

Abstract
Evading immune-mediated destruction is a critical step of tumor evolution and the immune system is one of the strongest selective pressures during tumorigenesis. Analyzing tumor immune evasion from a Darwinian perspective may provide critical insight into the mechanisms of primary immune escape and acquired resistance to immunotherapy. Here, we review the steps required to mount an anti-tumor immune response, describe how each of these steps is disrupted during tumorigenesis, list therapeutic strategies to restore anti-tumor immunity, and discuss each mechanism of immune and therapeutic evasion from a Darwinian perspective.
Keywords: breast cancer, immune evasion, tumor evolution
INTRODUCTION
Tumors are composed of mixed populations of cells residing within dynamic microenvironmental niches. They represent unique cellular ecosystems complete with a diversity of species (cellular heterogeneity), the reproduction of those species (proliferation), and an environment that imposes fluctuating selective pressures (microenvironmental variation in blood flow, extracellular matrix, and stromal cell interactions). Several models have been proposed to explain tumor progression and intratumor heterogeneity, including both Darwinian (selective) and neutral (stochastic) evolution [1–8]. However, pre-clinical studies as well as clinical data have described numerous examples of selection for subclones with particular features supporting the Darwinian model of tumor evolution [9, 10]. Data demonstrating that minor subclonal populations are capable of driving tumor growth and metastatic progression in a non-cell-autonomous manner exemplify the complexity of evolutionary dynamics and help explain the selective propagation of minor clones without their expansion [11]. Non-cell-autonomous drivers also maintain intratumor heterogeneity, thus, play key roles in tumorigenesis. Recent data indicates that even cancer driver genes thought to function in a cell autonomous manner (e.g., mutant TP53) have non-cell-autonomous functions highlighting the importance of cellular interactions in tumor development [12].
The microenvironment shapes tumor evolution by imposing a variety of selective pressures, including anti-tumor immunity [13, 14]. Immune-mediated destruction selects for tumor cell populations with evasive properties by eliminating cells vulnerable to immune killing. This strong selective pressure has been demonstrated in several experimental models and by clinical data in immunocompromised patients and is summarized in the cited review [13]. The immune system may eliminate malignant lesions throughout the human lifespan, with tumors growing into clinically detectable lesions only after immune escape [15, 16]. This transition from immune-mediated elimination to escape has been termed immunoediting [13, 17]. Immunoediting describes a three-step process in which selection by the immune system drives tumor evolution towards immune evasion (Figure 1). Immunoediting begins by the recognition of cancer cells and mounting of an immune response against them, in a phase termed elimination. If immune-resistant clones emerge before eradication of the tumor (through mechanisms such as genomic instability or epigenetic plasticity), these resistant clones can be sufficient to drive a tumor into an equilibrium phase, whereby immune-mediated killing is proportional to tumor cell proliferation and no net tumor outgrowth occurs. Killing of immune-sensitive clones during the equilibrium phase may then select for immune evasive clones, leading to immune escape and the outgrowth of an immune evasive tumor [11,14,15].
Figure 1. Schematics of immunoediting.
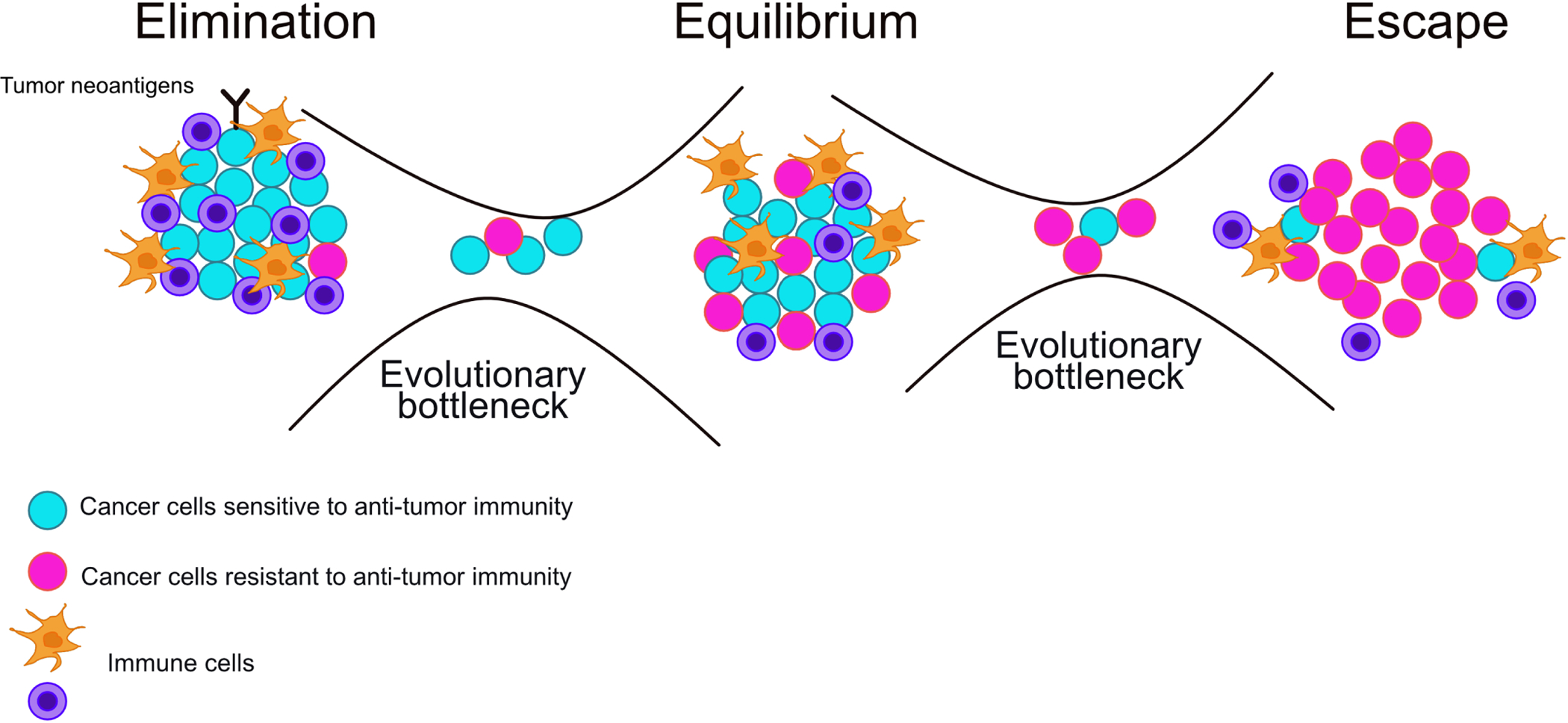
In the elimination phase, most malignant cells are sensitive to anti-tumor immunity and immunity steps are intact. The elimination of immune-sensitive cells selects for cells with perturbed immunity steps that are no longer sensitive to immune-mediated elimination. There is an evolutionary bottleneck that selects for cells that are either able to evade anti-tumor immunity or can proliferate quickly enough to sustain a high turnover, immune sensitive population. In the equilibrium phase, there is a mixture of immune-sensitive and insensitive cells and tumor cell elimination is proportional to rates of cell proliferation, leading to no net tumor outgrowth. Intact and perturbed immunity steps are both represented during this phase. For tumor outgrowth to occur, immune-insensitive cells are selected for during a second evolutionary bottleneck, allowing for tumor cell proliferation to occur without elimination by anti-tumor immunity. In the escape phase, most malignant cells are insensitive to anti-tumor immunity and immunity steps are perturbed. It is possible for immune-sensitive subclones with intact immunity steps to emerge and/or remain within this population if escaped clones maintain an immunosuppressive environment, thus shielding the sensitive clone from anti-tumor immunity.
Although immunoediting provides an important evolutionary framework for tumor progression towards immune evasion, it does not detail the mechanisms by which emergent tumor clones evade immune destruction, and therefore the fitness advantages that are selected for throughout this process. Understanding the mechanisms of tumor immune evasion has been critical in the development of novel immunotherapies, such as immune checkpoint inhibitors (ICI) and adoptive cell transfer (ACT). These immunotherapies are designed to restore anti-tumor immunity and with studies demonstrating complete responses in approximately 20% of patients with metastatic melanoma after receiving cell transfer therapy, long-term overall survival rates of up to ten years in approximately 20% of patients with advanced melanoma who received ipilimumab (anti-CTLA-4), and a confirmed response rate of 38% in patients with advanced melanoma after receiving lambrolizumab (anti-PD-1), the cancer curative potential of immunotherapy has generated widespread enthusiasm and interest [18–20]. Despite these advances, it has proven challenging to identify patients who would benefit from immunotherapy and even in those selected for treatment, only a minority of patients within a subset of malignancies show durable responses [21–23]. This lack of efficacy encompasses two important clinical obstacles: innate resistance (indicating alternative mechanisms of immune evasion) and acquired resistance (indicating selection for novel immune escape mechanisms during treatment). Understanding of the specific steps of anti-tumor immunity and mechanisms of immune escape is critical to our identification of novel immunotherapies and biomarkers for patient selection, as well as for the design of effective combination strategies to combat therapeutic resistance.
The steps of anti-tumor immunity have been outlined in several excellent reviews [24, 25]. Here, we streamline the process of anti-tumor immunity into four main steps: identification, activation, targeting, and elimination (Figure 1). We review the fundamental biology underlying each of these steps and synthesize mechanisms of immune evasion that can be selected for to circumvent them. We also discuss immunotherapies used in the clinic in the context of restoring each anti-tumor immunity step and the novel immunotherapeutic strategies under preclinical and clinical investigation. Importantly, we discuss each mechanism of primary immune evasion and immunotherapeutic resistance from a Darwinian perspective to conceptualize how biological and therapeutic selective pressures drive immune escape.
