

Abstract
Background
The chemokine CXCL12 and its two receptors (CXCR4 and CXCR7) are involved in inflammation and hematopoietic cell trafficking. This study was designed to investigate molecular docking interactions of four popular cardiovascular-active natural compounds; curcumin, resveratrol, quercetin, and eucalyptol; with these receptors and to predict their drug-like properties. We hypothesize that these compounds can modify CXCL12/CXCR4/CXCR7 pathway offering benefits for coronary artery disease patients.
Methods
Docking analyses were carried and characterized by Molecular Environment (MOE) software. Protein Data Bank (http://www.rcsb.org/) has been retrieved from protein structure generation and crystal structures of CXCR4 and CXCR7 receptors (PDB code = 3ODU and 6K3F). The active sites of these receptors were evaluated and extracted from full protein and molecular docking protocol was done for compounds against them. The presented parameters included docking scores, ligand binding efficiency, and hydrogen bonding. The pharmacokinetic/toxic properties (ADME/T) were calculated using SwissADME, ProTox-II, and Pred-hERG softwares to predict drug-like properties of the compounds. The thermochemical and molecular orbital analysis, and molecular dynamics simulations were also done.
Results
All compounds showed efficient interactions with the CXCR4 and CXCR7 receptors. The docking scores toward proteins 3ODU of CXCR4 and 6K3F of CXCR7 were − 7.71 and − 7.17 for curcumin, − 5.97 and − 6.03 for quercetin, − 5.68 and − 5.49 for trans-resveratrol, and − 4.88 and − 4.70 for (1 s,4 s)-eucalyptol respectively indicating that all compounds, except quercetin, have more interactions with CXCR4 than with CXCR7. The structurally and functionally important residues in the interactive sites of docked CXCR4-complex and CXCR7-complex were identified. The ADME analysis showed that the compounds have drug-like properties. Only (1 s,4 s)-Eucalyptol has potential weak cardiotoxicity. The results of thermochemical and molecular orbital analysis and molecular dynamics simulation validated outcomes of molecular docking study.
Conclusions
Curcumin showed the top binding interaction against active sites of CXCR4 and CXCR7 receptors, with the best safety profile, followed by quercetin, resveratrol, and eucalyptol. All compounds demonstrated drug-like properties. Eucalyptol has promising potential because it can be used by inhalation or skin massage. To our knowledge, this is the first attempt to find binding interactions of these natural agents with CXCR4 and CXCR7 receptors and to predict their druggability.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12906-021-03488-8.
Keywords: ADME, Coronary artery disease, Curcumin, CXCR4, CXCR7, Docking, Eucalyptol, HOMO–LUMO, Quercetin, Resveratrol
Background
The homeostatic chemokine stromal-cell-derived factor-1 (SDF-1) or CXCL12 and its two receptors CXCR4 and CXCR7 are considered important therapeutic targets that control inflammation. CXCR4 and CXCR7 are G protein coupled receptors (GPCR) that have similar trans-membrane helices and distinct conformation of the N- and C-terminals as well as of three extracellular loops [1, 2]. CXCL12 is abundant in many normal tissues. Blood vascular endothelial cells express both CXCL12 and its receptors [3]. The CXCL12/CXCR4/CXCR7 pathway induces migration of hematopoietic progenitor cells, endothelial cells, and leukocytes [4]. When CXCL12 interacts with CXCR4, the classical GPCR signaling occurs through phosphoinositide-3 kinase (PI3K/Akt), phospholipase C/inositol 1,4,5, triphosphate (PLC/IP3), and extracellular regulated kinase 1/2 (ERK1/2) pathways, and Ca2+ mobilization from endoplasmic reticulum through inhibition of adenylyl cyclase mediated c-AMP production. On the other hand, when CXCL12 interacts with CXCR7, this GPCR mobilization of Ca2+ does not happen, but β-arrestin is activated leading to scavenging of CXCL12 and internalization of CXCR4. Also, CXCR4 and CXCR7 can form either homodimers or heterodimers and the later induce conformational changes of the CXCR4/G proteins and block signaling [5]. Therapeutics targeting this axis can block the CXCL12/CXCR4 interaction or suppress the downstream intracellular signaling [6].
Coronary artery disease (CAD) is characterized by inadequate vasculogenic reaction to ischemia and dysfunction of endothelial outgrowth cells (EOCs). CXCR7 is a crucial modulator of neovascularization of EOCs to the perfusion defect area. In CAD patients, CXCR7 expression of EOCs was downregulated and phosphorylation of its downstream signaling (ERK) was reduced. CXCR7 upregulation increased phosphorylation of ERK and improved vasculogenic function of EOCs. Thus, CXCR7 signaling may be a new target for therapeutic vasculogenesis in CAD patients [7]. Effective improving of cardiac blood flow and controlling the atherosclerotic plaque are fundamental points in management of CAD. Angiogenesis (which is an important pathway for revascularization of ischemic tissues in CAD) improves blood flow but unfortunately, it may destabilize the atherosclerotic plaque. Thus, adjusting the balance of angiogenesis can be a main target for CAD treatment. Recently, herbal medicines are reported to be effective in adjusting such balance [8]. Poly-pharmacological compounds act on multiple targets, thus useful against complex (multifactorial) diseases. Also, through molecular docking, complexes formed between ligands and interesting targets (often many) can be observed for a particular disease [9].
Curcumin ((1E,6E)-1,7-bis (4-hydroxy- 3-methoxyphenyl) -1,6- heptadiene-3,5-dione), a polyphenol compound separated from turmeric, has antioxidant, anti-inflammatory, anticancer, anticoagulant, and lipid regulation effects. The underlying mechanisms of these effects are diverse and involve suppression of numerous molecular targets including nuclear factor-κB (NF-κB), vascular endothelial cell growth factor, pro-inflammatory cytokines, protein kinases (such as mitogen-activated protein kinases; MAPK; and Akt), enzymes (such as cyclooxygenase (COX) 1 & 2 and 5-lipoxygenase) and recently CXCR 4. Curcumin is quite safe in humans with few side effects [10]. Moreover, curcumin has displayed beneficial preventive and therapeutic effects in many neurological diseases including Alzheimer’s disease through several molecular mechanisms such as “antioxidant, amyloid β-binding, anti-inflammatory, tau inhibition, metal chelation, neurogenesis activity, and synaptogenesis promotion”. To overcome its low bioavailability, new nanocurcumin formulations with no toxicity for other body cells are being developed as therapeutic alternatives [11]. The curcumin’s anti-inflammatory effects have been linked to its phenyl and methoxy groups in the ortho position, which strongly inhibit nuclear factor kappa B and hence stop production of TNF-α and IL-6. Also, curcumin can inhibit cyclooxygenase-2, lipoxygenase, and inducible nitric oxide synthase which are key enzymes mediating inflammation. Therefore, curcumin was found to have anti-oral mucosal lesion activity due to its anti-inflammatory, antioxidant, antifungal, antiseptic, analgesic, and anti-neoplastic effects [12].
Quercetin (2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4H-1-benzopyran-4-one) and resveratrol (5-[(E)-2-(4-Hydroxyphenyl)ethen-1-yl]benzene-1,3-diol) are natural polyphenolic compounds found in the grapes, red wine, peanuts, and other food products. Trans-resveratrol is the active form which has antioxidant, anti-inflammatory, antiplatelet, anti-carcinogenic, and DNA protective effects. It inhibits pro-inflammatory factors such as platelet-activating factor, tumor necrosis factor-α (TNF-α) and interleukin 1b (IL-1b)-induced NF-kB activation, and enzymes like COX-2. No significant side effect has been reported with it, but due to its experimental anti-platelet effect, caution should be taken with any concomitant use of anti-platelet agents [13]. Quercetin is found in all plant foods. It has antioxidant, anti-inflammatory, cardiovascular protective, anticancer, and antiviral effects. Quercetin suppressed the non-typeable Haemophilus influenzae-induced CXCR4 expression levels in cases of otitis media suggesting a potential therapeutic benefit [14]. Quercetin is effective as a preventive and therapeutic agent against “different neuro degenerative brain disorders such as Alzheimer’s disease, Amyloid β peptide, Parkinson’s disease, Huntington’s disease, multiple sclerosis, and amyotrophic lateral sclerosis”. It decreases stress, inflammation, and enhances brain growth. Therefore, quercetin can be considered as a powerful dietary supplement with negligible toxicity. Due to its low bioavailability, studies on drug delivery mechanisms are needed. Also, more clinical trials to assess its effective dose for treatment of the neurodegenerative disorders are recommended [15].
Aromatherapy (or essential oil therapy) is use of essential oils extracted from plants as a complementary health approach. The essential oils are most often used by inhalation or by skin application [16]. Patients with chronic diseases such as CV diseases, respiratory diseases, GI diseases, cancer, and neurological diseases can benefit from aromatherapy in addition to their routine medications to combat chronic inflammation and oxidative stress and improve side effects such as insomnia and nausea. The 1,8-cineole or (1 s,4 s)-eucalyptol (1,3,3-Trimethyl-2-oxabicyclo[2.2.2]octane), a terpenoid oxide present in several plant essential oils, has anti-inflammatory and antioxidant effects. In women with acute coronary syndrome, aromatherapy massage with lavender essential oil (in addition to the routine care) significantly decreased levels of anxiety and depression and reduced blood pressure and heart rate but did not affect the respiratory rate [17]. In coronary care units, inhalation of lavender essential oil for 2 weeks improved sleep quality and alleviated anxiety [18].
Taken together, this study was designed to investigate the molecular docking interactions of four of the most researched anti-inflammatory natural compounds (three polyphenols; curcumin, resveratrol, quercetin; and a terpene; eucalyptol) with the CXCR4 and CXCR7 receptors, and to predict their pharmacokinetic/toxic properties. The thermochemical and molecular orbital analysis and molecular dynamics simulations were done to predict their drug-like properties. We hypothesize that these compounds can modify the CXCL12/CXCR4/CXCR7 pathway commonly involved in regulation of hematopoietic cell trafficking suggesting potential additional benefits of these compounds for CAD patients.
