

Abstract
Translocator protein 18 kDa (TSPO) is a biomarker of neuroinflammation. [11C]ER176 robustly quantifies TSPO in the human brain with positron emission tomography (PET), irrespective of subject genotype. We aimed to develop an ER176 analog with potential for labeling with longer-lived fluorine-18 (t1/2 = 109.8 min). New fluoro and trifluoromethyl analogs of ER176 were prepared through a concise synthetic strategy. These ligands showed high TSPO affinity and low human genotype sensitivity. Each ligand was initially labeled by a generic 11C-methylation procedure, thereby enabling speedy screening in mice. Each radioligand was rapidly taken up and well retained in the mouse brain at baseline after intravenous injection. Preblocking of TSPO showed that high proportions of brain uptake were specifically bound to TSPO at baseline. Overall, the 3-fluoro analog of [11C]ER176 ([11C]3b) displayed the most promising imaging properties. Therefore, a method was developed to label 3b with [18F]fluoride ion. [18F]3b gave similarly promising PET imaging results and deserves evaluation in higher species.
Graphical Abstract

INTRODUCTION
There is growing evidence for the involvement of central inflammation in some major neuropsychiatric disorders, including depression1 and schizophrenia.2 Much of this evidence has been gained from positron emission tomography (PET) with selective radioligands to image translocator protein 18 kDa (TSPO).1,2 TSPO3 is expressed on the outer mitochondrial membrane in activated microglia, astrocytes, and macrophages4 and is implicated in several functions, including cholesterol transport5 and steroidogenesis.6 TSPO levels are very low in healthy human brains but are vastly upregulated in several human pathologies involving inflammation.1,2 Thus, TSPO is considered to be an important biomarker of neuroinflammation.
Several classes of radioligand have been developed for PET imaging of TSPO.7-9 The prototypical radioligand, [11C]PK11195, first as racemate10 and later as (R)-enantiomer (Figure 1),11 has been in use for almost four decades to image TSPO in neurological and psychiatric disorders. However, this radioligand suffers from severe limitations, including low TSPO sensitivity and poor amenability to quantification.12,13 Therefore, considerable effort has been expended on the development of “second-generation” radioligands with improved properties. These radioligands include [11C]PBR28, 14-16 [11C]DPA-713,17 [18F]FBR,18 and [18F]FEPPA19 (Figure 1). Many of these second-generation radioligands show improved TSPO-specific signals but suffer from sensitivity to a single nucleotide polymorphism (SNP), namely, rs6971,20-22 which has an allelic frequency of about 30% in humans of European ancestry. This SNP is co-dominantly expressed, giving rise to three subject genotypes now called high-affinity binders (HABs), low-affinity binders (LABs), and mixed-affinity binders (MABs).20 The ratio of affinity for the two alleles can be considerable for many second-generation radioligands and notably reaches about 50-fold for [11C]-PBR28.23,24 For this radioligand, brain uptake is negligible in LABs and they must be excluded from PET studies by prescreening of all subjects for TSPO genotype.25 PET data from MAB subjects must also be corrected for the lowering effect (50%) of genotype on total specific binding.25
Figure 1.

Structures of prominent first- and second-generation TSPO PET radioligands.
PK11195 shows low sensitivity to SNP rs6971 when tested in vitro.23 Therefore, this chemotype has been used as a template for the design of new radioligands that might provide higher TSPO imaging performance while showing low to negligible genotype sensitivity. Notably, a small structural modification to PK11195, namely, the conversion of its core isoquinolinyl ring into a quinazolinyl ring, has led to the discovery of a new high-affinity and less lipophilic TSPO ligand, (R)-N-(sec-butyl)-4-(2-chlorophenyl)-N-methylquina-zoline-2-carboxamide,26 now dubbed ER176, which retains low genotype sensitivity in vitro.27
ER176, when labeled with carbon-11 (t1/2 = 20.4 min), shows excellent brain kinetics and high TSPO-specific signal in PET studies of monkey27 and human brains.28,29 Moreover, and unlike most-second generation TSPO PET radioligands, [11C]ER176 (Figure 1) gives quantifiable PET imaging results in all three human subject genotypes.29 For these reasons, [11C]ER176 is now regarded as one of the best performing radioligands for imaging and quantifying TSPO in human subjects.28 The production of this radioligand for this purpose has now been described in detail.30
Whereas [11C]ER176 is an excellent PET radioligand, analogs that can be labeled with longer-lived fluorine-18 (t1/2 = 109.8 min) and provide comparable imaging performance could be more broadly useful, especially to the large number of imaging centers lacking access to short-lived carbon-11.
The initial aims of this study were (i) to replace the 2-chloro substituent of ER176 with either a ring fluoro or trifluoromethyl substituent, (ii) to assess the effect of such a replacement on binding affinity to TSPO, (iii) to radiolabel each new ligand with carbon-11 in the common N-methyl position matching that in [11C]ER176 by taking advantage of a 11C-labeling procedure already well established in our laboratory,30 and (iv) to compare the behaviors of the new PET 11C-labeled ligands in mice. This study with 11C-labeled ligands would serve as a speedy screen to identify which, if any, of the new ligands would merit labeling with fluorine-18 and further evaluation in higher species. Therefore, the final aims of this study were to label the most promising ligand with fluorine-18 and to confirm mouse brain imaging behavior matching that of the 11C-labeled version.
RESULTS AND DISCUSSION
Ligand Design.
We considered modifications to the structure of ER176 that might provide possibilities for eventual labeling with fluorine-18 (t1/2 = 109.8 min). Replacement of the 2-chloro substituent in PK11195 with a 2-fluoro substituent is not detrimental to binding affinity for TSPO.31 A 2-fluoro substituent is also well tolerated in 4-phenyl-quinazoline-2-carboxamide TSPO ligands related to ER176.32 Moreover, such a change is calculated with the Pallas for Windows software to reduce the lipophilicity of ER176 (clogD = 4.12) to a value (clogD = 3.55) that would be more desirable for a brain PET radioligand (preferred clogD ~ 2.2)33 without altering the acceptably moderate total polar surface area (tPSA = 45 Å2). The pendant phenyl ring in ER176 is quite electron-rich and therefore is not activated for classical aromatic nucleophilic substitution of a good leaving group with [18F]fluoride ion.34 Nonetheless, labeling might be feasible through modern methods of aryl ring radiofluorination based on, for example, the use of hypervalent iodine35 or boronic acid ester precursors.36 Therefore, we aimed to explore the replacement of the chloro substituent in ER176 with a fluoro substituent as well as the effects of the fluoro substituent ring position.
Several methods for labeling aryl trifluoromethyl groups with fluorine-18 have emerged, mostly based on the use of [18F]trifluoromethyl-copper(I) derived from [18F]fluoroform. 37-41 They are generally quite efficient, albeit with molar activity outcomes constrained to moderate levels. An aryl trifluoromethyl group is expected to be metabolically stable and only marginally increases lipophilicity over an aryl chloro substituent (by ~0.17 logD units) without increasing tPSA. Moreover, an aryl trifluoromethyl group can provide an alternative site for labeling with carbon-11 at high molar activity by the use of readily prepared [11C]fluoroform as its [11C]trifluoromethyl-copper(I) derivative.42-44 Therefore, we also aimed to prepare radioligands in which the 2-chloro substituent of the pendant phenyl ring in ER176 is replaced with a single trifluoromethyl group at the 2-, 3-, or 4- position.
Synthesis of Fluorine-Containing Ligands.
ER176 and many close analogs have been prepared by multistep pathways from appropriately substituted 2-aminobenzophenones.26 The benzophenones are treated with glyoxylic acid followed by an amidation (Scheme 1A). We devised a new synthetic route to have faster access to our target ER176 derivatives. This route involves the synthesis of a chiral quinazoline amide synthon for rapid coupling to commercially available fluoro-containing aryl boronic acids (Scheme 1B).
Scheme 1.

Original Pathway for Preparing ER176 (A) and a New Pathway for Preparing Fluorine-Containing 4-Phenylquinazoline-2-carboxamides as Ligands for TSPO (B)
Several tautomerizable 2-quinoxalinones have been shown to couple directly with aryl boronic acids upon treatment with phosphonium salt activators, such as bromo-tris-pyrrolidinophosphonium hexafluorophosphate (PyBroP).45 We considered that this approach might be applied to a homochiral tautomerizable oxoquinazoline amide that might then be used as a common synthon for the preparation of a variety of fluorine-containing ER176 analogs. The new pathway would combine mild phosphonium salt activated coupling (Et3N, 1,4-dioxane, RT) with classical Suzuki–Miyaura cross-coupling conditions (Pd(PPh3)4, Na2CO3, water, heating) in a single pot (Scheme 2). The required homochiral synthon, (R)-N-(sec-butyl)-N-methyl-4-oxo-3,4-dihydroquinazoline-2-carboxamide (2), was obtained as follows. First, (R)-N-methylbutan-2-aminium chloride was prepared as previously reported46 and stored under vacuum until ready for use. The ester, ethyl 4-oxo-3,4-dihydroquinazoline-2-carboxylate (1), was then treated with the aminium salt in a Weinreb reaction47 in the presence of trimethylaluminum to give 2 in very high yield. The presence of a tertiary carboxamide in 2 leads to rotamers, with the dominant E and Z rotamer types clearly detected by solution 1H and 13C NMR. The ratio and significance of such rotamers in structurally similar TSPO ligands, such as PK11195, have been discussed thoroughly.48
Scheme 2.

Synthesis and Structures of TSPO ligands 3a–3f
Treatment of the synthon 2 with PyBroP followed by coupling in the same pot with variously selected fluorinated aryl boronic acids under palladium catalysis led to the target fluorine-containing compounds 3a–3f in overall yields between 35 and 50%. Reaction yields were limited by the emergence of several byproducts, including some from homocoupling and protodeboronation of the aryl boronic acid. We therefore investigated the conditions for each step to improve yields. From 31P NMR and 19F NMR, we found that the coupling of PyBroP with 2 in 1,4-dioxane was almost complete after 2 h of stirring at RT. Decreasing the amount of PyBroP to either 1.5 or 1.8 equiv instead of 2.4 equiv and extending the reaction time to 12 h greatly increased the yields. After the intermediate phosphonium salt had formed and precipitated, the aryl boronic acid, palladium catalyst, and sodium carbonate were added with water and heated to 100 °C until reaction was complete (2–4 h). A decrease of the ratio of oxoquinazoline to aryl boronic acid from 4 to 1.8 almost completely avoided byproducts to give reproducible yields of 3a–3f in the 66–81% range.
The new ligands were assayed in rat kidney and human U87MG cells for inhibition of [3H]PK11195 binding to TSPO. Each ligand was found to have high affinity with a Ki value in the low nanomolar range (Table 1). These assays showed that substitution of the 2-chloro substituent in ER176 with either a 2-fluoro or 2-trifluoromethyl substituent was negligibly detrimental to the binding affinity for TSPO. The 3- and 4-isomers also exhibited high affinity to TSPO, with the 3-trifluoromethyl compound 3e having the highest affinity (rat Ki = 1.44 nM; human U87MG, 1.19 nM) (Table 1). Notably, for each substituent position, the trifluoromethyl compound showed a lower Ki value than the fluoro compound. Affinities for rat kidney TSPO and human U87MG TSPO were highly comparable across the whole set of ligands, consistent with the highly conserved TSPO sequence homology across mammalian species (Table 1).
Table 1.
Pharmacological and Physiochemical Properties of Fluorine-Containing Ligands, 3a–3f (Compared with PK11195 and ER176)a
| ligand | rat kidney Ki (nM)b |
human U87MG Ki (nM)b |
HAB Ki (nM)b |
LAB Ki (nM)b |
LAB to HAB Ki ratio |
logD7.4c | rat kidney |
lipEd human U87MG |
HAB |
|---|---|---|---|---|---|---|---|---|---|
| PK11195 | 3.40 ± 0.35 | 2.90 ± 0.14 | 26.1 ± 18.5 | 17.8 ± 20.9 | 0.68 | 3.97 ± 0.02 | 4.50 | 4.57 | 3.78 |
| ER176e | 3.10 ± 0.30 | 1.48 | 1.89 | 1.28 | 3.55 ± 0.02 | 4.96 | 5.28 | ||
| 3a | 4.19 ± 0.35 | 5.64 ± 0.66 | 9.0 ± 9.5 | 25.0 ± 11.6 | 2.78 | 3.04 ± 0.03 | 5.34 | 5.21 | 5.01 |
| 3b | 5.17 ± 0.63 | 5.09 ± 0.65 | 9.1 ± 8.1 | 24.9 ± 12.7 | 2.74 | 3.09 ± 0.01 | 5.20 | 5.20 | 4.95 |
| 3c | 7.41 ± 0.82 | 7.03 ± 0.66 | 15.3 ± 10.3 | 44.5 ± 34.4 | 2.91 | 3.01 ± 0.02 | 5.12 | 5.14 | 4.81 |
| 3d | 3.74 ± 0.10 | 3.18 ± 0.17 | 6.8 ± 8.1 | 37.1 ± 30.2 | 5.46 | 3.25 ± 0.03 | 5.18 | 5.25 | 4.97 |
| 3e | 1.44 ± 0.21 | 1.19 ± 0.17 | 18.6 ± 15.7 | 36.9 ± 38.5 | 1.98 | 3.71 ± 0.03 | 5.14 | 5.21 | 4.02 |
| 3f | 4.39 ± 0.14 | 2.42 ± 0.66 | 5.0 ± 3.9 | 4.0 ± 5.0 | 0.80 | 3.93 ± 0.01 | 4.43 | 4.69 | 4.37 |
Human genotype sensitivity in vitro would be an important consideration in any eventual selection of a TSPO radioligand for labeling with fluorine-18 and evaluation in human. Therefore, we tested each ligand in brain cells from human HAB and LAB genotypes. Each ligand exhibited low sensitivity to TSPO genotype in vitro with HAB/LAB affinity ratios (as represented by LAB/HAB Ki ratios) between 0.8 and 5.46. Compound 3f showed the lowest in vitro sensitivity with an affinity ratio of 0.8, which is close to values already reported23,27 for PK11195 (0.8 and 0.9). In this study, we found a slightly lower value (0.68) for PK11195. The value for 3f is lower than that (1.28) reported27 for ER176. Other values were equal to or greater than 2.0. Thus, the set of six new ligands showed overall encouraging in vitro properties for development as PET radioligands.
