

Abstract
Due to the rapidly growing number of older people worldwide and the concomitant increase in cardiovascular complications, there is an urgent need for age-related cardiac disease modeling and drug screening platforms. In the present study, we developed a cardiac tissue chip model that incorporates hemodynamic loading and mimics essential aspects of the infarcted aging heart. We induced cellular senescence in H9c2 myoblasts using low-dose doxorubicin treatment. These senescent cells were then used to engineer cardiac tissue fibers, which were subjected to hemodynamic stresses associated with pressure-volume changes in the heart. Myocardial ischemia was modeled in the engineered cardiac tissue via hypoxic treatment. Our results clearly show that acute low dose doxorubicin treatment-induced senescence, as evidenced by morphological and molecular markers, including enlarged and flattened nuclei, DNA damage response foci, and increased expression of cell cycle inhibitor p16INK4a, p53, and ROS. Under normal hemodynamic load, the engineered cardiac tissues demonstrated cell alignment and retained cardiac cell characteristics. Our senescent cardiac tissue model of hypoxia-induced myocardial infarction recapitulated the pathological disease hallmarks such as increased cell death and upregulated expression of ANP and BNP. In summary, the described methodology provides a novel approach to generate stress-induced aging cardiac cell phenotypes and engineer cardiac tissue chip models to study the cardiovascular disease pathologies associated with aging.
Keywords: Aging, Biomimetic Cardiac Tissue Chip Models, Cellular Senescence, Hypoxia, DNA damage
Introduction
Cardiovascular diseases are the foremost causes of death in the elderly, as those older than 65 years account for higher than 80% of patients with ischemic heart disease [Rosamond et al., 2007]. Additionally, the exponential rise in mortality rate related to cardiovascular diseases in the geriatric population signifies that cardiac aging may be a significant risk factor for cardiovascular pathology such as Myocardial infarction (MI). Numerous studies indicate aging results in decreased cardiac function and increased myocardial apoptosis in normal humans. [Oxenham and Sharpe, 2003; Triposkiadis et al., 2019].
Cellular senescence is a complex and common observation in the aged organism that involves a permanent state of cell cycle arrest accompanied by metabolic traits like elevated secretion of pro-inflammatory and other factors. Increased cellular senescence can cause organismal aging manifested by impaired tissue function and reduced stem cell number. In vitro, morphologically senescent cells are large, flat, vacuolized, and, sometimes, multinucleated. [Muñoz-Espín and Serrano, 2014] The distinct molecular features of senescent cells are chronic DNA damage response (DDR), accumulated DDR foci, increased expression of cell cycle inhibitors like p26, p21, and p53, growth factors like TGFΒ, expression of the senescence-associated marker p16INK4a, accumulation of reactive oxygen species (ROS) [Kortlever et al., 2006; Kortlever et al., 2008; Elzi et al., 2012; Malaquin et al., 2015; Kannappan et al., 2017; Kannappan et al., 2019].
Although aging animal models of MI provide a valuable resource to study disease progression and discover potential therapeutic targets, they possess significant limitations like low throughput, labor-intensive surgical techniques, and exhausting histopathology assays. Besides, diversity in genetics and physiology amongst the small-animal models and humans narrow the application of the data for modeling the progression of cardiac disease in humans. In vitro senescence model incorporated with in-vivo like physiological condition could be developed into a plausible alternative to study the aging-mediated progression of cardiac diseases. Cellular senescence can be induced by stimuli such as ionizing radiation, oxidative stress, mechanical stress, heat shock, heavy metals, and genotoxic drugs [Fridlyanskaya et al., 2015]. These stimuli damage the DNA of the cell. Unrepaired DNA double-strand break is one of the triggers of cellular senescence. Doxorubicin (Doxo), an anthracycline antibiotic and a DNA intercalating agent that causes DSBs, is a commonly used anticancer drug and can create stress-induced premature cellular senescence.
Engineered human cardiac tissues are evolving as preferred models for drug screening [Smith et al., 2017; Mittal et al., 2019], tissue maturation and regeneration [Weinberger et al., 2017; Rogers et al., 2018], and cellular interactions models recapitulating the infarcted aging heart integrated with hemodynamic loading are lacking. Recently our group has successfully used the Biomimetic Cardiac Tissue Model (BCTM) to recreate hemodynamic loading associated with normal cardiac physiological as well as pathophysiological conditions. Our disease model precisely recapitulated the major structural and molecular changes associated with pressure and volume overload [Rogers et al., 2018; Rogers et al., 2019]. With our expertise to engineer cardiac tissue chip models and generate stress-induced cardiac cellular senescence, we developed an aging cardiac tissue chip model to study age-related cardiovascular disease.
Materials and Methods
Generating Senescent Cardiac Phenotype
H9c2 Rat myoblasts derived from rat embryonic heart tissue [Kimes and Brandt, 1976], referred to as cardiac cells in the manuscript, were purchased from ATCC (H9c2(2–1), ATCC; CRL-1446) and cultured in Dulbecco’s Modified Eagle’s Medium (ATCC; 30–2002) supplemented with 10% Fetal Bovine Serum (FBS)(Fisher; FB12999102) in 5% CO2 at 37 °C. 1 mM Doxo (Sigma-Aldrich; 44583) was freshly diluted to 0.5 μM in the culture medium. The cardiac cells in their exponential growth phase were treated with 0.5 μM Doxo. After 24 hours, the treatment medium was removed, the cells were washed twice with PBS and maintained in a complete culture medium for 48 hours. After 48 hrs of recovery and confirming the cellular senescence, the cells were used to create senescent cardiac tissue fibers.
Fibroblast Cell Culture
Primary human dermal fibroblasts (ATCC PCS-201–010) were cultured in DMEM supplemented with 10% FBS in a humidified incubator containing 5% CO2 at 37 °C.
