

Abstract
PTOV1 is an oncogenic protein, initially identified in prostate cancer, that promotes proliferation, cell motility and invasiveness. However, the mechanisms that regulate PTOV1 remain unclear. In an effort to understand these mechanisms, we identify 14-3-3 as a PTOV1 interactor and show that high levels of 14-3-3 expression, like PTOV1, correlate with prostate cancer progression. Further, we discover an SGK2-mediated phosphorylation of PTOV1 at S36, which is required for 14-3-3 binding. Disruption of the PTOV1-14-3-3 interaction results in an accumulation of PTOV1 in the nucleus and a proteasome-dependent reduction in PTOV1 protein levels. To understand the effect of 14-3-3 on PTOV1 stability, we find that loss of 14-3-3 binding leads to an increase in PTOV1 binding to the E3 ubiquitin ligase HUWE1, which promotes proteasomal degradation of PTOV1. Conversely, our data suggest that 14-3-3 stabilizes PTOV1 protein by sequestering PTOV1 in the cytosol and inhibiting its interaction with HUWE1. Finally, our data suggest that stabilization of the 14-3-3-bound form of PTOV1 promotes PTOV1-mediated expression of cJun, which drives cell cycle progression in cancer. Together, these data provide a first mechanism to understand the regulation of the oncoprotein PTOV1.
INTRODUCTION
Prostate tumor-overexpressed gene 1 protein (PTOV1) was initially identified as a highly expressed mRNA transcript in primary prostate tumor samples (1). Subsequent studies demonstrated that PTOV1 overexpression in prostate and other cancers correlates with metastasis, drug resistance and poor clinical outcomes (2–10). The overexpression of PTOV1 has been shown to promote the proliferation of cultured cells, tumor growth in mouse xenograft models, and increased motility in cancer cell lines (reviewed in (11)). Accordingly, PTOV1 is essential for cell growth and its depletion by siRNA results in G2/M arrest and cell death (9, 12, 13). Thus, PTOV1 has been proposed as a potential therapeutic target in cancer (11). However, the mechanisms that regulate PTOV1 function are still not understood.
PTOV1 protein is primarily composed of two homologous regions that sit adjacent to each other, referred to as the A (amino acids 88–234) and B domains (amino acids 253–336). The A and B domains are unusual in sequence, but bear resemblance to a domain within the Mediator of RNA polymerase II transcription subunit 25 (MED25). Accordingly, it has been proposed that PTOV1 may competitively inhibit MED25 by vying for interacting partners (14). In addition, the A and B domains each contain a putative nuclear localization sequence (NLS), which may play a role in nucleo-cytoplasmic shuttling of PTOV1 (11). Also, at the N-terminus is a stretch of 43 amino acids that forms a putative nucleic acid-binding eAT hook domain that has an affinity for RNA (15). Deletion of this domain results in an accumulation of PTOV1 in the nucleus (15).
PTOV1 has been shown to shuttle between the nucleus and cytoplasm in a cell cycle-dependent manner, with nuclear import occurring in early S phase and a shift back to the cytoplasm at G2/M phase (13, 16). These nuclear and cytosolic pools of PTOV1 are thought to have different functions. In the nucleus, PTOV1 regulates transcription, including the repression of NOTCH gene transcription, which is associated with increased deacetylase activity (17). In the cytosol, PTOV1 associates with the Receptor of activated protein C kinase 1 (RACK1), a component of ribosomes, to promote Proto-oncogene cJun translation and a consequent increase in cell motility (18). This nucleo-cytoplasmic shuttling also correlates with fluctuations in PTOV1 protein levels (13). Nevertheless, the mechanisms that regulate the nucleo-cytoplasmic shuttling and turnover of PTOV1 are not understood.
Our data implicate 14-3-3 as a key regulator of PTOV1 function. The human 14-3-3 protein family consists of seven structurally similar isoforms, some of which are associated with aggressive cancer phenotypes (reviewed in (19)). The ζisoform, in particular, drives oncogenic transformation, suppresses cell death, promotes epithelial-to-mesenchymal transition and strongly correlates with poor clinical outcomes in a variety of cancer types (20–25). 14-3-3s have no enzymatic activity, but instead exert their effects by binding to and modulating the function of a large network of binding partners. Importantly, the binding of 14-3-3 is dependent on one or two serine (S) or threonine (T) phosphorylations within loosely conserved motifs on the binding partner. In this manner, 14-3-3s integrate upstream kinase signaling to exert a specific effect on their partners. This effect can vary from the sequestration of proteins, positive or negative regulation of enzyme activity, or even scaffolding of protein-protein interactions—any of which depends on the binding partner in question.
In this study, we find that the understudied serum and glucocorticoid-induced kinase-2 (SGK2) phosphorylates PTOV1 at S36. SGK2 is a member of the SGK family, which includes SGK1, SGK2, and SGK3 (26–29). In contrast to the better-studied SGK1 and SGK3, little is known about SGK2. In general, the function of SGK2 appears to be pro-survival, as it was identified as a target for synthetic lethality in p53 −/− cells and its depletion sensitizes a variety of cancer lines to apoptosis (30–34). In addition, a recent study identified a VTPase subunit as an SGK2 substrate, implicating SGK2 in the control of autophagy and lysosomal acidification (30). Sequence and structural analyses place SGKs within the AGC serine/threonine kinase superfamily, which includes well-known 14-3-3 docking site kinases, such as PKC and AKT (35). SGK1 has even been shown to phosphorylate 14-3-3 binding sites (36, 37). In particular, SGKs share a high degree of homology with AKT, and both kinases can recognize the same substrates in vitro (27, 38). In addition, SGK1 and SGK3 (and potentially SGK2), like AKT, are activated downstream of PI3K activity (reviewed in (39)). However, the limited studies on SGK2 make it difficult to draw any conclusions on its unique function and regulation.
Here we uncover a mechanism by which SGK2, 14-3-3 and the E3 ubiquitin-protein ligase HUWE1, cooperate to regulate the localization and degradation of PTOV1, providing an explanation for the cellular partitioning and regulation of PTOV1 stability at the protein level. Furthermore, these data identify pathways (e.g., SGK2, 14-3-3) that could be exploited to inhibit PTOV1 in cancer.
MATERIALS AND METHODS
Plasmids and cloning
Point mutations, as described in the text, were cloned using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) per manufacturer’s protocol using primer sequences indicated in the supplemental methods. Clones were confirmed with sequencing by Eton Bioscience (San Diego, CA, USA). Plasmids were maxiprepped using GeneJET Plasmid Maxiprep Kit (Thermo Fischer Scientific, Waltham, MA, USA) per manufacturer’s protocol.
Confocal Microscopy
Cells were seeded onto acid-etched coverslips and incubated for 48 hours before fixation. Cells were fixed for 10 minutes with 1% or 4% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100/PBS for 10–15 minutes. Samples were then blocked with SEA BLOCK Blocking Buffer (Thermo Scientific, Waltham, MA, USA) for 1 hour at room temperature (RT) and subsequently incubated with primary antibodies at 4°C overnight. Cells were washed with 0.1% Tween/PBS (PBS-T) and incubated with secondary antibodies in PBS for 45 minutes to 1 hour at RT. Cells were washed with PBS-T and subsequently stained with 1.43 μM DAPI for 5 minutes. The coverslips were mounted with Prolong Diamond Antifade Mountant (Thermo Scientific, Waltham, MA, USA) and allowed to cure overnight at RT while protected from light. Images were acquired on a LEICA TCS SP8 confocal microscope fitted with a HC PL APO 63X/1.40 Oil CS2 objective and a HyD detection system (Leica Microsystems, Wetzlar, Germany).
For data integrity, samples for each set were seeded, fixed, and stained concurrently for consistent conditions. Furthermore, laser power and image resolution were kept constant for each set. All images were processed using Huygens Essential express deconvolution tool. Pearson’s coefficients were calculated using the colocalization analyzer tool in the same software. For the colocalization calculation, threshold intensity values were set at 10% of the highest intensity value for each image. Furthermore, the Pearson’s calculation was limited to individual cells using a trace tool within the Huygens software. Pearson’s coefficient averages and significance were calculated in GraphPad Prism 8 with the Welch’s t-test correction.
Cell culture and gene expression
HEK-293T (RRID:CVCL_0063), PC3 (RRID:CVCL_0035), and LnCAP (RRID:CVCL_0395) cells were purchased ATCC (Atlanta, GA, USA). Lenti-X 293T cells were purchased from Clontech (Mountainview, CA, USA). Cells purchased from ATCC and Clontech were authenticated and mycoplasma-tested by the supplier, but were not further tested in our laboratory. Cell lines were frozen at early passage numbers (~2–5) and, once thawed, limited to approximately 15 passages before disposal. HEK-293T and Lenti-X 293T cells were maintained in DMEM and PC3 cells in DMEM/F12 media, each supplemented with 10% fetal bovine serum and 1% penicillin-strepomycin. Cells were incubated at 37°C and 5% CO2.
