

Abstract
Synovial sarcoma (SyS) is an aggressive neoplasm driven by the SS18-SSX fusion, and characterized by low T cell infiltration. Here, we studied the cancer-immune interplay in SyS using an integrative approach that combines single-cell RNA-seq (scRNA-seq), spatial profiling, genetic and pharmacological perturbations. scRNA-Seq of 16,872 cells from 12 human SyS tumors uncovered a malignant subpopulation that marks immune deprived niches in situ and is predictive of poor clinical outcomes in two independent cohorts. Functional analyses revealed that this malignant cell state is controlled by the SS18-SSX fusion, repressed by cytokines secreted by macrophages and T cells, and can be synergistically targeted with a combination of HDAC and CDK4/6 inhibitors. This drug combination enhanced malignant cell immunogenicity in SyS models, leading to induced T cell reactivity and T-cell-mediated killing. Our study provides a blueprint for investigating heterogeneity in fusion-driven malignancies and demonstrates an interplay between immune evasion and oncogenic processes that can be co-targeted in SyS and potentially in other malignancies.
INTRODUCTION
Therapeutic strategies harnessing the cytotoxic capacity of the adaptive immune response to target tumor cells have radically changed clinical practice, but responses vary dramatically across patients and tumor types1,2. Studying malignancies with defined genetics and exceptionally low T cell infiltration levels could help provide clues to some of the immune escape mechanisms underlying lack of response to immune therapies.
One such cancer type is synovial sarcoma (SyS)3, an aggressive mesenchymal neoplasm that accounts for 10–20% of all soft-tissue sarcomas in young adults4. SyS tumors homogeneously express several immunogenic cancer-testis antigens (CTAs)5,6, which are recognized by circulating T cells in the peripheral blood of SyS patients5. Nonetheless, T cell infiltration remains exceptionally low in these tumors, suggestive of yet unidentified immune evasion mechanisms.
The cellular plasticity4, stem-like features7,8, and unique genetics of SyS may explain its escape of immune surveillance despite expressed immunogenic antigens. SyS is driven by the SS18-SSX fusion protein – where the BAF subunit SS18 is fused to SSX1, SSX2 or, rarely, SSX49. The BAF complex is a major chromatin regulator9, which can mediate resistance to immune checkpoint blockade in melanoma and renal cancer10,11. SSX genes are a family of CTAs involved in transcriptional repression12–15. The SS18-SSX oncoprotein dysregulates chromatin architecture and transcriptional processes9,16–18, generating a spectrum of malignant cell phenotypes4, including mesenchymal spindle cells and epithelial-like cells (in biphasic tumors), suggestive of pluripotential differentiation or mesenchymal to epithelial transitions.
The failure of clinical trials in SyS and lack of effective treatments for advanced disease, may partly stem from our partial understanding of this disease. Patients presenting with localized tumors undergo multi-modality therapy with surgery, radiation, and often chemotherapy; but despite this approach, almost half of patients will develop incurable metastatic disease, underscoring the need for new therapeutic strategies. Genomic studies of human SyS have either relied on bulk tissue profiling19,20 or on established cellular models9,16,17, masking important aspects of the tumor ecosystem. Because SyS is a rare tumor type, even concerted, large-scale sequencing efforts profiled only limited numbers of SyS tumors19–21. Only 10 SyS tumors were profiled by The Cancer Genome Atlas (TCGA)21 consortium, and other SyS-specific bulk gene expression cohorts were assembled from no more than a few dozen patients19,20.
To tackle this challenge we devised an integrative, data-driven approach that combined single-cell RNA-Seq (scRNA-Seq) and spatial profiling of human tumors with genetic and pharmacological perturbations in cellular models. First, we mapped the SyS ecosystem by scRNA-seq profiling of 16,872 cells from 12 human SyS tumors, along with spatial transcriptomic and multiplex immunofluorescence of tumors. We identify a malignant cellular state in all SyS tumors that is predictive of poor prognosis and immune evasion. The unique features of this cell state are driven by the SS18-SSX fusion, and repressed by immune cells, specifically through T cell- and macrophage-secreted cytokines. Modulating SS18-SSX targets and cell proliferation with a subcytotoxic combination of HDAC and CDK4/6 inhibitors selectively targeted this malignant cell state and increased the immunogenicity of SyS cells, resulting in enhanced T cell reactivity and T-cell-mediated killing in cell co-culture models. Taken together, our work provides a framework for studying fusion-driven tumors, uncovers a tight interplay between immune evasion and oncogenic processes, and suggests potential new therapeutic strategies for the management of SyS.
RESULTS
A SyS cellular map from expression and genetic features in tumor scRNA-seq
To comprehensively interrogate the SyS ecosystem, we used full-length22 and droplet-based23 scRNA-Seq to profile 16,872 high quality malignant, immune, and stromal cells from 12 human SyS tumors (Fig. 1a,b, Extended Data Fig. 1a,b, Supplementary Table 1, Online Methods), including four biphasic, three poorly differentiated, and five monophasic tumors (clinical characteristics are provided in Supplementary Table 1).
Fig. 1. Single-cell map of the cellular ecosystem of synovial sarcoma tumors.
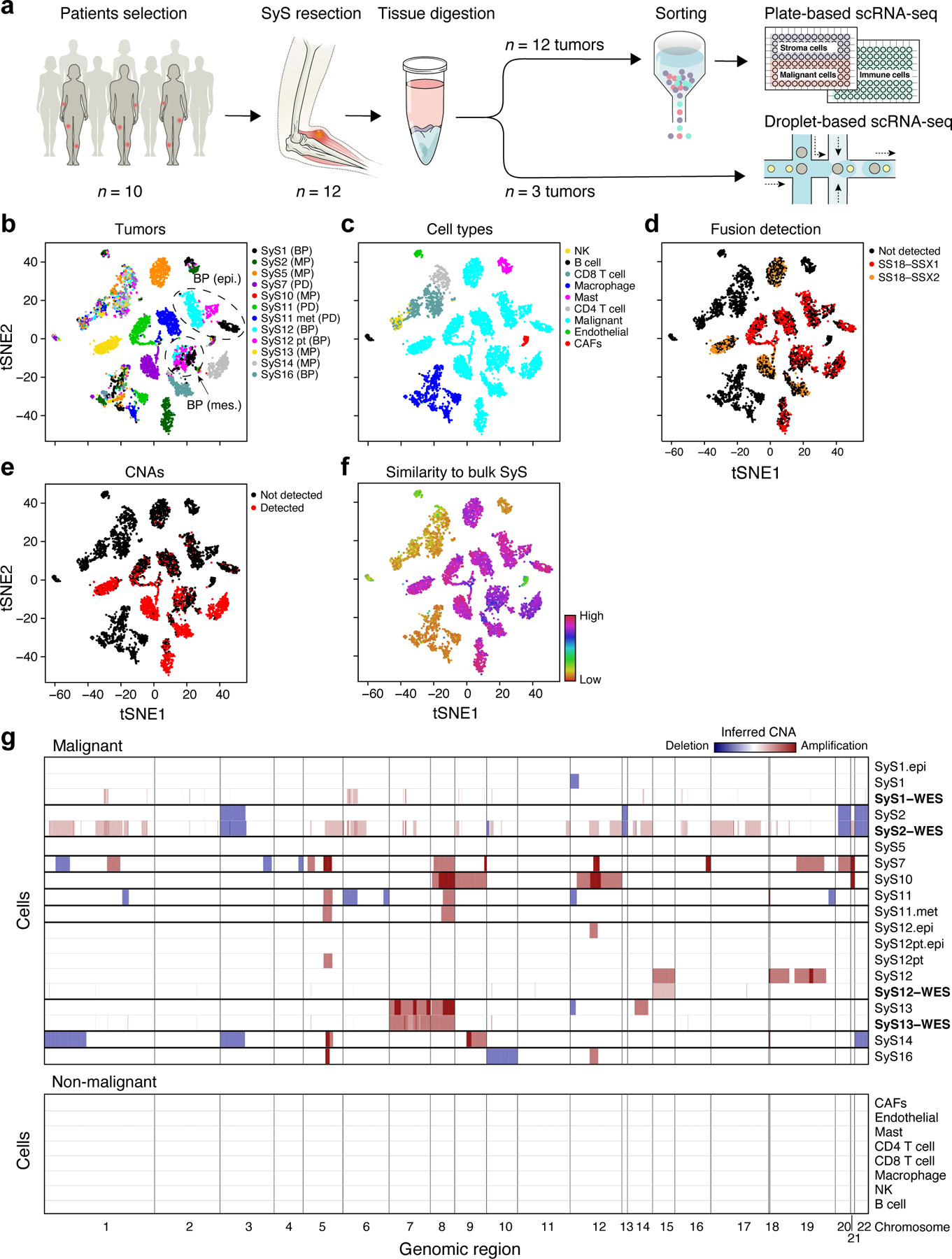
(a) Study workflow. (b-e) Consistent assignment of cell identity. t-SNE plots of scRNA-Seq profiles (dots), colored by either (b) tumor sample, (c) inferred cell type, (d) SS18-SSX1/2 fusion detection, (e) CNA detection, and (f) differential similarity to SyS compared to other sarcomas (Online Methods). Dashed ovals (b): mesenchymal and epithelial malignant subpopulations of biphasic (BP) tumors. (g) Inferred large-scale CNAs distinguish malignant (top) from non-malignant (bottom) cells, and are concordant with WES data (bold). The CNAs (red: amplifications, blue: deletions) are shown along the chromosomes (x axis) for each cell (y axis).
We assigned cells to different cell types according to both transcriptional and inferred genetic features (Fig. 1b-g, Extended Data Fig. 1, Online Methods): (1) expression-based clustering and annotation of non-malignant clusters by canonical markers (Fig. 1c, Extended Data Fig. 1a, Supplementary Table 2); (2) detection of the SS18-SSX fusion transcripts24 (Fig. 1d); (3) inference of copy number alterations (CNAs) from scRNA-Seq profiles25 (Fig. 1e), which we validated in four tumors using bulk whole-exome sequencing (WES) (Fig. 1g); and (4) similarity to bulk profiles of SyS tumors21 (Fig. 1f, Online Methods). The four approaches were highly congruent (Extended Data Fig. 1a; Supplementary Information).
We assigned the cells to nine subsets (Fig. 1c): malignant cells, endothelial cells, Cancer Associated Fibroblasts (CAFs), CD8 and CD4 T cells, B cells, Natural Killer (NK) cells, macrophages, and mastocytes, and generated signatures for each (Supplementary Table 2, Extended Data Fig. 1d). Malignant cells primarily grouped by their tumor of origin, while non-malignant immune and stroma cells grouped by cell type (Fig. 1b,c), as observed in other tumors26–30. Malignant cells from each of the biphasic (BP) tumors (SyS1 and SyS12) clustered first according to their differentiation state into an epithelial and a mesenchymal BP cluster, and within each clusters into sub-clusters by patient (Fig. 1b,c, black, cyan and magenta dots, Online Methods).
Cellular differentiation programs and a core oncogenic program characterize synovial sarcoma cells
Interrogating the malignant cell profiles for gene programs, we identified three co-regulated gene modules consistent across multiple tumors (Fig. 2a-d, Supplementary Table 3, Online Methods). Two modules reflected the expected mesenchymal and epithelial cell states (Fig. 2b, Extended Data Fig. 2a; Supplementary Information), with canonical mesenchymal (ZEB1, ZEB2, PDGFRA and SNAI2) or epithelial (MUC1 and EPCAM) markers31,32 (P < 1.55*10−10, hypergeometric test), and increase in antigen presentation and interferon (IFN) γ responses in epithelial cells (P < 8.49*10−6, hypergeometric test).
Fig. 2. Cellular plasticity and a core oncogenic program characterize synovial sarcoma cells.
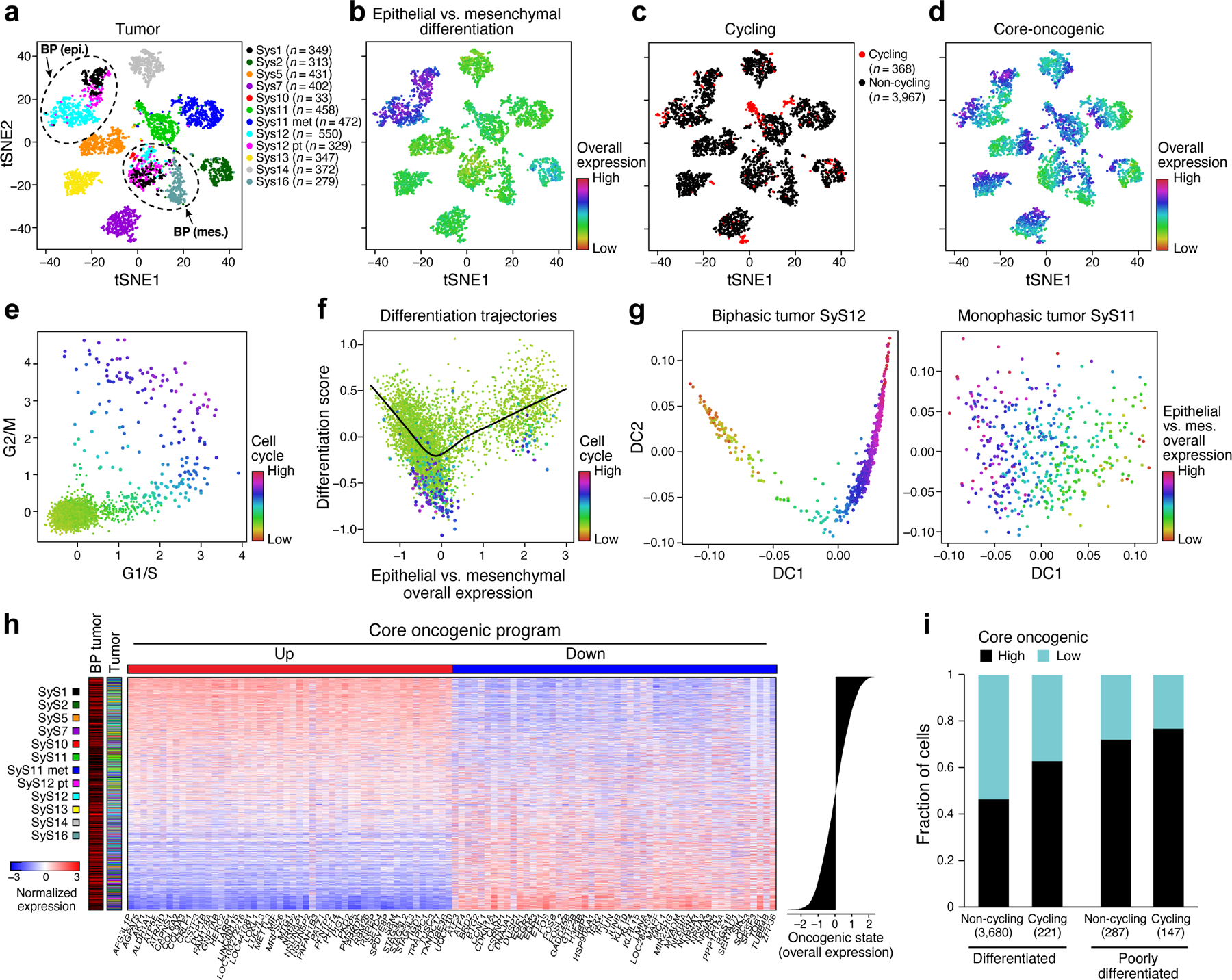
(a-d) De-differentiation, cell cycle, and the core oncogenic programs across malignant cells. t-SNE plots of malignant cell profiles (dots), colored by: (a) sample, (b) Overall Expression of the epithelial vs. mesenchymal differentiation program, (c) cell cycle status, or (d) Overall Expression of the core oncogenic program. Dashed ovals (A): mesenchymal and epithelial malignant subpopulations of biphasic (BP) tumors or poorly differentiated (PD) tumor. (e, f) Association between cell cycle and poor differentiation. (e) G1/S (x axis) and G2/M (y axis) phase signature scores for each cell. (f) Epithelial and mesenchymal-like differentiation. Scatter plots of the malignant cells’ (dots) scores for the epithelial vs. mesenchymal program (x axis) and for overall differentiation (y axis). Color: expression of cell cycle program (see also Extended Data Fig. 2b, c). (g) Distinct differentiation pattern in biphasic tumors. Single cell profiles dots arranged by the first two diffusion-map components (DCs) for representative examples of a biphasic (SyS12, left) and monophasic (SyS11, right) tumors, and colored by the Overall Expression of the epithelial vs. mesenchymal programs (colorbar). (h) Core oncogenic program genes. Normalized expression (centered TPM values, colorbar) of the top 100 genes in the core oncogenic program (columns) across the malignant cells (rows), sorted according to the Overall Expression of the program (bar plot, right). Leftmost color bars: biphasic tumor and sample ID. (i) The program is expressed in a higher proportion of cycling and poorly differentiated cells. Fraction of malignant cells (y axis) with a high (above median, black) and low (below median, blue) Overall Expression of the core oncogenic program, in cells stratified by cycling and differentiation status (x axis).
One subset of mesenchymal cells with a relatively low Overall Expression of the mesenchymal program (Online Methods) also expressed epithelial markers, suggesting a transition from a mesenchymal to an epithelial state, while another under-expressed both programs, suggesting a poorly differentiated state. These poorly differentiated cells were enriched with cycling cells (P = 2.44*10−60, mixed effects), suggesting they might function as tumor progenitors (Fig. 2e,f, Extended Data Fig. 2b,c). Diffusion map analysis identified differentiation patterns only in the biphasic tumors (Fig. 2g, Online Methods).
Both Principal Component Analysis (PCA)33,34 and Non-Negative Matrix Factorization (NMF)35,36 based approaches (Online Methods) revealed a novel module that was present in a subset of cells in each tumor, which we named the core oncogenic program (25.2–84.7% cells per tumor, Fig. 2d,h, Extended Data Fig. 3). The program includes induction of genes from respiratory carbon metabolism (oxidative phosphorylation, citric acid cycle, and carbohydrate/protein metabolism, P < 1*10−8, hypergeometric test, Supplementary Table 3), and repression of genes in the TNF signaling, apoptosis, p53 signaling, and hypoxia pre-annotated gene sets (P < 1*10−10, hypergeometric test, Supplementary Table 3), including known tumor suppressors, such as p21 (CDKN1A) and KLF4. The program was expressed in a higher proportion of cycling and poorly differentiated cells (P < 2.94*10−4, mixed-effects, Fig. 2i), and was heterogeneous in tumors in situ (P < 1*10−10, combined probability test, Methods, Fig. 3a-c, Extended Data Fig. 3d).
Fig. 3. The core oncogenic program is associated with poor prognosis and aggressive disease.