Methods
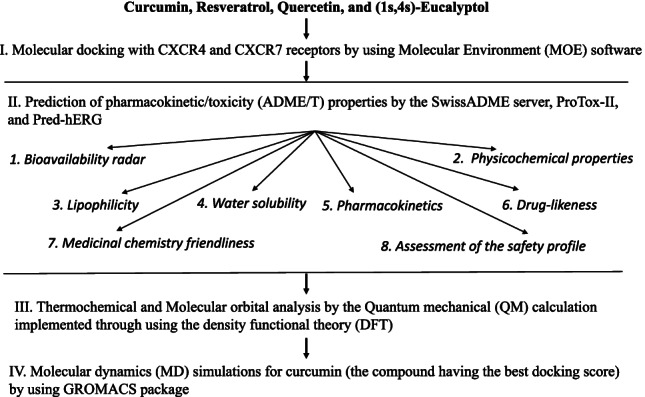
The manuscript’s design is summarized in this scheme followed by detailed description. A supplementary material file is provided which contains all figures presented in high quality.
Molecular docking analysis
The docking analyses were carried and characterized by the Molecular Environment (MOE) software. Chemdraw compound preparation, Chem 3d structures, Chem 3D ultra 12.0 (Molecular Modeling and Analysis; Cambridge Soft Corporation) software, and MOPAC were enhanced and deposited in MDL MolFile (*.mol). All calculations were performed on a (1.80-1.99) GHz-based MS Windows 10 pro 64-bit operating system Intel (R) Core (TM) i7-8550 CPU [19–22]. A Protein Data Bank (http://www.rcsb.org/) has been retrieved from protein structure generation and the crystal structures of the CXCR4 and CXCR7 receptors (PDB codes = 3ODU and 6K3F). The proteins were separated from all bound waters, ligands, and cofactors and then hydrogen atoms were applied to the optimization. Validation of the docking method was done by using the same crystalline compound “(6,6-dimethyl-5,6-dihydroimidazo(2,1-b) thiazol-3-yl)methyl (E)-N,N’-dicyclohexylcarbamimidothioate” with 3ODU protein with the same protocol and the resulting RMSD “root-mean-square deviation” value was 1.28 Å and by comparing our test compounds with the validated compounds which have a docking score of - 5.17 kcal/mol.
The molecular docking protocol was done for the four natural product compounds against the active sites of CXCR4 and CXCR7 receptors (PDB codes = 3ODU and 6K3F) [2]. The active sites were evaluated and extracted from the full protein. The energy minimization was done using MMFF94x force field [19]. London dG scoring was used as a function in the calculated docking score to rank the binding affinity of the synthesized compounds to the protein molecule [20]. The RMSD of the selected compounds position compared to the docking pose was used in the ranking.
All energy interaction and pi-pi stacking calculation were done in MOE program by using the Triangle Matcher where Poses are generated by aligning the ligand triplets of atoms on the triplets of alpha spheres in a more systematic way than in the Alpha Triangle method. The options for this method were: (1) Timeout (seconds): Amount of time allowed for each ligand placement, and (2) Number of Returned Poses: Maximum number of poses returned by each ligand placement. The scoring for high 30 pose was done using London dG Scoring function which estimates the free energy of binding of the ligand from a given pose. The functional form is a sum of terms:
where c represents the average gain/loss of rotational and translational entropy; Eflex is the energy due to loss of flexibility of the ligand (calculated from ligand topology only); fHB measures geometric imperfections of hydrogen bonds and takes a value in [0,1]; cHB is the energy of an ideal hydrogen bond; fM measures geometric imperfections of metal ligations and takes a value in [0,1]; cM is the energy of an ideal metal ligation; and Di is the desolvation energy of atom i. The difference in desolvation energies was calculated according to the formula:
where A and B are the protein and/or ligand volumes with atom i belonging to volume B; Ri is the solvation radius of atom i (taken as the OPLS-AA van der Waals sigma parameter plus 0.5 Å); and ci is the desolvation coefficient of atom i. The coefficients {c, cHB, ci} were fitted from approximately 400 X-ray crystal structures of protein-ligand complexes with available experimental pKi data. Atoms are categorized into about a dozen atom types for the assignment of the ci coefficients. The triple integrals were approximated using Generalized Born integral formulas.
The highest best five score is calculated using GBVI/WSA dG Scoring: The GBVI/WSA ΔG is a forcefield-based scoring function which estimates the free energy of binding of the ligand from a given pose. It has been trained using the MMFF94x and AMBER99 forcefield on the 99 protein-ligand complexes of the SIE training set [Naim]. The functional form is a sum of terms:
where: c represents the average gain/loss of rotational and translational entropy; α, β are constants which were determined during training (along with c and are forcefield-dependent. If not using an AMBER forcefield, the parameters were set by default to the MMFF trained parameters; ECoul is the coulombic electrostatic term which is calculated using currently loaded charges, using a constant dielectric of εi = 1; Esol is the solvation electrostatic term which is calculated using the GB/VI solvation model; EvdW is the van der Waals contribution to binding; Aweighted is the surface area, weighted by exposure. This weighting scheme penalizes exposed surface area.
The hydrogen bonding pattern and hydrophobic interaction pattern between the ligand and the receptor were considered. A pair of ligand-receptor atoms of the appropriate types are considered to have a hydrogen bond interaction if they are less than 3.5 Å apart. A hydrophobic atom on the ligand is considered to have a hydrophobic interaction with a receptor residue if that atom is under 4.5 Å away from a hydrophobic atom of that residue. Poses are considered as duplicates if the same set of ligand-receptor atom pairs are involved in hydrogen bond interactions and the same set of ligand atom receptor residue pairs are involved in hydrophobic interactions.
Use the enthalpy-based Affinity dG scoring function to score generated poses. The following options may be adjusted: Hydrogen Bond: The ideal hydrogen bond affinity in energy units; Ionic Contact: The ionic Coulomb coefficient; Metal Ligation: The metal ligation affinity; Hydrophobic Contact: The hydrophobic-hydrophobic contact energy; Hydrophobic-Polar: The hydrophobic-polar contact energy; Atom-Atom: The general atom-atom contact energy.
Prediction of pharmacokinetic/toxicity (ADME/T) properties
It was done using the SwissADME server [23, 24], ProTox-II [25], and Pred-hERG as follows [26, 27]:
Bioavailability radar
It is a tool for rapid appraisal of drug-likeness of a molecule. Six physicochemical properties (lipophilicity, size, polarity, solubility, flexibility, and saturation) were considered. On each axis, a physicochemical range was determined by descriptors as previously explained [28, 29] and illustrated as a pink area in which the radar plot of the compound has to fall wholly to be considered drug-like.
Physicochemical properties
These include simple molecular and physicochemical descriptors such as molecular weight (MW), number of specific atom types, Fraction Csp3 (carbon bond saturation as defined by fraction sp3) which measures of complexity of the molecule, number of specific bond types, and molecular refractivity (MR). Also the topological polar surface area (TPSA) was used to calculate the polar surface area (PSA) which quickly estimated some ADME properties, especially in relation to passing through the biological barriers like absorption and brain entry [29, 30].
Lipophilicity
It was described by calculating the partition coefficient between n-octanol and water (log Po/w). Five free predictors; iLOGP, XLOGP3, WLOGP, MLOGP, and SILICOS-IT are given access by SwissADME to generate the consensus log Po/w which is the mean of the values predicted by them [31, 32].
Water solubility
The SwissADME uses three methods to predict water solubility (ESOL (Estimated Solubility) model [33], that of Ali et al. [34], and SILICOS-IT). The output is the Log S values which are the decimal log of the molar solubility in water. The water solubility was provided in mg/ml and mol/l along with the qualitative solubility classes.
Pharmacokinetics
SwissADME uses specialized models to evaluate ADME behaviors of the test compound. The first model is prediction of passive gastrointestinal absorption and blood-brain barrier (BBB) penetration [30]. The second model is prediction of being substrate or non-substrate of the permeability glycoprotein (P-gp) which is essential to evaluate active efflux through membranes e.g., from the gastrointestinal wall to the lumen or from the brain [35]. The third model is prediction of interaction of compounds with cytochrome P450 (CYP) major isoenzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) which is an important contributor in drug elimination through metabolic biotransformation. Also, inhibition of these isoenzymes is a cause of drug interactions [36] leading to toxic or other adverse effects. The fourth model is prediction of the skin permeability coefficient (Kp) which is linearly correlated with molecular size and lipophilicity [37]. The more negative the log Kp (cm/s), the less skin permeation.
Drug-likeness
It is the qualitative evaluation of the opportunity for a molecule to become an oral drug with respect to oral bioavailability. It relates the physicochemical characters of a compound with its biopharmaceutical aspect in human body [38]. SwissADME uses five commonly used drug-likeness rules or filters with different ranges of properties inside of which the molecule is defined as a drug-like. These are the Lipinski [39], Ghose [40], Veber [41], Egan [42], and Muegge [43] methods. Any violation of any rule shows clearly in the output panel. Also the bioavailability score [44] is used which defines four classes of compounds with probabilities of oral bioavailability of 11, 17, 56%, or 85%.
Medicinal chemistry friendliness
SwissADME uses two pattern recognition filters for removal of PAINS (pan assay interference compounds) that appear as promiscuous compounds which cause potential problems in assays regardless of the protein target and can give false positive biological output [45]. In addition, “Structural Alert” is concerned with problematical functional groups which could cause toxicity, metabolic imbalance, or poor pharmacokinetics. Moreover, “leadlikeness” is similar to drug-likeness, but it focuses on physicochemical limits that define a good lead [46]. The “Synthetic accessibility” score; which ranges from 1.0 (very easy) to 10.0 (very difficult); guides selection of the most promising virtually-tested molecules to be synthetized and subjected to biological assays [47].