Synthesis of N-Desmethyl Compounds as Precursors for 11C-Labeling.
To allow a quick comparison of new radioligands with [11C]ER176, we decided to label all six new ligands by a generic 11C-N-methylation procedure, similar to that in place in our laboratory for [11C]ER176 synthesis. The N-desmethyl compounds 5a–5f that were required as precursors for labeling were prepared by the same route as the ligands, except that (R)-2-aminobutane was deployed to synthesize the desired desmethyl amide synthon 4 (Scheme 3). As expected, palladium-catalyzed coupling of 4 with the set of six fluorine-containing aryl boronic acids gave the corresponding precursors 5a–5f in good to high yields (65–89%).
Scheme 3.

Synthesis of N-Desmethyl Precursors, 5a–5f, for Use in 11C-Labeling
11C-Radiochemistry.
The desmethyl precursor 5e was selected as a substrate for optimizing radiolabeling conditions. After dissolution of 5e in DMSO at RT, treatment with powdered KOH (1.2 equiv) gave a yellow solution. Treatment of the precursor solution with [11C]iodomethane followed by heating at 85 °C for 2 min gave the targeted radioligand [11C]3e. However, we observed a wide variability in labeling yield that we attributed to the variation in ambient moisture levels during base preparation.
A series of conditions, including solvents, bases, and reaction times, and also labeling platforms, were tested to optimize labeling yields. Bases in solutions, such as t-BuOK or TBAH in THF, were less reliable than the solid bases NaH, KOH, or t-BuOK, with the latter giving the best yield. The reaction did not proceed in DMF or THF and performed best in very dry DMSO. The radioligand was produced in higher yields from 5 min reaction times than from 2 min reaction times. Additionally, a synthesis using the “loop method”50 failed to provide any labeled product. Overall, we found that reproducible and acceptably high yields were obtained when the precursor and solid t-BuOK (1.0 and 1.2 equiv, respectively) were loaded into an oven-dried vial within a glove-box under nitrogen followed by treatment with [11C]iodomethane in DMSO at 85 °C for 5 min. Under these conditions, precursors 5a–5f gave the corresponding radioligands [11C]3a–[11C]3f in moderate decay-corrected formulated yields (8–15%) from cyclotron-produced [11C] carbon dioxide (Scheme 4).
Scheme 4.

Radiosyntheses of Radioligands [11C]3a–[11C]3f
TSPO radioligands [11C]3a–[11C]3f were obtained in excellent radiochemical purities (>99%) and with molar activities in the range of 229–351 GBq/μmol. The formulated radioligands were free of appreciable chemical impurities. Radiosyntheses required 40–45 min for the trifluoromethyl radioligands and 52–55 min for the fluoro radioligands. Each radioligand was formulated for intravenous injection in saline (0.9%, w/v)-ethanol (9:1 v/v). Radioligand identity was verified by coinjection with the corresponding reference compound and by LC-MS analysis of the associated carrier. All the radioligands were greater than 97% radiochemically stable in the phosphate buffer for at least 2.5 h. They were also stable for at least 60 min at 37 °C in brain homogenate tissues from rats, monkeys, and humans (~100%).
LogD values, as indices of lipophilicity, were obtained by measuring the distribution coefficient of each radioligand between the sodium phosphate buffer (pH 7.4) and 1-octanol at RT. All six compounds exhibited logD values between 3.01 and 3.93, values that fall within the range that is considered compatible with good blood-brain barrier permeability (1.5 < logD < 4). High lipophilic efficiency (lipE), a log ratio of affinity (pKi) to lipophilicity (logD), is generally desirable in a PET radioligand for promoting a high ratio of specific to nonspecific signal. Four of the new radioligands [11C]3a–[11C]3d have lipE values closer to the value of ER176 than to the lower and less desirable value of PK11195 (Table 1). [11C]3e–[11C]3f also have higher lipE values than PK11195.
The plasma free fraction (fP) of a PET radioligand is a key factor with respect to the delivery of a PET radioligand into the brain from blood. Only free radioligand is able to cross the blood-brain barrier. High radioligand fP values are readily measurable with precision and accuracy and are therefore desirable for quantitative PET determination of target radioligand binding sites with compartmental modeling. With radioligands [11C]3a–[11C]3f available, opportunity was taken to measure fP in monkey and human plasma. Each of these radioligands showed high fP values in both species (Supporting Information, Table S1). Free fractions tended to decrease with increasing lipophilicity (logD) value.
In summary, radioligands [11C]3a–[11C]3f showed in vitro properties, such as high TSPO affinity, moderate lipophilicity, high lipE, and high fP, that warranted further evaluation of these radioligands with PET in vivo.
PET Imaging of 11C-Labeled Radioligands in the Mouse Brain.
The TSPO protein homology is highly conserved across the mammalian species. Mouse TSPO is 96% homologous with rat TSPO.51 TSPO density has been reported as 34 nM in mouse brain membranes52 and as 104 nM in mouse cerebral cortex.53 Moreover, we found that TSPO is over twice as abundant in the mouse brain (1242 fmol/mg protein, equivalent to 124 nM) as in the rat brain (580 fmol/mg protein, equivalent to 58 nM) (Supporting Information, Figure S1 and Table S2). Because mouse brain TSPO is dense and has high structural homology with TSPO in other mammalian species and because we are able to image groups of six mice readily and simultaneously with PET in a single camera, mouse offered a convenient animal model for screening the new radioligands.
Figure 2 shows the mean time–activity curves for the whole mouse brain at baseline and after TSPO blockade with PK11195 (5 mg/kg, i.v.) for each of the six intravenously administered radioligands, [11C]3a–[11C]3f, and for [11C]ER176 to allow direct comparison. The radioligands were administered at high molar activities with mean injected masses varying from 0.39 to 1.22 μg/kg. We estimate that brain TSPO occupancy would vary between 0.8 and 2.5% for [11C]3a–[11C]3f and would be 2.6% for [11C]ER176 at baseline (Supporting Information, Table S3), and would therefore have had little impact on recorded whole brain radioactivity curves.
Figure 2.

Mean brain time-activity curves for each of the radioligands [11C]3a–[11C]3f and [11C]ER176 in mice at baseline and after blocking of TSPO with PK11195 at 5 mg/kg i.v. Error bars are S.D. values for n ≥ 3 or half range for n = 2 and are within symbol size if not shown.
At baseline, all three mono-fluoro radioligands, [11C]3a–[11C]3c, gave a peak radioactivity uptake in the brain of about 0.8, 0.85, and 0.95 SUV at 22.5, 55, and 14 min, respectively, which were each followed by a slow radioactivity decline for [11C]3a and [11C]3b and a somewhat faster decline for [11C]3c. These three radioligands have very similar rat TSPO affinities and lipophilicities (logD values) (Table 1). Concerning the three trifluoromethyl radioligands, the meta trifluoromethyl radioligand [11C]3e showed similar brain radioactivity kinetics to the meta fluoro radioligand [11C]3b, with a peak radioactivity uptake of 1.06 SUV at 45 min. The two other trifluoromethyl radioligands, [11C]3d and [11C]3f, gave slightly lower peak radioactivity brain uptakes of 0.66 and 0.73 SUV at 8 and 17.5 min, respectively, each followed by slow radioactivity decline. Their brain radioactivity kinetics were therefore quite similar to those of the ortho and para fluoro radioligands [11C]3a and [11C]3c, respectively. The baseline kinetics of brain radioactivity for [11C]ER176 were broadly similar to those of the six new radioligands in terms of peak radioactivity uptake around 1.0 SUV and showed a subsequent slow decline like most of the new radioligands (Figure 2).
Preblocking of TSPO with PK11195 had strong effects on brain time-activity curves for all six radioligands that were broadly comparable to that seen for [11C]ER176 (Figure 2). Relative to the baseline curves, these curves obtained under TSPO preblock conditions showed the same pattern, namely, an early fast radioactivity uptake into brain to a much higher peak followed by a subsequent rapid decline to a lower terminal level. This pattern is attributed to (1) the blockade of radioligand binding to TSPO in peripheral TSPO-rich organs (e.g., kidney, heart, and lung), causing a higher initial input of radioligand into the brain from an increased level of free radioligand in plasma, and (2) the presence of predominantly nonspecific binding at any time in the brain, unlike the baseline curve that has predominantly both specific and nonspecific binding. Some free radioligand will also be in the brain under all conditions. The ratio of the area under the curve in the baseline scan to that in the preblock scan at a late time interval that avoids early radioligand distribution may therefore be taken as a crude estimate of total to nonspecific binding. Percentage decreases from the areas under the curve at baseline to those under preblock condition over the late interval of 80 to 100 min were therefore used to compare radioligand performances (Figure 3). Blocking effects ranged from 71 to 88% for the three mono-fluoro radioligands [11C]3a–[11C]3c and the meta-trifluoromethyl radioligand [11C]3e. These values compared well with that of the established TSPO radioligand [11C]ER176 (72%). Blocking effects were much lower for [11C]3d (33%) and [11C]3f (48%). Terminal SUV values for these two radioligands in preblocked scans were higher than for the other radioligands.
Figure 3.

Blocking effect (%) of PK11195 (5 mg/kg, i.v.), as indicated by the mean difference of mouse brain uptake in the last 20 min (80–100 min), between baseline and TSPO-preblocked conditions. Error bars are S.D. values for n ≥ 3 or half range for n = 2.
Based on blocking effects from these mouse imaging results, [11C]3b appeared to be the marginally preferred radioligand (over [11C]3c and [11C]3e). Moreover, we considered that 3b would be amenable to straightforward labeling direct from cyclotron-produced [18F]fluoride on a suitable precursor in high molar activity and that this could be achieved in already available automated radiosynthesis apparatus that would be suitable for eventual production of [18F]3b in a CGMP environment. The labeling of 3e with fluorine-18 would be possible from [18F]fluoroform, but, by contrast, this approach would require considerable extra effort on optimizing molar activity and on achieving automated production. For all these reasons, we selected 3b for labeling with fluorine-18 and for comparative evaluation as a radioligand with PET in mice.
Synthesis of Precursor for Labeling 3b with Fluorine-18.
Different methods were considered for labeling 3b with no-carrier-added (NCA) cyclotron-produced [18F]fluoride ion. These included methods based on a diaryliodonium salt or an iodonium ylide as precursor because such methods might be expected to allow the introduction of nucleophilic [18F]fluorine into the electron-neutral ring of 3b. A pilot investigation showed that an aryl(mesityl)iodonium triflate might be readily accessible as a precursor for copper-mediated radiofluorination.54 Thus, treatment of 2 with a 3-N-Boc-amino-aryl boronic acid gave compound 6 in high yield with no other major byproducts (Scheme 5). Brief sonication of 6 in TFA/DCM gave the free amine 7, which was readily diazotized with sodium nitrite in the presence of potassium iodide to give the 3-iodo compound 8 in good yield. The desired aryl(mesityl)iodonium salt 9 was obtained from 8 by modifying a recently reported oxidation-purification procedure.55 Specifically, treatment of 8 with m-chloroperbenzoic acid (mCPBA) and mesitylene in the presence of a large excess of triflic acid gave a di-cation salt that was then purified on a short column of basic alumina to provide 9 in good yield.
Scheme 5.