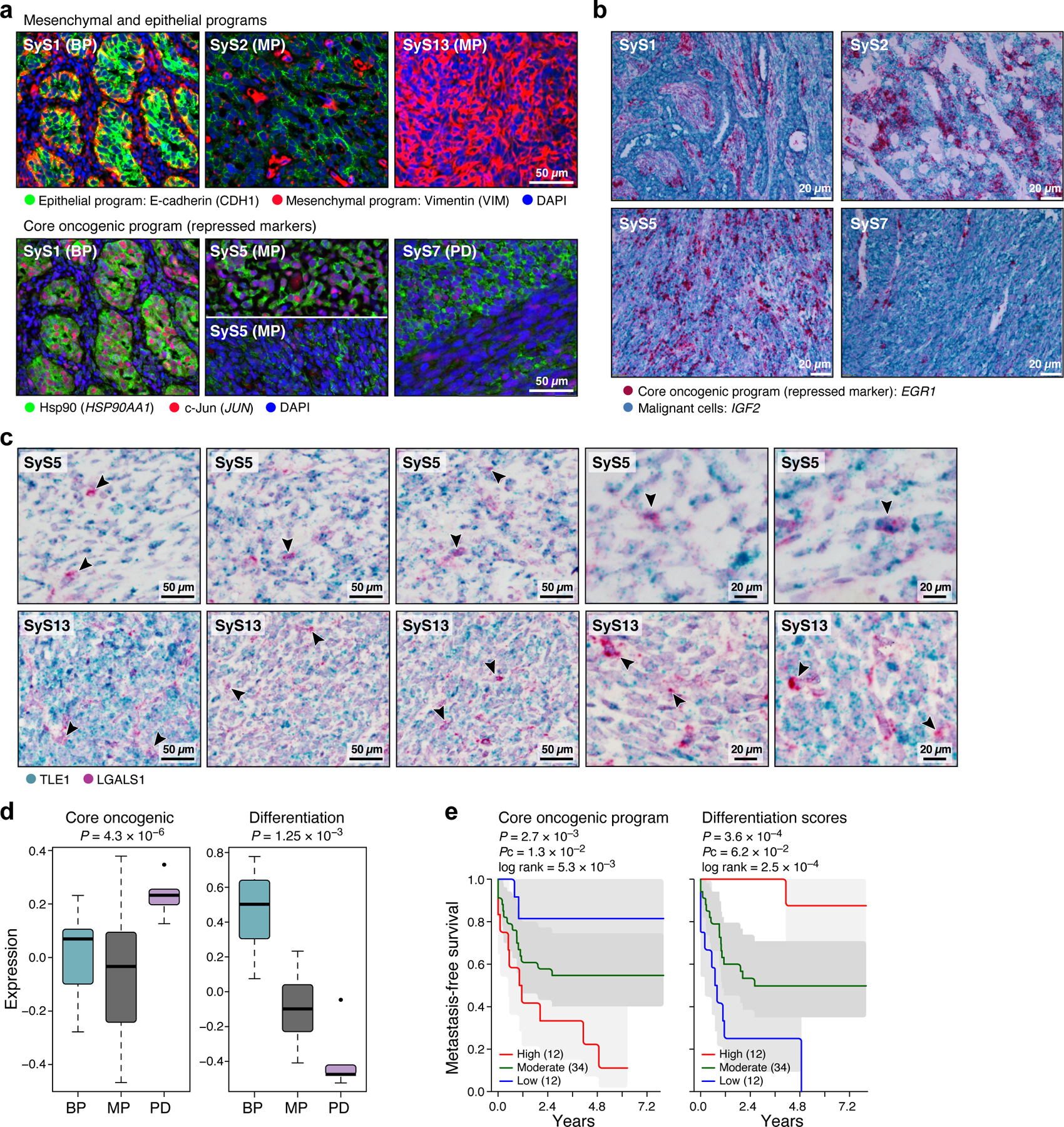
(a-c) In situ validation of programs. Detection of core oncogenic (induced: Hsp90, c-Jun and EGR1; repressed: LGALS1), epithelial (E-cadherin) and mesenchymal (Vimentin) markers, using immunofluorescence (t-CyCIF) (a) and in situ hybridization (ISH) (b,c). Arrows (c): LGALS1+ SyS cells. These patterns repeatedly appeared across tens of different fields of view (see also Extended Data Fig. 3d). (d) The core-oncogenic program and de-differentiation mark the aggressive poorly differentiated (PD) subtype. Overall expression of the core oncogenic or differentiation (both mesenchymal and epithelial) programs scores (y axis) across 34 SyS tumors19, including 7 biphasic (BP), 21 monophasic (MP), and 6 poorly differentiated (PD) (x axis). Middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually; one-sided t-test. (e) The core oncogenic program and differentiation scores (overall expression of both differentiation programs) are predictive of metastatic disease in an independent cohort of 58 SyS patients20. Kaplan-Meier (KM) curves of metastasis free survival (x axis, years), when stratifying the patients by high (top 25%), low (bottom 25%), or intermediate (remainder) expression of the respective program. P: COX regression p-value; Pc: COX regression p-value when controlling for fusion type and patient age group.
To test the clinical relevance of these programs, we analyzed bulk expression profiles from two published cohorts19,20. Both the de-differentiation score (Online Methods) and the core oncogenic program were more pronounced in more aggressive, poorly differentiated SyS tumors (P = 4.30*10−6, one-sided t-test, Fig. 3d) and were associated with increased risk of metastatic disease (P = 2.7*10−3, Cox regression, Fig. 3e). In another cohort of 64 SyS tumors37, genes up-regulated in the core oncogenic program were frequently amplified by CNAs (P = 3.78*10−7, Mann–Whitney test), especially in metastatic/recurrent tumors (P = 2.65*10−8), and in the primary tumors of patients who developed metastases/local recurrences (P = 2.3*10−6).
Evidence of antitumor immune activity despite low immune infiltration
T cell infiltration is exceptionally low in SyS, but it is unknown whether the lack of antitumor immunity results from the inability of immune cells to recognize and respond to malignant cells, from active tumor-driven inhibition of immune cell infiltration into the tumor, or both. We set out to explore these hypotheses by combining our data with a pan-cancer analysis approach.
To test the first possibility, we examined CD8 T cell states (Fig. 4a, Supplementary Table 4), and found hallmarks of antitumor immunity and recognition. T cell subsets spanned naïve, cytotoxic, exhausted, and regulatory states (Fig. 4b; Online Methods), with expansion based on TCR reconstruction38 (72 observed clones, all patient-specific, with shared clones between matched samples from the same patient; Extended Data Fig. 4a), and unique transcriptional features of an effector-like non-exhausted state (Fig. 4b, Extended Data Fig. 4b, Supplementary Table 4). Notably, SyS-specific CTAs were expressed in large fractions of malignant cells (Extended Data Fig. 4c). Compared to CD8 T cells from melanoma30, CD8 T cells in SyS (a) overexpressed a program characterizing T cells in melanoma tumors that were responsive to immune checkpoint blockade39 (Fig. 4c bottom, P = 1.22*10−10, mixed-effects), (b) overexpressed effector and cytotoxic gene modules40,41 (e.g., GZMB, CX3CR1, P = 6.36*10−9, mixed-effects); and (c) under-expressed exhaustion markers (P = 6.36*10−3, mixed-effects) and checkpoint genes (CTLA4, HAVCR2, LAG3, PDCD1, TIGIT, and LAYN40 (P = 7.69*10−7, mixed-effects, Fig. 4c, top).
Fig. 4. Limited immune infiltration and features of anti-tumor immunity in SyS tumors.
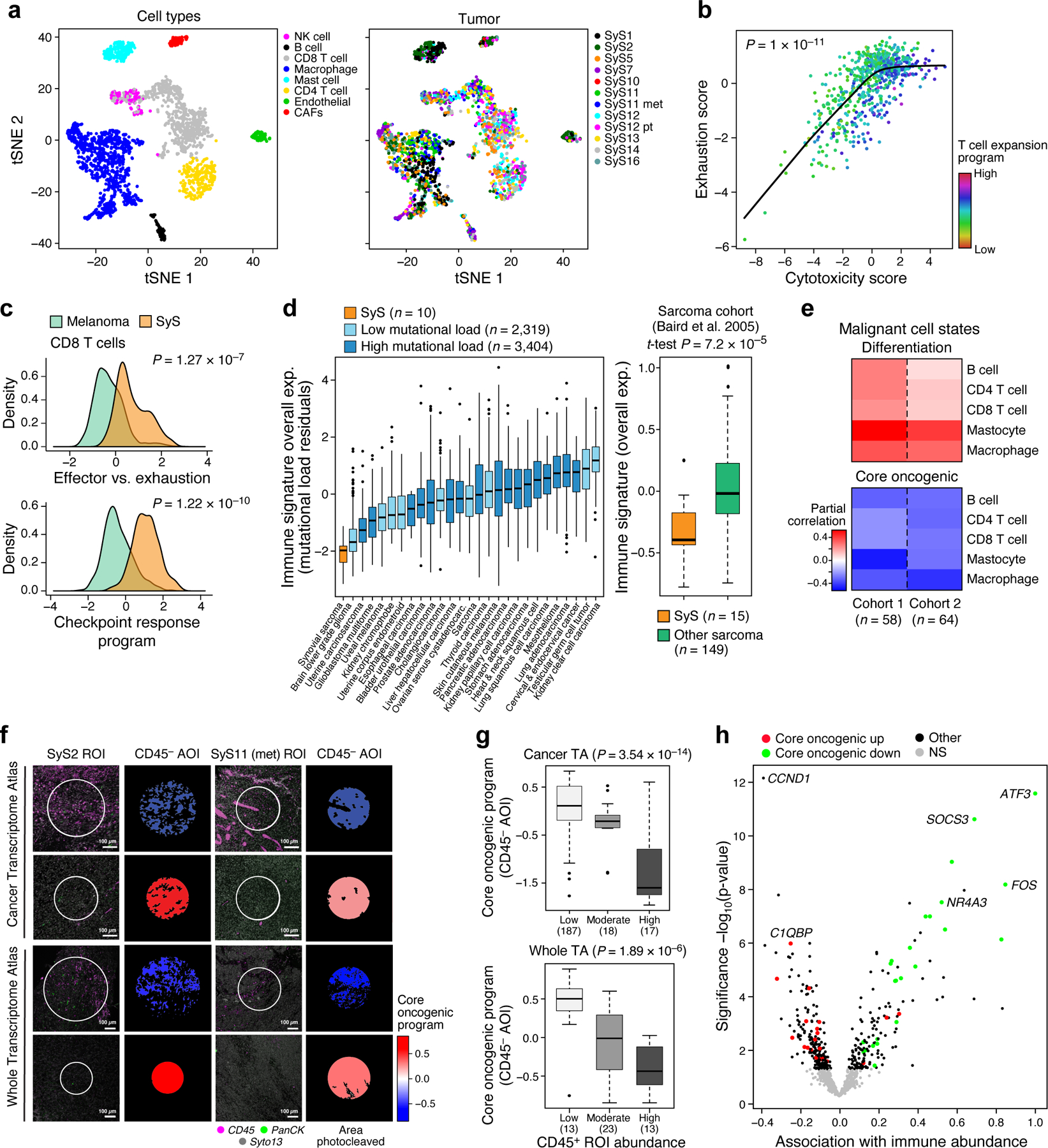
(a) t-SNE of immune and stroma cell profiles (dots), colored by inferred cell type (left) or sample (right). (b) Cytotoxicity (x axis) and exhaustion (y axis) scores of SyS CD8 T cells, colored by the T cell expansion program score. The latter is associated with high cytotoxicity and lower than expected exhaustion (P < 1*10−11, mixed-effects). (c) Distribution of effector vs. exhaustion scores (top) or an immune checkpoint blockade responsiveness program39 (bottom) in CD8 T cells from SyS (orange) and melanoma (green); p-value: mixed-effects test. (d) Overall Expression of the immune signatures (y axis) in SyS tumors (orange) and other cancer types (controlling for variation in the mutational load, left panel) or other sarcomas (right panel). (e) Inferred level of immune cell types is associated with the malignant programs in bulk SyS tumors, when controlling for tumor purity. Partial correlation (colorbar) between the inferred level of each immune subset (rows) and the core oncogenic and differentiation levels (columns). (f-h) GeoMx Cancer Transcriptome Atlas (TA) and Whole Transcriptome Atlas in situ profiling reveals that the core oncogenic program (COP) is associated with reduced immune infiltrates. (f) Representative CD45+ staining in COP-high and COP-low tumor niches; the trend was observed across 244 ROIs in 9 SyS tumors, as shown in (g) and (h); (g) the expression of the COP in malignant CD45− AOIs stratified according to the immune cell abundance in the pertaining ROI, with no. of ROIs in parenthesis; p-values: mixed-effects test. (h) association gene expression in malignant CD45− AOIs with immune abundance in the pertaining ROI. (d) and (g) middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually.
Among other immune cells, macrophages spanned M1-like and M2-like states42, with pro- and anti-inflammatory features, respectively (Extended Data Fig. 4d-f; Online Methods, Supplementary Table 4), and expressed relatively high levels of TNF (P = 1.13*10−7, mixed-effects, >4 fold higher than melanoma macrophages), while mastocytes showed regulatory features (39% expressing PD-L1 vs. 2% PD-L1 expressing malignant cells).
We next compared the immune composition (Online Methods) inferred from bulk profiles of SyS to those of 30 other cancer and sarcoma types. SyS tumors showed extremely low levels of immune cells, which could not be explained by variation in the mutational load (Fig. 4d, Extended Data Fig. 4g; P = 2.58*10−11, mixed-effects accounting for mutational load), and despite malignant-cell expression of CTAs (Extended Data Fig. 4c). Unlike melanoma (Extended Data Fig. 4h, left), T cell levels were not correlated with prognosis in SyS (Extended Data Fig. 4h, right), perhaps because they do not cross the critical threshold for clinical impact. Only mastocytes had a moderate positive association with improved prognosis (P = 0.012, Cox regression). These findings support the hypothesis that insufficient immune cell infiltration is key to SyS immune evasion.
The core oncogenic program is associated with immune deprived tumors and spatial niches
We next explored potential mechanisms of immune evasion by cancer cells, especially the connection between the malignant cells’ state and the tumor’s microenvironment. First, from our inferred composition of SyS tumors in published cohorts16,20 (Online Methods), we found that the levels of immune infiltrates were correlated with lower core oncogenic and cell cycle program scores and higher differentiation scores (P < 5.34*10−3, r = −0.44, −0.36 and 0.48, respectively, partial Pearson correlation, conditioning on inferred tumor purity, Online Methods; Fig. 4e).
Next, we spatially profiled the expression of 1,412 transcripts in situ across 9 tumors in our cohort with the GeoMx® Cancer Transcriptome Atlas RNA Assay (Online Methods)43. Two of the tumors (SyS11 and SyS2) were also profiled in situ for >18,000 genes using the GeoMx Whole Transcriptome Atlas (Online Methods; Figs. 4f,g). We first stained tumor sections with markers for immune (CD45) and epithelial (PanCK) compartments, defined immune (CD45+) and non-immune (CD45−) cells, classified CD45− cells as malignant based on cytonuclear atypia, and distinguished them as epithelial (PanCK+) and non-epithelial (PanCK−) (Fig. 4f). Based on this information, we distinguished multiple Regions of Interest (ROI) in each sample and Areas of Illumination (AOI) within each ROI, separately profiling the RNA from CD45+, malignant CD45−/PanCK−, and malignant CD45−/PanCK+ AOIs to a total number of 306 spatially distinct areas (to account for variation in AOI size in each AOI the counts were normalized to obtain transcript per million (TPM) values; Online Methods).
We observed an inverse spatial correlation between the expression of the core oncogenic program in the malignant cells (CD45− AOIs) in a given ROI and CD45+ cell abundance in the same ROI (P = 3.54*10−14, mixed-effects, r = −0.67, P < 1*10−6, partial Spearman correlation for Overall Expression of the program, Fig. 4f,g; and P < 1*10−6, hypergeometric test at the single-gene level, Fig. 4h). No other gene signature showed such associations, including both the programs defined here and >9,000 annotated gene sets44.
Cell type signatures (Supplementary Table 2), including the overall SyS malignant cell signature and immune (T, B, etc.) and stromal cell signatures, were not differentially expressed in the CD45− AOIs from immune-rich vs. poor ROIs (P > 0.1, hypergeometric test). Thus, negative spatial correlation between the core oncogenic program and CD45+ cell abundance in an ROI is unlikely to be due to undetected non-malignant ‘contamination’ in the CD45− AOIs. We confirmed these findings with multiplexed immunofluorescence (t-CyCIF)45 (Extended data Fig. 4i, P < 1*10−10, mixed-effects, Online Methods).
SS18-SSX sustains the core oncogenic program and blocks differentiation
To examine whether the SS18-SSX fusion regulates the programs identified in SyS tumors we depleted SS18-SSX in two SyS cell lines (SYO1 and Aska) using shRNA, and profiled 12,263 cells by scRNA-Seq. The fusion knock-down (KD) caused extensive and consistent transcriptional changes in both cell lines (Fig. 5a, Extended Data Fig. 5a,b, Supplementary Table 5), where it repressed the core oncogenic program and cell cycle genes (P < 8.05*10−107, and 5.2*10−71, t-test, respectively, Fig. 5a-c), and induced mesenchymal differentiation genes, including ZEB1 and VIM (P < 1*10−50, t-test and likelihood-ratio test Fig. 5a,b, Extended Data Fig. 5a,b).
Fig. 5. Impact of the genetic driver and immune cells on SyS malignant cells.