For transient expression experiments, HEK-293T cells were seeded at 15–20% confluence and grown overnight. Cells were then transfected in complete media with 8 μg plasmid/10 cm dish as indicated using 40 μg PEI-MAX transfection reagent (Polysciences, Warrington, PA, USA) using standard protocols. Media was changed 6–12 hours post-transfection. Cells were harvested 48 hours following transfection for downstream applications.
Lentiviral constructs were produced in Lenti-X 293T cells via transfection of the indicated transfer vectors for either FLAG-PTOV1 or GFP-PTOV1, along with psPAX2 and pMD2.G in a 4:2:1 ratio. To generate PC3 cells stably expressing the indicated PTOV1 constructs, PC3 cells were seeded at 20% confluence and grown overnight. Cells were then transduced with the appropriate lentiviral supernatants in the presence of polybrene. The media was changed after 24 hours, and cells were selected by FACS for GFP-expression 2–3 days post-transduction.
For RNAi depletion experiments, PC3 cells were transfected with pooled ON-TARGETplus siRNA reagents (Horizon Discovery, Cambridge, UK) for each indicated target using Lipofectamine™ RNAiMAX transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). Cells were seeded at 20% confluence and grown overnight. The cells were washed 2X with PBS, after which OPTI-MEM (Thermo Fisher Scientific, Waltham, MA, USA) was added to the cells. The siRNAs were incubated at 100 nM with the RNAiMAX reagent at RT for 20 minutes prior to addition to cells. The cells incubated with the siRNA-RNAiMAX complex in the OPTI-MEM for 4 hours. FBS was then added to the cells, and they incubated another 8 hours before the media was changed for complete DMEM/F12. Cells were harvested for downstream applications 48 hours later.
HUWE1 ubiquitination assay
In vitro ubiquitination assays were performed as previously described (40, 41). In brief, recombinant human HUWE1 HECT domain was produced and purified from BL21 bacterial cells. Recombinant E1, UbcH7 (E2), and ubiquitin proteins were purchased from R&D Systems. GFP-PTOV1 was overexpression in HEK-293T cells by transiently transfecting 16 ug of pcDNA3 encoding GFP-PTOV1 into 1×106 cells in a 10cm dish. 48 hours later, cells were lysed and GFP-PTOV1 was retrieved with GFP-TRAP resin (Chromotek, Planegg, Germany). After washing, GFP-PTOV1 on beads was incubated at 30°C for 3 hours with 10 ng of recombinant E1, 100 ng of recombinant UbcH7, 100 μg of ubiquitin, and 1 μg of a purified HECT domain of HUWE1 in 40 μl of reaction buffer [50 mM tris (pH 7.5), 5 mM MgCl2, 2 mM ATP, 2 mM DTT]. After the incubation, the beads were washed 3X with a buffer containing 10 mM Hepes (pH 7.4), 150 mM KCl, 1% NP-40, and 400 mM NaCl. GFP-PTOV1 protein was eluted with Laemmli SDS sample buffer and subjected to Western blot.
CoIP, immunoblotting, and antibodies
Antibodies are listed in the supplemental methods. Cells were harvested on ice and washed with cold PBS. Cells were scraped from the dishes in the cold PBS and transferred to 15 mL centrifuge tubes. Dishes were washed an additional time with PBS to collect residual cells. Cells were pelleted at 1200xg at 4°C. Cell pellets were resuspended in Amanda’s CoIP Buffer (10 mM HEPES KOH pH 7.5, 150 mM KCl, 0.1% Igepal CA-630, 1X Pierce protease inhibitors (EDTA free), 1X Pierce phosphatase inhibitors) and transferred to 1.5 mL microcentrifuge tubes. Samples were rotated at 4°C for ≥15 minutes, passaged through a 25-gauge needle, and then pelleted to remove debris. The clarified lysates were incubated with pre-washed beads (HA, FLAG, or GFP-Trap, as indicated) at 4°C for 1 hour to overnight. Beads were washed 3X with PBS.
For immunoblot analysis, the beads were resuspended in 1XSDS sample buffer and boiled for 5 min to elute. Samples were loaded onto Criterion TGX 4–15% pre-cast polyacrylamide gels (BIO-RAD, Hercules, CA, USA) and run at 150V for 1 hour. Following electrophoresis, the gels were rinsed with distilled water, and soaked in 20% EtOH. The samples were transferred to nitrocellulose membranes using the iBlot transfer system (Thermo Fisher Scientific, Waltham, MA, USA) at 20V for 6 minutes. The membranes were blocked in 1:1 PBST: Intercept Blocking Buffer for 1 hour at RT. Primary antibodies were diluted from 1:500 to 1:5k in 1:1 PBST: Intercept Blocking Buffer and incubated overnight at 4°C. Secondary antibodies were diluted 1:10k in 1:4 PBST: Intercept Blocking Buffer and incubated for 1 hour at RT. Blots were imaged and quantitated using a LI-COR Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
The custom pS36 PTOV1 antibody was developed in rabbits by Pacific Immunology (Ramona, CA) against a synthetic peptide RAVRSRpSWPASPRGC targeting the S36 phosphorylation site. Phospho-specific antibody was column-purified as described previously for custom PTM-specific antibodies (42).
Protein-protein interaction proteomics
For LC-MS/MS analysis, samples were eluted from beads with 6M guanidine and boiled for 5 min. Eluted samples were transferred to fresh microcentrifuge tubes twice to minimize bead carry-over. Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol for the microplate assay. Samples were then reduced with 5mM DTT at 55°C for 15 min. After cooling, samples were alkylated with 15 mM iodoacetamide (MilliporeSigma, St. Louis, MO, USA) for one hour in the dark. Samples were then loaded onto 30kD centrifugal filters (VWR, Radnor, PA, USA) and washed twice with 6M guanidine and twice with 10 mM ammonium bicarbonate. Protein samples were then digested with mass spectrometry grade trypsin on the filter at 1:50 (w/w) ratio at 37°C with shaking overnight. The next day, the digested protein samples were eluted from the filter and washed down with 10 mM ammonium bicarbonate. The eluate was transferred to mass spec vials, and vacuum dried. The samples were then resuspended in OrbiA solvent (3% acetonitrile, 0.1% formic acid). Quantitative LC-MS/MS was performed on 1 μg of each sample using a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA).
The LC-MS/MS data were analyzed using PEAKS analysis software (Bioinformatics Solutions, Waterlook ON, Canada) using the SwissProt Homo sapiens database. The database search included a fixed carbamidomethylation modification on Cys and variable modifications, including Meth oxidation, Asn/Gln deamination, Lys acetylation, and Ser/Thr/Tyr phosphorylation. Relative peptide abundance was calculated using area under the curve (AUC) analysis for the indicated targets for each sample.
Imaging flow cytometry
To assess PTOV1 nuclear localization, GFP-PTOV1 expressing PC3 cells were trypsinized, washed, and fixed in 1% PFA for 10 min at 4°C. Cells were then permeabilized in 70% ethanol at 4°C overnight. Prior to acquisition, cells were washed and incubated in PBS containing 1 μg/ml propidium iodide (PI) and 100 μg/ml RNase for 30 min at RT. Cells were transferred to ice prior to acquisition on the ImageStream MKII (Luminex Corporation, Austin, TX, USA). Sample preparation was performed identically for experiments involving NES-GFP-PTOV1 transfected HEK-293T cells. For MG132 and HUWE1 knockdown experiments, data was acquired using live PC3 cells expressing GFP-PTOV1 resuspended in PBS+2% FBS without fixation, permeabilization, or staining.
All analyses were performed in IDEAS software (Luminex Corporation). Nuclear localization was assessed using the integrated nuclear localization tool. PC3 cell images were spectrally compensated using single color controls prior to analysis. Cell images were gated by gradient RMS, and then by size and aspect ratio to include only single, in-focus cells. To measure nuclear localization, a log transformed Pearson’s correlation coefficient (i.e. similarity score) was calculated for each image using the appropriate channels for PI and GFP.
Drug treatments
Lysosomal and proteasomal degradation experiments utilized MG132 (Selleckchem, Houston, TX, USA cat. S2619) and Bafilomycin (Cayman Chemical Company, Ann Arbor, Michigan, USA cat. 11038). PC3 cells stably expressing FLAG-PTOV1 constructs or HEK-293T cells transiently transfected with GFP-PTOV1 constructs were treated (2 days after transfection) with either 10 μM MG132, 100 nM Bafilomycin, or a similar volume of vehicle control (DMSO) for 2 hours prior to harvest.
PTOV1 degradation rates were assessed using cycloheximide (CHX, Cayman Chemical Company, cat 14126). PC3 cells stably expressing FLAG-PTOV1 WT or S36A were treated with 50 μg/ml CHX or an equal volume of vehicle control (DMSO). Alternatively, HEK-293T cells were transiently transfected with FLAG-PTOV1 S36A or FLAG-NES-PTOV1 S36A and treated with 50 μg/ml CHX two days post-transfection. Cells were harvested at the indicated timepoints and lysed in RIPA lysis buffer.