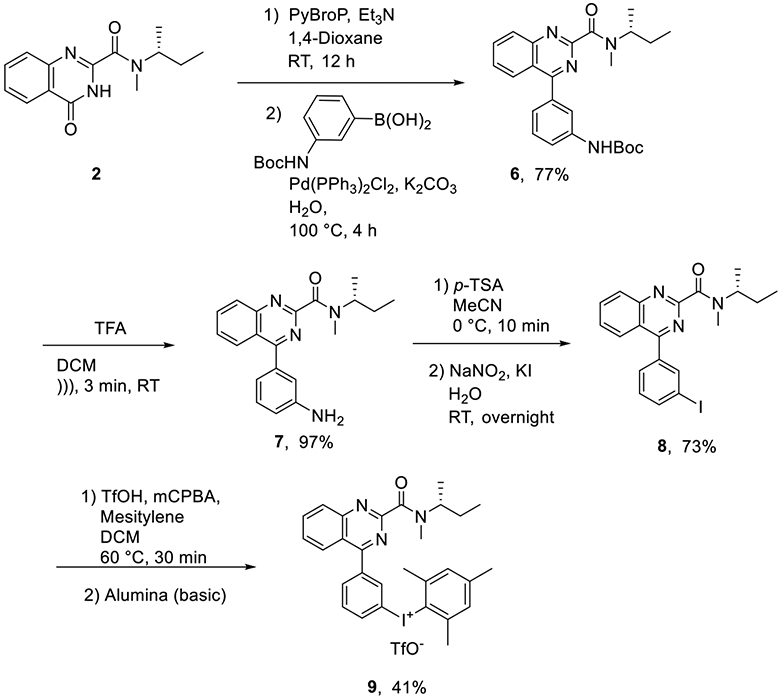
Synthesis of Aryl(mesityl)iodonium Salt Precursor 9 for 18F-Labeling of 3b
18F-Labeling of 3b.
For the labeling of 3b with fluorine-18, the precursor 9 was dissolved in DMF and mixed with a solution of tetrakis(acetonitrile)-copper(I) triflate. The resulting solution was treated with a mixture of [18F]fluoride ion and 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (K 2.2.2)-K2CO3 solution at 95 °C. The radiochemical yield reached 26% within 20 min. When a similar reaction mixture was heated by microwave irradiation for 3 min, the yield nearly doubled to 51% (Scheme 6). We therefore used microwave irradiation to prepare [18F]3b for imaging in mice. Overall, the formulated [18F]3b was obtained in 21% yield (n = 3), with a radiochemical purity higher than 99%.
Scheme 6.

Radiosynthesis of [18F]3b
PET Imaging of [18F]3b in the Mouse Brain.
Radioligand [18F]3b was evaluated with PET in mice in the same way as [11C]3b. The mean time–activity curves for the whole brain in three mice at baseline and another three mice after TSPO blockade with PK11195 (5 mg/kg, i.v.) are shown in Figure 4A. At baseline, [18F]3b gave a peak radioactivity uptake in the brain of about 0.8–0.9 SUV at 17 min followed by a slow but gradual radioactivity decline. As observed with [11C]3b, preblocking with PK11195 had a strong effect on brain time-activity curves. The blocking effect for [18F]3b was 82% (Figure 4B), which compares well to the 88% observed with [11C]3b. These data confirm that [18F]3b behaves as expected and very similar to its 11C-labeled analogue, [11C]3b.
Figure 4.

PET imaging with [18F]3b in mice. (A) Mean baseline and TSPO blocked (with PK11195 at 5 mg/kg, i.v.) brain time-activity curves. (B) Blocking effect (%) of PK11195 (5 mg/kg, i.v.) as indicated by the mean difference between baseline and blocked conditions for brain radioactivity concentrations in the last 20 min (100–120 min) after intravenous injections of [18F]3b. Error bars are the S.D. value and are within symbol size if not showing.
Metabolism of small drug-like molecules is generally considered to occur faster in mice than in higher species and also by possibly different routes. In this study, we did not investigate the metabolism of any of the radioligands. Nor did we directly assess whether radiometabolites accumulated in the brain, either by entry from plasma or through formation in brain. Nonetheless, the similarity of brain time-activity curves at baseline and after TSPO preblock for 3b that has been labeled in different positions strongly suggests that activity in mouse brain is not contaminated with radiometabolites to any great extent. Evaluation of [18F]3b in a higher species more relevant to human will need to assess its metabolism in more detail. [11C]ER176 does not have problems from radiometabolites for quantifying TSPO in human. The PET data obtained here on [18F]3b, and their similarity with those from [11C]ER176, augur well for advancing the further evaluation of [18F]3b into higher species.
CONCLUSIONS
Mono-fluoro and trifluoromethyl analogs of ER176 were readily accessible from a new homochiral synthon 2. Similarly, the desmethyl precursors for radiolabeling were readily obtained from an analogous homochiral synthon 4. All six fluorine-containing ligands showed excellent properties in vitro for PET radioligand development and were amenable to labeling by a generic 11C-methylation protocol. PET imaging in mice showed that each 11C-labeled ligand entered the brain and provided a sizeable TSPO-specific signal. In this simple and rapid screen in mice, radioligands [11C]3a–[11C]3c and [11C]3e showed TSPO imaging performance similar to or better than that of [11C]ER176. [11C]3b, the 3-fluoro analog of [11C]ER176, marginally displayed the most promising imaging behavior. Therefore, a method was devised for labeling 3b with fluorine-18 based on treating a synthesized aryl(mesityl)iodonium salt with no-carrier-added cyclotron-produced [18F]fluoride ion. [18F]3b gave comparably promising imaging results in the mouse brain to those of [11C]3b and therefore warrants further evaluation in nonhuman primates.
EXPERIMENTAL SECTION
General Materials and Methods.
[11C]ER176 was produced for intravenous injection as described previously.27 (R)-N-Methyl-2-butanamine hydrochloride was prepared freshly from (R)-(−)-2-aminobutane as reported46 and obtained as colorless needles: mp = 118–119 °C, [α]D = +6.1° (c = 5.2, EtOH) (Lit.46 m.p = 120 °C; [α]D = +6.2° (5.2, EtOH). This reagent was stored under argon until ready for use. All other reagents were purchased from MilliporeSigma (Burlington, MA). Water from a purification apparatus (Milli-Q; Waters Corp., Columbia, MD) was used in all syntheses and radiosyntheses. DMSO on molecular sieves was obtained from Acros Organics BVBA (Geel, Belgium), and stored and used in a glove-box under nitrogen. All other solvents were purchased with the highest purity available from Alfa Aesar (Tewkesbury, MA) and used as purchased. All reactions requiring inert atmosphere were performed under argon. Thin layer chromatography (TLC) was performed on silica gel layers (type 60 F254; 400–630 mesh) with compounds visualized under UV light (λ = 254 nm). Column chromatography was performed on silica gel with hexane/DCM-EtOAc mixtures as eluents. Flash chromatography was performed on a semi-automated apparatus (CombiFlash Rf + UV; Teledyne ISCO Inc., Lincoln, NE). The purities of new compound were >95% as determined with high-performance liquid chromatography (HPLC) on a Luna C18 column (5 μm, 250 mm × 4.6 mm; Phenomenex Torrance, CA) eluted isocratically with aq. NH4OH (10 mM)-MeCN and with eluate monitored for absorbance at 254 nm. Yields are recorded for chromatographically pure materials (>98%).
Melting points were measured on a digital melting point apparatus (Stuart SMP 20). Optical rotations were measured at RT using the wavelength of the sodium D line at 589.3 nm on a Model P-1010 polarimeter (Jasco, Easton, MD).
1H (400 MHz), 13C (100 MHz), and 19F NMR (376.49 MHz) spectra were recorded in CDCl3 at RT on an Avance-400 spectrometer (Bruker, Billerica, MA) using the chemical shift of residual deuterated solvent as the internal standard. 1H and 13C chemical shifts are reported in δ units (ppm) downfield relative to the chemical shift for tetramethylsilane (δ = 0) and 19F chemical shifts relative to that for CFCl3. Abbreviations br, d, dd, m, q, quin, s, st, and t denote broad, doublet, double of doublets, multiplet, quartet, quintuplet, singlet, sextuplet, and triplet, respectively. NMR spectra are shown in the Supporting Information. Note that NMR spectra for tertiary amides show the presence of two amide bond rotamers in equilibrium, as previously observed in related compounds.32
γ-Radioactivity from carbon-11 and fluorine-18 was measured with a calibrated dose calibrator (Atomlab 300; Biodex Medical Systems, USA) or for low levels (<40 kBq) with a well-type γ-counter (model 1080 Wizard; Perkin Elmer, Boston, MA) having an electronic window set between 360 and 1800 keV. All radioactivity measurements were corrected for background and physical decay.
All radiochemistry was performed in lead-shielded hot-cells to ensure radiation protection to personnel. The main apparatus for radiochemistry consisted of a PETtrace MeI Process Module (GE Medical Systems, Severna Park, MD) for [11C]iodomethane production and a semi-robotic Synthia instrument56 (Synthia AB, Uppsala, Sweden) upgraded57 with PLC-control for labeling reactions. Dedicated recipes were created in the Autorad software and followed step by step for each radiosynthesis. The HPLC apparatus for radioactive compound separation comprised a pump (P4.1S; Knauer, Berlin, Germany), a UV absorbance detector (UVD2.1S; Knauer), and a radioactivity detector (flow-count; Eckert & Ziegler, Berlin, Germany). The Clarity Chromatography Station software (DataApex, Prague, Czech Republic) was used to record the chromatograms. Equipment for radio-HPLC analyses comprised a pump (DGU-20A3R; Shimadzu, Columbia, MD), a UV absorbance detector (CBM-20A; Shimadzu), and a radioactivity detector (flow-count; Eckert & Ziegler).
All animals used in this study were handled in accordance with the Guide for the Care and Use of Laboratory Animals58 and the requirements of the National Institute of Mental Health Animal Care and Use Committee.
Results of statistical analyses are presented as mean ± SD for n ≥ 3.
Chemistry.
General Method 1: Synthesis of Amides by Weinreb Amidation.
A solution of AlMe3 (2.0 M, 6.0 mL, 12.0 mmol) in hexanes was added dropwise to a solution of amine or ammonium salt (12.0 mmol) in dry 1,2-dichloroethane (DCE; 20 mL). The mixture was stirred for 30 min at RT. Then a suspension of ethyl 4-oxo-3,4-dihydro-2-quinazolinecarboxylate 1 (1.1 g, 5 mmol) in DCE (10 mL) was added in one portion. The mixture was refluxed for 2 h, cooled to 0 °C, quenched by slow addition of 2 M HCl (10 mL) followed by water (50 mL), and shaken in a separatory funnel. The organic layer was collected, and the resulting solution was extracted with dichloromethane (DCM; 50 mL × 2). The organic phases were combined and then washed with water (50 mL), dried over MgSO4, and evaporated to dryness. The crude product was purified with chromatography on silica gel eluted with hexane/DCM/AcOEt/NEt3 (35:25:40:0.01 by vol).
1H NMR in CDCl3 revealed that each tertiary amide exists with at least two rotamers in a ratio of about 2:1 mainly due to amide bond rotation. Notably, the tertiary amide 3d showed four rotamers in the solution, whereas its N-desmethyl analogue 5d showed only two rotamers in 13C NMR. For rotamers, chemical shifts in the 1H, 13C, and 19F NMR data are reported first for the major rotamer (M) and then the minor rotamer (m), separated by an ampersand (&). Coupling constants JM and Jm are specified for each rotamer when distinct values are observed. When more than two rotamers are observed, peaks are reported in decreasing order of intensity.
(R)-N-sec-Butyl-N-methyl-4-oxo-3,4-dihydroquinazoline-2-carboxamide (2).
General method 1 was used with (R)-N-methylbutan-2-aminium chloride (1.48 g; 12.0 mmol) to give 2 as a white powder (1.15 g, yield 88%). [α]D = −118 (c = 0.25, CHCl3). Mp 84–86 °C. 1H NMR: δ 0.91 & 0.94 (t, 3H, JM = 7.36 Hz, Jm = 7.40 Hz), 1.33 & 1.23 (d, 3H, JM = 6.60 Hz, Jm = 6.76 Hz), 1.56–1.68 (m, 2H), 2.89 & 3.40 (s, 3H), 5.25 & 4.75 (st, 1H, JM = 6.44 Hz, Jm = 6.72 Hz), 7.57 (t, 1H, J = 6.44 Hz), 7.79 (m, 2H), 8.35 (d, 1H J = 8.24 Hz), 10.26 (br s, 1H). 13C NMR: δ 11.03 & 10.94, 18.61 & 17.23, 26.40 & 27.85, 27.48 & 30.74, 55.18 & 52.72, 122.69 & 122.65, 126.72, 128.35 & 128.39, 128.54 & 128.52, 134.62 & 134.64, 145.61 & 145.67, 147.45 & 147.51, 161.09 & 160.94, 161.28 & 161.71. HRMS: calcd for C14H18N3O2 260.1399 [M + H]+, found 260.1398.
