

Abstract
Ipilimumab improves clinical outcomes when combined with nivolumab in metastatic non-small cell lung cancer (NSCLC), but its efficacy and impact on the immune microenvironment in operable NSCLC remain unclear. We report the results of the phase 2 randomized NEOSTAR trial (NCT03158129) of neoadjuvant nivolumab or nivolumab + ipilimumab followed by surgery in 44 patients with operable NSCLC, using major pathologic response (MPR) as the primary endpoint. The MPR rate for each treatment arm was tested against historical controls of neoadjuvant chemotherapy. The nivolumab + ipilimumab arm met the prespecified primary endpoint threshold of 6 MPRs in 21 patients, achieving a 38% MPR rate (8/21). We observed a 22% MPR rate (5/23) in the nivolumab arm. In 37 patients resected on trial, nivolumab and nivolumab + ipilimumab produced MPR rates of 24% (5/21) and 50% (8/16), respectively. Compared with nivolumab, nivolumab + ipilimumab resulted in higher pathologic complete response rates (10% versus 38%), less viable tumor (median 50% versus 9%), and greater frequencies of effector, tissue-resident memory and effector memory T cells. Increased abundance of gut Ruminococcus and Akkermansia spp. was associated with MPR to dual therapy. Our data indicate that neoadjuvant nivolumab + ipilimumab-based therapy enhances pathologic responses, tumor immune infiltrates and immunologic memory, and merits further investigation in operable NSCLC.
Introduction
More than 50% of patients with operable NSCLC will experience recurrence after surgery alone1. Perioperative (neoadjuvant or adjuvant) chemotherapy provides only a modest (approximately 5%) improvement in 5-year overall survival (OS) and is a source of toxicity2,3, illustrating the need for new treatment approaches in this setting. The realization that a positive association exists between an MPR (≤10% viable tumor in resected tumor specimens) to neoadjuvant chemotherapy and improved survival outcomes in patients with operable NSCLC4–7 has stimulated efforts to test new neoadjuvant treatment strategies in this setting, including immune checkpoint inhibitors (ICIs).
ICIs that target cytotoxic T-lymphocyte antigen-4 (CTLA-4), programmed cell death protein 1 (PD-1) and its ligand (PD-L1), have changed the standard of care for patients with advanced NSCLC. Studies demonstrating that nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) augment antitumor immunity through distinct, nonredundant cellular mechanisms8 provided a rationale for adding ipilimumab to nivolumab for the treatment of patients with advanced NSCLC. Nivolumab + ipilimumab produced greater response rates compared to nivolumab monotherapy in patients with advanced NSCLC9. First-line nivolumab + ipilimumab treatment also produced longer OS than chemotherapy in patients with advanced NSCLC10. Although the addition of ipilimumab to nivolumab therapy has dramatically altered care of patients with advanced NSCLC, the impact of this dual ICI therapy on the immune microenvironment of resectable NSCLC has not been established. In the present study, we tested the clinical activity of and evaluated the immune responses to neoadjuvant nivolumab and nivolumab + ipilimumab, using MPR as the primary endpoint.
NEOSTAR is a phase 2 randomized clinical trial (NCT03158129) that, after completion of the arms reported in the present study, evolved into a modular platform design. The first portion of the trial reported included two completed, parallel, randomized arms testing neoadjuvant nivolumab monotherapy (arm A, 3 mgkg−1 intravenously (i.v.), every 14 d on day 1 (D1), D15 and D29) and nivolumab + ipilimumab (arm B, nivolumab 3 mg kg−1 i.v. every 14 d on D1, D15 and D29 and ipilimumab 1 mg kg−1 i.v. on D1 only), followed by surgical resection of the primary tumor with lymphadenectomy and adjuvant standard-of-care therapy (Extended Data Fig. 1). The primary endpoint of the trial was an MPR rate in the intention-to-treat (ITT) population in each study arm. The prespecified boundary for a treatment arm to be considered promising for further testing was ≥6 MPRs in 21 evaluable patients for each arm, based on historical controls for neoadjuvant platinum-based chemotherapy that yielded a response rate of approximately 15%4. Secondary and exploratory endpoints are listed in Extended Data Fig. 1. Primary and secondary endpoints are presented in the ITT population and in the resected subgroups in each arm. Correlative endpoints are presented and described in patients with available samples for biomarker analyses in each arm. The results of the reported comparisons are exploratory in nature and hypothesis generating.
Results
Participants.
From June 2017 to November 2018, 53 patients were consented on the trial and screened for eligibility. All patients provided written informed consent before treatment and this trial adhered to all relevant ethical considerations. Nine patients were deemed screen failures, but 44 eligible patients were treated on the study, with 23 randomized to nivolumab monotherapy and 21 randomized to nivolumab + ipilimumab (Extended Data Fig. 2). Patient characteristics and treatment disposition are shown in Table 1. Overall, 41 (93%) patients completed the planned neoadjuvant therapy regimen (nivolumab arm 22 (96%), dual therapy 19 (90%)). Three patients did not complete planned neoadjuvant therapy: one in the nivolumab group and two in the nivolumab + ipilimumab arm (Extended Data Fig. 2). In the nivolumab group, one patient developed grade 3 treatment-related hypoxia and a large nonmalignant pleural effusion requiring hospitalization after one dose of nivolumab (no surgery). In the nivolumab + ipilimumab arm, one patient developed treatment-related grade 3 diarrhea/colitis requiring hospitalization after one dose of combination therapy (surgery off trial). Another patient developed treatment-related grade 2 pneumonitis requiring hospitalization after two doses of therapy on nivolumab + ipilimumab arm; the third planned dose was withheld and the patient underwent surgery on trial. Postoperative chemotherapy was administered to 17 (46%) patients and postoperative radiation therapy (PORT) was administered to 4 (11%) patients (Table 1).
Table 1.
Patient characteristics and treatment disposition
| Factor | Category | Overall | Nivo n=23 |
Nivo + Ipi n=21 |
P value |
|---|---|---|---|---|---|
| mean (SD) | mean (SD) | mean (SD) | |||
| Age at randomization | 65.6 (8.3) | 66.1 (8.5) | 65.0 (8.3) | 0.680 | |
| n (%) | n (%) | n (%) | |||
| Gender | Female | 16 (36) | 8 (35) | 8 (38) | 0.820 |
| Male | 28 (64) | 15 (65) | 13 (62) | ||
| Race | Asian | 2 (5) | 1 (4) | 1 (5) | 0.491 |
| Black | 4 (9) | 1 (4) | 3 (14) | ||
| Other | 1 (2) | 0 (0) | 1 (5) | ||
| White | 37 (84) | 21 (91) | 16 (76) | ||
| Smoking status | Never smoker | 8 (18) | 5 (22) | 3 (14) | 0.767 |
| Former smoker | 26 (59) | 14 (61) | 12 (57) | ||
| Current smoker | 10 (23) | 4 (17) | 6 (29) | ||
| Stage | Stage IA | 8 (18) | 4 (17) | 4 (19) | 0.175 |
| Stage IB | 15 (34) | 7 (30) | 8 (38) | ||
| Stage IIA | 7 (16) | 2 (9) | 5 (24) | ||
| Stage IIB | 5 (11) | 5 (22) | 0 (0) | ||
| Stage IIIA | 9 (20) | 5 (22) | 4 (19) | ||
| Histology | Squamous cell carcinoma | 17 (39) | 10 (43) | 7 (33) | 0.641 |
| Adenosquamous carcinoma | 1 (2) | 0 (0) | 1 (5) | ||
| Adenocarcinoma | 26 (59) | 13 (57) | 13 (62) | ||
| ECOG | 0 | 26 (59) | 16 (70) | 10 (48) | 0.139 |
| 1 | 18 (41) | 7 (30) | 11 (52) | ||
| Baseline T | 1a | 5 (11) | 3 (13) | 2 (10) | 0.625 |
| 1b | 7 (16) | 2 (9) | 5 (24) | ||
| 2a | 20 (45) | 10 (43) | 10 (48) | ||
| 2b | 5 (11) | 3 (13) | 2 (10) | ||
| 3 | 7 (16) | 5 (22) | 2 (10) | ||
| Baseline N | 0 | 30 (68) | 16 (70) | 14 (67) | 0.349 |
| 1 | 7 (16) | 2 (9) | 5 (24) | ||
| 2 | 7 (16) | 5 (22) | 2 (10) | ||
| Baseline M | 0 | 44 (100) | 23 (100) | 21 (100) | |
| Invasive mediastinal staging | EBUS | 40 (91) | 21 (91) | 19 (90) | 0.795 |
| Mediastinoscopy | 3 (7) | 1 (4) | 2 (10) | ||
| No invasive mediastinal staging* | 1 (2) | 1 (4) | 0 (0) | ||
| Adjuvant (postoperative) therapy | PORT | 4 (11) | 2 (10) | 2 (13) | |
| Chemotherapy | 17 (46) | 11 (52) | 6 (38) |
Stage T2N0M0, IB.