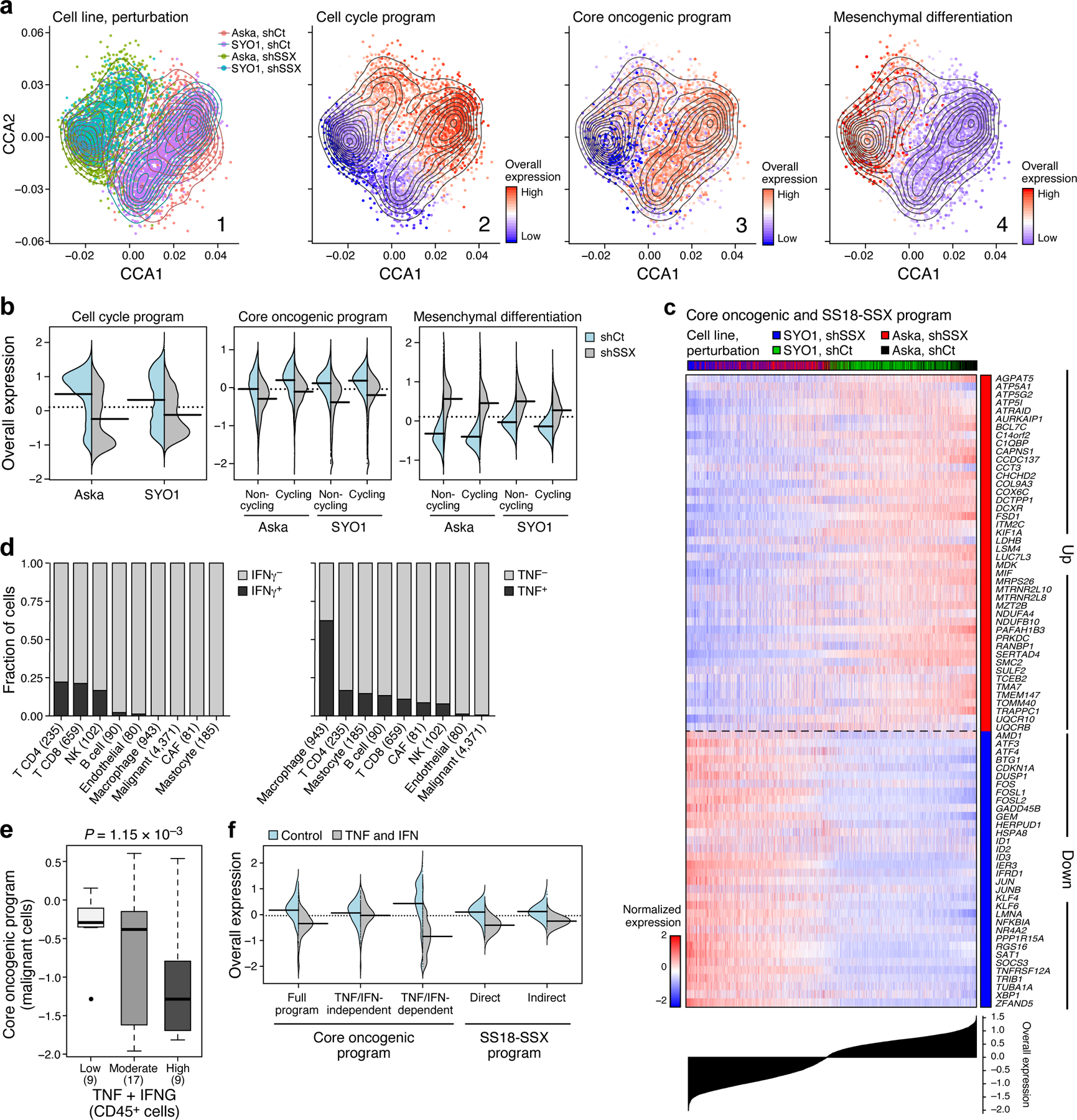
(a) scRNA-Seq following KD of SS18-SSX. Co-embedding of Aska and SYO1 cell profiles (dots), colored by: (1) cell line and perturbation; or the Overall Expression (colorbar) of the (2) cell cycle, (3) core oncogenic, or (4) mesenchymal differentiation31,32 programs. (b) SS18-SSX KD represses the core oncogenic program and induces the mesenchymal differentiation program irrespective of its repression of the cell cycle program. Distribution of Overall Expression scores (y axis) for each program in control (blue) and shSSX (grey) cells, for each cell line, where core oncogenic and mesenchymal program scores are shown separately for cycling and non-cycling cells. (c) Expression (centered TPM) of genes (rows) shared between the fusion and core oncogenic programs across the Aska and SYO1 cells (columns), with a control (shCt) or SSX (shSSX) shRNA. Cells are ordered by the Overall Expression of the SS18-SSX program (bottom plot) and labeled by type and condition (Color bar, top). (d) TNF and IFNγ are detected primarily in macrophages and T cells, respectively. Fraction of cell (y axis) of each subset in the tumor (x axis) that express (black) IFNγ (left) or TNF (right) by scRNA-seq. (e) The expression of TNF and IFNG in CD45+ cells is associated with the expression of the core oncogenic program in malignant cells according to the high-plex in situ RNA sequencing (P = 1.15*10−3, mixed-effects). Middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually. (f) TNF and IFNγ repress the core oncogenic and SS18-SSX programs. Distribution of Overall Expression score (y axis) of the core oncogenic (also stratified to its predicted and TNF/IFNγ-dependent and -independent components) and SS18-SSX programs (x axis) in control (blue) and TNF + IFNγ treated cells.
The KD impact on the core oncogenic and differentiation programs was decoupled from repression of cellular proliferation (Fig. 5b): the impact on these programs was observed also when controlling for cycling status (Online Methods), and when considering only cycling or non-cycling cells (P < 1.54*10−13, t-test, Fig. 5b). Thus, the fusion’s impact on cell cycle may be secondary or downstream to its impact on the core oncogenic program. The KD caused a cell autonomous overexpression of antigen presentation and immune response genes, such as TNF and IFN signaling (P < 1*10−30, mixed-effects, Extended Data Fig. 5a).
We next stratified the target genes affected by SS18-SSX KD (“SS18-SSX program”) to direct and indirect fusion targets based on SS18-SSX ChIP-Seq16,17 (Online Methods; Extended Data Fig. 5c, Supplementary Table 5). SS18-SSX directly dysregulates differentiation programs and promotes the core oncogenic program (P < 2.51*10−5, hypergeometric test, Extended Data Fig. 5c, Supplementary Table 5), while its impact on cell cycle genes is mostly indirect (P < 1.2*10−9, hypergeometric test, Supplementary Table 5, Extended Data Fig. 5c), and likely involves cyclin D2 (CCND2) and CDK6 – the only cell cycle genes that are direct SS18-SSX targets. As expected, the SS18-SSX program is exclusively active in SyS tumors compared to other cancer types (Extended Data Fig. 5d). Collectively, these findings support a model in which SS18-SSX directly promotes the core oncogenic program, blocks differentiation, and drives cell cycle progression.
TNF and IFNγ synergistically repress the core oncogenic and SS18-SSX programs
The negative correlation between the core oncogenic program and immune infiltration in situ suggests that the program may play a causal role in promoting T cell exclusion in SyS. Another (non-mutually exclusive) hypothesis is that, despite their low numbers, immune cells in the tumor microenvironment may nonetheless impact the malignant cells, for example, through the secretion of different cytokines. Indeed, the expression of IFNγ and TNF specifically from CD8 T cells and macrophages, respectively (Fig. 5d), was strongly associated with repression of the core oncogenic program in malignant cells, both by scRNA-Seq (P < 9.4*10−39, mixed-effects) and by in situ high-plex GeoMX profiles (P < 1*10−3, mixed-effects, Fig. 5e). We further predicted the TNF/IFNγ-dependent and -independent components of the core oncogenic program based on the association of each gene’s expression in malignant cells with TNF and IFNγ expression levels in corresponding macrophages and CD8 T cells, respectively (Online Methods, Supplementary Table 6).
To test these predictions, we treated primary SyS cells with TNF and IFNγ, separately and in combination, and profiled 1,050 cells by scRNA-Seq. As predicted, combined TNF and IFNγ treatment (a) repressed the core oncogenic program (P = 6.66*10−18, mixed-effects, Fig. 5f) in a synergistic manner (P = 9.49*10−4, interaction term, mixed-effects), impacting only the predicted TNF/IFNγ-dependent component (1.6*10−38, mixed-effects, Fig. 5f), (b) repressed both direct and indirect targets of the SS18-SSX program (P < 3.12*10−16, including TLE1; P = 1.23*10−4 for the interaction term, Fig. 5f, Supplementary Table 6) and (c) induced the epithelial program (P = 1.95*10−9, hypergeometric test, Supplementary Table 6). Short-term (4–6 hours) treatment with TNF alone: substantially repressed homeobox genes (e.g., MEOX2, Supplementary Table 6), which are directly bound by SS18-SSX16,17 (P < 1*10−17, hypergeometric test); repressed the core oncogenic program, but only temporarily (P = 8.73*10−18, mixed-effects; Extended Data Fig. 5e), suggesting that IFNγ is required for sustained effect; and induced TNF RNA expression in SyS cells (P < 5.57*10−8, mixed-effects), potentially leading to positive feedback through autocrine signaling. These findings demonstrate that TNF and IFNγ can suppress the SS18-SSX program, raising the possibility that their secretion by macrophages and T cells within the tumor might mediate a similar effect in vivo to counteract the transcriptional impact of SS18-SSX.
HDAC and CDK4/6 inhibitors synergistically repress the immune resistant features of SyS cells
Next, we examined whether the repression of the core oncogenic program in SyS cells could impact their interactions with surrounding T cells, by identifying compounds that can repress the core oncogenic program and potentially induce more immunogenic cell states in SyS cells. Modeling the core oncogenic regulatory network46–53(Online Methods) highlighted the SSX-SS18-HDAC1 complex18 as the program’s master regulator (Fig. 6a), and the tumor suppressor CDKN1A (p21) as its most repressed target. The latter indicates that the core oncogenic program regulates, but is not regulated by, cell cycle genes through the p21-CDK2/4/6 axis, potentially reinforcing the direct induction of cyclin D and CDK6 by SS18-SSX. In this model, modulators of cell cycle (e.g., CDK4/6 inhibitors) and SS18-SSX (e.g., HDAC inhibitors) could jointly target the immune resistance features of SyS cells, especially in the presence of cytokines such as TNF.
Fig. 6. HDAC and CDK4/6 inhibitors repress the core oncogenic program in SyS cells.
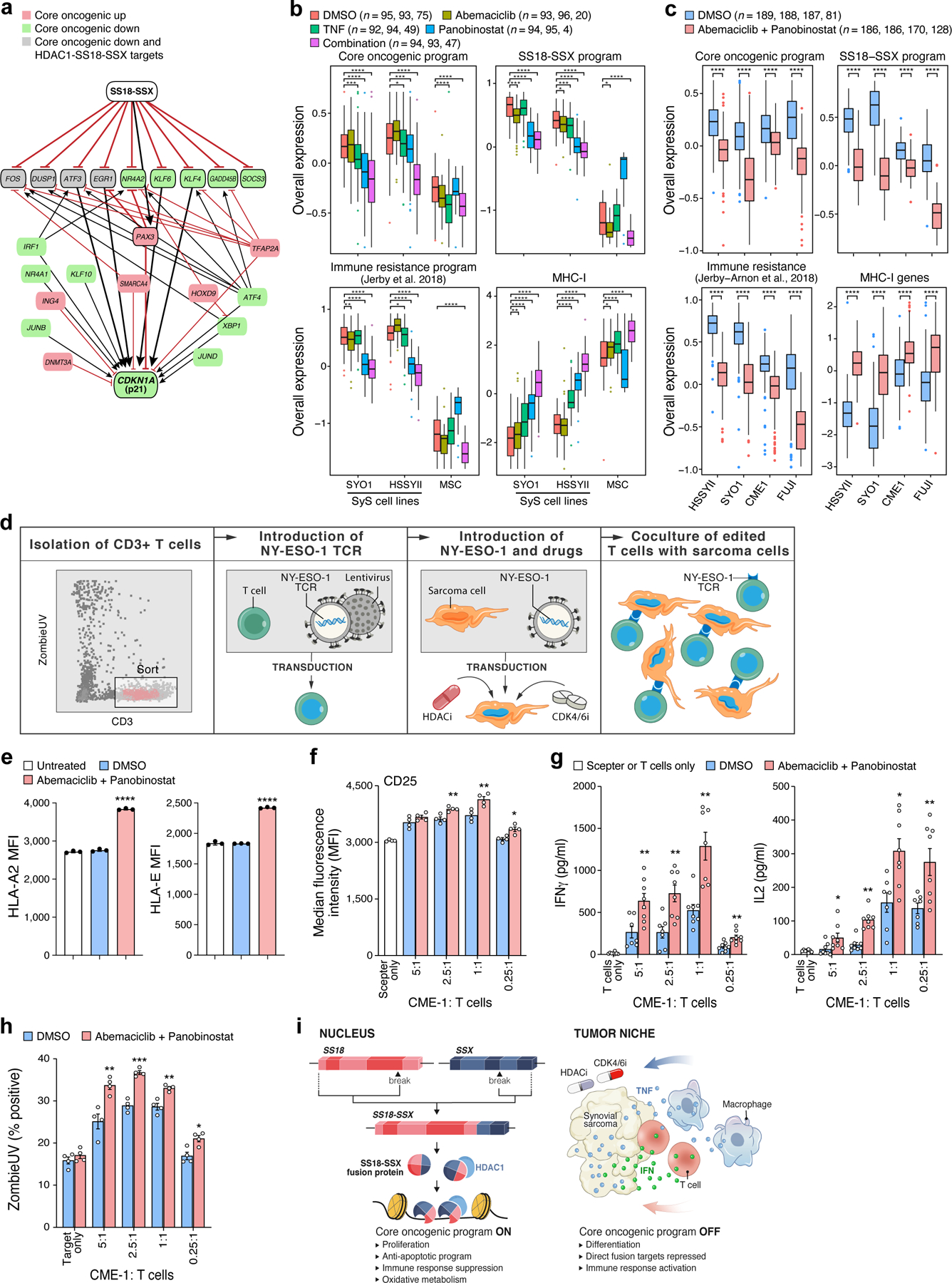
(a) Gene regulatory model links the core oncogenic program to SS18-SSX. Red/green: genes that are induced/repressed in the core oncogenic program. Grey: genes that are repressed in the core oncogenic program and directly repressed by HDAC1-SS18-SSX18. Red blunt arrows: repression; black pointy arrows: activation. Thick edges represent paths from SS18-SSX to p21. (b-c) TNF, abemaciclib and panobinostat suppress the core oncogenic program (n = no. of cells from each cell line, according to the order on the x axis). Overall Expression of the core oncogenic program, SS18-SSX program, an immune resistance program identified in melanoma30, and MHC-1 genes in SyS cells and MSCs (x axis) treated with different treatment regimens. *P<0.1,**P<0.01, ***P<1*10−3, ****P<1*10−4, one-sided t-test; middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually. (d) NY-ESO-1-based T-cell-sarcoma co-culture system. (e-h) Prior treatment of CME-1 cells with abemaciclib and panobinostat (e) increased HLA-A and HLA-E protein levels on the cell surface (P < 1*10−4, two-sided Mann-Whitney U test); (f) CD25 (activation marker) expression on the T cell surface (2.5:1 P = 0.0061, 1:1 P = 0.0082, 0.25:1 P = 0.0118, two-sided Mann-Whitney U test), (g) induces IFNγ and IL-2 secretion, and (h) improves T cell mediated killing (P = 0.0053, 2.5:1 P = 0.0009, 1:1 P = 0.0025, 0.25:1 P = 0.0122, two-sided Mann-Whitney U test). (e-h) Data are presented as mean values +/− SEM; each dot denotes one of 3 biologically independent experiments. (f-h) the results are shown for different malignant to T cell ratios. (i) Model of multifactorial SyS cell states. Left: The SS18-SSX oncoprotein sustains de-differentiation, proliferation and the core oncogenic program. Right: immune cells repress the core oncogenic and SS18-SSX programs through TNF and IFNγ secretion. Combined inhibition of HDAC and CDK4/6 mimics these effects in SyS cells.
To test these predictions, we treated SyS lines and primary mesenchymal stromal cells (MSCs) with low doses of HDAC and CDK4/6 inhibitors, and profiled only the viable cells by scRNA-Seq. Although most SyS cells were viable under the sub-cytotoxic drug concentrations we used, a small fraction underwent apoptosis (P = 3.48*10−4, mixed-effects), but not necrosis (P > 0.1, mixed-effects, Extended Data Fig. 6a), following treatment.
As predicted, the HDAC inhibitor panobinostat repressed the core oncogenic program (P = 3.34*10−14, mixed-effects; Fig. 6b), the SS18-SSX program (P = 5.32*10−72; Fig. 6b), cell cycle genes (P < 1.78*10−20), and an immune resistance program we previously identified30, and increased the expression of CDKN1A (P = 2.13*10−8, Extended Data Fig. 6b), antigen presentation and IFNγ response genes (P < 9.53*10−31, Fig. 6b, Extended Data Fig. 6c,d). The CDK4/6 inhibitor abemaciclib repressed cell cycle gene expression (P = 3.63*10−8), without impacting the core oncogenic program (P > 0.1, Fig. 6b), supporting the notion that cell cycle regulation is down-stream of the core oncogenic program.
A low dose combination of panobinostat, abemaciclib and TNF synergistically repressed the core oncogenic program (P = 1.72*10−37, Fig. 6b, Extended Data Fig. 6b) and multiple immune resistant features, while inducing antigen presentation, IFN responses, and self-antigens, such as MICA/B that can activate NK cells (P = 3.12*10−76; Fig. 6b, Extended Data Fig. 6c, d). It also repressed MIF (Macrophage Migration Inhibitory Factor), a member of the core oncogenic and SS18-SSX programs, which hampers T cell recruitment into tumors54. The less toxic and more clinically-relevant combination of HDAC and CDK4/6 inhibitors repressed the core oncogenic and SS18-SSX programs in four different SyS cell lines (CME-1, FUJI, SYO1, and HSSYII; P < 1*10−10, t-test, Fig. 6c), to an extent that significantly exceeded the drugs’ expected additive effect (P < 0.01, mixed-effects interaction term, Online Methods).
HDAC and CDK4/6 inhibitors enhance SyS cell immunogenicity and T cell-mediated-killing in co-culture
Finally, we examined whether the modulation of SyS cellular states by HDAC and CDK4/6 inhibitors can impact T cell mediated killing. We co-cultured the SyS cell line CME-1 engineered to express the cancer testis antigen NY-ESO1 with NY-ESO1-reactive human T lymphocytes (Fig. 6d; Extended Data Fig. 6e). Combined pre-treatment of the SyS cells with HDAC and CDK4/6 inhibitors before co-culture induced MHC-I cell surface protein expression (Fig. 6e, Extended Data Fig. 6f), and increased T cell activation in subsequent co-culture (Fig. 6e-h), as reflected by increased CD25 expression on the T cell surface (Fig. 6f), increased secretion of IFNγ and IL-2 (Fig. 6g), and increased T-cell-mediated killing (Fig. 6h). These effects were mediated only by malignant cell state modulation, as the T cells were not exposed to the drug combination (Fig. 6d). Thus, co-targeting CDK4/6 and HDAC in SyS cells sensitizes them to adaptive immunity.
DISCUSSION
Combining single-cell profiles from clinical specimens and functional experiments, we used integrative analysis to map the tumor ecosystem, uncover bi-directional cell-cell interactions, track the direct and indirect impact of the genetic driver on malignant and non-malignant cells, and decouple the intrinsic and extrinsic regulators of oncogenic cell states. Our results demonstrate that the genetic driver and tumor microenvironment coordinately shape cell states in SyS (Fig. 6i), and proposes therapeutic leads to target the intrinsic oncogenic mechanisms that actively repress SyS immunogenicity (Fig. 6i).
Our study also provides key resources that are particularly difficult to generate for such a rare cancer type, including the first scRNA-Seq atlas of a large cohort of primary human sarcomas, at similar size to the TCGA collection, spatial transcriptomic and multiplex in situ immunofluorescence for nine of the tumors in our single-cell cohort, functional scRNA-Seq data of SyS cells following different genetic and pharmacological perturbations, and new computational approaches to study regulatory circuits and cell-cell interactions based on these data.