(R)-N-sec-Butyl-4-oxo-3,4-dihydroquinazoline-2-carboxamide (4).
General method 1 was used with (R)-2-aminobutane (0.5 g; 6.83 mmol) to give 4 as a white powder (1.08 g, yield 88%). [α]D = −96° (c = 0.25, CHCl3). Mp 129–131 °C. 1H NMR: δ 0.88 (t, 3H, J = 12.0 Hz), 1.23 (d, 3H, J = 8.1 Hz), 1.58 (quin, 2H, J = 7.80 Hz), 4.01 (st, 1H, J = 4.52 Hz); 7.50 (m, 1H), 7.54 (br s, 1H), 7.71 (m, 2H), 8.28 (d, 1H, J = 7.81 Hz). 13C NMR: δ 10.44, 20.28, 29.58, 47.77, 122.95, 127.03, 128.03, 128.41, 134.83, 143.84, 147.38, 158.40, 160.69. HRMS: calcd for C13H16N3O2 [M + H]+, 246.1243; found, 246.1243.
General Methods for the Pd-Catalyzed Direct Arylation with PyBroP.
General method 2a below was used to prepare compounds 3a–3f on 0.5 mmol scale, and general method 2b below was used to prepare compounds 5a–5f on 1.0 mmol scale.
General Method 2a.
A mixture of the quinazoline amide (0.13 g, 0.5 mmol), PyBroP (0.49 g, 1.05 mmol), and Et3N (0.21 mL, 1.5 mmol) in 1,4-dioxane (4 mL) was stirred in a sealed tube at RT for 12 h. Then, aryl boronic acid (0.9 mmol), Na2CO3 (0.26 g, 2.5 mmol), Pd(PPh3)2Cl2 (17.5 mg, 5 mol %), and water (0.5 mL) were added, and the mixture was stirred at 100 °C for 4 h. The mixture was cooled to RT, diluted with EtOAc, washed with water and then with brine, and dried over MgSO4. The organic phase was evaporated under vacuum to give an oily residue. Chromatography of the residue on silica gel (hexane/EtOAc; 60:40, v/v) gave the desired compound.
General Method 2b.
Method 2b was similar to method 2a except that all scales were doubled to use the quinazoline desmethyl amide (0.245 g, 1.0 mmol).
(R)-(N-sec-Butyl)-4-(2-fluorophenyl)-N-methylquinazoline-2-carboxamide (3a).
General method 2a was used with 2-fluorophenyl boronic acid (0.125 g, 0.9 mmol) to give 3a as an off-white powder (0.13 g, yield 75%). Mp: 116–118 °C. 1H NMR (CDCl3): δ 0.88 & 1.04 (t, 3H, J = 7.40 Hz), 1.23 & 1.26 (d, 3H, JM = 6.56 Hz, Jm = 6.84 Hz), 1.41–1.73 (m, 2H), 2.81 & 3.04 (s, 3H), 3.56 & 4.85 (st, 1H, J = 6.84 Hz), 7.70 (m, 2H), 7.85 (d, 1H, J = 6.76 Hz), 7.98 (m, 2H), 8.01 (m, 2H), 8.18 (m, 1H). 13C NMR: δ 11.05 & 11.02, 18.40 & 17.20, 25.75 & 29.74, 27.24 & 26.63, 55.76 & 50.03, 116.19 & 115.98, 116.25 &116.03 (d, J = 22.0 Hz), 123.08, 124.60 & 124.03 and 124.71 & 124.80 (d, J = 7.50 Hz), 126.92, 128.39, 129.12 & 129.09, 131.93 & 131.77, 131.90 (d, J = 22.00 Hz), 132.06, 134.44, 150.62 & 150.56, 158.82, 159.86 (d, J = 250.0 Hz), 165.12, 167.65 & 167.36. 19F NMR: δ −113.33 & 113.07. HRMS calcd for C20H21FN3O [M + H]+, 338.1667; found, 338.1669.
(R)-(N-sec-Butyl)-4-(3-fluorophenyl)-N-methylquinazoline-2-carboxamide (3b).
General method 2a was used with 3-fluorophenyl boronic acid (0.125 g, 0.9 mmol) to give 3b as an off-white powder (0.11 g, yield 66%). Mp: 97–99 °C. 1H NMR (CDCl3): δ 0.87 & 1.04 (t, 3H, J = 7.40 Hz), 1.24 & 1.26 (d, 3H, JM = 6.60 Hz, Jm = 6.84 Hz), 1.40–1.72 (m, 2H), 3.03 & 2.82 (s, 3H), 3.54 & 4.85 (st, 1H, JM = 6.40 Hz, Jm = 6.96 Hz), 7.28, (m, 1H), 7.54 (m, 3H), 7.67 (t, 1H, J = 8.16 Hz), 7.96 (t, 1H, J = 8.08 Hz), 8.15 (m, 2H). 13C NMR: δ 11.07 & 11.03, 18.46 & 17.21, 26.64 & 25.74, 27.23 & 29.76, 50.02 &5 5.74, 117.10 & 117.26 (d, J = 25.15 Hz), 118.5 (d, J = 25.1 Hz), 122.07, 125.94 (d, J = 7.04 Hz), 126.00, 126.67, 128.48, 129.40 & 129.36, 130.28 & 130.21, 134.29, 138.86 & 138.78, 151.21, 158.71, 162.71 (d, J = 247.50 Hz), 167.39 & 167.52, 167. 65. 19F NMR: δ −112.07 & −112.06. HRMS calcd for C20H21FN3O [M + H]+, 338.1669; found, 338.1665.
(R)-(N-sec-Butyl)-4-(4-fluorophenyl)-N-methylquinazoline-2-carboxamide (3c).
General method 2a was used with 4-fluorophenyl boronic acid (0.125 g, 0.9 mmol) to give 3c as a white powder (0.14 g, yield 81%). Mp: 133–135 °C. 1H NMR (CDCl3): δ 0.87 & 1.04 (t, 3H, J = 7.40 Hz), 1.24 & 1.27 (d, 3H, JM = 6.80 Hz, Jm = 6.84 Hz), 1.44–1.68 (m, 2H), 3.03 & 2.82 (s, 3H), 3.56 & 4.85 (st, 1H, JM = 6.32 Hz, Jm = 6.80 Hz), 7.26 (t, 2H, J = 8.64 Hz), 7.66 (t, 1H, J = 7.24 Hz), 7.83 (m, 2H), 7.95 (t, 1H, J = 8.32 Hz), 8.14 (m, 2H). 13C NMR: δ 11.06 & 11.02, 18.46 & 17.20, 25.72 & 26.63, 27.21 & 29.75, 55.71 & 49.99, 115.78 (d, J = 22.13 Hz), 122.15, 126.77, 128.35 & 128.36, 129.30 & 129.34, 132.26 (d, J = 8.04 Hz), 132.28, 132.90 & 132.87, 134.16, 151.18 & 151.15, 158.68, 164.12 (dd, J = 251.53 Hz), 167.49 & 167.90, 167.76 & 167.81. 19F NMR: δ −110.04 and −110.05. HRMS calcd for C20H21FN3O [M + H]+, 338.1667; found, 338.1666.
(R)-N-(sec-Butyl)-N-methyl-4-(2-(trifluoromethyl)phenyl)-quinazoline-2-carboxamide (3d).
General method 2a was used with 2-(trifluoromethyl)phenyl boronic acid (0.171 g, 0.9 mmol) to give 3d as a white powder (0.14 g, yield 75%). Mp: 122–124 °C. 1H NMR (CDCl3): δ 0.85 & 0.80 and 0.98 & 1.03 (t, 3H, J = 7.40 Hz), 1.25 & 1.18 and 1.25 (d, 3H, J = 6.60 Hz), 1.40–1.65 (m, 2H), 3.01 & 3.00 and 2.75 & 2.74 (s, 3H), 3.48 & 4.82 (m, 1H), 7.45 (m, 1H), 7.57 (m, 2H), 7.71 (m, 2H), 7.88 (m, 1H), 7.95 (m, 1H), 8.16 (m, 1H). 13C NMR: δ 11.02 & 10.59, 18.19 & 17.14, 27.28 & 27.14 and 25.68 & 25.77, 26.59 and 30.04 & 29.36, 55.72 & 56.66 and 49.95, 122.42, 123.75 (qd, J = 271 Hz), 126.70 (m), 128.34, 128.70 and 128.66, 129.05, 129.61, 130.73 (qd, J = 24.15 Hz), 131.66, 134.60, 135.00, 150.55, 158.49 & 158.32, 167.51 & 167.45, 168.11. 19F NMR: δ −57.72, −57.75, −57.86, −57.67. HRMS: calcd for C21H21F3N3O [M + H]+ 388.1637; found, 388.1643.
(R)-N-(sec-Butyl)-N-methyl-4-(3-(trifluoromethyl)phenyl)-quinazoline-2-carboxamide (3e).
General method 2a was used with 3-(trifluoromethyl)phenyl boronic acid (0.171 g, 0.9 mmol) to give 3e as an off-white powder (0.138 g, yield 5%). Mp: 110–111 °C. 1H NMR (CDCl3): δ 0.87 & 1.04 (t, 3H, J = 7.40 Hz), 1.25 & 1,27 (d, 3H, JM = 6.60 Hz, Jm = 6.80 Hz), 1.42–1.73 (m, 2H), 3.04 & 2.82 (s, 3H), 3.57 & 4.85 (st, 1H, JM = 6.36 Hz, Jm = 6.76 Hz), 7.69 (m, 2H), 7.85 (d, 1H, J = 7.36 Hz), 7.98 (m, 2H), 8.06 (m, 2H), 8.19 (m, 1H). 13C NMR: δ 11.02 & 11.04, 18.46 & 17.22, 25.77 & 27.22, 26.63 & 29.79, 55.79 & 50.05, 122.04 & 122.07, 123.80 (qd, J = 274.6 Hz), 125.19, 126.38 & 126.35, 126.97 (m), 128.75, 129.22 & 129.20, 129.46 & 129.52, 131.24 (q, J = 31.10 Hz), 133.41 & 133.51, 134.44, 137.52, 151.25 & 151.17, 158.71, 167.33 & 167.51, 167.59. 19F NMR: δ −62.68 & −62.61. HRMS: calcd for C21H21F3N3O [M + H]+, 388.1637; found, 388.1637.
(R)-N-(sec-Butyl)-N-methyl-4-(4-(trifluoromethyl)phenyl)-quinazoline-2-carboxamide (3f).
General method 2a was used with 4-(trifluoromethyl)phenyl boronic acid (0.171 g, 0.9 mmol) to give 3f as a white powder (0.127 g, yield 69%). Mp: 135–136 °C. 1H NMR (CDCl3): δ 0.87 & 1.04 (t, 3H, J = 7.36 Hz), 1.25 & 1.27 (d, 3H, JM = 6.56 Hz, Jm = 6.84 Hz), 1.68–1.72 (m, 2H), 3.03 & 2.83 (s, 3H), 3.56 & 4.85 (st, 1H, JM = 6.36 Hz, Jm = 8.12 Hz), 7.68 (t, 1H, J = 7.61 Hz), 7.84 (d, 2H, J = 7.60 Hz), 7.92 (m, 2H), 7.96 (m, 1H), 8.08 (m, 1H), 8.20 (t, 1H, J = 7.38 Hz). 13C NMR: δ 11.08 & 11.02, 18.48 & 17.23, 25.75 & 29.78, 27.22 & 26.63, 55.78 & 50.07, 122.09, 123.89 (qd, J = 272.66 Hz), 125.58 (qd, J = 4.62 Hz), 126.46, 128.66, 129.49, 129.66, 130.52 & 130.60, 132.13 (q, J = 32.19 Hz), 134.44, 140.20, 151.20 & 152.17, 158.73, 167.53 & 167.34, 167.61. 19F N NMR: δ −62.79 & −62.80. HRMS: calcd for C21H21F3N3O [M + H]+, 388.1637; found, 388.1644.
(R)-N-(sec-Butyl)-4-(2-fluorophenyl)quinazoline-2-carboxamide (5a).
General method 2b was used with 2-fluorophenyl boronic acid (0.251 g, 1.8 mmol) to give 5a as a white powder (0.249 g, yield 7%). Mp: 94–96 °C. 1H NMR (CDCl3): δ 0.92 (t, 3H, J = 7.15 Hz), 1.22 (d, 3H, J = 12.5 Hz), 1.57 (m, 2H), 4.17 (qt, 1H, J = 5.01 Hz), 7.23 (t, 1H, J = 6.33 Hz), 7.31 (m, 1H), 7.57 (m, 3H), 7.80 (m, 1H), 7.89 (t, 1H, J = 7.44 Hz), 8.02 (br s, 1H), 8.27 (d, 1H, J = 5.78 Hz). 13C NMR: δ 10.47, 20.48, 29.68, 47.12, 116.12 (d, J = 21.12 Hz), 123.68 (d, J = 6.55 Hz), 126.90 (d, J = 11.20 Hz), 129.16, 129.89 & 131.89 (d, J = 12.00 Hz), 132.06, 132.14, 134.56, 150.76, 153.58, 158.64, 161.13; 162.06, 164.98. 19F NMR: δ −113.11. HRMS calcd for C19H19FN3O [M + H]+, 324.1512; found, 324.1509.
