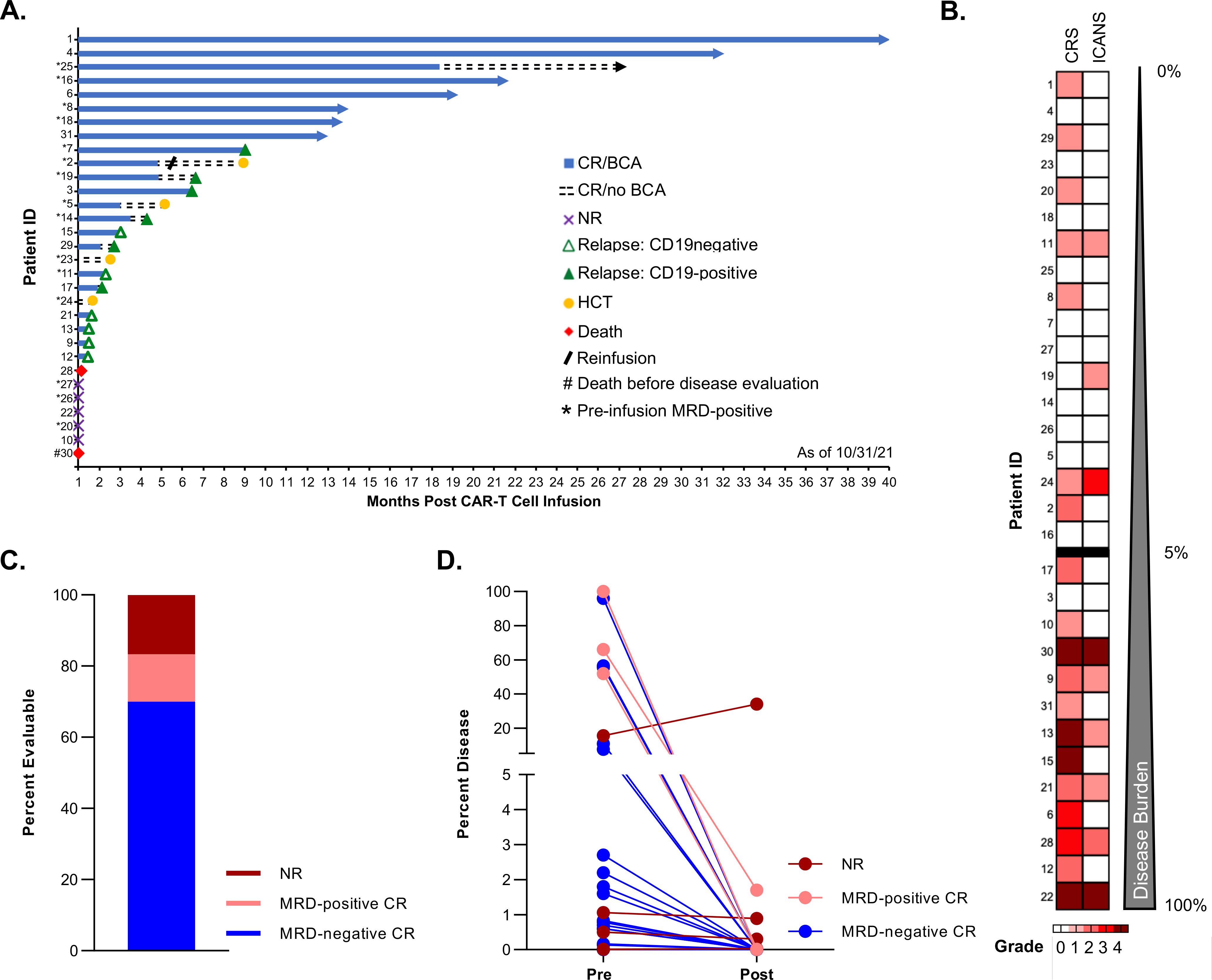
A. Swimmer plot depicting the longitudinal outcomes of the entire patient cohort (n=31, each lane represents a single patient), including the initial response at 4 weeks post CAR T-cell infusion (CR: complete response; NR: no response), the duration of B-cell aplasia (BCA) and subsequent events (relapse [CD19-positive or CD19-negative]), the time of planned consolidative allogeneic hematopoietic cell transplantation (HCT), and death. For each patient, the data end at the time of the first event (NR, relapse, HCT, or death). Ongoing remission without any event is indicated by an arrow. B. Heatmap depicting the maximum grade of cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) for each treated patient. Patients are ordered by increasing burden of leukemia (%) as measured by flow cytometry in the bone marrow before tisagenlecleucel therapy. C. Treatment response at approximately 4 weeks post infusion among the 30 evaluable patients. D. Change in bone marrow disease burden (%) measured using pretreatment and post tisagenlecleucel therapy evaluations. Each line represents a single patient and is color coded to indicate the response.
Median age at diagnosis of B-ALL was 6.5 years (range: 0.3–21.0 years) and median age at the time of cellular infusion was 7.9 years (range: 0.8–23.6 years; three patients aged ≤3 years). Most patients had high-risk disease characteristics, with 19 having high-risk genetic features, 4 having undergone prior AlloHCT, and 8 having received prior CD19-directed therapy (blinatumomab or CD19-CAR) and/or CD22-directed therapy (inotuzumab). Most patients received CAR T-cell therapy for primary refractory disease (35.5%) or in first relapse (45.2%) (Table 1). Pretreatment disease burden was assessed a median of 7 days (range: 1–23 days) prior to cellular infusion. Marrow results included a median morphologic blast percentage of 2% (range: 0–98%; n = 30) and a median MRD (flow cytometry) of 1.6% (range: 0–96%; n = 29) (Table 1). One of the three MRD-negative patients had disease detectable, but below our cutoff for definition of MRD positivity by NGS (3 clones/million). Pretreatment CSF samples were obtained for 29 patients, one of whom had detectable CNS disease (CNS3). The presence of non-CNS extramedullary disease was assessed by PET scan in seven patients, three of whom had abnormal metabolic activity concerning for leukemic disease (Table 1).
