

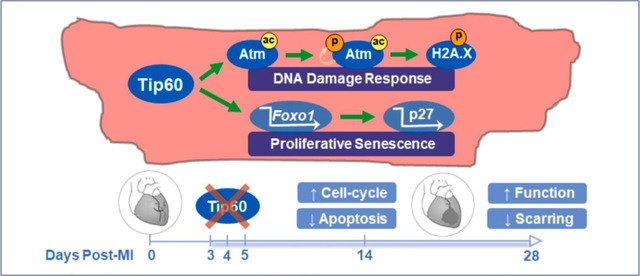
Abstract
Injury from myocardial infarction (MI) and consequent post-MI remodeling is accompanied by massive loss of cardiomyocytes (CM), a cell type critical for contractile function that is for all practical purposes non-regenerable due to its profound state of proliferative senescence. Identification of factors that limit CM survival and/or constrain CM renewal provides potential therapeutic targets. Tip60, a pan-acetyltransferase encoded by the Kat5 gene, has been reported to activate apoptosis as well as multiple anti-proliferative pathways in non-cardiac cells; however, its role in CMs, wherein it is abundantly expressed, remains unknown. Here, using mice containing floxed Kat5 alleles and a tamoxifen-activated Myh6-MerCreMer recombinase transgene, we report that conditional depletion of Tip60 in CMs three days after MI induced by permanent coronary artery ligation greatly improves functional recovery for up to 28 days. This is accompanied by diminished scarring, activation of cell-cycle transit markers in CMs within the infarct border and remote zones, reduced expression of cell-cycle inhibitors pAtm and p27, and reduced apoptosis in the remote regions. These findings implicate Tip60 as a novel, multifactorial target for limiting the damaging effects of ischemic heart disease.
Keywords: Apoptosis, Cardioprotection, Cardiomyocyte Proliferation, Cell-Cycle, Myocardial Infarction, Regeneration, Tip60
Graphical Abstract
1. Introduction
Contractile dysfunction and mortality associated with myocardial infarction (MI) are caused by the loss of cardiomyocytes (CMs), a cell-type that is essentially non-regenerable. As recently reviewed [1], the most promising therapeutic interventions for re-muscularizing the myocardium are via (i) transplantation of CMs derived from pluripotent stem cells, (ii) induction of new CMs by transcriptionally re-programming non-CMs, and, (iii) protection and expansion of pre-existing CMs. Strategies addressing the third approach have met with variable success during the past decade [2]. Thus, the goal of preventing CM loss and restoring lost CMs to a level sufficient to maintain normal cardiac function remains extremely challenging. In comparison with the ability of skeletal muscle to regenerate [3], heart regeneration is limited by massive CM loss during ischemia combined with the inability of remaining CMs to regenerate due to inhibitory factors that become established during the onset of CM proliferative senescence at neonatal stages of heart development [4]. It is therefore reasonable to consider that CM regeneration can be improved by inhibiting upstream factor(s) that activate apoptosis and anti-proliferative factors in the CM.
Proteins that activate apoptosis and inhibit the cell-cycle are reversibly regulated by enzymes that induce post-translational modifications (PTMs), most prominently phosphorylation, and the acetylation of ε-amino groups in conserved lysine (K) residues. As recently reviewed, lysine deacetylases (KDACs) have emerged as therapeutic targets for the management of cardiovascular disease, while in comparison the therapeutic targeting of lysine acetyltransferases (KATs) is receiving relatively little attention [5]. Within the context of CM protection and regeneration, the potential role of Kat5, also known as Tip60 (Tat-interactive protein U60U kD), should be of interest based on multiple reports from the cancer biology field that Tip60 acetylates non-histone proteins to induce cellular functions including apoptosis, and the DNA damage response (DDR) which culminates in cell-cycle inhibition. For example, it is well-documented that Tip60 acetylation activates p53 [6, 7] and Atm (Ataxia–telangiectasia mutated [8–10]), which respectively activate apoptosis and DDR signaling. These findings suggest an important role for Tip60 in the heart, wherein CM apoptosis is a debilitating consequence of MI [11], and activation of Atm inaugurates the onset of CM proliferative senescence during neonatal stages of heart development [12]. Moreover, evidence is accumulating that Tip60 regulates cell proliferation in other cell types by acetylating downstream targets including p21 [13], thereby inhibiting cell-cycle transit, and inhibiting Aurora kinase B [14], preventing cytokinesis.
These findings suggest that Tip60 may promote apoptosis and inhibit the CM cell-cycle in the adult heart, wherein it is robustly expressed [15, 16]. Here, we address this question in mice containing loxP-flanked Kat5 alleles by assessing whether tamoxifen-induced depletion of Tip60 after myocardial infarction releases CMs from proliferative senescence and protects CMs from apoptosis, resulting in cardio protection and regeneration. We previously reported a Tip60 isoprotein shift [16] that changes during heart development, from Tip60α (60 kD) enrichment during embryonic stages, to Tip60β (53 kD) enrichment at adult stages, created by alternative splicing of mRNA transcribed from the Kat5 gene (there is only one Kat5 gene in mice and humans). These data indicate that Tip60β is the only isoprotein present in the adult myocardium. We report that Tip60 depletion beginning 3 days post-MI remarkably improves cardiac function by 10 days post-MI, which is sustained until at least 28 days post-MI. This is accompanied by decreased apoptosis and scarring, and by increased numbers of cycling CMs at the 14-day as well as the 28-day post-MI timepoint, when numbers of cycling CMs are still increasing. Among potential cell-cycle regulators that are de-inhibited by Tip60 depletion, we show that percentages of pAtm-positive CMs are reduced, as well as levels of the cell-cycle inhibitor p27. These findings nominate Tip60 for inclusion among KATs that are under consideration for cardiovascular therapeutic targeting [5].
2. Materials and Methods
2.1. Animal Care & Experimentation:
This investigation adhered to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Pub. Nos. 85-23, Revised 1996). All protocols described in the authors’ Animal Use Application (AUA #000225), which were approved by the Medical College of Wisconsin’s Institutional Animal Care and Use Committee (IACUC), were adhered to in this study. The IACUC has Animal Welfare Assurance status from the Office of Laboratory Animal Welfare (A3102-01).
Preparation of mice containing floxed Kat5 alleles, wherein exons 3–11 comprising two-thirds of the Tip60 coding sequence including the chromo and acetyltransferase domains are removed by Cre-recombinase (hereafter Cre), was previously described [17]. For these experiments, floxed mice were mated with a line (Jackson Laboratory #005650) containing an α-myosin heavy chain (Myh6)-driven MerCreMer-recombinase transgene, the product of which (Cre), upon administration of tamoxifen, enters the nucleus to recombine floxed alleles [18]. All mice were on a mixed B6/sv129 genetic background. Experimental groups contained equal numbers of adult (10–14 weeks-old) males and females.
For the MI experiments, beginning on the third day after inducing MI by left main coronary artery ligation, mice received daily intraperitoneal injections of tamoxifen (40 mg/kg [Sigma #T5648] suspended with 5% ethanol in sunflower oil), for three consecutive days. Echocardiography was performed under general anesthesia with inhaled isoflurane on a subset of mice at intervals following MI. On the day before harvest (14 or 28 days post-MI), mice were injected (1 mg) intraperitoneally with BrdU. On the day of harvest, mice were euthanized with CO2 and hearts were prepared for immunostaining (tissue from the apex to ~1 mm below the suture) and qPCR (tissue from the base of the heart to ~1 mm above the suture), as described below.
For qPCR and western blotting determinations, non-infarcted adult Kat5 floxed mice were given the three-day regimen of tamoxifen (40 mg/kg/day × 3 days) and, 3–11 days after the 1st dose of tamoxifen, hearts were harvested and apportioned for RNA and protein isolation by respectively placing samples in TRIzol (Thermo-Fisher #15596026) and RIPA buffer (Thermo-Fisher #89901) containing Halt’s anti-protease/anti-phosphatase cocktail (Thermo-Fisher #78440). Samples were minced and homogenized with a teflon pestle and stored at −80° C until further processing.
