

Abstract
Introduction
The European Risk Management Plan (EU-RMP) is a proactive planning tool for identification, characterisation and management of important risks and missing information throughout the lifecycle of a medicinal product. Over the past 15 years the EU-RMP has been a part of the pharmacovigilance practice in Europe, but there are no published studies assessing impact of the growing experience and evolving regulatory framework on the content and focus of the EU-RMP.
Objectives
The objectives were to study the real-world impact of evolving pharmacovigilance guidelines on the proactive lifecycle management of important risks and missing information through EU-RMPs, and to further explore the impact of different resources on the management of the benefit-risk profile.
Methods
A retrospective study based on the review of 64 EU-RMPs dated between 01 January 2006 and 01 October 2020 for seven human medicinal products for which Boehringer Ingelheim holds the Marketing Authorisation in the European Union. Data on the timing and rational behind changes (i.e., inclusion, reclassification, removal) to the safety concerns (Important Identified Risks, Important Potential Risks, Missing Information) and associated additional Pharmacovigilance activities and/or Risk Minimisation measures were collected and assessed.
Results
The analysed EU-RMPs included a total of 197 safety concerns, 129 of which were removed and 19 were reclassified during the observation period. The implementation of the Guidelines on Good Pharmacovigilance Practices Module V in 2012 and Revision 2 in 2017 resulted in a noticeable decrease in the number of safety concerns. Clinical trial, non-clinical and routine post-marketing data were common sources that influenced the safety concern dynamics, and results from dedicated post-authorisation studies lead to the removal of 21 important risks and missing information. Many safety concerns were related to pharmacological class effect (n = 55) and target population characteristics (n = 37).
Conclusions
This study demonstrated that the growing knowledge regarding benefit-risk of approved products and the introduction of new or revised regulatory guidelines influenced the EU-RMP lifecycle of safety concerns, and moreover, the results emphasise that exchange of knowledge about the pharmacological class and target population between stakeholders are important for keeping an up-to-date understanding of a medicinal product’s safety profile. The aim of improving the efficiency of risk management has leveraged the accumulation of knowledge leading to revision of regulatory guidelines and increasingly, proactive Risk Management Plans focused on safety concerns that are important for patients and public health.
Key Points
| Changing regulatory definitions of safety concerns has had a key role in determining the focus of the Risk Management Plans. |
| The implementation of regulatory guidelines has led to consistent trends/patterns across different therapeutic areas. |
Introduction
The European Risk Management Plan (EU-RMP) became mandatory for all innovative medicinal products or upon request by the European Medicine Agency (EMA) in 2005 [1, 2] and was first described in a guideline [3, 4] that was later incorporated into Volume 9A of The Rules Governing Medicinal Products in the European Union—Guidelines on Pharmacovigilance for Medicinal Products for Human Use (Volume 9A) in 2008 [5]. Since 2012 and the implementation of the Guidelines on Good Pharmacovigilance Practices (GVP), the EU-RMP is an obligatory part of the market authorisation application dossier in the EU for any medicinal products [6, 7].
In this context, it is acknowledged that medicinal products are authorised in the EU on the basis that the benefit-risk profile is judged to be favourable for the target population. However, it is recognised that at the time of authorisation the information on the safety profile is limited and that some risks may only be identified during the post-authorisation phase. Consequently, to ensure the safe use of the medicinal products throughout the lifecycle of a marketed product, Marketing Authorisation Holders (MAHs) are obliged to have systems in place that facilitate risk identification, characterisation, and minimisation. MAHs are required to submit the description of the Risk Management System (RMS) in the form of a Risk Management Plan (RMP) for all medicinal products authorised in the EU [7]. The role of the RMP as a tool for risk management has evolved in parallel with updated regulatory guidelines reflecting the increased experience among stakeholders that is within the competent authorities in the EU as well as the MAHs.
The RMP is primarily a proactive planning tool with focus on the management of important risks throughout the product’s lifecycle with the aim to ensure that the benefits of a particular medicinal product exceed the risks by the greatest achievable margin. In principle, the EU-RMP consists of three parts: (i) The safety specification summarises what the MAH knows about the product safety profile at a defined time point with focus on safety concerns (SCs) (Box 1) that require further characterisation or risk minimisation activities. (ii) The pharmacovigilance (PV) plan that describes what activities a MAH needs to perform in order to increase knowledge of the SCs associated with the medicine. (iii) The risk minimisation (RM) plan presents what a MAH needs to do to minimise the risks associated with the medicinal product.
Routine PV activities (e.g., the collection, assessment and reporting of spontaneous reports of adverse reactions) [6] and routine RM measures (e.g., information on the product labelling and the legal status of a medicine) are mandatory for all medicinal products. For some safety topics it may be necessary to perform additional PV activities [8] (e.g., post-authorisation safety studies [PASS]) or additional RM measures [9] (e.g., educational material for healthcare professionals and patients) to balance the benefits versus the risks.
The classification of important risks relevant for inclusion in the RMP has undergone re-evaluation with time from being an adverse event, including topics from the Undesirable Effects section of the Summary of Product Characteristics (SmPC) from 2005 to 2012 [3], to an untoward occurrence that is likely to be included in the contraindications or warnings and precautions section of the product information (from 2012 to 2017) [7]. The risk classification was further revised with the introduction of the GVP Module V Revision 2 in March 2017 to SCs having an important clinical outcome and usually warranting additional PV activities and/or RM measures as a part of the RMP [10]. Revision 2 also included details on how to manage changes (i.e., inclusion, reclassification or removal) to SCs listed in the RMP as the safety profile is further refined throughout the product’s lifecycle. According to GVP Revision 2 principles (Box 2), the number of SCs in the EU-RMP is expected to decrease over time as risks become well-characterised and effectively managed via routine clinical practice, and neither additional PV activities nor RM measures continue to be warranted. Furthermore, as learnt from the Harmonisation of RMP Project (HaRP), and in agreement with Revision 2, mature products may have RMPs with “empty” important risks and missing information (MI) sections since all proposed SCs are well-managed with routine activities or measures [11]. There are also recent examples of approved EU-RMPs from the initial filing that have no safety concerns, showing that the updated guidelines have led to more precise considerations among industry stakeholders during initial EU-RMP compilation whether there are safety concerns relevant for patient and public health [12].