(R)-N-(sec-Butyl)-4-(3-fluorophenyl)-quinazoline-2-carboxamide (5b).
General method 2b was used with 3-fluorophenyl boronic acid (0.251 g, 1.8 mmol) to give 5b as a white powder (0.28 g, yield 86%). Mp: 90–91 °C. 1H NMR (CDCl3): δ 1.00 (t, 3H, J = 7.10 Hz), 1.31 (d, 3H, J = 12.2 Hz), 1.67 (m, 2H), 4.25 (qt, 1H, J = 5.05 Hz), 7.35 (t, 1H, J = 6.23 Hz), 7.55 (m, 3H), 7.72 (m, 1H), 7.98 (t, 1H, J = 6.51 Hz), 8.00 (br s, 1H), 8.01 (d, 1H, J = 7.50 Hz), 8.36 (d, 1H, J = 6.0 Hz). 13C NMR: δ 10.50, 20.49, 29.69, 47.17, 117.15, 117.37 (d, J = 12.45 Hz), 122.81, 125.97 (d, J = 11.82 Hz), 129.29, 130.19; 130.29, 130.37, 134.49, 138.76, 151.39, 153.40, 152.39 (d, J = 245.0 Hz), 161.54, 162.06, 164.01, 167.38. 19F NMR: δ −111.89. HRMS: calcd for C19H19FN3O [M + H]+, 324.1512; found, 324.1518.
(R)-(N-sec-Butyl)-4-(4-fluorophenyl)-quinazoline-2-carboxamide (5c).
General method 2b was used with 4-fluorophenyl boronic acid (0.251 g, 1.8 mmol) to give 5c as a white powder (0.26 g, yield 82%). Mp 96–97 °C. 1H NMR: δ 0.94 (t, 3H, J = 7.4 Hz), 1.23 (d, 3H, J = 6.5 Hz), 1.59 (m, 2H), 4.18 (qt, 1H J = 7.2 Hz), 7.22 (t, 2H, J = 7.5 Hz), 7.63 (t, 1H, J = 7.6 Hz), 7.77 (m, 2H), 7.88 (t, 1 H, J = 7.5 Hz), 8.01 (br. s, 1H), 8.01 (d, 1H, J = 7.6 Hz), 8.27 (d, 1H, J = 8.46 Hz). 13C NMR: δ 10.48, 20.49, 29.69, 47.12, 115.88 (d, J = 2.1 Hz), 122.85, 126.46, 126.92, 127.02, 122.46 (d, J = 27.1 Hz), 130.15, 131.31 (d, J = 33.2 Hz), 134.34, 152.38 (d, J = 203.1 Hz), 162.15, 162.91, 165.41, 167.68. 19F NMR: δ −109.84. HRMS: calcd for C19H19FN3O [M + H]+, 324.1512; found, 324.1509.
(R)-(N-sec-Butyl)-4-(2-(trifluoromethyl)phenyl)quinazoline-2-carboxamide (5d).
General method 2b was used with 2-(trifluoromethyl)phenyl boronic acid (0.342 g, 1.8 mmol) to give 5d as a white powder (0.23 g, yield 65%). Mp 86–88 °C. 1H NMR: δ 0.89 (t, 3H, J = 7.15 Hz), 1.19 (d, 3H, J = 12.5 Hz), 1.54 (m, 2H), 4.16 (qt, 1H, J = 5.01 Hz), 7.43 (t, 1H, J = 6.33 Hz), 7.58 (m, 2H), 7.66 (t, 2H, J = 7.01 Hz), 7.87 (d, 1H, J = 6.41 Hz), 7.92 (t, 1H, J = 7.44 Hz), 8.04 (br s, 1H), 8.32 (d, 1H, J = 5.78 Hz). 13C NMR: δ 10.18 & 10.41, 20.28 & 20.47, 29.55 & 29.64, 47.07, 123.68, 123.69 (qd, J = 273.66 Hz), 126.58, 127.14, 129.14, 129.59 (qd, J = 31.8 Hz), 129.81, 130.15 & 130.19, 131.19 & 131.46, 134.63, 134.95, 151.04 & 151.10, 152.93 & 152.99, 161.90, 167.39. 19F NMR δ −57.53 & −57.55. HRMS: calcd for C20H19F3N3O [M + H]+, 374.1480; found, 374.1473.
(R)-(N-sec-Butyl)-4-(3-(trifluoromethyl)phenyl)quinazoline-2-carboxamide (5e).
General method 2b was used with 3-(trifluoromethyl)phenyl boronic acid (0.342 g, 1.8 mmol) to give 5e as a white powder (0.29 g, yield 78%). Mp 91–93 °C. 1H NMR: δ 0.93 (t, 3H, J = 7.2 Hz), 1.24 (d, 3H, J = 12.5 Hz), 1.59 (m, 2H), 4.18 (m 1H), 7.65 (m, 2H), 7.80 (d, 2H, J = 7.5 Hz), 7.92 (d, 2H, J = 7.33 Hz), 7.95 (d, 1H, J = 7.1 Hz), 7.96 (br s, 1H), 8.01 (m, 1H), 8.28 (d, 1H, J = 7.25 Hz). 13C NMR: δ 10.45, 20.46, 29.67, 47.19, 122.82, 123.81 (qd, J = 272.65 Hz), 126.43, 126.88, 126.92 (qd, J = 3.01 Hz), 129.3, 129.51, 130.24, 131.29 (qd, J = 33.20 Hz), 133.40, 134.62, 137.55, 151.33, 153.47, 161.98; 167.35. 19F NMR: δ −63.72. HRMS: calcd for C20H19F3N3O [M + H]+, 374.1480; found, 374.1477.
(R)-(N-sec-Butyl)-4-(4-(trifluoromethyl)phenyl)quinazoline-2-carboxamide (5f).
General method 2b was used with 4-(trifluoromethyl)phenyl boronic acid (0.342 g, 1.8 mmol) to give 5f as a white powder (0.33 g, yield 89%). Mp 81–82 °C. 1H NMR: δ 0.93 (t, 3H, J = 7.10 Hz), 1.23 (d, 3H, J = 11.63 Hz), 1.59 (m, 2H), 4.18 (m 1H), 7.65 (m, 1H), 7.81 (d, 2H, J = 7.21 Hz), 7.87 (d, 2H, J = 7.11 Hz), 7.95 (dt, 2H, J1 = 7.33 Hz, J2 = 5.41 Hz), 8.01 (br s, 1H), 8.02 (d, 2H, J = 6.98 Hz), 8.29 (d, 1H, J = 7.07 Hz). 13C NMR: δ 10.48, 20.48, 29.69, 47.17, 122.65 (qd, J = 271.29 Hz), 125.22, 125.61 (qd, J = 3.10 Hz), 126.50, 129.44, 130.40 (qd, J = 31.10 Hz), 131.28, 132.22, 134.62, 140.19, 151.28, 152.40, 161.96, 167.42. 19F NMR: δ −63.72. HRMS: calcd for C20H19F3N3O [M + H]+, 374.1480; found, 374.1475.
tert-Butyl (R)-(3-(2-(sec-Butyl(methyl)carbamoyl)quinazolin-4-yl)phenyl)carbamate (6).
General method 2b was used with 3-(N-Boc-amino)phenyl boronic acid (0.259 g, 1.8 mmol) to give 6 as an off-white powder (0.336 g, yield 77%). Mp: 147–150 °C. 1H NMR: δ 0.87 and 1.03 (t, 3H, J = 7.40 Hz), 1.25 (d, 3H, J = 7.33 Hz), 1.43–1.71 (m, 2H), 1.52 & 1.67 (s, 9H), 2.82 & 3.02 (s, 3H), 3.55 & 4.84 (m, 1H), 6.64 (s, 1H), 7.46 (m, 2H), 7.64 (m, 2H), 7.78 (d, 1H, J = 13.52 Hz), 7.95 (m, 1H), 8.13 (m, 2H). 13C NMR (acetone-d6): δ 11.05 & 11.09, 18.46 & 17.19, 25.76, 26.64 & 27.25, 29.80, 55.74 & 50.02, 80.89, 119.82, 121.97 & 122.02, 122.98, 127.14, 128.29, 129.14 & 129.20, 134.13, 137.53 & 137.50, 138.82 & 138.78, 151.09 & 151.04, 152.65, 158.62, 167.53 & 167.82, 168.69 & 168.77. HRMS: calcd for C25H35N4O3 [M + H]+, 435.2396; found, 435.2394.
(R)-4-(3-Aminophenyl)-N-(sec-butyl)-N-methylquinazoline-2-carboxamide (7).
Compound 6 (300 mg, 0.70 mmol) was dissolved in DCM (2 mL) in a 25 mL flask. TFA (2 mL) was added, and the resulting mixture was sonicated for 3 min at RT, at which point TLC showed no remaining starting material. The solution was quenched with potassium carbonate slowly until the pH became greater than 8. Then DCM (10 mL) was added. The mixture was washed with water (2 × 10 mL) and then brine (10 mL). The organic phase was dried over MgSO4, evaporated to dryness, and purified over silica gel hexane/DCM/AcOEt/NEt3 (25:15:60:0.01 by vol) to afford 7 as a yellow powder (226 mg, yield 97%). Mp: 161–163 °C. 1H NMR: δ 0.87 & 1.04 (t, 3H, J = 7.38 Hz), 1.24 & 1.27 (d, 3H, JM = 6.26, Jm = 6.79 Hz), 1.40–1.62 (m, 2H), 3.04 & 2.82 (s, 3H), 3.54 & 4.84 (m, 1H), 6.87, (m, 1H), 7.11 (m, 2H), 7.33 (t, 1H, J = 7.4 Hz), 7.63 (t, 1H, J = 7.01 Hz), 7.92 (t, 1H, J = 7.92 Hz), 8.16 (m, 1H), 8.22 (m, 1H). 13C NMR: δ 11.11 & 11.07, 18.45 & 17.18, 25.74, 26.65 & 27.26, 29.76, 55.67 & 49.94, 116.46 & 116.49, 116.79, 120.60 & 120.56, 122.35, 127.38 & 128.00, 129.09, 129.12 & 129.35, 134.01, 137.81, 146.82 & 151.73, 167.88, 169.37. HRMS: calcd for C20H23N4O [M + H]+, 335.1872; found, 335.1866.
(R)-N-(sec-Butyl)-4-(3-iodophenyl)-N-methylquinazoline-2-carboxamide (8).
To a solution of amino compound 7 (200 mg, 0.45 mmol) in MeCN (3 mL) at 0 °C was added p-toluenesulfonic acid monohydrate (327 mg, 1.2 mmol), and the mixture was stirred at 0 °C for 10 min. A solution containing NaNO2 (289 mg, 4.19 mmol) and KI (146 mg, 0.88 mmol) in water (0.3 mL) was then added dropwise over 10 min. The mixture was then allowed to warm to RT and stirred overnight. The resulting dark solution was diluted with water (5 mL), treated with a solution of saturated sodium metabisulfite (5 mL), and then extracted with DCM (3 × 15 mL). The organic phases were combined, dried over MgSO4, and evaporated to dryness. The residue was purified by chromatography on silica gel with hexane/DCM/AcOEt/NEt3 (35:25:40:0.01 by vol) to give 8 as an off-white powder (195 mg, yield 73%). Mp: 175–177 °C. 1H-NMR (CDCl3): δ 0.87 & 1.04 (t, 3H, J = 7.37 Hz), 1.24 & 1.27 (d, 3H, JM = 6.60 Hz, Jm = 6.81 Hz), 1.43–1.70 (m, 2H), 2.82 & 3.03 (s, 3H), 3.54 & 4.84 (m, 1H), 7.31 (t, 1H, J = 7.82 Hz), 7.67 (m, 1H), 7.76 (m, 1H), 7.95 (m, 2H), 8.10 (m, 1H), 8.15 (m, 2H). 13C NMR: δ 11.09 & 11.05, 18.46 & 17.21, 27.24 & 25.78, 26.64 & 29.79, 55.75 & 50.03, 94.29 & 94.25, 122.07 & 122.10, 126.60 & 126.63, 128.55, 129.35, 129.40, 129.43, 130.16, 134.32, 138.72, 138.77 & 139.12, 151.11 & 151.16, 158.66 & 158.69, 167.38 & 167.23, 167.63. HRMS: calcd for C20H21N3OI [M + H]+, 446.0729; found, 446.0726.
(R)-(3-(2-(sec-Butyl(methyl)carbamoyl)quinazolin-4-yl)phenyl)(mesityl)iodonium Trifluoromethanesulfonate (9).