Treatment characteristics
For four infused patients, two apheresis attempts were required because of insufficient collection (n=3) or bacterial contamination of the apheresis product due to concurrent bacteremia (n=1). During product manufacturing, 29 patients received bridging therapy. Lymphodepleting chemotherapy began at a median of 39.5 days after apheresis (range: 25–284 days; date of apheresis was unavailable for two patients). All patients received fludarabine and cyclophosphamide, with two receiving additional agents because of their high disease burden; tisagenlecleucel dosing was in accordance with manufacturer guidance (Table 2). Patients received the cellular infusion while inpatient, with a median duration of hospitalization of 10 days (range: 1–35 days). All repeat infusions used product from the initial manufacture. De-identified peripheral blood samples were obtained from a subset of patients at various post infusion time-points for PCR-based detection of circulating CAR T cells (Supplemental Methods); all but one of these samples had detectable CAR T cells, and aggregation of the data showed the expected post infusion expansion (Supplementary Fig. 1). Limited availability of specimens precluded relating clinical factors to the amplitude or timing of in vivo CAR T-cell expansion.
Table 2:
Treatment Characteristics (n = 31)
| Lymphodepletion* (number [%]) | |
| Flu (120 mg/m2)/Cy (1000 mg/m2) | 19 (61.3) |
| Flu (75 mg/m2)/Cy (900 mg/m2) | 10 (32.3) |
| Flu (75 mg/m2)/Cy (900 mg/m2)/Eto (500 mg/m2) | 1 (3.2) |
| Flu (75 mg/m2)/Cy (900 mg/m2)/Ara-C (4 g/m2) | 1 (3.2) |
|
| |
| CAR T-cell dose administered (median [range]) | |
| Patients ≤ 50 kg (n = 21) | 2.1 [0.9–4.5] ** |
| Patients > 50 kg (n = 10) | 1.1 [0.5–1.6] *** |
Flu: fludarabine; Cy: cyclophosphamide; Eto: etoposide; Ara-C: cytarabine
cumulative doses
×106 CAR-positive T cells/kg patient weight
×108 CAR-positive T cells.
Treatment-related toxicities
Treatment was well tolerated, with the expected side effect profile (16). After initial infusion, 19 patients (61.3%) developed CRS at a median of 4 days (range: 0–9 days) post infusion. Among these patients, six (31.6%) had grade ≥3 CRS. Nine patients (29%) experienced ICANS, with three cases being grade ≥3, at a median of 6 days (range: 1–15 days) post infusion. Eight patients had concurrent CRS and ICANS (Fig. 1B). Two patients (6.5%) developed CAR T-cell therapy-associated hemophagocytic lymphohistiocytosis (carHLH)(17) at a median of 9.5 days post infusion; both patients were recovering from high-grade CRS at the time of carHLH onset (18). Treatment for immune-mediated side effects included tocilizumab (n=11 patients), corticosteroids (n=4), siltuximab (n=3) and, in those patients with carHLH, anakinra (n=2) and ruxolitinib (n=1). With the exception of one patient with carHLH, all patients recovered. As expected, the incidence and severity of immune-mediated toxicities correlated with pre-infusion leukemic burden (Fig. 1B). Seven patients required hospital readmission within 30 days (median: 8 days; range: 4–28 days) post infusion, five for CRS and two for late-onset bacteremia. Nine patients required care in the pediatric intensive care unit (ICU), 3 were pre-emptive due to presumed high risk of developing complications and the remainder for CRS management. ICU transfer occurred at a median of 5 days (range: 0–15 days) after infusion, with a median duration of ICU admission of 8 days (range: 1–20 days).
Disease response
Thirty patients were evaluable for disease response, at a median of 28 days (range: 22–35 days) post infusion, with one death occurring before disease evaluation. Twenty-five patients (83.3%) had a CR, 21 (84%) of whom were MRD-negative (Fig. 1C; 3 with available NGS testing). Four patients had an MRD-positive CR, including two who were positive by NGS (at 10 and 154 clones/million) and two by flow cytometry (at 0.01% and 1.7%) (Fig. 1D). Five patients had no response to treatment, including two with identified changes in CD19 expression (CD19-negative or CD19-dim). Post infusion CSF analysis was performed for 26 patients, none of whom had leukemia cells. The patient with pretreatment CNS leukemia was not reassessed post infusion because of no response to therapy, with persistent peripheral blasts. Of the three patients with pre-infusion PET positivity, one had a repeat PET scan that was negative for extramedullary disease and the other two were not re-evaluated either because of early death or no response in the marrow. Of the two patients who received intensified lymphodepletion, one had no response and the other had a MRD-positive CR.
Follow-up evaluations and outcomes
All 25 patients with a CR had concurrent autologous B-cell aplasia (BCA). Of these 25 patients, 10 (40%) had autologous B-cell recovery between 0.9 and 18.4 months (median: 4.1 months) post infusion. The remaining 15 patients had ongoing BCA at their last follow-up for a median of 2.8 months (range: 0.7–31.1 months) post infusion (Fig. 1A). Notably, although they did not experience B-cell recovery, two patients had early events (treatment-related death and consolidative HCT) that precluded extended monitoring for ongoing BCA. Similarly, continued monitoring for BCA was not performed in patients after leukemic relapse.