Identification: Cancer Cell Antigens
The process of identification
The anti-tumor immune response begins with the identification of malignant cells via antigenic peptides. There are two broad classes of tumor antigens, tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs). TAAs are peptides that are either highly overexpressed or aberrantly expressed in cancer cells [26, 27]. TAAs include carcinoembryonic antigens and cancer/testis antigens, which are found in tumors, at low levels in certain normal tissues, and during embryonic development or in germline cells of the testis, respectively [26, 28]. Alternatively, TSAs are characterized by unique tumor specificity, as they are generated by novel mutations not found in normal tissues (tumor neoantigens) or by viral peptides in viral-associated tumors [29]. For immune cells to identify tumors, cancer cells must express antigens that are both immunogenic and specific, so that immune responses can be elicited without interference by immune tolerance.
Identification of malignant cells also requires immune cell access to tumor antigens. To efficiently proceed to immune cell activation (discussed below) antigens must be taken up by antigen presenting cells (APCs). Dendritic cells (DC) are a subset of APC that are most frequently associated with anti-tumor immunity; for simplicity we will utilize the term APC in this review. A robust mechanism for APC access to tumor antigen is via antigen release following tumor cell death [24, 30]. An important caveat of this mechanism is that cell death must be immunogenic. Historically, it had been thought that only necrosis was an immunogenic form of cell death and that apoptosis was tolerogenic since it accounts for much of the natural cell turnover within normal tissues [30]. However, it is now accepted that both necrosis and apoptosis can elicit robust immune responses and that immunogenic forms of cell death are at least in part distinguished by the release of damage associated molecular patterns (DAMPs) [30, 31]. In summary, identification of malignant cells requires 1. the expression of immunogenic and specific cancer cell antigens and 2. the availability of those antigens for APC uptake following immunogenic cell death (Figure 2A).
Figure 2. Steps of immune recognition and escape.
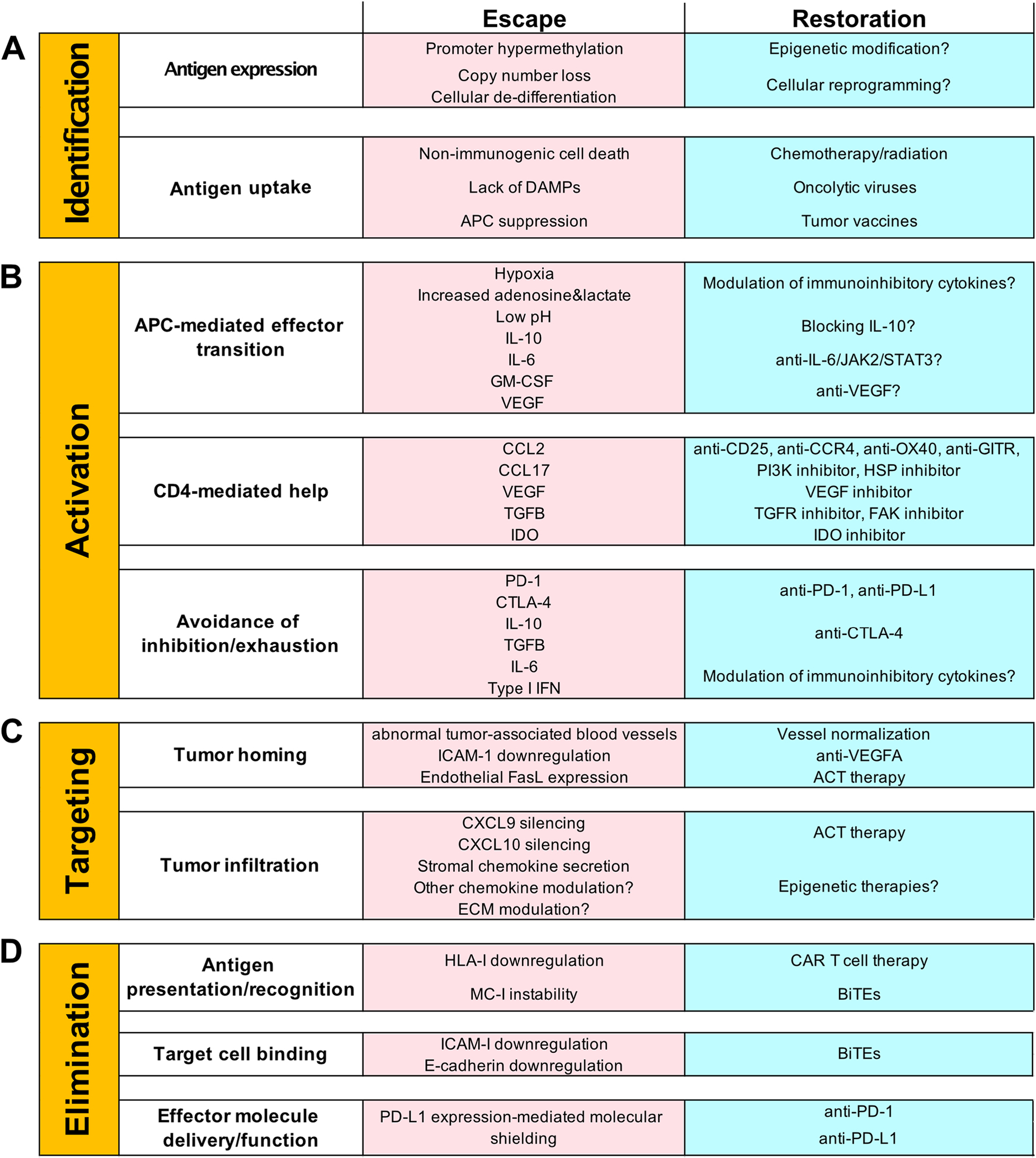
A, Requirements, escape mechanisms, and restoration options of immune identification. The successful identification of malignant cells requires cancer cell antigen expression, followed by antigen uptake by APCs. At each requirement, there is an opportunity for immune escape via the mechanisms listed. Possible methods of therapeutic restoration are shown and therapeutic success would allow for identification to continue. B, Requirements, escape mechanisms and restoration options of activation. Optimal activation of T cells requires an APC-mediated effector state transition, CD4+ T cell-mediated help, and avoidance of inhibition/exhaustion. At each requirement, there is an opportunity for immune escape via the mechanisms listed. Possible methods of therapeutic restoration are shown, and therapeutic success would allow for activation to continue. C, Requirements, escape mechanisms and restoration options of targeting. Successful targeting of immune cells to tumors requires appropriate tumor homing, followed by tumor infiltration. At each requirement, there is an opportunity for immune escape via the mechanisms listed. Possible methods of therapeutic restoration are shown, and therapeutic success would allow for targeting to continue. D, Requirements, escape mechanisms and restoration options of elimination. CTL-mediated cancer cell elimination requires antigen presentation and recognition, target cell binding, and effector molecule delivery and function. At each requirement, there is an opportunity for immune escape via the mechanisms listed. Possible methods of therapeutic restoration are shown, and therapeutic success would allow for elimination to continue.
Evading immune identification
Tumors evolve to evade immune identification. If cancer cells that express highly immunogenic antigens are eliminated by the immune system, a selective pressure favoring cancer cells that downregulate immunogenic antigen expression could drive tumor evolution. A recent elegant study described an example for this mechanism in non-small cell lung cancer (NSCLC) through neoantigen copy number loss and promoter hypermethylation [32]. In agreement with these findings, recent work in glioblastoma confirmed that reduction in neoantigen expression can facilitate tumor progression [33]. Work in a melanoma mouse model demonstrated that a reversible dedifferentiation program led to a loss of melanocytic antigen expression [34]. These studies suggest that downregulation of immunogenic antigens can occur through a variety of mechanisms (Figure 2A) and the prevalence of this downregulation among different malignancies suggests a robust selective pressure driving tumor evolution towards evasion of identification.
Clinical strategies to restore identification
Several clinical strategies have emerged to restore tumor identification. Perhaps the oldest of these strategies are the abscopal effects of radiotherapy. For decades it had been noted that focal radiation could lead to the regression of secondary tumors at distant sites [35]. Now, these effects are attributed to radiation-mediated cell death leading to the release of tumor antigens and DAMPs into the microenvironment, thus eliciting a robust immune response in some patients [35]. Although these effects had been noted in certain subsets of patients, it is now appreciated that more robust abscopal effects were likely not seen due to immunosuppressive signals such as PD-1 and CTLA-4 [35]. To explore this hypothesis, recent clinical studies and case reports have examined the effects of combining radio and immunotherapy and have led to promising results [35–37]. Another central cancer therapy that could be used to restore identification is chemotherapy. It has been reported that certain chemotherapeutic agents such as cyclophosphamide, doxorubicin, epirubicin, idarubicin, mitoxantrone, and oxaliplatin can induce immunogenic cell death, which would allow for the release of tumor antigens [38]. These chemotherapeutic agents can also increase MHC-I expression on tumor cells and stimulate a variety of immune cells, leading to even more robust anti-tumor immune responses [38]. The use of oncolytic viruses is another therapeutic strategy to induce immunogenic cell death and tumor antigen release [39]. Due to either innate or engineered tumor tropism, oncolytic viruses can demonstrate selective replication in cancer cells, leading to their destruction [39]. Along with tumor antigen release, oncolytic viruses bolster tumor immunogenicity via release of DAMPs and pathogen-associated molecular patterns (PAMPs) which bind pattern recognition receptors (PRR) on APCs [39].
Tumor vaccines are another immunotherapeutic strategy that can be used to restore identification by directly providing tumor antigens to immune cells. Several approaches including dendritic cell (DC), synthetic long peptide (SLP), and RNA vaccines have been proposed as antigen delivery mechanisms [40, 41]. While the superior delivery mechanism is still under investigation, it has become clear that neoantigen-based vaccines (as opposed to TAA based vaccines) have a clear advantage in promoting specific anti-tumor immunity with reduced autoimmunity or immune tolerance [26, 41]. While several clinical trials have evaluated the effectiveness of vaccines targeting TAAs due to their limited expression in normal tissues and the immune-privileged state of the testis (a common tissue expressing TAAs), the clinical effectiveness of TAA vaccines has been limited, potentially due to self-tolerance [26, 41]. Alternatively, TSAs provide unique tumor specificity and decreased risk of autoimmunity. For these reasons TSAs, and particularly neoantigens, have been targeted for use in vaccine and ACT clinical studies [26, 42, 43]. As neoantigen vaccines have resulted in promising phase I melanoma and glioblastoma clinical trial results, several neoantigen-based cancer vaccine phase I and II clinical trials are underway in a variety of malignancies and in combination with other therapies [29]. The potential for neoantigen vaccines to be combined with ACT may also yield promising results, as it would allow for the generation and expansion of a T cell population with a diverse T cell repertoire and high tumor specificity [43]. To date, clinical studies have shown that identification may be restored via radiation, chemotherapy, virus-mediated immunogenic cell death or by vaccine-mediated antigen delivery (Figure 2A).