Iodoarene 8 (112 mg, 0.25 mmol) was stirred with triflic acid (4 equiv) in DCM (1 mL) for 5 min at RT within a 25 mL flask. Mesitylene (33.0 mg, 0.275 mmol) and mCPBA (75.5 mg, 0.44 mmol) were added, and the solution was stirred at 60 °C for 30 min. The crude mixture was cooled to RT, concentrated in vacuo, and diluted with Et2O (1–3 mL). The resultant precipitate was collected by filtration, washed with Et2O, and then subjected to chromatography on a small column packed with basic alumina eluted with DCM/MeOH (20:1 v/v). The eluted product was concentrated in vacuo to give the deprotonated product 9 as a beige powder (82 mg, yield 41%). Mp: 122–125 °C. [α]23D = −4.2 (c = 0.1). 1H-NMR (CDCl3): δ 0.86 & 1.04 (t, 3H, J = 7.37 Hz), 1.24 & 1.27 (d, 3H, JM = 6.60 Hz, Jm = 6.81 Hz), 1.41–1.63 (m, 2H), 2.35 (s, 3H), 2.65 & 2.64 (s, 6H), 2.80 & 3.01 (s, 3H), 3.51 & 4.83 (m, 1H), 7.10 (s, 2H), 7.62 (t, 1H, J = 7.82 Hz), 7.70 (m, 1H), 7.90 (m, 1H), 8.00 (m, 3H), 8.15 (m, 2H). 13C NMR: δ 10.04 & 10.00, 17.48 & 16.22, 20.14 & 25.55, 24.72 & 28.76, 26.10, 16.17, 26.20, 54.78 & 49.10, 111.10, 117.64, 119.55 & 119.46, 120.70 & 120.82, 125.18, 128.34 & 128.32, 129.50, 131.36 & 131.31, 132.46 & 132.52, 133.25, 133.52, 133.81 & 133.77, 139.61, 141.42 & 141.49, 143.69, 150.12, 157.42, 164.83, 166.46 & 166.18. 19F NMR: δ −78.30 (s). HRMS: calcd for C29H31N3OI [M + H]+, 564.1512; found, 564.1508.
Determination of TSPO Density in the Mouse and Rat Brain.
The brain tissue was cut into small pieces (50–100 mg) and placed in a 2 mL tube containing 1.4 mm ceramic beads and cold (4 °C) buffer (5 mM Tris-Base, 0.32 mM sucrose, 1 mM MgCl2, pH 7.4; 10 times the weight of the tissue). The sample was processed in a bead mill homogenizer (Bead Ruptor Elite; Omni International, USA) at 4.85 m/s for 20 s. Membranes were suspended in a cold (4 °C) buffer (50 mM Tris-Base, 140 mM NaCl, 1.5 mM MgCl2, 5 mM KCl, 1.5 mM CaCl2, pH 7.4), and protein concentrations were determined in a spectrophotometer (Nanodrop; Marshall Scientific, Hampton, NH) from an absorbance of 280 nm. Then the aliquots were stored at −80 °C until use.
Binding assays were performed as reported.24 Briefly, aliquots of membrane suspension (about 250 mg protein/mL) were prepared using an assay buffer (50 mM Tris-Base, 140 mM NaCl, 1.5 mM MgCl2, 5 mM KCl, 1.5 mM CaCl2, pH 7.4, 37 °C) and incubated with [3H]PK11195 (molar activity = 3.11 GBq/μmol, radioactivity concentration = 37 MBq/mL, KD = 29 nM; Perkin Elmer) in a final volume of 250 μL for 60 min. Each assay vial contained 1 mg/mL of protein and [3H]PK11195 (5.9 nM) with unlabeled PK11195 (eight concentrations ranging from 0.01 nM to 10 μM). Incubations were terminated by filtration through GF/C filters (Whatman, Maidstone, UK) followed by three washes with 1.4 mM MgCl2 in 50 mM Tris-Base (pH 7.4). Whatman GF/C filters were preincubated with 0.05% polyethyleneimine at 60 min before filtration. Scintillation fluid (3 mL/vial; Ultima Gold MV; Perkin Elmer) was added, and vials were counted on a liquid scintillation counter (Tri-carb 2900; Perkin Elmer). Each point was performed in triplicate. Bmax (fmol/mg protein) was determined using the Prism 5.0 software (GraphPad Software Inc., La Jolla, CA) by the following equation:59
where [L] = radioligand concentration.
Assay of Ligands for Affinity to TSPO in Rat Kidney and Human U87MG Cell Membranes.
Inhibitory constants (Ki’s) for the test ligands 3a–3f at the PK11195 binding site of TSPO were determined by radioligand binding displacement assays, as previously described.60,61 Such experiments were performed using the rat kidney and human U87MG cell line, chosen as TSPO-rich sources. Briefly, membrane homogenates from rat kidney or U87MG cells (30 and 20 μg of protein, respectively) were incubated with increasing concentrations of the test ligand and [3H]PK11195 (1 nM; molar activity, 3.17 MBq/nmol; Perkin-Elmer Life Sciences) in an assay buffer (Tris–HCl 50 mM, pH 7.4; final volume 500 μL) for 90 min at 0 °C. To establish the nonspecific binding of [3H]PK11195, samples were incubated in the presence of PK11195 (1 μM; solubilized with ethanol); the solvent concentration was less than 1% and did not interfere with the specific binding of the radioligand. At the end of incubation, samples were filtered rapidly under vacuum through GF/C glass fiber filters (Sigma Aldrich). After being washed three times with a buffer (Tris–HCl 50 mM, pH 7.4; 3 mL), the radioactivity trapped on the filter was measured by a liquid scintillation counter (TopCount; Perkin-Elmer Life and Analytical Sciences; 65% counting efficiency). Competition displacement binding was fitted to sigmoidal (variable slope) curves using Prism 5.0 (GraphPad Software Inc., San Diego, CA). The concentration of test compounds that inhibited [3H]PK11195 binding to kidney and U87MG cell membranes by 50% (IC50 values) obtained from the inhibition curves were converted to Ki values by the method of Cheng and Prusoff.62 In parallel experiments, PK11195 was tested as a positive control. The obtained Ki values of PK11195 for rat kidney and U87MG cells (3.40 ± 0.35 and 2.90 ± 0.14 nM, respectively) agreed with those reported previously.51,52
Assay of Ligands for Affinity to Human Cerebellum TSPO Genotypes.
Ligands 3a–3f were subjected to receptor binding assays in postmortem healthy human cerebellum homogenates with [3H]PK11195 as the radioligand to characterize ligand binding sensitivities to TSPO polymorphism. Frozen human brain samples from three HAB subjects and three LAB subjects, whose affinity status had been predetermined by genotyping, were obtained from the Human Brain Collection Core (National Institute of Mental Health, NIMH) and used for measuring Ki values in triplicate from each subject. Binding assays with incubations for 1 h at 37 °C were performed as described previously.27 As a control in each assay, a self-displacement assay was used to calculate the KD of PK11195. A mean KD value of 4.35 nM was then used as the dissociation constant for PK11195 to calculate the Ki value for the test ligand.51 A nonlinear regression curve-fitting software (GraphPad Prism 5; GraphPad Software, San Diego, CA) was used to fit data to a one-site model to provide ligand Ki values.51
Radiochemistry.
Production of NCA [11C]Carbon Dioxide.
NCA [11C]carbon dioxide (~85 GBq) was produced with a PETtrace cyclotron (GE Medical Systems, Milwaukee, WI) according to the 14N(p,α)11C reaction63 by irradiation of nitrogen gas (initial pressure 160 psi; 75 mL volume) containing 1% oxygen with a proton beam (16.5 MeV, 45 μA) for 40 min.
Production of [11C]Iodomethane.
NCA [11C]iodomethane was produced within a hot cell from NCA [11C]carbon dioxide via reduction to [11C]methane and then vapor phase iodination.64 Thus, at the end of radionuclide production, [11C]carbon dioxide was delivered in the irradiated gas to a PETtrace MeI Process Module through a stainless tubing (OD 1/8 in., ID 1/16 in.) over 2 min, trapped on molecular sieve (13X), and reduced to [11C]methane with hydrogen over nickel at 360 °C. The [11C]methane was recirculated over iodine at 720 °C to generate [11C]iodomethane, which was trapped on Porapak Q held in the recirculation path.
Radiosynthesis of 11C-Labeled TSPO Ligands.
Each precursor (2.0 μmol; 0.646 mg for 5a–5c and 0.746 mg for 5d–5f) was placed in a reaction vial (1 mL neck vial, Waters Corp.) with t-BuOK (0.269 mg, 2.4 μmol) under nitrogen, and the vial was septum-sealed. At the end of cyclotron production of carbon-11 and preceding the production of [11C]iodomethane, DMSO (0.40 mL) was added to the vial and the mixture was shaken to give a light yellow solution. The vial was loaded into the hot-cell Synthia apparatus.
[11C]Iodomethane (>37 GBq) was then swept into the vial with a stream of helium (15 mL/min) until radioactivity was maximal. The vial was placed in the oven of the Synthia apparatus and heated at 85 °C for 5 min. The 11C-labeled ligand was separated with HPLC on an XTerra RP18 column (7.8 × 300 mm, 10 μm; Waters Corp., Columbia, MD) eluted at 6.5 mL/min with NH4OH (10 mM)/MeCN (58:42 v/v for [11C]3a, [11C]3c, and [11C]3d; 56:44 v/v for [11C]3b and [11C]3e; and 57:43 v/v for [11C]3f). The eluate was monitored for radioactivity and absorbance at 254 nm. Retention times were 18.8, 9.5, 10.8, 13.6, 15.1, and 17.1 min for [11C]3a–[11C]3f, respectively (see Supporting Information Figure S2 for representative radiochromatograms). The radioligand fraction was collected and then rotary evaporated to dryness (80 °C, water bath). The residue was dissolved in sterile saline for injection (USP; 10 mL) containing ethanol (USP; 1 mL). This solution was then passed through a sterile filter (0.2 μm pore size, 25 mm; Millex-MP, Millipore, Billerica, MA) into a sterile and pyrogen-free dose vial. Radiochemical yields of formulated radioligand from cyclotron-produced [11C]carbon dioxide were estimated to be 8, 9, 15, 12, 10, and 14%, respectively.
Production of [18F]Fluoride Ion.
NCA [18F]fluoride ion was obtained through the 18O(p,n)18F nuclear reaction by irradiating [18O]water (95 atom %) for 90 to 120 min with a proton beam (17 MeV; 20 μA) generated with a PETtrace cyclotron. The aqueous [18F]fluoride ion solution was taken to the hot-cell and dried in a modified65 Synthia apparatus. Thus, a solution of K2CO3 (0.28 mg, 2 μmol) with K 2.2.2 (1.50 mg, 4 μmol) in MeCN/H2O (100 μL, 9:1 v/v) and MeCN (200–400 μL) was added to a V-vial (5 mL; Alltech) containing [18F]fluoride ion (5.9–11 GBq) in [18O]water (200–500 μL). The vial was then placed in an oven at 110 °C and heated for 5 min under vacuum with a N2 gas flow (100 mL/min) to remove the water-MeCN azeotrope. Three further cycles of MeCN (250 μL) addition and evaporation were then performed. The dried [18F]fluoride ion-K+-K 2.2.2 reagent was dissolved in DMF (250 μL) in readiness for the labeling reaction.
Radiosynthesis of [18F]3b.
The aryl(mesityl)iodonium salt 9 (4.25 mg, 6 μmol) was weighed into a 4 mL glass vial containing a stirrer bar and then dissolved in DMF (350 μL). A stock solution of tetrakis(acetonitrile)copper(I) triflate was prepared (14.3 mg in 1 mL of DMF; 0.04 M). An aliquot of this solution (150 μL 6 μmol) was added to the vial containing 9. The reaction vial, having an atmosphere of ambient air, was sealed with a Teflon-silicone septum cap, and then the solution was thoroughly vortexed.
This solution of precursor 9 was then taken to the hot-cell and added from a syringe to the 5 mL vial containing the solution of the [18F]fluoride ion-K 2.2.2-K2CO3 complex in DMF (250 μL). The stirred vial was then heated in an aluminum block at 95 °C for 20 min. Alternatively, the 5 mL vial was heated in the hot-cell with microwave irradiation (2 × 90 s, 75–80 W). The reaction was allowed to cool off. Then the mixture was dissolved in water (1.0 mL) and injected onto an XTerra RP18 column (10 μm, 7.8 × 300 mm; Waters) for HPLC purification eluted at 6.5 mL/min with aq. NH4OH (10 mM)/MeCN (70:30 v/v). [18F]3b eluted with a retention time of 40.7 min (see Supporting Information Figure S3 for the representative chromatogram). This radioactive fraction was collected and then rotary evaporated to dryness (80 °C, water-bath). The residue was dissolved in ethanol (USP; 0.2 mL) and diluted with saline for injection (USP, 1.8 mL). This solution was then passed through a sterile filter (0.2 μm pore size, 25 mm; Millex-MP) into a sterile and pyrogen-free dose vial.