With a median follow-up time of 386 days (range: 11–1187 days), the EFS for the entire cohort (n = 31) at 6- and 12-months post infusion was 46.9% (95% confidence interval [CI]: 28.4–63.4%) and 35.2% (CI, 18.4–52.5%), respectively (Fig. 2A). Among the 25 patients with a CR after tisagenlecleucel treatment, the duration of remission at the time of data cutoff ranged from 1.2 to 39.2 months (median: 5.2 months). Twelve patients (48%) experienced subsequent disease recurrence (Fig. 2B), six with CD19-positive leukemia, all in the setting of preceding or concurrent loss of BCA, and six with CD19-dim to negative disease, all in the setting of ongoing BCA. Notably, CD19-negative recurrence occurred significantly earlier than CD19-positive relapse, at a median of 1.6 months versus 5.4 months post infusion (P = 0.0125) (Fig. 2C). Of note, of the four patients that achieved an MRD-positive CR, three relapsed wiith CD19-negative disease (at 45-, 45-, and 50-days post infusion) and one died secondary to infection. The OS was 80.6% (CI: 61.9–90.8%) at 6 months and 67.4% (CI: 47.9–81.0%) at 12 months (Fig. 2A). Of the 31 infused patients, 13 (41.9%) died at a median of 227 days (range: 11–865 days) post infusion. Two patients died of treatment-related complications. The first patient developed CRS and subsequent carHLH (18), with associated multi-organ failure and diffuse neuronal injury, and died 11 days post infusion. The second patient had disseminated multi-organism infection, including bacteremia, bacterial meningitis, and fungal rhinosinusitis with cerebral extension, and died 35 days post infusion. The remainder died of refractory leukemia (n = 4), relapsed leukemia (n = 5) or toxicities related to subsequent treatments (n = 2). Among survivors (n=18), median length of follow-up is 760 days (range: 318–1187 days).
Figure 2. High rates of initial remission and relapse in pediatric patients treated with tisagenlecleucel.
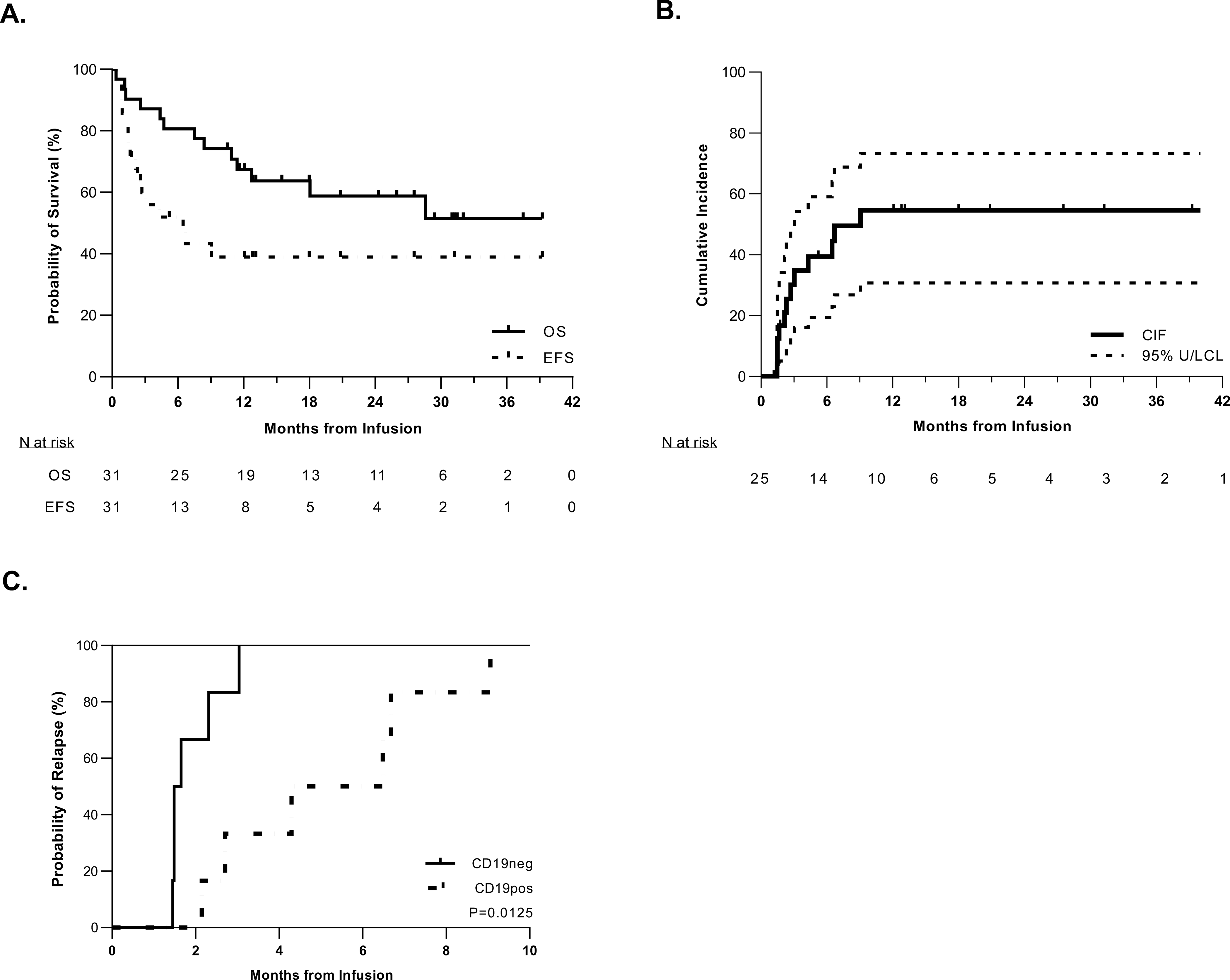
A. Kaplan-Meier curve of event-free survival (EFS) and overall survival (OS) in the total cohort of 31 patients. The median time of follow-up was 329 days (range: 11–1046 days). The median EFS time was 4.3 months (95%CI: 1.5-NA) (NA = not able to be calculated); the median OS has not been reached but the last death occurred at 28.9 months giving a 51.5% OS rate. B. Cumulative incidence of relapse among patients who experienced an initial CR. Relapse was defined as recurrent detectable disease after initial CR, including minimal residual disease (MRD-positive: ≥0.01% blasts by flow cytometry, ≥10−4 by PCR, and/or ≥10−5 by NGS). Death was considered as a competing risk. C. Cumulative incidence of relapse defined by antigen subtype: CD19-positive (n = 6) vs. CD19-negative (n = 6). CD19-negative relapse occurred sooner than CD19-positive relapse, with the median times to occurrence being 1.6 months and 5.4 months post infusion, respectively (P = 0.0125).