2.2. Myocardial Infarction and Echocardiography:
To induce MI, mice were intubated under anesthesia with etomidate (10 mg/kg by intraperitoneal injection), mechanically respirated (model 845, Harvard Apparatus) via an endotracheal tube with room air supplemented with 100% oxygen, and delivered isoflurane (1.5–2.0%) via the ventilator to maintain anesthesia. The electrocardiogram (ECG; limb lead II configuration) was continuously recorded (Powerlab) using needle electrodes and rectal temperature was maintained at 37°C throughout the experiments using a servo-controlled heating pad. Once the mice were anesthetized and prepared for surgery, thoracotomy was performed to the left of the sternum to expose the heart, followed by opening of the pericardium and placement of an 8.0 nylon suture beneath the left main coronary artery at a level below the tip of the left atrium to target the lower half of the ventricle, with the aid of a microscope. Ischemia was induced by carefully tying the suture with a double knot, after which coronary occlusion was verified by visual observation of blanching of the myocardium distal to the ligature and by ST segment elevation on the ECG. After ligation, the chest wall was closed with polypropylene suture and recovery was monitored until mice became fully ambulatory. Immediately prior to initiating the surgical procedure to produce MI, mice were injected subcutaneously with sustained release meloxicam (4 mg/kg) to limit post-operative pain.
At scheduled intervals, echocardiographic assessment (VisualSonics 3100 high-frequency ultrasound imaging systems) was performed on mice lightly anesthetized with isoflurane delivered via a nose cone (1.0–1.5%) in the parasternal long-axis, short-axis, and apical 4-chamber views using a transducer (MX550D) operating at 30–40 mHz. Short-axis views in M-mode were used to measure left ventricular anteroposterior internal diameter (LVID), anterior wall thickness (LVAW), and posterior wall thickness (LVPW) at end-diastole (d) and end-systole (s) at the mid-ventricular level. Long-axis views in B-mode were used to measure left ventricular internal area (LVA) and length (L) at end-diastole and end-systole. Left ventricular systolic function was assessed by: fractional shortening (FS %) = ([LVIDd – LVIDs] / LVIDd) * 100, fractional area change (FAC %) = ([LVAd – LVAs] / LVA d) * 100, and ejection fraction (EF %) = (end-diastolic volume – end-systolic volume) / end-diastolic volume whereby volumes were estimated by: 4π/3 * L/2 * (LVA * π(L/2)). In addition, global left ventricular function was assessed by calculating the myocardial performance index: MPI = (isovolumic contraction time + isovolumic relaxation time) / ejection time [19–22]. Time intervals were obtained from pulsed Doppler waveforms of mitral valve inflow and aortic valve outflow from apical 4-chamber views.
2.3. Quantitative Assessment of Myocardial Scarring:
Paraffinized hearts were transversely sectioned from the apex to ~1 mm above the suture site, after which eight 4 μm thick sections from equidistant (~0.34–0.80 mm) intervals were placed on microscope slides and stained with Masson trichrome to quantitatively assess scar size [23]. Trichrome-stained sections were photographed at 10x magnification on a Nikon SMZ800 microscope, and MIQuant software was used to quantitate infarct size in sections between the apex and the ligation site as previously described [23]. Results were expressed as the average percentage of area and the midline length around the left ventricle.
2.4. Genotyping:
Genotyping was performed by PCR in 20 μl reactions that included 2x GoTaq Green Master Mix (Promega #M7123), 1.1 mM MgCl2, 0.5 μM each primer, 0.5 μM internal control primers, and 4.0 μl template. Templates consisted of supernatants of ear tissue samples that had been boiled for 10 minutes in 0.3 mL 10 mM NaOH/1 mM EDTA. Sequences of primer pairs used for PCR are listed in Supplemental Table 1 (https://figshare.com/s/c9a0c4fa1f7fae925c95). PCR products were amplified in an AB Applied Biosystems GeneAmp PCR System 9700 using the following programs: for LoxP, one 5-minute cycle at 95° C, thirty-five cycles at 94° C 30 sec/61° C 45 sec/72° C 45 sec, followed by one 10-minute cycle at 72° C; for Myh6-MerCreMer, one 5-minute cycle at 95° C, thirty-five cycles at 94° C 30 sec/54° C 45 sec/72° C 45 sec, followed by one 10-minute cycle at 72° C. Amplicons were separated at 100–110 V for one hour in 1% agarose with ethidium bromide and imaged.
2.5. Quantitative RT-PCR (qPCR):
Heart tissue, previously disrupted by homogenization with a motorized (Kimble 749540–0000) Teflon pestle and stored at −80° C in TRIzol reagent, was thawed. RNA was immediately purified using PureLink RNA Mini-Kits (ThermoFisher #12183018A), including a genomic DNA removal step (PureLink DNase kit for on-column protocol, Thermo-Fisher #12185-010), according to the manufacturer’s instructions. RNA yield & quality were determined via 260/280 ratio using Eppendorf Biophotometer Plus Instrument.
cDNA was synthesized as follows. After diluting an RNA sample from each heart so that precisely 1.0 μg was suspended in 14 μl nuclease-free distilled water (NFDW), 4.0 μl 5x VILO reaction mixture (ThermoFisher #11754050) were added. To start the reverse-transcription reaction, 2.0 μl 10x SuperScript Enzyme Mix (ThermoFisher #11754050) were added, followed by transfer to an Applied Biosystems Veriti 96-well Thermocycler programmed as follows: 10 minutes at 25° C → 60 minutes at 42° C → 5 minutes at 85° C. cDNA templates were diluted with NFDW to a concentration of 3.125 ng/μl and stored at −20° C.
qPCR was carried-out by subjecting each biological replicate (i.e., sample from each individual heart) to triplicate determinations. Each reaction was performed in a total volume of 20 μl in 96-well arrays, each well containing 1x Taqman Fast-Advanced Master Mix (ThermoFisher #4444557), 1x Taqman Probe Kit (Supp. Table 1), and 12.5 ng cDNA as template. The arrayed samples were amplified in a Bio-Rad CFX96 Real Time System (C1000 Touch) programmed as follows: 2 minutes at 50° C → 20 seconds at 95° C → 3 seconds at 95° C → 30 seconds at 60° C; the last two steps were repeated 39 times. Results were processed using Bio-Rad CFX Manager 3.1 software.
2.6. Western Blotting:
Upon harvesting in ice-cold RIPA Lysis and Extraction buffer (ThermoFisher #89900) fortified with Halt Protease and Inhibitor Cocktail (ThermoFisher #78440), samples were finely minced, homogenized, and stored at −80° C. Prior to electrophoresis, tissues were thawed at 0° C and homogenized with a motorized Teflon pestle, followed by determination of total protein concentration using a Standard Bradford Assay (Bio-Rad #500–0006) and dilution in Laemmli Sample Buffer (Bio-Rad #161–0747) to a concentration of 2.5 mg/mL. For electrophoresis, 20 μg of each sample were loaded into each lane of a precast Bio-Rad 4–20% acrylamide gel, and separated proteins were transferred (60 min at 100 V) onto 0.45μm nitrocellulose membrane (Bio-Rad #162–0145). The blots were blocked with 5% non-fat dry milk/10 mM Tris-HCl (pH 7.6)/150 mM NaCl/Tween-20 (5% NFDM/TBST) or 5% BSA in TBST. Primary and secondary antibodies and dilutions are listed in Supplemental Table 2. Blots were reacted with primary antibody in 5% NFDM/TBST or 5% BSA blocking buffer overnight at 4°C. Secondary antibodies were diluted in 5% NFDM/TBST and applied for 60 minutes at RT. Reacted blots were covered with horseradish peroxidase substrate (ThermoFisher #34580) for 5 min at room temperature, followed by chemiluminescence imaging and densitometry using Bio-Rad ChemiDoc and ImageJ software, respectively.