Analysis of Radioligands.
A sample of each formulated radioligand was analyzed for radiochemical and chemical purity with HPLC on a Luna C18 column (4.6 mm × 250 mm, 5 μm; Phenomenex, Torrance, CA) eluted for 15 min with aq. NH4OH (10 mM) in MeCN (50: 50 v/v) at 2.0 mL/min. The eluate was monitored for absorbance at 254 nm and for radioactivity (pin diode detector; Bioscan Inc., Washington, DC). A sample was analyzed similarly after being spiked with the reference ligand to check for coelution. Retention times were 13.5, 8.6, 8.0, 10.1, 10.7, and 10.15 min for [11C]3a–[11C]3f, respectively ([18F]3b naturally has the same retention time as [11C]3b) (see Supporting Information Figures S4 and S5 for representative chromatograms). Each formulated radioligand had greater than 98% radiochemical purity. In each case, the response of the analytical system had been calibrated for the mass of the reference ligand to allow molar activity to be calculated. The stability of each formulated radioligand was also assessed with radio-HPLC at 1 and 2 h after radiosynthesis. Product identity was further affirmed after full radioactive decay by LC-MS/MS of the associated carrier on a Finnigan LCQ DECA instrument (ThermoFisher Scientific, Waltham, MD) fitted with a Luna C18 column (3 μm, 50 × 2 mm; Phenomenex) eluted with a gradient of binary solvents (A/B; 200 μL/min) composed of water/methanol/acetic acid (90:10:0.5 by vol; A) and methanol/acetic acid (100:0.5, v/v; B). Following electrospray ionization (positive ion), the MS acquired ions ranging between m/z 150 and 750.
Computation of cLogD.
cLogD (at pH 7.4) values for ligands were computed with the Pallas for Windows software version 3.8 in default mode (CompuDrug International, Bal Harbor, FL).
Measurement of LogD.
LogD (at pH 7.4) was measured on ligands 3a–3f by partition between n-octanol and the sodium phosphate buffer (pH 7.4) at room temperature, as described in detail previously.14
Stability of Radioligands in Buffer and in Whole Blood and Plasma In Vitro.
The stability of radioligands to incubation in the sodium phosphate buffer (0.15 M, pH 7.4) for 1 and 4 h at RT was assessed by reversed-phase HPLC on an Xterra column (7.8 × 300 mm, 10 μm; Waters Corp.) eluted at 4.0 mL/min with methanol/water/triethylamine (85:20:0.1, by vol).
Plasma Free Fraction Determinations.
Free fractions (fp) of [11C]3a–[11C]3f in monkey and human plasma were determined by an ultrafiltration method, as previously described.66
PET Imaging of Radioligands in Mice.
PET Image Acquisition.
Wild-type FVB mice were purchased from Taconic Farm (Germantown, NY) for this study. For each scanning session, a group of six mice was set up to be imaged simultaneously in the same microPET Focus 120 camera (Siemens Medical Solution, Knoxville, TN), three mice at baseline and three mice pretreated with PK11195 (5 mg/kg) at 10 min before radioligand injection. Mice were anesthetized with 1.5% isoflurane and injected with the formulated radioligand (100–150 μL) via a tail vein catheter at an infusion rate of 0.3 mL/min from a six-syringe infusion pump (Braintree Scientific Inc., Braintree, MA). Body temperature was controlled using a heat lamp. Table S3 (Supplementary Information) summarizes the PET study parameter information, such as administered doses. For three of the six 11C-labeled ligands, a second imaging session was necessary to make up the animal loss due to death or failed tail vein catheter so that all groups have two or more animals. PET scans were acquired without blood sampling for 100 min for 11C-labeled ligands and 120 min for [18F]3b. Durations of acquisition frames were in the following sequence: 6 × 20 s, 5 × 60 s, 4 × 120 s, 3 × 300 s, 3 × 600 s, and 3 × 1200 s. Images were reconstructed using Fourier rebinning plus a two-dimensional OSEM algorithm without scatter and attenuation correction.
Data Analysis.
A two-dimensional whole brain ROI was drawn manually on five contiguous slices for each animal in the coronal image using PMOD 3.9 (PMOD Technology, Zurich, Switzerland). Decay corrected radioactivity in the ROI was calculated at standardized uptake value (SUV), which normalizes for the amount of injected radioactivity and animal body weight, according to the following equation:
The time–activity curve of the whole-brain uptake was obtained as the mean SUV within the ROIs of each mouse and compared (midpoints of timeframes are plotted in time-activity curves). The blocking effect was calculated from the means of the brain uptake for the 80–100 min interval of imaging for baseline and blocked conditions. Negligible occupancy of TSPO by the carrier present in the radioligand at baseline was confirmed by estimating occupancy from the brain concentration and radioligand molar activity, assuming a Bmax value of 341 fmol per mg of protein for TSPO in mice.52
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH Clinical PET Center (Director: Dr. Peter Herscovitch) for carbon-11 and fluorine-18 production, the Human Brain Collection Core (NIMH, http://www.nimh.nih.gov/hbcc) for providing human brain tissue samples, and Dr. John Lloyd (NIDDK) for HRMS measurements.
Funding
F.G.S., J.-H.L., C.L.M, I.S., S.S.Z., L.S.M., J.-S.L., R.L.G., R.M.D., X.Y., R.B.I., and V.W.P. were supported by the Intramural Research Program of NIH (National Institute of Mental Health; projects ZIA-MH002793 and ZIA-MH002795). B.C. and S.C. were supported by the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Progetti di Ricerca di Interesse Nazionale (PRIN 2017MT3993).
ABBREVIATIONS
- A m
molar activity
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- f p
plasma free fraction
- HABs
high-affinity binders
- HPLC
high-performance liquid chromatography
- K 2.2.2
4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane
- LABs
low-affinity binders
- MABs
mixed-affinity binders
- mCPBA
m-chloroperbenzoic acid
- MIP
maximum intensity projection
- MRI
magnetic resonance image
- MW
microwave
- NCA
no-carrier-added
- PET
positron emission tomography
- NIDDK
National Institute of Diabetes and Digestive and Kidney Diseases
- NIMH
National Institute of Mental Health
- PyBroP
bromo-tris-pyrrolidino-phosphonium hexafluorophosphate
- RCY
radiochemical yield
- RT
room temperature
- SNP
single nucleotide polymorphism
- SUV
standardized uptake value
- TLC
thin layer chromatography
- TSPO
translocator protein 18 kDa
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01562.
Plasma free fractions; binding assay results; NMR spectra; chromatograms for radioligand separations and analyses; Molecular Formula Strings; and mouse PET parameters (PDF)
Compounds’ CSV files (CSV)
The authors declare no competing financial interest.
Contributor Information
Fabrice G. Siméon, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Jae-Hoon Lee, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States; Department of Nuclear Medicine, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul 03772, South Korea.
Cheryl L. Morse, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Ian Stukes, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States.
Sami S. Zoghbi, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Lester S. Manly, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Jeih-San Liow, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States.
Robert L. Gladding, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Rachel M. Dick, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Xuefeng Yan, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States.
Sabrina Taliani, Department of Pharmacy, University of Pisa, 56126 Pisa, Italy.
Barbara Costa, Department of Pharmacy, University of Pisa, 56126 Pisa, Italy.
Claudia Martini, Department of Pharmacy, University of Pisa, 56126 Pisa, Italy.
Federico Da Settimo, Department of Pharmacy, University of Pisa, 56126 Pisa, Italy.
Sabrina Castellano, Department of Pharmacy, University of Salerno, 84084 Fisciano, SA, Italy.
Robert B. Innis, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States
Victor W. Pike, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892, United States.
REFERENCES
- (1).Meyer JH; Cervenka S; Kim M-J; Kreisl WC; Henter ID; Innis RB Neuroinflammation in psychiatric disorders: PET imaging and promising new targets. Lancet Psychiatry 2020, 7, 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kreisl WC; Kim M-J; Coughlin JM; Henter ID; Owen DR; Innis RB PET imaging of neuroinflammation in neurological disorders. Lancet Neurology 2020, 19, 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Papadopoulos V; Baraldi M; Guilarte TR; Knudsen TB; Lacapere J-J; Lindemann P; Norenberg MD; Nutt D; Weizman A; Zhang M-R; Gavish M Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci 2006, 27, 402–409. [DOI] [PubMed] [Google Scholar]
- (4).Doble A; Malgouris C; Daniel M; Daniel N; Imbault F; Basbaum A; Uzan A; Guérémy C; Le Fur G Labelling of peripheral-type benzodiazepine binding sites in human brain with [3H]PK 11195: anatomical and subcellular distribution. Brain Res. Bull 1987, 18, 49–61. [DOI] [PubMed] [Google Scholar]
- (5).Rone MB; Ran J; Papadopoulos V Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim. Biophys. Acta 2009, 1791, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Papadopoulos V; Lecanu L; Brown RC; Han Z; Yao Z-X Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience 2006, 138, 749–756. [DOI] [PubMed] [Google Scholar]
- (7).Chaveau F; Boutin H; Van Camp N; Dollé F; Tavitian B Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2304–2319. [DOI] [PubMed] [Google Scholar]
- (8).Dollé F; Luus C; Reynolds A; Kassiou M Radiolabelled molecules for imaging the translocator protein (18 kDa) using positron emission tomography. Curr. Med. Chem 2009, 16, 2899–2923. [DOI] [PubMed] [Google Scholar]
- (9).Schweitzer PJ; Fallon BA; Mann JJ; Kumar JSD PET tracers for the peripheral benzodiazepine receptor and uses thereof. Drug Discovery Today 2010, 15, 933–942. [DOI] [PubMed] [Google Scholar]
- (10).Camsonne R; Moulin MA; Crouzel C; Syrota A; Maziere M; Comar D 11C-Labeling of PK 11195 and visualization of peripheral receptors of benzodiazepines by positron emission tomography. J. Pharmacol 1986, 17, 383. [Google Scholar]
- (11).Shah F; Hume SP; Pike VW; Ashworth S; McDermott J Synthesis of the enantiomers of [N-methyl-11C]PK 11195 and comparison of their behaviours as radioligands for PK binding sites in rats. Nucl. Med. Biol 1994, 21, 573–581. [DOI] [PubMed] [Google Scholar]
- (12).Petit-Taboué MC; Baron JC; Barré L; Travere JM; Speckel D; Camsonne R; MacKenzie ET Brain kinetics and specific binding of [11C]PK11195 to ω3 sites in baboon. Eur. J. Pharmacol 1991, 200, 347–351. [DOI] [PubMed] [Google Scholar]
- (13).Kreisl WC; Fujita M; Fujimura Y; Kimura N; Jenko KJ; Kannan P; Hong J; Morse CL; Zoghbi SS; Gladding RL; Jacobson S; Oh U; Pike VW; Innis RB Comparison of [11C](R)-PK 11195 and [11C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. NeuroImage 2010, 49, 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Briard E; Zoghbi SS; Imaizumi M; Gourley JP; Shetty HU; Hong J; Cropley V; Fujita M; Innis RB; Pike VW Synthesis and evaluation in monkey of two sensitive 11C-labeled aryloxyanilide ligands for imaging brain peripheral benzodiazepine receptors in vivo. J. Med. Chem 2008, 51, 17–30. [DOI] [PubMed] [Google Scholar]
- (15).Imaizumi M; Briard E; Zoghbi SS; Gourley JP; Hong J; Fujimura Y; Pike VW; Innis RB; Fujita M Brain and whole-body imaging in nonhuman primates of [11C]PBR28, a promising PET radioligand for peripheral benzodiazepine receptors. NeuroImage 2008, 39, 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fujita M; Imaizumi M; Zoghbi SS; Fujimura Y; Farris AG; Suhara T; Hong J; Pike VW; Innis RB Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. NeuroImage 2008, 40, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Endres CJ; Pomper MG; James M; Uzuner O; Hammoud DA; Watkins CC; Reynolds A; Hilton J; Dannals RF; Kassiou M Initial evaluation of [11C]DPA-713, a novel TSPO PET ligand, in humans. J. Nucl. Med 2009, 50, 1276–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Briard E; Zoghbi SS; Siméon F; Imaizumi M; Gourley JP; Shetty HU; Lu S-Y; Fujita M; Innis RB; Pike VW Singlestep high-yield radiosynthesis and evaluation of a selective 18F-labeled ligand for imaging brain peripheral benzodiazepine receptors with PET. J. Med. Chem 2009, 52, 688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wilson AA; Garcia A; Parkes J; McCormick P; Stephenson KA; Houle S; Vasdev N Radiosynthesis and initial evaluation of [18F]FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl. Med. Biol 2008, 35, 305–314. [DOI] [PubMed] [Google Scholar]