SGK2 inhibition experiments were performed using GSK 650394 (Tocris, Minneapolis, MN, USA cat. 3572). Cells were treated with 10 μM GSK 650394 for 48 hours prior to harvest.
Kinase screening (ProQinase)
245 purified Ser/Thr kinases were evaluated for activity against peptides encompassing S36 (amino acids 30–42) and S53 (amino acids 47–59) of PTOV1 via the radiometric KinaseFinder assay (ProQinase GmbH). In short, the peptides were reconstituted in 50 nM HEPES pH 7.5 at 200 μM stock solution. Reaction buffer (60 mM HEPES-NaOH pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 μM Na-orthovanadate, 1.2 mM DTT, 1 μM ATP/[γ−33P]-ATP), protein kinase (1–400 ng/50 μL) and PTOV1 peptides (1 μM) were distributed into 96-well, V-shaped polypropylene microtiter plates (assay plate). All PKC assays (except the PKC-mu and the PKC-nu assay) additionally contained 1 mM CaCl2, 4 mM EDTA, 5 μg/ml phosphatidylserine and 1 μg/ml 1.2-dioleyl-glycerol. The MYLK2, CAMK1D, CAMK2A, CAMK2B, CAMK2D, CAMK4, CAMKK2, and DAPK2 assays additionally contained 1 μg/ml calmodulin and 0.5 mM CaCl2. The PRKG1 and PRKG2 assays additionally contained 1 μM cGMP. One well of each assay plate was used for a buffer/substrate control containing no enzyme.
The assay plates were incubated at 30°C for 60 minutes. Subsequently, the reaction cocktails were stopped with 20 μl of 4.7 M NaCl/35 mM EDTA. The reaction cocktails were transferred into 96-well streptavidin-coated FlashPlate® HTS PLUS plates (PerkinElmer, Boston MA), followed by 30 min incubation at RT on a shaker to allow for binding of the biotinylated peptides to the streptavidin-coated plate surface. Subsequently, the plates were aspirated and washed three times with 250 μl of 0.9% NaCl. Incorporation of radioactive 33Pi was determined with a microplate scintillation counter (Microbeta, Perkin Elmer). For evaluation of the results of the FlashPlate® PLUS-based assays, the background signal of each kinase (w/o biotinylated peptide) was determined in parallel. Kinases of interest were selected from the screen described above to repeat at three peptide concentrations (1 μM, 0.5 μM and 0.25 μM) in triplicate.
Gene-expression data analysis
We downloaded RNA sequencing data for prostate-cancer patients from The Cancer Genome Atlas (43). These data had previously been aligned to version hg19 of the human reference genome (44) and summarized as gene-level read counts using the Rsubread software (45, 46). In addition, we downloaded clinical data for these patients and extracted Gleason scores. After identifying patients for whom we had both gene-expression data and Gleason scores, data for 485 patients remained. Using these data, we evaluated relationships among Gleason scores and log2-transformed expression levels of PTOV1 and YWHAZ. To download and parse the data, we wrote a script in the Python programming language (https://python.org). To generate the graphics, we used the R statistical software (version 3.6) and the ggplot2 package (version 3.3.1). The scatter plots use regression lines to show correlation trends and 95% confidence intervals to indicate uncertainty.
Data availability
Data generated in this study are available in the figures and supplementary data files.
RESULTS
Phosphorylation of PTOV1 at S36 and S53 are required for binding to 14-3-3.
14-3-3ζ promotes aggressive cancer phenotypes by interacting with a network of phosphorylated binding partners (19–25, 42). Thus, a major focus of our laboratory is to use proteomics and molecular approaches to identify cancer-driving mechanisms through elucidation of the 14-3-3ζ interactome. To identify phosphorylation-dependent 14-3-3-binding partners, we performed coimmunoprecipitation (coIP) LC-MS/MS proteomics in cells expressing HA-14-3-3ζ WT or the non-phosphobinding mutant HA-14-3-3ζ K49E (Supplemental data table 1). Among the interactors that pulled down with 14-3-3ζ WT, but not the K49E mutant, was PTOV1. A previous 14-3-3 LC-MS/MS study had also found PTOV1 among many proteins in the mass spectra, but its significance/regulation remained unexplored (47). To begin to validate these data, we also performed the converse LC-MS/MS experiment with GFP-PTOV1 as bait, which demonstrated a PTOV1 interaction with 6 of the 7 endogenous 14-3-3 isoforms (all but the σ isoform)—most notably with the ε and ζ isoforms (Supplemental data table 1). We further validated the PTOV1-14-3-3 interaction by coIP immunoblot (Figure 1A). Interestingly, like PTOV1, high expression of 14-3-3ζ (YWHAZ) correlates with higher risk prostate cancers (Figure 1B).
Figure 1. Phosphorylation of PTOV1 at S36 and S53 are required for binding to 14-3-3.
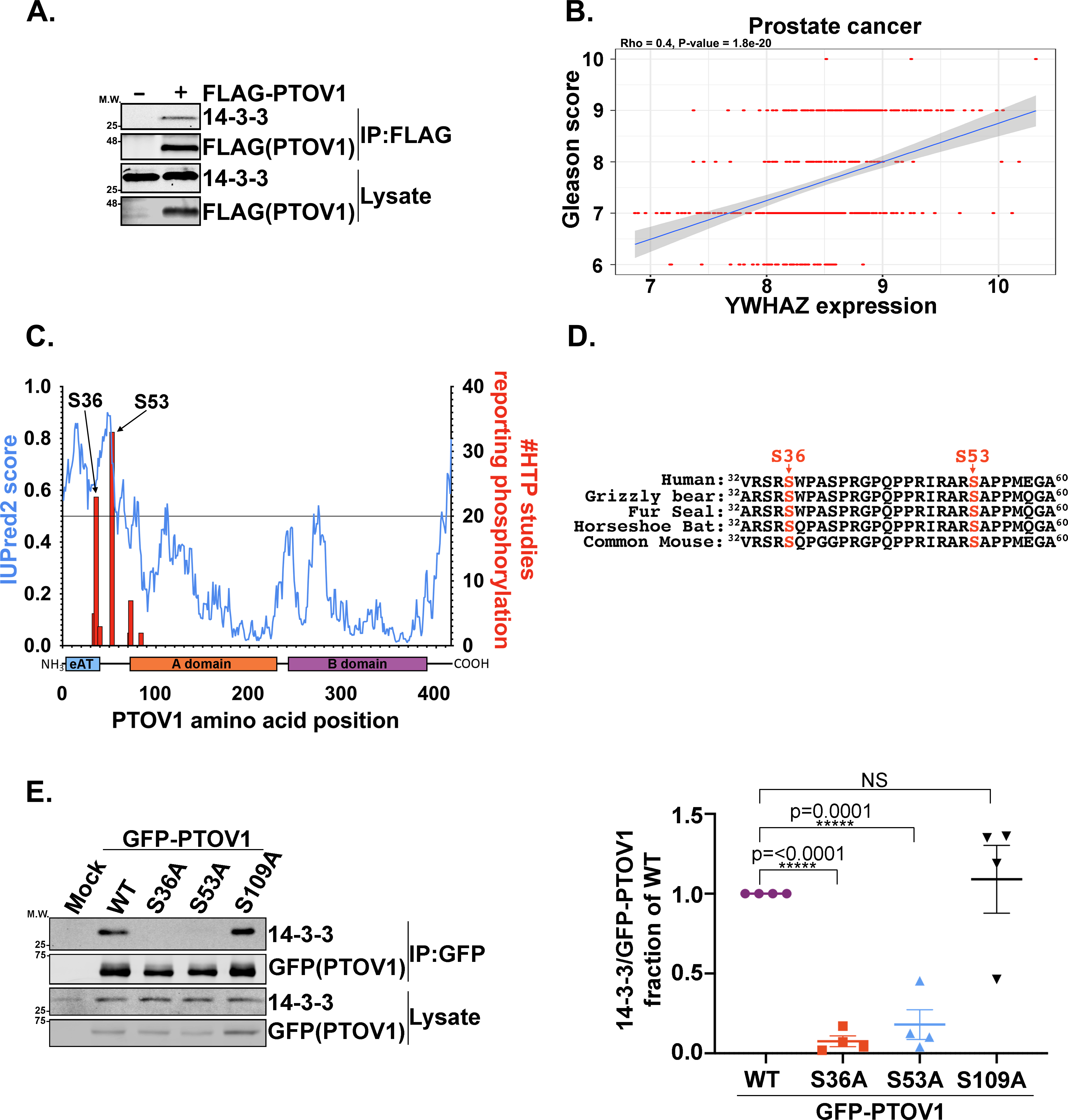
A) FLAG-PTOV1 was expressed in HEK-293T cells, followed by IP on FLAG resin and immunoblotting for 14-3-3 and FLAG. A representative image from three biological replicates is shown. B) Positive correlation between RNA expression levels of the YWHAZ gene and Gleason scores for 485 patients from The Cancer Genome Atlas. The regression line (blue) shows the correlation trend and the gray shaded area around the line represents the 95% confidence interval. C) Composite graph of IUPRED2 disorder score and the high-throughput identification frequency of phosphorylations (phosphosite.org, accessed 24 Feb 2020) across the PTOV1 amino acid sequence. D) Alignments of mammalian PTOV1 sequences surrounding S36 and S53. E) GFP-PTOV1 was expressed in HEK-293T cells, followed by IP on GFP-Trap resin and immunoblotting for 14-3-3 and GFP. Right panel shows quantification (LI-COR infrared imaging) of 14-3-3 coIP signal normalized to GFP (coIP) and expressed as a fraction of the GFP-PTOV1 WT normalized coIP signal from four biological replicates. Error bars represent SEM; p-values were calculated using a two-tailed Student’s t-test.