In general, the regulatory changes were introduced with the intention of making the EU-RMP more fit for its purpose as a proactive planning tool for the management of important risks. However, there are no published data that have evaluated the impact of new or revised regulatory requirements and their influence on lifecycle dynamics of SCs in the EU-RMPs in a real-world setting. Thus, it is not evident to what extent different stakeholders have adapted their approaches to comply with the regulatory changes. Did the number of SCs change following the implementation of new or revised regulation, e.g., was there a decrease in the number of SCs for mature products after 2017? Has the willingness among stakeholders to remove resolved SCs shifted along with the evolving regulations and, if so, was the removal of SCs primary prompted by Health Authorities (HAs) or proactively initiated by the MAHs? Furthermore, the EU-RMP is a dynamic document requiring a revision at any time point during post-authorisation with the learning about the safety profile relevant to the benefit risk. However, it is also not clear to what extent knowledge gained from various sources, such as routine post-marketing surveillance or dedicated post-authorisation studies, have contributed to the gradually increased understanding of the product safety profile.
Therefore, we reviewed the lifecycle of EU-RMP safety concerns in the context of the evolving European PV regulatory landscape for a selection of human medicinal products for which Boehringer Ingelheim holds the Marketing Authorisation (MA) in the EU with the aim to continue to ensure that the EU-RMPs are fit for purpose to support the management of the benefit-risk profile for medicinal products.
Box 1. Definitions of EU-RMP safety concerns extracted from the Good Pharmacovigilance Practices (GVP) Module V* [6] and the GVP Module V Revision 2** [10].
|
Identified risk: “An untoward occurrence for which there is sufficient evidence of an association with the medicinal product of interest” * Potential risk: “An untoward occurrence for which there is some basis for suspicion of an association with the medicinal product of interest but where this association has not been confirmed” * Missing information: “Gaps in knowledge about the safety for a medicinal product for certain anticipated utilisations or for use in a particular patient population, which could be clinically significant” *, ** Important risk: “An identified risk or potential risk that could have an impact on the risk-benefit balance of the product or have implications for public health” *, ** *GVP Module V **GVP Module V Revision 2 |
Box 2. Extracted statements on important risks to be included in the Risk Management Plan (RMP) from the Good Pharmacovigilance Practices (GVP) Module V Revision 2 Guideline [10].
|
(1) “ …. where the risk is fully characterised and appropriately managed, important identified risks may be removed from the safety specification…” (2) “…From the identified risks and potential risks …. the RMP should address only the risks that are undesirable clinical outcomes...” (3) “…An important identified risk to be included in the RMP would usually warrant further evaluation as a part of the pharmacovigilance plan, or risk minimisation activities” (4) “…Important potential risks can be removed from the RMP when accumulating scientific and clinical data do not support the initial supposition or when the impact of the potential risk is less than anticipated resulting in the potential risk not being considered important” (5) “Important potential risks included in the RMP would usually require further evaluation as part of the pharmacovigilance plan” (6) “...the classification as missing information might not be appropriate anymore once new data become available…” (7) “...the RMP should focus on those risks that are relevant for the risk management activities for the authorised product” |
Methods
Study Design
A retrospective study that was based on the qualitative review of data extracted from EU-RMPs approved between 01 January 2006 and 01 October 2020 for seven human medicinal products (Table 1) authorised in the EU, by Centrally Authorised Procedure (CAP) or Mutual Recognition Procedure (MRP), for which Boehringer Ingelheim (hereafter referred to as the MAH) holds the Market Authorisation (MA) in the EU. These products either had an initial EU-RMP submitted to the European Medicines Agency (EMA), or for MRP to a National Competent Agency (NCA), as a part of the Market Authorisation Application (MAA) dossier (Products C, D, E, F and G) or in the post-authorisation phase (Product A and Product B). Other product selection requirement was that the product should have received a favourable opinion from the Committee for Medicinal Products for Human Use (CHMP) on the renewal of the initial MA. The purpose of the study was to obtain an overview of patterns across different therapeutic areas, hence an upper limit of ≤ 2 products from the same therapeutic area (as defined by Medical Subjects Headings [MeSH]) was applied to ensure a diverse representation). The RMP versions were classified according to the date of the report, which meant that in this study ongoing variations were classified according to the legislation applicable as of the date of the report.
Table 1.
Medicinal Products and EU-RMPs included in the study
| Product name | Rare disease | Type of product | Year of MA valid in the EU for first authorised indication and type of procedure | Year for the renewal of the initial MA in the EU | Initial EU-RMP: year of report | Total number of HA-approved EU-RMP versions |
|---|---|---|---|---|---|---|
| Product A | No | NCE | 2002 (MRP) | Prior to the study timeframe | 2007 | 9 |
| Product B | No | NCE | 1997 (CAP) | Prior to the study timeframe | 2006 | 6a |
| Product C | No | NCE | 2011 (CAP) | 2016 | 2010 | 10 |
| Product D | No | NCE | 2014 (CAP) | 2018 | 2013 | 11 |
| Product E | No | NCE | 2013 (CAP) | 2018 | 2012 | 5 |
| Product F | No | NCE | 2014 (CAP) | 2019 | 2013 | 6 |
| Product G | Yes | NCE | 2015 (CAP) | 2019 | 2015 | 10 |
CAP Centrally Authorised Procedure, EU European Union, MA Market Authorisation, MRP Mutual Recognition Procedure, NCA National Competent Authority, NCE New Chemical Entity
aProduct B EU-RMP versions 2.0 to 4.0 could not be electronically retrieved and thus were excluded from this study
Data Sources and Collection
For each analysed medicinal product, the first EU-RMP version submitted to the EMA or an NCA (hereafter both will be referred to as Health Authority [HA]), along with all subsequently HA-approved EU-RMP versions up until study Data Lock Point (DLP) (01 October 2020), were retrieved from an internal business intelligence (BI) document management system. All data on the product-specific SCs throughout the period covered by this study were extracted from the EU-RMPs as described in Table 2.
Table 2.
