

Abstract
Aberrant activation of Wnt/β-catenin signaling is strongly associated with many diseases including cancer invasion and metastasis. Small-molecule targeting of the central signaling node of this pathway, β-catenin, is a biologically rational approach to abolish hyperactivation of β-catenin signaling but has been demonstrated to be a difficult task. Herein, we report a drug-like small molecule, ZW4864, that binds with β-catenin and selectively disrupts the protein–protein interaction (PPI) between B-cell lymphoma 9 (BCL9) and β-catenin while sparing the β-catenin/E-cadherin PPI. ZW4864 dose-dependently suppresses β-catenin signaling activation, downregulates oncogenic β-catenin target genes, and abrogates invasiveness of β-catenin-dependent cancer cells. More importantly, ZW4864 shows good pharmacokinetic properties and effectively suppresses β-catenin target gene expression in the patient-derived xenograft mouse model. This study offers a selective chemical probe to explore β-catenin-related biology and a drug-like small-molecule β-catenin/BCL9 disruptor for future drug development.
Graphical Abstract
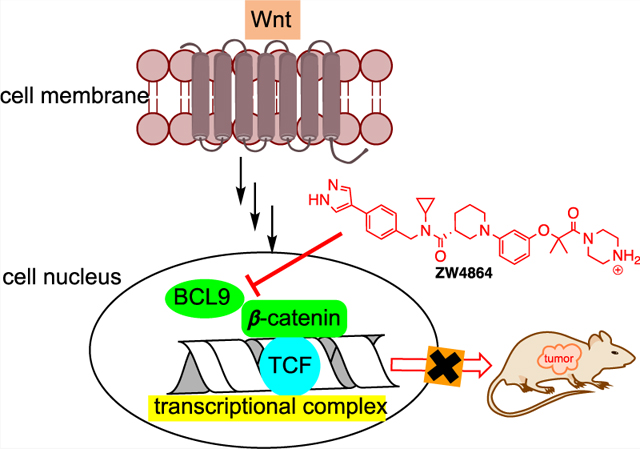
INTRODUCTION
The hyperactivation of the Wnt/β-catenin signaling pathway has a profound connection with initiation and progression of many cancers.1 When the suppressor genes of this pathway, such as APC and Axin, suffer from loss-of-function mutations or the N-terminal phosphorylation sites of CTNNB1 (β-catenin gene) are mutated inappropriately, β-catenin is stabilized into the dephosphorylated active form and translocated into the cell nucleus. Nuclear active β-catenin then binds with the DNA-binding lymphoid enhancer-binding factor (LEF)/T-cell factor (TCF) family of transcriptional factors and recruits CREB-binding protein (CBP)/p300, B-cell lymphoma 9 (BCL9)/BCL9-like (BCL9L), Pygo 1/Pygo2, etc., as coactivators to transcribe β-catenin target genes. These target genes induce the epithelial-to-mesenchymal transition (EMT), sustain stem-like cancer cells or cancer stem cells, and promote tumor immune evasion. β-Catenin signaling can also be activated by autocrine activation of frizzled (Fzd), Wnt ligands, and disheveled (Dvl), epigenetic silencing of Wnt suppressors, and/or crosstalk with other signaling pathways.
Substantial work has been conducted to search for inhibitors of the upstream effectors of the Wnt/β-catenin pathway,2 with porcupine inhibitors LGK974,3 ETC-159,4 CGX1321,5 and RXC004,6 soluble Wnt receptor OMP-54F28,7,8 and Fzd receptor blocker OMP-18R59 being tested in clinical trials. However, inhibition of these upstream Wnt targets (1) cannot produce efficacy against disease-causing loss-of-function mutations of APC and Axin and activation mutations of CTNNB1; (2) is ineffective against cancers owing to the crosstalk with the other signaling pathways to upregulate β-catenin; and (3) increases the risk of undesirable off-pathway effects by inhibiting noncanonical Wnt signaling pathways. The most appealing target for inhibitor development might be nuclear β-catenin-containing transcriptional complex because the penultimate step of the signaling cascade is the formation of this complex that confers activation of the β-catenin signaling circuit. However, direct targeting of β-catenin has proven to be a difficult task.1,10–12 Most β-catenin-targeting efforts were focused on antagonizing β-catenin interaction with TCF/LEF,12 but none of the compounds were advanced beyond the early stage of inhibitor development, probably due to two challenges: (1) β-catenin and TCF have a very large protein–protein interaction (PPI) area (3500 Å2) and strong binding affinity (KD = 7–10 nM)13 and (2) β-catenin adopts the same PPI area to interact with APC, E-cadherin, and TCF.13–20
The β-catenin/BCL9 PPI is a promising alternative target for inhibitor development. BCL9 or BCL9L is the scaffolding protein of the Wnt enhanceosome that captures newly stabilized, nuclear-localized β-catenin, facilitating β-catenin access to LEF/TCF and activating the transcription complex.21,22 The homology domain 2 (HD2) of BCL9 or BCL9L adopts a single α-helical structure to bind with β-catenin.13 The β-catenin/BCL9 PPI interface is much smaller (1450 Å2), and this PPI displays a moderate KD of 0.47 μM. The surface area of β-catenin for interacting with BCL9 has little overlap with that for other β-catenin partners, and E-cadherin (region V) is the only other known protein that binds this PPI interface.13 BCL9 and BCL9L are significantly overexpressed in many cancers, possibly through chromosomal 1q21 amplification and downregulation of BCL9-suppressing microRNAs.23–25 The β-catenin/BCL9 complex can also be upregulated through downregulation of endogenous negative regulators of this PPI.26 Dominant negative constructs of BCL9/BCL9L prohibit Wnt/β-catenin signaling.27,28 Knock-down of BCL9/BCL9L using siRNA or shRNA suppresses β-catenin signaling, blocks transcription of β-catenin target genes, and impedes migration and invasion of Wnt-activated cancer cells.23,24,27–30 Deletion of BCL9 and BCL9L suppresses Wnt-driven tumorigenesis, inhibits invasion and metastasis of Wnt-active tumors, and increases disease-free survival in mouse models that authentically mimic human cancer.31–33
In addition to peptide-based inhibitors that have been reported to disrupt the β-catenin/BCL9 PPI,34–37 Bienz and co-workers discovered carnosic acid and its analogues as small-molecule β-catenin/BCL9 PPI inhibitors through compound screening.38,39 We designed 3-(4-fluorophenyl)-N-phenylbenzamide (PNPB) derivatives as small-molecule β-catenin/BCL9 disruptors based on PPI hot-spot interactions.40–43 Through modifying a screening hit CP-868388 discovered in AlphaScreen assays,44 we obtained 2-(3-(3-carbamoylpiperidin-1-yl)phenoxy)acetic acid (CPPAA) derivatives as β-catenin/BCL9 PPI inhibitors (Figure 1).45 The best compound CPPAA-30 exhibited a Ki of 3.6 μM for β-catenin/BCL9 disruption and showed on-target activities in cell-based experiments. In this study, we set out to perform alternative optimization on CP-868388, which yields a drug-like small-molecule β-catenin/BCL9 PPI inhibitor ZW4864 that shows in vivo on-target effects (Figure 1).
Figure 1.

Design of new β-catenin/BCL9 PPI inhibitor ZW4864.
RESULTS
Inhibitor Design and Biochemical Characterizations.
We first synthesized both R- and S-isomers of the reported inhibitor CPPAA-2, resulting in compounds 1 and 2 in Table 1, respectively. The AlphaScreen assays indicated that 1 displayed a Ki of 46 μM for β-catenin/BCL9 PPI disruption, while 2 was less active (Ki > 90 μM). Therefore, compound 1 was employed as a starting point for further optimization. Within the BCL9-binding surface area of β-catenin, there are a few acidic residues, including the acidic knobs (D162, E163, and D164), D144, D145, E147, and E155. Our previous structure-based design of PNPB derivatives demonstrated that introduction of positively charged groups increased the binding affinity.40,41 Indeed, addition of a piperazine moiety led to 3, which disrupted the β-catenin/BCL9 PPI with a Ki of 5.8 μM and is 8 times more potent than 1 (Table 1). Another modification was the introduction of small substituents to the amide group (4–7 in Table 1). The cyclopropyl group was identified as the optimal group to yield 7, which disrupted the β-catenin/BCL9 interaction with a Ki of 2.7 μM and is 2-fold more potent than 3.
Table 1.
AlphaScreen Competitive Inhibition Assay Results of 1–20a
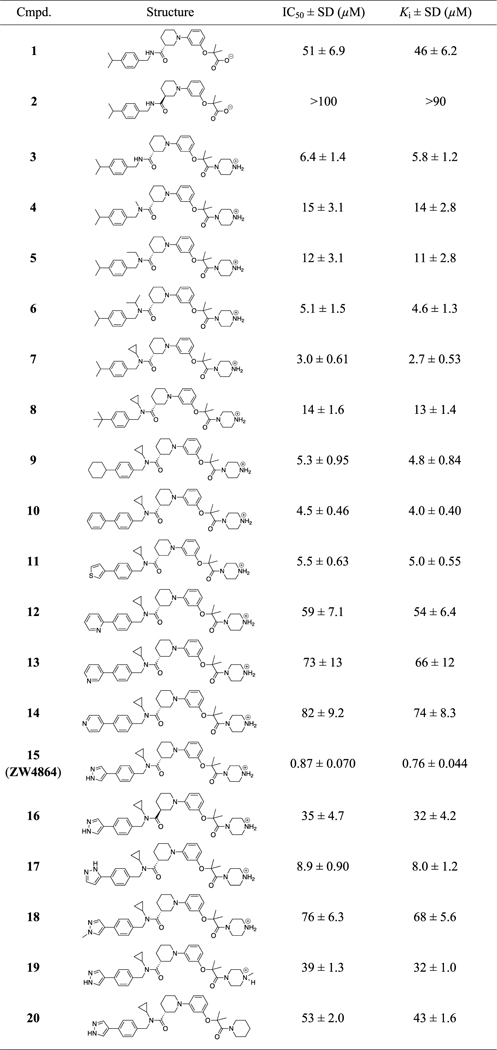
|
In AlphaScreen assays, the BCL9 peptide (residues 350–375) and full-length β-catenin (residues 1–781) were used. Each set of data was expressed as mean ± standard deviation (SD) (n = 3). The dose–response curves of the representative compounds are shown in Figure S1.
Further optimization was focused on the isopropyl group on the benzyl ring in Table 1. Different groups (8–18) including heterocycles were investigated, and 1H-pyrazol was found to be most suitable for this position. Compound 15 (ZW4864) inhibited β-catenin/BCL9 PPI with a Ki of 0.76 μM and is 3.5-fold more potent than 7. The importance of the 1H-pyrazol group was also demonstrated by analogues 17 and 18, which displayed the Kis of 8.0 and 68 μM and were 10- and 89-fold less potent than ZW4864, respectively. The enantiomer (16) of ZW4864 was synthesized. Consistent with the 1/2 pair, compound 16 disrupted β-catenin/BCL9 PPI with a Ki of 32 μM and is much less potent than ZW4864. The importance of the piperazine moiety was investigated by designing analogues 19 and 20, both of which displayed low inhibitory activities with the Kis of 32 and 43 μM, respectively.
We also developed an orthogonal surface plasmon resonance (SPR) assay to assess β-catenin/BCL9 competitive inhibition. As shown in Figure 2A, ZW4864 disrupted the β-catenin/BCL9 PPI with an IC50 of 1.2 μM. Our structure–activity relationship (SAR) studies revealed that compounds with the linear N-alkyl substituents, such as 21 with an N-ethyl group and 22 with an N-ethoxyethyl group in Figure 2B, maintained low μM inhibitory affinities. Biotinylated ZW4864 (Biotin-ZW4864 in Figure 2C) was synthesized and its binding affinity with the purified full-length β-catenin was determined by AlphaScreen saturation binding assays. As shown in Figure S3, the AlphaScreen KD between Biotin-ZW4864 and β-catenin is 0.77 ± 0.063 μM and comparable with AlphaScreen competitive inhibition assay results of ZW4864 in Table 1. Biotin-ZW4864 was incubated with the purified full-length β-catenin or SW480 cell lysates. The proteins that bound with Biotin-ZW4864 were “pulled down” by streptavidin-conjugated beads and examined by Western blot using the β-catenin-specific antibody. As shown in Figure 2D, Biotin-ZW4864 bound with the purified full-length β-catenin in a concentration-dependent manner starting at 1 μM. Biotin-ZW4864 can also effectively bind with β-catenin in SW480 cell lysates at 10 μM (Figure 2D, lower panel). These pull-down experiments demonstrated that ZW4864 can directly bind with full-length β-catenin. On the other hand, Biotin-ZW4864 did not pull down β-catenin R1C (residues 138–781) at 10 μM, indicating that the N-terminal disordered region of β-catenin participated in ZW4864 binding (Figure 2E, upper panel). Biotin-ZW4864 pulled down β-catenin R1C when its concentration was increased to 50 μM (Figure 2E, lower panel). This result was confirmed by AlphaScreen competitive inhibition assays (Figure 2F). ZW4864 disrupted the β-catenin R1C/BCL9 PPI with a Ki of 8.9 μM, which is 10-fold higher than that of the full-length β-catenin/BCL9 PPI.
Figure 2.

Biochemical characterizations of ZW4864 and derivatives. (A) SPR competitive inhibition assay results of ZW4864. The dose–response curves of SPR assays of ZW4864 are shown in Figure S2. (B) Chemical structures of 21 and 22, and their IC50 and Ki values for disrupting β-catenin/BCL9 PPI in AlphaScreen assays. The dose–response curves are provided in Figure S1. (C) Chemical structure of Biotin-ZW4864. TFA: trifluoroacetic acid. (D) Protein pull-down experiment results of Biotin-ZW4864 with β-catenin. Purified full-length β-catenin (upper panel) and SW480 cell lysates (lower panel) were incubated with Biotin-ZW4864, followed by streptavidin pull-down. Input: 5% β-catenin or cell lysate. Vehicle: 0.1% dimethyl sulfoxide (DMSO) in buffer. Uncropped Western blot gels are shown in Figure S4. (E) Protein pull-down experiment results of Biotin-ZW4864 with β-catenin R1C truncate (residues 138–781). Input: 5% β-catenin RIC. Uncropped Western blot gels are provided in Figure S5. (F) AlphaScreen assay results of ZW4864 to disrupt β-catenin R1C/BCL9 PPI. (G) AlphaScreen selectivity results of ZW4864 between β-catenin/BCL9 and β-catenin/E-cadherin PPIs. The KDs of wild-type full-length β-catenin with wild-type E-cadherin peptide (residues 824–877) in fluorescence anisotropy (FA) binding and AlphaScreen competitive binding assays are shown in Figure S6. The dose–response curves of AlphaScreen selectivity assays of ZW4864 are shown in Figure S7. Each experiment was performed in triplicate. Each set of the inhibitory activity data was expressed as mean ± standard deviation (n = 3).
Because the PPI interface of β-catenin for interacting with BCL9 also binds with E-cadherin region V and the formation of β-catenin/E-cadherin complex is essential for cell–cell adhesion, ZW4864 was assessed for its inhibitory selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs using AlphaScreen selectivity assays.44 Noting the importance of β-catenin N-terminal disordered region (residues 1–137) in developing β-catenin/BCL9 inhibitors, we have modified our AlphaScreen selectivity assays44 to include this β-catenin region. The KD values between full-length wild-type β-catenin and wild-type BCL9 HD2 peptide (residues 350–375) in fluorescence anisotropy (FA) and AlphaScreen competitive binding experiments are 0.27 and 0.23 μM, respectively, which match the result of ITC studies13 and are consistent with the KD values we previously obtained using two β-catenin truncates (residues 138–686 or residues 138–781).44 The KD values between wild-type β-catenin (residues 1–686) and wild-type E-cadherin peptide (residues 824–877) in FA binding and AlphaScreen competitive binding assays are 80 and 67 nM, respectively, which are consistent with the KD values obtained from ITC studies46,47 or the previous FA and AlphaScreen studies using β-catenin truncates (residues 138–686 or residues 138–781).44 This AlphaScreen selectivity assay system was employed to examine the selectivity of β-catenin/BCL9 inhibitors. The data in Figure 2G demonstrated that ZW4864 exhibited a 229-fold selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs.
Cell-Based Activities of ZW4864 for Inhibition of β-catenin/BCL9 Interaction and β-Catenin-Dependent Transcription.
Both BCL9 and BCL9L use their HD2 domain to bind with the N-terminal domain of β-catenin armadillo repeats, as illustrated in Figure 3A. The homology domain 1 (HD1) of BCL9 or BCL9L binds with the PHD finger of Pygo, which regulates transcription of β-catenin target genes. Cell-based coimmunoprecipitation (co-IP) assays were performed to evaluate effects of ZW4864 (15) on disruption of the β-catenin/BCL9 interaction in HCT116 cells. As shown in Figure 3B, ZW4864 disrupted the β-catenin/BCL9 interaction in a concentration-dependent manner, while input and immunoprecipitation controls were unchanged in the experiments. The parallel study revealed that the β-catenin/E-cadherin interaction was not affected by ZW4864 at the highest concentrations used, demonstrating its cell-based selectivity for β-catenin/BCL9 over β-catenin/E-cadherin PPIs. Cell-based co-IP experiments in Figure 3C showed that ZW4864 did not affect the interaction between BCL9 and Pygo but disrupted the association of β-catenin with Pygo, suggesting that ZW4864 selectively disrupts the β-catenin/BCL9 interaction in the β-catenin–BCL9–Pygo complex.
Figure 3.

(A) Illustration of β-catenin/TCF/BCL9/Pygo complex. (B) Cell-based co-IP assays to assess effects of ZW4864 for β-catenin/BCL9 PPI disruption and the inhibitory selectivity between β-catenin/BCL9 over β-catenin/E-cadherin PPIs. (C) Cell-based co-IP assays to evaluate effects of ZW4864 for disruption of BCL9/Pygo and β-catenin/Pygo interactions after a 24 h incubation. IB, immunoblotting; IP, immunoprecipitation; and input, 10% amount of cell lysate. (D) TOPFlash luciferase reporter assay results of ZW4864, its derivatives, and ICG-001 after a 24 h incubation. (E) Results of FOPFlash luciferase reporter assays of ZW4864 and ICG-001 after a 24 h incubation. (F) Quantitative real-time polymerase chain reaction (qPCR) to examine changes of mRNA expression of AXIN2, cyclin D1, LEF1, and BCL9L when treated with various concentrations of ZW4864 (24 h incubation). House-keeper gene HPRT was used as the negative control. (G) Western blot to monitor the expression changes of protein Axin2 and cyclin D1 when treated with various concentrations of ZW4864 (24 h incubation). β-Tubulin was used as the internal reference. (H) 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) IC50 (μM) results of ZW4864, the CBP/β-catenin inhibitor ICG-001, and the porcupine inhibitor LGK974 on β-catenin-hyperactive cancer cells and normal breast epithelial cells after a 72 h treatment. (I) Results of β-catenin rescue experiments. The effects of ZW4864 on growth inhibition of TNBC cells after a 72 h treatment were determined by MTS assays. The vector with constitutively active β-catenin or an empty vector was transfected in MDA-MB-231 and MDA-MB-468 cells. (J) Effects of ZW4864 on the clonogenic growth of TNBC cells. (K) The percent of apoptosis and (L) the effects on apoptosis by fluorescein isothiocyanate (FITC) annexin V/propidium iodide (PI) fluorescence-activated cell sorting (FACS) after 72 h treatment with the indicated doses of ZW4864. Each experiment was performed in triplicate. Each set of quantitative data was expressed as mean ± standard deviation (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001 determined by the unpaired, two-tailed Student’s t test. Uncropped Western blot gels and dose–response curves are provided in Figures S8–S15. The original FACS reports are included in the Supporting Information.
Wnt-specific TOPFlash/FOPFlash luciferase reporter assays were performed to evaluate effects of ZW4864 on β-catenin-dependent transcriptional activity using three cell lines, HEK293 cells transfected with pcDNA3.1–β-catenin to activate β-catenin signaling, colorectal cancer SW480 cells, and Wnt 3a-stimulated triple-negative breast cancer (TNBC) MDA-MB-468 cells. The firefly luciferase reporter gene was placed downstream of three wild-type TCF binding sites in the TOPFlash reporter construct and of three mutant TCF binding sites in the FOPFlash reporter construct. The firefly luciferase expression in TOPFlash assays is controlled by three tandem TCF binding sites. The Wnt/β-catenin signaling independent renilla luciferase reporter (pCMV-RL) was applied as the internal control to eliminate systematic errors, such as cell viability and transfection effects, and normalize luciferase reporter signals. As shown in Figure 3D, ZW4864 suppressed TOPFlash luciferase activities in β-catenin-expressing HEK293 cells in a dose-dependent manner with an IC50 of 11 μM. The negative control, compound 19, did not show any activity at 200 μM. ZW4864 also dose-dependently suppressed the TOPFlash luciferase activities in SW480 and Wnt 3a-activated MDA-MB-468 cells with the IC50s of 7.0 and 6.3 μM, respectively. The inhibitory activities of ZW4864 in TOPFlash luciferase reporter assays were comparable with that of ICG-001, a compound that was reported to bind CBP and disrupt CBP/β-catenin PPI48 (the IC50s of ICG-001 were 4.9 and 11 μM for SW480 and Wnt 3a-activated MDA-MB-468 cells, respectively). FOPFlash luciferase reporter assays revealed that ZW4864 did not show obvious inhibitory activity up to 25 μM in all these three cell lines (Figure 3E), indicating that ZW4864 selectively suppresses transactivation of β-catenin signaling.