Cellular Senescence
The cellular senescence parameters were measured in baseline conditions and following senescence induction using the above method. Senescence was induced in the cardiac cells seeded in an 8-well chamber (15,000 cells per well). Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences; 15710), and immunofluorescence staining was performed. Cells were stained with anti-Phospho-Histone H2A.X (Ser139) Antibody (Cell Signaling Technology; 2577) and immunofluorescence images were taken from 20 randomly selected, non-overlapping fields to spot DNA damage response foci using a fluorescence microscope (Nikon; Eclipse Ti-E). CellProfiler software was employed for the recognition of the γH2A.X-positive DDR foci and rendering of the data[Miller et al., 2019]. The same software was used to count the number of foci per nucleus. The cells that reached senescence and irreversible growth arrest were evaluated by the expression of the senescence-associated protein p16INK4a (Abcam; ab54210), transcription factor p53 (Cell Signaling Technology; 2527), and ser 15-phosphorylated p53 (Cell Signaling Technology; 9286).
Reactive Oxygen Species Quantification
Cardiac cells were seeded in a 96-well plate (3000 cells per well), and Doxo-mediated senescence was induced. The cells were treated with 10 μM of 5- (and 6-) chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-DCFH) (Invitrogen; C6827). The intracellular reactive oxygen species produced by the senescent cells convert CM-DCFH to a fluorescent CM-DCF [Xie et al., 1999], which can be quantified. After 60 minutes of incubation at 37°C, ROS activity was captured using fluorescence microscopy, and the fluorescence intensity was quantified using CellProfiler Software. ROS fluorescence intensity was normalized to the number of cells using the 4,6-diamidino-2-phenylindole (DAPI) count.
Cardiac tissue device Fabrication
The cardiac tissue devices used in this study were fabricated via soft lithography using polydimethylsiloxane (PDMS) (QSil 216, Quantum Silicones, Richmond, VA, USA) mixed with a 10:1 base-to-crosslinker ratio. First, a PDMS frame containing a 2 × 4 cm centrally-located rectangular cutout was fabricated from an 8.5 mm-thick PDMS slab (cured at 125°C for 2 hours in an oven). Cylindrical posts measuring 1 mm in diameter were also punched from this slab and trimmed to a 6 mm height. Next, 10 mL of PDMS was spin-coated onto an approximately 78.5 cm2 circular silicon wafer, which was then placed atop a hot plate. Immediately thereafter, 3 pairs of posts were placed onto the PDMS-coated wafer with the aid of a lightweight removable PDMS guide, which fit snugly around the posts. With the posts and stencil in place, the wafer was cured for 10 minutes at 125°C, resulting in a 1 mm-thick membrane with 3 pairs of posts. The posts in each pair measured 8 mm center-to-center, and each pair was spaced approximately 8 mm apart from the neighboring pair(s). Following this, the frame and post-laden membrane were cleaned and bonded with oxygen plasma (Harrick Plasma Systems, Ithaca, NY, USA) to create a durable, water-tight seal using 700 mTorr pressure, 30 W, and 30 seconds of exposure. The device was then carefully peeled off of the silicon wafer and the excess membrane trimmed. After autoclaving, 3.5% molten agarose was pipetted into the device chamber up to 1 mm from the tops of the posts. Following cooling and solidification, circles around the posts and a connecting central lane for each pair were punched out of the agarose, leaving 3 dumbbell-shaped troughs in each device chamber. After rinsing with PBS to remove any agarose debris from the PDMS membrane at the bottom of the troughs, the device chamber was filled with a 1% w/v solution of bovine serum albumin (BSA) (Sigma-Aldrich; A7906) in PBS and stored at 4°C overnight. This BSA coating aided in preventing the cells and fibrin/Matrigel from adhering to either the membrane or the agarose sidewalls during fiber seeding. 30 minutes prior to cell seeding, the device was moved to a 37°C incubator to warm, and immediately prior to seeding, the BSA solution was removed, and the device was washed once more with PBS.
Fibrin/Matrigel Gel Encapsulation
The cardiac cells were dissociated with 0.25% Trypsin/EDTA (Gibco; 25200–056) and intermixed with the human dermal fibroblasts, which were dissociated with 0.05% Trypsin/EDTA (Thermo Fisher Scientific; 25300–054). Each 200 μL fiber contained 2.4 × 106 H9c2s and 0.267 × 106 human dermal fibroblasts and was generated via a 3-step process. First, a 0.048 units/μL solution of thrombin (Millipore; 605195) in 40 mM CaCl2 was prepared, and 10 μL was dispensed into microcentrifuge tubes (one for each fiber). After trypsinizing, combining, and pelleting the H9c2s and fibroblasts, they were resuspended in a 10% v/v solution of either growth factor reduced Matrigel (Corning; 354230) or hESC-qualified Matrigel (Corning; 354277) dissolved in DMEM supplemented with 10% FBS, 1% antibiotic-antimycotic (Gibco; 15240–062), and 2 mg/mL aminocaproic acid (Acros; 103301000). (The growth factor reduced Matrigel was employed in the low-pressure dynamic studies, whereas the hESC-qualified Matrigel was used in the static normoxia and hypoxia studies.) Next, 180 μL of the cell suspension was added to each microcentrifuge tube. Then, 10 μL of 40 mg/mL fibrinogen (Millipore; 341576) was added, the cell suspension was gently pipetted to mix, and finally, the 200 μL suspension was pipetted into a dumbbell-shaped trough in the device chamber. This last step was done 1 tube/fiber at a time to prevent premature gelling in the tube or pipet tip. Each fiber had a final concentration of 2 mg/mL fibrin, 1.2 units thrombin/mg of fibrin, and 2 mM CaCl2. Following seeding, the devices were placed in a humidified 5% CO2 incubator for 45 min at 37°C to ensure complete fibrin polymerization. Following this, maintenance media (DMEM supplemented with 10% FBS, 2 mg/mL aminocaproic acid, and antibiotic-antimycotic) was added to the device. After 6 hours, this media was aspirated, and the agarose was carefully removed from the device. Following one PBS wash to remove any agarose debris, fresh maintenance media was added to the device. Each fiber was then maintained with daily media changes prior to experimentation.