Potential for resistance to therapeutic identification
Just as selective pressures by the immune system drive evasion of identification by tumor cells, clinical interventions restoring identification may select for therapeutic resistance. To predict potential mechanisms of immunotherapeutic resistance, it is important to conceptualize the requirements and thus potential points of escape from these therapies. For example, to stimulate an anti-tumor immune response, immunogenic cell death requires appropriate APC function. Interestingly, cytokines such as IL-10 have been linked to both chemoresistance and APC suppression, and thus represent a potential mechanism for acquired resistance [44, 45]. In this scenario, chemotherapy could select for IL-10 expressing tumor clones that are both chemo-resistant and capable of dampening the anti-tumor immune response despite the antigen release by chemo-sensitive clones. Therefore, perhaps therapeutic strategies such as DC vaccines, which remove DCs from the tumor microenvironment and load DCs with antigen ex vivo, may avoid these avenues of therapeutic evasion and increase odds of success.
The potential success of anti-tumor vaccines can be assessed through a similar evolutionary framework. In this case, intratumor heterogeneity presents a substantial obstacle and an opportunity for therapeutic escape. For example, mono-antigenic vaccines may selectively eliminate specific tumor clones, providing a selective advantage to clones not expressing the targeted antigen. This possibility should therefore promote the generation of poly-antigenic vaccines. However, while poly-antigenic vaccines may broaden the repertoire of targeted tumor clones, the antigens targeted by these vaccines are limited to what can be identified from a given tumor sample. Therefore, in a highly heterogeneous tumor, the tumor sample used for Next Generation DNA and/or RNA Sequencing and subsequent vaccine generation may not be representative of the whole tumor, allowing for the possibility of therapeutic escape. This knowledge calls for the inclusion of mutational lineage tracing and neoantigen selection targeted towards truncal mutations to increase the probability of targeting common neoantigens between subclones. While these are just a few potential examples of therapeutic resistance to identification, the conceptual framework they provide highlights the utility of Darwinian evolutionary theories in guiding the design of novel immunotherapies.
Activation: Immune Cell Coordination
The process of activation
The second step of anti-tumor immunity is the activation of immune cells. Here, we will primarily focus on T cells due to the groundbreaking drug developments that have resulted from their characterization. T cells are lymphocytes whose progenitors originate in the bone marrow and travel to the thymus to complete their development [46]. In the thymus, T cells become committed to a CD4+ or CD8+ lineage in a process that is tightly regulated by thymic architecture, stromal cell engagement, notch signaling, transcription factor activity, and binding to MHC-I or II [46, 47]. T cells also undergo stringent selection for self-reactivity in the thymus to ensure autoimmunity is avoided [46, 48]. After lineage commitment, CD4+ and CD8+ T cells are considered naïve until they engage with their T cell receptor (TCR) specific antigen and are co-stimulated by APCs, leading to an effector cell state transition [48, 49]. Effector T cell population expansion occurs after activation, with large-scale apoptosis occurring shortly thereafter, to regulate immune activity and avoid damage to normal tissues [48]. However, a small portion of effector T cells transition into memory T cells to enable rapid immune responses if an antigen is reencountered [50, 51]. There are several functional differences between naïve and memory T cells including their localization and activation requirements. While memory T cells can reside in non-lymphoid tissues and readily transition into effector cells, naïve T cells are primarily found in the circulation and secondary lymphoid tissues and require further co-stimulatory signals for activation ([49–53].
APCs play a crucial part in T cell activation as they express MHC-I and II and can therefore proficiently present antigen to CD8+ and CD4+ T cells, respectively [54]. Additionally, APCs provide the co-stimulatory signals required for T cell activation (such as B7 molecule engagement of CD28) [54, 55]. APCs are activated by antigen uptake and co-stimulation of PRR by DAMPs or PAMPs, and their subsequent maturation leads to increased expression of MHC and B7 molecules [54, 56, 57]. While APCs can locally activate tissue resident memory T cells, naïve T cells are not commonly found within non-lymphoid tissues and require APCs to travel to lymph nodes for their activation [49, 54]. In the lymph node, naïve CD4+ and CD8+ T cells systematically scan APCs for antigen [58]. It has been suggested that the directed migration of T cells (guided by chemokine and ECM gradients in the lymph node) allows for the efficient and rapid scanning of APCs, thus increasing the odds of encountering a TCR-specific antigen [58]. Upon TCR engagement and co-stimulation by APCs, CD4+ and CD8+ T cells activate and transition into an effector state [48, 49].
Helper CD4+ T cells (Th cells) function by releasing a variety of cytokines into the local microenvironment to regulate immune responses [59]. These cytokines include IL-2, which can promote the recruitment and cytolytic function of CD8+ cytotoxic T cells (CTL) [59, 60]. It has also been shown that in the absence of Th cells, effector CTL are more likely to undergo apoptosis upon re-exposure to antigen, thus promoting antigenic tolerance [61]. But not all CD4+ T cells serve to stimulate immunity. A subset of CD4+ T cells known as regulatory T cells (Treg) suppress immunity and have been shown to correlate with poor patient survival in a variety of malignancies [62]. Additionally, it has been suggested that along with activating Th cells, certain APCs can also drive CD4+ T cell differentiation into the inhibitory Treg subtype [57]. Thus, APCs and CD4+ T cells can promote and suppress immune activity.
CTL are responsible for tumor cell killing and their infiltration into tumors is correlated with improved patient prognosis in a variety of malignancies [63]. To avoid tissue damage, CTLs are tightly regulated by immunomodulatory signals that can induce CTL apoptosis, memory transition or exhaustion [50, 64–66]. It is thought that chronic antigen presentation and inflammation in tumors can lead to CTL exhaustion and loss of function [65, 66]. A hallmark of CTL exhaustion is expression of the immunoinhibitory receptor PD-1, which although expressed at low levels upon T cell activation, is significantly upregulated via promoter demethylation upon CTL exhaustion [65, 66]. In summary, the optimal activation of T cells requires (1) antigen presentation and co-stimulation by mature APCs, (2) immune cell coordination and ‘help’, and (3) avoidance of inhibitory signals and exhaustion (Figure 2B).
Evading activation
Tumors evolve to disrupt immune activation. This can be accomplished at any step of activation including disrupting APC maturation and therefore stimulation of T cells, promoting a Treg dominant CD4+ population, and driving T cell exhaustion and inhibitory signaling (Figure 2B). Tumors have been shown to functionally impair APCs via a suppressive tumor microenvironment (including hypoxia, increased levels of adenosine and lactate, and low pH) and by the secretion of inhibitory cytokines (such as IL-10, IL-6, GM-CSF, and VEGF) [56, 57] Tumors have also demonstrated an ability to recruit Treg cells via the secretion of cytokines such as CCL2, CCL17, and VEGF, and promote Treg population expansion via TGFβ and IDO secretion [67, 68]. The secretion of immunosuppressive and inflammatory cytokines (such as IL-10, TGFβ, IL-6, and type I IFNs) within tumors also promotes T cell exhaustion [65, 66]. Additionally, tumor-mediated APC dysfunction and Treg promotion may further promote T cell exhaustion [65, 66]. Expression of inhibitory receptors such as PD-1 and CTLA-4 increases during T cell exhaustion, with the level and number of these receptors influencing exhaustion severity [65, 66]. B7 molecules found on APCs are ligands for CTLA-4 and due to differences in affinity, T cell expression of CTLA-4 can outcompete CD28 binding to B7, thus disrupting T cell activation [69]. Alternatively, PD-L1 is the primary ligand for PD-1, and it is expressed in both tumor-associated stromal and cancer cells in a variety of malignancies [70, 71]. Unlike CTLA-4, PD-L1 binding to PD-1 disrupts T cell activation via attenuation of TCR signaling [71]. Although there is still much to learn about inhibitory receptor signaling and function, it is clear that these receptors play a major role in immune exhaustion and tumor immune escape.
Ageing, a major risk factor for cancer, may further enable evasion of immune activation. In particular, ageing has been associated with a decrease in naïve T cell production, a loss of T cell CD28 surface expression, an increase in exhausted T cells and impaired cytokine production by CD4+ and CD8+ T cells [72–74]. Age and infectious disease history may also interact to regulate TCR repertoire diversity, which has been shown to limit tumor development in pre-clinical models and improve responses to immunotherapy in a clinical study of NSCLC [75, 76]. Although immunological challenges usually serve to diversify TCR repertoires, chronic infections in aged adults may limit TCR diversification. For example, aged adults with chronic cytomegalovirus (CMV) infection demonstrate a decrease in TCR diversity, as CMV-responsive T cells become the dominant population in these individuals [72, 77]. Overall, a wide variety of mechanisms of perturbed immune activation and exhaustion are observed in tumors[78–80], and the diversity of these pathways suggests a strong selective pressure driving tumor evolution towards evasion of activation.
Clinical strategies to restore activation
The development of ICI has ushered in a new era of tumor immunology by demonstrating the therapeutic potential of restoring immune activation. The success of ICI in a subset of patients has led to FDA approval of anti-CTLA-4 (e.g., ipilimumab), anti-PD-1 (e.g., nivolumab, pembrolizumab, cemiplimab), and anti-PD-L1 (e.g., atezolizumab, avelumab, durvalumab), and has made these agents first-line therapies in certain malignancies [69, 70, 81, 82]. However, due to the relatively small subset of patients with durable responses from single agent ICI treatment, hundreds of clinical trials are underway to explore potential combination therapies and ICI uses in different malignancies [69–71]. As the name implies, ICI function by blocking the inhibitory receptors that limit the activation of T cells, thus ‘releasing the brakes’ on immune activation. Importantly, work with ICI has shown that T cell exhaustion is a reversible state, with ICI treatment being able to reinvigorate the effector functions of previously exhausted T cells [65, 66]. However, releasing the brakes on immunity can come with significant consequences, as immune checkpoints serve important roles in immune-homeostasis and avoidance of autoimmunity. Tumor progression and some severe cases of autoimmunity have been seen following ICI treatment in subsets of patients thus, identification of patients who would benefit from ICI and have low risk for immune-related adverse events, as well as developing clinical strategies to manage immune related adverse events is critical for extending ICI to larger patient populations and earlier stages [70, 83].