2.7. Immunostaining & Cell Counting:
On the day before harvest, mice were injected with 1 mg 5’-bromo-2’-deoxyuridine (BrdU; Sigma #B9285). Following removal, hearts were perfused with ice-cold cardioplegic solution and atria were removed. Ventricles were fixed overnight in fresh ice-cold 4% paraformaldehyde/PBS, processed through ethanol series, and embedded in paraffin. Sections (4 μm thick) mounted on microscope slides were de-waxed, subjected to antigen retrieval (100°C in 10 mM trisodium citrate pH6.0/0.05% Tween-20 for 20 minutes) followed by 30 minutes’ cooling at RT, and blocked with 2% goat serum/0.1% Triton-X-100 in PBS. Primary antibodies were diluted in blocking buffer and applied overnight at 4°C; secondary antibodies were applied for one hour in the dark. Combinations and dilutions of primary and secondary antibodies employed to immunostain each target antigen is provided in Supplementary Table 2.
Microscopy was performed on a Nikon Eclipse 50i microscope equipped with a Nikon DSU3 digital camera. During counting, at least 1,000 CMs were evaluated in 5–6 random 200x photomicrographic fields, identity of which were confirmed by immunostaining the cytoplasmic marker cardiac troponin-T (cTnT), α-actinin, or the nuclear marker pericentriolar material 1 (PCM-1). Percentages of CMs expressing Ki67, BrdU, phosphorylated histone H3 (pH3), and phosphorylated H2A.X were assessed by monitoring fluorescent signal in the Texas Red channel, after confirming the identity of each stained CM in the FITC channel. To identify nuclei as phosphorylated ATM (pATM)-positive, only those at least half-filled with pATM fluorescence were counted; pAtm-positive nuclei were also confirmed to be DAPI-positive. To enumerate CMs exhibiting phosphorylated histone H2A.X signal, α-actinin was used to identify CMs, and, the total number of double-positive cells in each section was manually counted; pH2A.X signals were counted only if confined to DAPI-positive nuclei. To quantitate CMs undergoing cytokinesis as indicated by the presence of Aurora kinase B (AurkB)-positive furrows between adjacent CMs, the entirety of each of three sections from equidistant (0.3 mm) intervals from each heart was manually scanned at 1,000x magnification by a blinded investigator; the identification of AurkB-positive CMs was restricted to images wherein punctate Texas Red signal was clearly located between two adjacent nuclei that were embedded within CM (i.e. cTnT-positive) cytoplasm. For cleaved caspase-3 determinations, WGA was employed as described below to identify CMs based on cellular outlines, and, the total number of cleaved caspase-3 positive CMs present in each section was manually enumerated at 400x magnification. In infarcted hearts, CM cell-cycle marker and pATM, immunoreactivity were assessed in both the remote zone, specifically the area of myocardium ~2 mm distal to the infarct boundary, and in the border zone, specifically the area immediately adjacent to the infarct zone.
2.8. Wheat Germ Agglutinin (WGA) Staining:
WGA staining was performed using Thermo-Fisher #W11261 Alexa Fluor 488 conjugate and Thermo-Fisher #W11263 Alexa Fluor 350 conjugate. Sections mounted on microscope slides were stained with 50 μg/ml WGA in PBS for 10 minutes at room temperature, followed by thorough washing. Images of CMs in transverse orientation were photographed at 400x magnification and processed using ImageJ software to determine numbers of CMs and average numbers of pixels per CM as indicative of CM density and size. Briefly, the FITC (488) or DAPI (350) channel displaying CMs outlined in cross-section was isolated, followed by thresholding to fill-in spaces occupied by CM cytoplasm, then adjusting settings to acquire particle sizes in the 600-infinity range having a circularity of 0.25–1. After results (which were set to “include holes”) were obtained, particles representing CMs that were non-transversely sectioned, or blood vessels, were removed.
2.9. Terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL):
Apoptosis was assessed using the DeadEnd Fluorometric TUNEL System (Promega #G3250) per the manufacturer’s instructions. The total number of TUNEL-positive cells within sections representing the border and remote zones were manually counted while scanning at 400x magnification. TUNEL signal was counted only if confined to a DAPI-positive nucleus. Nuclei were scored as TUNEL-positive only if at least 50% of the nucleus contained fluorescent signal. Attempts to immunofluorescently co-stain TUNEL-stained sections for markers of CM identity were unsuccessful due to removal of antigen during proteinase-K digestion. Apoptosis was also determined by immunostaining cleaved caspase-3 as described above.
2.10. Statistics:
All determinations were performed in blinded fashion and are reported as means ± SEM. Echocardiography data were analyzed by a two-way repeated measures ANOVA (time and genotype) to determine whether there was a main effect of time, genotype, or a time-genotype interaction. If global tests showed an effect, post hoc contrasts between baseline and subsequent timepoints within experimental groups were compared by a Dunnett’s multiple comparison t test; differences between genotypes at each timepoint were compared by a Student’s t test with the Bonferroni correction for multiple comparisons. Survival data were analyzed by Kaplan-Maier analysis. All other data were compared by an unpaired, two-tailed Student’s t test. P < 0.05 was considered statistically significant.
3. Results
3.1. Experimental Scheme
Our objective was to assess whether effects of MI could be minimized by subsequent depletion of Tip60. Experimentally, we assessed the effect of administering tamoxifen on days 3–5 post-MI to control mice containing floxed Kat5 alleles (Kat5f/f), in comparison with mice containing the Myh6-MerCreMer-recombinase Cre transgene (Kat5flox/flox;Myh6-MerCreMer, hereafter denoted Kat5Δ/Δ) [18]. To control for effects, if any, of Cre per se, mice bearing the genotypes Kat5+/+ and Kat5+/+;MerCreMer were compared. The experimental timeline consisted of a 28-day post-MI follow-up period to permit regeneration and healing to manifest.
Prior to performing MI experiments, the effect of administering three consecutive daily doses of tamoxifen (40 mg/kg) to non-infarcted adult mice was assessed to determine the extent of conditional Tip60 depletion, and whether depletion compromised cardiac function. As shown in Figure 1, levels of Kat5 transcripts (Fig. 1A) and Tip60 protein (Fig. 1B) were depleted ≥50% in hearts of Kat5Δ/Δ mice as early as 3 days after the first dose of tamoxifen. It is likely that depletion in CMs was even more extensive, considering that Tip60 should not be depleted in non-CMs comprising the majority of cells in whole heart samples [24]. Echocardiographic data in Supplemental Table 3 shows that Tip60 depletion had no untoward effects on non-injured mice at the 28-day experimental endpoint; also, Supplemental Figure 1 shows that Tip60 depletion did not affect survival until approximately 20 weeks post-tamoxifen.
Figure 1. Tamoxifen induces depletion of Tip60 in hearts with floxed Kat5 alleles.