Hemodynamic Cardiac Tissue Chip Experiments
A modified version of the Biomimetic Cardiac Tissue Model (BCTM) designed by our lab [Rogers et al., 2018; Rogers et al., 2019] was used in these experiments. To set up the modified BCTM, which was comprised of the seeded cardiac device connected to a flow loop, the device was first sandwiched between an upper 5 × 5 × 1 cm polycarbonate piece, which contained inlet and outlet ports for media, and a lower polycarbonate piece of the same size, which had a 3 × 3 × 0.5 cm centrally-located cavity bored into the upper half, as well as an inlet and outlet port on opposing sides of the cavity. To integrate the sandwiched device into the flow circuit, a media reservoir was attached to the top polycarbonate piece with flexible tubing containing one-way valves to ensure anterograde media flow, and the bottom piece was connected to a pneumatic pump (LB Engineering GbR, Berlin, Germany). In addition, a 4.5 mm diameter piece of tubing was situated in the bottom chamber to provide a rounded obstacle for the membrane to deform around. The “end-diastolic pressure”, which deformed the flexible PDMS membrane downward around the tubing and induced strain in the cardiac fibers, depended upon the height of the media reservoir and the “systolic” pressure, which returned the membrane and fibers to their original positions, was produced by the pneumatic pump injecting air into the bottom chamber at a regular frequency.
For these experiments, the cardiac fibers were rested in static conditions for 2 days following seeding and then were integrated into the circuit for 72 hours in a humidified 5% CO2 incubator at 37°C. The height of the media reservoir was adjusted such that an “end-diastolic pressure” of ~5 mmHg was produced, and the pneumatic pump was set to produce ~30 mmHg of pressure for 0.4 seconds at 1 Hz.
Hypoxia Experiments
Engineered cardiac fibers, after 5 days of static culture, were placed into a humidified Hypoxia Incubator Chamber (STEMCELL Technologies, Inc., Cambridge, MA, USA) kept at 37°C, and the oxygen within the chamber was purged with 5% CO2 in 95% N2. These fibers were exposed to hypoxia for 3 hours while the normoxic controls remained in the humidified 5% CO2 incubator at 37°C. The resting period for these fibers was extended to 5 days to match the total duration of the low-pressure fiber experiments (2 day recovery period plus 3 days of hemodynamic loading).
Histology
Engineered cardiac tissues were removed from the devices and fixed in 1:10 buffered formalin (Fisherbrand; 245–684) for 24 hours before further processing. Formalin-fixed paraffin-embedded engineered cardiac sections were stained with Masson’s trichrome staining or labeled with Phalloidin (Thermo Fisher Scientific; A34055) and Wheat germ agglutinin (Thermo Fisher Scientific; W11261). Nuclei were stained by DAPI.
qRT-PCR
Engineered cardiac tissues were bead milled (BeadBug; D1030) in tubes containing 0.5 mm zirconium beads (Benchmark; D1032–05) and 700 μL TRIzol (Invitrogen; 15596018). Total RNA was extracted and employed to measure the transcript quantities of Ankyrin D1, ANP, BNP, PKCα, βMHC, SOD1, TGFβ1, TGFβ2, and VEGF. cDNA for mRNAs was obtained from 2 μg total RNA in a 20 μl reaction using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems; 4368814). Quantitative RT-PCR was performed with primers specific to the target genes (Supplement Table 1). 20 ng synthesized cDNA was combined with Fast SYBR Green Master Mix (Applied Biosystems; 4385612) and 0.5 μM each of forward and reverse primers. Transcripts were quantified using BioRad 4000 Real-Time PCR system. Cycling conditions were as follows: 95 °C for 10 minutes followed by 40 cycles of amplification (95 °C denaturation for 15 seconds, and 60 °C annealing 30 seconds, 72 °C extension for 30 seconds). The melting curve was then obtained. To avoid the influence of genomic contamination, forward and reverse primers were designed to flank exons separated by at least an intron. Reactions with primers alone were also included as negative controls. Quantified values were normalized against the input determined by the housekeeping gene β2-microglobulin.
Cell-viability Assay
Live/Dead Viability/Cytotoxicity Kit (Invitrogen; L3224) was used to determine the viability of cells in the post-hypoxic and normoxic engineered tissues. 0.5 uL of Calcein AM and 2 uL of Ethidium homodimer-1 were diluted in 1 mL of PBS to obtain a manufacturer-suggested working concentration. The tissue was incubated in the reagent for 30 minutes at 37 °C then cell viability was assessed immediately using a fluorescence microscope. With this assay, dead cells give off bright red fluorescence as EthD-1 enters through the damaged cell membrane and binds to nucleic acids. Through intracellular esterase activity, live cells metabolically convert Calcein-AM into Calcein, giving off green fluorescence.
Results
Oxidative stress promotes cardiac cell aging
Cellular senescence has been implicated in the deterioration of organ function in aging organisms [Burton, 2009; Signer and Morrison, 2013]. Cells displaying the aged phenotype are identified by the expression of senescence-associated biomarkers [Cesselli et al., 2011; Bernardes de Jesus and Blasco, 2012]. Strategies to isolate live senescent cells remain to be developed, requiring the implementation of ex vivo models of cellular aging. An in vitro protocol for Doxo-mediated, oxidative-stress-induced senescence is introduced here that successfully recapitulates the aged phenotype in cardiac muscle cells (Fig. 1A). The senescence of cardiac cells was documented with several parameters: accumulation DDR foci, nucleus area and perimeter enlargement, excessive production of ROS, elevated expression and activation of the transcription factor p53, and upregulated expression of senescence-associated protein p16INK4a.