The two-sided P value is from the Student’s t-test for the continuous factor, Chi-square test and Fisher’s exact test for the categorical factors. EBUS, endobronchial ultrasound; ECOG, Eastern Cooperative Oncology Group.
Among the 44 patients (ITT) who received at least one dose of neoadjuvant ICIs on trial, 39 patients underwent curative-intent surgery on or off trial and five patients did not undergo surgery (11%, n = 1 nivolumab; n = 4 nivolumab + ipilimumab) (Extended Data Fig. 2). Among the overall 39 resected patients, 37 (95%) underwent surgery on trial (n = 21 nivolumab; n = 16 nivolumab + ipilimumab) and two (5%) were resected after receiving additional systemic therapies off trial (n = 1 nivolumab; n = 1 nivolumab + ipilimumab). Therefore, a total of seven patients were considered not to have undergone surgery on trial (no MPR) in the ITT analysis.
Clinical activity of neoadjuvant nivolumab and nivolumab + ipilimumab.
In the ITT population of all 44 randomized patients, the primary endpoint of MPR, as assessed by two independent and trained pathologists, was observed in 22% (5/23, 95% confidence interval (CI) = 7-44%) of patients in the nivolumab arm and 38% of patients in the dual therapy arm (8/21, 95% CI = 18-62%; P = 0.235; Fig. 1a). Therefore, the nivolumab + ipilimumab arm met the prespecified boundary of ≥6 MPRs in 21 patients to be considered promising for further evaluation. Two patients achieved pathologic complete response (pCR) after nivolumab monotherapy (2/23, 9%, 95% CI = 1-28%) compared with six patients treated with nivolumab + ipilimumab (6/21, 29%, 95% CI = 11-52%; P = 0.126; Fig. 1a). The radiographic objective response rate (ORR, partial responses (PRs) plus CR) in the ITT population was 22% (5/23, 95% CI = 7-44%) after nivolumab monotherapy (5 PRs; Fig. 1b), and 19% (4/21, 95% CI = 5-42%) after nivolumab + ipilimumab (1 CR and 3 PRs; one treated patient was not evaluable; P = 1.0; Fig. 1c and Supplementary Table 1).
Fig. 1. Pathologic and radiographic responses to neoadjuvant nivolumab and nivolumab + ipilimumab in ITT and resected patients on trial.
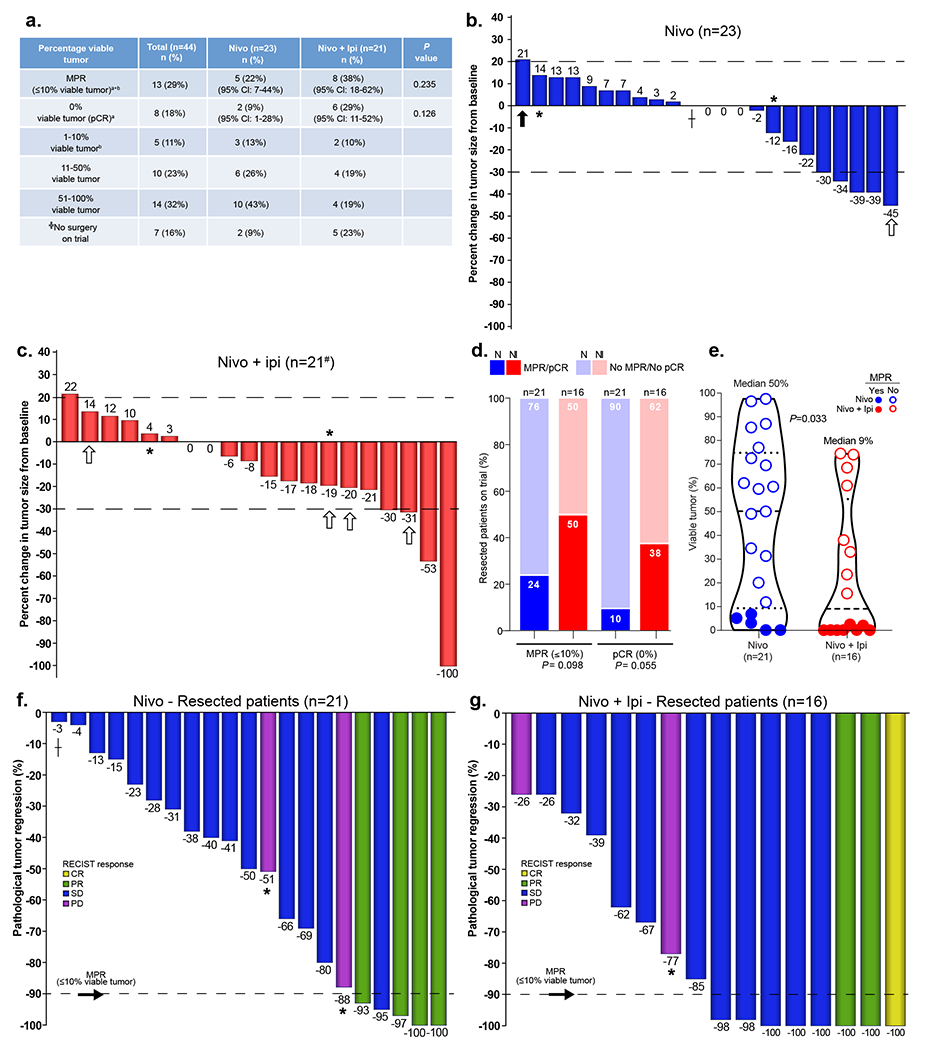
a, Pathologic responses in ITT population (resected and not resected patients, nivolumab, n = 23, nivolumab + ipilimumab, n = 21). Primary endpoint: MPR (≤ 10% viable tumor cells) consists of pCR, that is, 0% viable tumor, + 1-10% viable tumor. The MPR and pCR rates are estimated with exact 95% CIs from the binomial distribution. The two-sided P value is from Chi-square test for MPR and Fisher’s exact test for pCR. ╬Seven patients did not undergo surgery after neoadjuvant therapy on trial. Of these, five patients (one nivolumab, four nivolumab + ipilimumab) had no surgery; two patients had surgery off trial after additional systemic therapies (one nivolumab; one nivolumab + ipilimumab). a0% viable tumor (pCR); b1–10% viable tumor. b,c, Waterfall plots of radiographic percentage change in overall tumor size from baseline at least 14 d after the last dose of neoadjuvant therapy in nivolumab (b) and nivolumab + ipilimumab (c) groups (nivolumab, n = 23, 22% ORR; nivolumab + ipilimumab, n = 21, 19% ORR; #one patient was not evaluable on the study). Dashed black line at 20% point depicts cutoff for PD. Dashed black line at −30% point depicts cutoff for PR. *Indicates overall response of PD due to presence of enlarging and/or new lesions; ┼Indicates that the solid lesion was <1 cm, considered to be SD. The white arrows indicate the patients who did not undergo surgery; the black arrow indicates one patient who underwent surgery off trial after chemoimmunotherapy. #One patient was not radiographically evaluable on the study due to development of grade 3 diarrhea/colitis after one dose of nivolumab + ipilimumab (restaging scans were performed during and after neoadjuvant platinum doublet chemotherapy followed by surgery, both administered off trial). d, Proportion of MPR/no MPR (≤10% viable tumor/>10% viable tumor) and pCR/no pCR (0% viable tumor/>0% viable tumor) in resected patients on trial after neoadjuvant nivolumab (n = 21, bright blue/light blue) and nivolumab + ipilimumab (n = 16, bright red/light red). The two-sided P value is from Chi-square test for MPR and Fisher’s exact test for pCR. e, Percentage viable tumor in resected tumor specimens after nivolumab (n = 21, blue) and nivolumab + ipilimumab (red, n = 16). Median percentage viable tumor: nivolumab 50% (range 0-97.5%, n = 21), nivolumab + ipilimumab 9% (range 0-74.5%, n = 16). Data are presented as the median with minima, lower and upper quartiles, and maxima. The dashed line indicates the median; the dotted lines indicate the lower quartile and upper quartile values; the top and bottom of the violin plots indicate the minima and maxima. The two-sided P value is from Wilcoxon’s rank-sum test. f,g, Waterfall plots of pathologic tumor regression (% viable tumor – 100%) in resected patients after neoadjuvant nivolumab (f; n = 21) and nivolumab + ipilimumab (g; n = 16). Dashed black line represents MPR.
Due to the lack of tissue specimens in patients treated but not resected on trial (n = 7 in ITT analysis), we performed a sensitivity analysis to evaluate MPR and pCR rates in patients resected on study. Among the 37 patients resected on study, nivolumab monotherapy induced an MPR rate of 24% (5/21, 95% CI = 8-47%), whereas the MPR rate was 50% (8/16, 95% CI = 25-75%) after nivolumab + ipilimumab (P = 0.098; Fig. 1d). The pCR rate was 10% (2/21, 95% CI = 1-30%) after nivolumab alone compared with 38% (6/16, 95% CI = 15-65%) after dual therapy (P = 0.055; Fig. 1d). In an exploratory analysis, tumor specimens resected after nivolumab + ipilimumab contained fewer viable tumor cells as compared with that of nivolumab-treated tumors (P = 0.033; Fig. 1e).