Modulation of immunoinhibitory cytokines has also been an attractive therapeutic strategy. However, even cytokines with well documented immunosuppressive functions such as IL-10, IL-6 and TGFβ have shown both pro and anti-tumor effects in certain contexts, making them challenging therapeutic targets [84–86]. Interestingly, anti-VEGF-A therapy combined with anti-PD1/PD-L1 has proven to be beneficial in clinical studies [87]. However, the precise mechanism of this synergy requires further mechanistic characterization. Overall, further studies will be needed to elucidate the appropriate timing and context needed for cytokine manipulation.
Therapies targeting Tregs are also an area of intense interest in immunotherapy. Several approaches including anti-CD25, anti-CCR4, anti-OX40 and anti-GITR antibodies, as well as inhibitors of IDO-1, VEGF, PI3K, HSP, TGFβ, and FAK have been proposed to modulate Treg activation, function, and tumor infiltration [68, 88–90]. However, the detailed mechanism of action for these therapies is still under investigation in various clinical studies [68, 88–90]. In addition to consequences of increased autoimmunity with systemic Treg inhibition, Treg therapies are also confounded by the expression of similar receptors (such as CD25) on the surface of effector T cells [68, 88–90]. Therefore, efforts are underway to selectively target tumor associated Treg cells while maintaining effector T cell and peripheral Treg function. It has been suggested that differences in the kinetics and expression of target molecules may be key to this strategy [90]. To date, the greatest clinical success in restoring immune activation has been shown with ICI. However, several intriguing strategies are being investigated including combination ICI treatments, modulation of immunoinhibitory cytokines and inhibition of tumor associated Treg cells (Figure 2B).
Potential for resistance to therapeutic activation
Clinical interventions restoring activation can function as a selective pressure for therapeutic resistance. Although widespread usage of ICI in the clinic is still in relatively early stages, reports of acquired resistance to ICI therapy have begun to emerge. In fact, it is now estimated that 25–33% of melanoma patients initially responsive to ICI will experience disease progression even with continued usage [82, 91]. This significant relapse rate suggests robust therapeutically imposed selection and immunoediting during ICI treatment. In agreement with this model, studies done in NSCLC patients suggest that immunoediting drives acquired ICI resistance [82]. Additionally, a mouse model of colorectal cancer demonstrated that even in cancers that may follow neutral evolution, treatment with anti-PD-L1 imposed a sufficient selective pressure to alter dynamics consistent with Darwinian evolution [92]. Several studies in various cancer types have described multiple different mechanisms of acquired resistance to ICI including upregulation of alternative immune checkpoints, defective IFN signaling, downregulation of antigen expression and neoantigen loss, loss of MHC-I surface expression, and upregulation of VEGF [21, 82, 91, 93, 94]. Although additional mechanisms of acquired ICI resistance have yet to be characterized, it widely considered that there will be significant overlap with mechanisms of innate immune resistance as specified above. These similarities are intuitive from a Darwinian perspective, as the selective pressure of ICI treatment may initiate a new cycle of immunoediting (just as immunoediting shaped the primary immune escape), leading to the emergence of newly escaped clones [82, 91].
Targeting: Homing and Infiltration
The process of targeting
The third step of anti-tumor immunity is the targeting of immune cells to tumors. After activation, T cells exit the lymph node and enter the circulation in search of their TCR specific antigen. Homing of leukocytes to inflamed tissues requires several coordinated steps, beginning with rolling and tethering along blood vasculature. Rolling and tethering describes the process by which leukocytes can slowly survey appropriate exit cites along venule walls by making transient adhesions with endothelial cells via selectin/ligand binding [52]. Once rolling leukocytes encounter appropriate endothelial presented chemoattractants, a rapid inside-out activation of integrin heterodimers occurs, causing their arrest and firm adhesion to the venule wall [52]. In addition to the increased affinity and avidity of integrin bonds, this activation process serves to repolarize the leukocyte, leading to remodeling of the actin cytoskeleton [52, 95]. This actin remodeling event is key in allowing for either the immediate para/trans-endothelial migration of leukocytes or the migration (‘crawling’) of leukocytes along venule walls in search of an exit [52, 95]. Like other forms of directed cell migration, leukocyte crawling is mediated by a protrusive leading edge and a contractile cell rear, and is directed by intravascular chemotactic gradients [95]. Once chemotactic and/or haptotactic exit cues are detected, leukocytes arrest and begin weakening endothelial barrier function [95]. Leukocytes have been shown to perform both transendothelial and paraendothelial migration, with the latter occurring more frequently [95]. To fully exit the venule, leukocytes must also pass through the pericyte sheath and venular basement membrane. Although this process is not fully defined, it is thought that pericyte assistance and areas of low matrix density in the basement membrane contribute to leukocyte crossing [95].
Although leukocytes have been shown to perform both random and patterned movements during migration, chemotactic gradients are thought to play a significant role in directing T cell migration towards tumor beds, as CXCL9 and CXCL10 expression consistently correlate with T cell infiltration [96–99]. However, leukocytes are also capable of haptotactic and mechanotactic migration, which raises the question as to whether these forms of directed cell migration are also at play [95, 100]. Stromal cells in the tumor microenvironment are also capable of secreting chemoattractants (such as CXCL12) and may therefore redirect T cells away from tumor masses and toward stromal areas [98]. Overall, immune targeting requires a two-step process of tumor homing and infiltration (Figure 2C). Depending on the ability of immune cells to properly home and infiltrate, three broad tumor immune targeting phenotypes may arise: (1) ‘inflamed’, tumor cells are intermixed with immune cells (suggests successful homing and infiltration), (2) ‘stroma-restricted’, immune cells are found within tumor stroma but not intermixed with tumor cells (suggests successful homing but failed infiltration), and (3) ‘immune cold’, tumors in which immune cells are not found (suggests failed homing with infiltration capacity remaining unknown). Tumors with these three distinct patterns of leukocyte homing and infiltration have differences in response treatment and clinical outcomes [101]
Evading targeting
Tumors evolve to disrupt immune targeting. The ability of T cells to infiltrate tumors is a major determinant of tumor cell killing and patient survival [98, 99, 102]. Therefore, a survival advantage is conferred to tumors that evolve mechanisms to perturb this process. T cell trafficking to tumors is thought to be negatively affected by leaky tumor-associated blood vessels, due to abnormal blood flow [98, 103]. Additionally, tumor secreted molecules such as VEGF-A, can downregulate the expression of endothelial adhesion molecules such as ICAM-1, thus inhibiting T cell adhesion to the endothelium and exit from the vasculature [98]. Tumor secreted VEGF-A, prostaglandin E2, and IL-10 have been shown to induce expression of FASL on endothelial cells, leading to CD8+ T cell death in preclinical models [98]. Tumor cells can also silence their expression of T cell chemoattractants such as CXCL9 and CXCL10 via promoter methylation, and thus limit necessary directional cues for T cell tumor infiltration [98, 99]. It has also been proposed that tumor cells can secrete Galectin-3, which sequesters IFNγ in the extracellular matrix and thus limits IFNγ-mediated chemokine production [104]. Collectively, these studies suggest that tumors evolve several mechanisms to disrupt both the homing and infiltration aspects of leukocyte targeting (Figure 2C).
Clinical strategies to restore targeting
The mechanisms regulating immune targeting to tumors still require extensive characterization. However, the significantly better patient survival conferred by T cell infiltration of solid tumors has spurred interest in developing therapeutic strategies to restore targeting [98, 102]. Vessel normalization via treatment with anti-VEGF-A therapies has been proposed as one such strategy [98, 99, 103, 105] (Figure 2C). Encouragingly, when anti-VEGF-A was given in conjunction with ACT therapy in a mouse model of melanoma, anti-VEGF-A treatment was shown to increase the tumor infiltration of transferred T cells [103]. Importantly, several other pre-clinical and clinical studies have shown an increase in T cell recruitment and infiltration with combined anti-angiogenic and immunotherapy treatment regimens [106]. ACT therapies have also attempted to increase T cell targeting by collecting T cells that have already infiltrated tumors (tumor-infiltrating lymphocytes; TILs) for expansion and re-introduction, as well as by engineering T cells to express receptors for specific tumor secreted cytokines [97, 103] (Figure 2C). Usage of epigenetic modifiers has also been proposed to restore expression of silenced chemokines such as CXCL9 and CXCL10 and have shown promising results in preclinical models [98, 99] (Figure 2C).
Potential for resistance to therapeutic targeting
The mechanisms regulating immune targeting, its evasion and clinical restoration are still in their early stages of characterization. Immune targeting may be a unique immunity step as it seems to be regulated more by the collective tumor microenvironment than the individual tumor cell. Thus far, there have been few widely accepted clinical strategies for therapeutic modulation of the tumor microenvironment, apart from vessel normalization [98, 99, 107]. However, an important lesson can be learned from the history of anti-VEGF-A therapy. Anti-VEGF-A was initially conceived as an anti-angiogenic strategy, designed to be used at high doses to ‘starve’ tumors of oxygen and nutrients [98] [107]. However, instead of eradication, the hypoxic conditions resulting from this treatment strategy led to the emergence of highly aggressive tumor subclones. Thus, the concept of vessel normalization using low-dose anti-VEGF-A emerged as an alternative approach [107]. Vessel normalization uses the same drug (at lower doses or for shorter periods of time) to improve the function of abnormal tumor-associated vessels and has thus far led to promising clinical results [98, 99, 105, 107]. From an ecological perspective, microenvironmental modulation as opposed to targeted therapy, would be the equivalent difference between killing one species of animal in an ecosystem (targeted therapy) and transforming the environment the ecosystem resides within (such as converting a rainforest into a desert; microenvironmental modulation). Although there could be sweeping and multifaceted benefits to this strategy, the evolutionary outcomes of microenvironmental modulation are far less predictable and therefore warrant extensive characterization.