Fig. 1:

Generation of senescent cell phenotypes. (A) Schematics showing senescent-cardiac cell generation. (B) Representative fluorescence images of Doxo untreated (control, Ctrl) and treated (senescent, Sen) rat cardiac H9c2 myoblasts. H9c2 cells were treated with 0.5 μM Doxo for 24hr, washed with PBS, and cultured normally for 48hr to produce the senescent phenotype. Formalin-fixed cells were immunolabeled using anti-γH2A.X antibody (red, left, and middle panels) and imaged using an fluorescent microscope. DDR foci were rendered (right panel) and quantified from the fluorescence images using CellProfiler 4.0.6. (C) Quantification of DDR foci showing the fraction of cells exhibiting less than and more than 5 DDR foci per nucleus (n = 21047 control nucleus; n = 16519 senescent nuclei, *p<0.0001). (D&E) Quantification of nucleus morphometries area (D) and perimeter (E) in the control (Ctrl) and senescent (Sen) cells (21047 control nuclei, 16519 senescent nuclei, *p<0.0001).
DDR foci are characterized by the localization of γH2A.X at DNA injury sites. [Rodier and Campisi, 2011] Treatment of cells with Doxo is known to induce DNA damage and oxidative stress and promote cell death. We [Kannappan et al., 2017; Miller et al., 2019; Miller et al., 2020] and others [Sliwinska et al., 2009] have shown that a subtherapeutic dose of Doxo induces DNA damage and lead to an increased number of DDR foci per nucleus, a marker of cellular senescence. The cardiac cells that underwent cellular stress were analyzed for the presence of DDR foci, which were found in nearly all the cells—both Doxo treated and untreated cells. DDR foci per nucleus were counted and classified into cells with <5 DDR foci and cells with >5 DDR foci. While the cells with <5 foci were considered normal cells, the cells that expressed >5 DDR foci were considered as senescent [Miller et al., 2019]. The fraction of cells that had >5 DDR foci per nucleus increased significantly (baseline: 14.7%, Doxo: 22.5%) due to Doxo treatment (Fig. 1B, C). The “flattened” appearance of senescent cells correlates with increased nucleus area and perimeter [Cristofalo and Pignolo, 1993]. The cells were stained with DAPI, and the nuclei morphologies were captured and quantified. Average nucleus area (baseline: mean 2771 a.u., Doxo: mean 2975 a.u.) and perimeter (baseline: mean 201.4 a.u., Doxo: mean 207.3 a.u.) increased markedly in the Doxo treated cells (Fig. 1D, E).
Oxidative stress produced by the accumulation of ROS can lead to protein, DNA, lipid, and organelle damage, consequently leading to the manifestation of cellular senescence, which is one of the core mechanisms mediating aging [Birch-Machin and Bowman, 2016]. ROS levels increase after different types of stresses, including chemotherapeutic drug administration, aging, DNA damage, and oncogene activation [Passos et al., 2009]. ROS’s basal level increased significantly in the Doxo treated cardiac cells (baseline: 78.71 a.u., Doxo: 114.3 a.u.) as shown in Figures 2A and B. The tumor suppressor protein p53, a redox-active transcription factor, organizes and directs cellular responses to a variety of stresses. The cellular generation of ROS is central to redox signaling, and ROS accumulation triggers p53 expression and activation [Liu et al., 2008]. The Doxo treated cardiac cells were characterized by a significant increase in p53 expression (baseline: 185 a.u., Doxo: 227.5 a.u.) and activation (baseline: 155.4 a.u., Doxo: 212.5 a.u.) as shown in Figures 2C and D, respectively. Upregulated p16INK4a expression drives cells to enter senescence, an irreversible cell-cycle arrest that in part serves to prevent the proliferation of potentially malignant cells and contributes to cellular aging [Ce et al., 2014]. Thus, the expression of p16INK4a is a hallmark of cellular senescence. After Doxo treatment and 48 h of the recovery period, p16INK4a expression was barely detected in non-treated control cells by immunolabeling and fluorescence microscopy, but numerous treated cells expressed p16INK4a, as shown in Figure 2C. Thus, following oxidative stress and recovery, the cardiac cells expressed senescence phenotype markers, including persistent DDR foci, accumulation of ROS, enlargement of nuclear area and perimeter, upregulation of p53 and p53 activation, and the senescence-associated protein p16INK4a.
Fig. 2:

Molecular characterization of the senescent phenotype. (A) Representative fluorescence images showing ROS activity in control and senescent cells. H9c2 cells were treated with 10 μM CM-H2DCFDA and incubated for 60 minutes. Green fluorescence produced by the free radicals was captured by fluorescence microscopy. (B) Quantification of ROS fluorescence images showing ROS generation in control (Ctrl) and senescent (Sen) cells (n = 5681 control nuclei, n = 4094 senescent nuclei, *p<0.0001). (C&D) Quantification of total and phosphorylated p53 in control and senescent cells. H9c2 cells were fixed in formalin and immunostained using anti-p53 and anti-p53 (Ser15) antibodies, and the expression level was assessed with a fluorescent microscope (n= 11914 control cells; n = 10418 senescent cells, *p<0.0001). (E) Fluorescence micrograph of control and senescent cells immunolabelled for the senescent marker p16INK4a (green) and F-actin filaments (red).
Engineered senescent cardiac tissue
Following the generation of senescent cardiac cells, we attempted to engineer a senescent cardiac tissue chip model that mimics the aging cardiac tissues using our state-of-the-art Cardiac Tissue Model (BCTM) technology [Rogers et al., 2018; Rogers et al., 2019]. As shown in Figure 3A, a modified form of our established BCTM cell culture chamber was fabricated. As mentioned in the Methods section, senescent and control cardiac fibrin/Matrigel fibers were fabricated (Fig. 3B) and subjected to low pressure and strain hemodynamic load. The fabricated fibers were evaluated for cell alignment and general tissue morphology using Masson’s trichrome staining of paraffin-embedded sections (Fig. 3C). From the higher magnification images, as shown in Figure 3D, it is evident that contraction-induced alignment of the extracellular matrix leads to cell alignment. Significantly, the cell alignment process is not affected in senescent cardiac tissue. However, a noticeable increase in collagen deposition can be observed near the posts of the Doxo treated, senescent fibers. As shown in Figures 3E and F, immunofluorescence microscopy confirmed the fibers’ cell alignment. Micrographs show no observable difference between control and senescent cardiac fibers concerning cell density and alignment.