Assessment of the safety profile
The ProTox-II software predicts different toxicity endpoints such as acute toxicity, hepatotoxicity, carcinogenicity, mutagenicity, and others [25]. The Pred-hERG (human Ether-à-go-go-Related Gene) software was used to assess cardiotoxicity. It depends on statistically significant and externally predictive quantitative structure-activity relationship (QSAR) models of hERG blockage which is closely associated with severe and potentially fatal cardiac dysrhythmia. The SDF (structure data file) and SMILES (simplified molecular-input line-entry system) strings were used throughout the generation process [26, 27].
Thermochemical and molecular orbital analysis
Quantum mechanical (QM) methods keep an important role for the calculation of thermal and molecular orbital properties. In this study, optimization of compounds was done using the density functional theory (DFT). The QM calculation was implemented through using the DFT by Gaussian 09 program package for all compounds [48]. People’s 6-31G basis set was used to optimize the compounds and other calculations for global and absolute chemical reactivity parameter were done as previously described [49, 50].
Molecular dynamics (MD) simulations
Due to huge time consumption and limitation of our server, the 100 ns scale molecular dynamics simulation has been done for the compound having the best docking score (curcumin). The best docked complex was selected for 100 ns MD simulations using GROMACS package [51] in the explicit water conditions. The protein and ligand were separated and the topology of protein was generated using GROMOS96 53a6 force field [52]. Afterwards, the PRODRG server [53] was used for the ligand structure and the partial charges were corrected using of DFT based methods available in GAUSSIAN software suite in combination with B3LYP 6-31G (d, p) basis set and CHELPG program. Consequently, the solvation was performed using the SPC/E water model [54] and then neutralization step was proceeded by adding appropriate number of counter ions. Thereafter, the energy minimization was performed using steepest descent algorithm followed by positions restraining and then equilibration in NVT (constant volume) and NPT (constant pressure) ensemble conditions, each at 100 ps time scale. The temperature of 300 K was maintained for the system using Berendsen weak coupling method, and the pressure of 1 bar was maintained utilizing Parrinello-Rahman barostat in the equilibration stage. Furthermore, the final production stage was carried out using the LINCS algorithm and the generated trajectories were analysed for the changes in the pattern of protein-inhibitor distances, Hydrogen bonds (H-bonds), Root Mean Square Deviations (RMSD), and Radius of Gyration (Rg). The Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) protocols implemented in g_mmpbsa package [55–57] were used for the calculation of free energy of binding between the docked protein and inhibitor.
Results
Molecular docking
In this study, virtual screening via molecular docking was performed on four natural compounds with anti-inflammatory effects. These compounds were screened based on their interactions with the CXCR4 and CXCR7 receptor to find the most predicted compound–ligand interactions. The presented parameters include the docking scores, ligand binding efficiency and hydrogen bonding interactions. The docking scores toward the proteins 3ODU of CXCR4 and 6K3F of CXCR7 were − 7.71 and − 7.17 for curcumin, − 5.97 and − 6.03 for quercetin, − 5.68 and − 5.49 for trans-resveratrol, and − 4.88 and − 4.70 for (1 s,4 s)-eucalyptol respectively (Tables 1 and 3) indicating that all compounds, except quercetin, have more interactions with CXCR4 than with CXCR7. The curcumin docking scores were shown to be the highest compared to the other three drugs.
Table 1.
Docking scores (kcal/mol) of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol with protein 3ODU of CXCR4 receptor
| Compound | Score | rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|
| Curcumin | − 7.71 | 2.79 | −31.72 | − 59.10 | −10.67 | −42.34 | − 7.71 |
| Trans-resveratrol | −5.68 | 2.95 | −44.35 | −64.70 | −10.42 | −28.16 | −5.68 |
| Quercetin | −5.97 | 1.37 | 4.83 | −70.31 | −12.61 | −33.13 | −5.97 |
| (1 s,4 s)-Eucalyptol | −4.88 | 1.26 | −28.53 | −37.41 | −7.55 | −15.62 | −4.88 |
Table 3.
Docking scores (kcal/mol) of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol with protein 6K3F of CXCR 7 receptor
| Compound | Score | rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|
| Curcumin | −7.17 | 1.05 | −33.09 | − 81.95 | −10.17 | − 33.42 | − 7.17 |
| Trans-resveratrol | −5.49 | 1.71 | −46.85 | −67.60 | −9.93 | −27.15 | − 5.49 |
| Quercetin | −6.03 | 1.20 | 5.11 | −102.43 | −11.59 | −36.25 | −6.03 |
| (1 s,4 s)-Eucalyptol | −4.70 | 2.15 | −29.44 | −29.89 | −6.55 | − 20.54 | −4.70 |
Molecular docking with CXCR4 receptor protein
The docking studies of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol compounds have been screened to “3ODU” of CXCR4 receptor protein [2]. Curcumin was found to have the best activity compared with others in docking with 6K3F protein with scores of − 7.71, − 5.68, − 5.97, and − 4.88 respectively (Table 1).
The main structure activity relationship is contributed to the orientation of curcumin compound inside the cavity of the corresponding protein. In case of curcumin, it oriented inside the (His203-Gln200-Tyr116-Trp94-Asp97-His113-Val112-Arg188-Leu120-Lle259-Lle204-Tyr255-Leu208-Gly207) cavity due to the H-Acceptor of O1 atom of curcumin with NH(1) of Arg188 with bond length of 3.21 Å and − 2.40 Kcal/Mol, the H-Acceptor of O27 atom of curcumin with NH(2) of Arg188 with bond length of 3.13 Å and − 0.50 Kcal/Mol, and π- π stacking of 6-membered ring of curcumin with 5-membered ring of His113 with bond length of 3.88 Å and − 0.00 Kcal/Mol. In case of trans-resveratrol, it oriented inside the cavity of (His203-Lle204-Leu208-Gly207-Leu120-Tyr116-Glu288-Arg188-Tyr255-Lle259-Tyr256) by the H- π stacking interaction of 6-membered ring of trans-resveratrol with C atom of Ile204 with bond length of 4.33 Å and − 0.80 Kcal/Mol. In case of quercetin the compound oriented inside the cavity of (Tyr256-Lle204-Gin200-Tyr255-Leu120-Lle259-Ser260) by the π-H stacking interaction of 6-membered ring of quercetin with C atom of Ile204 with bond length of 4.39 Å and − 0.80 Kcal/Mol, but in case of (1 s,4 s)-eucalyptol the compound oriented inside the cavity of (Asp187-Cys186-His113-Trp102-Asp97) by the H- π stacking interaction between 5 membered ring of (1 s,4 s)-eucalyptol with His113 with bond length of 4.08 Å and − 0.70 kcal/mol (Figs. 1 and 2) and (Table 2).
Fig. 1.

The 2D and 3D docking interaction of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol with protein 3ODU of CXCR4 receptor (A 3D high quality figure, S1, is presented in the supplementary material)
Fig. 2.

The docking sites of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol with protein 3ODU of CXCR4 receptor
Table 2.
Docking interaction of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol with protein 3ODU of CXCR4 receptor
| Compound | Ligand | Receptor | Interaction | Distance (Å) | E (Kcal/Mol) |
|---|---|---|---|---|---|
| Curcumin | O 1 | NH1 ARG 188 (A) | H-acceptor | 3.21 | −2.40 |
| O 27 | NH2 ARG 188 (A) | H-acceptor | 3.13 | −0.50 | |
| 6-ring | 5-ring HIS 113 (A) | pi-pi | 3.88 | 0.00 | |
| Trans-resveratrol | 6-ring | CA ILE 204 (A) | pi-H | 4.33 | −0.80 |
| Quercetin | 6-ring | CA ILE 204 (A) | pi-H | 4.39 | −0.80 |
| (1 s,4 s)-eucalyptol | 5-ring | HIS 113 (A) | H-pi | 4.08 | −0.70 |
Molecular docking with CXCR7 receptor protein
The docking studies of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol compounds have been screened to “6K3F” of CXCR7 receptor protein [58]. Curcumin was found to have the highest activity compared with others in docking with 3ODU protein with scores of − 7.17, − 5.49, and − 6.03 and − 4.70 respectively (Table 3).
The main structure activity relationship is contributed to the orientation of the curcumin inside the cavity of the corresponding protein. In case of curcumin, the compound oriented inside the (Gln294-Asp292-Thr129-Gln131-Leu130-Lle243-Thr251-Lys139-Ala140-Pro132-Cys141-Arg63) cavity due to the H-Acceptor of O16 atom of curcumin with N atom of Gln294 with bond length of 3.28 Å and − 0.60 Kcal/Mol. In case of trans-resveratrol, the compound oriented inside the cavity of (Gly286-Arg287-Cys253-Tyr251-Ala140-Cys141-Pro132-Lys132-Leu130) by the H-Donor interaction of O28 atom of trans-resveratrol with O atom of Ala140 with bond length of 2.99 Å and − 1.40 Kcal/Mol. Moreover, in case of quercetin the compound oriented inside the cavity of (Gly288-Arg287-Cys253-Tyr251-Ala140-Cys141-Pro132-Lys139-Leu130) by the H-Donor interaction of O29 atom of quercetin with O atom of Gly288 with bond length of 3.29 Å and − 0.50 Kcal/Mol, but in case of (1 s,4 s)-eucalyptol the compound oriented inside the cavity of (Leu130-Pro132-Lie243-Tyr251-Gly133-Gln131-Asp136-Lys139-Arg287) by the H- π stacking interaction between C6 atom of (1 s,4 s)-eucalyptol with 6-membered ring of Tyr251 with bond length of 4.29 Å and − 0.60 kcal/mol (Figs. 3 and 4) and (Table 4).
Fig. 3.