To identify the phosphorylation sites on PTOV1 responsible for 14-3-3 binding, we first narrowed down a list of S and T residues using a combination of consensus sequence analysis via 14-3-3 site prediction algorithms (48), disorder prediction with IUPRED (49, 50), and the frequency of high-throughput detection of site-specific phosphorylations using phosphosite.org (51) (Figure 1C). From these analyses, we selected S36, S53, and S109 as candidate 14-3-3-docking-site phosphorylations. Both S36 and S53 are conserved from mouse to human (Figure 1D). CoIP immunoblotting of S-to-A mutants confirmed that S36 and S53, but not S109, are necessary for PTOV1 binding to 14-3-3 (Figure 1E).
SGK2 regulates phosphorylation of PTOV1 at S36.
To identify the PTOV1-targeted kinase, we attempted to generate phospho-specific antibodies for pS36 and pS53, but were only able to recover specific phospho-antibody for S36. Thus, we focused our efforts on Serine 36—importantly, phospho-null mutation of this site alone completely abrogates 14-3-3 binding (Figure 1E). Our initial biased efforts to identify the kinase(s) targeting the S36 and S53 sites focused on common 14-3-3-docking site kinases, including CAMK2, AKT, and PKC, but failed to reveal any compelling leads. Therefore, we took an unbiased approach in which we generated biotin-tagged peptides encompassing the S36 and S53 sites and performed in vitro radiometric kinase assays with 245 individual human kinases that span the majority of Ser/Thr kinase families (Figures S1–S2). These assays revealed a small subset of candidate direct kinases, including PBK, PKCD, SGK2 and DYRK1A. However, only SGK2 emerged as a common hit between the S36 and S53 sites.
Follow-up validation of a subset of these kinases demonstrated that recombinant SGK2, but not SGK1, PBK or PKCD, phosphorylates both sites on PTOV1 in vitro (Figure 2A–B). We found that inhibition of SGK2 led to a drop in pS36 signal and 14-3-3 binding (Figure 2C–D). Furthermore, we found that siRNA depletion of SGK2 decreased pS36, but also caused a corresponding loss of total PTOV1 protein levels (Figure 2E). This was consistent with the decrease in protein expression levels that we saw with the S36A or S53A PTOV1 mutants, suggesting that loss of these phosphorylations and 14-3-3 binding may destabilize the protein, which we revisit further below. Together, our data suggest that SGK2 phosphorylates PTOV1 at S36 (and likely S53, based on in vitro data) to promote 14-3-3 binding (Figure 2F).
Figure 2. SGK2 phosphorylates PTOV1 to promote 14-3-3 binding.
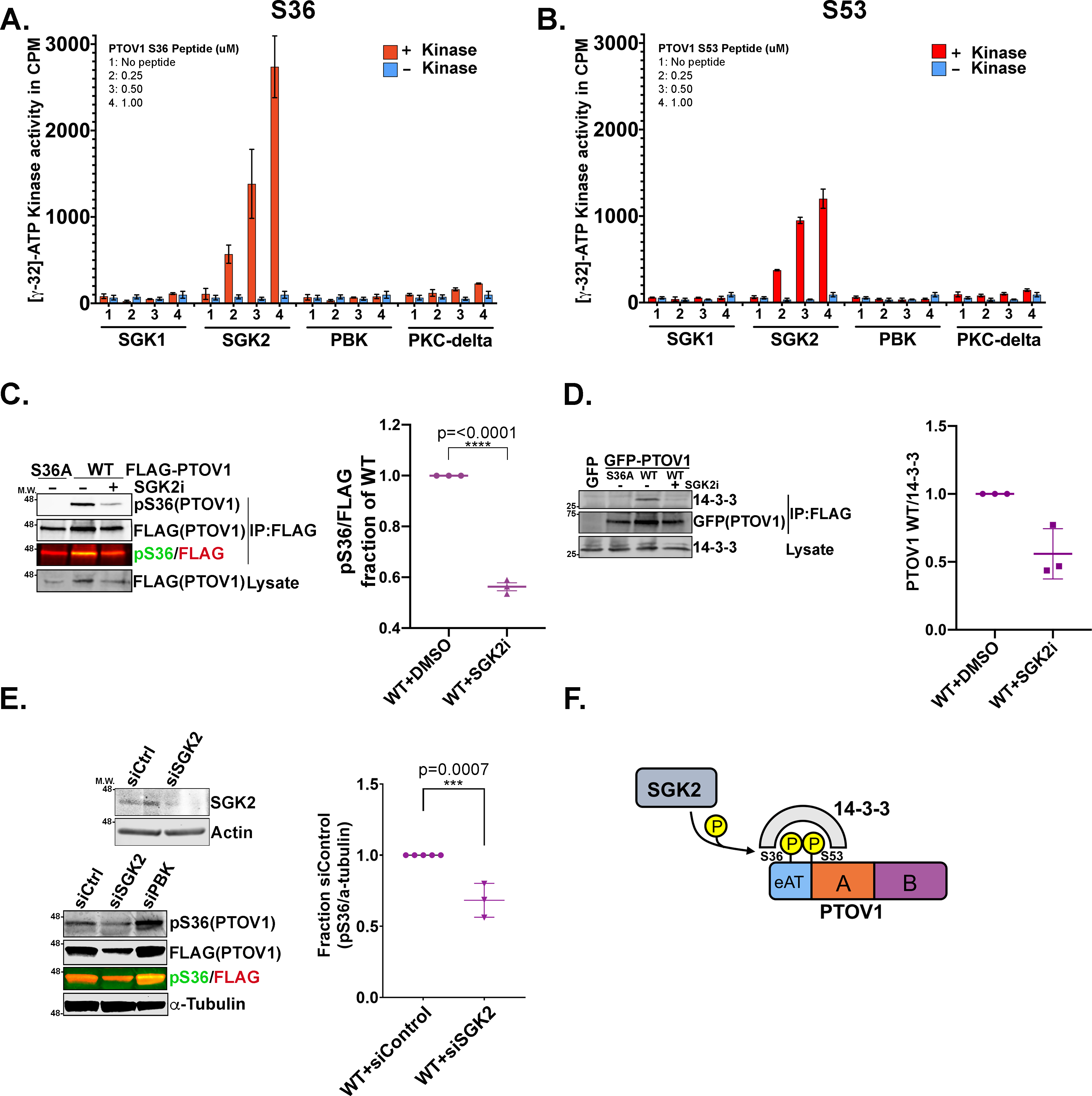
A) Radiometric assays were performed with the indicated kinases incubated with a biotin-tagged PTOV1 peptide encompassing S36. Corrected kinase activity (raw value minus sample peptide background) was measured in biological triplicate. Graph shows mean kinase activity in counts per minute (cpm) with error bars indicating standard deviations (SD). B) Radiometric kinase assays were performed as in panel A but against a peptide encompassing S53. C) PC3 cells stably expressing FLAG-PTOV1 WT or S36A were treated with 10 μM of the SGK2 inhibitor (SGK2i) GSK 650394 for 48 hours, followed by IP on FLAG resin and immunoblotting for pS36 PTOV1 and FLAG. Right panel shows quantification (LI-COR infrared imaging) of pS36 signal normalized to FLAG (coIP) and expressed as a fraction of normalized WT from three biological replicates. Error bars represent SEM; p-values were calculated using a two-tailed Student’s t-test. D) PC3 cells stably expressing GFP-PTOV1 (WT or S36A) were treated with SGK2i as in panel C, followed by immunoprecipitation of GFP-PTOV1 on GFP-trap resin and immunoblotting for 14-3-3. E) Upper panel shows an immunoblot validation of SGK2 siRNA (signal shown is endogenous SGK2) in PC3 cells. Lower panel shows pS36 PTOV1 immunoblot signal from PC3 cells stably expressing FLAG-PTOV1 and transfected with the indicated siRNAs for 48 hours. Right graph shows quantitation of pS36 signal normalized to loading control from 3 biological replicates. Error bars represent SEM; p-values were calculated using a two-tailed Student’s t-test. F) A model describing the relationship between SGK2, PTOV1 and 14-3-3.
Loss of 14-3-3 binding leads to an accumulation of PTOV1 in the nucleus.