Adult Kat5f/f and Kat5Δ/Δ mice were injected with 40 mg/kg tamoxifen on three consecutive days, after which hearts were collected and processed for assessment of Tip60 mRNA and protein. Panel A shows depletion of Kat5 mRNA in Kat5Δ/Δ hearts assessed by qRT-PCR 3–8 days after the first of three daily tamoxifen injections. Each biological replicate (n=4 mice for each timepoint per group) was subjected to three technical replicates. Panel B (upper) is a representative western blot showing depleted levels of Tip60 protein (55 kD) in Kat5Δ/Δ hearts collected 8–9 days after the first tamoxifen injection; the bar graph (lower) shows the extent of Tip60 depletion as assessed by quantitative densitometry. Each biological replicate (n=7 mice per group) was subjected to two technical replicates. Data are reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
3.2. Tip60 Depletion reduces percentages of pAtm-positive CMs, and levels of p27.
Based on previous findings indicating that phosphorylated Atm (pAtm) inhibits the cell-cycle in adult CMs [25], plus our recently reported finding that Tip60 maintains pAtm levels in the neonatal heart [26], we asked whether depletion of Tip60 reduces percentages of pAtm-positive CMs in the non-injured adult heart. As shown in Figure 2A, immunostaining revealed that percentages of pAtm CMs in the adult heart were reduced by the eleventh day after the first of three tamoxifen injections to deplete Tip60. This finding is consistent with significantly reduced levels of pAtm protein in Tip60-depleted hearts detected by western blotting (Supp. Fig. 2A), which was accompanied by the curious finding of increased levels of bulk Atm protein in Tip60-depleted hearts (Supp. Fig. 2B).
Figure 2. Reduced expression of pAtm and p27 following tamoxifen-induced depletion of Tip60 in non-injured hearts with floxed Kat5 alleles.

Non-injured adult Kat5f/f and Kat5Δ/Δ mice were injected with 40 mg/kg tamoxifen on three consecutive days, after which hearts were collected eleven days after the first injection and processed for immunostaining, western blotting, and qRT-PCR as indicated. Panel A shows immunostaining revealing reduced percentages of pAtm-positive CMs in Tip60-depleted (Kat5Δ/Δ) hearts. At least 1,000 CMs were evaluated in each biological replicate (n=4 mice per group). Panel B (lower) depicts qRT-PCR analyses showing reduced expression of genes encoding Foxo family members in Kat5Δ/Δ hearts, among which Foxo1 transcriptionally activates the gene (Cdkn1b) encoding p27. The upper bar graph depicts qRT-PCR showing reduced expression of p27 in Tip60-depleted adult hearts. Each biological replicate (n=4 mice per group) was subjected to three technical replicates. Panel C is a representative western blot showing reduced levels of p27 protein in Tip60-depleted adult hearts. Each biological replicate (n=4 mice per group) was subjected to two technical replicates. Data are reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
In addition, to follow-up our finding that activation of the cell-cycle in Tip60-depleted neonatal CMs is accompanied by reduced expression of the cell-cycle inhibitor p27 [26], we asked whether depletion of Tip60 in the non-injured adult heart has similar effects. Because Tip60 reportedly increases the expression of genes in the family of Foxo transcription factors [27], among which Foxo1 is well-documented to activate the gene encoding p27 (Cdkn1b; review [28]), we assessed the effects of Tip60 depletion on expression of Foxo1, Foxo3, and Cdkn1b (p27). As shown in Figure 2B, expression of these genes was significantly depressed in Tip60-depleted adult hearts, and was accordingly accompanied by significantly reduced levels of p27 protein (Fig. 2C); considering Tip60’s pro-apoptotic function it is interesting that reduced Foxo1 levels have also been associated with reduced levels of apoptosis in CMs [29, 30]. The expression of other cell-cycle inhibitors, including Meis1 (which is reduced in Tip60-depleted neonatal CMs [26]) and p21 (Supp. Fig. 3A, right), as well as genes associated with the DDR and the senescence-associated secretory phenotype (SASP, [31]; Supp. Fig. 3B), which are depressed in the neonatal Kat5Δ/Δ heart [26], was unaffected by Tip60 depletion. However, the expression of Wee1, which becomes activated in the neonatal heart as CMs undergo proliferative senescence [12], was significantly depressed (P=0.019; Supp. Fig. 3B). Control experiments revealed that none of these effects associated with Tip60 depletion were attributed to Cre alone (Supp. Fig. 4). Based on the above findings that expression of pAtm, p27, and Wee1 are depressed in the non-injured/Tip60-depleted adult heart, it was of interest to assess whether, as we recently demonstrated in the neonatal heart [26] and in accord with our hypothesis, depletion of Tip60 in the infarcted adult heart is associated with activation of the CM cell-cycle, concomitant with reduction of apoptosis and preservation of cardiac function.
3.3. Tip60 depletion preserves cardiac function and reduces myocardial scarring after MI.
The impact of Tip60 depletion on the effects of MI was therefore addressed. The method of MI employed in this study was designed to generate uniform infarctions by permanently ligating the left main coronary artery at a position below the tip of the left atrium to target the distal half of the ventricle. As indicated by the experimental timeline shown in Figure 3A, echocardiography was performed on infarcted Kat5f/f and Kat5Δ/Δ mice at intervals up to 28 days post-MI, during which time tamoxifen was injected to deplete Tip60 on days 3–5 post-MI. Remarkably, one week after the first tamoxifen injection -- at day 10 post-MI -- all indices of left ventricular function assessed by echocardiography were substantially improved in Kat5Δ/Δ mice, in comparison with Kat5f/f controls (Fig. 3B–D; Supp. Table 4). These indices included fractional shortening (FS; Fig. 3B), fractional area change (FAC; Fig. 3C), ejection fraction (EF; Fig 3D), and the myocardial performance index (MPI; Supp. Table 4; [19]). All measures between the 10- and 28-day post-MI time-points were maintained near the pre-MI baseline (day 0) values (Fig 3B–D). A summary of all echocardiographic data is provided in Supplemental Table 4. Preservation of function in Tip60-depleted hearts was consistent with survival data (Fig. 3E), which indicated a clear trend toward improved post-MI survival of Kat5Δ/Δ mice. Functional improvement with Tip60 depletion was evident in both male and female mice (Supp. Fig. 5). Control experiments in which echocardiography was performed on infarcted Kat5+/+ and Kat5+/+;MerCreMer mice that were identically treated with tamoxifen revealed that none of the functional benefits shown in Figure 3 could be attributed to Cre alone; by contrast, activation of Cre in the absence of Tip60 depletion caused dysfunction at all points along the experimental timeline (Supp. Fig. 6 and also [25]).
Figure 3. Tip60 depletion preserves function and reduces scarring in infarcted mouse hearts.

A, experimental timeline. Mice were injected with tamoxifen on three consecutive days beginning three days after induction of MI on day 0. Echocardiography was performed at the indicated intervals up to 28 days post-MI when hearts were removed for histological assessment. B-D, indices of LV function. Fractional shortening (FS), fractional area change (FAC), and ejection fraction (EF) were preserved in Tip60-depleted (Kat5Δ/Δ) hearts (N=8) but not in Kat5f/f controls (N=5); additional parameters are shown in Supplemental Table 4. *P<0.05 vs. Kat5f/f and †P<0.05 vs baseline value on day 0. Echocardiography data were analyzed by two-way repeated measures ANOVA followed by Dunnett’s (effect of time) and Bonferroni’s (effect of genotype) multiple comparisons. E, Kaplan-Maier curves indicating a trend (P=0.072) toward improved survival of Tip60-depleted mice. F, representative trichrome-stained cross-sections taken at 0.5 mm intervals; blue stain shows area of the scar. Infarct scar size (right) was quantified by measuring area and midline length below the ligation site. N=10 for Kat5f/f and N=12 for Kat5Δ/Δ. Data are reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
In agreement with improved function observed by echocardiography, Tip60 depletion protected the myocardium from scarring at the 28-day post-MI timepoint (Fig. 3F) assessed by Masson trichrome staining (Fig. 3F, left). The percentage of left ventricle occupied by scarring, and the scar midline length, were quantitatively assessed by digitizing areas occupied by blue staining between the apex and the site of ligation (Fig. 3F, right). This revealed that scarring, as evaluated by both parameters, was significantly diminished by 25–30% in the myocardium of Tip60-depleted Kat5Δ/Δ mice.