Effects of ZW4864 on the transcription of Wnt-specific target gene Axin2 and other Wnt target genes CCND1 (cyclin D1 gene), LEF1, and BCL9L were assessed. Quantitative realtime PCR (qPCR) experiments showed that ZW4864 suppressed the transcription of these β-catenin target genes in a concentration-dependent manner without affecting the expression of HPRT, a house-keeper gene, in both SW480 and Wnt 3a-activated MDA-MB-231 cells (Figure 3F). Consistent with the results of qPCR studies, Western blot experiments showed that the expression levels of proteins Axin2 and cyclin D1 were also substantially decreased after ZW4864 treatment without affecting the internal control β-tubulin in SW480 and Wnt 3a-activated MDA-MB-231 cells (Figure 3G).
Figure 3H shows the IC50 values of the 3-day MTS cell growth inhibition assay results of ZW4864 and control compounds. ZW4864 inhibited the growth of TNBC cells with hyperactive β-catenin signaling, while transfection of constitutively active β-catenin to these TNBC cells rescued the inhibitory effects of ZW4864 (Figure 3I), demonstrating on-target effects of ZW4864.49 ZW4846 also inhibited colony formation in TNBC cells (Figure 3J). Fluorescence-activated cell sorting (FACS) analyses revealed that ZW4864 treatment selectively triggered rapid apoptosis of TNBC cells with hyperactive β-catenin signaling while sparing normal mammary epithelial MCF10A cells, as shown in Figure 3K,L.
Drug Metabolism and Pharmacokinetic (DMPK) Properties of ZW4864.
ZW4864 (15) displays great aqueous solubility >3 mM, and ZW4864 at 2 mg/mL was completely soluble in saline (0.9% NaCl), EtOH/saline (5/95), or DMSO/Tween-80/H2O (10/10/80). Hepatic microsomal stability of ZW4864 and 21 was evaluated. As shown in Table 2A, these two compounds have moderate microsomal stability similar to the marketed drug, sunitinib. ZW4864 was then advanced for PK studies using C57BL/6 mice. As shown in Table 2B, ZW4864 exhibited good PK properties with an oral bioavailability (F) of 83%.
Table 2.
Results of DMPK Studies: (A) Hepatic Microsome Stability of ZW4864 (15), 21, and Positive Control Sunitinib; and (B) Mouse PK Data of ZW4864a
| (A) | ||||
|---|---|---|---|---|
|
t1/2 (min) |
CLint (μM/(min mg)) |
|||
| cmpd. | human | mouse | human | mouse |
| sunitinib | 27.8 | 13.0 | 25 | 53 |
| ZW4864 (15) | 38.4 | 9.6 | 18 | 72 |
| 21 | 28.3 | 7.7 | 24 | 90 |
| (B) | ||||||||
|---|---|---|---|---|---|---|---|---|
| cmpd. | route | dose (mg/kg) | Cmax (μM) | AUC (μM h) | Vd (L/kg) | CL (mL/(min kg)) | t1/2 (h) | %F |
| ZW4864 | i.v. | 1 | 0.79 | 0.71 | 8.0 | 44.20 | 3.51 | |
| ZW4864 | p.o. | 20 | 3.21 | 11.71 | 51.10 | 3.07 | 83 | |
The original data plots of PK studies are shown in Figure S16
Inhibitory Effects of ZW4864 on Migration and Invasion of Cancer Cells Induced by β-Catenin Signaling.
β-Catenin signaling plays a key role in inducing and maintaining invasion and metastasis of cancer cells including TNBC cells. β-Catenin signaling activates the expression of the EMT-promoting genes, such as Twist50 and Snail,51,52 to induce and maintain breast cells in the mesenchymal and stem cell states53 and cause breast cancer cell invasion and metastasis.54,55 β-Catenin signaling also upregulates the expression of metastasis-associated genes such as tenascin C.56,57 TNBC cells express tenascin C as a metastatic niche component to establish lung metastasis.57 LGR5 is a marker of adult stem cells and the target gene of β-catenin signaling.58,59 It maintains stem-like properties of cancer cells including TNBC cells.60–62 As shown in Figure 4A, disruption of β-catenin/BCL9 interaction with ZW4864 dramatically suppressed the expression of β-catenin target genes that are associated with TNBC metastasis and EMT. ZW4864 suppressed MDA-MB-231 cell migration in a dose-dependent manner in scratch wound healing assays (Figure 4B). As shown in Figure 4C, ZW4864 at 20 μM reduced the Matrigel-coated transwell invasion of MDA-MB-231 cells to 13% of the DMSO-treated control. Figure 4D shows the 96-well three-dimensional (3D) spheroid cell invasion assay of ZW4864. The use of basement membrane extracts (BMEs) resulted in a larger invasion area of MDA-MB-231 cells on day 6. The use of ZW4864 suppressed MDA-MB-231 cell invasion in a dose-dependent manner.
Figure 4.

(A) Changes of mRNA expression of Twist, Snail, tenascin C, and LGR5 were determined by qPCR studies when SW480 and Wnt 3a-activated MDA-MB-231 cells were treated with various concentrations of ZW4864. House-keeper gene HPRT was used as the negative control. (B) Scratch wound healing assays demonstrated that ZW4864 suppressed the migration of TNBC MDA-MB-231 cells that was induced by serum (10% in media). Mitomycin (10 μg/mL) was used to suppress cell proliferation, which allows examination of the effects on cell migration. Vehicle control: 0.2% DMSO in 10% fetal bovine serum (FBS). (C) Matrigel invasion assays showed that ZW4864 (20 μM) impeded the invasion of MDA-MB-231 cells. Control, 0.2% DMSO in 10% FBS. (D) Cultrex 3D spheroid BME cell invasion assay results of ZW4864 on day 6 using MDA-MB-231 cells. Left panel, representative images. Right panel, area of invasion when compared with that of the control spheroids. Each experiment was performed in triplicate. *p < 0.05, **p < 0.01, and ***p < 0.001 determined by the unpaired, two-tailed Student’s t test. All the original images are shown in Figures S17 and S18.
Pharmacodynamic (PD) Properties of ZW4864 in the Patient-Derived TNBC Xenograft Mouse Model.
To test the in vivo antitumor effects of ZW4864, we used a patient-derived xenograft (PDX) model, PDX 4013, derived from a TNBC patient that had limited response to treatment with dasatinib and docetaxel.63 The 4013 cells were implanted into the mammary fat pad of immunocompromised SCID/Beige mice. The hyperactive β-catenin signaling circuit drives the metastatic cascade of this clinically relevant TNBC model.64 We tested 90 mg/kg dose given orally in saline daily to mice with PDX 4013 tumors (~200 mm3, n = 4) grown orthotopically for 5 days. The tumor tissue was collected 3 h after the last dose to assess the PD properties of the compound. The results showed a variation in tumor growth in mice that were treated with ZW4864 compared to those treated only with vehicle, while no significant decrease in weight or major toxicity issues were observed over the treatment (Figure 5A,B). Moreover, we observed significant decreases in the expression of key β-catenin target genes in tumors treated with ZW4864 vs vehicle-treated tumors (Figure 5C). Collectively, these data support on-target effects and the promising antitumor effect of ZW4864 and grant development of specific antitumor efficacy studies with our β-catenin inhibitors.
Figure 5.

(A) Weight analysis on mice treated with ZW4864 at 90 mg/kg or saline normalized against their weight before treatment. (B) Percent change in tumor size in the 5 day course of treatment as indicated. (C) qRT-PCR analyses of the expression of the indicated genes in tumors from mice treated with ZW4864 at 90 mg/kg or vehicle performed in triplicates. All data are presented as mean values ± SD (n = 4). *p < 0.05, **p < 0.01 ***p < 0.001 by t-test.
Chemistry.
Scheme 1 shows the synthetic route for 1–2. The key intermediate 23 was obtained by the nucleophilic substitution reaction between 3-bromophenol and tert-butyl 2-bromo-2-methylpropanoate. The amide bond coupling reaction between (R)- or (S)-1-(tert-butoxycarbonyl)piperidine-3-carboxylic acid and (4-isopropylphenyl)methanamine generated intermediates 24a and 24b, which underwent Boc deprotection and Buchwald–Hartwig amination reactions with 23 to yield 25a and 25b. Removal of the tert-butyl group of 25a and 25b by TFA in CH2Cl2 solution offered final products 1 and 2.
Scheme 1.

Synthetic Route of 1 and 2
Scheme 2 shows the synthetic route for 3–7. The amide bond coupling between (R)-1-(tert-butoxycarbonyl)piperidine-3-carboxylic acid and (4-isopropylphenyl)methanamine derivatives offered intermediates 24a and 26. Compounds 24a and 26 went through Boc deprotection and Buchwald–Hartwig amination reactions with 23 to yield 25a and 27. Removal of the tert-butyl groups in 25a and 27 by TFA in CH2Cl2 solution and then condensation with tert-butyl piperazine-1-carboxylate provided 28, Boc deprotection of which under the acidic condition offered final products 3–7.
Scheme 2.

Synthesis of 3–7
The synthetic route for 8–15, 17, and 18 is illustrated in Scheme 3. The Buchwald–Hartwig amination reaction between compound 23 and ethyl (R)-piperidine-3-carboxylate produced intermediate 29. Removal of the tert-butyl group of 29 and then coupling with tert-butyl piperazine-1-carboxylate produced 30. Hydrolysis of 30 and condensation with N-benzylcyclopropanamine derivatives yielded 31, which went through the Boc deprotection reaction to afford final compounds 8–10 and 12–14.
Scheme 3.

Synthesis of 8–15, 17, and 18
Hydrolysis of 30 and then condensation with N-(4-bromobenzyl)cyclopropanamine yielded 32, which went through the Suzuki coupling reaction with various boronic acids to afford 33. Deprotection of the Boc protecting group of 33 produced final products 11, 15, 17, and 18.
The route for synthesis of 19 and 20 is described in Scheme 4. Removal of the tert-butyl in 29 and then condensation with 1-methylpiperazine or piperidine produced intermediate 34. Hydrolysis of 34 and then condensation with N-(4-bromobenzyl)cyclopropanamine yielded 35, which underwent the Suzuki coupling reaction with (1H-pyrazol-4-yl)boronic acid to afford final products 19 and 20.
Scheme 4.

Synthesis of 19 and 20
The route for synthesis of 21 and 22 is shown in Scheme 5. Hydrolysis of 30 and then condensation with (4-bromophenyl)methanamine derivatives yielded 36, which was coupled with (1H-pyrazol-4-yl)boronic acid by the Suzuki reaction to produce intermediate 37. Removal of the Boc protecting group of 37 under the acidic condition afforded final compounds 21 and 22.
Scheme 5.

Synthesis of 21 and 22
The route for the synthesis of 16 is described in Scheme 6. The Buchwald–Hartwig amination reaction between 23 and ethyl (S)-piperidine-3-carboxylate produced 38. Removal of tert-butyl of 38 and then condensation with tert-butyl piperazine-1-carboxylate produced 39. Hydrolysis of 39 and condensation with N-benzylcyclopropanamine derivatives yielded 40, which went through Suzuki coupling with (1-pyrazol-4-yl)boronic acid and the Boc deprotection reaction to afford the final compound 16.
Scheme 6.

Synthesis of 16
Scheme 7 shows the synthetic route for Biotin-ZW4864. The reductive-amination reaction between 4-bromobenzaldehyde and benzyl (2-(2-(2-aminoethoxy)ethoxy)ethyl)carbamate produced intermediate 42, which was condensed with (R)-1-(3-((1-(4-(tert-butoxycarbonyl)piperazin-1-yl)-2-methyl-1-oxopropan-2-yl)oxy)phenyl)piperidine-3-carboxylic acid to yield 43. Compound 43 was coupled with (1-trityl-1H-pyrazol-4-yl)boronic acid using the Suzuki reaction to give 44, which went through the Cbz deprotection and then the coupling reaction with Biotin-N-hydroxysuccinimide (Biotin-NHS) to yield 45. Deprotection of Boc and trityl protecting groups of 45 under the acidic condition and then putting back the Boc protecting group resulted in 46, which afforded the final product after applying 10% TFA in CH2Cl2.
Scheme 7.

Synthesis of Biotin-ZW4864
DISCUSSION AND CONCLUSIONS
There is compelling evidence that dysregulation of the β-catenin signaling circuit plays a key role in initiation and progression of many cancers. However, development of small-molecule PPI disruptors that directly bind with β-catenin has been highly challenging. Herein, we described ZW4864, a small molecule that selectively disrupts the β-catenin/BCL9 PPI without interfering the β-catenin/E-cadherin PPI. Using pull-down assays, co-IP experiments, and β-catenin rescue experiments, we provide evidence that ZW4864 binds directly with β-catenin and selectively disrupts the β-catenin/BCL9 PPI in both the protein level and the cellular context. ZW4864 also suppresses transactivation of β-catenin signaling and downregulates the transcription and expression of oncogenic β-catenin target genes in vitro and in vivo in a dose-dependent manner. This compound selectively suppresses growth and promotes apoptosis of cancer cells with hyperactive Wnt/β-catenin signaling. It is worth noting that the only other β-catenin/BCL9 inhibitor that has been characterized in vivo is the natural product carnosic acid, although its catechol group is known as a substructure of pan-assay interference compounds (PAINS). ZW4864 is orally bioavailable with high tolerability in mice. In a therapy-resistant TNBC PDX model, ZW4864 showed promising therapeutic effects. Importantly, ZW4864 displayed on-target activity in vivo in our 5 day assay and in the long efficacy experiment, as observed in the reduced expression of β-catenin target genes. Therefore, ZW4864 represents a better chemical probe to explore β-catenin-related biology and a drug-like small-molecule β-catenin/BCL9 inhibitor for further drug development.
It is worth noting that the N-terminal disordered region of β-catenin (residues 1–137) was found to be important for ZW4864 binding. Biotin-ZW4864 pulled down the full-length β-catenin at 1 μM, and ZW4864 displayed a Ki of 0.76 μM in the full-length β-catenin/BCL9 AlphaScreen assay. However, Biotin-ZW4864 was not able to pull down β-catenin R1C (residues 138–781) even at 10 μM, and ZW4864 showed an increased Ki of 8.9 μM to disrupt β-catenin R1C/BCL9 PPI in AlphaScreen assays. These data indicate that full-length β-catenin should be taken into consideration when developing β-catenin/BCL9 inhibitors or biochemical assays to evaluate β-catenin/BCL9 inhibitors.
EXPERIMENTAL SECTION
Chemical Synthesis.
All reagents were purchased from commercial sources and used as received. 1H NMR and 13C NMR spectra were recorded on Bruker AVANCE III HD 500 (500 MHz) spectrometers (125.7 MHz for 13C NMR spectra) in CDCl3, DMSO-d6, acetone-d6, and CD3OD. Chemical shifts were reported as values in parts per million (ppm), and the reference resonance peaks were set at 7.26 ppm (CHCl3), 3.31 ppm (CD2HOD), 2.50 ppm [(CD2H)2SO], and 2.05 ppm [(CD2H)2CO] for 1H NMR spectra and 77.23 ppm (CDCl3), 49.00 ppm (CD3OD), 39.52 ppm (DMSO-d6), and 29.84 ppm (acetone-d6) for 13C NMR spectra. Low-resolution mass spectra were determined on an Agilent 6120 single quadrupole MS with a 1220 infinity LC system (high-performance liquid chromatography-mass spectrometry (HPLC-MS)) and an electrospray ionization (ESI) source. High-resolution mass spectra were determined on an Agilent G6230BA TOF LC-MS Mass Spectrometer with a TOF mass detector. Thin-layer chromatography was carried out on E. Merck precoated silica gel 60 F254 plates with a UV–visible lamp. Column chromatography was performed with SilicaFlash@F60 (230–400 mesh). The purity of the final compounds was determined by HPLC analyses. The instrument was an Agilent 1260 Infinity II HPLC system with a quaternary pump, a vial sampler, and a DAD detector. A Kromasil 300–5-C18 column (4.6 × 250 mm2) was used. The DAD detector was set to 254 nm. The purity of all tested compounds was >95%.
General Procedure for Amide Bond Coupling Reaction.
At 0 °C, to a suspension of carboxylic acid (1 mmol), amine (1.1 mmol), and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) (1.5 mmol) in dichloromethane (CH2Cl2) (10 mL) was added N,N-diisopropylethylamine (DIPEA) (2 mmol) dropwise. The reaction mixture was warmed to room temperature and stirred overnight. After completion of the reaction, more CH2Cl2 was added. The organic phase was washed with 1 M HCl, saturated NaHCO3, and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. Column chromatography was used to purify the target compound.
General Procedure for Suzuki Coupling Reaction.
To a solution of bromobenzene derivatives (1 mmol) in dioxane/water (12/4 mL) were added boronic acid (1.2 mmol), Pd(PPh3)4 (0.1 mmol), and K2CO3 (2 mmol). The reaction mixture was heated to 90 °C under argon and stirred overnight. The reaction mixture was cooled to room temperature, concentrated under reduced pressure, then redissolved with ethyl acetate, washed with water and brine, and dried over Na2SO4. The solution obtained was concentrated under vacuum. The residue was purified by column chromatography to yield the target compound.
General Procedure for Buchwald–Hartwig Amination Reaction.
A solution of bromobenzene derivatives (1 mmol), amine (1.1 mmol), Pd2(dba)3 (0.1 mmol), Ruphos (0.2 mmol), and Cs2CO3 (2 mmol) in dioxane (10 mL) was heated to 80 °C under argon and stirred overnight. The reaction mixture was cooled to room temperature. The solid was removed, and the filtrate was concentrated under reduced pressure and purified by column chromatography to yield the target compound.
General Procedure for Deprotection of Boc Using 4 M HCl in Dioxane.
At 0 °C, 4 M HCl (8 mL) in dioxane was added to Boc-protected amine (1 mmol) in a 20 mL vial. The reaction was kept at 0 °C for 2 h. Upon completion, the solvent was removed under reduced pressure. Dioxane residue was removed by evaporation with additional methanol twice. Then, water (HPLC grade) was added and dried over a Labconco lyophilizer.
General Procedure for Deprotection of Boc Using 10% Trifluoroacetic Acid (TFA) in CH2Cl2.
To Boc-protected amine (1 mmol) in 10 mL of CH2Cl2 at room temperature was added TFA (1 mL). The reaction was stirred at room temperature for 6 h. TFA was removed by adding CH2Cl2 three times to afford the desired product. Then, water (HPLC grade) was added and dried over a Labconco lyophilizer.
General Procedure for Deprotection of tert-Butyl and Boc Using 50% TFA in CH2Cl2.
At 0 °C, to a solution of the tert-butyl ester or Boc-protected amine (1 mmol) in CH2Cl2 (5 mL) was added TFA (5 mL) dropwise. The reaction was stirred at 0 °C for 6 h. Upon completion, the solvent was removed under reduced pressure. TFA was removed by adding CH2Cl2 three times to afford the desired product, which was used directly in the next step.
General Procedure for Hydrolysis of Ethyl Ester.
To a solution of ethyl ester (1 mmol) in THF (4 mL) and methanol (2 mL) was added a solution of LiOH (2 mmol) in H2O (1 mL). The mixture was stirred at room temperature for 3 h. After completion of the reaction, the solvent was removed under reduced pressure. The residue was redissolved in H2O and acidified using 1 M HCl. Ethyl acetate was added, and the organic phase was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to yield the target compound, which was used directly in the next step.
General Procedure for Reductive-Amination Reaction.
A solution of aldehyde (1 mmol) and amine (2 mmol) in anhydrous methanol (5 mL) was stirred under Ar at room temperature overnight. Then, NaBH4 (1.5 mmol) was added portionwise to the mixture at 0 °C. The mixture was stirred at 0 °C for 1 h. Saturated NH4Cl was added, followed by addition of ethyl acetate. The organic phase was collected and washed with brine, dried over anhydrous Na2SO4, concentrated under reduced pressure, and purified by column chromatography to yield the target compound.
General Procedure for Boc-Protection Reaction.
To a solution of amine (1 mmol) in CH2Cl2 (10 mL) were added (Boc)2O (1.5 mmol) and triethylamine (Et3N) (2 mmol). The mixture was stirred at room temperature for 2 h. After completion of the reaction, the mixture was concentrated under reduced pressure and purified by column chromatography to yield the target compound.
General Procedure for Deprotection of the Cbz Group.
To the solution of the Cbz-protected amine (1 mmol) in methanol (10 mL) was added 10% Pd/C under Ar. The mixture was stirred overnight at room temperature under H2. The resulting product was collected by removal of the Pd/C catalyst and used directly in the next step.
General Procedure for Reaction between Amine and (+)-Biotin-N-Hydroxysuccinimide Ester (NHS-Biotin).
To a solution of amine (1 mmol) in dimethylformamide (DMF) (10 mL) were added NHS-Biotin (1 mmol) and DIPEA (2 mmol) under Ar. The mixture was stirred overnight. After completion of the reaction, DMF was evaporated and water and ethyl acetate were added. The organic phase was collected and washed with brine, dried over anhydrous Na2SO4, and purified by column chromatography to yield the target compound.
Synthesis of tert-Butyl 2-(3-Bromophenoxy)-2-methylpropanoate.
A solution of 3-bromophenol (1 mmol), tert-butyl 2-bromo-2-methylpropanoate (5 mmol), K2CO3 (4 mmol), and MgSO4 (1 mmol) in DMF was stirred at 100 °C overnight. The DMF was removed under reduced pressure. Water and ethyl acetate were added. The obtained organic phase was washed with brine, dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography to yield the target compound.