The 2D and 3D docking interaction of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol with protein 6K3F of CXCR7 receptor (A 3D high quality figure, S3, is presented in the supplementary material)
Fig. 4.

The docking sites of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol with protein 6K3F of CXCR7 receptor
Table 4.
Docking interaction of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol with protein 6K3F of CXCR7 receptor
| Compound | Ligand | Receptor | Interaction | Distance (Å) | E (Kcal/Mol) |
|---|---|---|---|---|---|
| Curcumin | O 16 | N GLN 294 (A) | H-acceptor | 3.28 | − 0.60 |
| Trans-resveratrol | O 28 | O ALA 140 (A) | H-donor | 2.99 | −1.40 |
| Quercetin | O 29 | O GLY 288 (A) | H-donor | 3.29 | −0.50 |
| (1 s,4 s)-eucalyptol | C 6 | 6-ring TYR 251 (A) | H-pi | 4.29 | −0.60 |
Conformation of ligands at the inhibition binding site of receptor protein
The conformation of ligands at the inhibition binding site of receptor protein and superimposed structures of docked ligands are shown in Figs. 5 and 6.
Fig. 5.

The docked conformation of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol at the inhibition binding site of receptor protein and superimposed structures of docked ligands with protein 3ODU of CXCR4 receptor
Fig. 6.

The docked conformation of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol at the inhibition binding site of receptor protein and superimposed structures of docked ligands with protein 6K3F of CXCR 7 receptor
Pharmacokinetic/toxic (ADME/T) properties
Bioavailability radar
The four compounds are predicted orally bioavailable (Fig. 7).
Fig. 7.

The bioavailability radar with the pink area. It represents the optimal range for physicochemical properties for oral bioavailability. Lipophilicity (− 0.7 < XlogP3 < + 0.5), size (150 < MW < 500 g/mol), polarity (20A2 <TPSA <130A2), Insolubility (0 < Log S (ESOL) < 6), Insaturation (0.25 < Fraction Csp3 < 1), and flexibility (number of rotatable bonds < 9). The compounds, A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol, are predicted orally bioavailable
Physicochemical properties
Simple molecular and physicochemical properties of the four compounds are shown in Table 5.
Table 5.
Molecular and physicochemical descriptors of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| Formula | C21H20O6 | C14H12O3 | C15H10O7 | C10H18O |
| Molecular weight (MW) | 368.38 g/mol | 228.24 g/mol | 302.24 g/mol | 154.25 g/mol |
| Number of heavy atoms | 27 | 17 | 22 | 11 |
| Number of aromatic heavy atoms | 12 | 12 | 16 | 0 |
| Carbon bond saturation (Fraction Csp3) | 0.14 | 0.00 | 0.00 | 1.00 |
| Number of rotatable bonds | 8 | 2 | 1 | 0 |
| Number of H-bond acceptors | 6 | 3 | 7 | 1 |
| Number of H-bond donors | 2 | 3 | 5 | 0 |
| Molar Refractivity (MR) | 102.80 | 67.88 | 78.04 | 47.12 |
| Topological polar surface area (TPSA) | 93.06 Å2 | 60.69 Å2 | 131.36 Å2 | 9.23 Å2 |
Lipophilicity
The values of the log Po/w of the five predictors of lipophilicity and the consensus (log Po/w) for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol are shown in Table 6.
Table 6.
Values of the partition coefficient between n-octanol and water (log Po/w) of the five predictors of lipophilicity and the consensus (log Po/w) of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| Log Po/w (iLOGP) | 3.27 | 1.71 | 1.63 | 2.58 |
| Log Po/w (XLOGP3) | 3.20 | 3.13 | 1.54 | 2.74 |
| Log Po/w (WLOGP) | 3.15 | 2.76 | 1.99 | 2.74 |
| Log Po/w (MLOGP) | 1.47 | 2.26 | −0.56 | 2.45 |
| Log Po/w (SILICOS-IT) | 4.04 | 2.57 | 1.54 | 2.86 |
| Consensus Log Po/w | 3.03 | 2.48 | 1.23 | 2.67 |
Water solubility
The predicted values are the decimal log of the molar solubility in water (log S) for the three methods which predict water solubility of the four compounds (ESOL, Ali, and SILICOS-IT) with qualitative solubility classes (Table 7).
Table 7.
Water Solubility (Log S) values of the three predictors for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol. The water solubility is provided in mg/ml and mol/l along with qualitative solubility classes
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| Log S (ESOL) | −3.94 | −3.62 | − 3.16 | − 2.52 |
| Solubility |
4.22e-02 mg/ml 1.15e-04 mol/l |
5.51e-02 mg/ml 2.41e-04 mol/l |
2.11e-01 mg/ml 6.98e-04 mol/l | 4.63e-01 mg/ml 3.00e-03 mol/l |
| Class | Soluble | Soluble | Soluble | Soluble |
| Log S (Ali) | −4.83 | − 4.07 | −3.91 | − 2.59 |
| Solubility |
5.50e-03 mg/ml 1.49e-05 mol/l |
1.93e-02 mg/ml 8.44e-05 mol/l |
3.74e-02 mg/ml 1.24e-04 mol/l | 3.98e-01 mg/ml 2.58e-03 mol/l |
| Class | Moderately soluble | Moderately soluble | Soluble | Soluble |
| Log S (SILICOS-IT) | −4.45 | −3.29 | − 3.24 | − 2.45 |
| Solubility |
1.31e-02 mg/ml 3.56e-05 mol/l |
1.18e-01 mg/ml 5.16e-04 mol/l |
1.73e-01 mg/ml 5.73e-04 mol/l | 5.45e-01 mg/ml 3.53e-03 mol/l |
| Class | Moderately soluble | Soluble | Soluble | Soluble |
Pharmacokinetics
Prediction of the gastrointestinal absorption, blood-brain barrier (BBB) penetration, effect on permeability glycoprotein (P-gp) and cytochromes P450 (CYP) major isoenzymes and skin permeation (log Kp) for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol are shown in Table 8.
Table 8.
Prediction of the main pharmacokinetics parameters for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol. These include gastrointestinal absorption, blood-brain barrier (BBB) penetration, effect on permeability glycoprotein (P-gp) and cytochromes P450 (CYP) major isoenzymes and skin permeation (log Kp)
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| GI absorption | High | High | High | High |
| Blood-brain barrier (BBB) permeant | No | Yes | No | Yes |
| P-gp substrate | No | No | No | No |
| CYP1A2 inhibitor | No | Yes | Yes | No |
| CYP2C19 inhibitor | No | No | No | No |
| CYP2C9 inhibitor | Yes | Yes | No | No |
| CYP2D6 inhibitor | No | No | Yes | No |
| CYP3A4 inhibitor | Yes | Yes | Yes | No |
| Log Kp (skin permeation) | −6.28 cm/s | −5.47 cm/s | −7.05 cm/s | − 5.30 cm/s |
Drug-likeness
Only (1 s,4 s)-Eucalyptol showed a violation of Ghose rule and two violations of Muegge rule. The four compounds reached a bioavailability score value of 0.55 (Table 9).
Table 9.
Prediction of the drug likeness for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol. Five rule-based filters and the bioavailability score were used
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| Lipinski rule | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose rule | Yes | Yes | Yes | No; 1 violation: MW < 160 |
| Veber rule | Yes | Yes | Yes | Yes |
| Egan rule | Yes | Yes | Yes | Yes |
| Muegge rule | Yes | Yes | Yes | No; 2 violations: MW < 200, Heteroatoms< 2 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 |
Medicinal chemistry friendliness
Regarding PAINS filter, only quercetin maintains undesirable moiety while regarding Brenk filter, all compounds, except (1 s,4 s)-Eucalyptol, maintain undesirable moieties. Regarding leadlikeness, all compounds, except quercetin, show violations (Table 10).
Table 10.
The medicinal chemistry friendliness filters for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| PAINS | 0 alert | 0 alert | 1 alert: catechol | 0 alert |
| Brenk | 2 alerts: beta_keto_anhydride, michael_acceptor_1 | 1 alert: stilbene | 1 alert: catechol | 0 alert |
| Leadlikeness | No; 2 violations: MW > 350, Rotors> 7 | No; 1 violation: MW < 250 | Yes | No; 1 violation: MW < 250 |
| Synthetic accessibility | 2.97 | 2.02 | 3.23 | 3.65 |
Safety profile
The ProTox-II showed that the four compounds are predicted to have oral LD50 value ranging from 159 to 2480 mg/kg in a rat model with (1 s,4 s)-Eucalyptol bearing the highest values and quercetin bearing the lowest one. The Pred-hERG showed that curcumin, trans-resveratrol, and quercetin are non-cardiotoxic while (1 s,4 s)-Eucalyptol may have cardiotoxicity. In addition, only curcumin has a good applicability domain (AD) (Table 11). The predicted probability maps are shown in Fig. 8.
Table 11.
The predicted toxicity for curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol using: (1) ProTox-II and (2) Pred-hERG softwares
| Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | |
|---|---|---|---|---|
| (1) ProTox-II | ||||
| Predicted LD50 (mg/kg) | 2000 | 1560 | 159 | 2480 |
| Predicted toxicity class | 4 | 4 | 3 | 5 |
| Average similarity (%) | 100 | 69.97 | 100 | 100 |
| Prediction accuracy (%) | 100 | 68.07 | 100 | 100 |
| (2) Pred-hERG | ||||
| Prediction/Potency | Non-cardiotoxic | Non-cardiotoxic | Non-cardiotoxic | Potential cardiotoxic (Weak or moderate) |
| Confidence (%) | 70 | 60 | 60 | 80 |
| Applicability domain (AD) | Yes (Value = 0.28 and limit = 0.26) | No (Value = 0.21 and limit = 0.26) | No (Value = 0.19 and limit = 0.26) | No (Value = 0.16 and limit = 0.26) |
Fig. 8.