3.4. Tip60 depletion after MI is accompanied by increased activation of the CM cell-cycle.
Observations shown in Figure 3 describing preserved cardiac function and muscle mass in infarcted/Tip60-depleted hearts could be explained by CM proliferation, protection from ischemia-induced cell death, hypertrophic growth of the myocardium, or by a combination of these factors. The possibility that Tip60 depletion caused activation of the CM cell-cycle in the post-MI adult heart, as we previously observed in the neonatal heart [26], was examined by evaluating percentages of Ki67-, BrdU-, and pH3-positive CMs. As shown in Figure 4, immunostaining revealed that each marker, which respectively monitors all cell-cycle phases, S-phase, and early M-phase, was significantly increased in Tip60-depleted CMs located within the border zone as well as the remote zone at both the 14-day and 28-day post-MI timepoints; cycling CMs were not detected in the infarct zone (not shown). Remarkably, in most instances the percentage of CMs exhibiting cell-cycle activation was further increased at 28 days post-MI, in comparison with the 14-day post-MI timepoint. CM identity in these determinations was assessed by cytoplasmic cTnT staining, employing rigorous criteria for inclusion (see Materials and Methods); comparable results were obtained when cardiac identity was verified using the CM-specific nuclear marker PCM-1 in 28-day post-MI hearts (Supp. Fig. 7). Control experiments in which immunostaining was performed on hearts of identically treated Kat5+/+ and Kat5+/+;MerCreMer mice at 14 days post-MI (Supp. Fig. 8), as well as at 28 days post-MI (Supp. Fig. 9), revealed that activation of the CM cell-cycle shown in Figure 4 was not due to Cre alone. To correlate cell-cycle activation detected by immunostaining with activity of cell-cycle phase-specific regulatory genes, qRT-PCR was performed on cardiac tissue dissected from the remote zone at 14 days post-MI. While expression of the G1-phase regulatory genes cyclins-D1/D2 and Cdk4 was unaffected, genes encoding the G2-phase regulators cyclins-A2/B1 and Cdk1 were significantly activated in Tip60-depleted hearts (Supp. Fig. 10A), an effect that was not attributable to an effect of Cre alone (Supp. Fig. 10B). qRT-PCR of 14-day post-MI samples also revealed that, similar to non-injured hearts that were Tip60-depleted (Fig. 2 & Supp. Fig.3), expression of p27 (Supp. Fig. 10A) and Wee1 (Supp. Fig. 11A) were also decreased in infarcted/Tip60-depleted hearts.
Figure 4. Increased CM cell-cycle activation in the border and remote zones of infarcted Tip60-depleted hearts at 14 and 28 days post-MI.

Left column, representative images photographed at 600x showing immunostaining of Ki67-, BrdU-, and pH3-positive nuclei, and co-staining with cTnT to assess CM identity. White/yellow arrows respectively denote positive CMs/non-CMs. Middle column, results of cell counting showing significantly increased percentages of Ki67-, BrdU-, and pH3-positive CMs on day 14 post-MI. The right column shows that each cell-cycle activation marker was further increased on day 28 post-MI. All counting was conducted in blind. At least 1,000 CMs were evaluated in each biological replicate (14 d post-MI: n=6 mice for Kat5f/f and n=5 mice for Kat5Δ/Δ; 28d post-MI: n=5 mice for Kat5f/f and n=6 mice for Kat5Δ/Δ). Data were reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
Because Tip60 was exclusively depleted in CMs, it was surprising that unknown cell types (ie non-CMs) devoid of cTnT or PCM-1 staining also displayed increased cell-cycle activation in Kat5Δ/Δ hearts at 28 days post-MI (non-CMs; Supp. Fig. 12). This unanticipated finding suggests involvement of a paracrine interaction, and/or perhaps compensated changes resultant from MI.
The findings shown in Figure 4 and supportive supplemental figures suggest that preserved cardiac function and muscle mass shown in Figure 3 may reflect, at least in part, de novo generation of CMs mediated by depletion of Tip60. To rule-out the possibility that CM hypertrophy contributed to preserved cardiac function in Tip60-depleted hearts, areas of myocardium containing CMs oriented in cross-section were stained with wheat germ agglutinin (WGA), to estimate CM size by quantitating pixels within WGA-stained CM perimeters (Fig. 5A). As shown in Figure 5B, this revealed that CM size was not increased in infarcted/Tip60-depleted hearts at the 14-day or 28-day post-MI timepoints, instead indicating that CMs within the border zone are smaller at the 14-day and perhaps the 28-day timepoints.
Figure 5. Estimation of CM size in infarcted/Tip60-depleted hearts at days 14 and 28 post-MI.

Panel A, representative 600x WGA-stained images from areas containing transversely oriented CMs. Panel B, pixels within WGA-stained CM perimeters in cross-sectional areas of myocardium were quantitated by ImageJ to estimate CM size in response to Tip60 depletion. At least 500 CMs were evaluated from each zone in each biological replicate (N=5 mice for each timepoint per group). Data were reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
Increased cell-cycle activation in the absence of CM hypertrophy suggested that depletion of Tip60 fosters proliferation-mediated CM regeneration. Because the initiation of CM proliferation requires de-differentiation [2, 32], we evaluated gap junction integrity, which becomes dis-organized in de-differentiating CMs by immunostaining Connexin-43. This revealed that Tip60 depletion was associated with gap junction dysmorphology in all hearts examined (Supp. Fig. 13A), a phenomenon accompanied by a ~2-fold increase in expression of the gene encoding Connexin-43 (Gja1; Supp. Fig. 13B), suggestive of a cellular response designed to restore gap junction integrity and/or enhanced cell-cell communication. And, because completion of CM proliferation requires cytokinesis, we immunostained Aurora kinase B (AurkB), expression of which is increased in cytoplasmic furrows of dividing adult CMs [25, 33]. As shown in Supplemental Figure 14, although the incidence of AurkB-positive CMs is low – as anticipated based on previous findings as well as on the stringency we applied when identifying AurkB-positive CMs – their numbers were significantly increased at the 14-day post-MI timepoint (Supp. Fig. 14B); we were unfortunately unable to evaluate AurkB-positive CMs at an earlier post-MI timepoint, when incidence should be higher, due to potential effects of Cre alone.
In summary, when taken together, the findings described in Figures 4–5 and associated supplemental figures are consistent with the possibility that improved function following Tip60 depletion is caused, in part, by increased CM proliferation. Importantly however, additional work is required to definitively assess generation of de novo CMs. Moreover, because Tip60 is known to activate apoptosis [6, 7] in addition to inhibiting the cell-cycle, we hypothesized that its depletion should also be cardioprotective, preventing apoptosis in the infarcted myocardium; this possibility is addressed in the following experiments.
3.5. Tip60 depletion after MI is accompanied by reduced apoptosis.
Because Tip60 is pro-apoptotic [6, 7], it was important to assess whether its depletion reduced apoptosis in infarcted myocardium. Levels of apoptosis at 14 and 28 days post-MI were assessed by TUNEL and cleaved caspase-3 staining. TUNEL staining revealed that numbers of apoptotic cells were equivalent in Kat5f/f and Kat5Δ/Δ hearts at the 14-day post-MI timepoint (Fig. 6A, upper), but were reduced in Tip60-depleted hearts at 28-days (Fig. 6A, lower), where a twofold decline was observed in the remote zone. Although protease treatment prior to TUNEL staining unfortunately prevented counter-staining to assess CM identity, the size of caspase-3-positive cells outlined by WGA in Figure 6B was consistent with their identity as CMs oriented in cross-section. Quantitation of caspase-3-positive CMs indicated that apoptosis was significantly reduced in infarcted/Tip60-depleted hearts at the 14-day timepoint (Fig. 6B, upper), and was consistent with TUNEL staining in that numbers of apoptotic cells in the remote zone were significantly decreased at 28-days (Fig. 6B, lower). Taken together, these findings indicate that apoptosis, which is known to be increased in the remote zone during post-infarction remodeling [11, 34–36], is reduced by Tip60 depletion, consistent with its pro-apoptotic function [6, 37]. These results were not affected by Cre (Supp. Fig. 15); in fact, in the absence of Tip60 depletion, Cre alone caused a highly significant increase in numbers of TUNEL-positive cells in the remote zone at the 28-day timepoint (Supp. Fig. 15B).