(R)-2-(3-(3-((4-Isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoic Acid (1).
1H NMR (500 MHz, chloroform-d) δ 7.17 (d, J = 1.5 Hz, 4H), 7.08 (t, J = 8.3 Hz, 1H), 6.91 (t, J = 5.8 Hz, 1H), 6.63 (d, J = 5.8 Hz, 2H), 6.50 (d, J = 8.1 Hz, 1H), 4.56−4.28 (m, 2H), 3.50−3.33 (m, 1H), 3.28 (dt, J = 10.3, 4.4 Hz, 1H), 3.19 (dd, J = 12.4, 8.6 Hz, 1H), 3.08−2.92 (m, 1H), 2.94−2.78 (m, 1H), 2.65 (dq, J = 9.5, 4.8, 4.3 Hz, 1H), 1.90−1.65 (m, 4H), 1.57 (d, J = 2.7 Hz, 6H), 1.22 (dd, J = 6.9, 1.5 Hz, 6H). 13C NMR (126 MHz, chloroform-d) δ 176.6, 173.9, 156.0, 148.2, 135.4, 129.7, 127.7, 126.8, 113.1, 112.1, 109.6, 79.7, 53.4, 51.4, 43.2, 41.9, 33.8, 26.9, 25.20, 25.16, 24.0, 23.1. HRMS (ESI) calcd for C26H34N2O4 (M − H)− 437.2446, found 437.2443. HPLC purity 98.6%, tR = 12.48 min.
(S)-2-(3-(3-((4-Isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoic Acid (2).
1H NMR (500 MHz, methanol-d4) δ 7.19 (d, J = 1.0 Hz, 4H), 7.09 (t, J = 8.2 Hz, 1H), 6.68−6.62 (m, 1H), 6.53 (t, J = 2.3 Hz, 1H), 6.38 (dd, J = 8.1, 2.2 Hz, 1H), 4.48−4.23 (m, 2H), 3.62 (ddt, J = 12.2, 3.5, 1.6 Hz, 1H), 3.53 (ddt, J = 12.5, 3.7, 2.1 Hz, 1H), 3.02−2.82 (m, 2H), 2.81−2.71 (m, 1H), 2.59 (ddd, J = 10.5, 6.7, 3.7 Hz, 1H), 2.00−1.90 (m, 1H), 1.80 (qd, J = 7.2, 3.4 Hz, 1H), 1.73−1.62 (m, 2H), 1.54 (s, 6H), 1.22 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 175.1, 156.4, 152.5, 147.7, 136.0, 129.0, 127.2, 126.1, 111.0, 110.6, 108.4, 78.8, 52.6, 50.0, 42.6, 42.3, 33.7, 27.4, 24.42, 24.37, 23.8, 23.1. HRMS (ESI) calcd for C26H34N2O4 (M − H)− 437.2446, found 437.2443. HPLC purity 98.9%, tR = 12.51 min.
(R)-N-(4-Isopropylbenzyl)-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (3 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.52 (t, J = 8.3 Hz, 1H), 7.43−7.33 (m, 1H), 7.32−7.12 (m, 5H), 6.99 (dd, J = 8.4, 2.2 Hz, 1H), 4.37 (q, J = 14.8 Hz, 2H), 4.02 (d, J = 117.9 Hz, 4H), 3.71(td, J = 11.0, 10.0, 5.5 Hz, 3H), 3.55 (d, J = 8.1 Hz, 1H), 3.25−2.82 (m, 6H), 2.13 (q, J = 12.4, 11.3 Hz, 3H), 1.92 (s, 1H), 1.68 (d, J = 1.0 Hz, 6H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 147.9, 143.9, 135.7, 131.4, 127.3, 126.2, 117.8, 114.2, 111.1, 81.7, 57.5, 43.1, 42.5, 39.6, 33.7, 25.3, 24.9, 24.7, 23.0, 21.9. HRMS (ESI) calcd for C30H42N4O3 (M + Na)+ 529.3149, found 529.3140. HPLC purity 97.6%, tR = 10.96 min.
(R)-N-(4-Isopropylbenzyl)-N-methyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (4 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.47 (dt, J = 25.8, 8.2 Hz, 1H), 7.38−7.01 (m, 6H), 6.90 (d, J = 30.1 Hz, 1H), 4.75−4.50 (m, 2H), 4.02 (d, J = 122.4 Hz, 4H), 3.78−3.41 (m, 5H), 3.25−2.87 (m, 8H), 2.20−1.79 (m, 4H), 1.70−1.65 (m, 6H), 1.24 (dd, J = 6.9, 3.5 Hz, 6H). 13C NMR (126 MHz, methanol-d4) δ 171.7, 171.6, 156.3, 148.5, 148.2, 134.1, 131.3, 127.6, 126.7, 126.5, 126.4, 81.6, 52.7, 50.3, 43.1, 34.1, 33.7 (d, J = 2.2 Hz), 33.2, 24.9, 24.8, 24.7, 23.01, 23.00. HRMS (ESI) calcd for C31H44N4O3 (M + H)+ 521.3486, found 521.3478. HPLC purity 100%, tR = 11.37 min.
(R)-N-Ethyl-N-(4-isopropylbenzyl)-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (5 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.48 (dt, J = 17.8, 8.1 Hz, 1H), 7.40−7.08 (m, 6H), 6.91 (s, 1H), 4.79−4.36 (m, 2H), 4.03 (d, J = 121.8 Hz, 4H), 3.78−3.34 (m, 7H), 3.27−2.79 (m, 5H), 2.30−1.74 (m, 4H), 1.68 (d, J = 3.3 Hz, 6H), 1.30−1.11 (m, 9H). 13C NMR (126 MHz, methanol-d4) δ 171.62, 171.61, 156.3, 148.5, 148.0, 134.6, 134.1, 131.3, 131.2, 127.4, 126.6, 126.6, 126.3, 113.9, 81.6, 81.5, 54.8, 50.3, 43.1, 42.7, 41.9, 41.2, 39.6, 33.7, 24.9, 24.80, 24.79, 23.04, 23.03, 21.9, 13.1, 11.4. HRMS (ESI) calcd for C32H46N4O3 (M + H)+ 535.3643, found 535.3640. HPLC purity 98.0%, tR = 11.55 min.
(R)-N-Isopropyl-N-(4-isopropylbenzyl)-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (6 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.46 (dt, J = 35.6, 8.2 Hz, 1H), 7.21 (d, J = 133.2 Hz, 6H), 7.00−6.79 (m, 1H), 4.73−4.57 (m, 2H), 4.50−4.35 (m, 1H), 4.03 (d, J = 124.8 Hz, 4H), 3.81−3.32 (m, 5H), 3.22−2.86 (m, 5H), 2.25−1.77 (m, 4H), 1.67 (t, J = 1.6 Hz, 6H), 1.27−1.13 (m, 12H). 13C NMR (126 MHz, methanol-d4) δ 171.7, 171.6, 156.3, 156.2, 148.1, 147.2, 136.2, 135.5, 131.3, 131.1, 126.6, 126.3, 126.0, 114.0, 113.4, 110.8, 81.6, 81.4, 54.8, 49.3, 46.2, 43.3, 43.1, 42.7, 39.5, 33.64, 33.62, 24.9, 24.83, 24.77, 23.10, 23.06, 20.6, 20.2, 19.1, 18.9. HRMS (ESI) calcd for C33H48N4O3 (M + H)+ 549.3799, found 549.3790. HPLC purity 100%, tR = 12.33 min.
(R)-N-Cyclopropyl-N-(4-isopropylbenzyl)-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (7 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.45 (d, J = 8.4 Hz, 1H), 7.34−7.02 (m, 6H), 6.87 (s, 1H), 4.47−4.38 (m, 1H), 4.21−3.84 (m, 5H), 3.76−3.38 (m, 5H), 3.26−2.67 (m, 6H), 2.22−1.73 (m, 4H), 1.67 (d, J = 2.4 Hz, 6H), 1.23 (d, J = 6.9 Hz, 6H), 1.09−0.64 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.71, 156.26, 147.81, 135.17, 131.09, 127.16, 126.63, 126.24, 81.45, 49.34, 43.13, 33.67, 29.81, 24.90, 24.78, 23.04, 9.17, 7.36. HRMS (ESI) calcd for C33H46N4O3 (M + H)+ 547.3643, found 547.3646. HPLC purity 100%, tR = 12.15 min.
(R)-N-(4-(tert-Butyl)benzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (8 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.54 (t, J = 8.3 Hz, 1H), 7.47−7.33 (m, 3H), 7.29 (s, 1H), 7.19 (d, J = 7.9 Hz, 2H), 7.00 (dd, J = 8.4, 2.3 Hz, 1H), 4.89 (s, 1H), 4.40 (d, J = 14.8 Hz, 1H), 4.02 (d, J = 116.9 Hz, 5H), 3.80−3.57 (m, 4H), 3.07 (d, J = 107.6 Hz, 4H), 2.84−2.74 (m, 1H), 2.38−1.79 (m, 4H), 1.68 (d, J = 2.7 Hz, 6H), 1.30 (s, 9H), 1.12−0.78 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 150.0, 143.9, 134.7, 131.4, 126.9, 125.1, 118.0, 114.2, 111.3, 81.7, 55.5, 49.3, 43.1, 42.6, 39.6, 33.9, 30.4, 29.8, 24.9, 24.7, 9.3, 7.2. HRMS (ESI) calcd for C34H48N4O3 (M + H)+ 561.3799, found 561.3791. HPLC purity 100%, tR = 12.49 min.
(R)-N-(4-Cyclohexylbenzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (9 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.46 (t, J = 8.2 Hz, 1H), 7.24 (d, J = 9.8 Hz, 1H), 7.17 (s, 5H), 6.87 (s, 1H), 4.80 (s, 1H), 4.42 (d, J = 14.8 Hz, 1H), 4.25−3.83 (m, 5H), 3.73 (dd, J = 12.2, 3.6 Hz, 2H), 3.68−3.40 (m, 2H), 3.24−2.88 (m, 4H), 2.82−2.73 (m, 1H), 2.49 (ddt, J = 11.8, 8.5, 3.2 Hz, 1H), 2.21−2.01 (m, 3H), 1.89−1.72 (m, 6H), 1.67 (d, J = 2.5 Hz, 6H), 1.47−1.39 (m, 4H), 1.34−1.25 (m, 1H), 1.09−0.84 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.7, 156.3, 147.0, 135.2, 131.1, 127.1, 126.7, 113.6, 81.5, 49.4, 44.3, 43.1, 42.7, 34.3, 29.8, 26.6, 25.8, 24.9, 24.8, 9.2, 7.4. HRMS (ESI) calcd for C36H50N4O3 (M + H)+ 587.3956, found 587.3948. HPLC purity 99.4%, tR = 13.42 min.
(R)-N-([1,1′-Biphenyl]-4-ylmethyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (10 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.64−7.55 (m, 4H), 7.43 (dd, J = 8.3, 7.1 Hz, 3H), 7.39−6.62 (m, 6H), 4.88 (d, J = 11.1 Hz, 1H), 4.55 (d, J = 14.9 Hz, 1H), 4.28−3.79 (m, 5H), 3.74 (dt, J = 11.1, 5.4 Hz, 2H), 3.68−3.35 (m, 2H), 3.23−2.82 (m, 5H), 2.23−1.79 (m, 4H), 1.67 (d, J = 2.6 Hz, 6H), 1.12−0.79 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.8, 156.3, 140.6, 140.1, 137.0, 131.0, 128.5, 127.7, 127.0, 126.8, 126.5, 81.4, 49.4, 43.1, 42.7, 39.6, 29.9, 24.9, 24.8, 9.2, 7.5. HRMS (ESI) calcd for C36H44N4O3 (M + Na)+ 603.3306, found 603.3293. HPLC purity 100%, tR = 12.29 min.
(R)-N-Cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)-N-(4-(thiophen-3-yl)benzyl)piperidine-3-carboxamide (11 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 7.68−7.55 (m, 3H), 7.51−7.41 (m, 3H), 7.30 (d, J = 7.9 Hz, 3H), 7.00 (d, J = 127.8 Hz, 2H), 4.87 (d, J = 12.4 Hz, 1H), 4.50 (d, J = 14.9 Hz, 1H), 4.20−3.82 (m, 5H), 3.74 (ddd, J = 12.4, 8.4, 4.0 Hz, 2H), 3.68−3.40 (m, 2H), 3.20−2.81 (m, 5H), 2.20−1.86 (m, 4H), 1.67 (d, J = 2.6 Hz, 6H), 1.10−0.88 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.7, 156.3, 141.7, 136.6, 134.9, 131.1, 127.7, 126.1, 126.0, 125.6, 119.9, 81.5, 49.4, 43.1, 42.7, 39.5, 29.9, 24.9, 24.8, 9.2, 7.4. HRMS (ESI) calcd for C34H42N4O3S (M + Na)+ 609.2870, found 609.2861. HPLC purity 99.4%, tR = 11.90 min.
(R)-N-Cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)-N-(4-(pyridin-2-yl)benzyl)piperidine-3-carboxamide (12 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.84 (dd, J = 6.0, 1.5 Hz, 1H), 8.68 (td, J = 8.0, 1.6 Hz, 1H), 8.41 (d, J = 8.2 Hz, 1H), 8.07−8.03 (m, 1H), 7.97 (d, J = 8.2 Hz, 2H), 7.61−7.50 (m, 3H), 7.50−7.27 (m, 2H), 7.02 (dd, J = 8.4, 2.3 Hz, 1H), 4.93 (s, 1H), 4.68 (d, J = 15.6 Hz, 1H), 4.36−3.85 (m, 5H), 3.84−3.66 (m, 4H), 3.27−2.88 (m, 5H), 2.36−1.83 (m, 4H), 1.68 (d, J = 2.0 Hz, 6H), 1.13−0.88 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 152.4, 146.9, 143.6, 143.2, 141.6, 131.5, 129.7, 128.4, 128.2, 125.9, 125.2, 118.3, 114.4, 111.5, 81.8, 57.8, 55.8, 49.7, 43.1, 42.6, 39.6, 30.3, 24.9, 24.7, 9.2, 7.3. HRMS (ESI) calcd for C35H43N5O3 (M + H)+ 582.3439, found 582.3432. HPLC purity 97.1%, tR = 8.05 min.
(R)-N-Cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)-N-(4-(pyridin-3-yl)benzyl)piperidine-3-carboxamide (13 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 9.20 (d, J = 2.1 Hz, 1H), 8.94 (dt, J = 8.3, 1.8 Hz, 1H), 8.84 (dt, J = 5.6, 1.0 Hz, 1H), 8.19 (dd, J = 8.3, 5.7 Hz, 1H), 7.91−7.75 (m, 2H), 7.65−7.23 (m, 5H), 7.03 (dd, J = 8.4, 2.3 Hz, 1H), 4.92 (d, J =15.8 Hz, 1H), 4.60 (d, J = 15.4 Hz, 1H), 4.35−3.83 (m, 5H), 3.82−3.63 (m, 4H), 3.27−2.73 (m, 5H), 2.43−1.76 (m, 4H), 1.68 (d, J = 2.1 Hz, 6H), 1.15−0.86 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 144.2, 143.5, 140.6, 140.3, 139.5, 139.4, 132.4, 131.5, 128.4, 127.4, 118.4, 114.4, 111.5, 81.8, 49.5, 43.1, 30.2, 24.9, 24.7, 9.2, 7.3. HRMS (ESI) calcd for C35H43N5O3 (M + Na)+ 604.3258, found 604.3249. HPLC purity 99.2%, tR = 8.21 min.
(R)-N-Cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)-N-(4-(pyridin-4-yl)benzyl)piperidine-3-carboxamide (14 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.90−8.83 (m, 2H), 8.45−8.38 (m, 2H), 8.00 (d, J = 8.3 Hz, 2H), 7.64−7.25 (m, 5H), 7.04 (dd, J = 8.4, 2.2 Hz, 1H), 4.93 (d, J = 15.7 Hz, 1H), 4.64 (d, J = 15.5 Hz, 1H), 4.35−3.85 (m, 5H), 3.83−3.65 (m, 4H), 3.27−2.87 (m, 5H), 2.43−1.85 (m, 4H), 1.68 (d, J = 2.0 Hz, 6H), 1.14−0.90 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 157.7, 156.3, 143.5, 142.8, 141.4, 133.2, 131.5, 128.4, 128.1, 124.0, 118.5, 114.4, 111.6, 81.8, 49.6, 43.1, 30.2, 24.9, 24.7, 9.2, 7.3. HRMS (ESI) calcd for C35H43N5O3 (M + Na)+ 604.3258, found 604.3251. HPLC purity 98.8%, tR = 13.07 min.
(R)-N-(4-(1H-Pyrazol-4-yl)benzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (15 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.55 (d, J = 8.3 Hz, 2H), 7.76−7.62 (m, 2H), 7.56 (t, J = 8.3 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 7.9 Hz, 3H), 7.04 (dd, J = 8.4, 2.3 Hz, 1H), 4.90 (s, 1H), 4.50 (d, J = 15.0 Hz, 1H), 4.04 (d, J = 128.1 Hz, 5H), 3.82−3.66 (m, 4H), 3.26−2.73 (m, 5H), 2.35−1.84 (m, 4H), 1.68 (d, J = 2.2 Hz, 6H), 1.13−0.85 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 143.4, 137.8, 131.5, 130.7, 128.6, 128.0, 125.9, 123.8, 118.6, 114.5, 111.6, 81.8, 58.2, 55.9, 49.5, 43.1, 42.7, 39.5, 30.0, 24.9, 24.7, 9.3, 7.2. HRMS (ESI) calcd for C33H42N6O3 (M + Na)+ 593.3211, found 593.3204. HPLC purity 96.8%, tR = 9.35 min.
(S)-N-(4-(1H-Pyrazol-4-yl)benzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (16 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.52 (s, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.55 (t, J = 8.2 Hz, 1H), 7.50 (dd, J = 8.1, 2.1 Hz, 1H), 7.40 (s, 1H), 7.33 (d, J = 7.9 Hz, 2H), 7.03 (dd, J = 8.4, 2.3 Hz, 1H), 4.85 (d, J = 15.1 Hz, 1H), 4.50 (d, J = 15.1 Hz, 1H), 4.36−3.85 (m, 5H), 3.82−3.67 (m, 4H), 3.28−2.75 (m, 5H), 2.40−2.08 (m, 3H), 1.95 (d, J = 12.2 Hz, 1H), 1.68 (d, J = 2.1 Hz, 6H), 1.15−0.84 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.4, 156.3, 143.4, 137.7, 131.5, 130.7, 128.9, 127.9, 125.9, 123.6, 118.5, 114.5, 111.6, 81.8, 58.0, 56.0, 49.5, 43.1, 42.7, 39.6, 37.1, 30.0, 25.0, 24.8, 23.9, 21.9, 9.3, 7.3. calcd for C33H42N6O3 (M + H)+ 571.3391, found 571.3389. HPLC purity 98.8%, tR = 9.34 min.
(R)-N-(4-(1H-Pyrazol-5-yl)benzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (17 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.23 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 8.2 Hz, 2H), 7.64 (ddt, J = 9.9, 6.7, 1.8 Hz, 1H), 7.56 (td, J = 7.8, 7.4, 2.4 Hz, 2H), 7.46 (d, J = 7.9 Hz, 3H), 7.37 (s, 1H), 7.09 (d, J = 2.7 Hz, 1H), 7.04 (dd, J = 8.4, 2.3 Hz, 1H), 4.91 (s, 1H), 4.59 (d, J = 15.4 Hz, 1H), 4.37−3.84 (m, 5H), 3.82−3.63 (m, 4H), 3.26−2.81 (m, 5H), 2.39−1.84 (m, 4H), 1.68 (d, J = 2.1 Hz, 6H), 1.14−0.85 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 147.6, 143.4, 141.0, 134.8, 132.4, 131.7, 131.6, 131.5, 128.6, 128.5, 128.1, 126.7, 125.8, 118.6, 114.5, 111.6, 104.4, 81.8, 55.9, 49.6, 43.1, 42.7, 39.6, 30.2, 24.9, 24.7, 9.3, 7.3. HRMS (ESI) calcd for C33H42N6O3 (M + Na)+ 593.3211, found 593.3203. HPLC purity 99.1%, tR = 9.41 min.
(R)-N-Cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)-N-(4-(1-methyl-1H-pyrazol-4-yl)benzyl)piperidine-3-carboxamide (18 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.11 (s, 1H), 8.00 (d, J = 0.9 Hz, 1H), 7.60−7.50 (m, 3H), 7.48−7.37 (m, 1H), 7.37−7.24 (m, 3H), 7.04 (dd, J = 8.3, 2.3 Hz, 1H), 4.89 (s, 1H), 4.45 (d, J = 14.9 Hz, 1H), 4.23−3.61 (m, 12H), 3.27−2.77 (m, 5H), 2.34−1.79 (m, 4H), 1.68 (d, J = 2.6 Hz, 6H), 1.12−0.86 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 143.4, 136.4, 134.8, 131.5, 130.7, 129.0, 127.8, 125.4, 123.1, 118.6, 114.4, 111.6, 81.8, 49.5, 43.1, 37.5, 29.9, 24.9, 24.7, 9.3, 7.2. HRMS (ESI) calcd for C34H44N6O3 (M + Na)+ 607.3367, found 607.3356. HPLC purity 98.0%, tR = 9.86 min.
(R)-N-(4-(1H-Pyrazol-4-yl)benzyl)-N-cyclopropyl-1-(3-((2-methyl-1-(4-methylpiperazin-1-yl)-1-oxopropan-2-yl)oxy)phenyl)piperidine-3-carboxamide (19).