In the resected population of 37 patients, ORRs were 19% in both arms (nivolumab, 4/21 CR/PR (95% CI = 5-42%); nivolumab + ipilimumab, 3/16 CR/PR (95% CI = 4-46%); P = 1.00; Supplementary Table 2). We found a positive association between radiographic tumor responses (CR/PR) and MPR in resected patients (Extended Data Fig. 3). All resected patients with CR/PR (7/7, 95% CI = 59-100%) achieved MPR at surgery, compared with 6 out of 30 (20%, 95% CI = 8-39%) resected patients with stable disease/progressive disease (SD/PD) (P = 0.0002; Supplementary Table 3 and Fig. 1f,g). However, there were cases in which patients achieved SD radiographically, but had MPR or achieved marked pathologic tumor regression at surgery (Fig. 1f,g). In some cases, we noted radiographic appearance of nodal disease progression with enlargement and/or increased fluorodeoxyglucose (FDG) uptake in nodes at computed tomography (CT) and positron emission tomography (PET)-CT restaging scans, respectively, after neoadjuvant ICIs. However, invasive pathologic examination of the flaring nodes revealed immune cell infiltration changes (noncaseating granulomas), but not malignancy, a phenomenon we refer to as nodal immune flare (NIF)11. Full details of the radiographic and pathologic features of the NIF phenomenon following ICIs will be reported separately.
Surgical resectability.
The overall resectability rate among the 44 patients (ITT) who received at least one dose of neoadjuvant ICIs on trial was 89% (39 patients underwent curative surgery on or off trial). The complete (R0) resection rate was 100%. Among the five patients who did not undergo surgery, one was treated in the nivolumab arm (4%; 1/23) and four in the nivolumab + ipilimumab arm (19%; 4/21, none related to treatment-related adverse events (TRAEs)). In the nivolumab arm, one patient developed grade 3 treatment-related hypoxia and a nonmalignant pleural effusion requiring hospitalization after one dose of therapy with subsequent elevated surgical risk. In the nivolumab + ipilimumab arm, one had PD, one had tumor involvement of the carina at restaging bronchoscopy and was deemed to be no longer resectable, one was considered at high surgical risk due to inadequate lung perfusion and active smoking, and one declined surgery. Of the two patients who underwent surgery off trial, one patient with stage IIIA disease appeared to have radiographic progression with new mediastinal involvement after three doses of nivolumab and the treatment was changed to platinum doublet chemotherapy + pembrolizumab off trial. It is interesting that biopsies after chemoimmunotherapy of the nodal sites of apparent radiographic disease progression failed to confirm metastases, but rather revealed granulomatous inflammation. This patient subsequently underwent surgery off trial. The second patient with stage IIA disease experienced TRAE grade 3 diarrhea/colitis following one dose of nivolumab + ipilimumab, so the regimen was changed to neoadjuvant platinum doublet chemotherapy followed by surgical resection off trial. The median time to surgical resection was 31 d (range 21-87 d) after the last dose of nivolumab. Eight operations performed on trial (8/37; 22%) were delayed past the recommended maximum 42 d after the last dose of immunotherapy.
Toxicity.
Toxicities were overall manageable, with no new safety concerns compared with known safety profiles of nivolumab and nivolumab + ipilimumab. Grade 3-5 TRAEs were reported in 13% (3/23) of patients treated with nivolumab (two events occurred in the same patient) and 10% (2/21) of patients treated with nivolumab + ipilimumab (Supplementary Table 4). One patient treated with nivolumab developed pneumonia and pneumonitis requiring steroids postoperatively and was diagnosed with bronchopleural fistula (BPF) and empyema, which ultimately resulted in respiratory failure and death. As a result of the sequence of complications, we attributed BPF and death to steroid-treated pneumonitis. Nine serious adverse events (five treatment related) were noted (Supplementary Table 5).
Survival outcomes after neoadjuvant nivolumab and nivolumab + ipilimumab.
All 44 patients were followed for recurrence and survival. At the last database lock (February 25, 2020), the median duration of follow-up after randomization was 22.2 months. Median OS and lung cancer-related recurrence-free survival (RFS) were not reached. One patient treated with nivolumab died within 90 d of surgery and 4.1 months after randomization (Fig. 2a). One patient treated with nivolumab + ipilimumab had PD 2.6 months after randomization (no surgery) (Fig. 2b) and died from the disease 17.1 months after randomization. Six patients experienced lung cancer-related recurrence (Fig. 2c,d). Pathologic and radiographic responses were not significantly different based on tumor histology, stage or smoking status in patients treated with neoadjuvant nivolumab (Supplementary Tables 6 and 7) or nivolumab + ipilimumab (Supplementary Tables 8 and 9). Tumor histology did not significantly affect lung cancer-related RFS (Extended Data Fig. 4a); however, we observed worse lung cancer-related RFS in patients with stage IIIA disease and in never smokers compared with that of patients with stage I/II disease (P = 0.040; Extended Data Fig. 4b), and current/former smokers (P = 0.003; Extended Data Fig. 4c). To address the impact of radiographic and pathologic responses, PORT and adjuvant chemotherapy on disease recurrence, we performed exploratory landmark analyses of lung cancer-related RFS, without formal statistical comparisons. These analyses revealed 1 death among 9 patients with CR/PR (1/9, 11%), and 6 recurrences among 34 patients with SD/PD (6/34, 18%) (Extended Data Fig. 4d). Among 13 patients with MPR, one death occurred (1/13, 8%), and among 24 patients with no MPR, 3 patients progressed (3/24, 13%) (Extended Data Fig. 4e). No meaningful differences in the number of events were noted in patients who underwent PORT and/or adjuvant chemotherapy compared with patients who did not (Extended Data Fig. 4f,g). Although the lower number of events seen in resected patients with MPR compared with those without MPR is encouraging, validation in larger cohorts and assessment at a longer follow up time are warranted.
Fig. 2. Survival outcomes in patients treated with neoadjuvant nivolumab and nivolumab + ipilimumab.
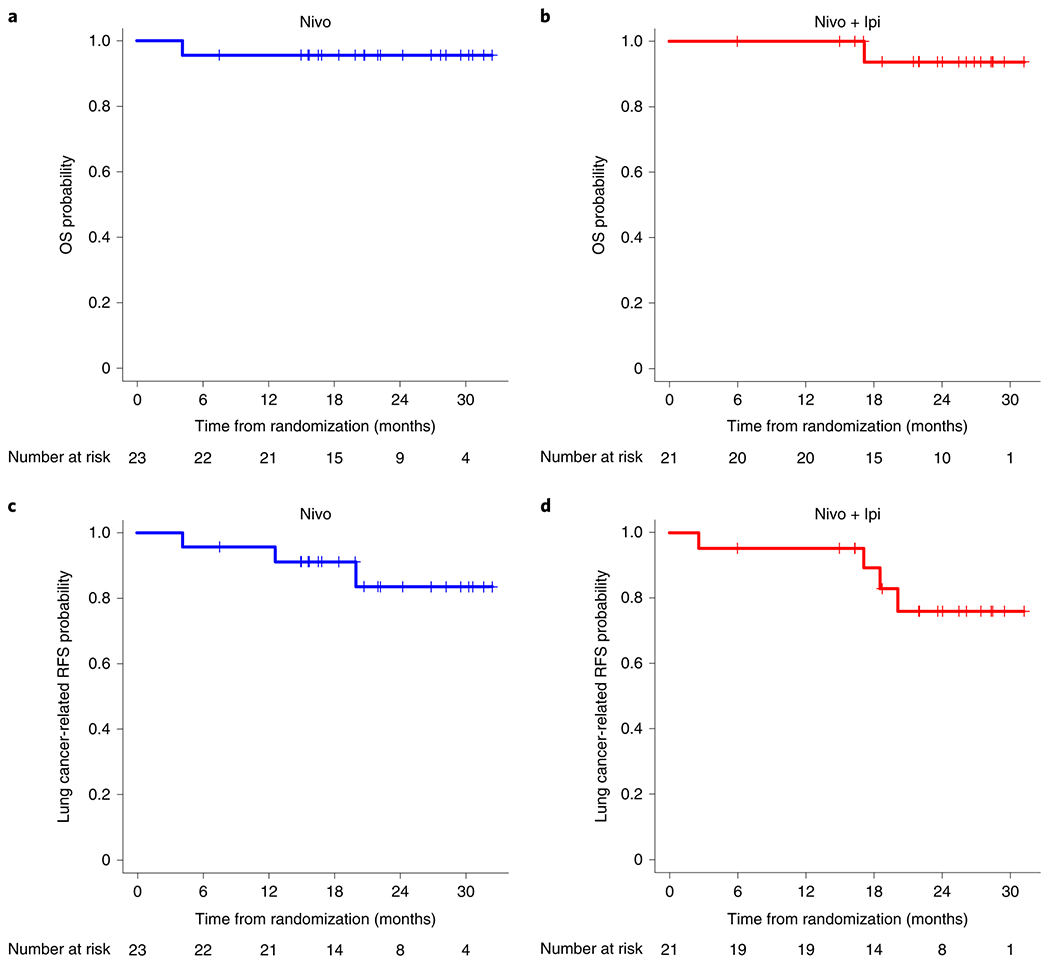
a, Kaplan-Meier curve of probability of OS in patients treated with neoadjuvant nivolumab (n = 23) from randomization to death. Median OS was not reached. One patient treated with neoadjuvant nivolumab and eventually diagnosed with pneumonitis requiring steroids, died within 90 d of surgery and 4.1 months after randomization due to BPF and empyema resulting in respiratory failure. b, Kaplan-Meier curve of probability of OS in patients treated with neoadjuvant nivolumab + ipilimumab (n = 21) from randomization to death. Median OS was not reached. One patient treated with combination therapy had PD 2.6 months after randomization (post-neoadjuvant therapy) and did not undergo surgery. This patient died from lung cancer 17.1 months after randomization following additional therapies. c, Kaplan-Meier curve of probability of lung cancer-related RFS in patients treated with neoadjuvant nivolumab (n = 23) from randomization to recurrence or death. Median lung cancer-related RFS was not reached. One patient died as reported above. Two patients experienced lung cancer-related recurrence due to local and/or distant metastatic disease after 20.0 months (n = 1) and 12.6 months (no surgery on trial, n = 1) after randomization. d, Kaplan-Meier curve of probability of lung cancer-related RFS in patients treated with neoadjuvant nivolumab + ipilimumab (n = 21) from randomization to recurrence or death. Median lung cancer-related RFS was not reached. Four patients experienced disease recurrence at 2.6 months (no surgery, n = 1, died 17.1 months after randomization), 18.5 months (n = 1), 20.1 months (no surgery, n = 1), and 17.1 months after randomization (n = 1).