Figure 6. Reduced numbers of TUNEL- and caspase-3-positive cells in the infarcted Tip60-depleted hearts at days 14 and 28 post-MI.

Panel A. TUNEL/WGA-stained sections. Upper: representative 600x image. Lower: total number of TUNEL-positive nuclei per section; cell types were not identifiable. 14 d post-MI: N=4 mice per group; 28d post-MI: N=6 mice per group. Panel B. Caspase-3/WGA-stained sections. Upper: representative 600x image. Lower: total numbers of caspase-3-positive CMs per section; CM identity was inferred from cellular size based on WGA outline. N=5 mice for each time point per group. Data were reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
3.6. DDR Markers pAtm and pH2A.X are reduced in Infarcted/Tip60-depleted CMs.
As described above, the expression of DDR components, pAtm (Fig. 2A, Supp. Fig. 2A), p27 (Fig. 2B,C) and Wee1 (Supp. Fig. 3B) was reduced in Tip60-depleted hearts (Kat5Δ/Δ) that were non-infarcted. It was important to assess whether these factors were also reduced in Tip60-depleted hearts that had been infarcted. In accord with reduced expression of the genes encoding p27 (Supp. Fig. 10A) and Wee1 (Supp. Fig. 11A) in infarcted/Tip60-depleted hearts, Figure 7, which shows results of immunostaining performed at the 14-day post-MI timepoint, revealed that numbers of pAtm-positive (Fig. 7A) as well as pH2A.X-positive (Fig. 7B) CMs were also reduced by Tip60 depletion; these results were not affected by Cre (Supp. Fig. 16).
Figure 7. Reduced numbers of pAtm- and pH2A.X-positive CMs in Infarcted/Tip60-depleted hearts at day 14 post-MI.

Panel A, Upper, representative pAtm/α-actinin double-stained image; yellow arrowheads denote Atm-positive nuclei identified according to the ground rules described in Materials and Methods. Lower, percentage of Atm-positive CMs, based on evaluating a minimum of 1,000 CMs in the border and remote zones. N=6 for Kat5f/f and N=5 for Kat5Δ/Δ. Panel B, Upper, representative pH2A.X/α-actinin double-stained image; yellow arrowheads denote pH2A.X-positive CMs. Lower, numbers of pH2A.X-positive CMs within each section of Kat5f/f and Kat5Δ/Δ hearts enumerated by scanning entire sections at 400x magnification. N=5 mice for Kat5f/f and N=4 mice for Kat5Δ/Δ. Data were reported as Mean ± SEM and compared by unpaired, two-tailed Student’s t tests with Welch’s correction.
4. Discussion
The goal of this study was to test the hypothesis that Tip60, which is known to inhibit the cell-cycle in cultured cells, maintains CMs within the in vivo heart in a state of apoptotic potential and proliferative senescence, preventing regeneration of the myocardium after injury. The findings described in Figures 3–7 are consistent with this hypothesis, indicating that genetic depletion of Tip60 after MI diminishes the DDR, inducing CM cell-cycle activation while inhibiting apoptosis, resulting in myocardial integrity and maintenance of cardiac function.
Reliability of the Tip60 Depletion Model
We previously reported that depletion of Tip60 from CMs using a constitutively active Myh6-Cre transgene [38], which commences robust expression at late embryonic stages of development, results in lethality due to CM fallout by three months of age [17]. Therefore, the experimental scheme employed here (Fig. 3A) was designed to cause only modest reduction of Tip60 in CMs by conditionally activating the MerCreMer product of the Myh6-driven MerCreMer recombinase transgene [18]. No untoward effects of conditional Tip60 depletion were observed during the 25-day period following tamoxifen injection into either non-injured (Supp. Table 3) or injured (Supp. Table 4) adult mice. In contradistinction, assessments comparing infarcted/tamoxifen-injected wild-type (Kat5+/+) and Kat5+/+;Myh6-MerCreMer hearts, which we performed to control for off-target effects of Cre, revealed cellular as well as organ dysfunction [39]. Therefore, we conclude that Tip60 depletion mediated the beneficial effects reported here, which occurred despite a background of deleterious effects caused by Cre-recombinase when using this model. It should also be noted that although the deleterious effects of Cre were transient, dissipating within two weeks post-MI [39], this has prevented us from investigating immediate and early effects of Tip60 depletion.
Is Preservation of Cardiac Function by Tip60 Depletion due to Protection and/or Regeneration of CMs?
Periodic echocardiographic evaluation of infarcted mice from which Tip60 was depleted beginning on day 3 post-MI revealed that cardiac function was markedly improved at 10 days, a condition that was sustained until termination of the experiment at 28 days post-MI (Fig. 3B–D; Supp. Table 4). While this remarkable recovery of function may in part reflect the ability to recuperate from a relatively small infarction, few previously reported interventions to improve regeneration after cardiac injury have attained a similar extent of functional preservation. Among these, Cre-mediated disruption of the Hippo component Salvador exhibited a similar level of functional recovery and scar resolution nine weeks after MI. Incredibly, this occurred even though the onset of Salvador depletion was not commenced until 21 days post-MI [23]; it will therefore be interesting to ascertain whether delaying the onset of Tip60 depletion after MI confers sustained functional improvement.
Preservation of function at 28 days post-MI was accompanied by significantly diminished myocardial scarring (Fig. 3F), concomitant with the expression of markers indicating CM cell-cycle re-entry including disrupted gap junction morphology (Supp. Fig. 13), increased cell-cycle activation (Fig. 4), and increased numbers of AurkB-positive CMs (Supp. Fig. 14). Activation of the CM cell-cycle consequent to Tip60 depletion is consistent with our previous observation of cell-cycle activation in CMs of hypertrophied Kat5+/− heterozygous adult hearts [16], and with our recently published findings in the neonatal heart [26]. Although the results shown in Figure 5B appear to rule out an increase in CM hypertrophy induced by cell-cycle activation, a definitive answer to the question of whether cell-cycle activation mediated by Tip60 depletion culminates in the generation of de novo CMs and myocardial re-muscularization will require further investigation. Although techniques involving direct counting and evaluation of CMs isolated from the adult heart would be informative, difficulties dis-aggregating adult CMs from scarred myocardium in this infarction model have precluded this approach.
As shown in Figure 6, preservation of function also correlated with significantly reduced numbers of apoptotic cells in Tip60-depleted post-MI hearts, particularly in remote regions. As in the instance of increased cell-cycle activation, this finding is consistent with the diminished numbers of apoptotic cells we previously noted in the myocardium of hypertrophied Kat5+/− heterozygous hearts [16], as well as with well-documented findings that Tip60 is pro-apoptotic [6, 7, 40–42]. Although we have been unable to conclusively ascertain the identity of TUNEL-labeled cells, diminished numbers of the cleaved caspase-3-positive cells outlined by WGA staining (Fig. 6B) in transversely sectioned myocardium suggest that Tip60 depletion mitigates CM losses in the remote zone during pathogenic post-infarction remodeling of the left ventricle [36, 43, 44]. This important point warrants further investigation.