Fig. 3:

Construction of engineered senescent cardiac tissue model. (A) Sketch of the Biomimetic Cardiac Tissue Model (BCTM) cell culture chamber. (B) BCTM cell culture chamber with senescent cell-laden fibrin gel suspended between the posts (left panel). The image was taken 48 hrs after the removal of the agarose mold. Magnified view of the area under the white box (right panel) (C) Control and senescent cardiac tissue sections stained by Masson’s trichrome stain. (D) Higher magnification of control and senescent cardiac sections displaying cell alignment. Cell enlargement and fibrosis are apparent in senescent tissues. (E) Fluorescence micrographs of control and senescent cardiac tissues (green: phalloidin, red: wheat germ agglutinin). (F) Higher magnified images displaying morphological characters of control and senescent phenotype.
To determine whether the fabrication process and hemodynamic loading affected the cardiac cells at a molecular level, several transcripts ANP, BNP, PKC-α, ANKD1, SOD1, TGFβ1, TGFβ2, and VEGF were quantified by qRT-PCR, and their relative expression was calculated after normalized to the housekeeping gene β2microglobulin. ANP, BNP, PKC-α, and ANKD1 transcript expression levels were comparable between the control and the senescent cardiac fibers (Fig. 4). Strikingly, SOD1, TGFβ1, TGFβ2, and VEGF expression were significantly reduced in the senescent cardiac fibers. This confirms that fabrication and hemodynamic loads do not affect the cardiac phenotype of senescent fibers. The changes in the cytokine, growth factor, and superoxide dismutase enzyme [Kondo et al., 2014] may be attributable to the senescent phenotype of the cardiac cells [Ingraham et al., 2008; Fan et al., 2019].
Fig. 4:
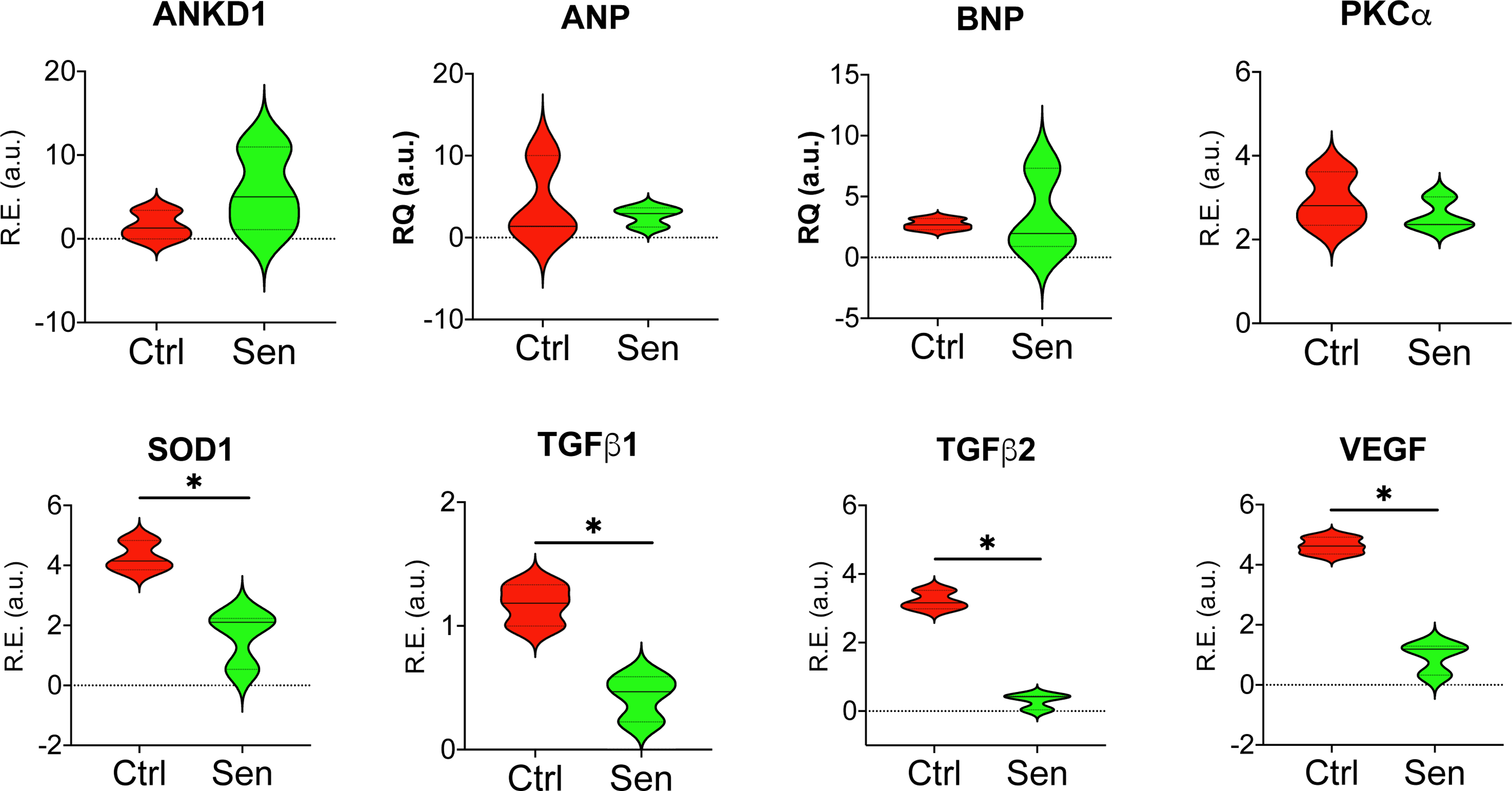
Molecular characterization of engineered senescent cardiac tissues. Total RNA from control (Ctrl) and senescent (Sen) cardiac tissue fibers were isolated, and transcript expression quantities were analysed by qRT-PCR. Superoxide dismutase1 (SOD1), TGFβ1, TGFβ2, and VEGF were significantly reduced in Sen compared to Ctrl. ANKD1, ANP, BNP, and PKCα transcript expression levels comparable. (n=3, *p<0.05)
Hypoxia induces myocardial ischemia in engineered cardiac fibers
Thus far, the experiments and the results suggest that senescent cardiac cell phenotype can be generated and fabricated to produce senescent cardiac fibers. Our next objective was to determine whether MI can be induced in these cardiac fibers that mimic ischemic heart attack. The engineered cardiac fibers were subjected to the hypoxic condition following the procedure outlined in the Methods section. The fibers were then analyzed for cell death using a live/dead staining assay. Both live and dead cells were found in all the cardiac fibers, regardless of oxygen tension (Fig. 5A). However, dead cells were more frequently observed in the hypoxia-treated cardiac fibers, as shown in Figure 5A (right panels), indicating hypoxia increased cell death in the cardiac fibers. To further understand whether hypoxia-induced cardiac cell death in the fibers mimics ischemic heart attack, the molecular transcript profile was analyzed by qRT-PCR. ANP, BNP, βMHC, PKC-α, SOD1, TGFβ1, TGFβ2, and VEGF transcripts were profiled.