Immune responses to neoadjuvant nivolumab and nivolumab + ipilimumab.
Tumor cell PD-L1 expression by immunohistochemistry (IHC) was overall higher in pretherapy tumor samples from patients who achieved radiographic responses (CR/PR) and MPR compared with that of tumors with SD/PD (P = 0.017; Fig. 3a,e–h) and no MPR (P = 0.037; Fig. 3b,e–h), respectively. However, responses were also noted in patients with pretherapy specimens lacking tumor PD-L1. Fewer viable tumor cells were also found in resected specimens with pretherapy tumor PD-L1 expression ≥1% compared to tumors with PD-L1 <1% (Extended Data Fig. 5a). In contrast, post-therapy tumor PD-L1 expression was not associated with responses (Fig. 3c–h and Extended Data Fig. 5b), and no significant differences were noted in the percentage of malignant cells expressing PD-L1 between pre- and post-therapy tumors treated with nivolumab or nivolumab + ipilimumab (Extended Data Fig. 6a). Immune profiling of resected tumor tissues by flow cytometry revealed higher frequencies of CD45+CD3+ tumor-infiltrating lymphocytes (TILs) (P = 0.021; Fig. 4a), CD3+CD4+CD103+ (non-T-regulatory (Treg) tissue-resident memory (TRM) T cells (P = 0.041; Fig. 4b), CD3+CD8+CD103+ effector TRM cells (P = 0.057; Fig. 4c), and CD3+CD4+CD27−CD28+ effector memory T cells (P = 0.034; Fig. 4d) after nivolumab + ipilimumab compared with those in tumors resected after nivolumab monotherapy. Multiplex immunofluorescence (mIF) staining of pre- and post-therapy tumor specimens revealed significant increases from pre- to post-therapy in CD3+ (P = 0.016; Fig. 4e,f) and CD3+CD8+ TILs (P = 0.016; Fig. 4e,g), and a trend toward higher densities of CD3+CD8+CD45RO+ memory TILs (P = 0.078; Fig. 4e,h) in tumors treated with nivolumab + ipilimumab. Nivolumab + ipilimumab also induced greater tumor infiltration of other immune cell populations, including CD3+CD8+PD-1+ T cells, CD68+ and CD68+PD-L1+ cells, CD3+CD8+GZB+ and CD3+CD8−FOXP3+ T cells compared with that of pretherapy tumors, although these increases were irrespective of MPR (Extended Data Fig. 6b–i). No substantial changes in immune cell subpopulations were detected from pre- to post-therapy in tumors treated with nivolumab alone (Fig. 4f–h, Extended Data Fig. 6), whereas the changes from pre- to post-therapy in CD3+ T lymphocytes were significantly higher in tumors treated with nivolumab + ipilimumab than in those treated with nivolumab alone (Supplementary Table 10).
Fig. 3. Association of tumor PD-L1 expression in malignant cells and response to neoadjuvant nivolumab and nivolumab + ipilimumab.
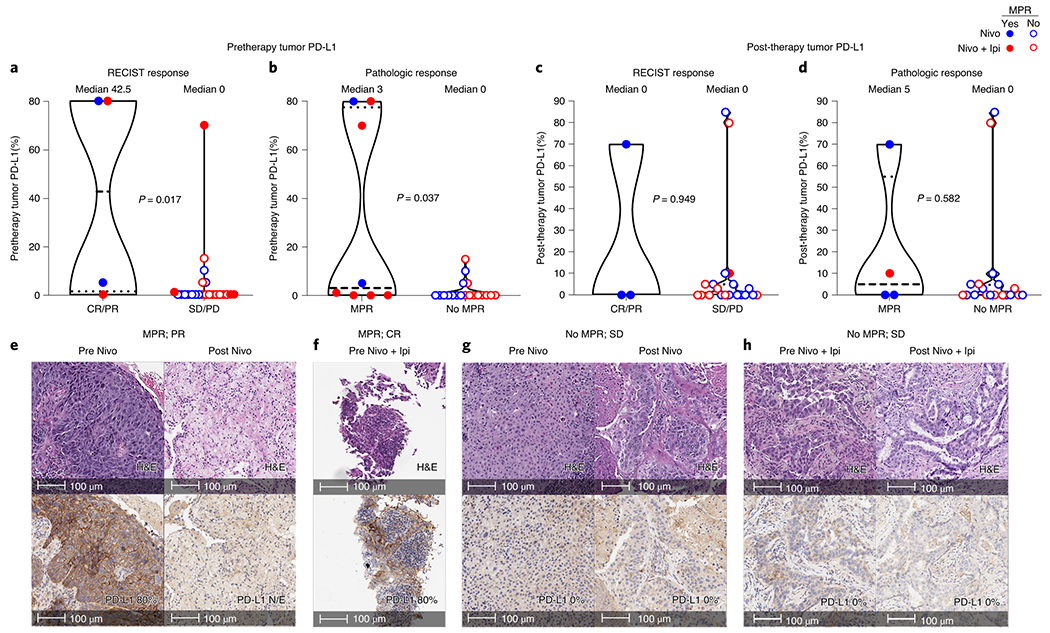
a,b, Pretherapy tumor PD-L1 IHC membranous expression (%) in malignant cells from responders and nonresponders treated with nivolumab and nivolumab + ipilimumab by RECIST (a; CR/PR versus SD/PD; n = 4 versus n = 23) and MPR status (b; n = 8 versus n = 19). Data are presented as the median with minima, lower and upper quartiles, and maxima. Individual data points are shown, dashed line shows the median value, and dotted lines show lower quartile and upper quartile values; the top and bottom of the violin plots indicate the minima and maxima. The two-sided P value is from Wilcoxon’s rank-sum test. c,d, Post-therapy (resected) tumor PD-L1 IHC expression in malignant cells from responders and nonresponders treated with nivolumab and nivolumab + ipilimumab by RECIST (c; n = 3 versus n = 20) and MPR status (d; n = 4 versus n = 19). Data are presented as the median with minima, lower and upper quartiles, and maxima. Individual data points are shown; the dashed line shows the median value, and dotted lines show the lower quartile and upper quartile values of the range; the top and bottom of the violin plots indicate the minima and maxima. The two-sided P value is from Wilcoxon’s rank-sum test. e,f, Examples of hematoxylin and eosin (H&E) micrographs (top panels) of pathologic response (MPR or pCR) in patients treated with nivolumab (e) or nivolumab + ipilimumab (f) with elevated pretherapy tumor PD-L1 expression in malignant cells (bottom panels). Experiments and scorings related to the presented micrographs were conducted once. g,h, Examples of H&E micrographs (top panels) of lack of pathologic response (no MPR) in patients treated with nivolumab (g) or nivolumab + ipilimumab (h) with lack of pretherapy tumor PD-L1 expression in malignant cells (bottom panels). Experiments and scorings related to the presented micrographs were conducted once. N/E, not evaluable PD-L1 due to lack of viable tumor cells in the analyzed tissue section.