Impact of disease burden on EFS and OS
Risk-factor analysis was conducted using the following variables: treating institution, receipt of prior CD19-directed and/or CD22-directed therapy, pretreatment disease burden (bone marrow MRD-negative, MRD-positive [>0-<5% blasts], or ≥5% blasts), development of severe CRS, and tisagenlecleucel dosing. Patients with high levels of pretreatment bone marrow disease (≥5% blasts) had significantly poorer 12-month EFS when compared to those who were MRD-positive or MRD-negative (15.4% [CI: 2.5–38.8%] vs. 46.2% [CI: 18.2–70.4%] vs. 66.7% [CI: 5.4–94.5%]; P = 0.0392) (Fig. 3A). This was also true for 12-month OS (38.5% [CI: 14.1–62.8%] vs. 86.2% [CI: 54.9–96.4%] vs. 100% [NA]; P = 0.0027) (Fig. 3B). Notably the effect of pre-treatment disease burden on both EFS and OS was minimized in patients who achieved an MRD-negative remission with treatment, though small overall numbers in the group of patients achieving an MRD-negative post treatment remission with ≥5% pre-treatment disease preclude informative statistical analysis (Supplemental Fig. 2). In univariate analysis, the presence of high-burden disease (≥5% blasts) was associated with worse EFS (hazard ratio [HR]: 3.1 [95% CI: 1.2–7.8], P = 0.0176) and OS (HR 7.3 [95% CI: 1.9–27.6], P = 0.0034) compared to those with lower pretreatment disease burden (<5% blasts). Additionally, severe CRS and prior CD19- or CD22-directed therapy was associated with worse OS (HR 5.32 [95% CI: 1.67–16.95], P = 0.0047 and HR 3.3 [95% CI: 1.1–10.3], P = 0.0403, respectively) (Fig. 4A). In multivariate analysis, higher pretreatment disease burden (≥5% blasts) remained an independent risk factor for worse EFS (HR 5.98 [95% CI, 1.10–32.4, P = 0.0380) and OS (HR 4.2 [95% CI, 1.33–13.39, P = 0.0148) (Fig. 4B). Among those patients who experienced an initial CR, higher pretreatment disease burden was associated with a higher cumulative incidence of relapse (P=0.0482) (Fig. 3C). None of the evaluated risk factors correlated significantly with initial disease response (no response vs. MRD-positive CR vs. MRD-negative CR) at 4 weeks post infusion (data not shown).
Figure 3. Disease burden of ≥5% before CAR T-cell therapy predicts worse outcomes after tisagenlecleucel treatment.