1H NMR (500 MHz, chloroform-d) δ 7.83 (s, 2H), 7.45 (d, J = 7.8 Hz, 2H), 7.24 (d, J = 7.8 Hz, 2H), 7.06 (t, J = 8.2 Hz, 1H), 6.54 (dd, J = 8.3, 2.3 Hz, 1H), 6.42 (t, J = 2.4 Hz, 1H), 6.28 (dd, J = 8.2, 2.3 Hz, 1H), 4.69 (d, J = 14.6 Hz, 1H), 4.54 (d, J = 14.7 Hz, 1H), 3.87 (s, 2H), 3.73−3.59 (m, 4H), 3.45 (tt, J = 11.0, 3.5 Hz, 1H), 2.99 (dd, J = 12.4, 11.0 Hz, 1H), 2.75 (td, J = 12.1, 3.0 Hz, 1H), 2.64 (ddd, J = 10.7, 6.9, 4.1 Hz, 1H), 2.31 (d, J = 6.1 Hz, 2H), 2.17 (s, 3H), 2.08 (s, 2H), 1.95 (d, J = 10.7 Hz, 1H), 1.85−1.70 (m, 3H), 1.63−1.60 (m, 6H), 0.93−0.80 (m, 4H). 13C NMR (126 MHz, chloroform-d) δ 176.9, 171.9, 156.3, 152.7, 136.7, 131.4, 129.6, 128.3, 125.9, 109.8, 107.7, 106.0, 80.7, 54.9, 54.7, 52.3, 49.9, 49.6, 45.9, 45.6, 42.8, 39.9, 29.8, 27.8, 26.23, 26.21, 24.5, 9.5, 9.1. HRMS (ESI) calcd for C34H44N6O3 (M + H)+ 585.3548, found 585.3546. HPLC purity 98.5%, tR = 9.23 min.
(R)-N-(4-(1H-Pyrazol-4-yl)benzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperidin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (20).
1H NMR (500 MHz, methanol-d4) δ 7.92 (s, 2H), 7.57−7.51 (m, 2H), 7.24 (d, J = 8.1 Hz, 2H), 7.09 (t, J = 8.2 Hz, 1H), 6.59 (dd, J = 8.3, 2.3 Hz, 1H), 6.44 (t, J = 2.3 Hz, 1H), 6.30 (dd, J = 8.1, 2.3 Hz, 1H), 4.66−4.56 (m, 2H), 3.81 (p, J = 7.2, 6.5 Hz, 2H), 3.72−3.63 (m, 2H), 3.59−3.49 (m, 3H), 2.88 (dd, J = 12.3, 10.9 Hz, 1H), 2.78−2.71 (m, 2H), 1.98 (d, J = 9.8 Hz, 1H), 1.83 (dq, J = 8.6, 2.8 Hz, 1H), 1.72 (qd, J = 12.1, 11.7, 3.4 Hz, 2H), 1.59−1.44 (m, 10H), 1.25 (dd, J = 7.2, 3.9 Hz, 2H), 0.89 (pd, J = 8.5, 7.7, 4.8 Hz, 4H). 13C NMR (126 MHz, methanol-d4) δ 177.6, 172.0, 156.4, 152.5, 136.2, 131.6, 129.3, 127.7, 125.3, 122.0, 109.7, 108.2, 105.5, 80.2, 52.5, 49.7, 49.2, 46.6, 44.0, 39.7, 29.8, 27.4, 25.8, 25.4, 25.3, 25.2, 24.1, 23.9, 8.5, 8.2. HRMS (ESI) calcd for C34H43N5O3 (M + H)+ 570.3439, found 570.3446. HPLC purity 99.7%, tR = 11.52 min.
(R)-N-(4-(1H-Pyrazol-4-yl)benzyl)-N-ethyl-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (21 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.45 (d, J = 4.4 Hz, 2H), 7.69 (dd, J = 29.3, 8.0 Hz, 2H), 7.55 (q, J = 8.1 Hz, 1H), 7.49−7.25 (m, 4H), 7.03 (ddd, J = 8.8, 6.7, 2.3 Hz, 1H), 4.84−4.47 (m, 2H), 4.31−3.83 (m, 4H), 3.84−3.62 (m, 5H), 3.60−3.41 (m, 2H), 3.28−2.77 (m, 4H), 2.37−1.80 (m, 4H), 1.68 (d, J = 3.4 Hz, 6H), 1.20 (dt, J = 63.2, 7.1 Hz, 3H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 143.3, 136.9, 136.1, 131.5, 130.7, 130.0, 129.4, 128.1, 127.5, 126.1, 125.9, 123.4, 118.5, 118.3, 114.5, 111.60, 111.56, 81.8, 58.3, 55.7, 50.3, 43.1, 42.2, 41.3, 39.6, 24.9, 24.7, 21.4, 13.2, 11.4. HRMS (ESI) calcd for C32H42N6O3 (M + Na)+ 581.3211, found 581.3204. HPLC purity 99.9%, tR = 8.99 min.
(R)-N-(4-(1H-Pyrazol-4-yl)benzyl)-N-(2-ethoxyethyl)-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (22 as HCl Salt).
1H NMR (500 MHz, methanol-d4) δ 8.43 (d, J = 4.1 Hz, 2H), 7.68 (dd, J = 31.4, 8.0 Hz, 2H), 7.57−7.52 (m, 1H), 7.46−7.28 (m, 4H), 7.02 (ddd, J = 7.3, 4.9, 2.2 Hz, 1H), 4.80 (d, J = 16.9 Hz, 1H), 4.57 (d, J = 15.3 Hz, 1H), 4.22−3.57 (m, 13H), 3.50−3.42 (m, 2H), 3.26−2.81 (m, 4H), 2.30−1.85 (m, 4H), 1.68 (d, J = 3.5 Hz, 6H), 1.15 (dt, J = 16.7, 7.0 Hz, 3H). 13C NMR (126 MHz, methanol-d4) δ 171.5, 156.3, 143.3, 136.9, 131.53, 131.49, 130.7, 129.4, 128.0, 127.4, 126.1, 125.8, 118.4, 118.3, 114.5, 114.4, 111.65, 111.56, 81.80, 81.77, 67.7, 67.3, 66.3, 66.1, 55.7, 43.1, 42.5, 39.5, 24.9, 24.7, 21.4, 14.2, 14.1. HRMS (ESI) calcd for C34H46N6O4 (M + H)+ 603.3653, found 603.3650. HPLC purity 99.7%, tR = 9.34 min.
tert-Butyl 2-(3-Bromophenoxy)-2-methylpropanoate (23).
1H NMR (500 MHz, chloroform-d) δ 7.16−7.04 (m, 2H), 7.01 (dd, J = 2.7, 1.3 Hz, 1H), 6.82−6.71 (m, 1H), 1.56 (s, 6H), 1.44 (s, 9H). MS (ESI) m/z = 315.1 [M + H]+.
tert-Butyl (R)-3-((4-Isopropylbenzyl)carbamoyl)piperidine-1-carboxylate (24a).
1H NMR (500 MHz, chloroform-d) δ 7.14 (s, 4H), 4.37 (d, J = 19.2 Hz, 2H), 4.05−3.61 (m, 2H), 3.15 (dd, J = 13.5, 9.3 Hz, 1H), 2.86−2.77 (m, 2H), 2.26 (dq, J = 9.8, 5.3, 4.9 Hz, 1H), 1.92−1.77 (m, 2H), 1.70−1.53 (m, 1H), 1.38 (s, 10H), 1.22 (d, J = 7.0 Hz, 6H). MS (ESI) m/z = 383.3 [M + Na]+.
tert-Butyl (S)-3-((4-Isopropylbenzyl)carbamoyl)piperidine-1-carboxylate (24b).
1H NMR (500 MHz, chloroform-d) δ 7.15 (s, 4H), 4.37 (d, J = 19.2 Hz, 2H), 4.03−3.60 (m, 2H), 3.14 (dd, J = 13.5, 9.3 Hz, 1H), 2.86 (hept, J = 6.8 Hz, 2H), 2.28 (dq, J = 9.8, 5.3, 4.9 Hz, 1H), 1.91−1.77 (m, 2H), 1.69−1.54 (m, 1H), 1.38 (s, 10H), 1.21 (d, J = 7.0 Hz, 6H). MS (ESI) m/z = 361.3 [M + H]+, 383.3 [M + Na]+.
tert-Butyl (R)-2-(3-(3-((4-Isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoate (25a).
1H NMR (500 MHz, chloroform-d) δ 7.18 (d, J = 1.6 Hz, 4H), 7.06 (t, J = 8.2 Hz, 1H), 6.91 (t, J = 5.7 Hz, 1H), 6.54 (dd, J = 8.3, 2.3 Hz, 1H), 6.48 (t, J = 2.3 Hz, 1H), 6.34 (dd, J = 8.2, 2.3 Hz, 1H), 4.43 (d, J = 5.6 Hz, 2H), 3.39 (dd, J = 12.5, 3.6 Hz, 1H), 3.30−3.15 (m, 2H), 3.02 (ddd, J = 11.8, 8.4, 3.1 Hz, 1H), 2.88 (p, J = 6.9 Hz, 1H), 2.55 (tt, J = 8.0, 4.1 Hz, 1H), 2.00−1.73 (m, 3H), 1.66 (tq, J = 8.7, 4.4 Hz, 1H), 1.56 (s, 6H), 1.42 (d, J = 0.9 Hz, 9H), 1.24 (d, J = 6.9 Hz, 6H). MS (ESI) m/z = 495.4 [M + H]+.
tert-Butyl (S)-2-(3-(3-((4-Isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoate (25b).
1H NMR (500 MHz, chloroform-d) δ 7.18 (d, J = 1.6 Hz, 4H), 7.06 (t, J = 8.2 Hz, 1H), 6.90 (t, J = 5.7 Hz, 1H), 6.54 (dd, J = 8.3, 2.3 Hz, 1H), 6.49 (t, J = 2.3 Hz, 1H), 6.34 (dd, J = 8.2, 2.3 Hz, 1H), 4.42 (d, J = 5.6 Hz, 2H), 3.39 (dd, J = 12.5, 3.6 Hz, 1H), 3.31−3.15 (m, 2H), 3.02 (ddd, J = 11.8, 8.4, 3.1 Hz, 1H), 2.88 (p, J = 6.9 Hz, 1H), 2.55 (tt, J = 8.0, 4.1 Hz, 1H), 2.02−1.72 (m, 3H), 1.66 (tq, J = 8.7, 4.4 Hz, 1H), 1.55 (s, 6H), 1.42 (d, J = 0.9 Hz, 9H), 1.23 (d, J = 6.9 Hz, 6H). MS (ESI) m/z = 495.4 [M + H]+.
tert-Butyl (R)-3-((4-Isopropylbenzyl)(methyl)carbamoyl)piperidine-1-carboxylate (26a).
1H NMR (500 MHz, chloroform-d) δ 7.24−7.12 (m, 2H), 7.11−6.89 (m, 2H), 4.51 (s, 2H), 4.16 (s, 1H), 4.06 (s, 1H), 2.95 (s, 2H), 2.90−2.79 (m, 3H), 2.70−2.45 (m, 2H), 2.02−1.54 (m, 3H), 1.53−1.30 (m, 10H), 1.21 (t, J = 6.9 Hz, 6H). MS (ESI) m/z = 375.3 [M + H]+.
tert-Butyl (R)-3-(Ethyl(4-isopropylbenzyl)carbamoyl)piperidine-1-carboxylate (26b).
1H NMR (500 MHz, chloroform-d) δ 7.20−7.14 (m, 1H), 7.14−6.90 (m, 3H), 4.85−3.96 (m, 4H), 3.26 (s, 2H), 2.84 (dp, J = 13.7, 6.9 Hz, 2H), 2.60 (tt, J = 31.9, 17.1 Hz, 2H), 1.97−1.55 (m, 3H), 1.38 (d, J = 30.3 Hz, 10H), 1.19 (dd, J = 8.0, 6.9 Hz, 6H), 1.08 (dt, J = 48.7, 7.1 Hz, 3H). MS (ESI) m/z = 389.3 [M + H]+.
tert-Butyl (R)-3-(Isopropyl(4-isopropylbenzyl)carbamoyl)piperidine-1-carboxylate (26c).
1H NMR (500 MHz, chloroform-d) δ 7.19−7.12 (m, 1H), 7.12−6.97 (m, 3H), 4.94−3.85 (m, 5H), 3.19−2.20 (m, 4H), 2.04−1.62 (m, 2H), 1.64−1.52 (m, 1H), 1.44 (s, 4H), 1.36 (s, 6H), 1.29−1.10 (m, 9H), 1.04 (d, J = 6.8 Hz, 3H). MS (ESI) m/z = 403.3 [M + H]+, 425.3 [M + Na]+.
tert-Butyl (R)-3-(Cyclopropyl(4-isopropylbenzyl)carbamoyl)piperidine-1-carboxylate (26d).
1H NMR (500 MHz, chloroform-d) δ 7.12 (t, J = 6.7 Hz, 4H), 4.91−3.94 (m, 4H), 3.19 (tt, J = 11.5, 3.8 Hz, 1H), 2.87 (dq, J = 13.8, 6.8 Hz, 2H), 2.65 (d, J = 69.8 Hz, 2H), 1.90 (d, J = 13.3 Hz, 1H), 1.85−1.63 (m, 2H), 1.45 (s, 10H), 1.22 (d, J = 6.9 Hz, 6H), 0.97−0.70 (m, 4H). MS (ESI) m/z = 401.4 [M + H]+, 423.3 [M + Na]+.
tert-Butyl (R)-2-(3-(3-((4-Isopropylbenzyl)(methyl)carbamoyl)piperidin-1-yl)phenoxy)-2- methylpropanoate (27a).
1H NMR (500 MHz, chloroform-d) δ 7.17−7.06 (m, 3H), 7.04−6.83 (m, 2H), 6.57−6.30 (m, 2H), 6.21 (ddd, J = 16.6, 8.1, 2.2 Hz, 1H), 4.62−4.28 (m, 2H), 3.87−3.51 (m, 2H), 3.00−2.76 (m, 6H), 2.67 (dtd, J = 18.5, 12.0, 2.7 Hz, 1H), 2.05−1.61 (m, 4H), 1.54−1.43 (m, 6H), 1.36 (d, J = 3.4 Hz, 9H), 1.17 (dd, J = 6.9, 3.2 Hz, 6H). MS (ESI) m/z = 509.4 [M + H]+.
tert-Butyl (R)-2-(3-(3-(Ethyl(4-isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoate (27b).
1H NMR (500 MHz, chloroform-d) δ 7.16−7.04 (m, 3H), 7.04−6.87 (m, 2H), 6.54−6.27 (m, 2H), 6.21 (ddd, J = 19.7, 7.9, 2.2 Hz, 1H), 4.62−4.31 (m, 2H), 3.75−3.49 (m, 2H), 3.42−3.16 (m, 2H), 3.03−2.52 (m, 4H), 1.96−1.57 (m, 4H), 1.50−1.41 (m, 6H), 1.35 (d, J = 3.3 Hz, 9H), 1.16 (dd, J = 6.9, 3.5 Hz, 6H), 1.05 (dt, J = 25.1, 7.1 Hz, 3H). MS (ESI) m/z = 523.4 [M + H]+.
tert-Butyl (R)-2-(3-(3-(Isopropyl(4-isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoate (27c).
1H NMR (500 MHz, chloroform-d) δ 7.19 (d, J = 8.2 Hz, 1H), 7.15−7.04 (m, 3H), 6.98 (t, J = 8.2 Hz, 1H), 6.63−6.21 (m, 3H), 5.07−4.21 (m, 3H), 3.71 (ddt, J = 16.2, 12.0, 2.0 Hz, 1H), 3.57 (td, J = 9.8, 7.6, 3.0 Hz, 1H), 3.18−2.56 (m, 4H), 2.05−1.63 (m, 4H), 1.59−1.52 (m, 6H), 1.43 (d, J = 7.8 Hz, 9H), 1.33−1.00 (m, 12H). MS (ESI) m/z = 537.4 [M + H]+.
tert-Butyl (R)-2-(3-(3-(Cyclopropyl(4-isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoate (27d).
1H NMR (500 MHz, chloroform-d) δ 7.20−7.12 (m, 4H), 7.07 (t, J = 8.2 Hz, 1H), 6.55 (dd, J = 8.2, 2.3 Hz, 1H), 6.49 (t, J = 2.4 Hz, 1H), 6.30 (dd, J = 8.1, 2.3 Hz, 1H), 4.71−4.37 (m, 2H), 3.85−3.61 (m, 2H), 3.43 (ddt, J = 11.0, 7.1, 3.4 Hz, 1H), 2.99 (dd, J = 12.4, 10.9 Hz, 1H), 2.88 (p, J = 6.9 Hz, 1H), 2.77 (td, J = 12.1, 2.6 Hz, 1H), 2.59 (tt, J = 6.9, 4.1 Hz, 1H), 2.00−1.89 (m, 1H), 1.88−1.65 (m, 3H), 1.55 (s, 6H), 1.44 (s, 9H), 1.24 (d, J = 6.9 Hz, 6H), 0.97−0.75 (m, 4H). MS (ESI) m/z = 535.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (28a).
1H NMR (500 MHz, chloroform-d) δ 7.14 (d, J = 8.2 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 6.99 (t, J = 8.2 Hz, 1H), 6.88 (s, 1H), 6.45 (dd, J = 8.3, 2.2 Hz, 1H), 6.33 (t, J = 2.3 Hz, 1H), 6.23 (dd, J = 8.1, 2.2 Hz, 1H), 4.37 (qd, J = 14.7, 5.6 Hz, 2H), 3.72 (d, J = 5.6 Hz, 2H), 3.50−3.36 (m, 3H), 3.33 (dt, J = 12.5, 4.4 Hz, 1H), 3.21 (s, 2H), 3.12 (dd, J = 12.8, 8.8 Hz, 1H), 2.94 (ddd, J = 22.2, 12.9, 4.5 Hz, 3H), 2.82 (p, J = 6.9 Hz, 1H), 2.54−2.32 (m, 1H), 1.82 (qd, J = 9.1, 3.9 Hz, 2H), 1.67 (dt, J = 13.3, 4.8 Hz, 1H), 1.55 (d, J = 5.0 Hz, 7H), 1.34 (s, 9H), 1.16 (d, J = 7.0 Hz, 6H). MS (ESI) m/z = 607.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Isopropylbenzyl)(methyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (28b).
1H NMR (500 MHz, chloroform-d) δ 7.24−7.10 (m, 3H), 7.09−6.88 (m, 2H), 6.55−6.32 (m, 2H), 6.24 (ddd, J = 19.9, 8.1, 2.3 Hz, 1H), 4.84−4.37 (m, 2H), 3.80 (dd, J = 10.7, 5.5 Hz, 2H), 3.72−3.53 (m, 4H), 3.32 (d, J = 6.0 Hz, 2H), 3.07 (d, J = 6.4 Hz, 2H), 3.02−2.92 (m, 4H), 2.92−2.84 (m, 2H), 2.72 (qd, J = 12.4, 2.6 Hz, 1H), 1.98−1.66 (m, 4H), 1.68−1.44 (m, 6H), 1.41 (s, 9H), 1.23 (dd, J = 6.9, 4.1 Hz, 6H). MS (ESI) m/z = 621.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-(Ethyl(4-isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (28c).
1H NMR (500 MHz, chloroform-d) δ 7.24−7.11 (m, 3H), 7.11−6.89 (m, 2H), 6.65−6.32 (m, 2H), 6.30−6.12 (m, 1H), 4.86−4.43 (m, 2H), 3.80 (dt, J = 11.2, 5.2 Hz, 2H), 3.71−3.55 (m, 4H), 3.48−3.24 (m, 4H), 3.17−2.66 (m, 6H), 1.99−1.67 (m, 4H), 1.66−1.52 (m, 7H), 1.42 (s, 9H), 1.24 (dd, J = 7.0, 4.2 Hz, 6H), 1.14 (dt, J = 27.5, 7.1 Hz, 3H). MS (ESI) m/z = 635.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-(Isopropyl(4-isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (28d).
1H NMR (500 MHz, chloroform-d) δ 7.22−6.90 (m, 5H), 6.62−6.14 (m, 3H), 4.95−4.19 (m, 3H), 3.90−3.45 (m, 6H), 3.32 (d, J = 6.9 Hz, 2H), 3.23−2.54 (m, 6H), 2.00−1.65 (m, 4H), 1.62 (dd, J = 7.7, 1.9 Hz, 6H), 1.52−1.39 (m, 10H), 1.34−1.05 (m, 12H). MS (ESI) m/z =649.5 [M + H]+, 671.5 [M + Na]+.
tert-Butyl (R)-4-(2-(3-(3-(Cyclopropyl(4-isopropylbenzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (28e).
1H NMR (500 MHz, chloroform-d) δ 7.21−7.13 (m, 4H), 7.07 (t, J = 8.2 Hz, 1H), 6.54 (dd, J = 8.3, 2.2 Hz, 1H), 6.43 (t, J = 2.4 Hz, 1H), 6.28 (dd, J = 8.1, 2.3 Hz, 1H), 4.68 (d, J = 14.5 Hz, 1H), 4.51 (d, J = 14.6 Hz, 1H), 3.83 (q, J = 5.5 Hz, 2H), 3.73−3.53 (m, 4H), 3.49−3.28 (m, 3H), 3.17−2.83 (m, 4H), 2.77 (td, J = 12.2, 3.0 Hz, 1H), 2.66−2.54 (m, 1H), 2.01−1.92 (m, 1H), 1.89−1.69 (m, 3H), 1.71−1.54 (m, 7H), 1.43 (s, 9H), 1.25 (d, J = 6.9 Hz, 6H), 1.02−0.74 (m, 4H). MS (ESI) m/z =647.4 [M + H]+.