Implications
As recently suggested [4], progress in the field of cardiac regeneration would be advanced by the identification, and ability to temporarily mitigate the function of, inhibitory factors that have evolved to maintain CMs in their profound state of proliferative senescence. Of course, regenerative approaches based on the relief of inhibitory factors would also mandate interventions designed to regulate CM proliferation, once unleashed, in order to prevent rhabdomyosarcoma formation [45] and/or mitotic catastrophe [46, 47] as observed following Gsk-3 depletion. (It may be notable that during Akt signaling, Gsk-3 resides immediately upstream of Tip60, which becomes activated consequent to Gsk-mediated phosphorylation [40, 48, 49].) Several inhibitory proteins, mostly tumor suppressors, have been identified that upon deletion result in activation of the CM cell-cycle; in addition to Gsk-3 [50] these include Retinoblastoma1 [51, 52], Meis1 [53] and Meis2 [51], and Hippo pathway components [23, 54]. Improved regenerative efficacy achieved by simultaneous depletion of two of these inhibitors was recently reported [51]. Although Tip60’s pleiotropic effects, which are mediated by the acetylation of multiple histone and non-histone targets, cause definitive resolution of its molecular mechanism in CMs to be beyond the scope of this report, we are planning to interrogate the acetylated status of multiple non-histone candidates in CMs, including Atm, p21, p27, p53, and Sp1. All of these proteins inhibit cell proliferation, and, with the exception of p27, have been shown to be directly regulated by Tip60-specific acetylation [6, 7, 13, 55, 56]. Perhaps most prominent among these, and in accord with our recent findings in the neonatal heart [26], we are considering the possibility that Tip60 maintains Atm activation, and the consequent DDR, in adult CMs in order to maintain proliferative senescence. Such a result would be consistent with findings that de-activation of Atm promotes CM proliferation in the adult heart [25], and that Atm depletion alleviates effects of DDR-induced heart failure [57]. The findings reported here justify the inclusion of Tip60 to the list of potential cardiac therapeutic targets.
Supplementary Material
Highlights.
Disruption of the Kat5 gene after myocardial infarction preserves heart function.
Improved cardiac function is accompanied by diminished apoptosis and scarring.
Cell-cycle inhibitors are reduced, and, the cardiomyocyte cell-cycle is activated.
Tip60 is a promising target for limiting damage caused by myocardial infarction.
Acknowledgements
The technical expertise of Taylor Tibbs, Mitchell Harrison, and Carri Lupton is gratefully acknowledged. Supported by NIH 5R01HL131788 (J.A.A. & JWL), NIH 1R01HL155085 (M.P.), NIH 1S10 OD025038 (J.A.A.), American Heart Association 18CDA34110240 (M.P.), Grant #FP00012308 (J.A.A.& J.W.L.) from the Medical College of Wisconsin Cardiovascular Center, and American Heart Association postdoctoral fellowship #828662 (X.W.).
Abbreviations:
- Atm
Ataxia-telangiectasia mutated
- AurkB
Aurora kinase B
- BrdU
5’-bromodeoxyuridine
- Cre
Myh6-driven cre-recombinase
- CM
cardiomyocyte
- cTnT
cardiac troponin-T
- Cxn-43
Connexin-43
- DDR
DNA damage response
- EF
ejection fraction
- FAC
fractional area change
- floxed
LoxP-flanked
- FS
fractional shortening
- Kat5
lysine acetyltransferase-5
- MI
myocardial infarction
- MPI
myocardial performance index
- PFA
paraformaldehyde
- pH3
phosphohistone H3
- Tip60
Tat-interactive protein 60 kD
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end-labeling
- WGA
wheat germ agglutinin
Footnotes
Conflict of interest: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sadek H, Olson EN, Toward the Goal of Human Heart Regeneration, Cell Stem Cell 26(1) (2020) 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhu Y, Do VD, Richards AM, Foo R, What we know about cardiomyocyte dedifferentiation, J Mol Cell Cardiol 152 (2021) 80–91. [DOI] [PubMed] [Google Scholar]
- [3].Almada AE, Wagers AJ, Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease, Nature reviews. Molecular cell biology 17(5) (2016) 267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Galdos FX, Guo Y, Paige SL, VanDusen NJ, Wu SM, Pu WT, Cardiac Regeneration: Lessons From Development, Circ Res 120(6) (2017) 941–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li P, Ge J, Li H, Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease, Nature reviews. Cardiology 17(2) (2020) 96–115. [DOI] [PubMed] [Google Scholar]
- [6].Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB, Acetylation of the p53 DNA-binding domain regulates apoptosis induction, Mol Cell 24(6) (2006) 841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tang Y, Luo J, Zhang W, Gu W, Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis, Mol Cell 24(6) (2006) 827–39. [DOI] [PubMed] [Google Scholar]
- [8].Sun Y, Jiang X, Price BD, Tip60: connecting chromatin to DNA damage signaling, Cell Cycle 9(5) (2010) 930–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD, Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60, Nat Cell Biol 11(11) (2009) 1376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun Y, Jiang X, Chen S, Fernandes N, Price BD, A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM, Proc Natl Acad Sci U S A 102(37) (2005) 13182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schwarz K, Simonis G, Yu X, Wiedemann S, Strasser RH, Apoptosis at a distance: remote activation of caspase-3 occurs early after myocardial infarction, Mol Cell Biochem 281(1–2) (2006) 45–54. [DOI] [PubMed] [Google Scholar]
- [12].Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA, The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response, Cell 157(3) (2014) 565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee MS, Seo J, Choi DY, Lee EW, Ko A, Ha NC, Yoon JB, Lee HW, Kim KP, Song J, Stabilization of p21 (Cip1/WAF1) following Tip60-dependent acetylation is required for p21-mediated DNA damage response, Cell death and differentiation 20(4) (2013) 620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bose A, Sudevan S, Rao VJ, Shima H, Trivedi AK, Igarashi K, Kundu TK, Haploinsufficient tumor suppressor Tip60 negatively regulates oncogenic Aurora B kinase, J Biosci 44(6) (2019). [PubMed] [Google Scholar]
- [15].McAllister D, Merlo X, Lough J, Characterization and expression of the mouse tat interactive protein 60 kD (TIP60) gene, Gene 289(1–2) (2002) 169–76. [DOI] [PubMed] [Google Scholar]
- [16].Fisher JB, Kim MS, Blinka S, Ge ZD, Wan T, Duris C, Christian D, Twaroski K, North P, Auchampach J, Lough J, Stress-induced cell-cycle activation in Tip60 haploinsufficient adult cardiomyocytes, PLoS One 7(2) (2012) e31569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fisher JB, Horst A, Wan T, Kim MS, Auchampach J, Lough J, Depletion of Tip60 from In Vivo Cardiomyocytes Increases Myocyte Density, Followed by Cardiac Dysfunction, Myocyte Fallout and Lethality, PLoS One 11(10) (2016) e0164855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD, Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein, Circ Res 89(1) (2001) 20–5. [DOI] [PubMed] [Google Scholar]
- [19].Broberg CS, Pantely GA, Barber BJ, Mack GK, Lee K, Thigpen T, Davis LE, Sahn D, Hohimer AR, Validation of the myocardial performance index by echocardiography in mice: a noninvasive measure of left ventricular function, J Am Soc Echocardiogr 16(8) (2003) 814–23. [DOI] [PubMed] [Google Scholar]
- [20].Tei C, Ling LH, Hodge DO, Bailey KR, Oh JK, Rodeheffer RJ, Tajik AJ, Seward JB, New index of combined systolic and diastolic myocardial performance: a simple and reproducible measure of cardiac function--a study in normals and dilated cardiomyopathy, J Cardiol 26(6) (1995) 357–66. [PubMed] [Google Scholar]
- [21].Poulsen SH, Jensen SE, Nielsen JC, Moller JE, Egstrup K, Serial changes and prognostic implications of a Doppler-derived index of combined left ventricular systolic and diastolic myocardial performance in acute myocardial infarction, Am J Cardiol 85(1) (2000) 19–25. [DOI] [PubMed] [Google Scholar]