The predicted probability maps of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol. The more contour lines and intense green color means a higher positive contribution of an atom or a fragment to the hERG blockage, while pink means that it contributes to decrease of hERG blockage, and gray means no contribution
Thermochemical and molecular orbital results
The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energies, gap, absolute electronegativities, absolute and global hardness, absolute and global softness, chemical potentials, global electrophilicity, and additional electronic charge of all compounds are presented in Tables 12 and 13 and Fig. 9.
Table 12.
Energy (eV) of HOMO, LUMO, and global chemical reactivity parameters of curcumin, trans-resveratrol, quercetin, and (1 s,4 s)-eucalyptol
| Compound | HOMO | LUMO | ∆ E | I | A | χ | ɳ | σ | Pi | S | H |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Curcumin | − 157.034 | −30.183 | 126.851 | 157.034 | 30.183 | 93.609 | 63.426 | 0.016 | −93.609 | 0.008 | 126.851 |
| Trans-resveratrol | − 124.950 | −32.022 | 92.928 | 124.950 | 32.022 | 78.486 | 46.464 | 0.022 | − 78.486 | 0.011 | 92.928 |
| Quercetin | −132.436 | −36.709 | 95.727 | 132.436 | 36.709 | 84.573 | 47.863 | 0.021 | − 84.573 | 0.010 | 95.727 |
| (1 s,4 s)-Eucalyptol | − 142.821 | −41.780 | 101.042 | 142.821 | 41.780 | 92.300 | 50.521 | 0.020 | −92.300 | 0.010 | 101.042 |
HOMO Highest occupied molecular orbital (EHOMO), LUMO Lowest unoccupied molecular orbital, ∆ E HOMO–LUMO energy gap, I Ionization potential, A Electron affinity, χ Absolute electronegativities, ɳ Absolute hardness, σ Absolute softness, Pi Chemical potentials, S Global softness, H Global hardness
Table 13.
Enthalpies and free energies of curcumin, trans-resveratrol, quercetin and (1 s,4 s)-eucalyptol
| Hartree × 103 |
Kcal/mol × 105 |
Hartree × 102 |
Kcal/mol × 105 |
Hartree × 103 |
Kcal/mol ×105 |
Hartree ×102 |
Kcal/mol × 105 |
|
|---|---|---|---|---|---|---|---|---|
| Compound | Curcumin | Trans-resveratrol | Quercetin | (1 s,4 s)-Eucalyptol | ||||
| Enthalpies (ΔH) | −1.26 | − 7.92 | − 7.66 | −4.81 | −1.10 | −6.92 | −4.67 | −2.93 |
| Free Energies (ΔG) | −1.26 | − 7.92 | − 7.66 | − 4.81 | −1.10 | −6.93 | −4.67 | − 2.93 |
Fig. 9.
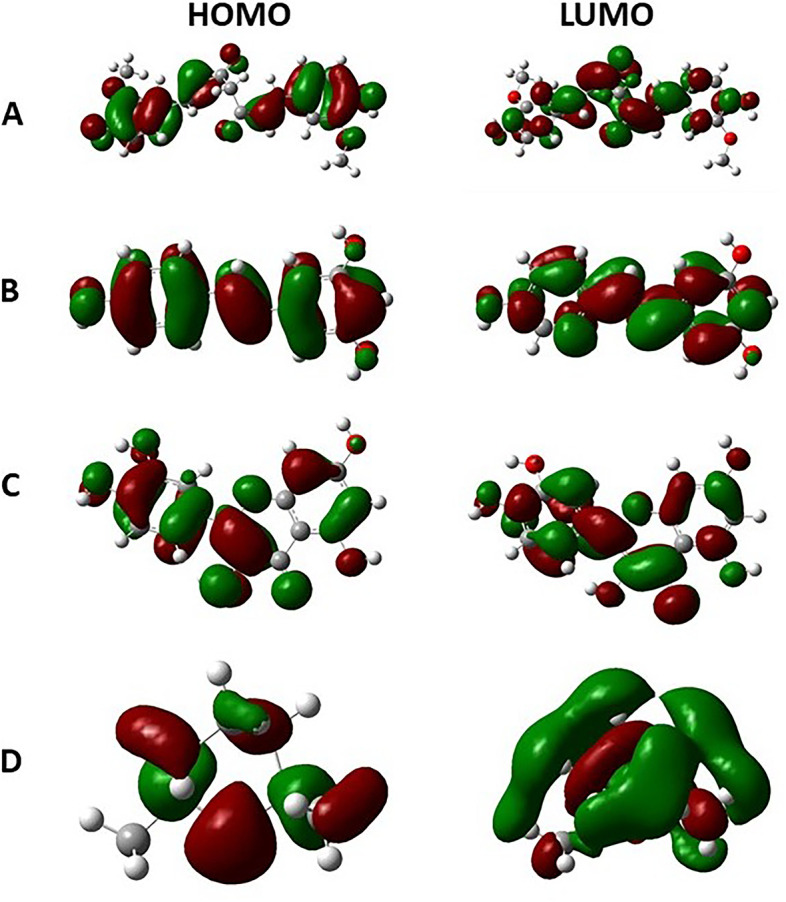
The 3D HOMO and LUMO frontier molecular orbital of A Curcumin, B Trans-resveratrol, C Quercetin, and D (1 s,4 s)-Eucalyptol
Molecular dynamics (MD) simulations
The protein complex with the highest binding affinities was selected for MD simulations, with the RMSD values provided the insight into the stability profile with the values for both the protein and docked system was fluctuating between 0.5 nm and 1.0 nm (Fig. 10A). There is a little perturbation in the RMSD values indicating the stability achieved by the binding of inhibitor in the docked complex. Furthermore, the compactness of the studied systems was analyzed in terms of Rg values which showed variation between 2.8 nm and 3.0 nm indicating the stability achieved in the system after 20 ns time period as indicated from the lower fluctuations in the graph (Fig. 10B). On the basis of the hydrogen bonds and calculated protein-inhibitor distances, it can be observed that the protein and ligand achieved a high degree of interactions (Fig. 10C & D). The docked complex showed up to five hydrogen bonds and the distance between the two molecules fluctuated between 0.15 nm and 0.25 nm.
Fig. 10.

The results obtained from 100 ns molecular dynamics (MD) simulations with: A Changes in the root mean square deviation (RMSD) values for free protein and docked system, B Changes in radius of gyration (Rg), C Changes in the pattern of hydrogen binding, D Calculated distance between the protein and inhibitor
Discussion
Molecular docking is an efficient tool widely used to know the molecular characteristics of protein-ligand interactions [59]. The SwissADME server is used to predict the pharmacokinetics of compounds and their probability to be drug candidates [23]. In addition to their known anti-inflammatory effects, our study shows that the three polyphenols; curcumin, resveratrol, quercetin; and the terpene eucalyptol interact efficiently with the CXCR4 and CXCR7 receptor, revealing different therapeutic modes of action through potentially regulating theses receptors. All compounds, except quercetin, have more interactions with CXCR4 than with CXCR7. Curcumin showed the highest docking scores followed by quercetin, resveratrol, and (1 s,4 s)-Eucalyptol. The study of docking to CXCR4 and CXCR7 has opened a novel aspect in this regard and its results should be validated through experimental studies. The structurally and functionally important residues such as inhibitor-binding residues in the interactive site of docked CXCR4-complex and CXCR7-complex were identified. Recently, it was reported that CXCR7 activation may be a possible therapeutic target in cases of ischemic myocardium. It protected ischemic cells in hypoxic endothelial cells and infarction model by stimulating angiogenesis and decreasing apoptosis in C57BL/6 J mice model of acute myocardial infarction [60].
The structure of curcumin makes it an interesting lead for more development of novel antagonist candidates. Its established efficacy, proven safety, and reduced cost make is a promising agent for prevention and treatment of various human diseases. However, clinical use of curcumin is limited by its low bioavailability and many studied tried to reformulate it to enhance its bioavailability, but full evaluation of efficacy and safety of reformulated curcumin is still lacking [61]. In rat coronary atherosclerosis heart disease model, the permeability of coronary artery was found to be high which may be attributed to up-regulation of matrix metalloproteinase-9 (MMP-9), TNF-α and C-reactive protein (CRP) expression. Administration of oral curcumin (100 mg/kg/d) for 4 weeks inhibited expression of MMP-9, TNF-α and CRP and improved the permeability of coronary artery [62]. In primary human retinal endothelial cell (HREC) culture, SDF-1α increased HREC migration while AMD3100 (an inhibitor of CXCR4) and EGTA (a Ca++ chelator) effectively decreased it. Similarly, curcumin decreased SDF-1α-induced HRECs migration. The mechanism of action could be upstream blockage of Ca2+ influx which is an important signal for HREC migration or reduction of downstream PI3-K/AKT signals. Thus, curcumin inhibits HREC migration by interfering with SDF-1alpha/CXCR4 signaling [63]. Moreover, docking studies revealed that curcumin inhibited HIV replication through binding with CXCR4 [64].