Elimination: Recognition and Killing
The process of elimination
The final step of anti-tumor immunity is the elimination of tumor cells, which is initiated by target cell recognition and binding by CTLs and completed by effector molecule-mediated killing [108, 109]. Once CTLs enter the tumor bed they must locate and engage with their TCR-specific antigen, which is presented on MHC-I molecules. Nearly all nucleated cells are capable of presenting peptides on MHC-I receptors, including tumor cells. Upon antigen recognition, CTLs bind target cells and establish an ‘immunological synapse’ to deliver death ligands and granules [108, 109]. Stable immunological synapses have been shown to provide long-lived contacts between CTLs and target cells, however recent in vivo evidence has suggested that highly dynamic contacts formed during the sustained motility of CTLs on target cells can also mediate cell killing [109]. Immunological synapses are formed around antigen-bound TCRs and are further facilitated by either CD103/E-cadherin or LFA-1/ICAM-1 adhesion [110, 111]. Actin cytoskeletal dynamics within CTLs, mediated by Arp2/3 and formins, are thought to cluster additional adhesion molecules and strengthen bonds, leading to the maturation of immunological synapses [111].
Synapse formation directs the repolarization of CTLs, leading to the directional exocytosis of cytolytic granules (such as granzymes) and/or presentation of death ligands (such as FASL and TRAIL) [109–111]. Delivery of cytolytic granules is reliant upon perforation of the target cell membrane, largely by perforin, which also has innate cytolytic ability [111]. Additionally, it has been proposed that the mechanical forces occurring at immunological synapses contribute to pore expansion within target cell membranes [109, 111]. It has been suggested that multiple CTLs and contacts are required for optimal target cell killing, which may speak to the heterogeneity of CTL effector molecule expression and variability in target cell susceptibility to specific death signals [109]. Ultimately, CTL-mediated target cell killing is a dynamic process mediated by antigen recognition, target cell binding, and delivery of effector molecules (Figure 2D).
Evading elimination
Tumors evolve to evade immune elimination. Escape from recognition via dysregulation of antigen presentation is a prominent mechanism seen in a variety of cancers. Data from NSCLC, colorectal, uterine, breast, gastric, cervical, and head and neck cancer have all shown genomic alterations in HLA and specifically, in MHC-I associated genes [112–115]. Although the functional consequences of specific genetic aberrations have not been fully characterized, decreases in HLA expression (via loss of heterozygosity) and mutations leading to MHC instability are frequently reported [112–115]. In addition to HLA class I (HLA-I) downregulation, several malignancies including breast, colorectal, lung, renal, ovarian cancer, and melanoma have shown increased HLA-G and/or HLA-E expression [116, 117]. Expression of HLA-G or HLA-E may promote tumor immunotolerance and is a biomarker of poor clinical outcome [116, 117]. Importantly, in a mouse model of sarcoma, it was suggested that loss of TSA expression or MHC-I presentation was necessary and sufficient to drive immunoediting, leading to the evolution of less immunogenic tumors [118].
In addition to aberrant antigen presentation, cancer cells can evade elimination by perturbing CTL binding and directly resisting CTL death signals (Figure 2D). As discussed above, CTLs require physical interaction with tumor cells to initiate killing. This interaction is mediated by CTL binding to E-cadherin or ICAM-1 on tumor cells. ICAM-1 expression is downregulated in some cancer cells, which confers resistance to CTL killing [119]. Similarly, E-cadherin expression is downregulated in some tumors as part of epithelial-mesenchymal transition (EMT) [120]. Finally, PD-L1 (also known as B7-H1) expression on tumor cells has also been linked to resistance of CTL-mediated apoptosis and FasL-mediated death, in a process termed ‘molecular shielding’ [121]. Therefore, PD-L1 expression can directly confer antiapoptotic signals to tumor cells. Collectively, these studies demonstrate the diverse mechanisms by which tumors evade immune elimination (Figure 2D).
Clinical strategies to restore elimination
Several of the successful immunotherapies already being used in the clinic have overlapping benefits for restoring immune-mediated elimination. Although anti-PD-1/PD-L1 drugs are largely discussed in the context of immune activation (discussed above), blocking PD-1/PD-L1 interaction may also inhibit molecular shielding and make cancer cells more susceptible to CTL-mediated killing [122] (Figure 2D). Additionally, chimeric antigen receptor (CAR) T cell therapy may be a key therapeutic strategy to restore T cell recognition of tumor cells that have downregulated MHC-I antigen presentation (Figure 2D). CAR T cells bypass the need for antigen presentation on MHC-I and instead utilize synthetic receptors to target specific tumor antigens [123, 124]. Additionally, next-generation CARs also contain co-stimulatory domains, which further enhance CAR T cell activity [124]. However, CAR T cell therapy has had limited success in solid tumors, potentially due to antigen heterogeneity and microenvironmental obstacles [123, 124].
Novel immunotherapies such as bispecific T cell engagers (BiTEs) are also being developed to restore immune-mediated elimination (Figure 2D). As the name implies, BiTEs are bispecific molecules that are engineered to bind T cells (usually via CD3) and tumor cells (via a TAA) [125, 126]. The tandem engagement of BiTEs to both of their target cell types physically links T cells to tumor cells, leading to immunological synapse formation and T cell-mediated killing [125]. Although the mechanisms regulating BiTE functions are not fully understood, clinical trials have shown promising results in hematological malignancies and successful preclinical studies have promoted phase I clinical trials for BiTEs targeted to solid tumors [125, 126]. However, several limitations including a short serum half-life and preferential cytotoxicity by memory T cells suggest that further optimization of this novel technology will be required [125].
Potential for resistance to therapeutic elimination
Clinical restoration of elimination can serve as a potent selective pressure. From an evolutionary perspective, a major flaw in the therapeutics discussed above (CAR T cell therapy and BiTEs) derives from the antigen specificity required by each of these strategies. Both utilize TAAs as tumor cell identifiers, which aside from issues with autoimmunity (due to low levels of TAA expression in normal tissues), will inherently select for tumor cells that do not express the targeted antigen. Tumor cell antigen expression is heterogeneous and subject to change via genetic and epigenetic mechanisms (discussed in the identification section above). Therefore, immunotherapies targeting specific TAAs will favor the propagation of preexisting tumor cells that do not express the TAA and further select for subclones that downregulate antigen expression. Importantly, therapeutics targeting antigens expressed in less aggressive cells (such as MT-110, a BiTE targeting EpCam) may select for highly aggressive subclones that have lost these antigens (such as cells that have undergone an EMT program). Therefore, current clinical strategies used to restore elimination may drive tumor evolution towards an aggressive and immune-resistant phenotype.
Concluding Remarks
Here, we streamline anti-tumor immunity into a four-step process and review the biological requirements of each step. We present a range of mechanisms that tumors evolve to disrupt each immunity step, and thus escape anti-tumor immunity (Figure 3). We then discuss potential strategies for the therapeutic restoration of each step, as well as possible avenues of acquired resistance to these restoration strategies. We discuss evasion and resistance from a Darwinian perspective to conceptualize how natural selection drives primary immunoediting and how therapy-induced selection may drive acquired resistance in the clinic.
Figure 3. Multiple escape mechanisms at each immunity step provide an opportunity for immune escape and selection of an immune-evasive tumor.
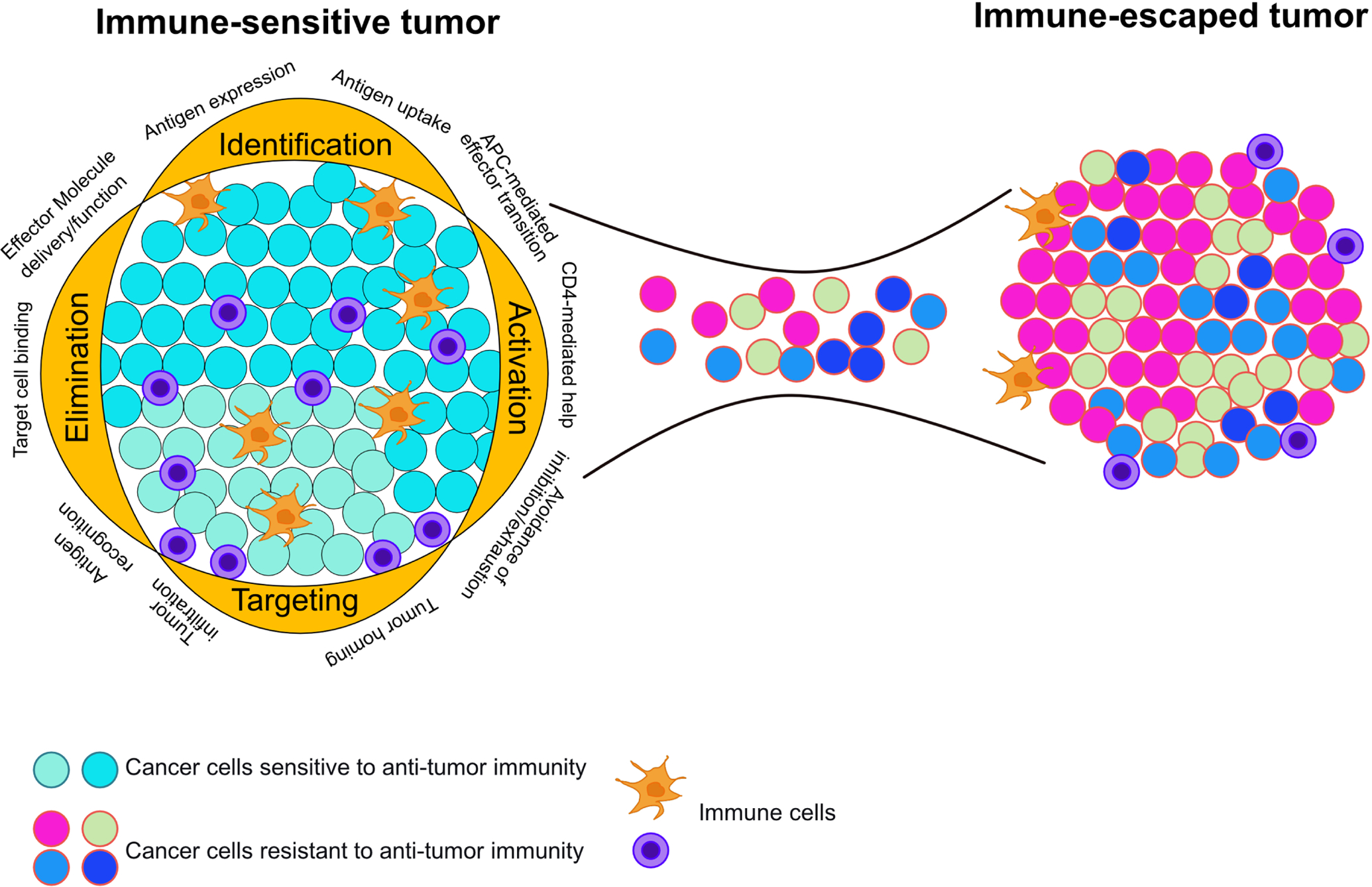
An immune-sensitive tumor contains intact immunity steps. At each step there are multiple opportunities for immune escape to occur, selecting for immune evasive cells. Elimination of immune-sensitive clones selects for cells with a variety of immune evasive properties. These cells are free to expand into immunologically heterogeneous, clinically detectable lesions. The diversity of immune escape mechanisms represented in this tumor reveals how immunotherapeutic approaches targeting only one mechanism of immune escape will select for clones with alternative escape mechanisms. Thus, a small biopsy may not reveal the heterogeneity of immune escape mechanisms present within a tumor.