- [22].Schaefer A, Meyer GP, Hilfiker-Kleiner D, Brand B, Drexler H, Klein G, Evaluation of Tissue Doppler Tei index for global left ventricular function in mice after myocardial infarction: comparison with Pulsed Doppler Tei index, Eur J Echocardiogr 6(5) (2005) 367–75. [DOI] [PubMed] [Google Scholar]
- [23].Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT, Martin JF, Hippo pathway deficiency reverses systolic heart failure after infarction, Nature 550(7675) (2017) 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD, Revisiting Cardiac Cellular Composition, Circ Res 118(3) (2016) 400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W, Sadek HA, Hypoxia induces heart regeneration in adult mice, Nature 541(7636) (2017) 222–227. [DOI] [PubMed] [Google Scholar]
- [26].Wang X, Lupton C, Lauth A, Wan TC, Foster P, Patterson M, Auchampach JA, Lough JW, Evidence that the acetyltransferase Tip60 induces the DNA damage response and cell-cycle arrest in neonatal cardiomyocytes, J Mol Cell Cardiol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ikeda T, Uno M, Honjoh S, Nishida E, The MYST family histone acetyltransferase complex regulates stress resistance and longevity through transcriptional control of DAF-16/FOXO transcription factors, EMBO Rep (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen J, Lu Y, Tian M, Huang Q, Molecular mechanisms of FOXO1 in adipocyte differentiation, J Mol Endocrinol 62(3) (2019) R239–R253. [DOI] [PubMed] [Google Scholar]
- [29].Chen CJ, Yu W, Fu YC, Wang X, Li JL, Wang W, Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway, Biochem Biophys Res Commun 378(3) (2009) 389–93. [DOI] [PubMed] [Google Scholar]
- [30].Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J, Silent information regulator 1 protects the heart from ischemia/reperfusion, Circulation 122(21) (2010) 2170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].van Deursen JM, The role of senescent cells in ageing, Nature 509(7501) (2014) 439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E, ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation, Nat Cell Biol 17(5) (2015) 627–38. [DOI] [PubMed] [Google Scholar]
- [33].Xiang FL, Guo M, Yutzey KE, Overexpression of Tbx20 in Adult Cardiomyocytes Promotes Proliferation and Improves Cardiac Function After Myocardial Infarction, Circulation 133(11) (2016) 1081–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Joki Y, Ohashi K, Yuasa D, Shibata R, Ito M, Matsuo K, Kambara T, Uemura Y, Hayakawa S, Hiramatsu-Ito M, Kanemura N, Ogawa H, Daida H, Murohara T, Ouchi N, FGF21 attenuates pathological myocardial remodeling following myocardial infarction through the adiponectin-dependent mechanism, Biochem Biophys Res Commun 459(1) (2015) 124–30. [DOI] [PubMed] [Google Scholar]
- [35].Dassanayaka S, Brittian KR, Jurkovic A, Higgins LA, Audam TN, Long BW, Harrison LT, Militello G, Riggs DW, Chitre MG, Uchida S, Muthusamy S, Gumpert AM, Jones SP, E2f1 deletion attenuates infarct-induced ventricular remodeling without affecting O-GlcNAcylation, Basic Res Cardiol 114(4) (2019) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang Y, Chen Y, Yan Y, Li X, Chen G, He N, Shen S, Chen G, Zhang C, Liao W, Liao Y, Bin J, Loss of CEACAM1, a Tumor-Associated Factor, Attenuates Post-infarction Cardiac Remodeling by Inhibiting Apoptosis, Sci Rep 6 (2016) 21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tang Y, Zhao W, Chen Y, Zhao Y, Gu W, Acetylation is indispensable for p53 activation, Cell 133(4) (2008) 612–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD, Cardiacspecific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability, Circ Res 98(6) (2006) 837–45. [DOI] [PubMed] [Google Scholar]
- [39].Wang X, Lauth A, Wan TC, Lough JW, Auchampach JA, Myh6-driven Cre-recombinase activates the DNA damage response and the cell-cycle in the myocardium in the absence of loxP sites, Dis Model Mech (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Charvet C, Wissler M, Brauns-Schubert P, Wang SJ, Tang Y, Sigloch FC, Mellert H, Brandenburg M, Lindner SE, Breit B, Green DR, McMahon SB, Borner C, Gu W, Maurer U, Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53, Mol Cell 42(5) (2011) 584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu Y, Liao R, Li N, Xiang R, Sun P, Phosphorylation of Tip60 by p38alpha regulates p53-mediated PUMA induction and apoptosis in response to DNA damage, Oncotarget 5(24) (2014) 12555–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y, Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis, Cell 102(4) (2000) 463–73. [DOI] [PubMed] [Google Scholar]
- [43].Dorn GW 2nd, Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling, Cardiovasc Res 81(3) (2009) 465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dorn GW 2nd, Novel pharmacotherapies to abrogate postinfarction ventricular remodeling, Nature reviews. Cardiology 6(4) (2009) 283–91. [DOI] [PubMed] [Google Scholar]
- [45].Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandona L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M, MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs, Nature 569(7756) (2019) 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhou J, Ahmad F, Parikh S, Hoffman NE, Rajan S, Verma VK, Song J, Yuan A, Shanmughapriya S, Guo Y, Gao E, Koch W, Woodgett JR, Madesh M, Kishore R, Lal H, Force T, Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy, Circ Res 118(8) (2016) 1208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Singh AP, Umbarkar P, Guo Y, Force T, Gupte M, Lal H, Inhibition of GSK-3 to induce cardiomyocyte proliferation: a recipe for in situ cardiac regeneration, Cardiovasc Res 115(1) (2019) 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cheng X, Ma X, Zhu Q, Song D, Ding X, Li L, Jiang X, Wang X, Tian R, Su H, Shen Z, Chen S, Liu T, Gong W, Liu W, Sun Q, Pacer Is a Mediator of mTORC1 and GSK3-TIP60 Signaling in Regulation of Autophagosome Maturation and Lipid Metabolism, Mol Cell 73(4) (2019) 788–802 e7. [DOI] [PubMed] [Google Scholar]
- [49].Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, Wang Z, Zhang CS, Chien KY, Wu J, Li Q, Han J, Lin SC, GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy, Science 336(6080) (2012) 477–81. [DOI] [PubMed] [Google Scholar]
- [50].Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, Epstein JA, Force T, Huggins GS, Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation, J Clin Invest 118(11) (2008) 3609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Alam P, Haile B, Arif M, Pandey R, Rokvic M, Nieman M, Maliken BD, Paul A, Wang YG, Sadayappan S, Ahmed RPH, Kanisicak O, Inhibition of Senescence-Associated Genes Rb1 and Meis2 in Adult Cardiomyocytes Results in Cell Cycle Reentry and Cardiac Repair Post-Myocardial Infarction, J Am Heart Assoc 8(15) (2019) e012089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sdek P, Zhao P, Wang Y, Huang CJ, Ko CY, Butler PC, Weiss JN, Maclellan WR, Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes, J Cell Biol 194(3) (2011) 407–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA, Meis1 regulates postnatal cardiomyocyte cell cycle arrest, Nature 497(7448) (2013) 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF, Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size, Science 332(6028) (2011) 458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sun Y, Xu Y, Roy K, Price BD, DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity, Mol Cell Biol 27(24) (2007) 8502–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rajagopalan D, Pandey AK, Xiuzhen MC, Lee KK, Hora S, Zhang Y, Chua BH, Kwok HS, Bhatia SS, Deng LW, Tenen DG, Kappei D, Jha S, TIP60 represses telomerase expression by inhibiting Sp1 binding to the TERT promoter, PLoS pathogens 13(10) (2017) e1006681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Higo T, Naito AT, Sumida T, Shibamoto M, Okada K, Nomura S, Nakagawa A, Yamaguchi T, Sakai T, Hashimoto A, Kuramoto Y, Ito M, Hikoso S, Akazawa H, Lee JK, Shiojima I, McKinnon PJ, Sakata Y, Komuro I, DNA single-strand break-induced DNA damage response causes heart failure, Nature communications 8 (2017) 15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.