Ethyl (R)-1-(3-((1-(tert-Butoxy)-2-methyl-1-oxopropan-2-yl)oxy)phenyl)piperidine-3-carboxylate (29).
1H NMR (500 MHz, chloroform-d) δ 7.04 (t, J = 8.2 Hz, 1H), 6.54 (dd, J = 8.1, 2.3 Hz, 1H), 6.45 (t, J = 2.4 Hz, 1H), 6.31−6.21 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.67 (ddt, J = 12.4, 3.5, 1.5 Hz, 1H), 3.42 (ddd, J = 12.3, 4.9, 3.1 Hz, 1H), 2.95 (dd, J = 12.4, 9.9 Hz, 1H), 2.80−2.68 (m, 1H), 2.60 (tt, J = 10.0, 3.9 Hz, 1H), 2.05−1.93 (m, 1H), 1.74 (th, J = 9.2, 3.1 Hz, 1H), 1.69−1.59 (m, 2H), 1.53 (s, 6H), 1.41 (s, 9H), 1.24 (t, J = 7.2 Hz, 3H). MS (ESI) m/z =392.3 [M + H]+, 414.3 [M + Na]+.
tert-Butyl (R)-4-(2-(3-(3-(Ethoxycarbonyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (30).
1H NMR (500 MHz, chloroform-d) δ 7.05 (t, J = 8.2 Hz, 1H), 6.55 (dd, J = 8.0, 2.3 Hz, 1H), 6.41 (t, J = 2.3 Hz, 1H), 6.27 (ddd, J = 8.2, 2.5, 0.7 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.81 (t, J = 5.2 Hz, 2H), 3.67 (ddt, J = 12.4, 3.5, 1.5 Hz, 1H), 3.59 (t, J = 5.2 Hz, 2H), 3.50−3.39 (m, 1H), 3.33 (q, J = 5.5 Hz, 2H), 3.07 (t, J = 5.2 Hz, 2H), 2.97 (dd, J = 12.4, 9.9 Hz, 1H), 2.81−2.70 (m, 1H), 2.62 (tt, J = 10.0, 3.9 Hz, 1H), 2.07−1.92 (m, 1H), 1.78 (qq, J = 4.9, 3.2, 2.2 Hz, 1H), 1.62 (s, 8H), 1.41 (s, 9H), 1.26 (t, J = 7.1 Hz, 3H). MS (ESI) m/z =504.3 [M + H]+, 526.3 [M + Na]+.
tert-Butyl (R)-4-(2-(3-(3-((4-(tert-Butyl)benzyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (31a).
1H NMR (500 MHz, chloroform-d) δ 7.35−7.29 (m, 2H), 7.18−7.12 (m, 2H), 7.06 (t, J = 8.2 Hz, 1H), 6.53 (dd, J = 8.3, 2.3 Hz, 1H), 6.42 (t, J = 2.3 Hz, 1H), 6.27 (dd, J = 8.1, 2.3 Hz, 1H), 4.67 (d, J = 14.6 Hz, 1H), 4.49 (d, J = 14.6 Hz, 1H), 3.88−3.74 (m, 2H), 3.74−3.49 (m, 4H), 3.43 (tt, J = 10.9, 3.5 Hz, 1H), 3.33 (s, 2H), 3.15−2.91 (m, 3H), 2.75 (td, J = 12.2, 2.9 Hz, 1H), 2.62 (tt, J = 6.6, 4.4 Hz, 1H), 2.00−1.89 (m, 1H), 1.87−1.67 (m, 3H), 1.63 (d, J = 1.4 Hz, 6H), 1.42 (s, 9H), 1.30 (s, 9H), 0.89−0.75 (m, 4H). MS (ESI) m/z = 661.5 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Cyclohexylbenzyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (31b).
1H NMR (500 MHz, chloroform-d) δ 7.13 (s, 4H), 7.05 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 8.3, 2.3 Hz, 1H), 6.41 (t, J = 2.3 Hz, 1H), 6.26 (dd, J = 8.2, 2.3 Hz, 1H), 4.65 (d, J = 14.6 Hz, 1H), 4.48 (d, J = 14.6 Hz, 1H), 3.86−3.72 (m, 2H), 3.71−3.50 (m, 4H), 3.47−3.20 (m, 3H), 3.15−2.92 (m, 3H), 2.75 (td, J = 12.2, 2.9 Hz, 1H), 2.66−2.54 (m, 1H), 2.53−2.33 (m, 1H), 1.95 (d, J = 11.2 Hz, 1H), 1.90−1.66 (m, 8H), 1.62 (d, J = 1.3 Hz, 6H), 1.41 (s, 12H), 1.29−1.18 (m, 2H), 0.91−0.75 (m, 4H). MS (ESI) m/z = 687.5 [M + H]+, 709.4 [M + Na]+.
tert-Butyl (R)-4-(2-(3-(3-(([1,1′-Biphenyl]-4-ylmethyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (31c).
1H NMR (500 MHz, chloroform-d) δ 7.63−7.52 (m, 4H), 7.46−7.39 (m, 2H), 7.37−7.28 (m, 3H), 7.06 (t, J = 8.2 Hz, 1H), 6.54 (dd, J = 8.3, 2.2 Hz, 1H), 6.43 (t, J = 2.4 Hz, 1H), 6.27 (dd, J = 8.2, 2.3 Hz, 1H), 4.74 (d, J = 14.7 Hz, 1H), 4.57 (d, J = 14.7 Hz, 1H), 3.81 (s, 2H), 3.72−3.50 (m, 4H), 3.45 (tt, J = 11.0, 3.6 Hz, 1H), 3.32 (s, 2H), 3.13−2.94 (m, 3H), 2.82−2.60 (m, 2H), 2.01−1.94 (m, 1H), 1.88−1.67 (m, 3H), 1.62 (s, 6H), 1.41 (s, 9H), 0.95−0.78 (m, 4H). MS (ESI) m/z = 681.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-(Cyclopropyl(4-(pyridin-2-yl)benzyl) carbamoyl) piperidin-1-yl) phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (31d).
1H NMR (500 MHz, chloroform-d) δ 8.67 (dt, J = 4.9, 1.3 Hz, 1H), 7.97−7.89 (m, 2H), 7.79−7.65 (m, 2H), 7.34 (d, J = 8.1 Hz, 2H), 7.21 (ddd, J = 6.6, 4.8, 1.5 Hz, 1H), 7.05 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 8.4, 2.3 Hz, 1H), 6.41 (t, J = 2.4 Hz, 1H), 6.26 (dd, J = 8.1, 2.3 Hz, 1H), 4.75 (d, J = 14.6 Hz, 1H), 4.58 (d, J = 14.6 Hz, 1H), 3.88−3.73 (m, 2H), 3.73−3.63 (m, 2H), 3.63−3.49 (m, 2H), 3.43 (tt, J = 11.0, 3.6 Hz, 1H), 3.32 (s, 2H), 3.14−2.93 (m, 3H), 2.76 (td, J = 12.2, 2.8 Hz, 1H), 2.61 (tt, J = 6.9, 3.2 Hz, 1H), 2.01−1.89 (m, 1H), 1.89−1.65 (m, 3H), 1.62 (d, J = 1.6 Hz, 6H), 1.41 (s, 9H), 0.91−0.77 (m, 4H). MS (ESI) m/z = 682.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-(Cyclopropyl(4-(pyridin-3-yl)benzyl) carbamoyl) piperidin-1-yl) phenoxy)–2-methylpropanoyl)piperazine-1-carboxylate (31e).
1H NMR (500 MHz, chloroform-d) δ 8.73−8.56 (m, 2H), 7.59 (dd, J = 8.1, 1.8 Hz, 2H), 7.51−7.45 (m, 2H), 7.37−7.29 (m, 2H), 7.05 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 8.3, 2.3 Hz, 1H), 6.41 (t, J = 2.3 Hz, 1H), 6.27 (dd, J = 8.2, 2.3 Hz, 1H), 4.74 (d, J = 15.1 Hz, 1H), 4.58 (d, J = 14.9 Hz, 1H), 3.80 (d, J = 6.3 Hz, 2H), 3.72−3.63 (m, 2H), 3.57 (p, J = 9.7, 7.7 Hz, 2H), 3.44 (tt, J = 11.1, 3.4 Hz, 1H), 3.31 (s, 2H), 3.13−2.95 (m, 3H), 2.81−2.64 (m, 2H), 1.96 (d, J = 10.7 Hz, 1H), 1.88−1.69 (m, 3H), 1.41 (d, J = 2.4 Hz, 9H), 0.94−0.76 (m, 4H). MS (ESI) m/z = 682.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-(Cyclopropyl(4-(pyridin-4-yl)benzyl) carbamoyl) piperidin-1-yl) phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (31f).
1H NMR (500 MHz, chloroform-d) δ 8.71−8.55 (m, 2H), 7.59 (dd, J = 8.1, 1.8 Hz, 2H), 7.52−7.45 (m, 2H), 7.38−7.29 (m, 2H), 7.05 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 8.3, 2.3 Hz, 1H), 6.41 (t, J = 2.3 Hz, 1H), 6.27 (dd, J = 8.2, 2.3 Hz, 1H), 4.74 (d, J = 15.1 Hz, 1H), 4.58 (d, J = 14.9 Hz, 1H), 3.80 (d, J = 6.3 Hz, 2H), 3.73−3.63 (m, 2H), 3.57 (p, J = 9.7, 7.7 Hz, 2H), 3.44 (tt, J = 11.1, 3.4 Hz, 1H), 3.31 (s, 2H), 3.14−2.94 (m, 3H), 2.75 (td, J = 12.1, 3.0 Hz, 1H), 2.67 (tt, J = 6.6, 4.0 Hz, 1H), 1.96 (d, J = 10.7 Hz, 1H), 1.87−1.68 (m, 3H), 1.62 (s, 6H), 1.41 (d, J = 2.4 Hz, 9H), 0.97−0.74 (m, 4H). MS (ESI) m/z = 682.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Bromobenzyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (32).
1H NMR (500 MHz, chloroform-d) δ 7.47−7.40 (m, 2H), 7.16−7.11 (m, 2H), 7.07 (t, J = 8.2 Hz, 1H), 6.53 (dd, J = 8.3, 2.2 Hz, 1H), 6.42 (t, J = 2.4 Hz, 1H), 6.29 (dd, J = 8.1, 2.3 Hz, 1H), 4.63 (d, J = 14.7 Hz, 1H), 4.49 (d, J = 14.7 Hz, 1H), 3.82 (q, J = 4.7 Hz, 2H), 3.72−3.49 (m, 4H), 3.42 (ddt, J = 10.9, 7.0, 3.5 Hz, 1H), 3.33 (s, 2H), 3.08 (d, J = 5.3 Hz, 2H), 3.02−2.92 (m, 1H), 2.80−2.72 (m, 1H), 2.62 (tt, J = 7.0, 3.9 Hz, 1H), 1.99−1.86 (m, 1H), 1.86−1.78 (m, 1H), 1.73 (td, J = 11.0, 5.6 Hz, 2H), 1.64 (d, J = 1.5 Hz, 6H), 1.43 (s, 9H), 0.96−0.76 (m, 4H). MS (ESI) m/z = 683.3 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-(Cyclopropyl(4-(thiophen-3-yl)benzyl) carbamoyl)piperidin-1-yl)phenoxy)–2-methylpropanoyl)piperazine-1-carboxylate (33a).
1H NMR (500 MHz, chloroform-d) δ 7.63−7.51 (m, 2H), 7.46 (t, J = 2.2 Hz, 1H), 7.40 (d, J = 2.1 Hz, 2H), 7.31−7.27 (m, 2H), 7.09 (t, J = 8.2 Hz, 1H), 6.56 (dd, J = 8.3, 2.3 Hz, 1H), 6.45 (t, J = 2.3 Hz, 1H), 6.30 (dd, J = 8.2, 2.3 Hz, 1H), 4.73 (d, J = 14.6 Hz, 1H), 4.57 (d, J = 14.6 Hz, 1H), 3.83 (s, 2H), 3.70 (t, J = 9.3 Hz, 2H), 3.66−3.53 (m, 2H), 3.46 (ddt, J = 10.9, 6.9, 3.5 Hz, 1H), 3.35 (s, 2H), 3.18−2.92 (m, 3H), 2.85−2.72 (m, 1H), 2.65 (tt, J = 6.5, 3.5 Hz, 1H), 2.03−1.92 (m, 1H), 1.88−1.67 (m, 3H), 1.65 (s, 6H), 1.44 (s, 9H), 0.98−0.79 (m, 4H). MS (ESI) m/z = 687.5 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-(1H-Pyrazol-4-yl)benzyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (33b).
1H NMR (500 MHz, chloroform-d) δ 7.80 (s, 2H), 7.43 (d, J = 7.9 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 7.05 (t, J = 8.2 Hz, 1H), 6.53 (dd, J = 8.4, 2.2 Hz, 1H), 6.42 (t, J = 2.3 Hz, 1H), 6.27 (dd, J = 8.2, 2.3 Hz, 1H), 4.69 (d, J = 14.7 Hz, 1H), 4.52 (d, J = 14.7 Hz, 1H), 3.88−3.72 (m, 2H), 3.72−3.63 (m, 2H), 3.63−3.52 (m, 2H), 3.44 (tt, J = 11.0, 3.5 Hz, 1H), 3.38−3.25 (m, 2H), 3.14−2.91 (m, 3H), 2.76 (td, J = 12.1, 2.8 Hz, 1H), 2.63 (td, J = 6.7, 3.2 Hz, 1H), 1.98−1.92 (m, 1H), 1.85−1.67 (m, 3H), 1.62 (s, 6H), 1.41 (s, 9H), 0.94−0.70 (m, 4H). MS (ESI) m/z = 671.5 [M + H]+, 669.3 [M − H]−.
tert-Butyl (R)-4-(2-(3-(3-((4-(1H-Pyrazol-5-yl)benzyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (33c).
1H NMR (500 MHz, chloroform-d) δ 7.76−7.66 (m, 2H), 7.61−7.53 (m, 1H), 7.47 (ddd, J = 8.4, 6.9, 2.9 Hz, 1H), 7.33−7.24 (m, 2H), 7.07 (t, J = 8.2 Hz, 1H), 6.63−6.50 (m, 2H), 6.43 (t, J = 2.3 Hz, 1H), 6.29 (dd, J = 8.1, 2.3 Hz, 1H), 4.71 (d, J = 14.7 Hz, 1H), 4.59 (d, J = 14.6 Hz, 1H), 3.89−3.51 (m, 6H), 3.51−3.24 (m, 3H), 3.18−2.89 (m, 3H), 2.77 (td, J = 12.1, 2.8 Hz, 1H), 2.63 (td, J = 6.8, 3.3 Hz, 1H), 2.05−1.91 (m, 1H), 1.89−1.67 (m, 3H), 1.64 (d, J = 1.8 Hz, 6H), 1.43 (s, 9H), 0.94−0.77 (m, 4H). MS (ESI) m/z = 671.5 [M + H]+, 669.4 [M − H]−.
tert-Butyl (R)-4-(2-(3-(3-(Cyclopropyl(4-(1-methyl-1H-pyrazol-4-yl)benzyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (33d).
1H NMR (500 MHz, chloroform-d) δ 7.76 (d, J = 0.9 Hz, 1H), 7.61 (s, 1H), 7.47−7.37 (m, 2H), 7.25 (d, J = 8.0 Hz, 2H), 7.08 (t, J = 8.2 Hz, 1H), 6.55 (dd, J = 8.3, 2.2 Hz, 1H), 6.44 (t, J = 2.3 Hz, 1H), 6.29 (dd, J = 8.2, 2.3 Hz, 1H), 4.70 (d, J = 14.6 Hz, 1H), 4.55 (d, J = 14.6 Hz, 1H), 3.96 (s, 3H), 3.83 (s, 2H), 3.75−3.52 (m, 4H), 3.51−3.24 (m, 3H), 3.15−2.94 (m, 3H), 2.78 (td, J = 12.1, 2.9 Hz, 1H), 2.69−2.56 (m, 1H), 1.97 (d, J = 11.1 Hz, 1H), 1.90−1.69 (m, 3H), 1.64 (s, 6H), 1.44 (s, 9H), 0.92−0.83 (m, 4H). MS (ESI) m/z = 685.4 [M + H]+.
Ethyl (R)-1-(3-((2-Methyl-1-(4-methylpiperazin-1-yl)-1-oxopropan-2-yl)oxy)phenyl)piperidine-3-carboxylate (34a).
1H NMR (500 MHz, acetone-d6) δ 6.95 (t, J = 8.2 Hz, 1H), 6.46 (ddd, J = 8.3, 2.3, 0.8 Hz, 1H), 6.31 (t, J = 2.3 Hz, 1H), 6.15 (ddd, J = 8.1, 2.4, 0.8 Hz, 1H), 4.00 (q, J = 7.1 Hz, 2H), 3.70 (s, 2H), 3.55−3.35 (m, 3H), 3.35−3.24 (m, 1H), 2.89 (dd, J = 12.4, 9.5 Hz, 1H), 2.69 (ddd, J = 12.3, 10.0, 3.2 Hz, 1H), 2.48 (tt, J = 9.6, 4.0 Hz, 1H), 2.13 (s, 2H), 1.98 (s, 3H), 1.93 (ddt, J = 8.8, 4.4, 2.4 Hz, 2H), 1.87−1.76 (m, 1H), 1.64 (dddd, J = 11.5, 6.8, 3.8, 2.0 Hz, 1H), 1.59−1.46 (m, 2H), 1.43 (s, 6H), 1.11 (t, J = 7.1 Hz, 3H). MS (ESI) m/z = 418.3 [M + H]+.
Ethyl (R)-1-(3-((2-Methyl-1-oxo-1-(piperidin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxylate (34b).
1H NMR (500 MHz, chloroform-d) δ 7.02 (t, J = 8.2 Hz, 1H), 6.51 (ddd, J = 8.2, 2.4, 0.8 Hz, 1H), 6.40 (t, J = 2.4 Hz, 1H), 6.28 (ddd, J = 8.2, 2.4, 0.7 Hz, 1H), 4.12 (q, J = 7.2 Hz, 2H), 3.73 (t, J = 5.4 Hz, 2H), 3.65 (ddt, J = 12.4, 3.6, 1.5 Hz, 1H), 3.53 (t, J = 4.9 Hz, 2H), 3.46−3.34 (m, 1H), 2.94 (dd, J = 12.4, 9.9 Hz, 1H), 2.80−2.65 (m, 1H), 2.60 (tt, J = 10.1, 4.0 Hz, 1H), 2.04−1.93 (m, 1H), 1.82−1.67 (m, 1H), 1.59 (s, 8H), 1.47 (t, J = 4.0 Hz, 4H), 1.24 (t, J = 7.1 Hz, 5H). MS (ESI) m/z = 403.3 [M + H]+.
(R)-N-(4-Bromobenzyl)-N-cyclopropyl-1-(3-((2-methyl-1-(4-methylpiperazin-1-yl)-1-oxopropan-2-yl)oxy)phenyl)piperidine-3-carboxamide (35a).
1H NMR (500 MHz, chloroform-d) δ 7.45−7.36 (m, 2H), 7.15−7.08 (m, 2H), 7.05 (t, J = 8.2 Hz, 1H), 6.51 (dd, J = 8.3, 2.3 Hz, 1H), 6.40 (t, J = 2.4 Hz, 1H), 6.28 (dd, J = 8.1, 2.3 Hz, 1H), 4.61 (d, J = 14.7 Hz, 1H), 4.47 (d, J = 14.7 Hz, 1H), 3.85 (s, 2H), 3.67−3.53 (m, 4H), 3.41 (tt, J = 11.0, 3.7 Hz, 1H), 2.94 (dd, J = 12.4, 10.9 Hz, 1H), 2.79−2.65 (m, 1H), 2.65−2.55 (m, 1H), 2.31 (t, J = 5.6 Hz, 2H), 2.17 (s, 3H), 2.08 (d, J = 6.5 Hz, 2H), 1.91 (d, J = 8.8 Hz, 1H), 1.80 (tt, J = 10.8, 4.1 Hz, 1H), 1.76−1.65 (m, 2H), 1.61 (d, J = 1.1 Hz, 6H), 0.93−0.74 (m, 4H). MS (ESI) m/z = 597.3, 599.3 [M + H]+.
(R)-N-(4-Bromobenzyl)-N-cyclopropyl-1-(3-((2-methyl-1-oxo-1-(piperidin-1-yl)propan-2-yl)oxy)phenyl)piperidine-3-carboxamide (35b).
1H NMR (500 MHz, chloroform-d) δ 7.42−7.33 (m, 2H), 7.08 (d, J = 8.1 Hz, 2H), 7.02 (t, J = 8.2 Hz, 1H), 6.48 (dd, J = 8.2, 2.3 Hz, 1H), 6.44 (s, 1H), 6.28 (dd, J = 8.2, 2.3 Hz, 1H), 4.57 (d, J = 14.7 Hz, 1H), 4.46 (d, J = 14.8 Hz, 1H), 3.72 (q, J = 5.7 Hz, 2H), 3.67−3.59 (m, 2H), 3.51 (qd, J = 13.4, 12.4, 5.7 Hz, 2H), 3.39 (tt, J = 10.8, 3.4 Hz, 1H), 2.92 (dd, J = 12.5, 10.9 Hz, 1H), 2.76−2.66 (m, 1H), 2.60 (tt, J = 7.0, 3.9 Hz, 1H), 1.89 (d, J = 9.6 Hz, 1H), 1.83−1.74 (m, 1H), 1.73−1.64 (m, 2H), 1.59 (d, J = 1.4 Hz, 6H), 1.51−1.40 (m, 4H), 1.26−1.19 (m, 2H), 0.92−0.72 (m, 4H). MS (ESI) m/z = 582.3, 584.2 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Bromobenzyl)(ethyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (36a).