Data extraction methodology for different EU-RMP formats
| Type of information | Location of the information in EU-RMP format Aa | Location of the information in EU-RMP format Bb | Location of the information in EU-RMP format Cc |
|---|---|---|---|
| Safety concerns (IIR, IPR, MI) | RMP part II, module SVIII “Summary of the safety concerns” | RMP Section 1. Safety Specification: Subsection 1.10 “Summary – Ongoing safety concerns” | RMP Section 2 Safety Specification Subsections: “Summary on Pre-clinical Safety” and “Summary of Clinical Safety” |
| Sources associated with the inclusion, removal and reclassification of the safety concerns | RMP part II, module SVII “Identified and potential risks” |
RMP Section 1 Safety Specification: Subsection 1.5.1 “Newly identified safety concerns” and Subsection 1.5.2 “Details of important identified and potential risks” |
RMP Section 2 Safety Specification Sub-sections: “Summary on Pre-clinical Safety” and “Summary of Clinical Safety” |
| Routine and additional PV activities | RMP part III “Pharmacovigilance plan (including post-authorisation safety studies)” | RMP Section 2 “Pharmacovigilance Plan” | RMP Section 3 “Pharmacovigilance plan” |
| Routine and additional RM measures | V “Risk minimisation measures (including evaluation of the effectiveness of risk minimisation activities)” | RMP Section 5. “Risk Minimisation Plan” | RMP Section 5 “The Risk Minimisation Action Plan” |
EU-RMP European Risk Management Plan, IIR important identified risk, IPR important potential risk, MI missing information, PV pharmacovigilance, RM risk minimisation
aEU-RMPs dated from July 2012 to October 2020 following the guidance on the format of the risk management plan (RMP) in the EU [6, 10, 18]
bEU-RMPs dated between 2005 to 2012 following to the “Guideline on Risk Management Systems for Medicinal Products for Human Use” [3] and Annex C: TEMPLATE FOR EU RISK MANAGEMENT PLAN (EU-RMP) [3, 4]
c“EMEA/192632/2006, Annex C: EU-RMP Template” [4] was not available at the time of submission of Product B EU-RMP version 1.0 (February 2006) and hence, Product B EU-RMP version 1.0 was prepared in a different format
Data Analysis
All figures and graphs were created using Microsoft Excel Office version 16. For Products A and B, single-product linear regressions (Fig. 1a–b) were plotted using the absolute numbers of different SCs (Important Identified Risk [IIR], Important Potential Risk [IPR], Important1 Missing Information [MI], Total = IIR + IPR + MI) per EU-RMP. Plotting using the absolute numbers better visualises the dynamic development and trends during the first phase of EU-RMPs (2005–2012).
Fig. 1.

Evolution of Product A and Product B EU-RMP SCs between 2006 and 2020 The y-axis shows the absolute number of product specific SCs (i.e., IIR, IPR, MI, total = IIR+IPR+MI) included in the first submitted EU-RMP and all subsequently HA-approved EU-RMPs for the indicated (Product A or Product B) medicinal product. The x-axis shows the EU-RMP date of report (Month/Year) and the corresponding EU-RMP version. Vertical black dotted lines illustrate the implementation date of a new or revised regulatory guidelines on Risk Management Systems. a. Product A: EU-RMP SCs from 2007 to 2020. b. Product B: EU-RMP SCs from 2006 to 2020. EU-RMP European Risk Management Plan, HA health authority, IIR important identified risk, IPR important potential risk, MI missing information, PV pharmacovigilance, RM risk minimisation, SCs safety concerns
The combined absolute number of SCs for Products C to G that were included and/or removed during different periods of the product lifecycle was visualised using a vertical, clustered bar chart (Fig. 2). In Fig. 2, each individual SC was counted only once as per product EU-RMP lifecycle even if it was reclassified between different categories (IIR, IPR or MI) of SCs.
Fig. 2.
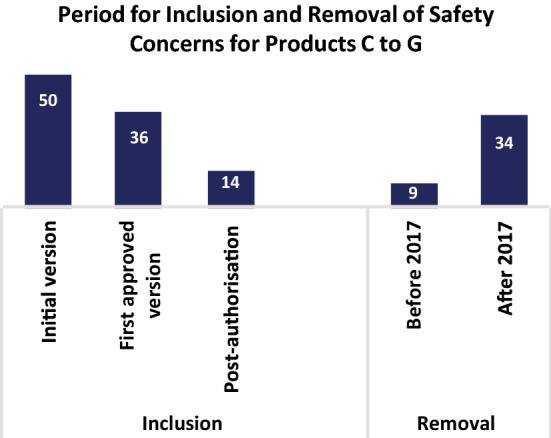
The number of EU-RMP SCs included or removed during various stages of the product lifecycle. Total number of SCs (i.e., IIR, IPR, MI for Product C, Product D, Product E, Product F and Product G that were included or removed as SCs during the indicated stages of the product lifecycles. The initial version corresponds to the number of SCs included in the initial product specific EU-RMPs submitted to HA: first approved corresponds to the number of new SCs that were included in the first HA approved but not in the initially submitted EU-RMPs; Post-authorisation corresponds to the number of new SCs that were added to the EU-RMPs in the post-marketing phase. Before 2017 corresponds to the number of SCs that were removed from the EU-RMPs prior to the implementation of the GVP Module V Revision 2; After 2017 corresponds to the number of SCs removed after the implementation of the GVP Module V Revision 2. EU-RMP European Risk Management Plan, GVP Guidelines on Good Pharmacovigilance Practices, HA health authority, IIR important identified risk, IPR important potential risk, MI missing information, PV pharmacovigilance, RM risk minimisation, SCs safety concerns
The EU-RMP lifecycle development for Products C to G, were co-plotted to allow for an effective comparison and identification of trends. In this case, to avoid that patterns gets washed out by the absolute numeric differences between Product C to G, the absolute numbers of SCs (IIR, IPR, MI, Total = IIR + IPR + MI) per EU-RMP were scaled between 0 to 1 using min-max normalisation (Eq. 1) before being co-plotted using linear regression (Fig. 3a–d). The number of SCs in the plotted linear regressions represent the cumulative number of product-specific SCs classified as either an IIR, IPR or MI at the indicated time point.
Fig. 3.

Evolution of the number of SCs for five medicinal products in the context of the introduction of GVP Module V Revision 2. The y-axis shows the min-max normalised (0 [min] to 1 [max]) number of product-specific SCs (i.e., IIR, IPR, MI, Total = IIR+IPR+MI) included in the first submitted EU-RMPs and in all subsequently approved EU-RMPs for the indicated medicinal products (Products C, D, E, F and G) between 2010 to 2020. The x-axis shows the period (Month/Year) for the reviewed EU-RMPs date of reports. The black dotted square illustrates the operational period for the GVP Module V Revision 2 Guideline on Risk Management Systems. a Total (IIR + IPR + MI) number of EU-RMP SCs per product over time; b number of EU-RMP IPRs per product over time; c number of EU-RMP MI topics per product over time; d number of EU-RMP IIRs per product over time. EU-RMP European Risk Management Plan, GVP Guidelines on Good Pharmacovigilance Practices, HA health authority, IIR important identified risk, IPR important potential risk, MI missing information, PV pharmacovigilance, RM risk minimisation, SCs safety concerns
The combined absolute number of IIR, IPR and MI topics for Products A to G that were associated with aPV activities or aRM measures was visualised using a vertical, clustered bar chart (Fig. 4a, b). In Fig. 4a, b, each individual SC was counted only once as per its initial SC category (IIR, IPR, MI) even if the SC was reclassified to another category.