Fig. 4. Immune correlates of response to neoadjuvant nivolumab and nivolumab + ipilimumab.
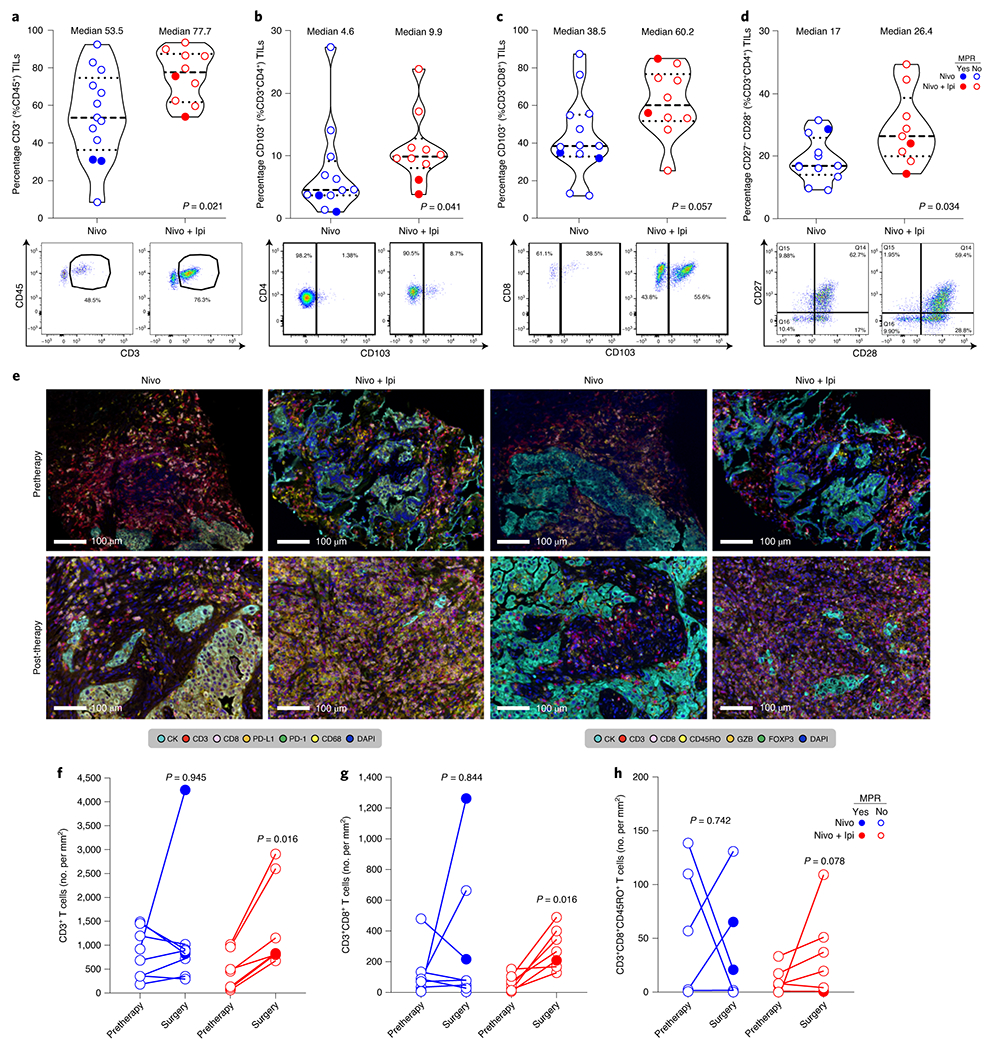
a-d, Frequencies (top panels) and representative gates (bottom panels) of CD3+ T cells (of CD45+) (a), CD103+ TRM cells (of CD3+ (from CD45+) CD4+) (b), CD103+ effector TRM cells (of CD3+ (from CD45+) CD8+) (c), and CD27−CD28+ effector memory T cells (of CD3+ (from CD45+) CD4+) (d) by flow cytometry in tumors resected after neoadjuvant nivolumab (blue; n = 13 (a); n = 12 (b); n = 13 (c); n = 12 (d)) and nivolumab + ipilimumab (red; n = 10 (a); n = 10 (b); n = 10 (c); and n = 9 (d)). Data are presented as the median with minima, lower and upper quartiles, and maxima. All violin plots show single data points; the dashed line shows the median value, and dotted lines show the lower quartile and upper quartile values of the range; the top and bottom of the violin plots indicate the minima and maxima. The two-sided P value is from Wilcoxon’s rank-sum test. Experiments and gating related to presented results were conducted once. Subgating was only performed when more than 100 events were present in parental gate. e, Examples of micrographs of mIF staining of immune markers (panels 1 and 2, respectively) in pretherapy and resected tumors after neoadjuvant nivolumab and nivolumab + ipilimumab. Experiments and scorings related to the presented micrographs were conducted once. f-h, Quantification of CD3+ T-cell (panel 1) (f), CD3+CD8+ T-cell (panel 2) (g), and CD3+CD8+CD45RO+ T-cell (h) densities (no. per mm2) by mIF staining in paired pretherapy and resected tumors after nivolumab (in blue; n = 8) and nivolumab + ipilimumab (in red; n = 7). The two-sided P value is from Wilcoxon’s signed-rank test.
Sequencing of T-cell receptors (TCRs) in a small number of samples demonstrated a positive correlation between the pretherapy peripheral (blood) TCR repertoire richness and richness in resected tumors after neoadjuvant ICIs (Spearman correlation coefficient, R = 0.82; P = 0.023; Fig. 5a). Richness and clonality were greater in resected tumors compared with tumor-adjacent, uninvolved lung samples (P = 0.0029 and P = 0.036, respectively; Fig. 5b,c). We observed that a greater proportion of resected tumors had higher TCR richness and clonality compared with their matched tumor-adjacent, uninvolved lung samples (richness, 83.3 % (10/12); clonality, 67% (8/12), respectively; Fig. 5d,e), and increases were observed in a greater number of tumors treated with nivolumab + ipilimumab (Fig. 5f,g). T-cell richness and clonality increased from pretherapy to surgery in three out of three tumors treated with dual therapy, but only in two out of four tumors treated with nivolumab (Fig. 5h,i). Blood T-cell richness increased in most samples treated with combination therapy, whereas blood clonality decreased in post-therapy compared with pretherapy blood samples (Extended Data Fig. 7a,b). Tumor and peripheral TCR clonality did not significantly correlate with pathologic tumor regression, despite trends in some cases showing inverse correlation between the two variables (Extended Data Fig. 7c–f), although only a single MPR sample was available for these analyses. We identified significantly expanded and contracted TCR clones between available pre- and post-therapy samples and determined that the greatest number of clones were expanded and contracted in resected tumors compared with pretherapy blood samples (Extended Data Fig. 7g,h). Collectively, these results suggest that nivolumab + ipilimumab induced a more profound tumor immune infiltration and enhanced immunologic memory compared with nivolumab, and, in a limited number of cases, was associated with increased T-cell richness and clonality in resected tumors compared with pretherapy samples.
Fig. 5. TCR changes in blood and tumors treated with neoadjuvant nivolumab and nivolumab + ipilimumab.