1H NMR (500 MHz, chloroform-d) δ 7.52−7.37 (m, 2H), 7.13−6.93 (m, 3H), 6.55−6.16 (m, 3H), 4.64−4.37 (m, 2H), 3.87−3.69 (m, 2H), 3.69−3.48 (m, 4H), 3.48−3.20 (m, 4H), 3.14−2.88 (m, 3H), 2.78 (s, 2H), 1.94−1.49 (m, 10H), 1.40 (s, 9H), 1.12 (dt, J = 27.6, 7.1 Hz, 3H). MS (ESI) m/z = 671.3 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Bromobenzyl)(2-ethoxyethyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (36b).
1H NMR (500 MHz, chloroform-d) δ 7.42−7.36 (m, 2H), 7.21−7.15 (m, 1H), 7.12−7.05 (m, 1H), 7.02 (dt, J = 8.2, 4.1 Hz, 1H), 6.54−6.34 (m, 1H), 6.34−6.06 (m, 2H), 4.72−4.41 (m, 2H), 3.82−3.22 (m, 14H), 3.11−2.61 (m, 5H), 1.95−1.64 (m, 4H), 1.62−1.57 (m, 6H), 1.39 (s, 9H), 1.12 (t, J = 7.0 Hz, 3H). MS (ESI) m/z = 715.4, 717.4 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-(1H-Pyrazol-4-yl)benzyl)(ethyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (37a).
1H NMR (500 MHz, chloroform-d) δ 7.83 (d, J = 7.6 Hz, 2H), 7.54−7.41 (m, 2H), 7.23 (d, J = 7.9 Hz, 1H), 7.17 (d, J = 8.0 Hz, 1H), 7.00 (dt, J = 58.6, 8.4 Hz, 1H), 6.57−6.19 (m, 3H), 4.74−4.42 (m, 2H), 3.88−3.54 (m, 6H), 3.54−3.16 (m, 4H), 3.16−2.60 (m, 5H), 1.99−1.66 (m, 4H), 1.65−1.56 (m, 6H), 1.41 (s, 9H), 1.16 (dt, J = 26.5, 7.1 Hz, 3H). MS (ESI) m/z = 659.5 [M + H]+, 657.5 [M − H]−.
tert-Butyl (R)-4-(2-(3-(3-((4-(1H-Pyrazol-4-yl)benzyl)(2-ethoxyethyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (37b).
1H NMR (500 MHz, chloroform-d) δ 7.83 (s, 2H), 7.46 (dd, J = 21.2, 7.9 Hz, 2H), 7.22 (d, J = 7.9 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 6.99 (dt, J = 56.4, 8.1 Hz, 1H), 6.61−6.40 (m, 1H), 6.37−6.30 (m, 1H), 6.23 (ddd, J = 26.8, 8.2, 2.3 Hz, 1H), 4.83−4.45 (m, 2H), 3.89−3.38 (m, 12H), 3.38−3.23 (m, 2H), 3.15−2.64 (m, 5H), 2.00−1.66 (m, 4H), 1.66−1.52 (m, 6H), 1.41 (s, 9H), 1.16 (td, J = 7.0, 4.2 Hz, 3H). MS (ESI) m/z = 703.5 [M + H]+, 701.5 [M − H]−.
Ethyl (S)-1-(3-((1-(tert-Butoxy)-2-methyl-1-oxopropan-2-yl)oxy)phenyl)piperidine-3-carboxylate (38).
1H NMR (500 MHz, chloroform-d) δ 7.07 (t, J = 8.2 Hz, 1H), 6.57 (ddd, J = 8.3, 2.4, 0.8 Hz, 1H), 6.48 (t, J = 2.4 Hz, 1H), 6.30 (ddd, J = 8.1, 2.3, 0.8 Hz, 1H), 4.20−4.11 (m, 2H), 3.70 (ddt, J = 12.3, 3.5, 1.5 Hz, 1H), 3.51−3.42 (m, 1H), 2.97 (dd, J = 12.4, 10.0 Hz, 1H), 2.77 (tdd, J = 10.2, 4.5, 3.1 Hz, 1H), 2.66−2.56 (m, 1H), 2.06−1.95 (m, 1H), 1.82−1.72 (m, 1H), 1.72−1.62 (m, 2H), 1.55 (s, 6H), 1.44 (s, 9H), 1.27 (t, J = 7.1 Hz, 3H). MS (ESI) m/z = 392.2 [M + H]+.
tert-Butyl (S)-4-(2-(3-(3-(Ethoxycarbonyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (39).
1H NMR (500 MHz, chloroform-d) δ 7.04 (t, J = 8.2 Hz, 1H), 6.54 (dd, J = 8.2, 2.3 Hz, 1H), 6.40 (t, J = 2.3 Hz, 1H), 6.29−6.21 (m, 1H), 4.14 (q, J = 7.1 Hz, 2H), 3.80 (t, J = 5.1 Hz, 2H), 3.66 (ddt, J = 12.4, 3.5, 1.5 Hz, 1H), 3.58 (d, J = 5.3 Hz, 2H), 3.46−3.37 (m, 1H), 3.32 (d, J = 5.4 Hz, 2H), 3.06 (t, J = 5.2 Hz, 2H), 2.97 (dd, J = 12.4, 9.9 Hz, 1H), 2.82−2.68 (m, 1H), 2.61 (tq, J = 12.2, 4.3 Hz, 1H), 2.05−1.94 (m, 1H), 1.77 (dddd, J = 10.5, 9.1, 5.3, 3.5 Hz, 1H), 1.61 (s, 8H), 1.40 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H). MS (ESI) m/z =504.3 [M + H]+.
tert-Butyl (S)-4-(2-(3-(3-((4-Bromobenzyl)(cyclopropyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (40).
1H NMR (500 MHz, chloroform-d) δ 7.42−7.34 (m, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.02 (t, J = 8.2 Hz, 1H), 6.48 (dd, J = 8.5, 2.3 Hz, 1H), 6.37 (t, J = 2.4 Hz, 1H), 6.23 (dd, J = 8.2, 2.3 Hz, 1H), 4.58 (d, J = 14.7 Hz, 1H), 4.44 (d, J = 14.7 Hz, 1H), 3.77 (d, J = 7.2 Hz, 2H), 3.61 (d, J = 12.5 Hz, 2H), 3.57−3.46 (m, 2H), 3.37 (tt, J = 10.9, 3.5 Hz, 1H), 3.31 (s, 2H), 3.07−2.96 (m, 2H), 2.96−2.84 (m, 1H), 2.69 (dd, J = 12.0, 3.0 Hz, 1H), 2.56 (td, J = 6.6, 3.2 Hz, 1H), 1.89 (d, J = 10.9 Hz, 1H), 1.77 (ddd, J = 11.6, 7.3, 4.2 Hz, 1H), 1.74−1.65 (m, 2H), 1.58 (d, J = 1.4 Hz, 6H), 1.37 (s, 9H), 0.90−0.72 (m, 4H). MS (ESI) m/z = 683.4, 685.3 [M + H]+.
tert-Butyl (S)-4-(2-(3-(3-((4-(1H-Pyrazol-4-yl)benzyl)(cyclopropyl)carbamoyl)piperidin-1-yl) phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (41).
1H NMR (500 MHz, chloroform-d) δ 7.80 (s, 2H), 7.49−7.38 (m, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.05 (t, J = 8.2 Hz, 1H), 6.53 (dd, J = 8.3, 2.2 Hz, 1H), 6.42 (t, J = 2.4 Hz, 1H), 6.27 (dd, J = 8.2, 2.3 Hz, 1H), 4.69 (d, J = 14.7 Hz, 1H), 4.53 (d, J = 14.7 Hz, 1H), 3.88−3.73 (m, 2H), 3.73−3.64 (m, 2H), 3.64−3.50 (m, 2H), 3.44 (ddt, J = 11.0, 7.0, 3.5 Hz, 1H), 3.32 (s, 2H), 3.14−2.95 (m, 3H), 2.76 (td, J = 12.1, 2.8 Hz, 1H), 2.63 (tt, J = 6.8, 3.2 Hz, 1H), 1.95 (d, J = 10.9 Hz, 1H), 1.85−1.71 (m, 3H), 1.62 (s, 6H), 1.41 (s, 9H), 0.92−0.78 (m, 4H). MS (ESI) m/z = 671.4 [M + H]+, 669.4 [M − H]−.
Benzyl (2-(2-(2-((4-Bromobenzyl)amino)ethoxy)ethoxy)ethyl)carbamate (42).
1H NMR (500 MHz, chloroform-d) δ 7.46−7.39 (m, 2H), 7.39−7.27 (m, 5H), 7.18 (d, J = 8.0 Hz, 2H), 5.60 (t, J = 5.7 Hz, 1H), 5.09 (s, 2H), 3.72 (s, 2H), 3.64−3.47 (m, 8H), 3.37 (p, J = 8.2, 6.8 Hz, 2H), 2.76 (t, J = 5.2 Hz, 2H), 2.16 (s, 1H). MS (ESI) m/z = 451.2, 453.1 [M + H]+.
tert-Butyl (R)-4-(2-(3-(3-((4-Bromobenzyl)(3-oxo-1-phenyl-2,7,10-trioxa-4-azadodecan-12-yl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (43).
1H NMR (500 MHz, chloroform-d) δ 7.53−7.47 (m, 1H), 7.47− 7.40 (m, 1H), 7.39−7.29 (m, 5H), 7.16−6.97 (m, 3H), 6.60−6.41 (m, 1H), 6.37−6.22 (m, 2H), 5.31 (s, 1H), 5.09 (d, J = 2.6 Hz, 2H), 4.77−4.36 (m, 2H), 3.81 (dt, J = 10.2, 4.7 Hz, 2H), 3.73−3.28 (m, 18H), 3.17−2.65 (m, 5H), 1.96−1.70 (m, 4H), 1.64 (d, J = 2.8 Hz, 6H), 1.44 (s, 9H). MS (ESI) m/z = 908.4, 910.4 [M + H]+.
tert-Butyl (R)-4-(2-Methyl-2-(3-(3-((3-oxo-1-phenyl-2,7,10-trioxa-4-azadodecan-12-yl)(4-(1-trityl-1H-pyrazol-4-yl)benzyl)carbamoyl)piperidin-1-yl)phenoxy)propanoyl)piperazine-1-carboxylate (44).
1H NMR (500 MHz, chloroform-d) δ 7.97−7.87 (m, 1H), 7.61 (dd, J = 9.5, 0.8 Hz, 1H), 7.43−7.26 (m, 16H), 7.23−7.14 (m, 7H), 7.14−6.90 (m, 2H), 6.57−6.17 (m, 3H), 5.29 (s, 1H), 5.07 (d, J = 3.8 Hz, 2H), 4.83−4.44 (m, 2H), 3.86−3.73 (m, 2H), 3.67−3.26 (m, 18H), 3.17−2.65 (m, 5H), 1.93−1.69 (m, 4H), 1.61 (dd, J = 13.3, 2.6 Hz, 6H), 1.42 (d, J = 1.3 Hz, 9H).
tert-Butyl 4-(2-Methyl-2-(3-((R)-3-((2-(2-(2-(5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d] imidazol-4-yl)pentanamido)ethoxy)ethoxy)ethyl)(4-(1-trityl-1H-pyrazol-4yl)benzyl)carbamoyl)piperidin-1-yl)phenoxy)propanoyl)piperazine-1-carboxylate (45).
1H NMR (500 MHz, chloroform-d) δ 7.93 (dd, J = 8.7, 0.8 Hz, 1H), 7.61 (dd, J = 11.7, 0.8 Hz, 1H), 7.46−7.40 (m, 1H), 7.40−7.35 (m, 1H), 7.32 (ddd, J = 4.5, 3.6, 1.9 Hz, 9H), 7.20−7.16 (m, 7H), 7.12 (d, J = 8.2 Hz, 1H), 6.99 (dt, J = 58.5, 8.2 Hz, 1H), 6.67−6.37 (m, 2H), 6.36−6.18 (m, 3H), 5.58 (d, J = 20.7 Hz, 1H), 4.80−4.40 (m, 3H), 4.32−4.19 (m, 1H), 3.80−3.30 (m, 17H), 3.16−2.89 (m, 5H), 2.86−2.81 (m, 1H), 2.75−2.60 (m, 4H), 2.25−2.12 (m, 2H), 1.97−1.53 (m, 15H), 1.41 (d, J = 1.8 Hz, 11H). 13C NMR (126 MHz, chloroform-d) δ 175.0, 174.7, 173.4, 172.1, 156.3, 156.2, 154.4, 154.3, 152.7, 152.4, 143.05, 143.01, 137.2, 135.8, 135.0, 132.1, 131.7, 130.1, 129.6, 129.0, 128.4, 127.85, 127.81, 127.79, 126.7, 126.0, 125.8, 121.2, 121.0, 109.9, 109.8, 105.7, 105.4, 80.6, 80.5, 80.14, 80.12, 78.93, 78.88, 70.6, 70.3, 70.10, 70.06, 69.9, 69.0, 68.7, 61.9, 60.3, 60.2, 55.5, 52.3, 52.1, 52.0, 49.9, 49.6, 48.3, 46.5, 46.0, 45.7, 42.9, 40.5, 39.20, 39.18, 39.10, 38.8, 35.9, 35.8, 28.4, 28.2, 28.1, 28.0, 26.24, 26.18, 25.5, 25.4, 24.4, 24.2.
tert-Butyl 4-(2-(3-((R)-3-((4-(1H-Pyrazol-4-yl)benzyl)(2-(2-(2(5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)ethoxy)ethoxy)ethyl)carbamoyl)piperidin-1-yl)phenoxy)-2-methylpropanoyl)piperazine-1-carboxylate (46).
1H NMR (500 MHz, chloroform-d) δ 8.31 (dd, J = 11.9, 0.9 Hz, 1H), 7.99 (dd, J = 8.5, 0.9 Hz, 1H), 7.59−7.52 (m, 1H), 7.52−7.44 (m, 1H), 7.26 (s, 1H), 7.23−7.13 (m, 2H), 7.01 (dt, J = 56.2, 8.2 Hz, 1H), 6.62−6.18 (m, 4H), 5.64 (d, J = 23.7 Hz, 1H), 4.92−4.50 (m, 3H), 4.47 (dt, J = 12.8, 6.4 Hz, 1H), 4.28 (dt, J = 13.4, 6.4 Hz, 1H), 3.84−3.25 (m, 18H), 3.19−2.63 (m, 8H), 2.24−2.15 (m, 2H), 1.99−1.54 (m, 15H), 1.42 (d, J = 1.7 Hz, 11H).
(R)-N-(4-(1H-Pyrazol-4-yl)benzyl)-1-(3-((2-methyl-1-oxo-1-(piperazin-1-yl)propan-2-yl)oxy)phenyl)-N-(2-(2-(2-(5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)ethoxy)ethoxy)ethyl)piperidine-3-carboxamide (Biotin-ZW4864 as TFA Salt).
1H NMR (500 MHz, methanol-d4) δ 7.98 (d, J = 16.4 Hz, 2H), 7.60 (dd, J = 38.8, 8.0 Hz, 2H), 7.26 (t, J = 7.7 Hz, 2H), 7.12 (dt, J = 88.7, 8.2 Hz, 1H), 6.82−6.58 (m, 1H), 6.54−6.18 (m, 2H), 4.85−4.53 (m, 4H), 4.45 (dd, J = 7.8, 4.9 Hz, 1H), 4.25 (dt, J = 7.8, 3.8 Hz, 1H), 4.11 (d, J = 36.6 Hz, 2H), 3.84 (s, 2H), 3.70 (ddd, J = 23.9, 12.2, 7.4 Hz, 3H), 3.62−3.51 (m, 8H), 3.46 (d, J = 5.5 Hz, 1H), 3.36 (d, J = 5.5 Hz, 1H), 3.29 (s, 1H), 3.19−2.64 (m, 10H), 2.19 (t, J = 7.4 Hz, 2H), 2.07−1.51 (m, 15H), 1.39 (p, J = 7.8 Hz, 2H). 13C NMR (126 MHz, methanol-d4) δ 175.5, 175.4, 174.7, 172.32, 172.28, 164.7, 156.15, 156.06, 151.4, 135.6, 135.4, 132.1, 131.8, 130.7, 130.0, 129.8, 127.9, 126.7, 125.7, 125.4, 121.8, 110.9, 110.4, 107.9, 106.2, 105.7, 80.6, 80.4, 70.3, 70.0, 69.9, 69.23, 69.21, 68.7, 68.2, 61.9, 60.2, 55.6, 53.0, 51.9, 50.7, 49.8, 46.3, 43.1, 42.7, 39.7, 39.5, 39.1, 39.0, 38.7, 38.2, 35.35, 35.33, 28.37, 28.36, 28.1, 27.4, 27.1, 25.5, 25.05, 25.01, 24.9, 23.6, 23.4. HRMS (ESI) calcd for C46H65N9O7S (M + Na)+ 910.4620, found 910.4616. HPLC purity 96.0%, tR = 8.83 min.
Protein Expression and Purification.
Full-length β-catenin (residues 1−781), β-catenin R1C (residues 138−781), and β-catenin (residues 1−686) were cloned into the pEHISTEV vector carrying an N-terminal 6× histidine tag and transformed into Escherichia coli BL21 DE3 (Novagen). Cells were cultured in Luria-Bertani (LB) medium with 50 μg/mL kanamycin until the OD600 was approximately 0.8, and then protein expression was induced with 400 μM of isopropyl β-D-1-thiogalactopyranoside (IPTG) at 20 °C overnight. Cells were lysed by sonication. The proteins were purified by three steps of chromatography, including Ni-NTA affinity chromatography (30210, Qiagen), HiTrap Q HP anion-exchange chromatography (17–1154-01, GE Healthcare Life Science), and size-exclusion chromatography with a HiLoad 26/600 Superdex 200 pg column (28–9893-36, GE Healthcare Life Science) using an AKTA Pure FPLC (GE Healthcare Life Science) system. Protein was eluted in a buffer containing 20 mM of Tris (pH 8.5), 100 mM of NaCl, and 2 mM of dithiothreitol (DTT). The purity of β-catenin was greater than 95% as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel analyses. Thermal shift assays were performed on a CFX96 Real-Time System (Bio-Rad) to monitor protein stability and detect protein aggregation. Protein unfolding was evaluated through measurement of the fluorescence changes of the fluorescent dye Sypro Orange when interacting with β-catenin proteins. A temperature increment of 1°/min was applied. One set of representative results is shown in Figure S19. All proteins were stable, and no aggregation was observed under storage or assay conditions. Proteins were aliquoted and stored at −80 °C.
BCL9 Peptide Synthesis and Purification.
Human BCL9 (residues 350−375), N-terminally biotinylated human BCL9 (residues 350−375), human E-cadherin (residues 824−877), and N-terminally biotinylated human E-cadherin (residues 824−877) were synthesized by InnoPep Inc. (San Diego, CA, www.innopep.com). All synthesized peptides were purified by HPLC with purity >95%. The structures were validated by LC/MS. The sequences of the peptides are shown in Table 3 (Ahx, 6-aminohexanoic acid).
Table 3.
Sequences of Peptides
| peptide | sequence | |
|---|---|---|
| AlphaScreen | BCL9 26-mer | H-350GLSQEQLEHRERSLQTLRDIQRMLFP375-NH2 |
| biotinylated BCL9 26-mer | Biotin-Ahx-350GLSQEQLEHRERSLQTLRDIQRMLFP375-NH2 | |
| E-cadherin 54-mer | H-824APPYDSLLVFDYEGSGSEAASLSSLNSSESDKDQDYDYLNEWGNRFKKLADMYG877-NH2 | |
| biotinylated E-cadherin 54-mer | Biotin-824APPYDSLLVFDYEGSGSEAASLSSLNSSESDKDQDYDYLNEWGNRFKKLADMYG877-NH2 | |
| SPR | wild-type BCL9 peptide | Biotin-(PEG)4-350GLSQEQLEHRERSLQTLRDIQRMLFP375-NH2 |
| mutant BCL9 peptide | Biotin-(PEG)4-350GLSQEQLEAAERSLQTARDAQRMAAP375-NH2 |
AlphaScreen Assays of β-Catenin and BCL9 Interaction.