Fig. 4.
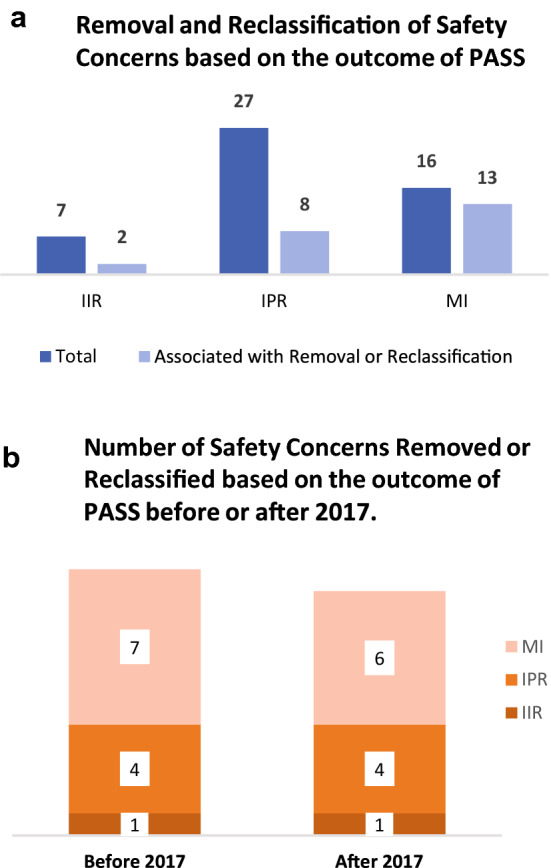
The influence of PASS on the removal and reclassification of SCs. Figure shows the absolute number of European Risk Management Plans (EU-RMPs) SCs (i.e., IIR, IPR and MI) with additional (a) Pharmacovigilance (PV) activities, such as PASS, for Products A, B, C, D, E, F, and G any time point between 2006 and 2020, and the number of SCs (i.e., IIR, IPR and MI) whose removal or reclassification were associated with results from an aPV activity. a The absolute number of SCs that were removed or reclassified based on results from at least one aPV activity compared to the total number of SCs with at least one aPV activity b The absolute number of SCs that were removed or reclassified based on results from completed aPV activities before or after the implementation of GVP Module V Revision 2 in 2017, respectively. aPV additional pharmacovigilance, EU-RMP European Risk Management Plan, GVP Guidelines on Good Pharmacovigilance Practices, HA health authority, IIR important identified risk, IPR important potential risk, MI missing information, PASS Post-authorisation Safety Studies, PV pharmacovigilance, RM risk minimisation, SCs safety concerns
In Fig. 5a–c, the current absolute number of unique IIR, IPR and MI for each analysed medicinal product in the latest HA-approved EU-RMP per product (A–G) as of study DLP (right y-axis) was co-visualised as a bar plot together with the number of SCs with PASS as a dot plot (left y-axis).
| 1 |
Fig. 5.

EU-RMPs SCs with ongoing or planned aPV activities. Data were obtained from the latest HA-approved EU-RMPs as of 01 October 2020 for each of the seven studied medicinal products. a The left y-axis shows the absolute number of SCs included in the EU-RMP for per indicated medicinal product (x-axis) and the sum of the SCs (i.e., IIR, IPR, MI) for all 7 studied medicinal products. Above each column are dots (right y-axis) representing the total number of SCs per product with ongoing or planned additional (a) PV activities or (a) RM measures (red); b The percentage of the total number of EU-RMP SCs with ongoing or planned aPV activities or aRM measures (YES) versus SCs having no ongoing or planned additional PV or RM activities (NO); c The absolute number of EU-RMP SCs per risk category (i.e., IIR, IPR, MI) with ongoing or planned aPV activities. aPV additional pharmacovigilance, EU-RMP European Risk Management Plan, GVP Guidelines on Good Pharmacovigilance Practices, HA health authority, IIR important identified risk, IPR important potential risk, MI missing information, PASS Post-authorisation Safety Studies, PV pharmacovigilance, RM risk management, SCs safety concerns
zi = rescaled value to between 1 to 0, xi = original value (the absolute number of safety concerns), min(x) = minimum value of x, max(x) = maximum value of x.
Based on the information provided in the EU-RMP Safety Specification, sources of information and reasons that had an impact on the evolution of EU-RMP SCs, in terms of their inclusion, removal or reclassification, were categorised into 7 classes (Box 3). In this analysis, each specific SC and related action (inclusion, removal, or reclassification) could be associated with one or multiple classes. The IIRs section in early EU-RMPs (versions 1.0 to 4.0) for Product A reflected all undesirable effects included in section 4.8 of the EU SmPC, with no further details given on the rationale for their respective inclusion, re-classification and/or removal. On this basis, the IIRs for Product A were excluded from the analysis of sources and reasons associated with the evolution of SCs (Fig. 6a–f). To visualise the proportion of SCs whose inclusion, removal and reclassification in the EU-RMP was based on different categories of information that formed the rationale for the inclusion, removal and reclassification of in the EU-RMP, the total absolute number of individual IIR, IPR and MI topics for Products A to G per category were plotted using bar charts (Fig. 6a–c). Pie charts (Fig. 6d–f) were used to visualise the percentage of SCs associated with certain indicated characteristics.
Fig. 6.