The core oncogenic program we uncovered is a cell state in SyS that is regulated by SS18-SSX, marks immune-deprived tumor regions, predicts patient prognosis and manifests the dynamic cancer-immune crosstalk, as it is repressed by cytokines secreted by immune cells and desensitizes malignant cells to T cell mediated killing. Future studies should chart the detailed mechanisms underlying the ability of SS18-SSX to modulate the expression of the core oncogenic program. Subsequent efforts should examine whether HDAC and CDK4/6 inhibitors could induce T cell priming and recruitment in SyS models in vivo55, and test potential synergies with different forms of cancer immunotherapies, such as immune checkpoint blockade, adoptive T cell therapies, or cancer vaccines. Several clinical trials evaluating the efficacy of these new therapeutic approaches are currently ongoing. Thus far, it has been reported that CTLA-4 and PD-1 inhibitors had minimal to no effect in SyS patients56, whereas trials with more targeted immunotherapies against tumor specific antigens have shown greater promise in SyS, in particular vaccines that trigger priming of NY-ESO-1-specific T cells5 (NCT03520959), as well as therapies based on autologous T cells transduced with a TCR directed against NY-ESO157 (NCT01343043).
Notably, our proposed combinatorial effects should first be tested in pre-clinical models. This requires identifying genetic mouse models that faithfully recapitulate the heterogeneity of human SyS. The rapid growth in current models may unfortunately limit the establishment of both the intra-tumoral heterogeneity and the tumor-stroma/immune crosstalk we identified in patient tumors. Our extensive single cell and spatial profiling should help adjudicate models with respect to patient tumors, identify the most suitable for pre-clinical testing, and maybe draw conclusions about the yet uncertain cell of origin of these tumors.
Finally, the high-resolution approach we applied here to human SyS can serve as a blueprint for studies of other fusion-driven malignancies. For example, efforts for single cell profiling of patient tumors are ongoing in two well-defined translocated sarcomas: Ewing (EWS-FLI1) and alveolar rhabdomyosarcoma (PAX3/7-FKHR), and may benefit from the approach presented here. It remains to be seen whether targeting the driving oncogenic processes of these cancers can simultaneously induce their immunogenicity as we demonstrate here in SyS.
ONLINE METHODS
Human tumor specimen collection and dissociation
All patient samples included in this study are covered according to their respective Institutional Review Boards. Patients at Massachusetts General Hospital were consented preoperatively on Dana-Farber/Harvard Cancer Center protocol DF/HCC 13–416. Patients at the University Hospital of Lausanne were consented preoperatively on protocol Comité Ethique de Recherche CER-VD 260/15. Fresh tumors were collected directly from the operating room at the time of surgery and presence of malignancy was confirmed by frozen section. Tumor tissues were mechanically and enzymatically dissociated using a human tumor dissociation kit (Miltenyi Biotec, Cat. No. 130–095-929), following the manufacturers recommendations. Clinical annotations are provided in Supplementary Table 1.
Fluorescence-activated cell sorting (FACS)
Tumor cells were kept in Phosphate Buffered Saline with 1% bovine serum albumin (PBS/BSA) while staining. Cells were stained using calcein AM (Life Technologies) and TO-PRO-3 iodide (Life Technologies) to identify viable cells. For all tumors, we used CD45-VioBlue (human antibody, clone REA747, Miltenyi Biotec) to identify immune cells and in few cases, we also used CD3-PE to specifically identify lymphocytes (human antibody, clone BW264/56, Miltenyi Biotec). For all the samples, we used unstained cells as control. Standard, strict forward scatter height versus area criteria were used to discriminate doublets and gate only single cells. Viable single cells were identified as calcein AM positive and TO-PRO-3 negative. Sorting was performed with the FACS Aria Fusion Special Order System (Becton Dickinson) using 488nm (calcein AM, 530/30 filter), 640nm (TO-PRO-3, 670/14 filter), 405nm (CD45-VioBlue, 450/50 filter) and 561nm (PE, 586/15 filter) lasers. We sorted individual, viable, immune and non-immune single cells into 96-well plates containing TCL buffer (Qiagen) with 1% beta-mercaptoethanol. Plates were snap frozen on dry ice right after sorting and stored at −80°C prior to whole transcriptome amplification, library preparation and sequencing.
Library construction and sequencing
For plate-based scRNA-seq, Whole transcriptome amplification was performed using the SMART-seq2 protocol22, with some modifications as previously described28,58. The Nextera XT Library Prep kit (Illumina) was used for library preparation, with custom barcode adapters (sequences available upon request). Libraries from 384 to 768 cells with unique barcodes were combined and sequenced using a NextSeq 500 sequencer (Illumina).
In addition to SMART-seq2, cells from three samples (SS12pT, SS13 and SS14) were also sequenced using droplet-based scRNA-Seq with the 10x genomics platform. The samples were partitioned for SMART-seq2 and 10x genomics after dissociation. For each tumor, approximately two thirds of the sample were used for SMART-seq2 and one third for droplet based scRNA-seq (10x genomics). We sorted viable cells using MACS (Dead Cell Removal Kit, Miltenyi Biotec) and ran up to 2 channels per sample with a targeted number of cell recovery of 2,000 cells per channel. The samples were processed using the 10x Genomics Chromium 3’ Gene Expression Solution (version 2) based on manufacturer instructions and sequenced using a NextSeq 500 sequencer (Illumina).
Whole exome sequencing (WES)
DNA and RNA were extracted from fresh frozen tissue or Formalin-Fixed Paraffin-Embedded (FFPE) blocks for each patient (obtained according to their respective Institutional Review Board-approved protocols) using the AllPrep DNA/RNA extraction kit (Qiagen). We used tumor tissue and matched normal muscle tissue from the same patient as reference. Library construction was performed as previously described58, with the following modifications: initial genomic DNA input into shearing was reduced from 3µg to 20–250ng in 50µL of solution. For adapter ligation, Illumina paired end adapters were replaced with palindromic forked adapters, purchased from Integrated DNA Technologies, with unique dual-indexed molecular barcode sequences to facilitate downstream pooling. Kapa HyperPrep reagents in 96-reaction kit format were used for end repair/A-tailing, adapter ligation, and library enrichment PCR. In addition, during the post-enrichment SPRI cleanup, elution volume was reduced to 30µL to maximize library concentration, and a vortexing step was added to maximize the amount of template eluted. After library construction, libraries were pooled into groups of up to 96 samples. Hybridization and capture were performed using the relevant components of Illumina’s Nextera Exome Kit and following the manufacturer’s suggested protocol, with the following exceptions: first, all libraries within a library construction plate were pooled prior to hybridization. Second, the Midi plate from Illumina’s Nextera Exome Kit was replaced with a skirted PCR plate to facilitate automation. All hybridization and capture steps were automated on the Agilent Bravo liquid handling system. After post-capture enrichment, library pools were quantified using qPCR (automated assay on the Agilent Bravo), using a kit purchased from KAPA Biosystems with probes specific to the ends of the adapters. Based on qPCR quantification, libraries were normalized to 2nM. Cluster amplification of DNA libraries was performed according to the manufacturer’s protocol (Illumina), using exclusion amplification chemistry and flowcells. Flowcells were sequenced using Sequencing-by-Synthesis chemistry. The flowcells are then analyzed using RTA v.2.7.3 or later. Each pool of whole exome libraries was sequenced on paired 76 cycle runs with two 8 cycle index reads across the number of lanes needed to meet coverage for all libraries in the pool.
In situ immunofluorescence imaging
Formalin-fixed, paraffin-embedded (FFPE) tissue slides, 5 µm in thickness, were generated at theMassachusetts General Hospital from tissue blocks collected from patients under IRB-approved protocols (DF/HCC 13–416). Multiplexed, tissue cyclic immunofluorescence (t-CyCIF) was performed as described recently45. For direct immunofluorescence, we used the following antibodies (manufacturer, clone, dilution): c-Jun-Alexa-488 (Abcam, Clone E254, 1:200), CD45-PE (R&D, Clone 2D1, 1:150), p21-Alexa-647 (CST, Clone 12D1, 1:200), Hes1-Alexa-488 (Abcam, Clone EPR4226, 1:500), FoxP3-Alexa-570 (eBioscience, Clone 236A/E7, 1:150), NF-κB (Abcam, Clone E379, 1:200), E-Cadherin-Alexa-488 (CST, Clone 24E10, 1:400), pRB-Alexa-555 (CST, Clone D20B12, 1:300), COXIV-Alexa-647 (CST, Clone 3E11, 1:300), β-catenin-Aleaxa-488 (CST, Clone L54E2, 1:400), HSP90-PE (Abcam, polyclonal, lot# GR3201402–2, 1:500) and vimentin-Alexa-647 (CST, Clone D21H3, 1:200). Stained slides from each round of t-CyCIF were imaged with a CyteFinder slide scanning fluorescence microscope (RareCyte Inc. Seattle WA) using either a 10X (NA=0.3) or 40X long-working distance objective (NA = 0.6). Imager5 software (RareCyte Inc.) was used to sequentially scan the region of interest in 4 fluorescence channels. Image processing, background subtraction, image registration, single-cell segmentation and quantification were performed as previously described45.
RNA in situ hybridization
Paraffin-embedded tissue sections from human tumors from Massachusetts General Hospital and University Hospital of Lausanne were obtained according to their respective Institutional Review Board-approved protocols. Sections were mounted on glass slides and stored at −80°C. Slides were stained using the RNAscope 2.5 HD Duplex Detection Kit (Advanced Cell Technologies, Cat. No. 322430), as previously described28,29: slides were baked for 1 hour at 60°C, deparaffinized and dehydrated with xylene and ethanol. The tissue was pretreated with RNAscope Hydrogen Peroxide (Cat. No. 322335) for 10 minutes at room temperature and RNAscope Target Retrieval Reagent (Cat. No. 322000) for 15 minutes at 98°C. RNAscope Protease Plus (Cat. No. 322331) was then applied to the tissue for 30 minutes at 40°C. Hybridization probes were prepared by diluting the C2 probe (red) 1:50 into the C1 probe (green). Advanced Cell Technologies RNAscope Target Probes used included Hs-EGR1 (Cat. No. 457671-C2), Hs-IGF2 (Cat. No. 594361), Hs-TLE1 (Cat. No. 409191) and Hs-LGALS1 (Cat. No. 486281). Probes were added to the tissue and hybridized for 2 hours at 40°C. A series of 10 amplification steps was performed using instructions and reagents provided in the RNAscope 2.5 HD Duplex Detection Kit. Tissue was counterstained with Gill’s hematoxylin for 25 seconds at room temperature followed by mounting with VectaMount mounting media (Vector Laboratories).
RNA profiling in situ hybridization (ISH)
Complete methods for GeoMx RNA assays can be found in Merritt et al 202059. DNA oligo probes were designed to bind mRNA targets. From 5’ to 3’, they each comprised of a 35–50 nt target complementary sequence, a UV photocleavable linker, and a 66 nt indexing oligo sequence containing a unique molecular identifier (UMI), RNA ID sequence, and primer binding sites. Up to 10 RNA detection probes were designed per target mRNA. Precommercial research-use-only versions of the GeoMx Cancer Transcriptome Atlas (CTA) and the Human Whole Transcriptome Atlas (WTA) were provided by Nanostring Technologies.
To perform the ISH, 5 µm FFPE tissue sections from nine patients were mounted on positively charged histology slides. Sections were baked at 65⁰C for 45 minutes in a Hyb EZ II hybridization oven (Advanced cell Diagnostics, Inc). Slides were deparaffinized using Citrisolv (Decon Labs, Inc., 1601), rehydrated in an ethanol gradient, and washed in 1x phosphate-buffered saline pH 7.4 (PBS: Invitrogen, AM9625). Slides were incubated for 15 minutes in 1X Tris-EDTA pH 9.0 buffer (Sigma Aldrich, SRE0063) at 100°C with low pressure in a TintoRetriever Pressure cooker (bioSB, 7008). Slides were washed, then incubated in 1 µg/mL proteinase K (Thermo Fisher Scientific, AM2546) in PBS for 15 minutes at 37°C and washed again in PBS. Tissues were then fixed in 10% neutral-buffered formalin (Thermo Fisher Scientific, 15740) for 5 minutes, incubated in NBF stop buffer (0.1M Tris Base, 0.1M Glycine, Sigma) for 5 minutes twice, then washed for 5 minutes in PBS. Tissues were then incubated overnight at 37°C with GeoMx™ RNA detection probes in Buffer R (Nanostring Technologies) using a Hyb EZ II hybridization oven (Advanced cell Diagnostics, Inc). During incubation, slides were covered with HybriSlip Hybridization Covers (Grace BioLabs, 714022). Following incubation, HybriSlip covers were gently removed and 25-minute stringent washes were performed twice in 50% formamide and 2X SSC at 37°C. Tissues were washed for 5 minutes in 2X SSC then blocked in Buffer W (Nanostring Technologies) for 30 minutes at room temperature in a humidity chamber. 500nM Syto13 and antibodies targeting PanCK and CD45 (Nanostring Technologies) in Buffer W were applied to each section for 1 hour at room temperature. Slides were washed twice in fresh 2X SSC then loaded on the GeoMx™ Digital Spatial Profiler (DSP)43.
In the process entire slides were imaged at 20x magnification and a total of 244 circular regions of interest (ROI) with 300–600 μm diameter were selected per sample and the GeoMx software was used to define areas of illumination (AOIs or segments) within each ROI as one segment containing positive immunofluorescent signal for CD45 and auto-fluorescence in the same channel (CD45+) and the inverse of that segment (CD45−). Segmentation thresholds for CD45- segments were adjusted to enrich for tumor regions with minimal immune signal. As a result, the entire tumor region was not photocleaved. The CD45− AOIs included only malignant cells based on morphological and histological examination. The CD45− segments in the biphasic sample (SyS1) were further segmented in the same manner also based on PanCK, separating the epithelial (CD45−/PanCK−) and mesenchymal (CD45−/PanCK+) malignant cells. In the WTA, a PanCK+ criterion was included in the CD45- segment, but the threshold was set to background levels resulting in segmentation comparable to a CD45-segment.
Once AOIs were defined, the DSP then exposed AOIs to 385 nm light (UV) releasing the indexing oligos and collecting them with a microcapillary. Indexing oligos were then deposited in a 96-well plate for subsequent processing. The indexing oligos were dried down overnight and resuspended in 10 μL of DEPC-treated water.
Sequencing libraries were generated by PCR from the photo-released indexing oligos and AOI-specific Illumina adapter sequences and unique i5 and i7 sample indices were added. Each PCR reaction used 4 μL of indexing oligos, 1 μL of indexing PCR primers, 2 μL of Nanostring 5X PCR Master Mix, and 3 μL PCR-grade water. Thermocycling conditions were 37°C for 30 min, 50°C for 10 min, 95°C for 3 min; 18 cycles of 95°C for 15sec, 65°C for 1min, 68°C for 30 sec; and 68°C 5 min. PCR reactions were pooled and purified twice using AMPure XP beads (Beckman Coulter, A63881) according to manufacturer’s protocol. Pooled libraries were sequenced at 2×75 base pairs and with the single-index workflow on an Illumina NextSeq to generate 458M raw reads.
Primary cell cultures and cell lines
Human primary Synovial Sarcoma (SyS) spherogenic cultures (SScul1, SScul2 and SScul3) were derived from patients undergoing surgery at Massachusetts General Hospital and University Hospital of Lausanne, according to their respective Institutional Review Board-approved protocols. Directly after dissociation (as above), the dissociated bulk tumor cells were put in culture and grown as spheres using ultra-low attachment cell culture flasks in IMDM 80% (Gibco, Cat. No. 1244053), Knock-Out Serum Replacement 20% (Gibco, Cat. No. 10828028), Recombinant Human EGF Protein 10 ng/mL (R&D systems, Cat. No. 236-EG-200), Recombinant Human FGF basic, 145 aa (TC Grade) Protein 10ng/mL (R&D systems, Cat. No. 4114-TC-01M) and 1% Penicillin-Streptomycin (Gibco, Cat. No. 15140122). Cells were expanded by mechanical and enzymatical dissociation every week using TrypLE Express Enzyme (ThermoFisher, Cat. No. 12605010).
The SyS cell lines used for the SS18-SSX KD experiments and the functional drug assays include: Aska (a generous gift from Kazuyuki Itoh, Norifumi Naka, and Satoshi Takenaka, Osaka University, Japan), SYO1 (a generous gift from Akira Kawai, National Cancer Center Hospital, Japan), HS-SY-II (purchased from RIKEN Bio Resource Center, 3–1-1 Koyadai, Tsukuba, Ibaraki 305–0074, Japan), CME-1 (a generous gift from Dr. Armando Bartolazzi, Pathology Research Laboratory, Cancer Center Karolinska, Karolinska Hospital, Stockholm, Sweden) and FUJI (a generous gift from Duan Zhenfeng and Francis J. Hornicek, Orthopaedic Institute for Children, Department of Orthopaedic Surgery, UCLA, US). All cell lines excepted CME-1 were cultured using standard protocols in DMEM medium (Gibco) supplemented with 10–20% fetal bovine serum, 1% Glutamax (Gibco), 1% Sodium Pyruvate (Gibco) and 1% Penicillin-Streptomycin (Gibco) and grown in a humidified incubator at 37°C with 5% CO2. The CME-1 line was cultured in RPMI containing 10% FBS and 1% Penicillin-Streptomycin.
Human primary pediatric mesenchymal stromal cells (MSCs) were isolated from healthy donors undergoing corrective surgery in agreement with the Institutional Review Board-approved protocol of the University Hospital of Lausanne (Protocol number 2017–0100). According to the Swiss ethic legislation no written consent was required since samples were anonymized prior to culture and analysis. Cells were expanded in 90% IMDM (Gibco, Cat. No. 1244053) containing 10% Fetal Bovine Serum (Gibco), 1% Penicillin-Streptomycin (Gibco) and 10ng/mL Platelet-Derived Growth Factor BB (PDGF-BB, PeproTech).
SS18-SSX knockdown in Aska and SYO1 cell lines
The SyS cell lines Aska and SYO1 were cultured using standard protocols in DMEM medium (Gibco) supplemented with 10–20% fetal bovine serum, 1% Glutamax (Gibco), 1% Sodium Pyruvate (Gibco) and 1% Penicillin-Streptomycin (Gibco) and grown in a humidified incubator at 37°C with 5% CO2. Cells expressing a pLKO.1 vector with a scrambled shRNA hairpin control (5’- CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAAC CTTAGG-3’) or a shSSX hairpin targeting SSX of the SS18-SSX fusion (5’-CAGTCACTGACAGTTAATAAA-3’) were prepared by lentiviral infection. Briefly, lentivirus was prepared by transfection of HEK293T cells with gene delivery vector and the packaging vectors pspax2 and pMD2.G, filtration of media followed by ultracentrifugation, and then resuspension of viral pellet in PBS. Aska and SYO1 cells were infected with lentivirus for 48 hours and then underwent 5 days of selection with puromycin (2 μg/mL) prior to collection for scRNA-seq.