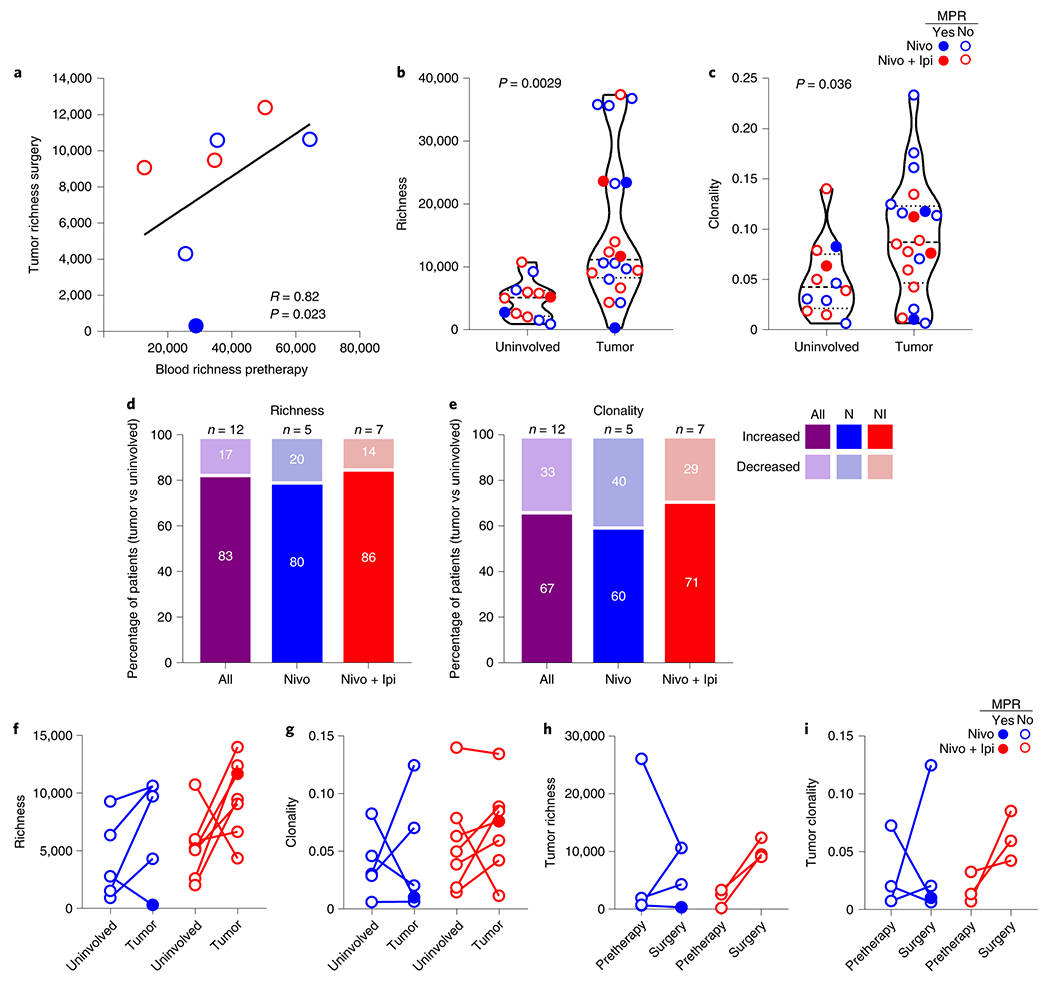
a, Correlation of TCR repertoire richness between the pretherapy (baseline) peripheral blood and resected (surgery) tumors after neoadjuvant nivolumab (n = 4, blue) and nivolumab + ipilimumab (n = 3, red). The two-sided P value is from Spearman’s rank-order correlation. b,c, TCR repertoire richness (b) and clonality (c) between resected tumor-adjacent, uninvolved lungs (n = 12) and resected tumors (n = 20) after neoadjuvant nivolumab (blue) and nivolumab + ipilimumab (red). Data are presented as the median with minima, lower and upper quartiles, and maxima. All violin plots show single data points; the dashed line shows the median value and dotted lines show lower quartile and upper quartile values of the range; the top and bottom of the violin plots indicate the minima and maxima. The two-sided P value is from a two-sample Student’s t-test. d,e, Proportion of patients with increased and decreased TCR repertoire richness (d) and clonality (e) in resected tumors compared with their matched resected, tumor-adjacent, uninvolved lungs (n = 12, purple), after nivolumab (N, n = 5, blue) or nivolumab + ipilimumab (NI, n = 7, red). f,g, Changes in TCR repertoire richness (f) and clonality (g) between matched resected tumors and tumor-adjacent, uninvolved lungs after neoadjuvant nivolumab (n = 5, blue) or nivolumab + ipilimumab (n = 7, red). h,i, Changes in TCR repertoire richness (h) and clonality (i) in matched pretherapy and resected (surgery) tumors after neoadjuvant nivolumab (n = 4, blue) or nivolumab + ipilimumab (n = 3, red). Closed dots: MPR; Open dots: No MPR.
Compositional differences in the gut microbiome and their association with treatment response and toxicity.
We performed exploratory analyses of the gut microbiome composition and diversity from fecal samples collected from patients treated on the study using targeted 16S ribosomal RNA gene sequencing. We questioned whether ICI therapy affected the composition of the gut microbiome. Weighted-UniFrac analysis showed similar variability across microbiome communities in both pre- and post-therapy samples, and no difference between both groups of samples (R = −0.032, P = 0.99), or by MPR or TRAE status (Extended Data Fig. 8a,b). We then compared the microbiome composition by arm and at the family level, and found similar distribution of the top ten most abundant bacterial families in patients treated with either nivolumab or nivolumab + ipilimumab. Microbiome diversity measured by the inverse Simpson index also appeared to be similar based on MPR status in both the nivolumab (P = 0.301; Extended Data Fig. 8c,d) and the nivolumab + ipilimumab arm (P = 0.336; Extended Data Fig. 8e,f), although these results were derived from a limited number of samples. Linear discriminant analysis effect size (LEfSe) studies at the genus level were performed to identify taxa associated with MPR. LEfSe analysis by treatment arm revealed that Paraprevotella and Akkermansia spp. were associated with MPR in the nivolumab and nivolumab + ipilimumab arms, respectively (Extended Data Fig. 8g). An unclassified Ruminococcus sp., a close relative to R. bromii in sequence identity, was associated with MPR in samples from the nivolumab + ipilimumab arm (Extended Data Fig. 8g, bottom panel). We also observed an association between Dialister sp. and decreased toxicity to nivolumab and between Bifidobacterium and Enterobacter spp., and an unclassified genus of Erysipelotrochaceae and reduced toxicity to dual therapy (Extended Data Fig. 8h). Last, we analyzed correlations between fecal microbiome and TCR clonality and richness in post-therapy tumors using Spearman’s correlations. We found that genera associated with MPR or decreased toxicity, including Akkermansia sp. (R = 0.44, P = 0.05) and Bifidobacterium sp. (R = 0.47, P = 0.04), had a positive correlation with TCR clonality, whereas genera associated with a lack of MPR or increased toxicity, including Coprococcus_3 sp. (R = −0.48, P = 0.04), Lachnospiraceae_UCG_004 (R = −0.66, P < 0.001) and Lachnospiraceae_unclass (R = −0.47, P = 0.04) were negatively correlated with TCR clonality Extended Data Fig. 9a,b). Bacteria associated with T-cell richness included Anaerofustis sp. (R = 0.58, P = 0.001), Faecalibaculum sp. (R = 0.61, P = 0.006), and Ruminococcus_1 sp. (R = −0.65, P = 0.003), among others (Extended Data Fig. 9c,d). Together these results suggest that abundance of different gut bacteria is associated with pathologic responses, reduced toxicity and, in some cases, higher T-cell clonality and richness after neoadjuvant ICIs.
Discussion
NEOSTAR is the first reported, randomized phase 2 trial testing neoadjuvant nivolumab and nivolumab + ipilimumab in patients with resectable NSCLC using MPR as the primary endpoint. Nivolumab + ipilimumab produced higher MPR rates than historical neoadjuvant chemotherapy controls. The combination therapy also produced higher MPR rates compared with nivolumab monotherapy in both the ITT and the resected populations. Moreover, 38% of resected patients had a pCR in the nivolumab + ipilimumab arm, compared with only 10% in the nivolumab arm. Tumors treated with combination therapy were much less viable compared with those treated with nivolumab monotherapy. The toxicity profile was overall manageable with no new safety concerns. Nivolumab + ipilimumab induced greater overall tumor infiltration of CD3+ and CD3+CD8+ T lymphocytes, TRM and effector memory T cells.
Historically, neoadjuvant chemotherapy has resulted in MPR rates between 7% and 27% in localized NSCLC4,6,7,12,13. Previous studies examining the clinical activity of single agent anti-PD-1/PD-L1 therapies in the neoadjuvant setting have reported MPR rates between 19% and 45% in patients with resectable NSCLC14–16. In the present study, nivolumab monotherapy produced an MPR of 22% in 23 treated patients and 24% in 21 resected tumors. The addition of ipilimumab to nivolumab produced twice as many MPRs in resected patients compared to nivolumab alone (50% versus 24%) and a greater proportion of pCRs compared with nivolumab (38% versus 10%). In comparison, several clinical trials of neoadjuvant platinum doublet chemotherapy have reported a median pCR rate of only of 4%5, indicating that a chemotherapy-sparing regimen produces complete tumor regression more frequently. However, the 95% CI of the MPR rate to neoadjuvant nivolumab + ipilimumab was relatively wide (18-62%), which indicates that further studies with a larger sample size could more precisely determine the magnitude of increment of the primary endpoint compared with chemotherapy alone.
The immune composition of tumors treated with nivolumab + ipilimumab was distinct from those treated with nivolumab monotherapy. We observed enhanced T-cell infiltration in tumors treated with combination therapy; however, immune cell infiltration of tumors was independent of MPR. Studies in melanoma using higher dosages of ipilimumab reported enhanced immune infiltration in responding tumors17. In our study, the extent of pathologic response in some tumors rendered them unavailable for analysis and the longer duration between ipilimumab dosing and surgery may not have coincided with the maximal immune cell response. We noted higher frequencies of CD4+ (non-Tregs) CD103+ and CD8+CD103+ TRM and effector memory T cells in tumors treated with nivolumab + ipilimumab compared with tumors treated with nivolumab alone. This observation is particularly encouraging given the tendency of early-stage tumors to recur following surgical resection. To our knowledge, this is the first report of neoadjuvant ICIs imparting immunologic memory in clinical specimens of resected NSCLC. Previous studies demonstrated that enhanced CD8+CD103+ TRM TILs correlate with improved survival in patients with early stage NSCLC18 and with other types of cancer19. Wei and colleagues20 have shown that combination therapy with CTLA-4 and PD-1 blockade mediates a switch from expansion of exhausted CD8+ T cells to expansion of activated effector CD8+ T cells. It is possible that the addition of ipilimumab to nivolumab may have enhanced tumor infiltration of effector T cells with immunologic memory phenotypes.