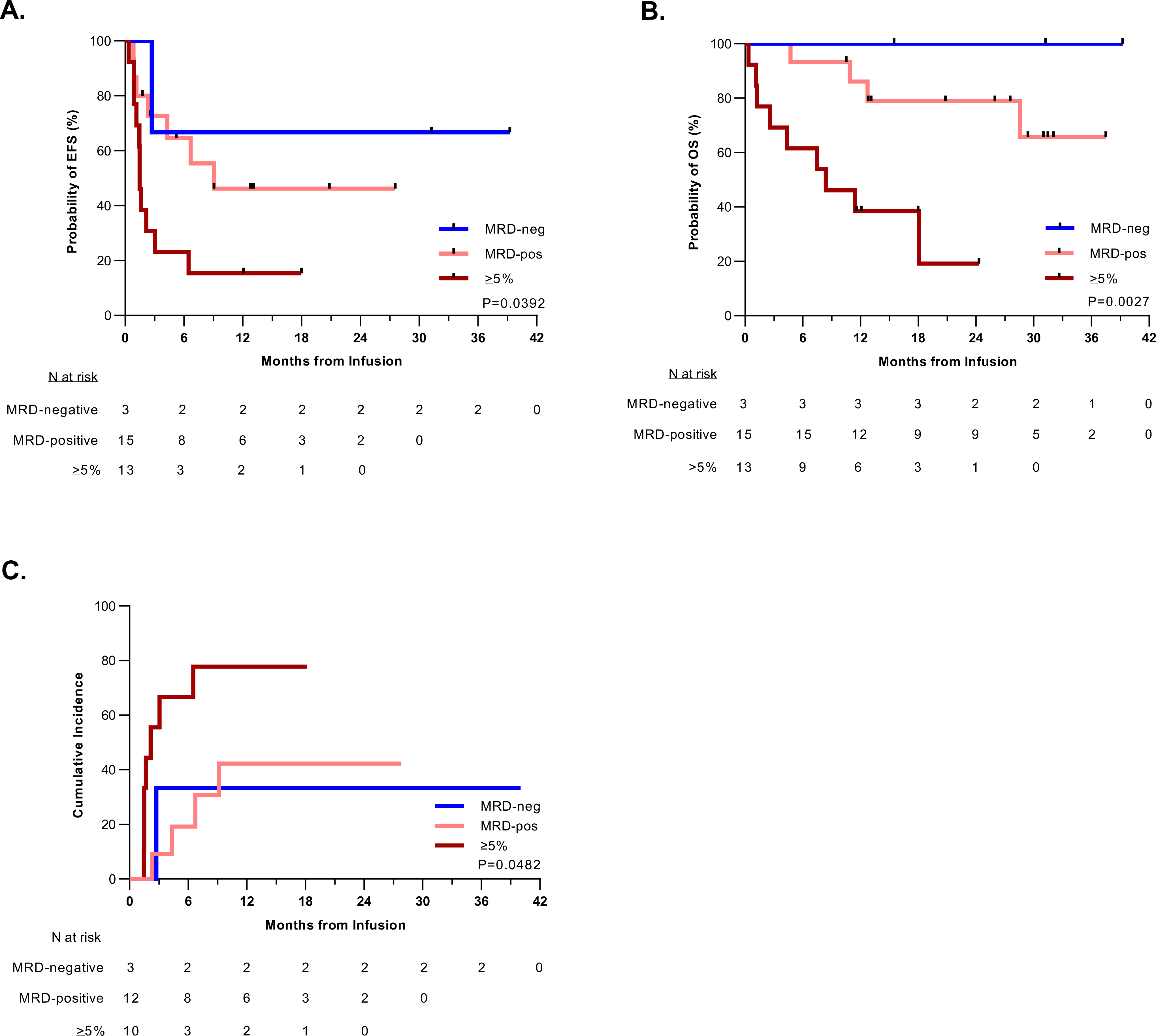
A. Kaplan-Meier estimation of the event-free survival (EFS) of patients based on their pre-infusion disease burden. Patients with higher pretreatment disease burden had worse EFS (P = 0.0392). Median EFS time based on disease burden: ≥5% blasts: 1.5 months (95% CI: 0.9–3.0); MRD-positive (>0-<5% blasts): 9.1 months (95% CI: 1.2-NA); MRD-negative: NA. B. Kaplan-Meier estimation of the overall survival (OS) of patients stratified by pre-infusion disease burden. Patients with higher pretreatment disease burden had worse OS (P = 0.0027). The median survival for patients with a disease burden of ≥5% was 8.4 months (95% CI: 1.3-NA) and unable to be calculated (NA) for patients with pretreatment MRD-positive or MRD-negative disease. C. Cumulative incidence of relapse after initial CR among patients by pre-infusion disease burden, such that patients with a disease burden of ≥5% had a higher rate of relapse (P = 0.048), with a median time to relapse of 2.2 months. The median time to relapse could not be calculated (NA) for patients with pretreatment MRD-positive or MRD-negative disease. Relapse included any recurrent detectable disease, including MRD-positive cases. Death in remission was considered as a competing risk.
Figure 4. Analysis of the impact of pretreatment and post treatment factors on event-free and overall survival.
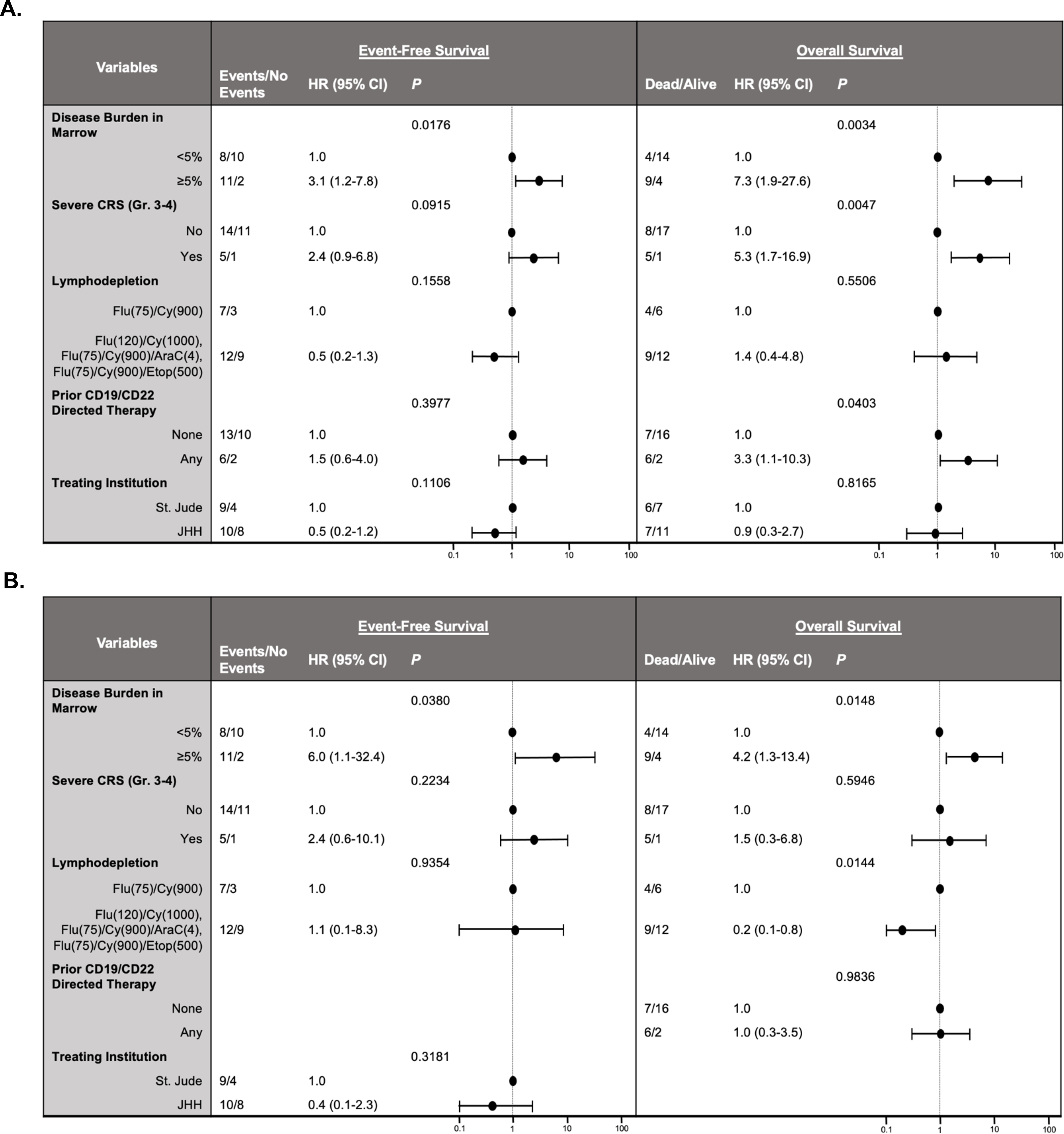
A. Univariate analyses of event-free survival (EFS) and overall survival (OS) performed by fitting Cox regression models. Higher pre-infusion disease burden (≥5% blasts) was significantly associated with worse EFS and OS (P = 0.0176 and 0.0034, respectively). Severe CRS and antigen-directed pre-treatment were also associated with worse OS (P = 0.0047 and 0.0403, respectively) but had no impact on EFS. B. Multivariate analysis of EFS and OS, including risk factors with univariate P values less than or equal to 0.20. Higher pre-infusion disease (≥5% blasts) burden remained significantly associated with poorer EFS (P = 0.0380) and OS (P = 0.0148). EFS: date from infusion to first event (NR at week 4 evaluation, relapse [recurrent detectable disease, including MRD-positive cases], or death; patients who underwent HCT before one of these events were censored at the time of HCT). OS: date from infusion to date of death.
Subsequent treatment
Four patients received a second infusion of their previously manufactured tisagenlecleucel product without other intervening therapy because of recurrent CD19-positive leukemia (n = 3) or early autologous B-cell recovery (n = 1). Median time between the infusions was 189 days (range: 90–223 days). All four patients received fludarabine and cyclophosphamide lymphodepletion before reinfusion, in two cases at higher doses than used with the first infusion. Reinfusion