In vitro IFN/TNF experiment
Cells were dissociated 12 hours before adding the drugs at the concentrations indicated directly to the growing media and cells were collected at different time point (ranging from 4 hours to 4 days) for SMART-seq2. Viability was determined by CellTiter-Glo Luminescent Cell Viability Assay (Promega) after 5 to 7 days of treatment. TNF-alpha (Miltenyi Biotec, Human TNF-α, Cat. No. 130–094-014) IFN-gamma (R&D systems, Recombinant Human IFN-gamma Protein, Cat. No. 285-IF-100) were suspended in deionized sterile-filtered water.
In vitro drug assay, cell proliferation and cell death measurements
For the functional drug assay, 200,000 SYO-1 cells and HSSYII cells, and 100,000 MSCs were seeded in 60 × 15 mm plates (Falcon). Cells were stimulated for five days with the following compounds: 100 or 200 nM Abemaciclib (Selleckchem, U.S.A.), 15 or 30 ng/ml TNF (Miltenyi Biotech, Germany) or a combination of the two. Compounds were refreshed at days three and four, and the solvent (DMSO) was used as control. At day 4, 12.5 or 25 nM Panobinostat (Selleckchem, U.S.A.) was added to the cultures, and the cells were harvested 24 hours later for proliferation scoring. To assess cellular proliferation, cells were detached with trypsin, washed in PBS, and re-suspended in 1 ml of complete medium. After diluting 1:2 with Trypan blue (Invitrogen) viable cells were counted using the Automated Cell Counter Countess II FL (Thermo Fisher Scientific). Each experimental condition was measured in triplicate.
To assess the rate of cell death cells were seeded in 100mm plates at a 10–15% confluency, stimulated for 5 days with daily refreshed Abemaciclib (100nM), TNF (15ng/ml), or both. DMSO was added to control cells. After 4 days, 12.5nM Panobinostat was added and cells were harvested 24 hrs later. Cell viability and cell number were controlled by trypan blue exclusion and using an automated cell counter Countess II (ThermoFisher Scientific). The number of apoptotic, necrotic and viable cells was determined by flow cytometry after fluorescent detection of annexin V and PI staining using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences). Data were analyzed using the FlowJo software (FlowJo, LLC, Ashland, Ore.)
scRNA-seq pre-processing and gene expression quantification
BAM files were converted to merged, demultiplexed FASTQ files. The paired-end reads obtained with SMART-Seq2 were mapped to the UCSC hg19 human transcriptome using Bowtie60, and transcript-per-million (TPM) values were calculated with RSEM v1.2.8 in paired-end mode61. The paired-end reads obtained with droplet scRNA-Seq (10x Genomics) were mapped to the UCSC hg19 human transcriptome using STAR62, and gene counts/TPM values were obtained using CellRanger (cellranger-2.1.0, 10x Genomics).
For bulk RNA-Seq, expression levels were quantified as E=log2(TPM+1). For scRNA-seq data, expression levels were quantified as E=log2(TPMi,j/10+1). TPM values were divided by 10 because the complexity of our single-cell libraries is estimated to be within the order of 100,000 transcripts63. The 10−1 factoring prevents counting each transcript ~10 times and overestimating differences between positive and zero TPM values. The average expression of a gene i across a population P of N cells, was defined as
For each cell, we quantified the number of genes with at least one mapped read, and the average expression level of a curated list of housekeeping genes30. We excluded all cells with either fewer than 1,700 detected genes or an average housekeeping expression (E, as defined above) below 3 (Supplementary Table 1). For the remaining cells, we calculated the average expression of each gene (Ep), and excluded genes with an average expression below 4, which defined a different set of genes in different analyses depending on the subset of cells included. In cases where we analyzed different cell subsets together, we removed genes only if they had an average Ep below 4 in each of the different cell subsets included in the analysis. Different cell types and malignant cells from different tumors were considered as different cell subsets in this regard.
For completeness we also provide extended gene signatures (Supplementary Tables 3,4, “low QC genes”), where we list genes that did not pass the cutoffs described above, yet show a significant association with the overall expression of the pertaining signature (mixed-effect models controlling for cell quality, see Multilevel mixed-effects models section).
WES data pre-processing
A BAM file was produced with the Picard pipeline (http://picard.sourceforge.net/), which aligns the tumor and normal sequences to the hg19 human genome build using Illumina sequencing reads. The BAM was uploaded into the Firehose pipeline (http://www.broadinstitute.org/cancer/cga/Firehose). Quality control modules within Firehose were applied to all sequencing data for comparison of the origin for tumor and normal genotypes and to assess fingerprinting concordance. Cross-contamination of samples was estimated using ContEst64.
Somatic alteration assessment
MuTect65 was applied to identify somatic single-nucleotide variants. Indelocator (http://www.broadinstitute.org/cancer/cga/indelocator), Strelka66, and MuTect2 (https://software.broadinstitute.org/gatk/documentation/tooldocs/current/org_broadinstitute_gatk_tools_walkers_cancer_m2_MuTect2) were applied to identify small insertions or deletions. A voting scheme was used with inferred indels requiring a call by at least 2 out of 3 algorithms.
Artifacts introduced by DNA oxidation during sequencing were computationally removed using a filter-based method67. In the analysis of primary tumors that are formalin-fixed, paraffin-embedded samples (FFPE) we further applied a filter to remove FFPE-related artifacts68. Reads around mutated sites were realigned with Novoalign (www.novocraft.com/products/novoalign/) to filter out false positive that are due to regions of low reliability in read alignment. At the last step, we filtered mutations that are present in a comprehensive WES panel of 8,334 normal samples (using the Agilent technology for WES capture) aiming to filter either germline sites or recurrent artifactual sites. We further used a smaller WES panel of 355 normal samples that are based on Illumina technology for WES capture, and another panel of 140 normal samples sequenced without our cohort69 to further capture possible batch-specific artifacts. Annotation of identified variants was done using Oncotator70 (http://www.broadinstitute.org/cancer/cga/oncotator).
Copy number and copy ratio analysis
To infer somatic copy number from WES, we used ReCapSeg (http://gatkforums.broadinstitute.org/categories/recapseg-documentation), calculating proportional coverage for each target region (i.e., reads in the target/total reads) followed by segment normalization using the median coverage in a panel of normal samples. The resulting copy ratios were segmented using the circular binary segmentation algorithm71. To infer allele-specific copy ratios, we mapped all germline heterozygous sites in the germline normal sample using GATK Haplotype Caller72 and then evaluated the read counts at the germline heterozygous sites in order to assess the copy profile of each homologous chromosome. The allele-specific copy profiles were segmented to produce allele specific copy ratios.
Gene sets Overall Expression
We used the following scheme to compute the Overall Expression (OE) of a gene set (signature). The OE metric30 filters technical variation and highlights biologically meaningful patterns. The procedure is based on the notion that the measured expression of a specific gene is correlated with its true expression (signal), but also contains a technical (noise) component. The latter may be due to various stochastic processes in the capture and amplification of the gene’s transcripts, sample quality, as well as variation in sequencing depth. The OE of a gene signature is computed in a way that accounts for the variation in the signal-to-noise ratio across genes and cells.
Given a gene signature and a gene expression matrix E (as defined above), we first binned the genes into 50 expression bins according to their average expression across the cells or samples. The average expression of a gene across a set of cells within a sample is Ei,p (see: scRNA-seq pre-processing and gene expression quantification) and the average expression of a gene across a set of N tumor samples was defined as: . Given a gene signature S that consists of K genes, with kb genes in bin b, we sample random S-compatible signatures for normalization. A random signature is S-compatible with a signature S if it consists of overall K genes, such that in each bin b it has exactly kb genes. The OE of signature S in cell or sample j is then defined as:
Where is a random S-compatible signature, and Cij is the centered expression of gene i in cell or sample j, defined as . Because the computation is based on the centered gene expression matrix C, genes that generally have a higher expression compared to other genes will not skew or dominate the signal. We found that 100 random S-compatible signatures are sufficient to yield a robust estimate of the expected value . The distribution of the OE values was normal or a mixture of normal distributions, facilitating subsequent analyses.
We use the term transcriptional program (e.g., the core oncogenic program) to denote cell states defined by a pair of signatures, such that one (S-up) is overexpressed and the other (S-down) is underexpressed. The OE of a program is then the OE of S-up minus the OE of S-down.
In cases where the OE of a given signature/program has a bimodal distribution across the cell population, it can be used to naturally separate the cells into two subsets. To this end, we applied the Expectation Maximization (EM) algorithm for mixtures of normal distributions to define the two underlying normal distributions. We then assigned cells to two subsets, depending on the distribution (high or low) they were assigned to.
Cell type assignments
Cell type assignments were performed based on genetic and transcriptional features, according to the following four analyses:
(1). Fusion detection.
Fusion detection was performed with STAR-Fusion24, to detect any transcript that indicates the fusion of two genes. The detection of gene-fusions in single cells is based on the detection of mRNA reads that either span or include the junction between the SS18 and SSX1/2 genes. It is therefore impacted by technical variabilities and stochasticity inherent to single-cell RNA-seq methods (e.g., the effective sequencing depth, drop-out rate and amplification noise of each cell). Indeed, within the cells that were identified as malignant by other approaches (listed below), the detection of the fusion is tightly linked to the number of genes and reads sequenced per cell (P < 1*10−10, mixed-effects). Because the fusion is detected in a subset of malignant cells in each expression cluster (Fig. 1d) and because cells with or without detected fusion do not form separate clusters, the detection of the fusion in a subset of cells in a cluster allowed us to confidently annotate clusters as either malignant or non-malignant.
(2). Copy Number Alterations (CNA) inference.
To infer CNAs from the scRNA-seq data we used the approach described in25, as implemented in the R code provided in https://github.com/broadinstitute/inferCNV with the default parameters. To avoid circularity, we first used only a small set of cells that were annotated as fibroblasts as the reference set. These cells formed a cluster that was completely deprived of fusion transcripts, and expressed multiple fibroblast markers. We used the resulting CNAs to examine the consistency between the different cell type annotation approaches. In the next iteration we inferred CNA using all the cells that were annotated as non-malignant (according to multiple other analyses) as reference cells, such that each cell type compromises and independent reference group. These two CNA-inference approaches resulted in the same CNA-based cell-type-annotations. In addition, when using only a subset of the non-malignant cells as reference, we do not find any CNA in the non-malignant cells that were not provided as reference, as shown in Fig. 1g (bottom).
To identify malignant cells based on CNA patterns, we defined the overall CNA level of a given cell as the sum of the absolute CNA estimates across all genomic windows. Within each tumor, we identified CD45− cells with the highest overall CNA level (top 10%), and considered their average CNA profile as the CNA profile of the pertaining tumor. For each cell we then computed a CNA-R-score defined as the Spearman correlation coefficient obtained when comparing its CNA profile to the inferred CNA profile of its tumor. Cells with a high CNA-R-score (greater than the 25% of the CD45− cell population) were considered as malignant according to the CNA criterion. As certain tumors/malignant cells have a stable genome, we did not use the CNA criterion to identify non-malignant cells. Large-scale CNAs were visualized (Fig. 1g) as described in https://github.com/broadinstitute/infercnv/wiki/infercnv-i6-HMM-type, such that the HMM was parameterized with a state transition probability of 1*10−6, and the Bayesian network was configured to use an uninformative Dirichlet prior, and run for 1000 iterations after a burn-in of 500 iterations.
(3). Differential similarity to bulk tumors.
We compared the scRNA-Seq profiles to those of bulk sarcoma tumors21. RNA-Seq of bulk sarcoma tumors was downloaded from TCGA (http://xena.ucsc.edu). For each cell in our scRNA-Seq cohort we: (i) computed the Spearman correlation between its expression profile and the expression profiles of the bulk sarcoma tumors, and (ii) examined if the rs coefficients obtained when comparing the cell to SyS tumors were higher than those obtained when comparing the cell to non-SyS sarcoma tumors, using a one-sided Wilcoxon ranksum test. Cells with a ranksum p-value < 0.05 were considered as potentially malignant, and as potentially non-malignant otherwise.
(4). Expression profile clustering.
We clustered the cells by applying a shared nearest neighbor (SNN) modularity optimization algorithm73, as implemented in the Seurat R package. First, Principle Component Analysis (PCA) was performed using the (2,000) topmost overdispersed genes. These genes were identified using the Seurat package FindVariableFeatures function. In this procedure local polynomial regression (LOESS) is used to estimate the expected variance given the average gene expression values across the cells, on a log-log scale. Deviation from the expected value is then used to identify overdispersed genes. Next, k-nearest neighbors (kNN) were calculated based on the top 25 PCs to construct a k-NN graph, which was then used to identify clusters that optimize the modularity function. Similar results were obtained when using different numbers of PCs and overdispersed genes (data not shown).
Next, clusters were assigned to cell types, such that clusters where the majority of cells had the SS18-SSX1/2 fusion (by the method in (1)) were considered as malignant clusters. Non-malignant clusters were assigned to cell types by computing the OE of well-established cell type markers across the non-malignant cells (Supplementary Table 2). The OE of each of these cell type signatures had a bimodal distribution across the cell population. Applying the Expectation Maximization (EM) algorithm for mixtures of normal distributions, we defined the two underlying normal distributions, and assigned cells to cell types. Each non-malignant cluster was enriched for cells of a particular cell type, and was assigned to that cell type.
We used these four converging criteria to assign the cells to nine cell subsets: malignant cells, epithelial cells, Cancer Associated Fibroblasts (CAFs), CD8 and CD4 T cells, B cells, NK cells, macrophages, and mastocytes. Specifically, a cell was labeled malignant if it was CD45− and classified as malignant according to analyses (3) and (4) above. A cell was labeled non-malignant if it was classified as non-malignant according to analyses (1–4) above. Non-malignant cells were then further assigned to cell types based on their cluster assignment by (4). Cells with inconsistent assignments (157 in the SMART-Seq dataset and 558 in the droplet-based dataset) were removed from further analyses. Lastly, within malignant cells we identified epithelial cells by clustering each of the biphasic tumors into two clusters.
Cell type assignments were preformed separately for the SMART-Seq2 and droplet scRNA-Seq datasets cohort. Fusion detection was performed only with the full-length SMART-Seq2 data.
Cell type signatures
Cell type signatures were generated based on pairwise comparisons between identified cell subtypes: malignant cells, epithelial cells, CAFs, CD8 and CD4 T cells, B cells, NK cells, macrophages, and mastocytes. For each pair of cell subtypes we identified differentially expressed genes using the likelihood-ratio test74, as implemented in the Seurat package (http://www.satijalab.org/seurat). Genes were considered as cell type specific if they were overexpressed in a particular cell subtype compared to all other cell subtypes (log-fold change > 0.25 and p-value < 0.05, following Bonferroni correction). We defined a general T cell signature for both CD4 and CD8 cells by identifying genes that were overexpressed in both CD4 and CD8 compared to all other (non T) cells. A more permissive version of this generic T cell signature includes genes which were overexpressed in CD4 or CD8 T cells compared to all other (non T) cells.
Inferring tumor composition
Tumor composition was assessed based on the Overall Expression of the different cell type specific signatures we identified from the scRNA-seq data (Supplementary Table 2). For example, the CD8 T cell signature was used to infer the level of CD8 T cells in the tumor, and likewise for other cell types. To estimate tumor purity, we used the malignant SyS signature identified here (Supplementary Table 2), which consists of genes that are exclusively expressed by malignant SyS cells compared to non-malignant cells in SyS tumors.
To evaluate the performance of this approach, we simulated 200 bulk RNA-Seq profiles. For each simulated bulk RNA-Seq profile we: (1) randomly chose one of the tumors in the cohort; (2) sampled 100 cells from different cell types profiled in this tumor – these cells include a mix of immune, stroma and malignant cells, at a randomly chosen composition; (3) summed the scRNA-Seq profiles of this randomly chosen population (P) of 100 cells, such that the bulk expression of gene i across this population was defined as
We also used cell type signatures we previously derived from melanoma scRNA-Seq data30 to predict the tumor composition of the simulated SyS bulk RNA-Seq profiles, and vice versa. We then compared the predictions to the known cell type composition. The predicted composition was highly correlated with the known composition (r > 0.9, P < 1*10−30, Spearman correlation) for all cell types.
Multilevel mixed-effects models
To examine the association between two cell features, denoted here as x and y, across different patients or experiments we used multilevel mixed-effects regression models (random intercepts models). The models include patient/experiment-specific intercepts to control for the dependency between the scRNA-seq profiles of cells that were obtained from the same patient/experiment. The models also control for data quality by providing the number of reads (log-transformed) that were detected in each cell as a covariate. To compute the association between features x and y we provided x as another covariate and used y as the dependent variable. The models were implemented using the lme4 and lmerTest R packages (https://CRAN.R-project.org/package=lme4, https://CRAN.R-project.org/package=lmerTest).
For example, to test if malignant cycling cells were more frequent in treatment naïve samples, we used a logistic mixed-effects model as described above. The dependent variable y was the cycling status of the malignant cells. The independent covariate x was a binary variable denoting if the sample was obtained before or after treatment. Only malignant cells were included in this model.
T Cell Receptor (TCR) reconstruction and T cell expansion program
TCR reconstruction was performed using TraCeR38, with the Python package in https://github.com/Teichlab/tracer. To characterize the transcriptional state of clonally expanded T cells, we first identified the clonality level of the T cells in our cohort. T cells that were obtained from tumors with a larger number of T cells with reconstructed TCRs were more likely to be defined as expanded. To control for this confounder, we performed the following down-sampling procedure. First, we removed T cells without a reconstructed alpha or beta TCR chain, and samples with less than 20 T cells with a reconstructed TCR. Next, we computed the probability that a given cell will be a part of a clone when subsampling 20 T cells from each tumor. T cells with a high probability to be a part of a clone (above the median) were considered expanded, and non-expanded otherwise. To identify the genes differentially expressed in expanded CD8 T cells we used mixed-effects models with a binary covariate, denoting if the cell was classified as expanded or not.
CD8 T cell analyses
The analysis of T cell exhaustion vs. T cell cytotoxicity was performed as previously described 75, with the exhaustion signature provided in75. First, we computed the cytotoxicity and exhaustion scores of each CD8 T cell. Next, to control for the association between the expression of exhaustion and cytotoxicity markers, we estimated the relationship between the cytotoxicity and exhaustion scores using locally-weighted polynomial regression (LOWESS, black line in Fig. 4b). Based on these values we classified the CD8 T cells into four groups: Cells with a low cytotoxicity score (below the 25th percentile) were classified as naïve or memory-like cells, while the others were considered effector or exhausted if their cytotoxicity scores were significantly higher or lower than expected given their exhaustion scores, respectively. According to this classification, we examined if the clonal expansion program was higher in the effector-like cells. In addition, we compared the SyS CD8 T cells to CD8 T cells from human melanoma tumors30 using mixed-effects models with a sample-level covariate denoting if the sample was obtained from a SyS or melanoma tumor.