Types of sources that resulted in the inclusion, removal or reclassification of SCs listed in EU-RMP. Data on the types of sources (categories defined in Box 3) influencing the lifecycle evolution of SCs were obtained from EU-RMPs for all seven medicinal (excluding the IIRs for Product B). Multiple types of sources could be associated with the inclusion, removal or reclassification of each unique SC. a The x-axis shows the total number of unique SCs included in the reviewed EU-RMPs at any point in time, and the number of SCs whose inclusion in the EU-RMP was associated with evidence from the specified
source category (y-axis); b The x-axis shows the total number of SCs that were removed from the EU-RMPs during the studied period, and the number of SCs whose removal from the EU-RMP was based on data from the indicated source category (y-axis); c The x-axis shows the total number of reclassified safety concerns, and the number of SCs whose reclassification from IPR to IIR or from MI to IPR in the EU-RMP was based on information from the indicated category (y-axis); d The proportion (%) of reclassified SCs associated with (YES) or not with (NO) Pharmacological Class Effect and/or Target Population characteristics; e shows the proportion (%) of removed SCs triggered by a HA versus initiated by BI; f shows the proportion (%) of SCs associated with Pharmacological Class Effect and/or Target Population Characteristics that was added in response to feedback from regulators during the market authorisation application process. aPV additional pharmacovigilance, BI Boehringer Ingelheim, EU-RMP European Risk Management Plan, GVP Guidelines on Good Pharmacovigilance Practices, HA health authority, IIR important identified risk, IPR important potential risk, MI missing information, PASS Post-authorisation Safety Studies, PV pharmacovigilance, RM risk management, SCs safety concerns
Box 3. Sources of information associated with the evolution (inclusion, removal or reclassification) of European Risk Management Plan (EU-RMP) safety concerns*.
|
Clinical trial data. Evidence generated from clinical trials with the medicinal product during development and post-authorisation (Phase I–IV) Non-clinical trial data. Data from non-clinical studies that constitutes an important safety finding, e.g., toxicity, safety pharmacology or other toxicity-related information Post-marketing data. Evidence generated from post-authorisation experience, e.g., Individual Case Safety Report (ICSR) and literature (excluding data from clinical trials and additional pharmacovigilance activities) Regulation. Introduction of new or revised European Pharmacovigilance Legislations, Regulations and/or Regulatory Guidelines Class effect. A risk which is common to the pharmacological class. Target population. Risk factors associated with disease population characteristics such as comorbidities and concomitant medications. PASS. Post-authorisation safety study irrespective of the design (interventional/non-interventional) included in the EU-RMP as an additional pharmacovigilance activity linked to a safety concern in the RMP *Based on the information provided in the EU-RMP Section: Safety Specification |
Results
The 64 reviewed EU-RMPs included a total of 197 unique SCs (including 70 IIRs, 78 IPRs, and 49 MI Topics) 129 of which were removed from their respective EU-RMPs, while 19 SCs were reclassified either from an IPR to an IIR (n = 15) or from MI to an IPR (n = 4) during the period covered by this review.
Of the seven medicinal products, five had an EU-RMP included in the MAA dossier in line with the applicable regulatory requirements, and two had the initial EU-RMP submitted in the post-authorisation phase. The initial EU-RMPs for the each included medicinal product were dated between 2006 to 2014.
Evolving Regulations have Resulted in Variance in Risk Presentation for Mature Products
To investigate whether the introduction of regulations have affected the number of EU-RMP SCs, we first studied the evolution of the SCs for the two products with the longest Market Authorisation (MA), Products A and B whose first EU-RMPs were submitted post-authorisation (Fig. 1a, b). Product A was authorised in European Member States via MRP in 2002 and its first EU-RMP was submitted in 2007, having a total of nine HA-approved EU-RMPs as of 01 October 2020 (Table 1). From the first submitted EU-RMP in 2007 to the EU-RMP version 4.0 in 2011, the number of IIRs ranged between 30 and 39 (Fig. 1a) reflecting all undesirable effects included in Section 4.8 of the Product A’s EU SmPC effective at the specified time points. The introduction of the GVP Module V in 2012 resulted in the demotion of all the IIRs for Product A as they did not fulfil the criteria as per this guideline [6]. After an initial decrease from 15 to 11 between 2007 and 2008, IPRs for Product A showed a stable development from 2008 until 2019. After which seven IPRs were removed in accordance with the principles outlined in GVP Module V Revision 2 [10].
Product B was authorised for use via CAP in 1997 and the first EU-RMP was submitted in 2006 in conjunction with the approval for use in an additional indication. For Product B, the period from 2006 to 2012 was characterised by a high degree of variability with the inclusion of new SCs as well as the removal, re-addition and re-classification of multiple SCs (Fig. 1b). This was followed by a period of stable development in the number of important risks. Following, the approval of RMP version 11.0 in 2019, one IIR and seven IPRs were removed to align with GVP Module V Revision 2. Furthermore, one IIR and one IPR were included in the RMP in 2019 based on new safety data from post-marketing data.
The first EU-RMP for Product A did not include any MI topics but four topics were added between 2013 to 2015 following the approval for use in a new indication. One MI was related to obligations outlined in the paediatric investigation plan (PIP) encompassing the new indication and was removed once those commitments had been fulfilled. The first EU-RMP for Product B submitted in 2006 included two MI topics and following re-evaluations to align with updated regulatory guidelines, they were first re-classified to IPRs in 2008 and 2009, respectively, and then finally removed from the EU-RMP in 2013 and 2019. In summary, these data showed that alignment with new or revised regulatory guidelines resulted in periodic variance in the risk presentation for the most mature products.
The Introduction of GVP Module V Revision 2 Resulted in a General Decrease in the Number of EU-RMP Safety Concerns
Next, we analysed the lifecycle evolution of SCs for the five more-recent products (Products C, D, E, F and G) with an initial EU-RMP submitted as a part of the MAA dossier (Fig. 2 and Fig. 3a–d). Figure 2 presents the number of SCs that were included or removed during the various stages of the product lifecycles. The majority (n = 50) of the SCs for these five products were included in the initially submitted RMP, while 36 SCs were included in the first HA-approved EU-RMPs as a results of the MAA evaluation process. Additionally, 14 SCs were added as an effect of new safety findings post-authorisation. Although, some (n = 9) SCs were removed from the EU-RMPs prior to the implementation of the GVP Module V Revision 2 in 2017, the implementation of this revision was followed by the removal of a larger number of SCs (n = 34).
The lifecycle evolution of the number of SCs is presented in Fig. 3a–d. Amongst the five products, three (Products C, D and E) saw an overall decrease in the number of SCs following the introduction of the GVP Module V Revision 2 in 2017. Product F had a stable development in the number of IPRs and MI from 2015 to 2020, whereas the number of IIRs increased in 2019. This increase in the number of IIRs was also observed for Product G, i.e., same active substance as in Product F but in a different indication in 2019 reflecting a new SC applicable for both products despite the different target population. Otherwise, for Product G one IPR was removed between 2016 to 2020 in conjunction with the renewal process to reflect that accumulated data did not support the initial categorisation. Furthermore, in 2018 one MI for Product G was removed to reflect the results from new safety data from a post-authorisation study and another MI was added in 2020 following the EMA approval for use in a new target population.