was well tolerated, with no CRS or ICANS. However, response to reinfusion was poorer than that for initial infusion. Two of three patients who were reinfused for active leukemia did not attain MRD negativity. The third patient experienced a second MRD-negative CR, but their CD19-positive disease relapsed after a remission shorter than that after the first infusion (63 vs. 196 days post infusion). The fourth patient infused for B-cell recovery did not experience BCA after reinfusion and ultimately proceeded to AlloHCT.
The decision to proceed with consolidative AlloHCT was patient/provider dependent. Of the 25 patients who had a CR after their first infusion, one underwent preemptive AlloHCT and three underwent AlloHCT because of early loss of BCA, 1 of whom had AlloHCT after CAR T-cell reinfusion (Fig. 1A). This was first HCT for these patients, three of whom remained alive and in remission at their last evaluation. Notably, nine additional patients were ultimately treated with AlloHCT as a component of the treatment for post-CAR T-cell therapy relapse. For seven of these patients, it was their first AlloHCT. Among these nine patients, five remained in remission at time of last follow-up, two died secondary to transplant related toxicities and two experienced leukemic relapse.
DISCUSSION
We have reported on a cohort of 31 patients treated with tisagenlecleucel at two treatment centers. In this study, we included some patients who would have been excluded from the tisagenlecleucel registration trial (8), such as children aged ≤ 3 years, patients with high-risk leukemic genetics, and those with CNS leukemia, prior treatment with CD19-directed therapies, and/or low pre-infusion disease burden (<5% blasts). However, our cohort is representative of the diversity of patients now being treated with tisagenlecleucel worldwide (9, 10).
Overall, the treatment was well tolerated, with the expected rates of immune-mediated side effects (9, 10). As anticipated, initial response rates were high, with 83.3% of evaluable patients experiencing a CR at approximately 4 weeks post infusion, the majority being MRD-negative. However, the EFS rates of 46.9% and 35.2% at 6 and 12 months, respectively, highlight a considerable limitation of single-agent autologous CD19-CAR T-cell therapy. Relapse was the most common event, with 48% of patients who experienced a CR after tisagenlecleucel treatment subsequently experiencing leukemic relapse. Notably, of patients that achieved a CR, those with MRD-positivity had a high rate of relapse, including 2 that were positive by NGS testing (3/3 evaluable; 1 deceased secondary to toxicity). However, not all patients in the MRD-negative cohort had NGS testing available at the time of disease assessment and therefore low-level disease post treatment may be underestimated.