Malignant epithelial and mesenchymal differentiation programs
The epithelial and mesenchymal signatures were obtained through intra-tumor differential expression analysis, using the likelihood-ratio test for single cell gene expression74, as implemented in the Seurat package (http://www.satijalab.org/seurat). We compared the mesenchymal to epithelial cells in each of the three biphasic tumor samples (SyS1, SyS12 and SyS12pt). The tumor SyS16 was not included in this analysis (although it was annotated as partially biphasic according to its histology), because its scRNA-Seq sample did not include any epithelial malignant cells, potentially due to misclassification of SyS16 as biphasic, biased tumor sampling, or less transcriptionally distinct epithelial cells in this tumor.
Genes that were up-regulated in the epithelial cells compared to the mesenchymal cells in all three samples were defined as epithelial genes, and likewise for mesenchymal genes. When using the epithelial and mesenchymal signatures in the analysis of bulk gene expression we removed from these signatures those genes that are also part of non-malignant cell type signatures.
Using these signatures, we defined: (1) the epithelial vs. mesenchymal differentiation score as the OE of the epithelial signature minus the OE of the mesenchymal signature, and (2) the differentiation score as the OE of the epithelial signature plus the OE of the mesenchymal signature. An alternative way to define the differentiation score of a particular cell is first to assign it to the epithelial or mesenchymal subset, and then use only the pertaining signature to estimate its differentiation level. However, this approach will not distinguish between poorly-differentiated mesenchymal cells, and mesenchymal cells which have begun to transition to an epithelial state. Hence, we used the inclusive definition of differentiation.
Based on the genes in the epithelial and mesenchymal signatures we then generated diffusion maps76 for each one of the tumors in our cohort, using the density R package (https://bioconductor.org/packages/release/bioc/html/destiny.html) with the default parameters.
Identifying co-regulated gene modules
To identify co-regulated gene modules that capture intra-tumor heterogeneity we applied both a PCA-based and an NMF-based approach. As this analysis was geared to identify new types of intratumor variation beyond the epithelial/mesenchymal one, the biphasic tumors (SyS1, SyS12, and SyS12pt) were represented by two “samples”, one of epithelial and anther of mesenchymal cells.
In our PCA-based approach we first identified overdispersed gene modules separately in each of the tumors in our cohort using PAGODA33 as implemented in https://github.com/hmsdbmi/scde. The number and size of the gene modules was determined based on the significance of their overdispersion across the cells. Redundant modules that represent the same co-variation across the cells were merged. To identify genes that were repeatedly co-regulated we then constructed a gene-gene co-regulation graph. In this graph, an edge between two genes denotes that the two genes appeared together in the same gene module in at least five samples (similar results were obtained with lower and higher cutoffs). Next, we identified dense clusters in the graph using the Newman-Girvan34 community clustering, and filtered out small gene clusters (< 20 genes). Lastly, for each gene cluster we identified the opposing gene module by identifying genes that were negatively correlated with its Overall Expression (OE) across the malignant cells. Correlation was computed using partial Spearman correlation, when controlling for the number of genes and (log-transformed) reads detected per cells, and correcting for multiple hypotheses testing using the Benjamini-Hochberg procedure77.
For comparison we applied another complementary NMF-based approach, LIGER35,36, which identifies repeating gene modules in the malignant cells using integrative non-negative matrix factorization (NMF)36. Integrative NMF learns a low-dimensional space, where cells are defined by one set of dataset-specific factors (denoted as Vi), and another set of shared factors (denoted as W). Each factor, or metagene, represents a distinct pattern of gene co-regulation. To find these metagenes it solves the following optimization problem
Where Ei denotes the expression matrix (log-transformed TPM) of the malignant cells in sample i, Vi denotes sample-specific metagenes and W denotes the shared metagenes across all samples. We used the top 100 genes of each metagene in W as the iNMF signatures, and then computed the overall expression of these signatures in the malignant cells. The resulting signatures and their expression across the malignant cells matched the core-oncogenic and cell cycle programs that were identified in the PCA-based approach (R > 0.93, P < 1*10−20, Pearson correlation; Extended Data Fig. 3a), but did not completely recapitulate the other PCA-based signatures.
Predicting patient prognosis
To test if a given program predicts metastasis free-survival or overall survival, we first computed the OE of the program in each tumor based on the bulk gene expression data. Next, we used a Cox regression model with censored data to compute the significance of the association between the expression values and survival. To visualize the predictions of a specific signature in a Kaplan Meier (KM) plot, we stratified the patients into three groups according to the program expression: high or low expression correspond to the top or bottom 20% of the population, respectively, and intermediate otherwise. We used a log-rank test to examine if there was a significant difference between the survival rates of the three patient groups.
Analysis of in situ immunofluorescence imaging
Immune cells were detected based on the protein level of CD45 (>7.5 log-transformed). Malignant cells were identified based on histological morphology, and high protein levels of Hes1. High protein expression was detected by applying the EM algorithm for mixtures of normal distributions. The core oncogenic program score was computed only in the malignant cells based on the combined expression of its repressed protein markers: Hsp90, p21, NFkB, and cJun (minus sum of centered log-transformed values). Each image – corresponding to a specific sample in the scRNA-Seq cohort – was divided to frames of 100 cells. The average expression of the core oncogenic program in the malignant cells and the fraction of immune cells in each frame was computed. Using these frame-level values we examined the association between the expression of the core oncogenic program in the malignant cells and the fraction of the immune cells, using a mixed-effects model, with a sample-level intercept (see Multilevel mixed-effects models). The mixed-effect model accounts for the nested structure of the data (frames are associated with samples), and ensures the pattern repeatedly appears across different samples.
Analysis of in situ RNA profiling
FASTQ files from multiple lanes were merged to generate single files for processing and insure proper removal of PCR duplicates later in the pipeline. Illumina adapter sequences were trimmed using Trim Galore (version 0.4.5) with a minimum base pair overlap stringency of four bases and a base quality threshold of 20. Paired end reads were stitched using Paired-End reAd mergeR (PEAR, version 0.9.10) specifying a minimum stitched read length of 24bp and a maximum stitched read length of 28bp. The 14bp UMI sequence was extracted from the stitched FASTQ files from the 5’ end of the sequence reads using umi tools (version 0.5.3). The FASTQ files with extracted UMIs were then aligned to a genome containing the 12bp reference sequence tags using bowtie2 (version 2.3.4.1) in end-to-end mode with a seed length of four. Using a custom python function, the generated SAM files were split into multiple SAM files based on the tag to which they aligned to limit memory usage when removing PCR duplicates. The split SAM files were converted to bam files, sorted, and indexed using samtools (version 1.9) with the import, sort, and index options respectively. PCR duplicates were removed from the sorted and indexed bam files using the dedup command from umi tools with an edit distance threshold of three. An edit distance threshold of three was used. Using custom python functions, the SAM files with PCR duplicates removed were merged for each sample and used to generate digital counts of the tags.
Outlier counts were removed before generating a consensus count for each target. Outlier tags were identified as those with counts 90% below the mean of the probe group in at least 20% of the AOIs analyzed and removed them from the analysis. Subsequently, we removed tags from the analysis if they were flagged as outliers in at least 20% of the AOIs analyzed. This was done using the Rosner Test if there were at least 10 probes for the target (k = 0.2 * Number of Probes, alpha = 0.01), or the Grubbs test if there were less than 10 probes for the target. Probes flagged as outliers in less than 20% of the AOIs analyzed were only removed from the analysis for the AOIs in which they were flagged. Count reported for each target transcript were calculated as the geometric mean of the remaining probes. The counts for each target transcript in each AOI were then normalized to the count of all genes in that AOI.
The normalized in situ RNA measures were used to compute the overall expression of the malignant programs in each of the malignant areas of interest (AOIs), as described in the Gene sets Overall Expression section. To examine whether the core oncogenic program captures more in situ intratumoral variation than expected, we compared its intratumor variance in the malignant cells to that of 1,000 other S-compatible signatures. Each tumor was considered separately, resulting in a set of empirical p-value (one per tumor), which we merged using the Fisher combined probability test. For each malignant AOI we also computed the abundance of CD45+ cells in the respective ROI based on the CD45 staining, and examine the association between the OE of the core oncogenic program in the malignant AOIs and CD45+ cell abundance as well as the expression of TNF and IFNG in the adjacent CD45+ AOIs, which reside in the same ROI. This was done using a multilevel model that accounts for the sample identify, technical variability (i.e., log-transformed total number of reads detected in each AOI), and the epithelial vs. mesenchymal status (i.e., PanCK+ or PanCK−)
Identifying SS18-SSX targets
The fusion program consists of genes that were differentially expressed in the Aska or SYO1 cells with the SS18-SSX shRNA (shSSX) compared to those with control shRNA (shCt) 3 or 7 days post-infection. Gene that were previously reported16,17 to be bound by the SS18-SSX oncoprotein in at least two SyS cell lines were defined as direct SS18-SSX targets, and were used to stratify the SS18-SSX program to direct and indirect targets.
Mapping cancer-immune interactions
The association between the core oncogenic program in the malignant cells and the expression of different ligands/cytokines in the immune cells was examined using the multilevel mixed-effects regression model described above, using the scRNA-Seq data collected from SyS tumors. The dependent variable y was the OE of the core oncogenic program and the covariate x was the average expression of a certain ligand/cytokine in a specific type of immune cells (e.g., macrophages) that were profiled from the same tumor. The model also corrected for inter-patient dependencies using the patient-specific intercepts and for cell complexity (log(number of reads)). We restricted the analysis to ligands/cytokines that can physically bind to proteins expressed by the malignant cells78. The immune cells were either macrophages or CD8 T cells, as other immune cell types were not sufficiently represented in the data.
We used a similar approach to further stratify the program to its TNF/IFN-dependent and independent components. We repeated the same analysis described above, using each one of the genes in the core oncogenic program as the dependent variable. Genes which were associated with both TNF and IFN (P < 0.05, following Bonferroni correction) were considered as TNF/IFN-dependent, and genes which were not associated with both cytokines (P > 0.05) were considered as TNF/IFN-independent.
TNF and IFNγ impact on SyS cell cultures
SyS cell cultures were treated with TNF and IFNγ, separately and in combination (see In vitro IFN/TNF experiment section), and profiled with scRNA-Seq. Given this data, differentially expressed genes and gene sets were identified using mixed-effects regression models (Multilevel mixed-effects models section), with experiment-specific intercepts. The dependent variable y was the expression of a gene or the OE of a gene set. The model included three treatment covariates: only TNF, only IFN, and a combination of TNF and IFN. Another binary covariate denoted the duration of the treatment (1 for < 24h duration and 0 otherwise). The model corrected for differences between the different SyS cultures and experiments, and identified patterns that repeatedly appeared across the different experiments. The effect-size and significance of the combination covariate denotes the effect of the combination, and not the synergy between the two cytokines.
To examine if the combined treatment with TNF and IFNγ had synergistic effects, we used only the control cells and the cells treated for 4 days with one or two of the cytokines. This model also included 3 binary treatment covariates (TNF, IFN, and the combination), but this time cells that were treated with the combination were positive for all three treatment covariates. The effect-size and significance of the combination covariate hence denotes the synergistic effect of the combination.
Reconstructing regulatory networks
To reconstruct the gene regulatory network controlling the core oncogenic program we assembled a database of transcription factor (TF) to target mapping based on four sources: JASPAR49, HTRIdb46, MSigDB50, and TRRUST47, and augmented it with the direct SS18-SSX targets identified here (Supplementary Table 5) and TF-target pairs we identified in a cis-regulatory motif analysis of the core oncogenic program. Specifically, for the cis-regulatory analysis, we used RcisTarget48 (a R/Bioconductor implementation of icisTarget52 and iRegulon53) to identify cis-regulatory elements significantly overrepresented in a window of 500bp around the transcription start site of the core oncogenic genes (normalized enrichment score > 3.0) along with their cognate TFs.
We pruned the resulting network to include only core oncogenic program genes (and SS18-SSX) (i.e., all TFs and targets aside from SS18-SSX are program genes). An edge in the network between a TF and its target denotes that: (1) the TF is regulating the target according to at least one of the sources described above, and (2) there is an association between their expression levels in the scRNA-Seq data of SyS tumors. Edges are weighted 1 and −1 to reflect positive and negative associations. We used pageRank51 (with the R implementation as provided in igraph (https://igraph.org/r/)) as a measure of TF and target importance in the network. To compute TF importance, we first flipped the direction of the edges in the network, going from target to TFs. Consistent with the network weights, targets from the up- or down-regulated side of the network were considered induced or repressed, respectively. Likewise, TFs from the up- or down-regulated side of the network were considered activators and repressors, respectively.
Selectivity and synergy in drug experiments
To evaluate the impact of each drug on the expression of a certain program or gene in different cell lines (SYO1, HSSYII, or MSCs), we used a regression model with four binary treatment covariates: abemaciclib, TNF, panobinostat, and the combination of all three drugs. As in the case of TNF/IFN analysis, to examine the synergy of the combination, the cells treated with the combination were positive for all four treatment covariates. The model also included the number of reads detected in each cell (log-transformed) to control for technical variation. When examining the impact on the two SyS cell lines together, we used a mixed-effects model with a cell line specific intercept, to control for cell line specific baseline states. Drug selectivity was examined by using a mixed-effects model that accounts for all three cell lines and has another covariate to denote if the treated cells were SyS or not.
Co-culture experiments: prior cell culture and HLA typing
CME-1 cells (a generous gift from Dr. Armando Bartolazzi, Pathology Research Laboratory, Cancer Center Karolinska, Karolinska Hospital, Stockholm, Sweden) were maintained in vitro with RPMI-1640 medium supplemented with 10% FBS, 2 mM Glutamax, and 100 units/ml penicillin/streptomycin. CME-1 cells were HLA typed to confirm the HLA-A*02:01 genotype by performing Sanger sequencing on genomic DNA amplified with primers for forward: ACCGTCCAGAGGATGTATGG and reverse: CCAGGTAGGCTCTCAACTGC to ensure cells were at minimum, heterozygous.
Plasmid cloning and lentiviral vectors
CME-1 cells were transduced with the lentivirus in a serial dilution to express the NY-ESO-1 cDNA that encoded the relevant NY-ESO-1 protein (NYEP, processed peptide sequence SLLMWITQC) that is recognized by the 1G4 TCR when presented by HLA-A*02:01. Briefly, the CME-1 NY-ESO-1+ tumor line was generated by joining the cDNA encoding NY-ESO-1 (Accession number: NM_139250.2) and luciferase through a 2A ribosomal skip sequence and cloned into the pHAGE-MCS lentiviral vector under the control of the EF-1ɑ promoter. The purified amplicon from a single colony was ligated into the vector backbone using the NotI and XbaI restriction enzymes. The plasmid was transfected with packaging plasmids pCMV-dR8.91 and pCMV-VSV-G (Addgene #8454) into HEK293FT cells. This lentiviral vector also encoded the non-functional human nerve growth factor receptor (NGFR) extracellular domain down stream of an IRES to enable isolation of transduced cells. CME-1 NGFR+ cells were purified by FACS to > 99% purity to select a population with uniform NY-ESO-1 expression. CME-1 NGFR+ tumor cells were utilized between 2 and 5 passages for all experiments, and NGFR marker expression was periodically verified to be expressed by > 99% of the cells.
To generate the NY-ESO-1 [1G4] T cell receptor cells, the NY-ESO-1 TCR (1G4 TCR) cDNA (Robbins, Paul F, JI 2008) was synthesized, gel purified, and inserted into the pHAGE-MCS lentiviral vector backbone under the control of the EF-1ɑ promoter using NheI and ClaI restriction enzymes. The DNA sequence of the construct was confirmed for a single bacterial colony. Lentivirus was packaged as described above and stored at −80°C.
Isolation of primary human T cells for co-culture experiments
Primary human T cells were isolated from fresh leukophoresis blood collars provided by the Brigham and Women’s Hospital blood bank. Briefly, PBMCs were isolated using SepMate PBMC isolation tubes (Stemcell Technologies; Vancouver, Canada). T cells were isolated using the Human T cell isolation kit (EasySep, cat#17951) following the manufacturer’s instructions. Human T cells were maintained in vitro in RPMI-1640 medium supplemented with 9% fetal bovine serum (FBS), 1% human serum, 50 units/ml penicillin/streptomycin (Pen/Strep), 5 mM HEPES, 2 mM Glutamax, 5 mM non-essential amino acids, 5 mM sodium pyruvate, 50 μM β-mercaptoethanol, and 30 units/ml of recombinant human IL-2 (Peprotech; Rocky Hill, NJ). T cells were subsequently stimulated with human dynabeads (Life Technologies) at a ratio of 1:1 in the presence of 30 U/ml IL-2 for 3 days. The endogenous T cell receptor (TCR) was inactivated as described79. Briefly, the guide RNA directed against the T cell receptor ɑ constant (TRAC) locus was electroporated into the human T cells using an Amaxa 4-D nucleofector with the sequence 5′- TGTCTATAGGTCTTGGGAC-3’. The T cells were cultured in IL-2 (30 U/ml) for 3 days following electroporation. Next, the T cells were transduced with NY-ESO-1 TCR lentivirus (described above) and expanded with dynabeads (1:1 ratio) and IL-2 for 10 days. Prior to co-culture assay, the T cells were rested for 3 days by removing the magnetic dynabeads following the manufacturer’s instructions and cultured in IL-2.
Preparation of CME-1 cells and setup of co-culture
CME-1 cells were collected and 1.5 × 105 cells were seeded into each well of 6-well plates and cultured overnight. The next day (day 1), the media was replaced containing 100nM Abemaciclib or vehicle control (DMSO) and refreshed again on day 4. The media was next refreshed on day 5 containing 100nM Abemaciclib and 12.5nM Panobinostat. Vehicle control groups were refreshed with media containing DMSO.
The cells were harvested on day 6, counted, and the drugs washed off two times. The vehicle control or drugged CME-1 cells were seeded into 96-well flat bottom plates at 2.5e4 cells per well and allowed to attach to the plate for 3 hours. The NY-ESO-1 TCR+ T cells (described above) were seeded into each well of the 96-well plate and cultured for 72 hours.
Analysis of co-culture
The supernatants from the co-culture plates were collected and used for the detection of IL-2 and IFNγ by enzyme linked immunosorbent assays (ELISA) according to the manufacturer’s instructions (Biolegend). ELISAs were performed in triplicate in Costar high binding 96-well assay plates (Corning; Corning, NY). Each well was coated with capture antibody (based on lot specific antibody concentration) that was diluted in carbonate buffer with pH of 9.5 and incubated overnight at 4°C. Plates were washed with PBS + 0.05% Tween-20 and subsequently blocked with 1% BSA in PBS at room temperature for 1 hour. Following a 2-hour incubation with experimental cell culture supernatants and the manufacturer provided standard, plates were washed and incubated with the manufacturer’s biotinylated detection antibody for 1 hour at room temperature. The secondary antibody was next detected with streptavidin-Europium (Perkin-Elmer; Waltham, MA) and DELFIA Enhancement Solution (Perkin-Elmer). Next, the Europium fluorescence was analyzed using an EnVision multimode plate reader (Perkin-Elmer).