Fig. 5.

Hypoxia treatment mimicked myocardial infarction. (A) Control and senescent cardiac tissues were subjected to hypoxic conditions and live/dead assays were performed. Cell death was induced in both control and senescent cardiac tissue fibers; however, more cell death can be observed in senescent cardiac tissue fibers. (B) Total RNA from control (Ctrl) and senescent (Sen) cardiac tissue fibers subjected to hypoxic (H) and normoxic conditions (N) were prepared, and transcript expression quantities were analyzed by qRT-PCR. Several transcripts normalized to the housekeeping gene b2Microglobulin were found to be differentially expressed. (normoxia n=3; hypoxia n=5, *p=<0.05)
Interestingly, the MI markers ANP, BNP, and βMHC were found to be significantly upregulated in the hypoxia-treated cardiac fibers compared to their counterparts. Hypoxia treatment significantly upregulated PKC-α control cardiac fibers. Together, these results suggest that the fabricated senescent cardiac fiber model of hypoxia-induced MI recapitulates major pathological hallmarks such as increased cell death and detrimental transcriptional changes.
Discussion/Conclusion
Evidence suggests that cellular senescence via its growth arrest phenotype and secretory factors is a substantial contributing component in developing age-associated diseases [Pole et al., 2016]. Through autocrine and paracrine activities, senescent cells can accelerate degeneration of surrounding cells in various tissue environments and contribute to age-related cardiovascular diseases [Fyhrquist et al., 2013]. We have verified the prevalence of cellular senescence and DNA damage, the two critical hallmarks of aging [López-Otín et al., 2013], in our engineered cardiac tissue model. Decreased cardiac function and increased myocardial apoptosis have been implicated in aging cardiomyopathy [Oxenham and Sharpe, 2003; Triposkiadis et al., 2019]. Thus, a viable model of age-related cardiovascular diseases must consider the contribution of cellular senescence to model cardiac tissue in older individuals more accurately.
Advanced tissue engineering techniques enable the fabrication of tissue models that recapitulate integral parameters associated with aging and age-related pathologies. Several engineered tissue models and platforms have been developed to investigate age-related diseases, such as Alzheimer’s disease in-a-dish and Barth syndrome on-a-chip [Choi et al., 2014; Wang et al., 2014]. In the present study, we have developed a first-of-its-kind cardiac tissue model with hemodynamic loading recapitulating an infarcted aging heart. We generated senescent cardiac cells, engineered cardiac tissue fibers using the senescent cells, and subjected them to hemodynamic stresses associated with pressure-volume changes in the heart. Notably, we also recreated important aspects of MI in the engineered aging cardiac fibers via hypoxic treatment.
An early cellular response to DNA damage is the phosphorylation of H2A.X [Mah et al., 2010], and it is well-established that this chromatin modification correlates with DNA damage and repair processes [Redon et al., 2002; Sedelnikova et al., 2002]. In addition to promoting tumorigenesis, persistent DNA damage can induce terminal cell cycle arrest and senescence [Sinclair and Oberdoerffer, 2009]. In this study, we determined the extent of DNA damage by counting the number of DDR foci and judged senescent status based upon this number. In our model, we found a significantly higher prevalence of senescent cells in the Doxo-treated group. The Doxo-treated cells were also significantly larger and “flattened,” another morphological marker of senescent cells. This finding corroborates the previous studies by ourselves and others that a lower dose of Doxo treatment is sufficient to induce cellular senescence [Sliwinska et al., 2009; Kannappan et al., 2017]. Tumor suppressor protein p53regulates a variety of cellular activities, including DNA damage repair, cell cycle arrest, apoptosis, and cellular senescence [Vurusaner et al., 2012]. A significant factor contributing to aging is elevated oxidative stress in the senescent cells [Cadenas and Davies, 2000; Terman and Brunk, 2006]. Increased levels of ROS can cause the accumulation of oxidative-damaged proteins, lipids, and DNA and promotes aging [Balaban et al., 2005; Rufini et al., 2013]. A high level of oxidative stress is also present in the ischemic myocardium [Acun et al., 2017]. Additionally, the critical tumor suppressor p16INK4a has been shown to be expressed in senescent cells [Alcorta et al., 1996; Hara et al., 1996]. Noxious stimuli like oxygen radicals and ionizing radiation have been found to induce p16INK4a expression in vitro and in vivo [Kim and Sharpless, 2006]. Our cellular senescence model is characterized by significantly increased levels of ROS and both basal and phosphorylated p53. Further, the Doxo treated cells express the cellular senescent marker p16INK4a. These findings clearly indicate the presence of senescent signaling and senescent phenotypic hallmarks, providing further confirmation that Doxo-induced oxidative stress promotes aging in cardiac cells within our model.