Our experiments up to this point consistently showed that loss of 14-3-3 binding—either by mutation of S36A/S53A or inhibition of SGK2—resulted in reduced recovery of PTOV1 protein from cells. Thus, we questioned whether this decreased recovery during lysis was due to either a reduction in total PTOV1 protein levels and/or a shift in PTOV1 localization.
To observe PTOV1 localization in the cell, we first used confocal microscopy and imaging flow cytometry on PC3 cells stably expressing either GFP- or FLAG-tagged PTOV1 WT or PTOV1 S36A. We found that PTOV1 WT is primarily distributed throughout the cytosol with relatively low levels of protein in the nucleus (Figure 3A–B). In contrast, the PTOV1 S36A mutant protein is primarily nuclear (Figure 3A–B). We also confirmed that loss of 14-3-3 binding by the S36A mutation resulted in nuclear accumulation of PTOV1 in LNCaP cells (Figure S3). Consistent with the idea that 14-3-3 binding controls PTOV1 localization, a phospho-null mutation at S53, but not at S109, causes a similar shift of PTOV1 protein into the nucleus (Figure 3B). Importantly, we also found that inhibition of SGK2 phenocopied the effect of the S36A mutation, resulting in an accumulation of PTOV1 WT in the nucleus (Figure 3C), further confirming SGK2 as the PTOV1-targeted kinase. Together, these data indicate that loss of 14-3-3 binding, either through mutation of the docking site phosphorylation or reduction in the phosphorylation via inhibition of SGK2, results in nuclear accumulation of PTOV1.
Figure 3. Loss of 14-3-3 binding leads to an accumulation of PTOV1 in the nucleus.

A) PC3 cells stably expressing FLAG-PTOV1 WT or S36A were analyzed by confocal imaging, deconvolved by Hyugens software and assessed for cytosolic and nuclear localization of PTOV1. Right panel shows Pearson coefficient (Hyugens colocalization software) of PTOV1 colocalization with DAPI. B) PC3 cells stably expressing GFP-PTOV1 (WT or indicated mutants) were analyzed by imaging flow cytometry for nuclear localization of PTOV1 as a function of overlap with propidium iodide (PI) nuclear stain. Right panel shows quantification of PTOV1/PI colocalization expressed as a log transformed Pearson coefficient. Each point represents the median similarity score from a separate population of cells. C) PC3 cells from panel A were treated with 10 μM SGK2 inhibitor (GSK 650393) as in Figure 2B. Right panel shows quantification as in panel A.
Loss of 14-3-3 binding destabilizes PTOV1 protein.
Our observation that loss of 14-3-3 binding shifts PTOV1 localization to the nucleus did not rule out the possibility that 14-3-3 may also control PTOV1 stability. Indeed, we noted that the PTOV1 S36A consistently showed lower levels of expression by either immunoblot or whole cell imaging. To address this question, we performed cycloheximide (CHX)-chase experiments using PC3 cells stably expressing FLAG-PTOV1 WT or FLAG-PTOV1 S36A. We found that degradation of the S36A mutant protein was significantly faster than PTOV1 WT, suggesting that 14-3-3 binding protects PTOV1 from degradation (Figure 4A).
Figure 4. Loss of 14-3-3 binding destabilizes PTOV1 protein.
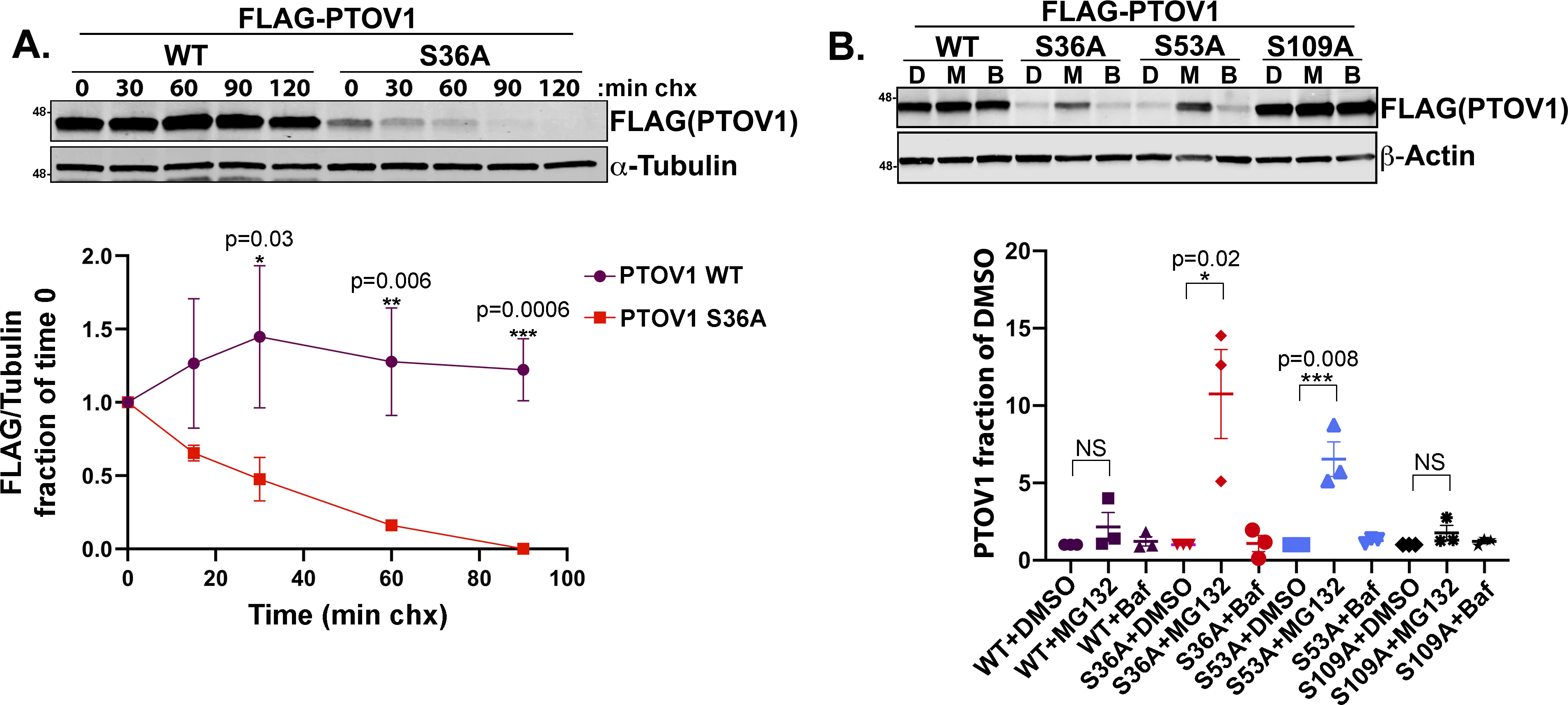
A) PC3 cells stably expressing FLAG-PTOV1 WT or S36A were treated with cycloheximide (CHX) over the indicated time course, followed by immunoblotting for FLAG (PTOV1) and α-Tubulin. Bottom panel shows quantification of immunoblot signal for FLAG normalized to α-Tubulin and expressed as a fraction of the signal at time 0. Error bars represent SEM and p-values were calculated with a Student’s t-test comparing WT and S36A signal at each timepoint from three biological replicates. B) PC3 cells stably expressing FLAG-PTOV1 WT or indicated mutants were treated with DMSO (D) 10 μM MG132 (M) or 100 nM bafilomycin (B) for 2 hours. Bottom panel shows quantification from three biological replicates of FLAG (PTOV1) immunoblot signal normalized to actin and expressed as fractions of the normalized signals for the associated DMSO-treated samples. Error bars represent SD of the mean; a two-tailed Student’s t-test was used to calculate p-values
To begin to understand the mechanism of PTOV1 degradation, we found that inhibition of the proteasome with MG132 stabilized the PTOV1 S36A and S53A mutant proteins. In contrast, the lysosome inhibitor bafilomycin had no effect, indicating that PTOV1 is degraded by the proteasome and not through bulk or targeted autophagy (Figure 4B). Imaging flow cytometry analysis of PC3 cells stably expressing GFP-PTOV1 WT and mutants confirmed the increase in total PTOV1 levels with proteasomal inhibition (Figure S4A). These data suggest that 14-3-3 binding stabilizes PTOV1, while loss of 14-3-3 binding accelerates the targeted degradation of PTOV1 through the proteasome.
HUWE1 interacts with PTOV1 and controls PTOV1 stability.