The cell pellets from the co-cultures were stained for analysis by flow cytometry (Extended Data Fig. 6g). Briefly, cells were washed with PBS to remove residual serum and incubated with Zombie UV (Biolegend; San Diega, CA) for 10 minutes at room temperature following the manufacturer’s instructions. Next, the cells were stained with antibodies against CD3-APC [HIT3a], CD25-BV785 [BC96], CD69-BV421 [FN50] in PBS containing 2% FBS for 15 minutes at 4°C, washed twice, and fixed using Fixation and Permeabilization solution (BD Bioscience; Franklin Lakes, NJ) for 20 minutes at 4°C. The samples were analyzed using an LSR Fortessa (BD Bioscience). Data were analyzed using FlowJo software (Tree Star). Antibodies were purchased from Biolegend. For additional information, see “Life Sciences Reporting Summary”.
Overview of statistical tests
Mixed-effects hierarchical models were used to examine changes in overall expression across cells from different samples and patients (see Multilevel mixed-effects models section). Hypergeometric enrichment tests were used to examine the enrichment of specific signatures with pre-annotated gene sets. Differential gene expression was tested using likelihood-ratio test74. Wilcoxon ranksum test and t-tests were used for pairwise comparisons when comparing protein or overall gene expression readouts, respectively.
Extended Data
Extended Data Fig. 1. Consistent classification of cells based on expression and genetic features.
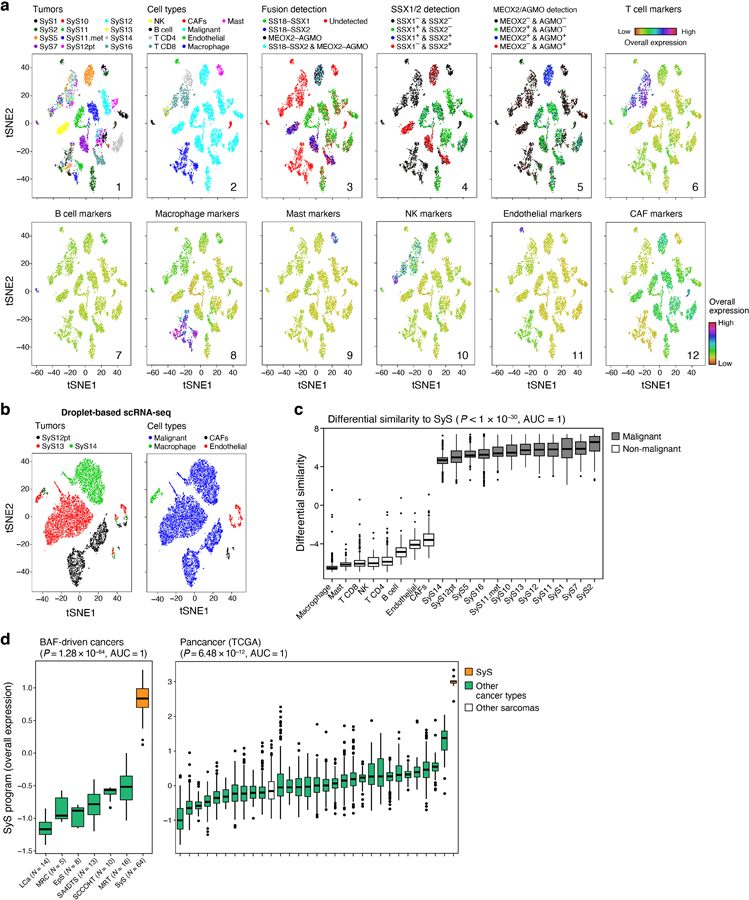
(a) Converging assignments of cell identity. tSNE of single-cell profiles (dots), colored by (1) tumor sample, (2) inferred cell type, (3) SS18-SSX1/2 and MEOX2-AGMO fusion detection, (4) SSX1/2 gene detection (mRNA level > 0), (5) MEOX2 and AGMO gene detection (mRNA level > 0), (6–12) overall expression of well-established cell type markers (Supplementary Table 2). (b) Droplet based scRNA-Seq of SyS. tSNE of single cells (dots), profiled with droplet-based scRNA-seq23, colored according to tumor sample (left) and inferred cell type (right). (c) Differential similarity to SyS compared to other sarcomas (Online Methods) distinguishes malignant (n = 4,371) from non-malignant (n = 2,375) cells. Differential similarity (y axis) to SyS shown for cells in each cell subset (x axis). (d) The SyS program distinguishes between SyS and non-SyS cancer types. Distribution of the SyS program Overall Expression (y axis) across BAF driven tumors (left, x axis) and in TCGA (right, x axis; n = 9,128; 253; and 10, for other, sarcoma, and SyS tumors). In (c-d) middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually; P-values: one-sided Wilcoxon-ranksum test; AUC: Area Under the receiver operating characteristic Curve.
Extended Data Fig. 2. Characterizing mesenchymal, epithelial and poorly differentiates malignant cells.
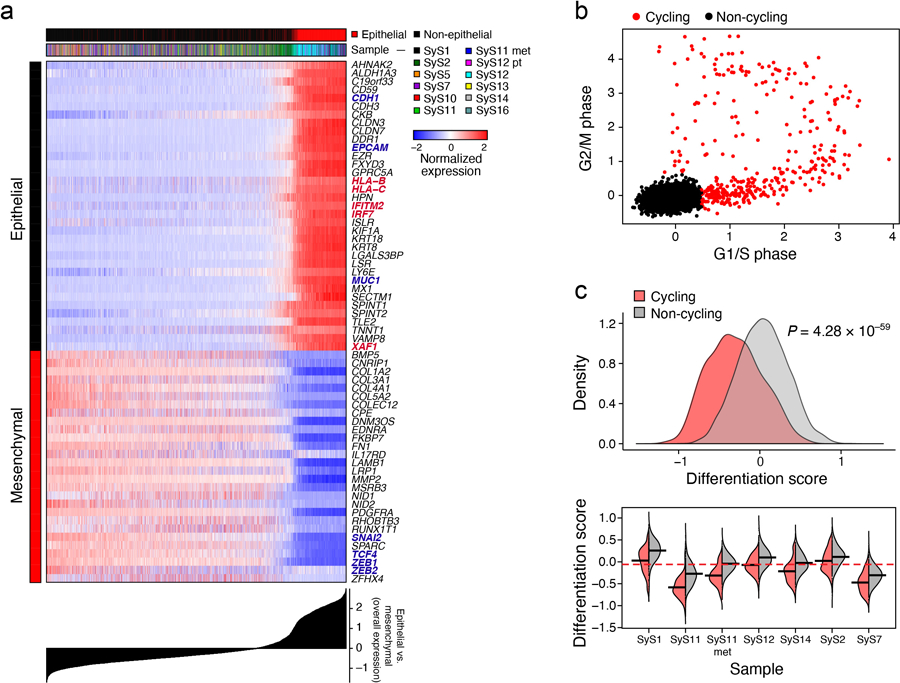
(a) Epithelial and mesenchymal program genes. The expression of the top epithelial and mesenchymal program genes (rows) across the malignant cells (columns), with cells sorted according to the difference in epithelial vs. mesenchymal OE scores (bottom plot). Topmost Color bar: epithelial vs. non-epithelial cell status, and sample. Canonical markers and immune-related genes are in red and blue, respectively. (b) Cell cycle signature. Overall Expression of the G2/M (y axis) and G1/S (x axis) phase signatures in each malignant cell, colored by their cycling status. (c) Cycling cells are less differentiated. The distribution of differentiation scores of cycling (red) and non-cycling (grey) malignant cells, across all tumors (top) and within each tumor (bottom; only tumors with at least 10 cycling cells are shown); p-value: mixed-effects test.
Extended Data Fig. 3. The core oncogenic program is detected using different approaches and datasets.
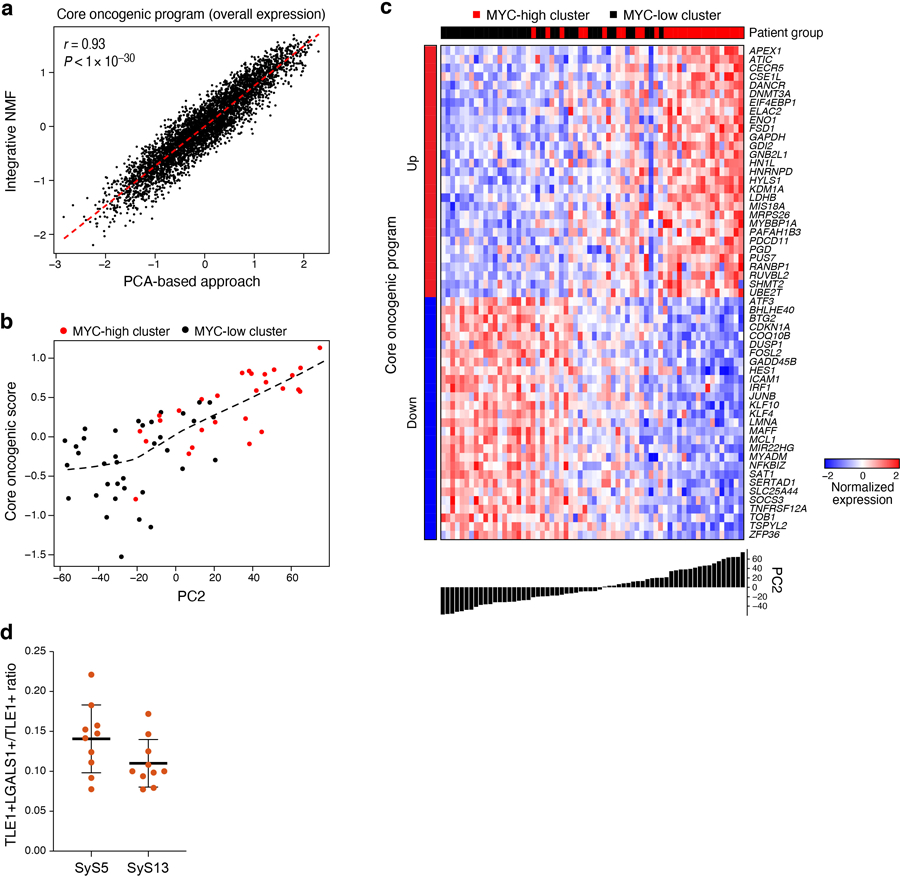
(a) Agreement between the core oncogenic program detected by a PCA and an iNMF approach36. Overall Expression (OE) of the core oncogenic program across malignant SyS cells, as identified in the PCA-based approach33 (x axis) and in the integrative-NMF approach36 (y axis) (Online Methods). (b-c) Program Overall Expression captures inter-tumor variation and the MYC-high cluster in 64 SyS tumors from an independent RNA-Seq cohort16. The tumors were previously classified into two transcriptionally distinct clusters16, denoted here as MYC-high and MYC-low. (b) For each tumor (dots), shown is the Overall Expression (OE) of the core oncogenic program (y axis) vs. the projection on the second Principle Component (PC2) of the data. (c) Normalized expression (centered log-transformed RPKM) of the core oncogenic program genes (columns) most correlated with PC2 across the tumors (columns). Tumors are sorted by their PC2 projection (bottom bar). (d) The fraction of TLE1+LGALS1+ cells out of TLE1+ ones based on ISH of tumors SyS5 and SyS13; Data are presented as mean values +/− SD, such that each dot corresponds to one high power field (HPF), with a total of 10 HPF per sample; TLE1 is a SyS cell marker and LGALS1 is a positive marker of the core oncogenic program.
Extended Data Fig. 4. Antitumor immunity and immune evasion in SyS.
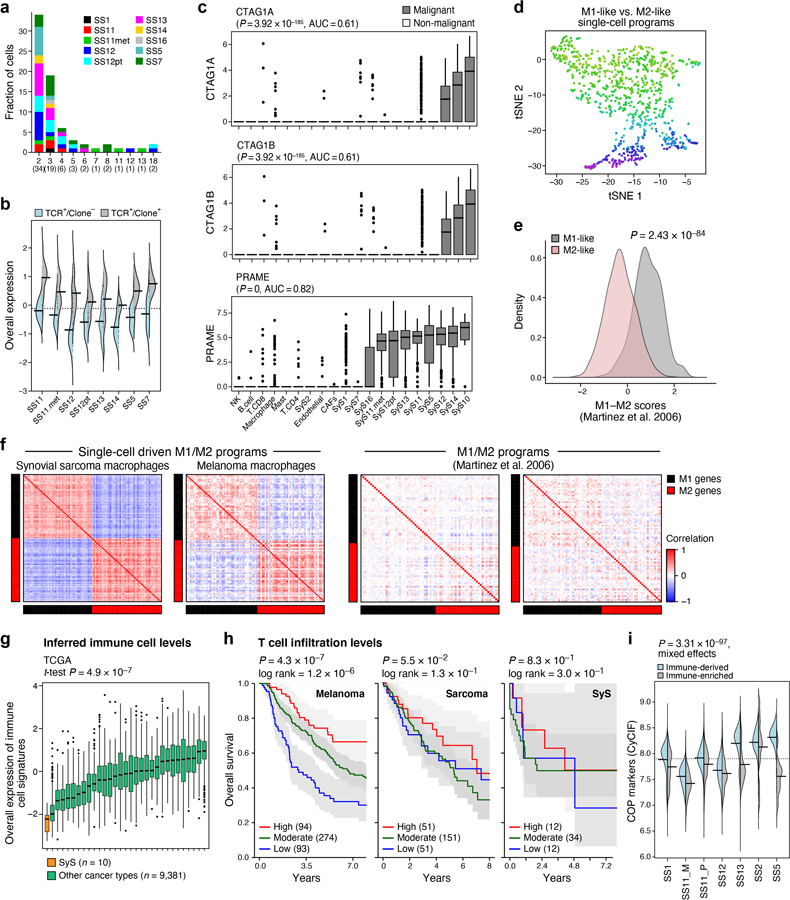
(a) CD8 T cell clones, stratified based on clone size (x axis) and tumor (color). (b) Overall expression of the T cell expansion program in CD8 T cells with a reconstructed TCR (TCR+), when stratified based on clonality (Clone+ and Clone−, denoting clone size greater or equal to 1, respectively). (c) The cancer testis antigens CTAG1A, CTAG1B (encoding for NY-ESO-1), and PRAME are exclusively expressed by SyS malignant (n = 4,371) cells compared to non-malignant ones (n = 2,375). Log-transformed TPM (y axis) in different cell subsets (x axis); p-values: one-sided Mann-Whitney test. (d) tSNE of macrophage profiles, colored by M1/M2 polarization scores, according to signatures defined here (Supplementary Table 4). (e) M1/M2 polarization scores (y axis) according to previously defined signatures42 in macrophages in our datasets partitioned to M1-like and M2-like subgroups (p-value: two-sided t-test). (f) Spearman correlation coefficient (color bar) between each pair of genes from M1 and M2 signatures defined here (top, Supplementary Table 4) or previously42 (bottom) across macrophages in SyS (left) and melanoma30 (right). (g) Overall Expression of the immune cell signatures (y axis, Online Methods) in SyS tumors (orange) and other cancer types (green); p-value: one-sided t-test. (c) and (g) middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually. (h) Prognostic value of T cell levels in different tumor types. Kaplan-Meier (KM) curves of survival in melanoma (left; TCGA), sarcoma (middle)21, and SyS (8) (right), stratified by high (top 25%, red), low (bottom 25%, blue), or intermediate (remainder, green) levels of inferred T cell infiltration levels; P: COX regression. (i) Protein expression (CyCIF) of core oncogenic program markers in immune-enriched and deprived niches.
Extended Data Fig. 5. Characterizing the transcriptional impact of SS18-SSX inhibition and tumor microenvironment cytokines on synovial sarcoma cells.
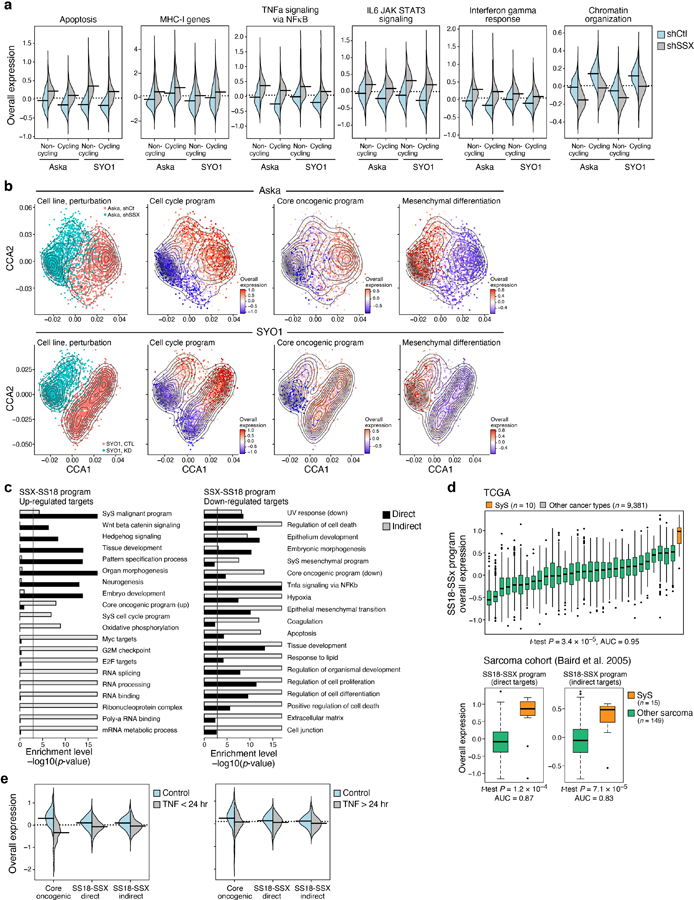
(a) The fusion KD induces innate immune programs. Distribution of Overall Expression scores (y axis) in the pathways most differentially expressed between SyS cells with SS18-SSX (shSSX, grey) vs. control (shCt, blue) shRNA, shown separately for non-cycling and cycling cells (x axis). (b) Co-embedding (using PCA and canonical correlation analyses80, Online Methods) of Aska (top) and SYO1 (bottom) cell profiles (dots), colored by: (1) perturbation; or the Overall Expression (colorbar) of the (2) cell cycle, (3) core oncogenic, or (4) mesenchymal differentiation31,32 programs. (c) Biological processes regulated in the SS18-SSX program. Gene sets (rows) most enriched (-log10(P-value), hypergeometric test, x axis) in induced (left) and repressed (right) SS18-SSX program genes, which are either direct (black bars) or indirect (grey bars) targets of SS18-SSX based on ChIP-Seq data16,17 and genetic perturbation. Vertical line denotes statistical significance following multiple hypotheses correction. (d) The SS18-SSX program distinguishes SyS from other cancer types and other sarcomas. Overall Expression of the SS18-SSX program (y axis) in either TCGA samples (n = 9,391, top), stratified by cancer types (x axis), or in another independent cohort of sarcoma tumors (n = 164, bottom) (48). Middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually. **P<0.01, ***P<1*10−3, ****P<1*10−4, one-sided t-test. (e) Repression of the core oncogenic and SS18-SSX programs by short term TNF treatment is not sustained long term. Distribution of Overall Expression scores (y axis) of the core oncogenic program and the direct and indirect SS18-SSX programs (x axis) in control cells (blue) and cells treated with TNF for 4–6 hours (left) or more than 24 hours (right).