Both resveratrol and quercetin inhibited angiogenesis in vitro where they inhibited growth of bovine aorta endothelial (BAE) cells, but resveratrol inhibited BAE cells migration more effectively than quercetin. Also, quercetin inhibited the tube formation of vascular endothelial cells. Thus, resveratrol and quercetin could be useful for tumor angiogenesis [65]. Resveratrol retarded development of cardiovascular disease by affecting certain risk factors such as diabetes or atherosclerosis. On human coronary arterial endothelial cells (HCAECs) resveratrol had anti-inflammatory effects via induction of autophagy which confers cardio-protection [66]. In retinal pigment epithelial cells, resveratrol effectively decreased phosphorylation of nuclear factor (NF)-κB (an upstream activator of CXCR4) resulting in suppression of CXCR4 expression and downregulation of vascular endothelial growth factor (VEGF) secretion. Resveratrol also suppressed VEGF secretion induced by SDF-1 [67]. In BTBR and C57BL/6 J mice, resveratrol treatment (20 and 40 mg/kg) significantly decreased CXCR production and expression in CD4+ T cells and decreased mRNA expression levels of CCR and CXCR in brain and spleen tissues [68]. Resveratrol and its combination with AMD3100 as a CXCR4 inhibitor significantly inhibited dihydrotestosterone (DHT)-induced progression of prostate cancer cell line via affecting the CXCR4 pathway [69]. Unfortunately, resveratrol has low bioavailability and solubility [70]. Quercetin, in cultured human endothelial cells, showed anti-oxidative and anti-inflammatory properties and protected against lipid peroxidation. Quercetin also decreased the transcriptional activity of NF-κB in human hepatocytes. In a humanized inflammation model (human CRP transgenic mice), quercetin inhibited IL-1β-induced CRP expression (CRP is a cardiovascular risk marker). In a humanized atherosclerosis model (ApoE*3Leiden transgenic mice), quercetin significantly attenuated atherosclerosis. Quercetin affected vascular cell proliferation thus reduced growth of atherosclerotic lesion. Quercetin systemically decreased expression of human CRP and cardiovascular risk factors and locally in aorta showed anti-proliferative effect resulting in attenuation of atherosclerosis [71]. Unfortunately, like curcumin and resveratrol, quercetin has low bioavailability, and its absorption can be affected by macronutrients [72].
Eucalyptol decreases lipopolysaccharide-induced cytokine production via action of NF-κB, TNF-α, IL-6, and IL-1β, and the ERK pathway. Thus, eucalyptol could be a potentially significant agent for treatment of chronic diseases [73]. In the Aβ (25-35) treated PC12 cells (a clonal cell line from an induced, transplantable rat pheochromocytoma), treatment with 1,8-cineole lowered the levels of TNF-α, IL-6, and IL-1β and the expression of nitric oxide synthase (NOS)-2, COX-2 and NF-κB indicating its protective effects and potential benefits in neurodegenerative disease [74]. In both anesthetized and conscious normotensive rats, intravenous 1,8-cineole dose-dependently decreased mean aortic pressure and only at the highest dose decreased heart rate. This hypotension is mostly due to an active vascular relaxation rather than inhibition of the sympathetic system [75].
Although all compounds, except quercetin, have more interactions with CXCR4 than with CXCR7, but the differences in the docking scores are minimal and do not imply great differences in the docking behavior. When a ligand is docked into a receptor protein, the interactions between them act between atoms which are not linked by covalent bonds i.e., there are non-bonded (non-covalent) interactions or bonds. The interactions between them could be hydrogen, electrostatic, van der Walls, polar-polar, and bi-bi staking and in some cases ionic bonds interactions which are all reversible and transient [76]. This means that the binding of the test natural compounds can be displaced by other antagonists or the endogenous ligands (CXCL11 and CXCL12) if their concentrations exceed those of the test natural compounds. Generally, based on the docking scores it can be decided which compound, compared with others, is the most effective toward the desired protein or receptor. In our study, we used a rigid protein and flexible ligands to evaluate the docking of the compounds inside the active site of protein so that the receptors remain unchanged and retain their original structure. The stability of compounds inside the cavity and determination of the duration of their binding to the receptors can be evaluated by using the molecular dynamics, which simulates the dynamic behavior of molecular systems as a function of time [61], but unfortunately, we do not have the programs which can do such study in addition to being time-consuming because it takes too much time to evaluate the stability of compounds inside the cavity to 500 ns or not. This is considered a limitation of our study. Moreover, detection of effect of the conformational rearrangements occurring in the CXCR4 and CXCR7 receptors as a result of their homodimerization and heterodimerization on binding to the test compounds is a second limitation of this study. However, these are good ideas for further studies.
The ADME prediction showed that the compounds possess several desirable ADME properties. The bioavailability radar showed that all compounds are predicted orally bioavailable. The consensus log Po/w, the classical descriptor for lipophilicity, showed that all the tested compounds are lipophilic because their values are above zero. Curcumin showed the highest lipophilicity and moderate solubility in water while quercetin, which showed the lowest lipophilicity, is soluble in water [77]. The four compounds have high GI absorption while only trans-resveratrol and (1 s,4 s)-Eucalyptol can penetrate the BBB. None of the compounds is a substrate of P-glycoprotein indicating absence of any active efflux for them from the gastrointestinal wall to the lumen or from the brain [35]. While (1 s,4 s)-Eucalyptol has no interactions with cytochromes P450 (CYP) major isoenzymes, other compounds can inhibit a certain CYP isoenzyme or another leading to adverse effects or drug interactions [36]. The skin permeability coefficient (Kp) is linearly correlated with molecular size and lipophilicity and the more negative the log Kp (cm/s), the less skin permeation [37]. Thus, quercetin has the lowest while (1 s,4 s)-Eucalyptol has the highest skin penetration which allows its application through skin massage. Moreover, the four compounds are considered to pass all the five commonly used drug-likeness rules and thus can be categorized as drug-like compounds and all of them reached a bioavailability score value of 0.55. All compounds, except (1 s,4 s)-Eucalyptol, maintain undesirable functional groups which could lead to carcinogenic, mutagenic, and hepatotoxic effects [78]. In agreement with previous studies, curcumin, trans-resveratrol, and quercetin were found non-cardiotoxic [79–81]. Although (1 s,4 s)-Eucalyptol showed a potential weak to moderate cardiotoxicity, but to our knowledge, there is no previous reports of cardiotoxicity of (1 s,4 s)-Eucalyptol. It was reported to cause hypotension [82], however this effect could be useful for hypertensive patients. “Leadlikeness” is similar to drug-likeness, but it focuses on physicochemical limits that define a good lead [46]. All compounds except quercetin have violations, but leads (by definition) can be subjected to chemical modifications which most likely increase their size and lipophilicity, thus leads are needed to be smaller and less hydrophobic than drug-like molecules [83]. All compounds have low “synthetic accessibility” score which is essential in the selection process of the most promising virtually-tested molecules that can be synthetized and provided to biological assays [47].
Based on the frontier molecular orbital theory, energies of HOMO and LUMO play an important role in chemical reactivity. The electronic absorption relates to the transition from the ground to the first excited state and mainly described by one electron excitation from HOMO to LUMO [84]. The HOMO-LUMO gap is related to the global chemical reactivity parameters. Large HOMO-LUMO gap is responsible for high kinetic stability [85]. In the current study, curcumin showed the highest values of ∆ E, ionization potential, absolute electronegativity, absolute and global hardness, global electrophilicity, and additional electronic charge. In another hand, curcumin showed the lowest values in electron affinity, and absolute and global softness which may be attributed to its high expected activity toward the CXCR4 and CXCR7 receptors.
The molecular dynamics simulation with molecular-mechanics-generalized born or Poisson-Boltzmann and surface area (MM/GBSA or MM/PBSA) methods are famous ways to evaluate free energy of binding of small ligands to biological macromolecules [86]. In the current study, MM/PBSA based methods was used for calculation of different interaction energies between the protein and inhibitor during the course of 100 ns MD simulations. It was observed that the free energy of Van der Waals was around − 218.226 kJ/mol, along with electrostatic component of − 27.723 kJ/mol. The free energy associated with the solvent accessibility was around − 18.424 kJ/mol and the free energy of binding was around − 282.797 kJ/mol validating the outcomes generated from the molecular docking. For molecular dynamics, we did that calculation to validate the probability of the compound with the highest docking score (curcumin) to be a good candidate to the active site. We did not make MD calculations for the other three compounds due to their low activities in addition to time, budget, and server limitations.
The current study provides novel visions into regulation of the CXCR4 and CXCR7 receptors by these natural compounds enabling possible explanations for their behavior. Use of these compounds either orally (for the three polyphenols) or by inhalation or skin massage (for eucalyptol) is considered non-invasive, cheap, easily applicable, and cost-effective approach. Therefore, repurposing of both curcumin and eucalyptol against coronary artery disease to combat chronic inflammation and oxidative stress and improve side effects such as insomnia and nausea is suggested for clinical trials.
Limitations of this study include: (1) The 100 ns scale molecular dynamics simulation has been done only for curcumin (the compound having the best docking score). (2) The stability of compounds inside the cavity and determination of the duration of their binding to the receptors can be evaluated by using the molecular dynamics, but unfortunately, we do not have the programs which can do such study in addition to time constraints because it takes too much time to evaluate the stability of compounds inside the cavity to 500 ns. (3) Detection of effect of the conformational rearrangements occurring in the CXCR4 and CXCR7 receptors as a result of their homodimerization and heterodimerization on binding to the test compounds. Further studies are recommended to address these limitations.
Strengths of this study include: (1) To our knowledge, this is the first attempt to find binding interactions of these natural agents with CXCR4 and CXCR7 receptors and to predict their druggability. Curcumin showed the top binding interaction against active sites of CXCR4 and CXCR7 receptors, with the best safety profile, followed by quercetin, resveratrol, and eucalyptol. All compounds demonstrated drug-like properties. Eucalyptol has promising potential because it can be used by inhalation or skin massage. (2) The current study provides novel visions into regulation of the CXCR4 and CXCR7 receptors by these natural compounds enabling possible explanations for their behavior. Recently, it was reported that CXCR7 activation may be a possible therapeutic target in cases of ischemic myocardium.
Some previous studies have reported relationships between CXCR4 and curcumin, quercetin, resveratrol as mentioned before but to the best of our knowledge, this is the first study that reports binding interactions of eucalyptol with CXCR4 and interactions of the four compounds with the CXCR7 receptors. But unfortunately, curcumin, resveratrol, and quercetin have low bioavailability. This is the main clinical gap facing these compounds; thus, studies and clinical trials are needed to overcome this issue.