Pretreatment leukemic disease burden in the bone marrow was associated with worse outcomes in this study, such that having ≥5% blasts portended an increased toxicity risk, lower EFS and OS, and a higher incidence of relapse. Importantly, higher disease burden was the only risk factor significantly associated with worse EFS and OS in multivariate analyses. Other groups have also found that a high pretreatment disease burden predicts an increased relapse risk and worse outcomes (10, 11, 19, 20). Although the ‘cut-off’ used to distinguish ‘high’ and ‘low’ leukemic burden has varied, the presence of some level of leukemic disease in the bone marrow before CAR T-cell treatment clearly affects outcomes. Such analysis was inherently lacking in earlier trials, given the disease-burden requirements for patients to receive protocol treatment (8). Furthermore, as more patients receive commercially available CAR T-cell products, patient characteristics not accounted for in early clinical trials need to be carefully considered in contemporary analysis. This is particularly important as there may be a correlation between disease features (such as disease status or high-risk genetics) and pretreatment disease burden, such that higher pretreatment disease burden represents a biologically higher risk patient population.
In our patient cohort, 48% of patients had recurrent leukemic disease after treatment with tisagenlecleucel. Relapses with retained CD19 expression occurred in the setting of CD19+ B-cell recovery, suggestive of a loss of functional CAR T cells resulting from limited persistence and/or T-cell exhaustion. This is consistent with previous reports (8). Interestingly, in our cohort, half of the relapses demonstrated loss of CD19 expression and these relapses occurred earlier than CD19-positive cases. CD19-negative relapses after targeted CAR T-cell therapy have been reported to result from i) expression of alternately spliced isoforms that mask expression of the targeted CD19 epitope or prevent surface protein expression (21, 22); ii) lineage switch to a myeloid phenotype (23–25); iii) genomic mutations causing frameshift transcripts with the loss of elements necessary for surface expression (26); or iv) intron retention predicted ultimately to cause nonsense-mediated decay (27, 28) It is unknown whether these relapses are due to pre-existing CD19-negative subclones or to mutations acquired under targeted immunological pressure. Not surprisingly, comparison of pretreatment disease specimens with those acquired at relapse has identified CD19-negative subclones predating CD19-CAR T-cell therapy (22, 28). Furthermore, two recent reports have highlighted the association of pre-tisagenlecleucel high disease burden (20) and the impact of prior Blinatumomab therapy on CD19-negative relapse (11). We did not analyze biological specimens collected from our patients but believe that the timing of the CD19-negative disease emergence supports a hypothesis that this represents pretreatment subclonal disease that is selected for with single-antigen targeting. We hope for the development of biomarker assays capable of identifying minute populations of CD19-negative disease with high accuracy. If such disease is detected, therapeutic consolidation with AlloHCT or alternate dual-antigen targeted therapy should be considered to prevent subclonal selection and subsequent disease relapse.
There is a real risk of relapse after tisagenlecleucel treatment. Establishing a consensus regarding the ‘best’ post CAR T-cell clinical management remains difficult, particularly with respect to using pre-emptive consolidative AlloHCT. Given that relapse and treatment-related morbidity remain significant threats after transplant, it is often desirable to avoid AlloHCT. At present, such a decision is based largely on institutional and family/patient preferences, with consideration of patient-specific history and clinical status (29–31). In our study, few patients proceeded directly to AlloHCT after tisagenlecleucel treatment. However, of the 12 patients with recurrent disease after tisagenlecleucel treatment, nine underwent AlloHCT as part of their relapse treatment regimen. Our experience suggests that a substantial subset of patients will ultimately receive AlloHCT after tisagenlecleucel treatment. Proceeding immediately to AlloHCT after CAR T-cell therapy–induced remission may offer the best chance of cure with the least treatment-related cumulative organ toxicity. Careful recording and analysis of patient-specific and disease-specific data that can inform such population-based treatment decisions is critical.
Our data highlight the poorer outcomes and high rates of relapse in patients with higher leukemic disease burden (≥5%) before CAR T-cell therapy, as well as the possibility that CD19-negative subclonal expansion is a mechanism of post infusion recurrent disease. This is of particular concern given the lack of available salvage therapies for such high-risk patients. Repeat CD19-CAR T-cell infusions are generally not as effective (32), as was the case in our cohort, where two of four patients treated with a second tisagenlecleucel infusion had no response to reinfusion. Rather than reinfusion with tisagenlecleucel, use of alternate CD19-CAR T-cell products can be considered. A recent study showed that treatment with humanized CD19-CAR T-cells led to an overall response rate of 64% at 1 month post infusion in patients who had refractory or relapsed B-ALL, and prior exposure to CD19-CAR T-cell therapy. This approach may therefore salvage some patients who relapse after tisagenlecleucel. However, subsequent post treatment relapse remained a concern (33). The use of alternate CAR T-cell therapies (e.g., with CD22-specific or CD19/CD22-bispecific CAR T-cells) may be another option for some patients, but these therapies are currently unavailable outside clinical trial enrollment. Dual-antigen targeting, allogeneic platforms, and/or gene-editing strategies are being actively explored and may ultimately offer more targeted, less toxic treatment alternatives. It is hoped that these ‘next-generation’ CAR T-cell therapies will mitigate the relapse risk and obviate the need for AlloHCT. However, with the tools currently at our disposal, AlloHCT should be strongly considered to extend immune surveillance and a durable remission state for high-risk patients who experience a CR after CD19-CAR T-cell therapy.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (NIH)/National Cancer Institute (NCI) grant P30CA021765, by the American Society of Transplantation and Cellular Therapy (AT), by a St. Baldrick’s Foundation Scholar Award (CLB), by a Johns Hopkins Summer Provost’s Undergraduate Research Award (JWR), by a Johns Hopkins Woodrow Wilson Fellowship (JWR), and by the American Lebanese Syrian Associated Charities (ALSAC). Part of the laboratory studies were performed by the Center for Translational Immunology and Immunotherapy (CeTI2), which is supported by St. Jude. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We would like to thank Keith A. Laycock, PhD, ELS, for scientific editing of the manuscript. We also thank the staff of our clinical research offices and clinical care teams for their assistance in providing excellent patient care.