- (20).Owen DR; Yeo AJ; Gunn RN; Song K; Wadsworth G; Lewis A; Rhodes C; Pulford DJ; Bennacef I; Parker CA; StJean PL; Cardon LR; Mooser VE; Matthews PM; Rabiner EA; Rubio JP An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab 2012, 32, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Guo Q; Colasanti A; Owen DR; Onega M; Kamalakaran A; Bennacef I; Matthews PM; Rabiner EA; Turkheimer FE; Gunn RN Quantification of the specific translocator protein signal of 18F-PBR111 in healthy humans: a genetic polymorphism effect on in vivo binding. J. Nucl. Med 2013, 54, 1915–1923. [DOI] [PubMed] [Google Scholar]
- (22).Mizrahi R; Rusjan PM; Kennedy J; Pollock B; Mulsant B; Suridjan I; De Luca V; Wilson AA; Houle S Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J. Cereb. Blood Flow Metab 2012, 32, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Owen DRJ; Gunn RN; Rabiner EA; Bennacef I; Fujita M; Kreisl WC; Innis RB; Pike VW; Reynolds R; Matthews PM; Parker CA Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J. Nucl. Med 2011, 52, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Owen DR; Howell OW; Tang S-P; Wells LA; Bennacef I; Bergstrom M; Gunn RN; Rabiner EA; Wilkins MR; Reynolds R; Matthews PM; Parker CA Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J. Cereb. Blood Flow Metab 2010, 30, 1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kreisl WC; Jenko KJ; Hines CS; Lyoo CH; Corona W; Morse CL; Zoghbi SS; Hyde T; Kleinman JE; Pike VW; McMahon FJ; Innis RB; Biomarkers Consortium PET Radioligand Project Team. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J. Cereb. Blood Flow Metab 2013, 33, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Castellano S; Taliani S; Milite C; Pugliesi I; Da Pozzo E; Rizzetto E; Bendinelli S; Costa B; Cosconati S; Greco G; Novellino E; Sbardella G; Stefancich G; Martini C; Da Settimo F Synthesis and biological evaluation of 4-phenylquinazoline-2-carboxamides designed as a novel class of potent ligands of the translocator protein. J. Med. Chem 2012, 55, 4506–4510. [DOI] [PubMed] [Google Scholar]
- (27).Zanotti-Fregonara P; Zhang Y; Jenko KJ; Gladding RL; Zoghbi SS; Fujita M; Sbardella G; Castellano S; Taliani S; Martini C; Innis RB; Da Settimo F; Pike VW Synthesis and evaluation of translocator18 kDa protein (TSPO) positron emission tomography (PET) radioligands with low binding sensitivity to human single nucleotide polymorphism rs6971. ACS Chem. Neurosci 2014, 5, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fujita M; Kobayashi M; Ikawa M; Gunn RN; Rabiner EA; Owen DR; Zoghbi SS; Haskali MB; Telu S; Pike VW; Innis RB Comparison of four 11C-labeled PET ligands to quantify translocator protein 18 kDa (TSPO) in human brain: (R)-PK11195, PBR28, DPA-713, and ER176-based on recent publications that measured specific-to-non-displaceable ratios. EJNMMI Res. 2017, 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ikawa M; Lohith TG; Shrestha S; Telu S; Zoghbi SS; Castellano S; Taliani S; Da Settimo F; Fujita M; Pike VW; Innis RB; Biomarkers Consortium Radioligand Project Team. 11C-ER176, a radioligand for 18-kDa translocator protein, has adequate sensitivity to robustly image all three affinity genotypes in human brain. J. Nucl. Med 2017, 58, 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hong J; Telu S; Zhang Y; Miller WH; Shetty HU; Morse CL; Pike VW Translation of 11C-labeled tracer synthesis to a CGMP environment as exemplified by [11C]ER176 for PET imaging of human TSPO. Nat. Protoc 2021, 16, 4419–4445. [DOI] [PubMed] [Google Scholar]
- (31).Dubroeucq M-C; Renault CLA; Le Fur GR Nouveaux dérivés d’arène et d’hétéroarènecarboxamides, leur procédé de préparation et médicaments les contenant. Eur. Patent 1983, 0094271B1. [Google Scholar]
- (32).Castellano S; Taliani S; Viviano M; Milite C; Da Pozzo E; Costa B; Barresi E; Bruno A; Cosconati S; Marinelli L; Greco G; Novellino E; Sbardella G; Da Settimo F; Martini C Structure-activity relationship refinement and further assessment of 4-phenyl-quinazoline-2-carboxamide translocator protein ligands as antiproliferative agents in human glioblastoma tumors. J. Med. Chem 2014, 57, 2413–2428. [DOI] [PubMed] [Google Scholar]
- (33).Pike VW Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr. Med. Chem 2016, 23, 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cai L; Lu S; Pike VW Chemistry with [18F]fluoride ion. Eur. J. Org. Chem 2008, 2853–2873. [Google Scholar]
- (35).Pike VW Hypervalent aryliodine compounds as precursors for radiofluorination. J. Labelled Compd. Radiopharm 2017, 61, 196–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tredwell M; Preshlock SM; Taylor NJ; Gruber S; Huiban M; Passchier J; Mercier J; Gnicot C; Gouverneur V A general copper-mediated 18F fluorination of arenes. Angew. Chem., Int. Ed 2014, 126, 7885–7889. [DOI] [PubMed] [Google Scholar]
- (37).Huiban M; Tredwell M; Mizuta S; Wan Z; Zhang X; Collier TL; Gouverneur V; Passchier J A broadly applicable [18F]trifluoromethylation of aryl and heteroaryl iodides for PET imaging. Nat. Chem 2013, 5, 941–944. [DOI] [PubMed] [Google Scholar]
- (38).van der Born D; Sewing C; Herscheid JDM; Windhorst AD; Orru RVA; Vugts DJ A universal procedure for the [18F]trifluoromethylation of aryl iodides and aryl boronic acids with highly improved specific activity. Angew. Chem., Int. Ed 2014, 53, 11046–11050. [DOI] [PubMed] [Google Scholar]
- (39).Ivashkin P; Lemonnier G; Cousin J; Grégoire V; Labar D; Jubault P; Pannecouck X [18F]CuCF3: A [18F]trifluoromethylating agent for arylboronic acids and aryl iodides. Chem.-A Eur. J 2014, 20, 9514–9518. [DOI] [PubMed] [Google Scholar]
- (40).Yang BY; Telu S; Haskali MB; Morse CL; Pike VW A gas phase route to [18F]fluoroform with limited molar activity dilution. Sci. Rep 2019, 9, 14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Pees A; Windhorst AD; Vosjan MJWD; Tadino V; Vugts DJ Synthesis of [18F]fluoroform with high molar activity. Eur. J. Org. Chem 2020, 1177–1185. [Google Scholar]
- (42).Haskali MB; Pike VW [11C]Fluoroform, a breakthrough for versatile labeling of PET radiotracer trifluoromethyl groups in high molar activity. Chem. - Eur. J 2017, 23, 8156–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Young NJ; Pike VW; Taddei C Rapid and efficient synthesis of [11C]trifluoromethylarenes from primary aromatic amines and [11C]CuCF3. ACS Omega 2020, 5, 19557–19564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ramos-Torres KM; Zhou Y-P; Yang BY; Guehl NJ; Hyun MS; Telu S; Normandin MD; Pike VW; Brugarolas P Syntheses of [11C]2- and [11C]3-trifluoromethyl-4-aminopyridine: potential PET radioligands for demyelinating diseases. RSC Med. Chem 2020, 11, 1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kang F-A; Sui Z; Murray WV Pd-Catalyzed direct arylation of tautomerizable heterocycles with aryl boronic acids via C-OH bond activation using phosphonium salts. J. Am. Chem. Soc 2008, 130, 11300–11302. [DOI] [PubMed] [Google Scholar]
- (46).Higashiyama K; Fujikura H; Takahashi H Diastereoselective addition of chiral aliphatic imines and 2-alkyl-1,3-oxazolidines to organometallic reagents. Chem. Pharm. Bull 1995, 43, 722–728. [Google Scholar]
- (47).Haider N; Nuß S Weinreb amidation as the cornerstone of an improved synthetic route to a ring-modified derivatives of luotonin A. Molecules 2012, 17, 11363–11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lee Y-S; Siméon FG; Briard E; Pike VW Solution structures of the prototypical 18 kDa translocator protein ligand, PK 11195, elucidated with 1H/13C NMR spectroscopy and quantum chemistry. ACS Chem. Neurosci 2012, 3, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Shultz MD Setting expectations in molecular optimizations: strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett 2013, 23, 5980–5991. [DOI] [PubMed] [Google Scholar]
- (50).Wilson AA; Garcia A; Jin L; Houle S Radiotracer synthesis from [11C]iodomethane: a remarkable simple captive solvent method. Nucl. Med. Biol 2000, 27, 529–532. [DOI] [PubMed] [Google Scholar]
- (51).Owen DR; Fan J; Campioli E; Venugopal S; Midzak A; Daly E; Harlay A; Issop L; Libri V; Kalogiannopoulou D; Oliver E; Gallego-Colon E; Colasanti A; Huson L; Rabiner EA; Suppiah P; Essagian C; Matthews PM; Papadopoulos V TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem. J 2017, 474, 3985–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Giannaccini G; Betti L; Palego L; Pirone A; Schmid L; Lanza M; Fabbrini L; Pelosini C; Maffe IM; Santini F; Pinchera A; Lucacchini A Serotonin transporter (SERT) and translocator protein (TSPO) expression in the obese ob/ob mouse. BMC Neurosci. [On line] 2011, 12: Article 18. http://www.biomedcentral.com/1471-2202/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chen M-K; Baidoo K; Verina T; Guilarte TR Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain 2004, 127, 1379–1392. [DOI] [PubMed] [Google Scholar]
- (54).Ichiishi N; Brooks AF; Topczewski JJ; Rodnick ME; Sanford MS; Scott PJH Copper-catalyzed [18F]fluorination of (mesityl)(aryl)iodoniun salts. Org. Lett 2014, 16, 3224–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bielawski M; Malmgren J; Pardo LM; Wikmark Y; Olofsson B One-pot synthesis and applications of N-heteroaryl iodonium salts. ChemistryOpen 2014, 3, 19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Bjurling P; Reineck R; Westerburg G; Gee AD; Sutcliffe J; Långström B Synthia, a compact radiochemistry system for automated production of radiopharmaceuticals, in Proceedings Sixth Workshop on Targetry and Target Chemistry (Link JM, Ruth TJ; Eds.) 1995. pp 282–284, TRIUMF, Vancouver. [Google Scholar]
- (57).Smith DM Synthia gets extreme makeover courtesy of National Instruments, NIWeek08—Worldwide Graphic System Design Conference, Austin, Texas, 2008; p 56. [Google Scholar]
- (58).Clark JD; Baldwin RL; Bayne KA; Brown MJ; Gebhart GF; Gonder JC; Gwathmey JK; Keeling ME; Kohn DF; Robb JW; Smith OA; Steggerda J-AD; VandeBer JL Guide for the Care and Use of Laboratory Animals, National Academy Press, Washington, DC.,1996. [Google Scholar]
- (59).Motulsky H; Neubig R Analyzing radioligand binding data. Curr. Protoc. Neurosci 2002, 19, Ch. 7: Unit 7.5. DOI: 10.1002/0471142301.ns0705s19. [DOI] [PubMed] [Google Scholar]
- (60).Costa B; Da Pozzo E; Giacomelli C; Barresi E; Taliani S; Da Settimo F; Martini C TSPO ligand residence time: a new parameter to predict compound neurosteroidogenic efficacy. Sci. Rep 2016, 6, 18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Costa B; Da Pozzo E; Giacomelli C; Taliani S; Bendinelli S; Barresi E; Da Settimo F; Martini C TSPO ligand residence time influences human glioblastoma multiforme cell death/life balance. Apoptosis 2015, 20, 383–398. [DOI] [PubMed] [Google Scholar]
- (62).Cheng Y; Prusoff WH Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (63).Christman DR; Finn RD; Karlström K; Wolf AP The production of ultra high specific activity 11C-labeled hydrogen cyanide, carbon dioxide, carbon monoxide and methane via the 14N(p,α)11C reaction. Int. J. Appl. Radiat. Isot 1975, 26, 435–442. [Google Scholar]
- (64).Larsen P; Ulin J; Dahlström K; Jensen M Synthesis of [11C]iodomethane by iodination of [11C]methane. Appl. Radiat. Isot 1997, 48, 153–157. [Google Scholar]
- (65).Lazarova N; Simćon FG; Musachio JL; Lu SY; Pike VW Integration of a microwave reactor with Synthia to provide a fully automated radiofluorination module. J. Labelled Compd. Radiopharm 2007, 50, 463–465. [Google Scholar]
- (66).Gandelman MS; Baldwin RM; Zoghbi SS; Zea-Ponce Y; Innis RB Evaluation of ultrafiltration for the free-fraction detection of single photon emission computed tomography (SPECT) radiotracers: β-CIT, IBF, and iomazenil. J. Pharm. Sci 1994, 83, 1014–1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.