Although we discuss the steps of anti-tumor immunity in a compartmentalized fashion, it is important to highlight that each step is innately interconnected with and reliant upon all previous steps (Figure 3). Therefore, tumors may perturb step four by disrupting any of the mechanisms of steps one through three, with the same being true for all steps. Although we largely focus on acquired therapeutic resistance (to model how selection drives tumor evolution towards resistance), innate therapeutic resistance is likely conferred by an escaped immunity step outside the one being clinically targeted. Additionally, acquired therapeutic resistance can also occur by disrupting other steps of immunity. Although this exponentially increases the avenues by which tumor cells can evade anti-tumor immunity, we propose that by improved understanding of the natural and therapy-imposed selective pressures and mechanisms driving cancer cell heterogeneity, we can begin to predict patient-specific dynamics of immune evasion.
Another critical point is that immunoediting describes the temporal accumulation of immune-evasion mechanisms (broken immunity steps). Therefore, we propose that earlier detection and treatment provides a clinical opportunity to restore anti-tumor immunity before a broad repertoire of evasion mechanisms are acquired, thus increasing the likelihood of immunotherapeutic success. Immune escape has been suggested to occur even before tumor invasion [127]. Thus, single-agent immunotherapies, which demonstrate lower levels of adverse events compared to combination treatments, may be the most successful during early-stage disease. There is also a significant need to develop a comprehensive, standardized panel of biomarkers for disrupted immunity steps. This panel would allow clinicians to select appropriate immunotherapeutic agents and determine the minimum number of non-redundant immune pathways in need of clinical restoration. A promising member of this panel may be Immunoscore, a method of quantifying anti-tumor immune infiltration [128]. However, markers for multiple steps of the anti-tumor immunity cascade should be incorporated. An additional consideration for immune escape diagnostics is the spatial heterogeneity of immune evasion phenotypes that may be present within a tumor (Figure 3). While single biopsies may not reveal the diversity of immune escape mechanisms within a tumor, multiple spatially distributed biopsies may be more representative of immune evasion phenotypes and should thus be considered.
Finally, cellular heterogeneity and plasticity are drivers of tumor evolution and thus, mediate immune escape. Therefore, epigenetic modulators that decrease these properties should be considered as combination treatments with immunotherapy to slow tumor evolution, immune escape and acquired immunotherapeutic resistance [129].
ACKNOWLEDGEMENTS
We thank members of our laboratory for their critical reading of our manuscript and useful suggestions. The authors were supported by the National Cancer Institute R35CA197623 (K.P.), DFCI Helen Gurley Brown Presidential Initiative Award (J.P.), and the Breast Cancer Research Foundation (K.P.). We apologize to all authors whose publication we may have omitted due to space limitations.
COMPETING FINANCIAL INTERESTS
K.P is a member of the scientific advisory board of Acrivon Therapeutics, Scorpion Therapeutics, and Vividion Therapeutics, has equity positions in Scorpion Therapeutics, receives research support from Novartis, and is a consultant to Aria Pharmaceuticals.
Declaration of interests
Kornelia Polyak reports financial support was provided by National Cancer Institute. Kornelia Polyak reports financial support was provided by Breast Cancer Research Foundation. Kornelia Polyak reports a relationship with Scorpion Therapeutics that includes: board membership, consulting or advisory, and equity or stocks. Kornelia Polyak reports a relationship with Vividion Therapeutics that includes: board membership, consulting or advisory, and equity or stocks. Kornelia Polyak reports a relationship with Acrivon Therapeutics that includes: board membership. Kornelia Polyak reports a relationship with Aria Pharmaceuticals that includes: consulting or advisory. Kornelia Polyak reports a relationship with AstraZeneca Pharmaceuticals LP that includes: consulting or advisory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A, Identification of neutral tumor evolution across cancer types, Nat Genet 48(3) (2016) 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Turajlic S, Sottoriva A, Graham T, Swanton C, Resolving genetic heterogeneity in cancer, Nat Rev Genet 20(7) (2019) 404–416. [DOI] [PubMed] [Google Scholar]
- [3].Greaves M, Maley CC, Clonal evolution in cancer, Nature 481(7381) (2012) 306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C, Intratumor heterogeneity and branched evolution revealed by multiregion sequencing, N Engl J Med 366(10) (2012) 883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dagogo-Jack I, Shaw AT, Tumour heterogeneity and resistance to cancer therapies, Nat Rev Clin Oncol 15(2) (2018) 81–94. [DOI] [PubMed] [Google Scholar]
- [6].Burrell RA, Swanton C, Re-Evaluating Clonal Dominance in Cancer Evolution, Trends Cancer 2(5) (2016) 263–276. [DOI] [PubMed] [Google Scholar]
- [7].Belkhir S, Thomas F, Roche B, Darwinian Approaches for Cancer Treatment: Benefits of Mathematical Modeling, Cancers (Basel) 13(17) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vendramin R, Litchfield K, Swanton C, Cancer evolution: Darwin and beyond, EMBO J 40(18) (2021) e108389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, Gelmon K, Chia S, Mar C, Wan A, Laks E, Biele J, Shumansky K, Rosner J, McPherson A, Nielsen C, Roth AJ, Lefebvre C, Bashashati A, de Souza C, Siu C, Aniba R, Brimhall J, Oloumi A, Osako T, Bruna A, Sandoval JL, Algara T, Greenwood W, Leung K, Cheng H, Xue H, Wang Y, Lin D, Mungall AJ, Moore R, Zhao Y, Lorette J, Nguyen L, Huntsman D, Eaves CJ, Hansen C, Marra MA, Caldas C, Shah SP, Aparicio S, Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution, Nature 518(7539) (2015) 422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, Matthews N, Rowan A, Chambers T, Moore DA, Turajlic S, Xu H, Lee SM, Forster MD, Ahmad T, Hiley CT, Abbosh C, Falzon M, Borg E, Marafioti T, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Shah R, Joseph L, Quinn AM, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Dentro S, Taniere P, O’Sullivan B, Lowe HL, Hartley JA, Iles N, Bell H, Ngai Y, Shaw JA, Herrero J, Szallasi Z, Schwarz RF, Stewart A, Quezada SA, Le Quesne J, Van Loo P, Dive C, Hackshaw A, Swanton C, Consortium TR, Tracking the Evolution of Non-Small-Cell Lung Cancer, N Engl J Med 376(22) (2017) 2109–2121. [DOI] [PubMed] [Google Scholar]
- [11].Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K, Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity, Nature 514(7520) (2014) 54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Capaci V, Mantovani F, Del Sal G, Amplifying Tumor-Stroma Communication: An Emerging Oncogenic Function of Mutant p53, Front Oncol 10 (2020) 614230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, Cancer immunoediting: from immunosurveillance to tumor escape, Nat Immunol 3(11) (2002) 991–8. [DOI] [PubMed] [Google Scholar]
- [14].Polyak K, Haviv I, Campbell IG, Co-evolution of tumor cells and their microenvironment, Trends Genet 25(1) (2009) 30–8. [DOI] [PubMed] [Google Scholar]
- [15].Mittal D, Gubin MM, Schreiber RD, Smyth MJ, New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape, Curr Opin Immunol 27 (2014) 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].O’Donnell JS, Teng MWL, Smyth MJ, Cancer immunoediting and resistance to T cell-based immunotherapy, Nat Rev Clin Oncol 16(3) (2019) 151–167. [DOI] [PubMed] [Google Scholar]
- [17].Dunn GP, Old LJ, Schreiber RD, The immunobiology of cancer immunosurveillance and immunoediting, Immunity 21(2) (2004) 137–48. [DOI] [PubMed] [Google Scholar]
- [18].Rosenberg SA, Raising the bar: the curative potential of human cancer immunotherapy, Sci Transl Med 4(127) (2012) 127ps8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A, Safety and Tumor Responses with Lambrolizumab (Anti-PD-1) in Melanoma, N Engl J Med (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD, Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma, J Clin Oncol 33(17) (2015) 1889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kelderman S, Schumacher TN, Haanen JB, Acquired and intrinsic resistance in cancer immunotherapy, Molecular oncology 8(6) (2014) 1132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, Allison JP, Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade, Cell 170(6) (2017) 1120–1133 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jackson CM, Choi J, Lim M, Mechanisms of immunotherapy resistance: lessons from glioblastoma, Nat Immunol 20(9) (2019) 1100–1109. [DOI] [PubMed] [Google Scholar]
- [24].Chen DS, Mellman I, Oncology meets immunology: the cancer-immunity cycle, Immunity 39(1) (2013) 1–10. [DOI] [PubMed] [Google Scholar]
- [25].Kim JM, Chen DS, Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure), Ann Oncol 27(8) (2016) 1492–504. [DOI] [PubMed] [Google Scholar]