Conclusions
Molecular modeling procedures were used to screen for the potential regulatory effects of four cardiovascular active natural compounds; three polyphenols and a terpene; on the CXCL12/CXCR4/CXCR7 pathway. After a virtual screening against the active sites of CXCR4 and CXCR7 receptor, curcumin showed the top binding interaction, with the best safety profile, followed by quercetin, resveratrol, and lastly eucalyptol. The ADME prediction using the SwissADME server showed that all compounds possess drug-like properties. Only (1 s,4 s)-Eucalyptol has potential weak cardiotoxicity. The results of the thermochemical and molecular orbital analysis and the molecular dynamics simulation validated the outcomes of the molecular docking study. The four compounds have low oral bioavailabilities and thus eucalyptol has a promising potential because it can be used by inhalation or skin massage. These results may provide novel visions into CXCR4 and CXCR7 regulation enabling a possible explanation of behavior shown by these natural compounds. Use of these compounds either orally (for the three polyphenols) or by inhalation or skin massage (for eucalyptol) is considered non-invasive, cheap, easily applicable, and cost-effective approach. Therefore, repurposing of both curcumin and eucalyptol against CAD disease to combat chronic inflammation and oxidative stress and improve side effects such as insomnia and nausea is suggested for clinical trials. To the best of our knowledge, this is the first structural attempt to find binding interactions of natural anti-inflammatory agents; curcumin, resveratrol, quercetin, and eucalyptol with the CXCR4 and CXCR7 receptors and to predict their drug-likeness properties.
Supplementary Information
Additional file 1: Fig. S1. The 2D and 3D docking interaction of Curcumin, Trans-resveratrol, Quercetin, and (1 s,4 s)-Eucalyptol with protein 3ODU of CXCR4 receptor. Fig. S2. The 2D and 3D docking interaction of Curcumin, Trans-resveratrol, Quercetin, and (1 s,4 s)-Eucalyptol with protein 6K3F of CXCR 7 receptor.
Acknowledgements
Not applicable.
Abbreviations
- ADME/T
Absorption, distribution, metabolism, and excretion/toxicity
- CXCL12
Chemokine (C-X-C motif) ligand 12
- SDF-1
Stromal-cell-derived factor-1
- GPCR
G protein coupled receptors
- PI3K
Phosphoinositide-3 kinase
- PLC/IP3
Phospholipase C/inositol 1,4,5, triphosphate
- ERK1/2
Extracellular regulated kinase 1/2
- CAD
Coronary artery disease
- EOCs
Endothelial outgrowth cells
- NF-κB
Nuclear factor-κB
- MAPK
Mitogen-activated protein kinases
- COX
Cyclooxygenase
- TNF-α
Tumor necrosis factor-α
- IL-1b
Interleukin 1b
- MOE
Molecular environment
- MW
Molecular weight
- MR
Molecular refractivity
- TPSA
Topological polar surface area
- PSA
Polar surface area
- Log Po/w
Log partition coefficient between n-octanol and water
- Log S
Log molar solubility in water
- ESOL
Estimated solubility
- hERG
Human Ether-à-go-go-Related Gene
- QSAR
Quantitative structure-activity relationship
- SDF
Structure data file
- SMILES
Simplified molecular-input line-entry system
- QM
Quantum mechanical
- DFT
Density functional theory
- MD
Molecular dynamics
- MM/GBSA or MM/PBSA
Molecular-mechanics-generalized born or Poisson-Boltzmann and surface area
- H-bonds
Hydrogen bonds
- RMSD
Root mean square deviations
- Rg
Radius of gyration
- AD
Applicability domain
- HOMO
Highest occupied molecular orbital
- LUMO
Lowest unoccupied molecular orbital
- ∆E
HOMO–LUMO energy gap
- I
Ionization potential
- A
Electron affinity
- χ
Absolute electronegativities
- ɳ
Absolute hardness
- σ
Absolute softness
- Pi
Chemical potentials
- S
Global softness
- H
Global hardness
- ΔH
Enthalpies
- ΔG
Free energies
- BBB
Blood-brain barrier
- P-gp
Permeability glycoprotein
- CYP
Cytochrome P450
- Log Kp
Log skin permeability coefficient
- MMP-9
Matrix metalloproteinase-9
- CRP
C-reactive protein
- HREC
Human retinal endothelial cell
- BAE cell
Bovine aorta endothelial cell
- HCAECs
Human coronary arterial endothelial cells
- NF-κB
Nuclear factor-κB
- NOS
Nitric oxide synthase
Authors’ contributions
H.M. designed the study. M.H. conducted the practical work. H.M., M.H., and T.A. collected and analyzed data. All authors interpreted data, wrote, and approved the final manuscript.
Funding
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (839). The funding body has no role in the study design, data collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
The data and materials are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Costantini S, et al. Common structural interactions between the receptors CXCR3, CXCR4 and CXCR7 complexed with their natural ligands, CXCL11 and CXCL12, by a modeling approach. Cytokine. 2013;64(1):316–321. doi: 10.1016/j.cyto.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Min K, et al. Crystal structure of β-Arrestin 2 in complex with CXCR7 phosphopeptide. Structure. 2020;28(9):1014–1023.e4. doi: 10.1016/j.str.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miao Z, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104(40):15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssens R, Struyf S, Proost P. The unique structural and functional features of CXCL12. Cell Mol Immunol. 2018;15(4):299–311. doi: 10.1038/cmi.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Würth R, et al. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front Cell Neurosci. 2014;8:144. doi: 10.3389/fncel.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23(1):43–52. doi: 10.1038/leu.2008.299. [DOI] [PubMed] [Google Scholar]
- 7.Cao Z, et al. CXCR7/p-ERK-signaling is a novel target for therapeutic vasculogenesis in patients with coronary artery disease. PLoS One. 2016;11(9):e0161255. doi: 10.1371/journal.pone.0161255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan R, et al. Holistic regulation of angiogenesis with Chinese herbal medicines as a new option for coronary artery disease. Evid Based Complement Alternat Med. 2018;2018:3725962. doi: 10.1155/2018/3725962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scotti L, et al. Docking studies for multi-target drugs. Curr Drug Targets. 2017;18(5):592–604. doi: 10.2174/1389450116666150825111818. [DOI] [PubMed] [Google Scholar]
- 10.Hewlings SJ, Kalman DS. Curcumin: a review of its effects on human health. Foods. 2017;6(10):92. doi: 10.3390/foods6100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salehi B, et al. Curcumin’s nanomedicine formulations for therapeutic application in neurological diseases. J Clin Med. 2020;9(2):430. doi: 10.3390/jcm9020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salehi B, et al. Plant-derived bioactives in oral mucosal lesions: a key emphasis to curcumin, lycopene, chamomile, aloe vera, green tea and coffee properties. Biomolecules. 2019;9(3):106. doi: 10.3390/biom9030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiciński M, et al. Beneficial effects of resveratrol administration-focus on potential biochemical mechanisms in cardiovascular conditions. Nutrients. 2018;10(11):1813. doi: 10.3390/nu10111813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma YK, Chen YB, Li P. Quercetin inhibits NTHi-triggered CXCR4 activation through suppressing IKKα/NF-κB and MAPK signaling pathways in otitis media. Int J Mol Med. 2018;42(1):248–258. doi: 10.3892/ijmm.2018.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Islam MS, et al. Neuropharmacological effects of quercetin: a literature-based review. Front Pharmacol. 2021;12:665031. doi: 10.3389/fphar.2021.665031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buckle J. Clinical aromatherapy-e-book: essential oils in practice. Amsterdam: Elsevier Health Sciences; 2014.