Although we found overall higher tumor PD-L1 expression in baseline samples from responding patients, we also observed responses in patients lacking pretreatment tumor PD-L1. Similarly, studies of neoadjuvant immunotherapy in patients with resectable NSCLC reported responses irrespective of PD-L114,21, illustrating that that studies in larger cohorts will be needed to define the relationship between tumor PD-L1 and responses.
The results of our TCR sequencing performed in a small subset of samples suggested potential homing of T cells from the periphery pretherapy to the resected tumors post-therapy. The higher TCR richness and clonality in a greater proportion of treated tumors as compared to tumor-adjacent, uninvolved lung samples indicate that resected tumors may have a more diverse T-cell repertoire after neoadjuvant immune checkpoint blockade, with a subset of dominant clones accounting for the higher clonality, although these results were irrespective of response. In contrast, in studies performed in patients with treatment-naïve and resected NSCLC, higher TCR clonality was observed in the uninvolved resected lung22. The expansion of T-cell clones from the periphery to the resected tumors may represent a recruitment of dominant clones after neoadjuvant therapy; however, further studies are needed in larger sets of samples. Studies in other cancers suggest that peripheral T-cell expansion to adjacent uninvolved tissues and tumors is a key determinant of clinical benefit from immunotherapy23. Amaria and colleagues17 demonstrated that a more clonal T-cell repertoire at baseline correlated with response to neoadjuvant ICI therapy in patients with melanoma, with further expansion of the pre-existing high-frequency clones with treatment. Blank et al.24 found that melanoma patients treated with neoadjuvant nivolumab + ipilimumab had a greater expansion of tumor-resident T-cell clones than patients treated with adjuvant combined therapy.
We found a positive association between radiographic and pathologic tumor responses; however, in some cases, imaging underestimated the degree of pathologic tumor regression, emphasizing the limitation of imaging to accurately capture responses to neoadjuvant therapies. We have previously shown that the histopathologic response to neoadjuvant chemotherapy is a stronger predictor of improved survival outcome, compared with responses at CT13,25. In the present study, we also noted that some patients developed a phenomenon we referred to as NIF. NIF is characterized by the radiographic appearance of disease progression in lymph nodes that were histologically negative at baseline and that became enlarged and/or FDG avid after neoadjuvant ICIs. Invasive pathologic examination of the enlarged/flaring nodes post-therapy revealed immune cell infiltration and granulomas without evidence of malignancy11. Invasive restaging allowed us to proceed with curative surgery in cases where false radiographic nodal progression may have prohibited surgery.
Our microbiome analyses revealed that administration of ICIs had no significant impact on the diversity, structure and composition of microbiomes as assessed by 16S V4 rRNA gene sequencing. However, qualitative biomarker discovery analyses demonstrated that higher abundance of Ruminococcus spp. in pretherapy samples was associated with MPR to nivolumab + ipilimumab. Recent studies have shown that higher diversity and relative abundance of fecal Ruminococcaceae are associated with response to ICIs in patients with melanoma26. We also found that higher abundance of Akkermansia spp. in baseline samples was associated with MPR to nivolumab + ipilimumab and positively correlated with TCR clonality in resected tumors. Similarly, Akkermansia muciniphila was associated with favorable clinical outcomes in patients with melanoma, NSCLC and renal cell carcinoma receiving ICIs in previous studies26,27. Our results support a role for the fecal microbiome in modulating tumor response to ICIs.
Our study has some limitations. It was not powered to formally test the differences in clinical activity and immune responses between the two treatment arms, but rather to compare tumor responses in both arms with historical controls of neoadjuvant chemotherapy. The results of the reported comparisons are exploratory and hypothesis generating, including the findings of our gut microbiome qualitative and correlative analyses, and require additional investigations, such as validation of proposed biomarker bacteria with quantitative measurements and deeper taxonomic and functional characterization. The number of pre- and post-therapy samples and the number of tissues derived from resected tumors with MPR for correlatives was limited. Despite these limitations, to our knowledge, this is the first completed randomized study comparing the impact of neoadjuvant nivolumab monotherapy and combined nivolumab + ipilimumab in resectable NSCLC. The MPR rate of 50% in resected patients after combination therapy is promising and provides a platform for further evaluation in larger studies. First-line nivolumab + ipilimumab resulted in a longer duration of OS than chemotherapy in patients with metastatic NSCLC, irrespective of the PD-L1 expression10,28. Those findings, along with results from NEOSTAR, suggest that combined nivolumab + ipilimumab may represent a chemotherapy-sparing regimen in both resectable and metastatic NSCLC.
We found the differences between the two arms compelling. Recent results from the NADIM29 and the neoadjuvant atezolizumab + chemotherapy21 studies suggest that combining chemotherapy with an ICI may offer an additional clinical benefit. Building on these results, and on the recent findings of the CheckMate 9LA study, showing that in patients with advanced NSCLC front-line nivolumab + ipilimumab combined with chemotherapy improved survival compared with chemotherapy alone30, our study sets the stage to evaluate the role of dual ICIs added to neoadjuvant chemotherapy, which is currently being tested (NCT03158129).
In conclusion, our findings provide evidence that the addition of neoadjuvant ipilimumab to nivolumab produces higher rates of MPR and pCR and enhances tumor immune infiltrates and immunologic memory. The combination of nivolumab and ipilimumab merits further investigation in the perioperative setting for patients with resectable NSCLC.
Methods
Trial design, hypotheses and endpoints.
This is a phase 2, open-label, single-institution, randomized study that after completion of the first two arms evolved into a modular platform design. The original study design required eligible patients to be equally randomized to two arms; however, after trial commencement, the trial design was changed to a platform of multiple, independent, single arm studies to follow the first two randomized arms and was expected to be analyzed and reported separately with the goal to expedite the investigation of novel ICI-based promising strategies in the neoadjuvant setting to be tested in larger, definitive studies. The first two randomized arms of the study have completed accrual and are reported herein. The patients included in this article were those randomly assigned at 1:1 ratio to the first two arms of the study and treated with either nivolumab monotherapy or combination of nivolumab and ipilimumab. The primary hypothesis to be tested in the present study is that, in patients with stage I-IIIA NSCLC amenable for surgical resection, induction immunotherapy with nivolumab alone and/or nivolumab + ipilimumab will produce MPR rates of at least 40%, a target response that is superior to the one observed after induction platinum-based chemotherapy alone of 15% (as observed in MD Anderson historical controls4). The prespecified boundary for a treatment arm to be considered promising for further testing was ≥6 MPRs in 21 evaluable patients. The secondary hypothesis to be tested in the present study was that immunotherapy with nivolumab and/or nivolumab + ipilimumab would induce immune responses (as assessed by CD8+ TILs), and tumor shrinkage (as assessed by radiographic imaging), and improve RFS and OS in a subset of patients. Analysis of correlative studies in these patients would assist in developing biomarkers predictive of response to immunotherapeutic agents in NSCLC and determining immune modulation by neoadjuvant immunotherapy. The primary endpoint of the trial was MPR, defined as ≤ 10% viable tumor cells in the original resected tumor bed after neoadjuvant therapy on trial. Secondary endpoints included treatment toxicity, perioperative morbidity and mortality, quantification of CD8+ T cells in resected tumor tissues from patients treated with neoadjuvant nivolumab alone and nivolumab + ipilimumab, ORR, OS, lung cancer-related RFS, completeness of surgical resection, pCR, correlation of radiographic responses and MPR with RFS and OS, and correlation of blood, tissue and stool biomarkers with efficacy and toxicity. Exploratory endpoints included identification and treatment modulation of radiographic, blood, tissue, and stool biomarkers, along with the association between the fecal microbiome and the responses and toxicity to neoadjuvant therapy in each study arm (Extended Data Fig. 1).
Sample size justification and toxicity monitoring guidelines.
Simon’s minimax two-stage design31 is applied to test the MPR rate for each of the treatment arms. We assumed a historical MPR rate of 15%4 under the null hypothesis versus an MPR rate of 40% under the alternative hypothesis. For each treatment arm, 15 patients were enrolled in the first stage. If only ≤2 of the 15 patients have experienced an MPR, enrollment to that treatment arm would be terminated and the treatment would be considered inefficacious. Otherwise, with at least 3 MPRs, an additional 6 patients were enrolled to reach a total of 21 patients. At the end of trial, if we observed six or more patients experiencing MPR, the treatment would be considered efficacious and inefficacious otherwise. The trial has 90% power when the MPR rate is 40%. When the MPR rate is 15%, the probability of early termination is 60%, with an average sample size of 17.4 and one-sided 10% type I error rate. From the above calculations, the study needs up to 21 evaluable patients in each arm. Assuming a nonevaluable rate of 5% (for example, patients drop out, are lost to follow-up or rescind the consent due to nontreatment-related reasons before endpoints can be evaluated), we would need to enroll up to a total of 22 patients per arm. Enrolled patients were monitored for adverse events (AEs). AEs were treated as detailed in the protocol algorithm of toxicity management. We applied a Bayesian method to formally monitor the toxicity in the perioperative phase within each treatment arm32.