Competing Interests Statement:
SG and CLB have patents/patent applications in the fields of T-cell therapy and/or gene therapy for cancer. CHP is a DSMB member of Novartis and serves on the scientific advisory board of Adaptive Biotechnology, Inc. CLB has received research funding from Merck, Sharpe, and Dohme, Kiadis Pharma, and Bristol Myers Squibb. SG consults for TESSA Therapeutics, is a DSMB member of Immatics, and serves on the scientific advisory board of Tidal. BMT has received travel support from Miltenyi Biotec.
REFERENCES
- 1.Bhatla T, Jones CL, Meyer JA, Vitanza NA, Raetz EA, Carroll WL. The biology of relapsed acute lymphoblastic leukemia: opportunities for therapeutic interventions. J Pediatr Hematol Oncol 2014;36(6):413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017;129(25):3322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 2019;134(26):2361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385(9967):517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah NN, Lee DW, Yates B, Yuan CM, Shalabi H, Martin S, et al. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J Clin Oncol 2021:JCO2002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Administration FaD. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome 2017. [Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome.
- 8.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasquini MC, Hu ZH, Curran K, Laetsch T, Locke F, Rouce R, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv 2020;4(21):5414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, et al. Risk-Adapted Preemptive Tocilizumab to Prevent Severe Cytokine Release Syndrome After CTL019 for Pediatric B-Cell Acute Lymphoblastic Leukemia: A Prospective Clinical Trial. J Clin Oncol 2021:JCO2002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers RM, Taraseviciute A, Steinberg SM, Lamble AJ, Sheppard J, Yates B, et al. Blinatumomab Nonresponse and High-Disease Burden Are Associated With Inferior Outcomes After CD19-CAR for B-ALL. J Clin Oncol 2021:Jco2101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol 2019;16(4):227–40. [DOI] [PubMed] [Google Scholar]
- 13.Pui CH. Precision medicine in acute lymphoblastic leukemia. Front Med 2020;14(6):689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014;371(11):1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant 2019;25(4):625–38. [DOI] [PubMed] [Google Scholar]
- 16.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 2016;3:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii K, Pouzolles M, Chien CD, Erwin-Cohen RA, Kohler ME, Qin H, et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest 2020;130(10):5425–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, et al. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J Haematol 2021;194(4):701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest 2019;129(5):2123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dourthe ME, Rabian F, Yakouben K, Chevillon F, Cabannes-Hamy A, Mechinaud F, et al. Determinants of CD19-positive vs CD19-negative relapse after tisagenlecleucel for B-cell acute lymphoblastic leukemia. Leukemia 2021. [DOI] [PubMed] [Google Scholar]
- 21.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 2015;5(12):1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer J, Paret C, ElMalki K, Alt F, Wingerter A, Neu MA, et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J Immunother 2017;40(5):187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun 2016;7:12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CART-cell therapy. Blood 2016;127(20):2406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mo G, Wang HW, Talleur AC, Shahani SA, Yates B, Shalabi H, et al. Diagnostic approach to the evaluation of myeloid malignancies following CAR T-cell therapy in B-cell acute lymphoblastic leukemia. J Immunother Cancer 2020;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 2018;24(10):1504–6. [DOI] [PubMed] [Google Scholar]
- 27.Asnani M, Hayer KE, Naqvi AS, Zheng S, Yang SY, Oldridge D, et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia 2020;34(4):1202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabilloud T, Potier D, Pankaew S, Nozais M, Loosveld M, Payet-Bornet D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat Commun 2021;12(1):865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dholaria B, Savani BN, Huang XJ, Nagler A, Perales MA, Mohty M. The evolving role of allogeneic haematopoietic cell transplantation in the era of chimaeric antigen receptor T-cell therapy. Br J Haematol 2021. [DOI] [PubMed] [Google Scholar]
- 30.Bouziana S, Bouzianas D. Exploring the Dilemma of Allogeneic Hematopoietic Cell Transplantation after Chimeric Antigen Receptor T Cell Therapy: To Transplant or Not? Biol Blood Marrow Transplant 2020;26(8):e183–e91. [DOI] [PubMed] [Google Scholar]
- 31.Fabrizio VA, Kernan NA, Boulad F, Cancio M, Allen J, Higman M, et al. Low toxicity and favorable overall survival in relapsed/refractory B-ALL following CAR T cells and CD34-selected T-cell depleted allogeneic hematopoietic cell transplant. Bone Marrow Transplant 2020;55(11):2160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauthier J, Bezerra ED, Hirayama AV, Fiorenza S, Sheih A, Chou CK, et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell malignancies. Blood 2021;137(3):323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers RM, Li YM, Leahy AB, Barrett DM, Teachey DT, Callahan C, et al. Humanized CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CAR-Naive and CAR-Exposed Children and Young Adults With Relapsed or Refractory Acute Lymphoblastic Leukemia. J Clin Oncol 2021;39(27):3044–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.