Following the authorisation of Product D in 2014, the number of SCs remained stable until 2016 when the number of IPRs for Product D varied due to parallel submissions of multiple EU-RMPs related to different variation processes. The total number of SCs for Product D, mainly IPRs and MI, decreased after 2016 and the availability of safety data from one large cardiovascular outcome trial. The renewal procedure in 2018 resulted in the removal of multiple IIRs and MI to align the RMP with the Revision 2 principles. The number of important potential and identified risks for Product E increased during the first three years post-authorisation, i.e., between 2012 and 2015, which was then followed by a period of stability until 2017, whereas for MI, this period was associated with sequential decreases and increases. The Product E renewal procedure in 2018 resulted in a decrease in the number of SCs. Product C had a stable development in the number of important risks and MI between 2011 and 2017, and the renewal in 2016 had no impact on the number of SCs. However, following the availability of safety data from two large cardiovascular outcome trials after 2017, several SCs could be removed according to the principles outlined in GVP Module V Revision 2.
Overall, most removals were prompted after the introduction of GVP Module V Revision 2 as shown by the general decrease in the number of EU-RMP safety concerns after 2017.
Mixed Impact of Post-authorisation Safety Studies (PASS)
We then examined the influence of PASS on the lifecycle dynamics of SCs for all seven studied products during the study period. In total, 50 EU-RMP SCs (seven IIRs, 27 IPRs, 16 MI) were associated with additional (a)PV activities such as PASS, whereas less than half (n = 23) of these SCs were removed or reclassified based on the result from PASS (Fig. 4a). Among the 23 aforementioned important risks and MI, 12 were removed or reclassified prior to the implementation of Revision 2 in 2017 (Fig. 4b). For the SCs associated with PASS, 16 had a PASS initiated prior to the introduction of the GVP Module VIII guidelines on post-authorisation studies in 2012 [8]. For IPRs, the outcome of aPV activities lead to the removal or reclassification of eight important potential risks. In contrast, almost all the aPV activities (13 of 16) associated with MI topics resulted in removal or reclassification. Overall, seven IIRs needed aPV activities while only the removal of two IIRs was related to the outcome of aPV activities. While dedicated post-authorisation studies have supported the characterisation of some SCs they did not have an impact on the risk presentation of many SCs.
Preceding Guidelines are Still Having an Influence on the Risk Presentation of Many EU-RMP Safety Concerns
Subsequently, we studied the number and characteristics of the SCs included in the latest HA-approved EU-RMP for each of the seven medicinal products as of 01 October 2020 (Fig. 5a). The two products, Products F and G, for which no decrease was observed after 2017 (Fig 3a–d) had the highest number of SCs (n = 20 and n = 18, respectively). The oldest products, Products A and B, both had five SCs, which is a significant decrease compared to the highest observed number of SCs for the respective products (n = 50 and n = 21) during the study period (Fig. 1a, b). Of the seven products, Product C had the lowest number of SCs (n = 3). The total number of IIRs and MI included in the EU-RMP for Product D had decreased from the maximum 18 to only 8. A similar trend was observed for Product E for which the number of SCs had decreased from a maximum of 17 to 8. None of the current 68 SCs needed any aRM measures (Fig. 5b), 13% (n = 9) were associated with an ongoing or planned aPV activity, whereas 87% (n = 59) did only require routine PV activities. Among the seven medicinal products just two, Products D and G, had SCs (n = 8 and n = 1, respectively) that required aPV activities 3 of which were associated with IIRs, four with IPRs and two with MI topics (Fig. 5c). Thus, while stakeholders have begun to implement the changes introduced by Revision 2, many SCs are still influenced by previous applicable guidelines.
Diverse Sources Influence Dynamics of EU-RMP Safety Concerns
Finally, one assessed how the knowledge gained from various sources (Box 3) has contributed to the characterisation of the safety profile (Fig. 6a–c). The sources of information that prompted the inclusion of SCs (Fig. 6a) were clinical trial data (n = 40), post-marketing data (n = 24) and non-clinical data (n = 9). Furthermore, the inclusion of 55 IIRs and MI was related to available data on the pharmacological class effect and 37 SCs were included based on target population characteristics. Information that triggered the removal (Fig. 6b) of SCs was derived from post-marketing data (n = 40), clinical trial data (n = 30) and PASS (n = 21). The most frequent sources associated with the reclassification of SCs were post-marketing data and clinical trial data, which influenced 15 and 11 reclassifications, respectively (Fig. 6c). Additionally, data from PASS formed the basis of the reclassification of one MI to IPR and one IPR to IIRs. In summary, diverse sources of data contributed to the increasing knowledge of the safety profile during the various stages of the lifecycle for marketed medicinal products.
The results also showed that the introduction of the new GVP Module V in 2012, as well as the revision in 2017, had an impact on the removal of 45 SCs. Note that the introduction of the GVP Module V in 2012 also prompted the removal of 37 identified risks for Product B (Fig. 1b) not included in Fig. 6b. The implementation of new or revised regulations (Volume 9A in 2008 and the GVP Module V in 2012) had an impact on the reclassification of six IPRs to IIRs and three MI to IPRs. Of the reclassified safety concerns, 47% (seven IPRs to IIRs and two MI to IPRs) were related to pharmacological class effect and/or target population characteristics (Fig. 6d).
Feedback During Assessment of the Market Authorisation Application
The last step was to assess whether the process to update (i.e., inclusion, removal, or re-classification) the risk presentations was prompted by HAs or by BI. HAs prompted the removal of 49 % (n = 45) of the SCs, whereas BI proactively proposed the removal of 51 % (n = 46) SCs (Fig. 6e). Feedback from HA during the assessment of the MA application dossier, resulted in the inclusion of 34 new SCs to the EU-RMPs. Of these SCs, 50 % (2 IIRs, 10 IPRs and 5 MI) were associated with pharmacological class effect and/or target population characteristics (Fig. 6f). Thus, the results showed that BI and HAs played an equal role in proposing the removal of important risks and MI topics, whereas HAs made a larger contribution to the inclusion of SCs, in particular those associated with class effect and target population.
Discussion
The past 15 years and the experience with the RMP as a proactive tool to minimise the impact of risks on patients, have had a profound impact on risk management, and ultimately on the safe use of marketed medicinal products by supporting the management of the benefit-risk profile for the respective products.
Evidently, the success of risk management relies upon having a comprehensive understanding of the evolving product’s safety profile and a focus on safety topics that need to be further characterised or mitigated. In this study, seven human medicinal products from the MAH were reviewed to better understand ways to best improve the risk management for medicinal products.