In heart failure and dilated cardiomyopathy, Ankyrin D1 forms a signalosome complex with PKC-α, and chronic PKC-α stress signaling is associated with heart failure [Lange et al., 2016; Ling et al., 2017]. Endogenously generated natriuretic peptides ANP and BNP, synthesized and secreted by cardiomyocytes [Sarzani et al., 2017], are typically expressed at elevated levels during cardiac dysfunction. In the present study, transcript analysis shows similar expression levels of ANKD1, ANP, BNP, and PKC-α (Fig 4) in both control and senescent cardiac fibers indicative of retaining cellular properties throughout the experimental procedure. However, the SOD1 responsible for obliterating free superoxide radicals was decreased in the senescent fibers. This decreased expression provides further evidence that senescence was truly induced in these fibers, as a lower expression of SOD1 correlates with increased ROS formation. TGFβ has been implicated in inducing and accelerating senescence in various cell types [Senturk et al., 2010]. However, it is surprising that TGFβ1 and TGFβ2 are decreased in our senescent model, which indicates that TGFβ signaling was affected but may not have been a senescence driving factor here. Interestingly, VEGF, an angiogenic factor affected by aging, was found to be down-regulated in our senescence model. Importantly, following hypoxic treatment, we found significant upregulation of MI markers like ANP, BNP, and βMHC, which indicates human myocardial ischemia can be induced within the engineered cardiac fibers.
In summary, this study highlights a novel approach for developing cardiac tissue chips for studying aging and aging-related cardiovascular pathologies. Our model is physiologically relevant as cellular aging and DNA damage have been observed in animal models and humans’ senescent tissue, our approach of inducing cellular senescence using Doxo, a DNA intercalating agent [Dimri et al., 1995; Herbig et al., 2006]. Finally, our results suggest that our engineered senescent cardiac fiber model of hypoxia-induced MI faithfully recapitulates specific disease pathological hallmarks, such as increased cell death, structural remodeling, and MI markers.
Supplementary Material
Acknowledgments
Funding Sources
This work was funded by NIH 1 R01 HL148462–01, and funds from the University of Alabama at Birmingham Comprehensive Cardiovascular Center.
Footnotes
Statements
Statement of Ethics
All authors followed standard ethical procedures. No studies involved the use of humans or animals.
Conflict of Interest Statement
The authors have no conflict of interest.
References
- Acun A, Vural DC, Zorlutuna P. A Tissue Engineered Model of Aging: Interdependence and Cooperative Effects in Failing Tissues. Scientific Reports. 2017;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proceedings of the National Academy of Sciences. 1996;93(24):13742–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. cell. 2005;120(4):483–95. Bernardes de Jesus B, Blasco MA. Assessing cell and organ senescence biomarkers. Circ Res. 2012. Jun 22;111(1):97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch-Machin MA, Bowman A. Oxidative stress and ageing. Br J Dermatol. 2016. Oct;175 Suppl 2:26–29. [DOI] [PubMed] [Google Scholar]
- Burton D. Cellular senescence, ageing and disease. Age. 2009;31(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free radical biology and medicine. 2000;29(3–4):222–30. [DOI] [PubMed] [Google Scholar]
- Ce MLK, Burd. The molecular balancing act of p16(INK4a) in cancer and aging. Molecular cancer research : MCR. 2014 2014. Feb;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesselli D, Beltrami AP, D’Aurizio F, Marcon P, Bergamin N, Toffoletto B, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011. Jul;179(1):349–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515(7526):274–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofalo VJ, Pignolo RJ. Replicative senescence of human fibroblast-like cells in culture. Physiol Rev. 1993. Jul;73(3):617–38. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences. 1995;92(20):9363–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Plasminogen activator inhibitor 1 - insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proceedings of the National Academy of Sciences. 2012;109(30):12052–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Ji Q, Zhang C, Xu S, Sun H, Li Z. TGF-β induces periodontal ligament stem cell senescence through increase of ROS production. Molecular Medicine Reports. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlyanskaya I, Alekseenko L, Nikolsky N. Senescence as a general cellular response to stress: A mini-review. Experimental Gerontology. 2015;72:124–28. [DOI] [PubMed] [Google Scholar]
- Fyhrquist F, Saijonmaa O, Strandberg T. The roles of senescence and telomere shortening in cardiovascular disease. Nature Reviews Cardiology. 2013;10(5):274–83. [DOI] [PubMed] [Google Scholar]
- Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Molecular and cellular biology. 1996;16(3):859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311(5765):1257–57. [DOI] [PubMed] [Google Scholar]
- Ingraham JP, Forbes ME, Riddle DR, Sonntag WE. Aging reduces hypoxia-induced microvascular growth in the rodent hippocampus. J Gerontol A Biol Sci Med Sci. 2008. Jan;63(1):12–20. [DOI] [PubMed] [Google Scholar]
- Kannappan R, Matsuda A, Ferreira-Martins J, Zhang E, Palano G, Czarna A, et al. p53 Modulates the Fate of Cardiac Progenitor Cells Ex Vivo and in the Diabetic Heart In Vivo . EBioMedicine. 2017. Feb;16:224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannappan R, Turner JF, Miller JM, Fan C, Rushdi AG, Rajasekaran NS, et al. Functionally Competent DNA Damage-Free Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Myocardial Repair. Circulation. 2019. Aug 6;140(6):520–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127(2):265–75. [DOI] [PubMed] [Google Scholar]