To identify the upstream cellular machinery that controls PTOV1 degradation, we searched our PTOV1 interactome data for E3 ligases (Supplemental data table 1). The E3 ligase HUWE1 emerged as the top overall PTOV1 interactor based on peptide count. We validated by coIP that PTOV1 interacts with endogenous HUWE1 (Figure 5A). To assess whether HUWE1 regulates PTOV1 protein stability, we depleted HUWE1 with siRNA in PC3 cells stably expressing GFP-PTOV1 S36A for imaging flow cytometry and FLAG-PTOV1 WT or S36A for immunoblotting. In both scenarios, for WT and mutant PTOV1, knockdown of HUWE1 results in a marked increase in steady-state PTOV1 protein (Figure 5B and S4B). We then confirmed that this increase in PTOV1 protein is due to slower kinetics of PTOV1 degradation in HUWE1-depeted cells (Figure 5C–D). We also questioned whether the decrease in global PTOV1 levels in SGK2-inhibited or -siRNA treated cells (Figure 2) could be rescued by HUWE1 depletion. As shown in Figure S4C, depletion of HUWE1 rescues the loss of PTOV1 induced by SGK2 knockdown. In addition, we see a loss of pS36 signal on the rescued PTOV1 protein in cells depleted of HUWE1 and SGK2, further validating SGK2 as the PTOV1 S36-targeted kinase (Figure S4C)
Figure 5. HUWE1 interacts with PTOV1 and controls PTOV1 stability.

A) HEK-293T cells overexpressing GFP or GFP-PTOV1 (or mock transfected) were subject to IP on GFP-Trap resin, followed by immunoblotting for HUWE1 and GFP. B) PC3 cells stably expressing FLAG-PTOV1 WT or S36A were transfected with siRNA against HUWE1 or a control sequence (non-specific), followed by immunoblotting for FLAG (PTOV1) and indicated proteins. Right panel shows quantification of FLAG (PTOV1) signal normalized to α-Tubulin and expressed as a fraction of control siRNA treatment. Error bars represent SD and p-values were calculated using a two-tailed Student’s t-test from 3 biological replicates. C) PC3 cells stably expressing FLAG-PTOV1 WT were transfected with siRNA against HUWE1 or control siRNA for 48 hours, then treated with CHX as in Figure 4A. Cells were harvested at timepoints that were determined empirically to visualize PTOV1 WT degradation. Lower panel shows quantification of signal from three biological replicates analyzed as in Figure 4A. Error bars represent SD. D) PC3 cells stably expressing FLAG-PTOV1 S36A were treated and analyzed as in panel C. Quantification represents three biological replicates and error bars represent SD. E) HEK-293T cells were transfected with GFP, GFP-PTOV1 WT or indicated GFP-tagged PTOV1 truncation mutants, followed by IP on GFP-Trap resin and immunoblotting for HUWE1 and indicated proteins. Right panel shows quantification of HUWE1 coIP signal normalized to the GFP(PTOV1) coIP signal for each mutant and expressed as a fraction of normalized HUWE1 coIP signal for GFP-PTOV1 WT. Error bars represent SEM and p-values were calculated using a two-tailed Student’s t-test from four biological replicates. F) GFP-PTOV1 was immunopurified from HEK-293T cells and incubated with ubiquitin and recombinant human HUWE1 HECT domain or a catalytically inactive version of the HECT domain (C/S), followed by immunoblotting for GFP and ubiquitin.
Deletion mapping of the binding site suggests that HUWE1 interacts with the B domain of PTOV1 (Figure 5E). Consistent with the idea that HUWE1 promotes PTOV1 degradation, we found that PTOV1 lacking the B domain (PTOV1 1–246) expressed at much higher levels than WT or other deletion mutants (see immunoblot in Figure 5E). LC-MS/MS examination of potential HUWE1-mediated ubiquitination sites on PTOV1 revealed a ubiquitination at K114, under MG132 treatment, that only appeared on the PTOV1 S36A mutant (Supplemental data table 1). However, single arginine substitutions at K114 and several other candidate lysines failed to fully stabilize PTOV1 S36A protein levels, perhaps due to the lysine promiscuity of E3 ligases. Nevertheless, we were able to see direct ubiquitination of PTOV1 by recombinant HUWE1, but not a catalytically inactive HUWE1 mutant (Figure 5F)
PTOV1 nuclear localization is required for degradation.
Our observation that the PTOV1 S36A mutant is destabilized and enriched in the nucleus raised the possibility that the sequestration of PTOV1 in the cytosol prevents PTOV1 degradation. Thus, we asked whether cytosolic sequestration itself—even in the absence of 14-3-3 binding— is sufficient to stabilize PTOV1 protein. To address this question, we appended an in-frame nuclear export sequence (NES) to the N-terminal end of both PTOV1 WT and the 14-3-3 binding-defective PTOV1 S36A mutant (Figure S5A). We verified that the NES sequence resulted in a predominately cytosolic pattern of PTOV1 S36A localization (Figure S5A–B) and found that it also increased steady-state levels of PTOV1 protein (Figure S5C). More importantly, CHX-chase experiments indicated that forcing the PTOV1 S36A mutant back into the cytosol inhibited PTOV1 degradation in the absence of 14-3-3 binding (Figure S5D). Thus, together our data suggest that the sequestration of PTOV1 in the cytosol, either by 14-3-3 or other means (e.g., an orthologous NES), protects PTOV1 from degradation.
14-3-3 and HUWE1 regulate PTOV1 localization and stability to control PTOV1 function.
To further explore the relationship between 14-3-3 and HUWE1 in regulating PTOV1 localization and function, we examined the potential inverse relationship between 14-3-3 and HUWE1 binding to PTOV1. As shown in Figure 6A, the 14-3-3 binding-defective mutants of PTOV1 (S36A and S53A) interact at significantly higher levels with HUWE1 than PTOV1 WT, suggesting that 14-3-3 sequesters PTOV1 away from HUWE1 or sterically inhibits HUWE1 binding. To understand how 14-3-3 and HUWE1 cooperate to control PTOV1 function, we focused on the role of PTOV1 in promoting cJun translation, which occurs through a direct interaction with RACK1 and ribosomes (18). We found that depletion of HUWE1 resulted in a PTOV1 WT-mediated increase in cJun expression not observed in PTOV1 S36A-expressing cells (Figure 6B). Furthermore, as we would predict, depletion of HUWE1 resulted in a compartmentalized accumulation depending on the PTOV1 genotype—with PTOV1 WT accumulating primarily in the cytosol and PTOV1 S36A primarily in the nucleus (Figure 6C). Therefore, the increase in cJun expression in PTOV1 WT cells depleted of HUWE1 requires 14-3-3 binding and is likely explained by the elevated level of PTOV1 in the cytosol.
Figure 6. 14-3-3 and HUWE1 regulate PTOV1 localization and stability to control PTOV1 function.

A) HEK-293T cells were transfected with GFP-PTOV1 WT or indicated mutants and subject to IP on GFP-Trap resin, followed by immunoblotting for HUWE1 and GFP (PTOV1). Right panel shows quantification of coIP signal for HUWE1 normalized to GFP and expressed as a fraction of the normalized HUWE1 coIP for GFP-PTOV1 WT. Error bars represent SD and p-values were calculated using a two-tailed Student’s t-test from four biological replicates. B) PC3 cells stably expressing FLAG-PTOV1 WT and S36A were transfected with siRNA against HUWE1 or control siRNA for 48 hours, followed by immunoblotting for cJun and indicated proteins. Panel on the right shows quantification of cJun signal normalized to α-Tubulin. Error bars represent SD and p-values were calculated using a two-tailed Student’s t-test from three biological replicates. C) HEK-293T cells overexpressing FLAG-PTOV1 WT or S36A were transfected with siRNA to HUWE1 as in panel B, followed by FLAG antibody staining and confocal imaging to determine nuclear localization of PTOV1. Right panel shows quantification as in Figure 3A.
All together, these data support a model in which an SGK2-mediated interaction between 14-3-3 and PTOV1 sequesters PTOV1 in the cytosol to promote the expression of cJun and potentially other targets (18). Conversely, the inhibition of SGK2, and the resulting loss of 14-3-3 binding, triggers an accumulation of PTOV1 in the nucleus, where we suspect the interaction between PTOV1 and HUWE1 occurs. In support of this idea, we see some overlap of PTOV1 and HUWE1 signal in the nucleus (Figure S6). The nuclear accumulation of PTOV1 would allow PTOV1 to carry out its nuclear functions (e.g., regulation of gene transcription). Once released from 14-3-3, PTOV1 is subject to HUWE1-mediated ubiquitination and proteasomal degradation (see model in Figure 7). PTOV1 degradation depends in part on translocation to the nucleus, as NES-mediated export from the nucleus stabilizes PTOV1 levels even in the absence of 14-3-3 interaction. This mechanism of PTOV1 degradation upon loss of 14-3-3 binding may serve to clear excess PTOV1 from the cytosol and/or limit the influx of PTOV1 into the nucleus.
Figure 7. Model of SGK2-, 14-3-3-, and HUWE1-mediated regulation of PTOV1 localization, stability, and function.