- 17.Bahrami T, et al. Effect of aromatherapy massage on anxiety, depression, and physiologic parameters in older patients with the acute coronary syndrome: a randomized clinical trial. Int J Nurs Pract. 2017;23(6). 10.1111/ijn.12601. [DOI] [PubMed]
- 18.Karadag E, et al. Effects of aromatherapy on sleep quality and anxiety of patients. Nurs Crit Care. 2017;22(2):105–112. doi: 10.1111/nicc.12198. [DOI] [PubMed] [Google Scholar]
- 19.Mubarak S, Khanday MA, Lone AUH. Mathematical analysis based on eigenvalue approach to study liver metastasis disease with applied drug therapy. Netw Model Anal Health Inform Bioinform. 2020;9(1):25. [Google Scholar]
- 20.Naik PA. Modeling the mechanics of calcium regulation in T lymphocyte: a finite element method approach. Int J Biomath. 2020;13(5):2050038–2050059. [Google Scholar]
- 21.Naik PA, Pardasani KR. Three-dimensional finite element model to study calcium distribution in oocytes. Netw Model Anal Health Inform Bioinform. 2017;6:1–11. [Google Scholar]
- 22.Naik PA, Zu J. Modeling and simulation of spatial-temporal calcium distribution in T lymphocyte cell by using a reaction-diffusion equation. J Bioinforma Comput Biol. 2020;18(2):2050013–2050033. doi: 10.1142/S0219720020500134. [DOI] [PubMed] [Google Scholar]
- 23.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daina A, Michielin O, Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model. 2014;54(12):3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee P, et al. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–w263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braga RC, et al. Pred-hERG: a novel web-accessible computational tool for predicting cardiac toxicity. Mol Inform. 2015;34(10):698–701. doi: 10.1002/minf.201500040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braga RC, et al. Tuning HERG out: antitarget QSAR models for drug development. Curr Top Med Chem. 2014;14(11):1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritchie TJ, Ertl P, Lewis R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov Today. 2011;16(1-2):65–72. doi: 10.1016/j.drudis.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52(21):6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 30.Daina A, Zoete V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnott JA, Planey SL. The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov. 2012;7(10):863–875. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- 32.Mannhold R, et al. Calculation of molecular lipophilicity: state-of-the-art and comparison of log P methods on more than 96,000 compounds. J Pharm Sci. 2009;98(3):861–893. doi: 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- 33.Delaney JS. ESOL: estimating aqueous solubility directly from molecular structure. J Chem Inf Comput Sci. 2004;44(3):1000–1005. doi: 10.1021/ci034243x. [DOI] [PubMed] [Google Scholar]
- 34.Ali J, et al. Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J Chem Inf Model. 2012;52(2):420–428. doi: 10.1021/ci200387c. [DOI] [PubMed] [Google Scholar]
- 35.Montanari F, Ecker GF. Prediction of drug-ABC-transporter interaction--recent advances and future challenges. Adv Drug Deliv Rev. 2015;86:17–26. doi: 10.1016/j.addr.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang SM, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48(6):662–670. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- 37.Pecoraro B, et al. Predicting skin permeability by means of computational approaches: reliability and caveats in pharmaceutical studies. J Chem Inf Model. 2019;59(5):1759–1771. doi: 10.1021/acs.jcim.8b00934. [DOI] [PubMed] [Google Scholar]
- 38.Bickerton GR, et al. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4(2):90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipinski CA, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 40.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1(1):55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 41.Veber DF, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 42.Egan WJ, Merz KM, Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43(21):3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 43.Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem. 2001;44(12):1841–1846. doi: 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- 44.Martin YC. A bioavailability score. J Med Chem. 2005;48(9):3164–3170. doi: 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- 45.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 46.Brenk R, et al. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem. 2008;3(3):435–444. doi: 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ertl P, Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform. 2009;1(1):8. doi: 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharfalddin AA, et al. Synthesis and theoretical calculations of metal-antibiotic chelation with thiamphenicol: in vitro DNA and HSA binding, molecular docking, and cytotoxicity studies. New J Chem. 2021;45(21):9598–9613. [Google Scholar]
- 49.Hoque MJ, Ahsan A, Hossain MB. Molecular docking, pharmacokinetic, and DFT calculation of naproxen and its degradants. Biomed J Sci Tech Res. 2018;9(5):7360–7365. [Google Scholar]
- 50.El-Ghamaz N, et al. Synthesis and optical properties studies of antipyrine derivatives thin films. J Saudi Chem Soc. 2017;21:S339–S348. [Google Scholar]
- 51.Van Der Spoel D, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 52.Oostenbrink C, et al. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25(13):1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 53.Schüttelkopf AW, Van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 54.Zielkiewicz J. Structural properties of water: comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J Chem Phys. 2005;123(10):104501. doi: 10.1063/1.2018637. [DOI] [PubMed] [Google Scholar]
- 55.Kumari R, et al. g_mmpbsa— a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 56.Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved peptide and protein torsional energetics with the OPLS-AA force field. J Chem Theory Comput. 2015;11(7):3499–3509. doi: 10.1021/acs.jctc.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dodda LS, et al. LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017;45(W1):W331–W336. doi: 10.1093/nar/gkx312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu B, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330(6007):1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitchen DB, et al. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 60.Zhang S, et al. Activation of CXCR7 alleviates cardiac insufficiency after myocardial infarction by promoting angiogenesis and reducing apoptosis. Biomed Pharmacother. 2020;127:110168. doi: 10.1016/j.biopha.2020.110168. [DOI] [PubMed] [Google Scholar]
- 61.Zhou H, Beevers CS, Huang S. The targets of curcumin. Curr Drug Targets. 2011;12(3):332–347. doi: 10.2174/138945011794815356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X, et al. Effect of curcumin on permeability of coronary artery and expression of related proteins in rat coronary atherosclerosis heart disease model. Int J Clin Exp Pathol. 2015;8(6):7247–7253. [PMC free article] [PubMed] [Google Scholar]
- 63.Sameermahmood Z, et al. Curcumin modulates SDF-1alpha/CXCR4-induced migration of human retinal endothelial cells (HRECs) Invest Ophthalmol Vis Sci. 2008;49(8):3305–3311. doi: 10.1167/iovs.07-0456. [DOI] [PubMed] [Google Scholar]
- 64.Singh S, Malik BK, Sharma DK. Targeting HIV-1 through molecular modeling and docking studies of CXCR4: leads for therapeutic development. Chem Biol Drug Des. 2007;69(3):191–203. doi: 10.1111/j.1747-0285.2007.00478.x. [DOI] [PubMed] [Google Scholar]
- 65.Igura K, et al. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett. 2001;171(1):11–16. doi: 10.1016/s0304-3835(01)00443-8. [DOI] [PubMed] [Google Scholar]
- 66.Huang FC, et al. Anti-inflammatory effect of resveratrol in human coronary arterial endothelial cells via induction of autophagy: implication for the treatment of Kawasaki disease. BMC Pharmacol Toxicol. 2017;18(1):3. doi: 10.1186/s40360-016-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seong H, et al. Resveratrol suppresses vascular endothelial growth factor secretion via inhibition of CXC-chemokine receptor 4 expression in ARPE-19 cells. Mol Med Rep. 2015;12(1):1479–1484. doi: 10.3892/mmr.2015.3518. [DOI] [PubMed] [Google Scholar]
- 68.Bakheet SA, et al. Resveratrol treatment attenuates chemokine receptor expression in the BTBR T+tf/J mouse model of autism. Mol Cell Neurosci. 2016;77:1–10. doi: 10.1016/j.mcn.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Jang YG, et al. Resveratrol inhibits DHT-induced progression of prostate cancer cell line through interfering with the AR and CXCR4 pathway. J Steroid Biochem Mol Biol. 2019;192:105406. doi: 10.1016/j.jsbmb.2019.105406. [DOI] [PubMed] [Google Scholar]
- 70.Galiniak S, Aebisher D, Bartusik-Aebisher D. Health benefits of resveratrol administration. Acta Biochim Pol. 2019;66(1):13–21. doi: 10.18388/abp.2018_2749. [DOI] [PubMed] [Google Scholar]
- 71.Kleemann R, et al. Anti-inflammatory, anti-proliferative and anti-atherosclerotic effects of quercetin in human in vitro and in vivo models. Atherosclerosis. 2011;218(1):44–52. doi: 10.1016/j.atherosclerosis.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 72.Rich GT, et al. Towards an understanding of the low bioavailability of quercetin: a study of its interaction with intestinal lipids. Nutrients. 2017;9(2):111. doi: 10.3390/nu9020111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seol GH, Kim KY. Eucalyptol and its role in chronic diseases. Adv Exp Med Biol. 2016;929:389–398. doi: 10.1007/978-3-319-41342-6_18. [DOI] [PubMed] [Google Scholar]
- 74.Khan A, et al. 1,8-cineole (eucalyptol) mitigates inflammation in amyloid Beta toxicated PC12 cells: relevance to Alzheimer’s disease. Neurochem Res. 2014;39(2):344–352. doi: 10.1007/s11064-013-1231-9. [DOI] [PubMed] [Google Scholar]
- 75.Lahlou S, et al. Cardiovascular effects of 1,8-cineole, a terpenoid oxide present in many plant essential oils, in normotensive rats. Can J Physiol Pharmacol. 2002;80(12):1125–1131. doi: 10.1139/y02-142. [DOI] [PubMed] [Google Scholar]
- 76.Rahman A, et al. Non-covalent interactions involving halogenated derivatives of capecitabine and thymidylate synthase: a computational approach. Springerplus. 2016;5:146. doi: 10.1186/s40064-016-1844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Czyrski A. Determination of the lipophilicity of ibuprofen, naproxen, ketoprofen, and flurbiprofen with thin-layer chromatography. J Chem. 2019;2019:3407091. [Google Scholar]
- 78.Park BK, et al. Managing the challenge of chemically reactive metabolites in drug development. Nat Rev Drug Discov. 2011;10(4):292–306. doi: 10.1038/nrd3408. [DOI] [PubMed] [Google Scholar]
- 79.Chakraborty M, Bhattacharjee A, Kamath JV. Cardioprotective effect of curcumin and piperine combination against cyclophosphamide-induced cardiotoxicity. Indian J Pharmacol. 2017;49(1):65–70. doi: 10.4103/0253-7613.201015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Monahan DS, et al. Resveratrol significantly improves cell survival in comparison to dexrazoxane and carvedilol in a h9c2 model of doxorubicin induced cardiotoxicity. Biomed Pharmacother. 2021;140:111702. doi: 10.1016/j.biopha.2021.111702. [DOI] [PubMed] [Google Scholar]
- 81.Daubney J, et al. Cardioprotective and cardiotoxic effects of quercetin and two of its in vivo metabolites on differentiated h9c2 cardiomyocytes. Basic Clin Pharmacol Toxicol. 2015;116(2):96–109. doi: 10.1111/bcpt.12319. [DOI] [PubMed] [Google Scholar]
- 82.Moon HK, et al. Effects of 1,8-cineole on hypertension induced by chronic exposure to nicotine in rats. J Pharm Pharmacol. 2014;66(5):688–693. doi: 10.1111/jphp.12195. [DOI] [PubMed] [Google Scholar]
- 83.Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov. 2012;11(5):355–365. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- 84.Uzzaman M, Hoque MJ. Physiochemical, molecular docking, and pharmacokinetic studies of naproxen and its modified derivatives based on DFT. Int J Sci Res Manag. 2018;6(9):C. [Google Scholar]
- 85.Alamri MA, et al. Biological activity evaluation and computational study of novel triazene derivatives containing benzothiazole rings. J Mol Struct. 2021;1227:129507. [Google Scholar]
- 86.Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Fig. S1. The 2D and 3D docking interaction of Curcumin, Trans-resveratrol, Quercetin, and (1 s,4 s)-Eucalyptol with protein 3ODU of CXCR4 receptor. Fig. S2. The 2D and 3D docking interaction of Curcumin, Trans-resveratrol, Quercetin, and (1 s,4 s)-Eucalyptol with protein 6K3F of CXCR 7 receptor.
Data Availability Statement
The data and materials are available from the corresponding author on reasonable request.