Experiments were performed in white opaque 384-well plates from PerkinElmer (Waltham, MA), and the samples were read on a Biotek Synergy 2 plate reader (Winooski, VT) with an excitation at 680 nm and an emission at 570 nm. The standard AlphaScreen protocol was used with a sensitivity setting of 200. All dilutions were made in a 1× assay buffer containing 25 mM N-(2-hydroxyethyl)piperazine-N′ethanesulfonic acid (Hepes) (pH 7.4), 100 mM NaCl, 0.01% Triton X-100, and 0.1% bovine serum albumin (BSA) to minimize nonspecific interactions. For the competitive inhibition assays of the β-catenin/BCL9 PPI, the negative control (equivalent to 0% inhibition) refers to 5.0 nM biotinylated BCL9, 40 nM His6-tagged β-catenin, and 10 μg/mL donor and acceptor beads in a final volume of a 25 μL assay buffer, but no tested inhibitor was present. The positive control (equivalent to 100% inhibition) refers to 5.0 nM biotinylated BCL9 and 10 μg/mL donor and acceptor beads in a final volume of a 25 μL assay buffer. For the competitive inhibition assays of the β-catenin/E-cadherin PPI, the negative control (equivalent to 0% inhibition) refers to 10 nM biotinylated E-cadherin, 40 nM His6tagged β-catenin, and 10 μg/mL donor and acceptor beads in a final volume of a 25 μL assay buffer. The positive control (equivalent to 100% inhibition) of the β-catenin/E-cadherin PPI refers to 10 nM biotinylated E-cadherin and 10 μg/mL donor and acceptor beads in a final volume of a 25 μL assay buffer.
For the β-catenin/BCL9 assay, 5 nM biotinylated BCL9 and 50 nM His6-tagged β-catenin were incubated in an assay buffer for 30 min. For the β-catenin/E-cadherin assay, 10 nM biotinylated human E-cadherin and 50 nM His6-tagged human β-catenin were added and incubated in an assay buffer for 30 min. Different concentrations of the tested inhibitor were added and incubated in a 20 μL assay buffer for another 1 h. All of the assay plates were covered and gently mixed on an orbital shaker. The donor and acceptor beads were then added to the plates to a final concentration of 10 μg/mL in a 25 μL assay buffer. The mixture was incubated for 1 h at 4 °C before detection. The IC50 value was determined by the nonlinear least-square analysis of GraphPad Prism 8.0. The Ki values were derived from the IC50 values. The equation used is Ki = [I]50/([L]50/Kd + [P]0/Kd + 1) (where [I]50 denotes the concentration of the free inhibitor at 50% inhibition, [L]50 is the concentration of the free labeled ligand at 50% inhibition, [P]0 is the concentration of the free protein at 0% inhibition, and Kd is the dissociation constant of the protein−ligand complex). This equation used free concentrations to derive Kis for competitive inhibition assays (https://bioinfo-abcc.ncifcrf.gov/IC50_Ki_Converter/index.php).65,66 All of the experiments were performed in triplicate and carried out in the presence of 1% DMSO for small-molecule inhibitors. Each compound was assayed at least by two independent experiments. The results were expressed as mean ± standard deviation. The inhibitor selectivity for β-catenin/BCL9 over β-catenin/E-cadherin interactions was defined as the ratio of the respective Ki value of β-catenin/E-cadherin interactions over that of β-catenin/BCL9 interactions.
Fluorescence Anisotropy Binding Assays to Determine the KD values.
The fluorescence anisotropy binding assays were performed in 96-well Microfluor 2 black plates (Thermo Fisher Scientific), and the sample signals were read by a Synergy 2 plate reader (Biotek). The experiments were performed in an assay buffer of 25 mM Hepes (pH 7.4), 100 mM NaCl, 0.01% Triton X-100, and 100 μg/mL γ-globulin. The concentration of human BCL9 or E-cadherin fluorescence tracer was fixed at 5 nM. Different concentrations of full-length human β-catenin were added to the assay buffer giving a final volume of 100 μL. After the addition, each assay plate was covered and gently mixed on an orbital shaker for 2 h before the data were recorded at room temperature with an excitation wavelength at 485 nm and an emission wavelength at 535 nm. Each experiment was repeated three times, and the results were expressed as mean ± standard deviation. The parallel fluorescence intensity (Is), the perpendicular fluorescence intensity (Ip), and the anisotropy (r) were recorded directly by the plate reader. The total intensity (I) and the fraction ligand bound (Lb) are calculated by eqs 1 and 2 shown below67,68
| (1) |
G is the G factor. It was 0.993 for the instrument used
| (2) |
rmin is the average anisotropy value for the fluorescently labeled peptide, and rmax is the average anisotropy value for the fluorescently labeled peptide with saturated β-catenin
Ibound is the average intensity value for the fluorescently labeled peptide with saturated β-catenin, and Iunbound is the average intensity value for the fluorescently labeled peptide
The above data were then imported to GraphPad Prism 8.0, and the KD value for the protein−protein interaction (PPI) between β-catenin and BCL9 or E-cadherin was analyzed by the nonlinear regression equation (eq 3) shown below
| (3) |
. [fluorescent-peptide], the concentration was 5 nM. X = [β-catenin].
AlphaScreen Competitive Binding Assays to Determine the KD Values.
AlphaScreen competitive binding experiments were performed to determine the apparent KD values of β-catenin/BCL9 and β-catenin/E-cadherin.44 In the competitive binding experiments to determine the KD value of full-length β-catenin/BCL9 interaction, 5 nM of N-terminally biotinylated BCL9, 0.5 nM of N-terminally full-length His6-tagged β-catenin, and different concentrations of unlabeled BCL9 peptide (0−50 μM) were incubated at 4 °C in a 20 μL assay buffer for 2 h. In the competitive binding experiments to determine the KD value of full-length β-catenin/E-cadherin interactions, 5 nM of N-terminally biotinylated E-cadherin, 0.5 nM of N-terminally full-length His6-tagged β-catenin, and different concentrations of unlabeled E-cadherin peptide (0−2000 nM) were incubated at 4 °C in a 20 μL assay buffer for 2 h. The streptavidin-coated donor beads and nickel chelate acceptor beads were added to a final concentration of 10 μg/mL in a 25 μL assay buffer. The mixture was covered black and incubated for 2 h before detection. The IC50 values, which were also the apparent KD values from the AlphaScreen assay, were determined by nonlinear least-squares analyses using GraphPad Prism 8.0. The IC50 values were determined by nonlinear regression (curve fit) by using a one-site competition binding model (Y = bottom + (top − bottom)/(1 + 10X − Log IC50)), X is the logarithm of concentration, and Y is the bound form. Each experiment was repeated three times. The results were expressed as mean ± standard deviation.
β-Catenin Pull-Down Experiments.
SW480 cancer cells with adherently hyperactive β-catenin signaling (70−80% confluency) in T75 flask were lysed first in 1 mL buffer A containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 2 mM ethylenediaminetetraacetic acid (EDTA), and protease inhibitors. Cell debris was removed by centrifugation at 10 000g for 20 min at 4 °C. In 500 μL SW480 cell lysates or 3 μg purified full-length β-catenin protein in 300 μL buffer B (20 mM Tris pH 7.4,150 mM NaCl, and 0.5% NP-40), 1 or 10 μM biotinylated inhibitor was added in and incubated at 4 °C for 3 h. Then, 25 μL of streptavidin sepharose beads (S-1638, Sigma) was added to the lysate mixture and rotated at 4 °C for 2 h. The lysate mixture was centrifuged at 4000 rpm for 2 min at 4 °C. The beads were washed with buffer B four times. The beads were resuspended in 60 μL of 2× SDS sample buffer. After boiling, the samples were loaded onto an 8% SDS polyacrylamide gel for electrophoretic analysis. Separated proteins were transferred onto nitrocellulose membranes for immunoblot analysis. The antibody against β-catenin (610153, BD Biosciences. Immunogen: mouse β-catenin residues 571−781) was incubated with the membranes. IRDye 800CW goat antirabbit IgG (926–32211, LiCOR) was used as the secondary antibody. The images were detected by the Odyssey Infrared Imaging System (LiCOR). Experiments were performed in triplicate.
SPR Experiments.
Experiments were performed on a Biacore T100 instrument (GE Healthcare). N-Terminally biotinylated human BCL9 (residues 350−375) was immobilized to the surface of a Series S Sensor Chip SA (GE Healthcare) by streptavidin−biotin capturing. Approximately 630 RUs of peptide were immobilized on flow cell 2 (Fc-2). N-Terminally biotinylated mutant human BCL9 (residues 350−375) was immobilized on flow cell 1 (Fc-1) with around 630 RU immobilization as reference surface. Serially diluted inhibitors (0.0013−30 μM) were mixed with full-length β-catenin (residues 1−781) 700 nM in running buffer (20 mM Tris·HCl, pH 7.5, 100 mM NaCl, 0.005% P-20, 1% DMSO) and incubated for 1 h at room temperature. Then, the mixtures were injected over surfaces at 25 °C at a flow rate of 20 μL/min for 600 s with a dissociation time of 420 s. Regeneration was a 75 s injection once with buffer (10 mM glycine· HCl and 5% glycerol, pH 3.0) at 30 μL/min. To subtract background noise from each data set, all samples were also run over reference surface, and random injections of running buffer were performed throughout every experiment (double referencing). Data were fit to a simple 1:1 interaction with the global data analysis program GraphPad Prism 8.0.
Co-IP Experiments.
HCT116 cancer cells with hyperactive β-catenin signaling at 1 × 106 cells/mL were treated with different concentrations of the inhibitor for 24 h. Cells were lysed in buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 2 mM EDTA, and protease inhibitors. For Pygo2-BCL9 and Pygo-β-catenin co-IP experiments, HCT116 cell nuclear extracts were used. The nuclear extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (78835, Thermo Scientific). The cell lysates or nuclear extracts were preadsorbed to A/G plus agarose (sc-2003, Santa Cruz Biotechnology) at 4 °C for 1 h. Preadsorbed lysates were incubated with a specific primary antibody against β-catenin (610153, BD Biosciences) or Pygo2 (MA524240, Invitrogen. Immunogen: human Pygopus-2 residues 160−336) overnight at 4 °C. A/G plus agarose was then added to the lysate mixture and incubated for 3 h. The beads were washed four times with the lysis buffer at 4 °C. The bound protein was eluted by boiling in the SDS sample buffer and loaded onto an 8% SDS polyacrylamide gel for electrophoretic analysis. Separated proteins were transferred onto nitrocellulose membranes for immunoblot analysis. The antibodies against BCL9 (ab37305, Abcam. Immunogen: human BCL9 residues 800−900), β-catenin (610153, BD Biosciences), E-cadherin (610404, BD Biosciences. Immunogen: human E-cadherin residues 735−883), Pygo2 (MA524240, Invitrogen), β-tubulin (sc-55529, Santa Cruz Biotechnology. Immunogen: human β-tubulin residues 210−444), and lamin B1 (sc-374015, Santa Cruz Biotechnology. Immunogen: mouse lamin B1 residues: 559−586) were incubated with the membranes. IRDye 680LT goat antimouse IgG (827–11080, LiCOR) and IRDye 800CW goat antirabbit IgG (926–32211, LiCOR) were used as the secondary antibodies. The images were detected by the Odyssey Infrared Imaging System (LiCOR). Experiments were performed in triplicate.
Cell Transfection and Luciferase Assays.
A FuGENE6 (E269A, Promega) 96-well plate format was used for the transfection of cells according to the manufacturer’s instruction. HEK293 cells were cotransfected with 45 ng of TOPFlash or FOPFlash reporter gene, 135 ng of pcDNA3.1−β-catenin, and 20 ng of pCMV-RL normalization reporter gene. SW480 or MDA-MB-468 cells were cotransfected with 60 ng of TOPFlash or FOPFlash firefly luciferase reporter gene and 40 ng of renilla luciferase pCMV-RL normalization reporter. HEK293 or SW480 cells were cultured in DMEM and 10% FBS at 37 °C for 24 h, and different concentrations of inhibitors or DMSO were added and incubated in DMEM with 5% FBS. MDA-MB-468 cells were treated with Wnt3a (100 ng/mL) (rmW3aL-010, Time Bioscience) for 30 min, and different concentrations of inhibitors or DMSO was added and incubated in DMEM with 5% FBS. After the 24-h incubation, the luciferase reporter activity was measured using the Dual-Glo system (E2940, Promega). Normalized luciferase activity in response to the treatment with inhibitors was compared with that obtained from the cells treated with DMSO. Experiments were performed in triplicate.
qPCR Analyses.
SW480 cells at 1 × 106 cells/mL were treated with inhibitors at different concentrations for 24 h. MDA-MB-231 cells at 1 × 106 cells/mL were treated with Wnt 3a (100 ng/mL) for 30 min, and then the inhibitors at different concentrations were added and incubated for 24 h. Total RNAs were extracted with TRIzol (15596026, Life Technologies), and the cDNA was synthesized with the superscript III first-strand kit (18080–051, Invitrogen). qPCR experiments were performed using the iQTM SYBR green supermix kit (170–8880, BIO-RAD) on a CFX96 Real-Time System (BIO-RAD). The threshold cycle (CT) values were normalized to that of internal reference GAPDH. Experiments were performed in triplicate. The primer pairs are human GAPDH forward: 5′-GAAGGTGAAGGTCGGAGTC-3′, reverse: 5′-GAAGATGGTGATGGGATTTC-3′; human HPRT forward: 5′-GCTATAAATTCTTTGCTGACCTGCTG-3′, reverse: 5′-AATTACTTTTATGTCCCCTGTTGACTGG-3′; human AXIN2 forward: 5′AGTGTGAGGTCCACGGAAAC-3′, reverse: 5′-CTTCACACTGCGATGCATTT-3′; human LEF1 forward: 5′-GACGAGATGATCCCCTTCAA-3′, reverse: 5′-AGGGCTCCTGAGAGGTTTGT-3′; human LEF1 forward: 5′-CCAATCTCAGCACCAGAATGTG-3′, reverse: 5′-TTGCTAGGCGAGATCTGGTTG3′; human cyclin D1 forward: 5′-ACAAACAGATCATCCGCAAACAC-3′, reverse: 5′-TGTTGGGGCTCCTCAGGTTC-3′; human TWIST forward: 5′-GTCCGCAGTCTTACGAGGAG-3′, reverse: 5′-TGGAGGACCTGGTAGAGGAA-3′; human SNAIL forward: 5′-GGTTCTTCTGCGCTACTGCT-3′, reverse: 5′-TAGGGCTGCTGGAAGGTAAA-3′; human TENASCIN C forward: 5′-GAGATTTAGCCGTGTCTGAGGTTG-3′, reverse: 5′-GCCATCCAGGAGAGATTGAAGC-3′; human LGR5 forward: 5′TGCTGGCTGGTGTGGATGCG-3′, reverse: 5′-GCCAGCAGGGCACAGAGCAA-3′; human VEGF-A forward: 5′ -CTTGCCTTGCTGCTCTACC-3′, reverse: 5′-CACACAGGATGGCTTGAAG-3′; human CD44 forward: 5′CGCTTTGCAGGTGTATTCCA-3′, reverse: 5′-ACCACGTGCCCTTCTATGAA-3′; human Slug forward: 5′-GGGGAGAAGCCTTTTTCTTG-3′, reverse, 5′-TCCTCATGTTTGTGCAGGAG-3′.
Western Blotting of Wnt Target Genes.
SW480 cells at 1 × 106 cells/mL were treated with different concentrations of inhibitors for 24 h. MDA-MB-231 cells at 1 × 106 cells/mL were treated with Wnt3a (100 ng/mL) for 30 min, then the inhibitors at different concentration were added and incubated for 24 h. Cells were lysed in buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitors. After centrifugation at 12 000 rpm for 20 min at 4 °C, the supernatant was loaded onto an 8% SDS polyacrylamide gel for electrophoretic analysis. Separated proteins were transferred onto nitrocellulose membranes for immunoblot analysis. The antibodies against Axin2 (MA5–15015, Thermo Fisher. Immunogen: residues surrounding Pro566 of human Axin2), cyclin D1 (sc-853, Santa Cruz Biotechnology. Immunogen: human cyclin D1 residues 1−295), and β-tubulin (sc-55529, Santa Cruz Biotechnology) were incubated with the membranes overnight at 4 °C. IRDye 680LT goat antimouse IgG (827–11080, LiCOR) or IRDye 800CW goat antirabbit IgG (827–08365, LiCOR) was used as the secondary antibody. The images were detected by the Odyssey Infrared Imaging System (LiCOR). Experiments were performed in triplicate.
MTS Cell Growth Inhibition Experiments.
Colorectal cancer cells (SW480 and HCT116) and TNBC cells (MDA-MB-231, MDA-MB-468) were seeded in 96-well plates at 5 × 103 cells/well in DMEM with 10% fetal calf serum (FCS), maintained overnight at 37 °C, and then incubated with the tested compounds at various concentrations in DMEM with 5% FBS. Human mammary epithelial cell MCF10A was seeded in 96-well plates at 1 × 104 cells/well in MEGM (CC-3150, Lonza) with 100 ng/mL cholera toxin, maintained overnight at 37 °C, and incubated with the tested compounds at various concentrations. Cell viability was monitored after 72 h using a freshly prepared mixture of 1 part phenazine methosulfate (PMS, Sigma-Aldrich) solution (0.92 mg/mL) and 19 parts 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS, Promega) solution (2 mg/mL). Cells were incubated in 10 μL of this solution at 37 °C for 3 h, and A490 was measured. The effect of each compound was expressed as the concentration required to reduce A490 by 50% (IC50) relative to DMSO-treated cells. Experiments were performed in triplicate.
β-Catenin Rescue Experiments.
TNBC MDA-MB-231 and MDA-MB-468 cells were seeded in 96-well plates at 5 × 103 cells/well in DMEM with 10% FCS, maintained overnight at 37 °C. Then, 250 ng of pcDNA3.1−β-catenin or empty vector pcDNA3.1 per well was transfected into the cells, and 1 h later, the tested compounds at various concentrations were added into the cells. Cell viability was monitored after 72 h using a freshly prepared mixture of 1 part phenazine methosulfate (PMS, Sigma-Aldrich) solution (0.92 mg/mL) and 19 parts 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS, Promega) solution (2 mg/mL). Cells were incubated in 10 μL of this solution at 37 °C for 3 h, and A490 was measured. The effect of each compound is expressed as the concentration required to reduce A490 by 50% (IC50) relative to DMSO-treated cells. Experiments were performed in triplicate.
Clonogenic Assays.
Cells were plated in six-well dishes in triplicate at a density of 1000 cells per well. After incubation overnight, inhibitor or vehicle (0.1% DMSO) was added to the medium (DMEM with 10% FCS) for 72 h, and cells were allowed to grow out for 7−10 days, during which the medium was changed every 2−3 days without adding the compound. Colonies were fixed in acetic acid/methanol (1:7, v/v) for 5 min, stained with 0.5% crystal violet for 2 h at room temperature, and destained with water. Colonies with more than 50 cells were counted using a low-magnification light microscope. Experiments were performed in triplicate.
FACS Experiments.
Cells (2.5 × 105) seeded in triplicate in six-well dishes were cultured with compounds or vehicle (0.1% DMSO) for 72 h. Cells were then harvested and stained with annexin V−fluorescein isothiocyanate (FITC) and propidium iodide (PI) using the FITC Annexin V Apoptosis Detection Kit (556547, BD Biosciences) as per the manufacturer’s instructions and analyzed with the FACSCanto II flow cytometer. Experiments were performed in triplicate.
Scratch Wound Healing Experiments.
To the confluent monolayer of MDA-MB-231 cells in 24-well plates, wounds will be made by scraping a sterile 200 μL pipette tip. Cells were maintained in DMEM containing 10% FBS with 10 μg/mL of mitomycin to inhibit cell proliferation and with different concentrations of inhibitors. Images of wounds were taken immediately and 14 h after wounding. Experiments were performed in triplicate.
Matrigel Invasion Experiments.
MDA-MB-231 cells (5 × 104 cells) suspended in a 200 μL starvation medium were added to the upper chamber of a Matrigel-coated insert with a 6.5 mm diameter and an 8 mm pore size (353097, Corning). The insert was placed in a 24-well plate containing 600 mL of DMEM medium with 10% FBS. Inhibitors were added to both the upper and the lower chambers. Invasion assays were performed for 24 h, and cells were fixed with 3.7% formaldehyde. Cells were stained with crystal violet staining solution. The cells on the upper side of the insert were removed with a cotton swab. Five randomly selected fields (10× objectives) on the lower side of the insert were photographed, and the invaded cells were counted. Experiments were performed in triplicate.
Three-Dimensional (3D) Spheroid BME Cell Invasion Assays.
The 3D invasion assays were performed using the 96-well 3D Spheroid BME Cell Invasion Assay kit (3500–096-K, Cultrex) following the manufacturer’s protocol. Briefly, MDA-MB-231 cells were mixed with the kit-supplied ECM and seeded into ultralow attachment plates (3,000 cells/well). The plates were then centrifuged at 300g for 5 min and incubated at 37 °C for 72 h for spheroid formation. After that, the kit-supplied BME matrix was added and preincubated on ice for 10 min, followed by centrifugation at 300g, 4 °C for 5 min. Then, the inhibitor at various concentrations was added. The plates were cultured for 6 days at 37 °C. The spheroid in each well was photographed every 24 h using the 4× objective. The images were analyzed using the image analysis software ImageJ to measure the changes in the area of the invasive structures to determine the extent of 3D culture BME cell invasion for each sample.
DMPK and PD Studies.
Microsome stability was evaluated by incubating 1 μM of compound with 1 mg/mL of hepatic microsomes (human, rat, or mouse) in 100 mM of potassium phosphate buffer, pH 7.4. The reactions were held at 37 °C with continuous shaking. The reaction was initiated by adding reduced nicotinamide adenine dinucleotide phosphate (NADPH) (1 mM final concentration). The final incubation volume was 300 μL, and 40 μL aliquots were removed at 0, 5, 10, 20, 40, and 60 min. The removed aliquot was added to 160 μL acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction was evaluated in parallel incubations without NADPH. At the end of the assay, the samples were centrifuged through a 0.45 μm filter plate (MSRLN0450, Millipore Multiscreen Solventer low binding poly(tetrafluoroethylene) (PTFE) hydrophilic plates) and analyzed by LC-MS/MS. The data were log-transformed, and the results are reported as half-life and intrinsic clearance.