The early EU-RMPs (2005–2009) prior to obligatory EU-RMPs, were characterised by the high degree of variability in the classification of risks, which was reflected in the evolution of the number of SCs (Fig. 1a, b). The less harmonised approach continued from 2009 to 2012 when the RMPs were prepared in accordance with the definitions outlined in Volume 9A. This may be a consequence of the limited experience among all involved stakeholders on the one hand in conjunction with the aspiration of transparency, diverse interpretations, and disharmonised classification of the safety topics on the other. Unfortunately, the inclusion of many SCs in RMPs in the early years of RMP requirements diluted the focus on the SCs that required proactive risk management in order to protect patients and the public health. The GVP Module V, which became operational in 2012, included changes in the definitions of risks: An important risk shifted from simply being an adverse event to an untoward occurrence considering the impact on the individual and seriousness of the risk. Thus, conceptual changes resulted in moving away from equating topics included in the undesirable effect section of the SmPC as risks and aiming at a clinically meaningful list of SCs. The impact of this conceptual change was reflected in the decrease and subsequent stabilisation in the number of SCs for Product A and Product B after 2012. Furthermore, a general decrease in the number of SCs was observed in the analysed EU-RMPs after the implementation of GVP Module V Revision 2 in 2017 (Fig. 3a–d) leading to removal of well-characterised and appropriately managed risks as the RMPs were revised to focus on unresolved risks with a clinically meaningful outcome. The two more recently approved products, Products F and G, did not follow this trend, which can be attributed to addition of new indications and the risks in the new patient populations not being fully evaluated. However, it is evident that the implementation of new and/or revised guidelines did result in a change in the number of important risks and MI topics with a general trend downwards.
Revision 2 of GVP Module V mandated that only important risks and MI requiring an aPV activity or aRM measure should be included as SCs in the EU-RMP. However, most of the SCs included in the latest HA-approved EU-RMPs at study DLP were not associated with aPV activities and none required aRM measures. While in some EU RMPs, the Revision 2 philosophy has been followed, others continue to list SCs despite them not fulfilling the criteria for inclusion. This reluctance among stakeholders to remove resolved SCs implies that previous guidelines are still having an influence on the risk presentation. This reaffirms the benefit of clear and unambiguous guidelines on how to manage previously included SCs where no future data are expected from post-authorisation studies.
Although clinical trial data as well as non-clinical data were the most common sources for pre-authorisation included SCs, diverse sources of information, including post-authorisation studies and information from the market, influenced the SC dynamics throughout the lifecycle of the medicinal products. This further emphasises that risk management is an iterative process that requires a continuous and effective utilisation of all available data sources and regular assessment. Furthermore, in summary 49 % of the risk removals were prompted by HA, while 51 % were proactively initiated by BI, which demonstrates that both HAs and companies are moving forward with the revision of safety concerns in RMPs. Moreover, the evidence presented supports that companies should be successful in proactively proposing and removing risks based on a mutual understanding of the scope and purpose of RMPs by the HAs and MAHs. The removal of some important risks and MI was the result of dedicated post-authorisation studies. However, not all post-authorisation studies resulted in the removal or reclassification of SCs. It also became clear that post-authorisation safety studies, which were initiated before the introduction of the GVP Module VIII on post-authorisation safety studies in 2012 [8], had a lower impact on the classification of important risks and MI. This further indicates the importance of a careful and appropriate study design, supported by clear guidance, to achieve the required quality and usefulness of post-authorisation safety studies [13].
The significant increase in the number of SCs between the first submitted and first HA-approved EU-RMP (Fig. 3a–d) prompted us to look for common features. Strikingly, more than half of these IIRs and MI topics were associated with pharmacological class effect and/or target population characteristics, emphasising that familiarity with class effect and the target population is important for understanding the safety profile. Another interesting observation was that most of these risks were added in response to feedback from HAs. Considering that HAs have access to broader data sets, these observations emphasise the importance of a continuous and close cooperation between HAs and applicants or holders of a marketing authorisation. One way to increase the collaborative efforts could be the establishment of a digital database of SCs for authorised products, which would help stakeholders to align on new safety data findings and facilitate the assessment of SCs. One step in this direction is the Harmonisation of RMP project (HaRP), which seeks to harmonise RMPs for the same active substances, launched by the Working Party on PV Procedures Work Sharing in 2018 [11].
Furthermore, the management of SCs is influenced by the complexity in the global regulatory environment, which has resulted in local RMPs, that follow different regulatory requirements compared to GVP Module V Revision 2, with SCs deviating from the EU-RMP and territory-specific additional pharmacovigilance activities and risk minimisation measures. This complexity is exemplified by the variation in the definition of risks between different regulatory safety documents [14–16]. To overcome this issue, members of the pharmaceutical industry have proposed a framework to support the documentation and presentation of risks across relevant safety documents throughout the product lifecycle [17]. Some companies use so-called core-RMPs corresponding to additional internal safety documents, e.g., the Company Core Safety Sheet, to manage SCs globally.
A more comprehensive global regulatory frameworks would help stakeholders to navigate through the diversity of regulatory approaches on risk identification, characterisation, evaluation, and minimisation, resulting in a benefit for patient safety.
This study demonstrates how cumulative knowledge regarding the safety profile and more stringent guidelines have changed the risk presentation in a sample of EU-RMPs over the past 15 years. The evidence presented supports that evolution of regulatory guidelines and updates to RMPs are more focused on safety concerns that require investment in pharmacovigilance activities and risk minimisation in order to protect patients and public health. Potential bias and limitations are acknowledged since only medicinal products from one MAH have been included. The reviewed products and their EU-RMPs represent only a small sample size and are restricted to small molecules. Hence, it cannot be excluded that a larger sample size, the inclusion of biological products or different target populations would have resulted in a change to the observed characteristics. Positively, the study covered EU-RMPs published over a period of 15 years (2006 to 2020) and a diverse spectrum of therapeutic areas including orphan diseases and mass diseases. Despite the study’s limitations, the results provide valuable information and a basis for discussions of proactive risk management strategies between stakeholders, and on how to appropriately address patient and health care system-related matters.