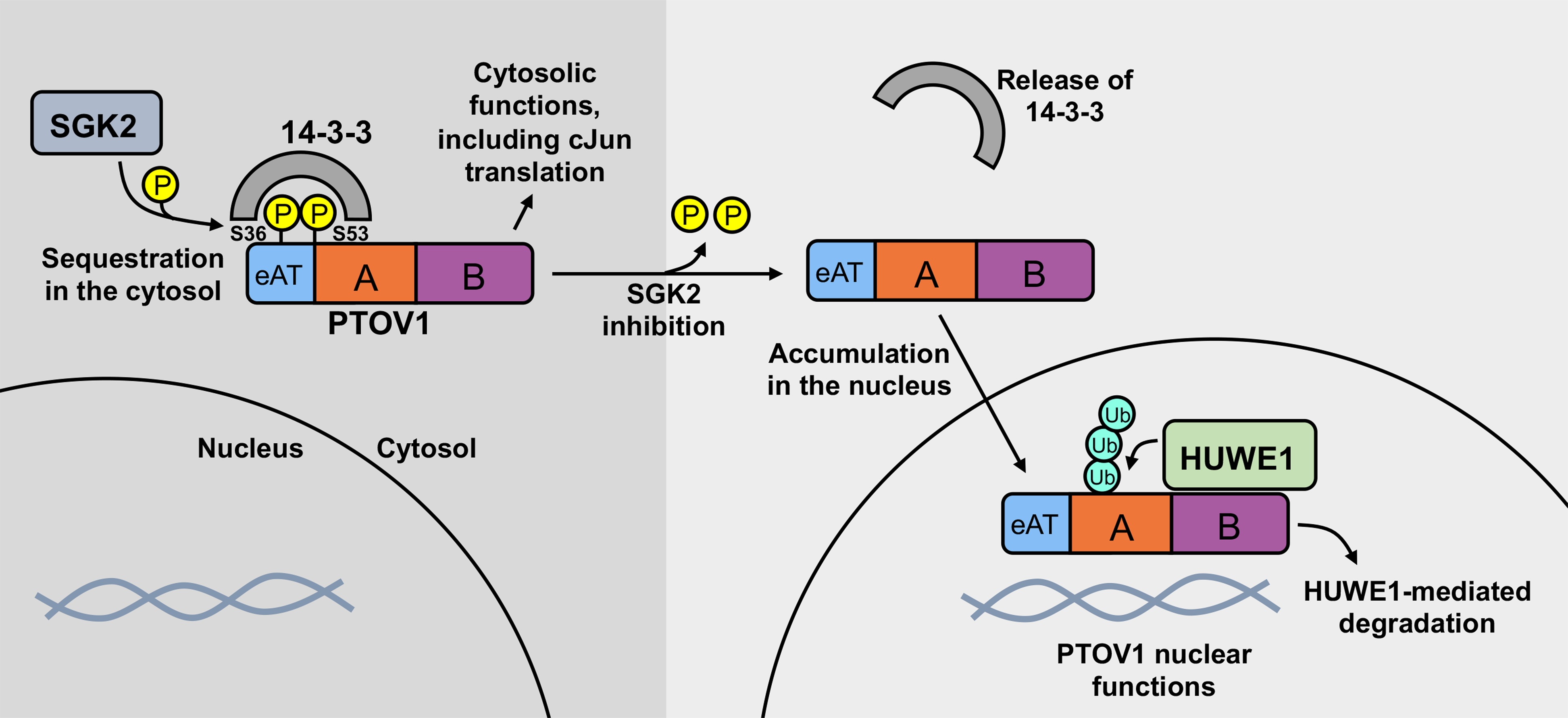
Our data suggest that an SGK2-governed interaction between 14-3-3 and PTOV1 sequesters PTOV1 in the cytosol, which promotes PTOV1-mediated expression of cJun. Upon loss of 14-3-3 binding, PTOV1 accumulates in the nucleus and is subject to HUWE1-dependent degradation via the proteasome.
DISCUSSION
We began this study with a focus on 14-3-3—a signaling hub that regulates a network of oncogenes and tumor suppressors to collectively promote a cellular program of growth, stress-adaptation and survival. Much of our laboratory’s effort has focused on expanding our understanding of the 14-3-3 signaling hub by identifying and studying binding partners. Because 14-3-3 interactions are dependent on phosphorylation, each new interaction is linked directly to an upstream kinase(s), which provides a handle to understand the interaction in the larger picture of cell signaling.
The identification of SGK2 as the PTOV1-targeted kinase places this mechanism within an understudied kinase signaling pathway. Indeed, remarkably little is known about SGK2. However, based on studies of SGK1 and SGK3 (36, 52), as well as some homology between SGK2 and AKT (28), it seems likely that SGK2 is activated downstream of PI3K signaling (27). Thus, mitogenic activation of PI3K during the cell cycle and the potential downstream activation of SGK2, together with the mechanism presented here, may explain the cell cycle-dependence of PTOV1 nucleo-cytoplasmic shuttling (13, 16). Future work will focus on understanding the upstream signaling that controls SGK2 and its temporal regulation over the cell cycle—an effort that will require the development of better tools than we currently have to study SGK2.
Given that earlier work showed that PTOV1 is cytosolic in G1 and enters the nucleus at S phase (16), we posit that the cytosolic sequestration of PTOV1 by 14-3-3 promotes cJun translation in G1 to allow for cell cycle progression. Indeed, cJun plays a critical role in progression through G1 by promoting the expression of cyclin D1 (53). Accordingly, the deletion of cJun results in a drop in cyclin D1 levels, Rb activation, and G1 arrest (54). This may also explain why, in our hands, we were unable to generate viable PTOV1 KO lines and others have shown that overexpression or depletion of PTOV1 causes a corresponding increase or decrease in cyclin D1 expression, respectively (12, 13, 16). Furthermore, the degradation of PTOV1 after release from 14-3-3 may serve to eliminate cytosolic PTOV1, which would otherwise promote inappropriate expression of cJun and cyclin D1 beyond G1.
We found that the loss of 14-3-3 binding increased the interaction between PTOV1 and HUWE1, which led to HUWE1-dependent degradation of PTOV1 via the proteasome. HUWE1 is a large (482 kDa) E3 ligase of the HECT domain family (reviewed in (55)). Numerous studies show that HUWE1 functions by directly interacting with and ubiquitinating a wide variety of substrates, including p53 (56), Mcl-1 (41), c-Myc (57), Chk1 (40), and H2AX (58). The prevailing thought is that HUWE1 is generally oncogenic, but its individual substrates reveal a more complex picture. For example, depletion of HUWE1 simultaneously upregulates p53 and Mcl-1—two proteins with opposite roles in cell growth. Therefore, as an oncogene, PTOV1 fits within the array of diverse HUWE1 substrates, adding another layer to the complexity of HUWE1 biology. Our data suggest that depletion of HUWE1 elevates PTOV1 protein levels, which, in turn, promote the expression of cJun, a pro-growth translational target of PTOV1 (18)
In our model, we propose that HUWE1 mediates the degradation of PTOV1 in the nucleus. However, this is still speculative. Our data suggest that HUWE1 can ubiquitinate PTOV1 in vitro and depletion of HUWE1 in cells increases the stability of PTOV1 S36A protein in the nucleus. Conversely, depletion of HUWE1 also increases the stability of WT PTOV1 protein in the cytosol, although WT PTOV1 is known to shuttle in and out of the nucleus and may get ubiquitinated while in the nucleus (13). We also found that forcing PTOV1 S36A out of the nucleus with an NES stabilized the protein, supporting the idea that PTOV1 nuclear localization is required for degradation. On the other hand, our imaging data suggest that HUWE1 is mostly cytosolic with only a fraction in the nucleus. We were also unable to see clear points of colocalization between PTOV1 and HUWE1 in the nucleus. Thus, it is possible that other nuclear E3 ligases may participate in regulating PTOV1 turnover in the nucleus. Toward this end, our LC-MS/MS data identified other E3 ligases as candidate interactors of PTOV1, including UBR5, which has nuclear functions (59, 60).
In conclusion, our data provide the first mechanism of regulation for the poorly understood oncogene, PTOV1, and shed new light on the poorly understood kinase SGK2. In addition, this mechanism adds to an expanding theme of 14-3-3 biology: 14-3-3s frequently serve to sequester binding partners away from a particular target or function. For example, 14-3-3 sequesters the pro-apoptotic Bcl-2 family protein Bad away from pro-survival Bcl-2 proteins to inhibit cell death. 14-3-3s sequester PRAS40 and TSC2 to allow for Rheb-mediated activation of mTORC1. 14-3-3 also sequesters FOXO and YAP/TAZ transcription factors in the cytoplasm to inhibit their nuclear function. In the case of PTOV1, which functions in both the cytosol and nucleus, its retention in the cytosol by 14-3-3 likely partitions its cytosolic and nuclear roles. Thus, PTOV1 expands the paradigm of 14-3-3 regulation and also illustrates the value of 14-3-3 as a tool to discover functional phosphorylations, kinase-substrate relationships and mechanisms that could be exploited therapeutically.
Supplementary Material
IMPLICATIONS.
These findings identify a potentially targetable mechanism that regulates the oncoprotein PTOV1
ACKNOWLEDGEMENTS
We thank the Fritz B. Burns Foundation for instrumentation, research and student support, and a postdoctoral fellowship for KLP. We thank the Simmons Center for Cancer Research for a year-round graduate fellowship to CMM. We thank the BYU graduate school for a HIDRA fellowship to CME. We thank all members of the Andersen laboratory for constructive discussion and critiques. JLA is supported by a National Cancer Institute/National Institutes of Health grant (2R15CA202618-02) and an American Cancer Society Research Scholar Grant (133550-RSG-19-006-01-CCG) (2019-current).