The procedures of mouse PK studies are covered under existing protocols and have been approved by the Scripps Florida IACUC (IACUC Protocol #: 15–022-02) to be conducted in the Scripps vivarium, which is fully AAALAC accredited. Pharmacokinetics were determined in n = 3 male C57BL/6 mice (Charles River, strain code #027). Compounds were dosed as indicated via intravenous (i.v.) injection via tail vein or oral (p.o.) gavage. Twenty-five microliters of blood was collected via a small nick in the tail using heparin-coated hematocrit capillary tubes, which were sealed with wax and kept on ice until plasma was generated by centrifugation using a refrigerated centrifuge equipped with a hematocrit rotor. Dose levels are provided as described. Time points for determination of PK parameters were 5 min (i.v. only), 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, and 24 h (p.o. only). Plasma concentrations were determined via LC-MS/MS using an eight-point standard curve between 2 and 2000 ng/mL prepared in mouse plasma. The PK analyses were done with WinNonLin, Pharsight Inc., using a noncompartmental model.
For PD analyses, female Fox Chase SCID-Beige mice (Charles River, strain code no. 250, n = 4) with orthotopically implanted PDX 4013 tumors were used. All animal studies were approved by the University of South Florida/Moffitt Cancer Center Animal Care and Use Committee (IACUC Protocol no. R IS00005598). ZW4864 was dosed as indicated in the text via oral gavage. Tumor was snap-frozen in liquid nitrogen and kept at −80 °C until processed. The PD studies were performed from tumor samples. RNA was extracted using Qiagen RNAeasy kit (74104, Qiagen) as per the manufacturer’s instructions, and qPCR experiments were performed as described above.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Susan G. Komen Career Catalyst Research Grant CCR16380693, the Floridian Breast Cancer Foundation Scientific Grant (19012901), and the 2020 Moffitt Team Science Grant. The H. Lee Moffitt Cancer Center & Research Institute is an NCI-designated Comprehensive Cancer Center, supported under NIH grant P30CA76292. The authors thank Dr. Michael Cameron and his team in the Drug Metabolism and Pharmacokinetics (DMPK) core facility at the Scripps Research Institute for microsome stability and mouse PK studies.
ABBREVIATIONS
- APC
adenomatous polyposis coli
- BCL9
B-cell lymphoma 9
- BCL9L
BCL9-like
- CBP
CREB-binding protein
- CK1α
casein kinase 1α
- co-IP
coimmunoprecipitation
- Dvl
disheveled
- FACS
fluorescence-activated cell sorting
- Fzd
frizzled
- GSK3β
glycogen synthase kinase 3β
- Ki
inhibition constant
- LEF
lymphoid enhancer-binding factor
- PAINS
pan-assay interference compounds
- PNPB
3-(4-fluorophenyl)-N-phenylbenzamide
- PP2A
protein phosphatase 2A
- PPI
protein–protein interaction
- Pygo
Pygopus
- qPCR
quantitative real-time PCR
- TCF
T-cell factor
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00742.
Dose–response curves for AlphaScreen competitive inhibition assays, SPR competitive inhibition assays, AlphaScreen competitive binding assays of full-length β-catenin and Biotin-ZW4864, fluorescence anisotropy binding assays, AlphaScreen competitive binding assays, TOPFlash luciferase reporter, and MTS growth inhibition assays; uncropped Western blot gels for protein pull-down, co-IP, and Western blot experiments; original data plots of C57BL/6 mouse PK studies; original images for clonogenic assays, matrigel invasion, and 3D spheroid BME cell invasion; HPLC condition and traces; NMR spectra; and original FACS reports (PDF)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c00742
The authors declare the following competing financial interest(s): A provisional patent application has been filed based on these results.
Contributor Information
Zhen Wang, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States.
Min Zhang, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States.
Victor Quereda, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States.
Sylvia M. Frydman, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States
Qianqian Ming, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States.
Vincent C. Luca, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States.
Derek R. Duckett, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States
Haitao Ji, Drug Discovery Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612-9497, United States.
REFERENCES
- (1).Nusse R; Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [DOI] [PubMed] [Google Scholar]
- (2).Liu Z; Wang P; Wold EA; Song Q; Zhao C; Wang C; Zhou J. Small-molecule inhibitors targeting the canonical WNT signaling pathway for the treatment of cancer. J. Med. Chem 2021, 64, 4257–4288. [DOI] [PubMed] [Google Scholar]
- (3).Liu J; Pan S; Hsieh MH; Ng N; Sun F; Wang T; Kasibhatla S; Schuller AG; Li AG; Cheng D; Li J; Tompkins C; Pferdekamper A; Steffy A; Cheng J; Kowal C; Phung V; Guo G; Wang Y; Graham MP; Flynn S; Brenner JC; Li C; Villarroel MC; Schultz PG; Wu X; McNamara P; Sellers WR; Petruzzelli L; Boral AL; Seidel HM; McLaughlin ME; Che J; Carey TE; Vanasse G; Harris JL Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Madan B; Ke Z; Harmston N; Ho SY; Frois AO; Alam J; Jeyaraj DA; Pendharkar V; Ghosh K; Virshup IH; Manoharan V; Ong EH; Sangthongpitag K; Hill J; Petretto E; Keller TH; Lee MA; Matter A; Virshup DM Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yang D; Fu W; Li L; Xia X; Liao Q; Yue R; Chen H; Chen X; An S; Zeng C; Wang WE Therapeutic effect of a novel Wnt pathway inhibitor on cardiac regeneration after myocardial infarction. Clin. Sci 2017, 131, 2919–2932. [DOI] [PubMed] [Google Scholar]
- (6).Bhamra I; Armer R; Bingham M; Eagle C; Cook AE; Phillips C; Woodcock S. Porcupine inhibitor RXC004 enhances immune response in pre-clinical models of cancer. Cancer Res. 2018, 78, No. 3764. [Google Scholar]
- (7).Le PN; McDermott JD; Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol. Ther 2015, 146, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jimeno A; Gordon M; Chugh R; Messersmith W; Mendelson D; Dupont J; Stagg R; Kapoun AM; Xu L; Uttamsingh S; Brachmann RK; Smith DC A first-in-human phase I study of the anticancer stem cell agent Ipafricept (OMP54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin. Cancer Res 2017, 23, 7490–7497. [DOI] [PubMed] [Google Scholar]
- (9).Gurney A; Axelrod F; Bond CJ; Cain J; Chartier C; Donigan L; Fischer M; Chaudhari A; Ji M; Kapoun AM; Lam A; Lazetic S; Ma S; Mitra S; Park IK; Pickell K; Sato A; Satyal S; Stroud M; Tran H; Yen W-C; Lewicki J; Hoey T. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 11717–11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lyou Y; Habowski AN; Chen GT; Waterman ML Inhibition of nuclear Wnt signalling: challenges of an elusive target for cancer therapy. Br. J. Pharmacol 2017, 174, 4589–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cui C; Zhou X; Zhang W; Qu Y; Ke X. Is β-catenin a druggable target for cancer therapy? Trends Biochem. Sci 2018, 43, 623–634. [DOI] [PubMed] [Google Scholar]
- (12).Wang Z; Li Z; Ji H. Direct targeting of β-catenin in the Wnt signaling pathway: Current progress and perspectives. Med. Res. Rev 2021, 41, 2109–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sampietro J; Dahlberg CL; Cho US; Hinds TR; Kimelman D; Xu W. Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 2006, 24, 293–300. [DOI] [PubMed] [Google Scholar]
- (14).Graham TA; Weaver C; Mao F; Kimelman D; Xu W. Crystal structure of a β-catenin/Tcf complex. Cell 2000, 103, 885–896. [DOI] [PubMed] [Google Scholar]
- (15).Poy F; Lepourcelet M; Shivdasani RA; Eck MJ Structure of a human Tcf4-β-catenin complex. Nat. Struct. Biol 2001, 8, 1053–1057. [DOI] [PubMed] [Google Scholar]
- (16).Graham TA; Ferkey DM; Mao F; Kimelman D; Xu W. Tcf4 can specifically recognize β-catenin using alternative conformations. Nat. Struct. Biol 2001, 8, 1048–1052. [DOI] [PubMed] [Google Scholar]
- (17).Sun J; Weis WI Biochemical and structural characterization of β-catenin interactions with nonphosphorylated and CK2phosphorylated Lef-1. J. Mol. Biol 2011, 405, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huber AH; Weis WI The structure of the β-catenin/Ecadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 2001, 105, 391–402. [DOI] [PubMed] [Google Scholar]
- (19).Ha NC; Tonozuka T; Stamos JL; Choi HJ; Weis WI Mechanism of phosphorylation-dependent binding of APC to βcatenin and its role in β-catenin degradation. Mol. Cell 2004, 15, 511–521. [DOI] [PubMed] [Google Scholar]
- (20).Xing Y; Clements WK; Le Trong I; Hinds TR; Stenkamp R; Kimelman D; Xu W. Crystal structure of a β-catenin/ APC complex reveals a critical role for APC phosphorylation in APC function. Mol. Cell 2004, 15, 523–533. [DOI] [PubMed] [Google Scholar]
- (21).Fiedler M; Graeb M; Mieszczanek J; Rutherford TJ; Johnson CM; Bienz M. An ancient Pygo-dependent Wnt enhanceosome integrated by Chip/LDB-SSDP. Elife 2015, 4, No. e09073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).van Tienen LM; Mieszczanek J; Fiedler M; Rutherford TJ; Bienz M. Constitutive scaffolding of multiple Wnt enhanceosome components by Legless/BCL9. Elife 2017, 6, No. e20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mani M; Carrasco DE; Zhang Y; Takada K; Gatt ME; Dutta-Simmons J; Ikeda H; Diaz-Griffero F; Pena-Cruz V; Bertagnolli M; Myeroff LL; Markowitz SD; Anderson KC; Carrasco DR BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 2009, 69, 7577–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Elsarraj HS; Hong Y; Valdez KE; Michaels W; Hook M; Smith WP; Chien J; Herschkowitz JI; Troester MA; Beck M; Inciardi M; Gatewood J; May L; Cusick T; McGinness M; Ricci L; Fan F; Tawfik O; Marks JR; Knapp JR; Yeh H-W; Thomas P; Carrasco DR; Fields TA; Godwin AK; Behbod F. Expression profiling of in vivo ductal carcinoma in situ progression models identified B cell lymphoma-9 as a molecular driver of breast cancer invasion. Breast Cancer Res. 2015, 17, No. 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huge N; Sandbothe M; Schröder AK; Stalke A; Eilers M; Schäffer V; Schlegelberger B; Illig T; Vajen B; Skawran B. Wnt status-dependent oncogenic role of BCL9 and BCL9L in hepatocellular carcinoma. Hepatol. Int 2020, 14, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li J; Chen X; Ding X; Cheng Y; Zhao B; Lai ZC; Al Hezaimi K; Hakem R; Guan KL; Wang CY LATS2 suppresses oncogenic Wnt signaling by disrupting β-catenin/BCL9 interaction. Cell Rep. 2013, 5, 1650–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Adachi S; Jigami T; Yasui T; Nakano T; Ohwada S; Omori Y; Sugano S; Ohkawara B; Shibuya H; Nakamura T; Akiyama T. Role of a BCL9-related β-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 2004, 64, 8496–8501. [DOI] [PubMed] [Google Scholar]
- (28).de la Roche M; Worm J; Bienz M. The function of BCL9 in Wnt/β-catenin signaling and colorectal cancer cells. BMC Cancer 2008, 8, No. 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brembeck FH; Schwarz-Romond T; Bakkers J; Wilhelm S; Hammerschmidt M; Birchmeier W. Essential role of BCL9–2 in the switch between β-catenin’s adhesive and transcriptional functions. Genes Dev. 2004, 18, 2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Brembeck FH; Wiese M; Zatula N; Grigoryan T; Dai Y; Fritzmann J; Birchmeier W. BCL9–2 promotes early stages of intestinal tumor progression. Gastroenterology 2011, 141, 1359.e3–1370.e3. [DOI] [PubMed] [Google Scholar]
- (31).Gay DM; Ridgway RA; Muller M; Hodder MC; Hedley A; Clark W; Leach JD; Jackstadt R; Nixon C; Huels DJ; Campbell AD; Bird TG; Sansom OJ Loss of BCL9/9l suppresses Wnt driven tumourigenesis in models that recapitulate human cancer. Nat. Commun 2019, 10, No. 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Moor AE; Anderle P; Cantú C; Rodriguez P; Wiedemann N; Baruthio F; Deka J; André S; Valenta T; Moor MB; Győrffy B; Barras D; Delorenzi M; Basler K; Aguet M. BCL9/ 9L-β-catenin signaling is associated with poor outcome in colorectal cancer. EBioMedicine 2015, 2, 1932–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mieszczanek J; van Tienen LM; Ibrahim AEK; Winton DJ; Bienz M. Bcl9 and Pygo synergise downstream of Apc to effect intestinal neoplasia in FAP mouse models. Nat. Commun 2019, 10, No. 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kawamoto SA; Coleska A; Ran X; Yi H; Yang CY; Wang S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem 2012, 55, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Takada K; Zhu D; Bird GH; Sukhdeo K; Zhao JJ; Mani M; Lemieux M; Carrasco DE; Ryan J; Horst D; Fulciniti M; Munshi NC; Xu W; Kung AL; Shivdasani RA; Walensky LD; Carrasco DR Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med 2012, 4, No. 148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Feng M; Jin JQ; Xia L; Xiao T; Mei S; Wang X; Huang X; Chen J; Liu M; Chen C; Rafi S; Zhu AX; Feng YX; Zhu D. Pharmacological inhibition of β-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci. Adv 2019, 5, No. eaau5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sang P; Zhang M; Shi Y; Li C; Abdulkadir S; Li Q; Ji H; Cai J. Inhibition of β-catenin/B cell lymphoma 9 protein-protein interaction using α-helix-mimicking sulfono-γ-AApeptide inhibitors. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 10757–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).de la Roche M; Rutherford TJ; Gupta D; Veprintsev DB; Saxty B; Freund SM; Bienz M. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun 2012, 3, No. 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).de la Roche M; Ibrahim AE; Mieszczanek J; Bienz M. LEF1 and B9L shield β-catenin from inactivation by Axin, desensitizing colorectal cancer cells to tankyrase inhibitors. Cancer Res. 2014, 74, 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hoggard LR; Zhang Y; Zhang M; Panic V; Wisniewski JA; Ji H. Rational design of selective small-molecule inhibitors for βcatenin/B-cell lymphoma 9 protein-protein interactions. J. Am. Chem. Soc 2015, 137, 12249–12260. [DOI] [PubMed] [Google Scholar]
- (41).Zhang M; Wang Z; Zhang Y; Guo W; Ji H. Structure-based optimization of small-molecule inhibitors for the β-catenin/Bcell lymphoma 9 protein–protein interaction. J. Med. Chem 2018, 61, 2989–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wisniewski JA; Yin J; Teuscher KB; Zhang M; Ji H. Structure-based design of 1,4-dibenzoylpiperazines as β-catenin/B-cell lymphoma 9 protein–protein interaction inhibitors. ACS Med. Chem. Lett 2016, 7, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Li Z; Zhang M; Teuscher KB; Ji H. Discovery of 1Benzoyl 4-Phenoxypiperidines as Small-Molecule Inhibitors of the β-Catenin/B-Cell Lymphoma 9 Protein–Protein Interaction. J. Med. Chem 2021, 64, 11195–11218. [DOI] [PubMed] [Google Scholar]
- (44).Zhang M; Wisniewski JA; Ji H. AlphaScreen selectivity assay for β-catenin/B-cell lymphoma 9 inhibitors. Anal. Biochem 2015, 469, 43–53. [DOI] [PubMed] [Google Scholar]
- (45).Wang Z; Zhang M; Luo W; Zhang Y; Ji H. Discovery of 2-(3-(3-carbamoylpiperidin-1-yl)phenoxy)acetic acid derivatives as novel small-molecule inhibitors of the β-catenin/B-cell lymphoma 9 protein-protein interaction. J. Med. Chem 2021, 64, 5886–5904. [DOI] [PubMed] [Google Scholar]
- (46).Choi H-J; Huber AH; Weis WI Thermodynamics of β-catenin-ligand interactions: the roles of the N- and C-terminal tails in modulating binding affinity. J. Biol. Chem 2006, 281, 1027–1038. [DOI] [PubMed] [Google Scholar]
- (47).Choi H-J; Gross JC; Pokutta S; Weis WI Interactions of plakoglobin and β-catenin with desmosomal cadherins: basis of selective exclusion of α- and β-catenin from desmosomes. J. Biol. Chem 2009, 284, 31776–31788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Emami KH; Nguyen C; Ma H; Kim DH; Jeong KW; Eguchi M; Moon RT; Teo JL; Oh SW; Kim HY; Moon SH; Ha JR; Kahn M. A small molecule inhibitor of β-catenin/ CREB-binding protein transcription. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kaelin WG Jr. Common pitfalls in preclinical cancer target validation. Nat. Rev. Cancer 2017, 17, 441–450. [DOI] [PubMed] [Google Scholar]
- (50).Howe LR; Watanabe O; Leonard J; Brown AM Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 2003, 63, 1906–1913. [PubMed] [Google Scholar]
- (51).Yook JI; Li X-Y; Ota I; Fearon ER; Weiss SJ Wnt-dependent regulation of the E-cadherin repressor snail. J. Biol. Chem 2005, 280, 11740–11748. [DOI] [PubMed] [Google Scholar]
- (52).Yook JI; Li X-Y; Ota I; Hu C; Kim HS; Kim NH; Cha SY; Ryu JK; Choi YJ; Kim J; Fearon ER; Weiss SJ A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol 2006, 8, 1398–1406. [DOI] [PubMed] [Google Scholar]
- (53).Scheel C; Eaton EN; Li SH-J; Chaffer CL; Reinhardt F; Kah K-J; Bell G; Guo W; Rubin J; Richardson AL; Weinberg RA Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).DiMeo TA; Anderson K; Phadke P; Fan C; Perou CM; Naber S; Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009, 69, 5364–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wu Y; Ginther C; Kim J; Mosher N; Chung S; Slamon D; Vadgama JV Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res 2012, 10, 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Beiter K; Hiendlmeyer E; Brabletz T; Hlubek F; Haynl A; Knoll C; Kirchner T; Jung A. β-Catenin regulates the expression of tenascin-C in human colorectal tumors. Oncogene 2005, 24, 8200–8204. [DOI] [PubMed] [Google Scholar]
- (57).Oskarsson T; Acharyya S; Zhang XH-F; Vanharanta S; Tavazoie SF; Morris PG; Downey RJ; Manova-Todorova K; Brogi E; Massagué J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med 2011, 17, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Barker N; van Es JH; Kuipers J; Kujala P; van den Born M; Cozijnsen M; Haegebarth A; Korving J; Begthel H; Peters PJ; Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- (59).Plaks V; Brenot A; Lawson DA; Linnemann JR; Van Kappel EC; Wong KC; de Sauvage F; Klein OD; Werb Z. Lgr5-expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell Rep. 2013, 3, 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Yang L; Tang H; Kong Y; Xie X; Chen J; Song C; Liu X; Ye F; Li N; Wang N; Xie X. LGR5 promotes breast cancer progression and maintains stem-like cells through activation of Wnt/β-catenin signaling. Stem Cells 2015, 33, 2913–2924. [DOI] [PubMed] [Google Scholar]
- (61).Hou M-F; Chen P-M; Chu P-Y LGR5 overexpression confers poor relapse-free survival in breast cancer patients. BMC Cancer 2018, 18, No. 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Hagerling C; Owyong M; Sitarama V; Wang C-Y; Lin C; van den Bijgaart RJE; Koopman CD; Brenot A; Nanjaraj A; Wärnberg F; Jirström K; Klein OD; Werb Z; Plaks V. LGR5 in breast cancer and ductal carcinoma in situ: a diagnostic and prognostic biomarker and a therapeutic target. BMC Cancer 2020, 20, No. 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zhang X; Claerhout S; Prat A; Dobrolecki LE; Petrovic I; Lai Q; Landis MD; Wiechmann L; Schiff R; Giuliano M; Wong H; Fuqua SW; Contreras A; Gutierrez C; Huang J; Mao S; Pavlick AC; Froehlich AM; Wu MF; Tsimelzon A; Hilsenbeck SG; Chen ES; Zuloaga P; Shaw CA; Rimawi MF; Perou CM; Mills GB; Chang JC; Lewis MT A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Rosenberg LH; Lafitte M; Quereda V; Grant W; Chen W; Bibian M; Noguchi Y; Fallahi M; Yang C; Chang JC; Roush WR; Cleveland JL; Duckett DR Therapeutic targeting of casein kinase 1δ in breast cancer. Sci. Transl. Med 2015, 7, No. 318ra202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem 2004, 332, 261–273. [DOI] [PubMed] [Google Scholar]
- (66).Cer RZ; Mudunuri U; Stephens R; Lebeda FJ IC50-toKi: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Plante JP; Burnley T; Malkova B; Webb ME; Warriner SL; Edwards TA; Wilson AJ Oligobenzamide proteomimetic inhibitors of the p53-hDM2 protein-protein interaction. Chem. Commun 2009, 5091–5093. [DOI] [PMC free article] [PubMed]
- (68).Yeo DJ; Warriner SL; Wilson AJ Monosubstituted alkenyl amino acids for peptide “stapling”. Chem. Commun 2013, 49, 9131–9133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.