- Kimes BW, Brandt BL. Properties of a clonal muscle cell line from rat heart. Exp Cell Res. 1976. Mar 15;98(2):367–81. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Masutomi H, Noda Y, Ozawa Y, Takahashi K, Handa S, et al. Senescence marker protein-30/superoxide dismutase 1 double knockout mice exhibit increased oxidative stress and hepatic steatosis. FEBS Open Bio. 2014;4:522–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nature Cell Biology. 2006;8(8):877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Nijwening JH, Bernards R. Transforming Growth Factor-β Requires Its Target Plasminogen Activator Inhibitor-1 for Cytostatic Activity. Journal of Biological Chemistry. 2008;283(36):24308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S, Gehmlich K, Lun AS, Blondelle J, Hooper C, Dalton ND, et al. MLP and CARP are linked to chronic PKCα signalling in dilated cardiomyopathy. Nature communications. 2016;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SSM, Chen Y-T, Wang J, Richards AM, Liew OW. Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential. International journal of molecular sciences. 2017;18(7):1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008. Apr 15;44(8):1529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah L-J, El-Osta A, Karagiannis TC. γH2AX as a molecular marker of aging and disease. Epigenetics. 2010;5(2):129–36. [DOI] [PubMed] [Google Scholar]
- Malaquin N, Carrier-Leclerc A, Dessureault M, Rodier F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Frontiers in Genetics. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JM, Mardhekar NM, Pretorius D, Krishnamurthy P, Rajasekaran NS, Zhang J, et al. DNA damage-free iPS cells exhibit potential to yield competent cardiomyocytes. Am J Physiol Heart Circ Physiol. 2020. Apr 1;318(4):H801–H15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JM, Mardhekar NM, Rajasekaran V, Zhang J, Kannappan R. Assessing Stem Cell DNA Integrity for Cardiac Cell Therapy. J Vis Exp. 2019. Jan 25(143). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal R, Woo FW, Castro CS, Cohen MA, Karanxha J, Mittal J, et al. Organ-on-chip models: Implications in drug discovery and clinical applications. J Cell Physiol. 2019. Jun;234(6):8352–80. [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nature Reviews Molecular Cell Biology. 2014;15(7):482–96. [DOI] [PubMed] [Google Scholar]
- Oxenham H, Sharpe N. Cardiovascular aging and heart failure. Eur J Heart Fail. 2003. Aug;5(4):427–34. [DOI] [PubMed] [Google Scholar]
- Passos JF, Simillion C, Hallinan J, Wipat A, von Zglinicki T. Cellular senescence: unravelling complexity. Age (Dordr). 2009. Dec;31(4):353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pole A, Dimri M, Dimri GP. Oxidative stress, cellular senescence and ageing. AIMS Molecular Science. 2016;3(3). [Google Scholar]
- Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2a variants H2AX and H2AZ. Current opinion in genetics & development. 2002;12(2):162–69. [DOI] [PubMed] [Google Scholar]
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011. Feb 21;192(4):547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AJ, Kannappan R, Abukhalifeh H, Ghazal M, Miller JM, El-Baz A, et al. Hemodynamic Stimulation Using the Biomimetic Cardiac Tissue Model (BCTM) Enhances Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cells Tissues Organs. 2018;206(1–2):82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers AJ, Miller JM, Kannappan R, Sethu P. Cardiac Tissue Chips (CTCs) for Modeling Cardiovascular Disease. IEEE Transactions on Biomedical Engineering. 2019;66(12):3436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, et al. Heart disease and stroke statistics−−2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007. Feb 6;115(5):e69–171. [DOI] [PubMed] [Google Scholar]
- Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32(43):5129–43. [DOI] [PubMed] [Google Scholar]
- Sarzani R, Spannella F, Giulietti F, Balietti P, Cocci G, Bordicchia M. Cardiac Natriuretic Peptides, Hypertension and Cardiovascular Risk. High Blood Press Cardiovasc Prev. 2017. Jun;24(2):115–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of 125IdU-induced DNA double-strand breaks with γ-H2AX antibody. Radiation research. 2002;158(4):486–92. [DOI] [PubMed] [Google Scholar]
- Senturk S, Mumcuoglu M, Gursoy-Yuzugullu O, Cingoz B, Akcali KC, Ozturk M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology. 2010;52(3):966–74. [DOI] [PubMed] [Google Scholar]
- Signer RA, Morrison SJ. Mechanisms that regulate stem cell aging and life span. Cell stem cell. 2013;12(2):152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Oberdoerffer P. The ageing epigenome: damaged beyond repair? Ageing research reviews. 2009;8(3):189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliwinska MA, Mosieniak G, Wolanin K, Babik A, Piwocka K, Magalska A, et al. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mechanisms of Ageing and Development. 2009;130(1–2):24–32. [DOI] [PubMed] [Google Scholar]
- Smith AS, Macadangdang J, Leung W, Laflamme MA, Kim DH. Human iPSC-derived cardiomyocytes and tissue engineering strategies for disease modeling and drug screening. Biotechnol Adv. 2017. Jan - Feb;35(1):77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Oxidative stress, accumulation of biological’garbage’, and aging. Antioxidants & redox signaling. 2006;8(1–2):197–204. [DOI] [PubMed] [Google Scholar]
- Triposkiadis F, Xanthopoulos A, Butler J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J Am Coll Cardiol. 2019. Aug 13;74(6):804–13. [DOI] [PubMed] [Google Scholar]
- Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radical Biology and Medicine. 2012;52(1):7–18. [DOI] [PubMed] [Google Scholar]
- Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nature medicine. 2014;20(6):616–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger F, Mannhardt I, Eschenhagen T. Engineering Cardiac Muscle Tissue: A Maturating Field of Research. Circ Res. 2017. Apr 28;120(9):1487–500. [DOI] [PubMed] [Google Scholar]
- Xie Z, Kometiani P, Liu J, Li J, Shapiro JI, Askari A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. Journal of Biological Chemistry. 1999;274(27):19323–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.