Financial support: A National Cancer Institute/National Institutes of Health grant (2R15CA202618-02) and an American Cancer Society Research Scholar Grant (133550-RSG-19-006-01-CCG) to JLA
Footnotes
COMPETING INTERESTS
The authors declare they have no conflict of interest.
REFERENCES
- 1.Benedit P et al. , PTOV1, a novel protein overexpressed in prostate cancer containing a new class of protein homology blocks. Oncogene 20, 1455–1464 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Fernandez S et al. , PTOV1 is overexpressed in human high-grade malignant tumors. Virchows Arch 458, 323–330 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Canovas V et al. , Prostate Tumor Overexpressed-1 (PTOV1) promotes docetaxel-resistance and survival of castration resistant prostate cancer cells. Oncotarget 8, 59165–59180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen YW, Hu ZY, Chai D, High expression of prostate tumor overexpressed 1 (PTOV1) is a potential prognostic biomarker for cervical cancer. Int J Clin Exp Patho 10, 11044–11050 (2017). [PMC free article] [PubMed] [Google Scholar]
- 5.Guo F et al. , Increased PTOV1 expression is related to poor prognosis in epithelial ovarian cancer. Tumor Biol 36, 453–458 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Jiang N, Li Y, PTOV1 Is a Novel Prognostic Marker for Hepatocellular Carcinoma Progression and Overall Patient Survival. Gastroenterology 148, S1022–S1023 (2015). [Google Scholar]
- 7.Li R et al. , Overexpressed PTOV1 associates with tumorigenesis and progression of esophageal squamous cell carcinoma. Tumor Biol 39 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Morote J et al. , PTOV1 expression predicts prostate cancer in men with isolated high-grade prostatic intraepithelial neoplasia in needle biopsy. Clinical Cancer Research 14, 2617–2622 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Wu ZQ et al. , Depleting PTOV1 sensitizes non-small cell lung cancer cells to chemotherapy through attenuating cancer stem cell traits. J Exp Clin Canc Res 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Q et al. , Prostate Tumor Overexpressed 1 (PTOV1) Is a Novel Prognostic Marker for Nasopharyngeal Carcinoma Progression and Poor Survival Outcomes. Plos One 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canovas V, Lleonart M, Morote J, Paciucci R, The role of prostate tumor overexpressed 1 in cancer progression. Oncotarget 8, 12451–12471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karna SKL, Ahmad F, Lone BA, Pokharel YR, Knockdown of PTOV1 and PIN1 exhibit common phenotypic anti-cancer effects in MDA-MB-231 cells. Plos One 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santamaria A et al. , PTOV1 enables the nuclear translocation and mitogenic activity of flotillin-1, a major protein of lipid rafts. Molecular and Cellular Biology 25, 1900–1911 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youn HS, Park UH, Kim EJ, Um SJ, PTOV1 antagonizes MED25 in RAR transcriptional activation. Biochemical and Biophysical Research Communications 404, 239–244 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Filarsky M et al. , The extended AT-hook is a novel RNA binding motif. RNA Biol 12, 864–876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santamaria A et al. , PTOV-1, a novel protein overexpressed in prostate cancer, shuttles between the cytoplasm and the nucleus and promotes entry into the S phase of the cell division cycle. Am J Pathol 162, 897–905 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alana L et al. , Prostate tumor OVerexpressed-1 (PTOV1) down-regulates HES1 and HEY1 notch targets genes and promotes prostate cancer progression. Molecular Cancer 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marques N et al. , Regulation of protein translation and c-Jun expression by prostate tumor overexpressed 1. Oncogene 33, 1124–1134 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Pennington KL, Chan TY, Torres MP, Andersen JL, The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein-protein interactions. Oncogene 10.1038/s41388-018-0348-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu J et al. , 14-3-3zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell 16, 195–207 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J et al. , 14-3-3zeta turns TGF-beta’s function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer Cell 27, 177–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi S et al. , 14-3-3zeta loss impedes oncogene-induced mammary tumorigenesis and metastasis by attenuating oncogenic signaling. Am J Cancer Res 7, 1654–1664 (2017). [PMC free article] [PubMed] [Google Scholar]
- 23.Andersen JL et al. , A biotin switch-based proteomics approach identifies 14-3-3zeta as a target of Sirt1 in the metabolic regulation of caspase-2. Molecular Cell 43, 834–842 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nutt LK et al. , Metabolic control of oocyte apoptosis mediated by 14-3-3zeta-regulated dephosphorylation of caspase-2. Dev Cell 16, 856–866 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z et al. , Down-regulation of 14-3-3zeta suppresses anchorage-independent growth of lung cancer cells through anoikis activation. Proc Natl Acad Sci U S A 105, 162–167 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waldegger S, Barth P, Forrest JN Jr., Greger R, Lang F, Cloning of sgk serine-threonine protein kinase from shark rectal gland - a gene induced by hypertonicity and secretagogues. Pflugers Arch 436, 575–580 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi T, Deak M, Morrice N, Cohen P, Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J 344 Pt 1, 189–197 (1999). [PMC free article] [PubMed] [Google Scholar]
- 28.Lang F, Cohen P, Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci STKE 2001, re17 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Firestone GL, Giampaolo JR, O’Keeffe BA, Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 13, 1–12 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Ranzuglia V et al. , Serum- and glucocorticoid- inducible kinase 2, SGK2, is a novel autophagy regulator and modulates platinum drugs response in cancer cells. Oncogene 10.1038/s41388-020-01433-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y et al. , SGK2 promotes renal cancer progression via enhancing ERK 1/2 and AKT phosphorylation. Eur Rev Med Pharmacol Sci 23, 2756–2767 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Chen JB et al. , Glucocorticoid-Inducible Kinase 2 Promotes Bladder Cancer Cell Proliferation, Migration and Invasion by Enhancing beta-catenin/c-Myc Signaling Pathway. J Cancer 9, 4774–4782 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J et al. , SGK2 promotes hepatocellular carcinoma progression and mediates GSK-3beta/beta-catenin signaling in HCC cells. Tumour Biol 39, 1010428317700408 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Baldwin A et al. , Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. Proc Natl Acad Sci U S A 107, 12463–12468 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson C et al. , Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem J 427, 69–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunet A et al. , Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21, 952–965 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhalla V et al. , Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4–2 by inducing interaction with 14-3-3. Mol Endocrinol 19, 3073–3084 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Tessier M, Woodgett JR, Serum and glucocorticoid-regulated protein kinases: variations on a theme. J Cell Biochem 98, 1391–1407 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Bruhn MA, Pearson RB, Hannan RD, Sheppard KE, Second AKT: the rise of SGK in cancer signalling. Growth factors (Chur, Switzerland) 28, 394–408 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Cassidy KB, Bang S, Kurokawa M, Gerber SA, Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. The FEBS journal 287, 1985–1999 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurokawa M et al. , A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci Signal 6, ra32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mortenson JB et al. , Histone deacetylase 6 (HDAC6) promotes the pro-survival activity of 14-3-3zeta via deacetylation of lysines within the 14-3-3zeta binding pocket. J Biol Chem 10.1074/jbc.M114.607580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research N et al. , The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujita PA et al. , The UCSC Genome Browser database: update 2011. Nucleic Acids Res 39, D876–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y, Smyth GK, Shi W, The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47, e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rahman M et al. , Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics 31, 3666–3672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benzinger A, Muster N, Koch HB, Yates JR 3rd, Hermeking H, Targeted proteomic analysis of 14-3-3 sigma, a p53 effector commonly silenced in cancer. Mol Cell Proteomics 4, 785–795 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Madeira F et al. , 14-3-3-Pred: improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 31, 2276–2283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dosztanyi Z, Prediction of protein disorder based on IUPred. Protein Sci 27, 331–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dosztanyi Z, Csizmok V, Tompa P, Simon I, IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Hornbeck PV et al. , PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bago R et al. , Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem J 463, 413–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wisdom R, Johnson RS, Moore C, c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J 18, 188–197 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schreiber M et al. , Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev 13, 607–619 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong XF et al. , The structure and regulation of the E3 ubiquitin ligase HUWE1 and its biological functions in cancer. Invest New Drug 38, 515–524 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Chen DL et al. , ARF-BP1/mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071–1083 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Adhikary S et al. , The ubiquitin ligase HectH9 regulates transcriptional activation by myc and is essential for tumor cell proliferation. Cell 123, 409–421 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Atsumi Y et al. , ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient gamma H2AX Foci Formation. Cell Reports 13, 2728–2740 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Henderson MJ et al. , EDD mediates DNA damage-induced activation of CHK2. J Biol Chem 281, 39990–40000 (2006). [DOI] [PubMed] [Google Scholar]
- 60.Gudjonsson T et al. , TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are available in the figures and supplementary data files.