Conclusions
In summary, this study revealed how diverse sources of data, including non-clinical data, clinical trial data, post-marketing experience, and familiarity with the class effect, contribute to the increasing knowledge of the safety profile and subsequently to the presentation of risks in EU-RMPs during the lifecycle of medicinal products. Additionally, our data showed how revised PV regulations have impacted RMPs through encouragement to continuously re-evaluate SCs and maintain an RMP that is fit-for-purpose based on the understanding of the benefit-risk profile of the product. This study also provided evidence that stakeholders have moved from an initial heterogenous approach towards an evidence-based evaluation of risks as their increased experience was reflected in the evolving regulatory environment. This emphasises the importance of clear and unambiguous guidelines for a harmonised scientific approach on risk classification and management. While both the HA and the MAH are moving forward with the revision of SCs presented in EU-RMPs according to Revision 2, preceding guidelines are still having an impact on many SCs. Moreover, this exercise highlighted the importance of close collaboration between regulators and industry to facilitate exchange of learnings and to take full advantage of all available sources of data throughout the product’s lifecycle. On this basis, we postulate that strong and harmonised regulatory guidelines covering the product lifecycle, along with continuous cooperation between stakeholders, are important for ensuring fit-for-purpose proactive risk management and thus, ultimately for protecting patient safety worldwide.
Acknowledgements
We would like to thank Martin Hüber from BfArM for his critical review of the manuscript. We acknowledge Sabine Jeck-Thole for her support and insightful comments on the manuscript. We thank Maria Weber for her input to the initial concept discussions and review of the manuscript. The views expressed in this article are those of the authors and should not be understood or quoted as being made on behalf of or reflecting the position of Boehringer Ingelheim. We acknowledge Sven Kohler for his constructive feedback on the manuscript.
Declarations
Funding
No honoraria or payments were made for authorship. Open access fee was paid by Boehringer Ingelheim International GmbH.
Conflict of interest
Julia E. J. Holm, Jan G. Ruppert and Samuel D. Ramsden are employed by Boehringer Ingelheim International GmbH. No other potential conflict of interest relevant to this article was reported.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data about specific products are available upon request and via European Public Assessment Reports published by the European Medicines Agency".
Code availability
Not applicable.
Authors’ contributions
All authors contributed to the study conception and design. The Literature review was performed by Julia E.J. Holm. Data collection and analysis were performed by Julia E.J. Holm for Products A, B, D, E, F and G. Jan G. Ruppert collected and analysed the data for Product C. Samuel D. Ramsden provided strategic direction for the data analysis and conclusions. The manuscript was written by Julia E.J. Holm. All authors reviewed the previous versions of the manuscript. All authors read and approved the final manuscript.
Footnotes
GVP V Revision 2 replaced the concept of “Important” Missing Information by Missing Information. In the following it is only referred to Missing Information (MI).
References
- 1.European Commission (EC). Directive 2001/82/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to veterinary medicinal products. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32001L0082&from=EN. Accessed 9 Feb 2021.
- 2.European Commission (EC). Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Union procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02004R0726-20190330. Accessed 10 Feb 2021.
- 3.European Medicines Agency (EMA). EMEA/CHMP/96268/2005. Committee for medicinal products for human use (CHMP) guideline on risk management systems for medicinal products for human use. (2005).
- 4.European Medicines Agency (EMA). EMEA/192632/2006. Annex C: template for EU risk management plan (EU–RMP). https://eudravigilance.ema.europa.eu/human/docs/19263206en.pdf. Accessed 5 Feb 2021.
- 5.European Commission (EC). VOLUME 9A—guidelines on pharmacovigilance for medicinal products for human use. 2008. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-9/pdf/vol9a_09-2008_en.pdf. Accessed 9 Feb 2021.
- 6.European Medicines Agency (EMA). EMA/838713/2011. Guideline on good pharmacovigilance practices (GVP) Module V—risk management systems superseded version. 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-module-v-risk-management-systems-superseded_en.pdf. Accessed 10 Feb 2021.
- 7.European Commission (EC). Directive 2010/84/EU of the European Parliament and of the Council of 15 December 2010 amending, as regards pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use. 2010. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2010_84/dir_2010_84_en.pdf. Accessed 10 Feb 2021.
- 8.European Medicines Agency (EMA). EMA/813938/2011. Guideline on good pharmacovigilance practices (GVP)—Module VIII—post-authorisation safety studies (Rev 3). 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-viii-post-authorisation-safety-studies-rev-3_en.pdf. Accessed 10 Feb 2021.
- 9.European Medicines Agency (EMA). EMA/204715/2012. Guideline on good pharmacovigilance practices (GVP)—Module XVI—risk minimisation measures: selection of tools and effectiveness indicators (Rev 2). 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-module-xvi-risk-minimisation-measures-selection-tools_en-3.pdf. Accessed 10 Feb 2021.
- 10.European Medicines Agency (EMA). EMA/838713/2011 Rev 2 guideline on good pharmacovigilance practices (GVP) Module V—risk management systems. 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-module-v-risk-management-systems-rev-2_en.pdf. Accessed 10 Feb 2021.
- 11.The Heads of Medicines Agencies. HaRP (Harmonisation of RMP Project). 2019. https://www.hma.eu/464.html. Accessed 25 Apr 2021.
- 12.European Public Assessment Report for Verquvo. 2021. https://www.ema.europa.eu/en/documents/rmp-summary/verquvo-epar-risk-management-plan-summary_en.pdf. Accessed 28 Nov 2021
- 13.Engel P, Almas MF, Bruin MLD, Starzyk K, Blackburn S, Dreyer NA. Lessons learned on the design and the conduct of Post-Authorisation Safety Studies: review of 3 years of PRAC oversight. Brit J Clin Pharmaco. 2017;83:884–893. doi: 10.1111/bcp.13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.European Medicines Agency (EMA). Harmonised tripartite ICH guideline development safety update report E2F. 2011. https://database.ich.org/sites/default/files/E2F_Guideline.pdf. Accessed 3 Mar 2021
- 15.The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Investigator Brochure (IB): ICH guideline for good clinical practice E6 (R2). 2016.
- 16.European Medicines Agency (EMA). Guideline on good pharmacovigilance practices (GVP) Module VII—periodic safety update report (Rev 1). 2013. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-vii-periodic-safety-update-report_en.pdf. Accessed 3 Mar 2021.
- 17.TransCelerate the Interpretation of Pharmacovigilance Guidances & Regulations Initiative. A framework on the presentation of the safety of risks throughout the product lifecycle (PORTL). Transcelerate BioPharma; 2020.
- 18.European Medicines Agency (EMA). EMA/164014/2018 Rev.2.0.1 accompanying GVP Module V Rev.2. Guidance on the format of the risk management plan (RMP) in the EU—in integrated format. 2017. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guidance-format-risk-management-plan-rmp-eu-integrated-format-rev-201_en.pdf. Accessed 5 Feb 2021.