Extended Data Fig. 6. HDAC and CDK4/6 inhibitors synergistically repress the core oncogenic program and induce cell autonomous immune responses.
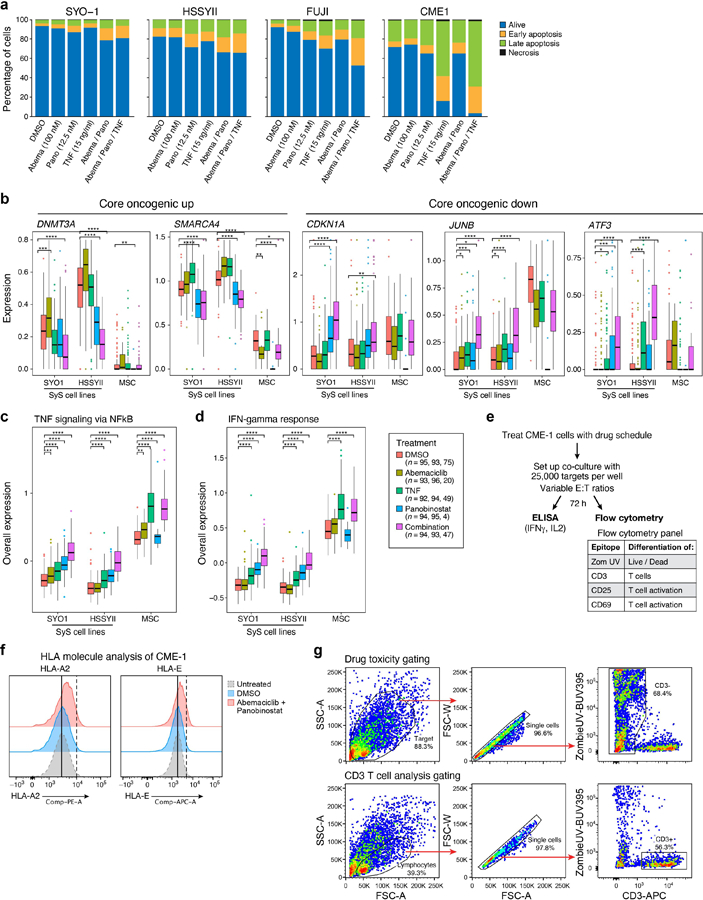
(a) The fraction of viable, necrotic, and apoptotic cells, showing four different SyS cell lines. (b-d) Distribution of the expression (y axis) of core oncogenic genes (b), as well as the Overall Expression of TNF (c) and IFN (d) signaling pathways in SyS cells and MSCs (x axis) under different treatments (color legend; n = no. of SYO1, HSSYII, and MSC cells). Middle line: median; box edges: 25th and 75th percentiles, whiskers: most extreme points that do not exceed ±IQR*1.5; further outliers are marked individually. **P<0.01, ***P<1*10−3, ****P<1*10−4, onesided t-test. (e) Workflow of the co-culture CME-1-T-cell experiment. (f) HLA-A2 and HLA-E protein levels on the cell surface of CME-1 cells under different treatments. (g) Standard, FSC vs. SSC gating was performed followed by strict FSC-width vs. FSC-area criteria to discriminate doublets and gate only single cells. Top: Singlets were gated upon the CD3- population to clearly identify the tumor cell population. The percentage of Zombie-UV+ cells were determined on the CD3- population. Bottom: Singlets were gated upon the Zombie-UV- (live) CD3+ population to clearly identify the viable T cell population.
Supplementary Material
The Supplemental Tables are provided in separate (Excel) files.
Supplementary Table 1. (A) Clinical characteristics of the patients and samples in the scRNA-seq cohort and (B) Quality measures of the scRNA-seq cohort.
Supplementary Table 2. Cell type signatures derived from the analysis of the SyS scRNA-seq cohort, as well as canonical cell type markers used for cell assignments.
Supplementary Table 3. Malignant programs: epithelial, mesenchymal, cell cycle and core oncogenic programs (A), and their enrichment with pre-defined gene sets50 (B).
Supplementary Table 4. The T cell expansion program (A), and M1-like and M2-like macrophage signatures (B).
Supplementary Table 5. The SS18-SSX program (A) and its enrichment with pre-defined gene sets50 (B).
Supplementary Table 6. TNF and IFNγ effects in synovial sarcoma: (A) The predicted TNF/IFNγ-dependent and independent components of the core oncogenic program according to the cell-cell interaction analyses (Online Methods); (B) differentially expressed genes following TNF and IFNγ treatment, and (C) their enrichment with pre-defined gene sets50.
ACKNOWLEDGMENTS
We thank Leslie Gaffney and Anna Hupalowska for help with artwork and Leslie Gaffney for help in figure preparation. We thank Kazuyuki Itoh, Norifumi Naka, and Satoshi Takenaka (Osaka University, Japan) for providing the Aska cell lines, and Akira Kawai (National Cancer Center Hospital, Japan) for providing the SYO1 cell line. We thank Maxwell Brown for help with CNA visualization. L.J.A. is a Chan Zuckerberg Biohub investigator and holds a Career Award at the Scientific Interface from BWF. L.J.A. was a fellow of the Eric and Wendy Schmidt postdoctoral program and a CRI Irvington Fellow supported by the CRI. Av.R. is an HHMI Investigator. Work was supported by the Klarman Cell Observatory, STARR cancer consortium, NCI grants 1U24CA180922, R33-CA202820, the Koch Institute NCI Support (core) grant P30-CA14051, Ludwig Centers at Harvard and MIT, AMRF and the Broad Institute (Av.R.). Work was also supported by grants from the Howard Goodman Fellowship at MGH (M.L.S.), the Merkin Institute Fellowship at the Broad Institute of MIT and Harvard (M.L.S.), R37CA245523 (M.L.S.), the Swiss National Science Foundation Sinergia grant CRSII5_177266 (M.L.S. and I.S.). Imaging CyCIF work was supported by a grant (CA225088) from the Center for Cancer Systems Pharmacology at Harvard Medical School (P.K.S.), K08CA222663 (B.I.), Burroughs Wellcome Fund Career Award for Medical Scientists (B.I.), Louis V. Gerstner, Jr. Scholars Program (B.I.), Velocity Fellow Program (B.I.). N.R. is supported by the Swiss National Science Foundation Professorship grant (PP00P3–157468/1 and PP00P3_183724), the Swiss Cancer League grant KFS-3973–08-2016, the Fond’Action Contre le Cancer grant and the FORCE grant. N.D.M. was supported by a postdoctoral fellowship from the American Cancer Society (PF-17–042-01-LIB) and the NIH education loan repayment program funded by the NCI (L30 CA231679–01). M.N.R. is supported by the Thomas and Diana Ryan MGH Research Scholar Award. Processed scRNA-Seq data are available at https://portals.broadinstitute.org/single_cell/study/synovial-sarcoma and GEO GSE131309 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131309). Raw scRNA-Seq data is deposited in the controlled access repository DUOS (https://duos.broadinstitute.org/#/hom) accession: DUOS-000123.
Footnotes
Editor summary: Single-cell transcriptional profiling of primary human synovial sarcoma tumors suggests that combinatorial treatment with HDAC and CDK4/6 inhibitors could enhance tumor immunogenicity.
Editor recognition statement: Saheli Sadanand was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
COMPETING INTERESTS STATEMENT
Av.R. is a founder of and equity holder in Celsius Therapeutics, an equity holder in Immunitas Therapeutics, and was a scientific advisory board member for ThermoFisher Scientific, Syros Pharmaceuticals and Neogene Therapeutics until August 1, 2020. From August 1, 2020, Av.R. is an employee of Genentech. M.L.S. is an equity holder, scientific co-founder and advisory board member of Immunitas Therapeutics. K.W.W. serves on the scientific advisory board of TCR2 Therapeutics, T-Scan Therapeutics, SQZ Biotech, Nextechinvest and receives sponsored research funding from Novartis. He is a co-founder of Immunitas Therapeutics. L.J.A, N.R., M.L.S. and Av.R. are co-inventors on US patent application filed by the Broad Institute relating to synovial sarcoma. O.R.-R. is an employee of Genentech and a co-inventor on patent applications filed by the Broad Institute for inventions relating to single cell genomics, such as in PCT/US2018/060860 and US provisional application no. 62/745,259. D.R.Z., N.O., and J.M.B. are employees of Nanostring which developed GeoMx. C.K. is the scientific founder, fiduciary Board of Directors member, Scientific Advisory Board member, shareholder, and consultant for Foghorn Therapeutics. E.C. reports support paid to his institution for the conduct of clinical trials from Amgen, Astra Zeneca, Novartis, Bayer, Merck, Exelixis, GSK, Adaptimmune, and Iterion. G.M.C. reports Advisory Board fees and support paid to his institution for the conduct of clinical trials from Agios, Epizyme, PharmaMar, Eisai; support paid to his institution for the conduct of clinical trials from Macrogenics, Boston Biomedical, Plexxicon, Merck KGaA / EMD Serono Research and Development Institute, CBA, SpringWorks Therapeutics, Bavarian-Nordic; compound for preclinical research and support paid to his institution for the conduct of clinical trials from Bayer. P.K.S. is a member of the SAB or Board of Directors of Applied Biomath, Glencoe Software and RareCyte and has equity in these companies. In the last five years the Sorger lab has received research funding from Novartis and Merck. The authors declare that these activities are not related to the research reported in this publication and have not influenced the conclusions in this manuscript. B.I. is a consultant for Merck and Volastra Therapeutics. N.W. is an equity holder and scientific advisory board member of Relay Therapeutics, a paid advisor to Eli Lilly and Co, and receives grant support from Puma Biotechnology. N.D.M. serves as a scientific advisor to Immunitas Therapeutics. C.N., M.E.S., H.R.W, M.J.M, B.H., B.I, A.V, G.B., L.C., A.R.Ri, L.C.B., J.M.G., C.C.L, R.M., L.N., S.M., J.C.M., C.G., O.C., J.E.B., A.S., M.S., M.S.C, D.L., S.G., G. P.N., I.C., T.N.N, M.M., E.C., I.L., S.C., A.B.H., J.T.M., I.S., and M.N.R declare no competing interests.
Data availability
Processed scRNA-seq data is available via the Gene Expression Omnibus (GEO), accession number GSE131309 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131309). Processed scRNA-seq data and interactive plots generated for this study are also provided through the Single Cell Portal at https://portals.broadinstitute.org/single_cell/study/synovial-sarcoma. Raw scRNA-Seq data are deposited in the controlled access repository DUOS (https://duos.broadinstitute.org/#/hom) accession: DUOS-000123. For additional information, see “Life Sciences Reporting Summary”.
REFERENCES
- 1.Trujillo JA, Sweis RF, Bao R & Luke JJ T Cell–Inflamed versus Non-T Cell–Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res 6, 990 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fridman WH, Pagès F, Sautès-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Pollack SM et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer 123, 3291–3304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen TO, Poulin NM & Ladanyi M Synovial sarcoma: recent discoveries as a roadmap to new avenues for therapy. Cancer Discov 5, 124–134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollack SM The potential of the CMB305 vaccine regimen to target NY-ESO-1 and improve outcomes for synovial sarcoma and myxoid/round cell liposarcoma patients. Expert Rev. Vaccines 17, 107–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iura K et al. Cancer-testis antigen expression in synovial sarcoma: NY-ESO-1, PRAME, MAGEA4, and MAGEA1. Hum. Pathol 61, 130–139 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Zhou Y et al. Evaluation of expression of cancer stem cell markers and fusion gene in synovial sarcoma: Insights into histogenesis and pathogenesis. Oncol. Rep 37, 3351–3360 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Naka N et al. Synovial sarcoma is a stem cell malignancy. Stem Cells Dayt. Ohio 28, 1119–1131 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Kadoch C & Crabtree GR Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan D et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miao D et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayyoub M et al. CD4+ T Cell Responses to SSX-4 in Melanoma Patients. J. Immunol 174, 5092 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Ayyoub M et al. Tumor-reactive, SSX-2-specific CD8+ T Cells Are Selectively Expanded during Immune Responses to Antigen-expressing Tumors in Melanoma Patients. Cancer Res 63, 5601 (2003). [PubMed] [Google Scholar]
- 14.Smith HA & McNeel DG The SSX Family of Cancer-Testis Antigens as Target Proteins for Tumor Therapy. Clin. Dev. Immunol 2010, 18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith HA & McNeel DG Vaccines targeting the cancer-testis antigen SSX-2 elicit HLA-A2 epitope-specific cytolytic T cells. J. Immunother. Hagerstown Md 1997 34, 569–580 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McBride MJ et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell (2018) doi: 10.1016/j.ccell.2018.05.002. [DOI] [PMC free article] [PubMed]
- 17.Banito A et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 33, 527–541.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su L et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21, 333–347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama R et al. Gene expression profiling of synovial sarcoma: distinct signature of poorly differentiated type. Am. J. Surg. Pathol 34, 1599–1607 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Lagarde P et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 31, 608–615 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 171, 950–965.e28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas B et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv (2017) doi: 10.1101/120295. [DOI]
- 25.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puram SV et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jerby-Arnon L et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taube JH et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. U. S. A 107, 15449–15454 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gröger CJ, Grubinger M, Waldhör T, Vierlinger K & Mikulits W Meta-Analysis of Gene Expression Signatures Defining the Epithelial to Mesenchymal Transition during Cancer Progression. PLOS ONE 7, e51136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fan J et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman MEJ & Girvan M Finding and evaluating community structure in networks. Phys. Rev. E 69, 026113 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Welch J et al. Integrative inference of brain cell similarities and differences from single-cell genomics. bioRxiv 459891 (2018) doi: 10.1101/459891. [DOI]
- 36.Yang Z & Michailidis G A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinforma. Oxf. Engl 32, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Przybyl J et al. Metastatic potential is determined early in synovial sarcoma development and reflected by tumor molecular features. Int. J. Biochem. Cell Biol 53, 505–513 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Stubbington MJT et al. T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods 13, 329–332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sade-Feldman M et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng C et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Böttcher JP et al. Functional classification of memory CD8+ T cells by CX3CR1 expression. Nat. Commun 6, 8306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez FO, Gordon S, Locati M & Mantovani A Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. Baltim. Md 1950 177, 7303–7311 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Merritt CR et al. High multiplex, digital spatial profiling of proteins and RNA in fixed tissue using genomic detection methods. bioRxiv 559021 (2019) doi: 10.1101/559021. [DOI] [PubMed]
- 44.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin J-R et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bovolenta LA, Acencio ML & Lemke N HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics 13, 405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han H et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci. Rep 5, 11432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan A et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46, D260–D266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brin S & Page L The Anatomy of a Large-Scale Hypertextual Web Search Engine in Seventh International World-Wide Web Conference (WWW 1998) (1998). [Google Scholar]
- 52.Herrmann C, Van de Sande B, Potier D & Aerts S i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res 40, e114 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janky R et al. iRegulon: From a Gene List to a Gene Regulatory Network Using Large Motif and Track Collections. PLOS Comput. Biol 10, e1003731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balogh KN, Templeton DJ & Cross JV Macrophage Migration Inhibitory Factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses. PLOS ONE 13, e0197702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones KB et al. Of mice and men: opportunities to use genetically engineered mouse models of synovial sarcoma for preclinical cancer therapeutic evaluation. Cancer Control J. Moffitt Cancer Cent 18, 196–203 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martín-Broto J, Moura DS & Van Tine BA Facts and Hopes in Immunotherapy of Soft-Tissue Sarcomas. Clin. Cancer Res 26, 5801 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robbins PF et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 29, 917–924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHOD REFERENCES
- 58.Fisher S et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol 12, R1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merritt CR et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol 38, 586–599 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cibulskis K et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinforma. Oxf. Engl 27, 2601–2602 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cibulskis K et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saunders CT et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinforma. Oxf. Engl 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Costello M et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 41, e67–e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Allen EM et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med 20, 682–688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cohen O et al. Abstract S1–01: Whole exome and transcriptome sequencing of resistant ER+ metastatic breast cancer. Cancer Res 77, S1–01 (2017). [Google Scholar]
- 70.Ramos AH et al. Oncotator: cancer variant annotation tool. Hum. Mutat 36, E2423–2429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olshen AB, Venkatraman ES, Lucito R & Wigler M Circular binary segmentation for the analysis of array-based DNA copy number data. Biostat. Oxf. Engl 5, 557–572 (2004). [DOI] [PubMed] [Google Scholar]
- 72.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Waltman L & Jan van Eck N A smart local moving algorithm for large-scale modularity-based community detection. Eur Phys J B 86, (2013). [Google Scholar]
- 74.McDavid A et al. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinforma. Oxf. Engl 29, 461–467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haghverdi L, Buettner F & Theis FJ Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinforma. Oxf. Engl 31, 2989–2998 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300 (1995). [Google Scholar]
- 78.Ramilowski JA et al. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun 6, 7866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Tables are provided in separate (Excel) files.
Supplementary Table 1. (A) Clinical characteristics of the patients and samples in the scRNA-seq cohort and (B) Quality measures of the scRNA-seq cohort.
Supplementary Table 2. Cell type signatures derived from the analysis of the SyS scRNA-seq cohort, as well as canonical cell type markers used for cell assignments.
Supplementary Table 3. Malignant programs: epithelial, mesenchymal, cell cycle and core oncogenic programs (A), and their enrichment with pre-defined gene sets50 (B).
Supplementary Table 4. The T cell expansion program (A), and M1-like and M2-like macrophage signatures (B).
Supplementary Table 5. The SS18-SSX program (A) and its enrichment with pre-defined gene sets50 (B).
Supplementary Table 6. TNF and IFNγ effects in synovial sarcoma: (A) The predicted TNF/IFNγ-dependent and independent components of the core oncogenic program according to the cell-cell interaction analyses (Online Methods); (B) differentially expressed genes following TNF and IFNγ treatment, and (C) their enrichment with pre-defined gene sets50.
Data Availability Statement
Processed scRNA-seq data is available via the Gene Expression Omnibus (GEO), accession number GSE131309 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131309). Processed scRNA-seq data and interactive plots generated for this study are also provided through the Single Cell Portal at https://portals.broadinstitute.org/single_cell/study/synovial-sarcoma. Raw scRNA-Seq data are deposited in the controlled access repository DUOS (https://duos.broadinstitute.org/#/hom) accession: DUOS-000123. For additional information, see “Life Sciences Reporting Summary”.