- [26].Wagner S, Mullins CS, Linnebacher M, Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens, World J Gastroenterol 24(48) (2018) 5418–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Havel JJ, Chowell D, Chan TA, The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy, Nat Rev Cancer 19(3) (2019) 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, Maher CA, Panji S, Schaefer U, Kruger A, Lehvaslaiho M, Carninci P, Hayashizaki Y, Jongeneel CV, Simpson AJ, Old LJ, Hide W, Genome-wide analysis of cancer/testis gene expression, Proc Natl Acad Sci U S A 105(51) (2008) 20422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, Ren S, Zhou C, Tumor neoantigens: from basic research to clinical applications, J Hematol Oncol 12(1) (2019) 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Green DR, Ferguson T, Zitvogel L, Kroemer G, Immunogenic and tolerogenic cell death, Nat Rev Immunol 9(5) (2009) 353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hernandez C, Huebener P, Schwabe RF, Damage-associated molecular patterns in cancer: a double-edged sword, Oncogene 35(46) (2016) 5931–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, Lund T, Tanic M, Reading JL, Joshi K, Henry JY, Ghorani E, Wilson GA, Birkbak NJ, Jamal-Hanjani M, Veeriah S, Szallasi Z, Loi S, Hellmann MD, Feber A, Chain B, Herrero J, Quezada SA, Demeulemeester J, Van Loo P, Beck S, McGranahan N, Swanton C, consortium TR, Neoantigen-directed immune escape in lung cancer evolution, Nature 567(7749) (2019) 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, Tanaka S, Takayanagi S, Hana T, Takahashi S, Kitagawa Y, Koike T, Kobayashi Y, Nagae G, Yamamoto S, Ueda H, Tatsuno K, Narita Y, Nagane M, Ueki K, Nishikawa R, Aburatani H, Mukasa A, Saito N, Kakimi K, Reduced Neoantigen Expression Revealed by Longitudinal Multiomics as a Possible Immune Evasion Mechanism in Glioma, Cancer Immunol Res 7(7) (2019) 1148–1161. [DOI] [PubMed] [Google Scholar]
- [34].Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, Fatho M, Lennerz V, Wolfel T, Holzel M, Tuting T, Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation, Nature 490(7420) (2012) 412–6. [DOI] [PubMed] [Google Scholar]
- [35].Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC, Using immunotherapy to boost the abscopal effect, Nat Rev Cancer 18(5) (2018) 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, Heguy A, Imai N, Gnjatic S, Emerson RO, Zhou XK, Zhang T, Chachoua A, Demaria S, Radiotherapy induces responses of lung cancer to CTLA-4 blockade, Nat Med 24(12) (2018) 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, Sedrak C, Jungbluth AA, Chua R, Yang AS, Roman RA, Rosner S, Benson B, Allison JP, Lesokhin AM, Gnjatic S, Wolchok JD, Immunologic correlates of the abscopal effect in a patient with melanoma, N Engl J Med 366(10) (2012) 925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wu J, Waxman DJ, Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy, Cancer Lett 419 (2018) 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bommareddy PK, Shettigar M, Kaufman HL, Integrating oncolytic viruses in combination cancer immunotherapy, Nat Rev Immunol 18(8) (2018) 498–513. [DOI] [PubMed] [Google Scholar]
- [40].Sabado RL, Balan S, Bhardwaj N, Dendritic cell-based immunotherapy, Cell Res 27(1) (2017) 74–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, Guo C, Wu X, Li Y, Li X, Li G, Xiong W, Zeng Z, Neoantigen vaccine: an emerging tumor immunotherapy, Mol Cancer 18(1) (2019) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS, Alternative tumour-specific antigens, Nat Rev Cancer 19(8) (2019) 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yamamoto TN, Kishton RJ, Restifo NP, Developing neoantigen-targeted T cell-based treatments for solid tumors, Nat Med 25(10) (2019) 1488–1499. [DOI] [PubMed] [Google Scholar]
- [44].Stassi G, Todaro M, Zerilli M, Ricci-Vitiani L, Di Liberto D, Patti M, Florena A, Di Gaudio F, Di Gesu G, De Maria R, Thyroid cancer resistance to chemotherapeutic drugs via autocrine production of interleukin-4 and interleukin-10, Cancer Res 63(20) (2003) 6784–90. [PubMed] [Google Scholar]
- [45].Mittal SK, Roche PA, Suppression of antigen presentation by IL-10, Curr Opin Immunol 34 (2015) 22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zuniga-Pflucker JC, T-cell development made simple, Nat Rev Immunol 4(1) (2004) 67–72. [DOI] [PubMed] [Google Scholar]
- [47].Yang Q, Jeremiah Bell J, Bhandoola A, T-cell lineage determination, Immunol Rev 238(1) (2010) 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kumar BV, Connors TJ, Farber DL, Human T Cell Development, Localization, and Function throughout Life, Immunity 48(2) (2018) 202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mempel TR, Henrickson SE, Von Andrian UH, T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases, Nature 427(6970) (2004) 154–9. [DOI] [PubMed] [Google Scholar]
- [50].Gerritsen B, Pandit A, The memory of a killer T cell: models of CD8(+) T cell differentiation, Immunol Cell Biol 94(3) (2016) 236–41. [DOI] [PubMed] [Google Scholar]
- [51].Nguyen QP, Deng TZ, Witherden DA, Goldrath AW, Origins of CD4(+) circulating and tissue-resident memory T-cells, Immunology 157(1) (2019) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Nolz JC, Starbeck-Miller GR, Harty JT, Naive, effector and memory CD8 T-cell trafficking: parallels and distinctions, Immunotherapy 3(10) (2011) 1223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Clark RA, Resident memory T cells in human health and disease, Sci Transl Med 7(269) (2015) 269rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Amon L, Lehmann CHK, Baranska A, Schoen J, Heger L, Dudziak D, Transcriptional control of dendritic cell development and functions, Int Rev Cell Mol Biol 349 (2019) 55–151. [DOI] [PubMed] [Google Scholar]
- [55].Sharma P, Allison JP, The future of immune checkpoint therapy, Science 348(6230) (2015) 56–61. [DOI] [PubMed] [Google Scholar]
- [56].Pinzon-Charry A, Maxwell T, Lopez JA, Dendritic cell dysfunction in cancer: a mechanism for immunosuppression, Immunol Cell Biol 83(5) (2005) 451–61. [DOI] [PubMed] [Google Scholar]
- [57].Veglia F, Gabrilovich DI, Dendritic cells in cancer: the role revisited, Curr Opin Immunol 45 (2017) 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gasteiger G, Ataide M, Kastenmuller W, Lymph node - an organ for T-cell activation and pathogen defense, Immunol Rev 271(1) (2016) 200–20. [DOI] [PubMed] [Google Scholar]
- [59].Luckheeram RV, Zhou R, Verma AD, Xia B, CD4(+)T cells: differentiation and functions, Clinical & developmental immunology 2012 (2012) 925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bos R, Sherman LA, CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes, Cancer Res 70(21) (2010) 8368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Melssen M, Slingluff CL Jr., Vaccines targeting helper T cells for cancer immunotherapy, Curr Opin Immunol 47 (2017) 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gallimore A, Quezada SA, Roychoudhuri R, Regulatory T cells in cancer: where are we now?, Immunology 157(3) (2019) 187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Amsen D, van Gisbergen K, Hombrink P, van Lier RAW, Tissue-resident memory T cells at the center of immunity to solid tumors, Nat Immunol 19(6) (2018) 538–546. [DOI] [PubMed] [Google Scholar]
- [64].Zhang N, Bevan MJ, CD8(+) T cells: foot soldiers of the immune system, Immunity 35(2) (2011) 161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion, Nat Rev Immunol 15(8) (2015) 486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kurachi M, CD8(+) T cell exhaustion, Semin Immunopathol 41(3) (2019) 327–337. [DOI] [PubMed] [Google Scholar]
- [67].Liu C, Workman CJ, Vignali DA, Targeting regulatory T cells in tumors, FEBS J 283(14) (2016) 2731–48. [DOI] [PubMed] [Google Scholar]
- [68].Shitara K, Nishikawa H, Regulatory T cells: a potential target in cancer immunotherapy, Ann N Y Acad Sci 1417(1) (2018) 104–115. [DOI] [PubMed] [Google Scholar]
- [69].Buchbinder EI, Desai A, CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition, Am J Clin Oncol 39(1) (2016) 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK, PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome, Front Pharmacol 8 (2017) 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cha JH, Chan LC, Li CW, Hsu JL, Hung MC, Mechanisms Controlling PD-L1 Expression in Cancer, Mol Cell 76(3) (2019) 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang X, Meng X, Chen Y, Leng SX, Zhang H, The Biology of Aging and Cancer: Frailty, Inflammation, and Immunity, Cancer J 23(4) (2017) 201–205. [DOI] [PubMed] [Google Scholar]
- [73].Derhovanessian E, Solana R, Larbi A, Pawelec G, Immunity, ageing and cancer, Immun Ageing 5 (2008) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Simon AK, Hollander GA, McMichael A, Evolution of the immune system in humans from infancy to old age, Proc Biol Sci 282(1821) (2015) 20143085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, Chen S, Tian Y, Wang D, Fei K, Yao Z, Wang S, Lu Z, Wang Z, Wang J, TCR Repertoire Diversity of Peripheral PD-1(+)CD8(+) T Cells Predicts Clinical Outcomes after Immunotherapy in Patients with Non-Small Cell Lung Cancer, Cancer Immunol Res 8(1) (2020) 146–154. [DOI] [PubMed] [Google Scholar]
- [76].Schreiber K, Karrison TG, Wolf SP, Kiyotani K, Steiner M, Littmann ER, Pamer EG, Kammertoens T, Schreiber H, Leisegang M, Impact of TCR Diversity on the Development of Transplanted or Chemically Induced Tumors, Cancer Immunol Res 8(2) (2020) 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, Schonitzer D, Grubeck-Loebenstein B, Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons, J Virol 79(6) (2005) 3675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y, T Cell Dysfunction and Exhaustion in Cancer, Frontiers in Cell and Developmental Biology 8(17) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dolina JS, Van Braeckel-Budimir N, Thomas GD, Salek-Ardakani S, CD8(+) T Cell Exhaustion in Cancer, Front Immunol 12 (2021) 715234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mendes F, Domingues C, Rodrigues-Santos P, Abrantes AM, Goncalves AC, Estrela J, Encarnacao J, Pires AS, Laranjo M, Alves V, Teixo R, Sarmento AB, Botelho MF, Rosa MS, The role of immune system exhaustion on cancer cell escape and anti-tumor immune induction after irradiation, Biochim Biophys Acta 1865(2) (2016) 168–75. [DOI] [PubMed] [Google Scholar]
- [81].Kythreotou A, Siddique A, Mauri FA, Bower M, Pinato DJ, Pd-L1, J Clin Pathol 71(3) (2018) 189–194. [DOI] [PubMed] [Google Scholar]
- [82].Syn NL, Teng MWL, Mok TSK, Soo RA, De-novo and acquired resistance to immune checkpoint targeting, Lancet Oncol 18(12) (2017) e731–e741. [DOI] [PubMed] [Google Scholar]
- [83].Mahoney KM, Freeman GJ, McDermott DF, The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma, Clin Ther 37(4) (2015) 764–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Colak S, Ten Dijke P, Targeting TGF-beta Signaling in Cancer, Trends Cancer 3(1) (2017) 56–71. [DOI] [PubMed] [Google Scholar]
- [85].Jones SA, Jenkins BJ, Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer, Nat Rev Immunol 18(12) (2018) 773–789. [DOI] [PubMed] [Google Scholar]
- [86].Ouyang W, O’Garra A, IL-10 Family Cytokines IL-10 and IL-22: from Basic Science to Clinical Translation, Immunity 50(4) (2019) 871–891. [DOI] [PubMed] [Google Scholar]
- [87].Apte RS, Chen DS, Ferrara N, VEGF in Signaling and Disease: Beyond Discovery and Development, Cell 176(6) (2019) 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Young A, O’Donnell JS, Allen S, Smyth MJ, Teng MW, Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease, Cancer Discov 6(12) (2016) 1382–1399. [DOI] [PubMed] [Google Scholar]
- [89].Anderson KG, Stromnes IM, Greenberg PD, Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies, Cancer Cell 31(3) (2017) 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Tanaka A, Sakaguchi S, Targeting Treg cells in cancer immunotherapy, Eur J Immunol 49(8) (2019) 1140–1146. [DOI] [PubMed] [Google Scholar]
- [91].Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A, Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy, Cell 168(4) (2017) 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, Hermann-Kleiter N, Lower M, Baier G, Krogsdam A, Trajanoski Z, Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution, Nat Commun 9(1) (2018) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TN, Lo RS, Ribas A, Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma, N Engl J Med 375(9) (2016) 819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bai J, Gao Z, Li X, Dong L, Han W, Nie J, Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade, Oncotarget 8(66) (2017) 110693–110707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nourshargh S, Alon R, Leukocyte migration into inflamed tissues, Immunity 41(5) (2014) 694–707. [DOI] [PubMed] [Google Scholar]
- [96].Masopust D, Schenkel JM, The integration of T cell migration, differentiation and function, Nat Rev Immunol 13(5) (2013) 309–20. [DOI] [PubMed] [Google Scholar]
- [97].Slaney CY, Kershaw MH, Darcy PK, Trafficking of T cells into tumors, Cancer Res 74(24) (2014) 7168–74. [DOI] [PubMed] [Google Scholar]
- [98].van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, de Vries IJM, Migrating into the Tumor: a Roadmap for T Cells, Trends Cancer 3(11) (2017) 797–808. [DOI] [PubMed] [Google Scholar]
- [99].Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, Depil S, Cold Tumors: A Therapeutic Challenge for Immunotherapy, Front Immunol 10 (2019) 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hornung A, Sbarrato T, Garcia-Seyda N, Aoun L, Luo X, Biarnes-Pelicot M, Theodoly O, Valignat MP, A Bistable Mechanism Mediated by Integrins Controls Mechanotaxis of Leukocytes, Biophys J 118(3) (2020) 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Badalamenti G, Fanale D, Incorvaia L, Barraco N, Listi A, Maragliano R, Vincenzi B, Calo V, Iovanna JL, Bazan V, Russo A, Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone?, Cell Immunol 343 (2019) 103753. [DOI] [PubMed] [Google Scholar]
- [102].Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, Sasso M, Bilocq AM, Kirilovsky A, Obenauf AC, Hamieh M, Berger A, Bruneval P, Tuech JJ, Sabourin JC, Le Pessot F, Mauillon J, Rafii A, Laurent-Puig P, Speicher MR, Trajanoski Z, Michel P, Sesboue R, Frebourg T, Pages F, Valge-Archer V, Latouche JB, Galon J, Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability, Immunity 44(3) (2016) 698–711. [DOI] [PubMed] [Google Scholar]
- [103].Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA, Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer, Cancer Res 70(15) (2010) 6171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Gordon-Alonso M, Hirsch T, Wildmann C, van der Bruggen P, Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration, Nat Commun 8(1) (2017) 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, Denker M, Leveque V, Canamero M, Babitski G, Koeppen H, Ziai J, Sharma N, Gaire F, Chen DS, Waterkamp D, Hegde PS, McDermott DF, Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma, Nat Commun 7 (2016) 12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Jain RK, Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia, Cancer Cell 26(5) (2014) 605–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK, Normalization of the vasculature for treatment of cancer and other diseases, Physiol Rev 91(3) (2011) 1071–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Martinez-Lostao L, Anel A, Pardo J, How Do Cytotoxic Lymphocytes Kill Cancer Cells?, Clin Cancer Res 21(22) (2015) 5047–56. [DOI] [PubMed] [Google Scholar]
- [109].Halle S, Halle O, Forster R, Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo, Trends Immunol 38(6) (2017) 432–443. [DOI] [PubMed] [Google Scholar]
- [110].Franciszkiewicz K, Le Floc’h A, Boutet M, Vergnon I, Schmitt A, Mami-Chouaib F, CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions, Cancer Res 73(2) (2013) 617–28. [DOI] [PubMed] [Google Scholar]
- [111].Basu R, Huse M, Mechanical Communication at the Immunological Synapse, Trends Cell Biol 27(4) (2017) 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N, Molecular and genetic properties of tumors associated with local immune cytolytic activity, Cell 160(1–2) (2015) 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T, The urgent need to recover MHC class I in cancers for effective immunotherapy, Curr Opin Immunol 39 (2016) 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, Swanton C, Consortium TR, Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution, Cell 171(6) (2017) 1259–1271 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Ozcan M, Janikovits J, von Knebel Doeberitz M, Kloor M, Complex pattern of immune evasion in MSI colorectal cancer, Oncoimmunology 7(7) (2018) e1445453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Rouas-Freiss N, Moreau P, Ferrone S, Carosella ED, HLA-G proteins in cancer: do they provide tumor cells with an escape mechanism?, Cancer Res 65(22) (2005) 10139–44. [DOI] [PubMed] [Google Scholar]
- [117].de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ, HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients, J Immunol 185(12) (2010) 7452–9. [DOI] [PubMed] [Google Scholar]
- [118].DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T, Expression of tumour-specific antigens underlies cancer immunoediting, Nature 482(7385) (2012) 405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ueda R, Kohanbash G, Sasaki K, Fujita M, Zhu X, Kastenhuber ER, McDonald HA, Potter DM, Hamilton RL, Lotze MT, Khan SA, Sobol RW, Okada H, Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1, Proc Natl Acad Sci U S A 106(26) (2009) 10746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kalluri R, Weinberg RA, The basics of epithelial-mesenchymal transition, J Clin Invest 119(6) (2009) 1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L, B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells, Blood 111(7) (2008) 3635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L, Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity, Cancer Res 65(3) (2005) 1089–96. [PubMed] [Google Scholar]
- [123].Newick K, O’Brien S, Moon E, Albelda SM, CAR T Cell Therapy for Solid Tumors, Annu Rev Med 68 (2017) 139–152. [DOI] [PubMed] [Google Scholar]
- [124].Met O, Jensen KM, Chamberlain CA, Donia M, Svane IM, Principles of adoptive T cell therapy in cancer, Semin Immunopathol 41(1) (2019) 49–58. [DOI] [PubMed] [Google Scholar]
- [125].Huehls AM, Coupet TA, Sentman CL, Bispecific T-cell engagers for cancer immunotherapy, Immunol Cell Biol 93(3) (2015) 290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Clynes RA, Desjarlais JR, Redirected T Cell Cytotoxicity in Cancer Therapy, Annu Rev Med 70 (2019) 437–450. [DOI] [PubMed] [Google Scholar]
- [127].Mascaux C, Angelova M, Vasaturo A, Beane J, Hijazi K, Anthoine G, Buttard B, Rothe F, Willard-Gallo K, Haller A, Ninane V, Burny A, Sculier JP, Spira A, Galon J, Immune evasion before tumour invasion in early lung squamous carcinogenesis, Nature 571(7766) (2019) 570–575. [DOI] [PubMed] [Google Scholar]
- [128].Mahoney KM, Rennert PD, Freeman GJ, Combination cancer immunotherapy and new immunomodulatory targets, Nat Rev Drug Discov 14(8) (2015) 561–84. [DOI] [PubMed] [Google Scholar]
- [129].Hinohara K, Polyak K, Intratumoral Heterogeneity: More Than Just Mutations, Trends Cell Biol 29(7) (2019) 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]