Study oversight, ethical approval and ethical standards.
Written informed consent was provided by all study participants or their legal representatives. The study was approved by the University of Texas MD Anderson Cancer Center’s Institutional Review Board. The Data and Safety Monitoring Board provided oversight until all participants randomized to arms A (nivolumab) and B (nivolumab + ipilimumab) completed treatment on study. Data were collected and analyzed by the investigators, and all authors approved and agreed to submit the final manuscript for publication. The authors vouch for the accuracy and completeness of the data and for the fidelity of the trial to the study protocol.
Participants.
Patients who met inclusion criteria for the study were aged 18 years and older and had stage IA-IIIA NSCLC according to American Joint Commission on Cancer (AJCC) 7th edition staging system. Only a single mediastinal N2 station was allowed for the enrollment. All patients had to have surgically resectable disease and Eastern Cooperative Group performance status 0-1, adequate organ function, and cardiopulmonary status. Patients were excluded from the study if they had autoimmune disease, immunodeficiency, or previously received immunotherapy for other disease, if they had active infectious disease requiring ongoing treatment or cancer within the last 2 years. The first and last patient were enrolled on study on 16 June 2017 and 15 November 2018, respectively. Patients were screened, enrolled and treated on the study at the MD Anderson Cancer Center.
Interventions.
The patients included in this manuscript were those randomly assigned at a 1:1 ratio to receive either nivolumab administered at the dose of 3 mg kg−1 i.v. on D1, D15, and D29 or nivolumab + ipilimumab with ipilimumab given at the dose of 1 mg kg−1 i.v. on D1 only. The treating physicians enrolled and consented patients on the study and the clinical trial coordinator assigned consented participants to the study intervention. The randomization was performed by the clinical trial coordinator using the database system in the Department of Biostatistics by applying the Pocock Simon minimization method with stage as a stratification factor. No blinding was done as part of this study. Before randomization, patients underwent clinical radiographic staging, which included PET-CT, contrast CT, and brain magnetic resonance imaging. Baseline invasive mediastinal staging was strongly recommended with endobronchial ultrasound or mediastinoscopy. CT and PET-CT imaging was repeated at least 14 d after completion of neoadjuvant immunotherapy and responses were evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST) v.1.133 by two experienced clinical radiologists. Surgical resection was planned at least 21 d after and within 42 d of the last dose of neoadjuvant therapy. Surgical resection of the primary tumor and mediastinal lymph nodes was performed based on surgeons’ discretion and specialty standards. The randomization part of the study reached its accrual goal and follow-up duration. Adjuvant chemotherapy and/or PORT was administered at the discretion of the treating physician.
Pathologic assessment.
Pathologic assessment consisted of gross and histopathologic examination of the lung resection specimens. After gross identification of the tumor or tumor bed, at least one section per centimeter of greatest tumor (bed) diameter was submitted for histopathologic evaluation, as previously reported4. In cases in which no residual viable tumor was identified microscopically on initial representative sections, the entire tumor bed was subsequently submitted for review. In total, the tumor (bed) was submitted entirely in 17 cases. Histopathologically, the mean percentage of viable tumor cells, averaged across all reviewed tumor slides, was assessed for each patient as previously reported4. Tumors with ≤ 10% of viable tumor cells were considered to have undergone MPR and tumors with 0% viable tumor were considered to have undergone pCR. After initial clinical reporting, pathologic responses were subsequently reviewed in a blinded manner by two pathologists experienced in the evaluation of tumor response after neoadjuvant therapy, and the average scores were used for final analysis. To maximize interobserver consistency, these two pathologists had undergone an initial training period consisting of co-examination of 5-10 slide samples followed by examining 5-10 slide samples independently, with subsequent review and consensus agreement on tumor response12. Mediastinal and peribronchial lymph nodes were submitted and processed in a routine fashion for microscopic assessment and examined for metastatic disease. Pathologic staging was performed based on tumor and lymph node assessment of the resection specimens according to the criteria for lung cancer staging of the AJCC 7th (cases before 2018) and 8th (cases from 2018) editions.
Statistical methods.
The Student’s t-test was applied to compare the mean between two treatment arms and Chi-square test or Fisher’s exact test34 was applied to assess the association between two treatment arms and categorical variables. As the primary analysis, the MPR rate was estimated with exact 95% CI within each study arm. At the end of the trial, the null hypothesis was rejected if the lower end of the CI is larger than the null MPR rate. TRAEs were summarized with frequencies and percentages. Subsequent analyses comparing the two treatments arms are considered to be exploratory in nature. For correlative analyses, Wilcoxon’s rank-sum test was used to compare continuous variables between two independent groups. Wilcoxon’s signed-rank test was used for comparison of paired data. Exact tests were performed where applicable. The Spearman’s correlation coefficient was used to assess the correlation between two continuous variables. The exact number of values used to calculate the statistics is reported in each figure and/or figure legend. OS was defined as the time from randomization to the time of death or to the time of last contact. Lung cancer-related RFS was defined as the time from randomization to the time of recurrence or death, whichever occurred first, or to the time of last contact. For the analysis of lung cancer-related RFS according to radiographic and pathologic responses, PORT and adjuvant chemotherapy, the landmark analysis method was used, and post-treatment radiographic tumor measurement date and surgery date were considered the starting point of lung cancer-related RFS. The distributions of OS and lung cancer-related RFS were estimated by the Kaplan-Meier method35. Logrank test was performed to test the difference in survival between groups36. A two-sided P value of 0.05 was considered to be significant. Clinical analyses were conducted using SAS (v.9.4), R v.3.6.1, Microsoft Excel v.2016 and GraphPad Prism v.8.00.
Tissue, blood and fecal microbiome sampling.
Pretherapy (baseline), on-therapy (longitudinal), post-therapy, at surgery and post-surgery specimen collections were performed, when available, for biomarker analysis. Pretherapy archival or fresh tumor tissues (when archival tumor tissue was not available) were obtained for diagnosis and, when available, for correlative analysis purposes. At surgery, tumor-adjacent, uninvolved lung and tumor tissues were collected for histopathologic and biomarker analyses. Blood specimens were collected, when available, for biomarker analysis pretherapy (pre-dose 1), before each dose of therapy (pre-dose 2 and dose 3), at least 14 d after the last dose of neoadjuvant therapy (post-therapy, before surgery) and within 8 weeks after surgery. Fecal samples were collected, when available, for microbiome 16S analysis pre- and post-therapy (pre-dose 1 and at least 14 d after the last dose, respectively).
Multiparameter flow cytometry.
Fresh tumor tissue was disaggregated using the BD Medimachine System (BD Biosciences) to make a single cell suspension for flow cytometry staining. Surface staining was performed in 1X DPBS with 1% bovine serum albumin for 30min on ice using fluorochrome-conjugated monoclonal antibodies37 against CD45 (BUV395, Clone HI30, catalog no. 563792), CD3 (PerCP-Cy5.5, Clone SK7, catalog no. 340949, BD Biosciences), CD8 (AF 700, Clone RPA-T8, catalog no. 557945, BD Biosciences), CD4 (BUV496, Clone SK3, catalog no. 564651, BD Biosciences), PD1 (BV650, Clone EH12, catalog no. 564104, BD Biosciences), TIM3 (BV605, Clone F38-2E2, catalog no. 345018, BioLegend), CD103 (BV711, Clone Ber-Act8, catalog no. 563162, BD Biosciences), CTLA-4 (BV786, Clone BNI3, catalog no. 563931, BD Biosciences), GITR (AF 488, Clone eBioAITR, catalog no. 53-5875-42, eBioscience), LAG3 (PE, Clone 3DS223H, catalog no. 12-2239-42, eBioscience), CD56 (PE-Cy7, Clone B159, catalog no. 557747, BD Biosciences), ICOS (BV421, Clone C398.A4, catalog no. 313524, BioLegend), and CD25 (APCFire/750, Clone BC96, catalog no. 302642, BioLegend). After surface staining, cells were fixed and permeabilized using eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (catalog no. 00-5523-00, ThermoFisher) according to the manufacturer’s instructions, and stained using FOXP3 (PE-eFluor610, catalog no. 61-4776-42, eBioscience) and Ki67 (APC, Clone 20Raj1, catalog no. 17-5699-42, eBioscience) anti-human antibodies. Samples were acquired using the BD Fortessa X20 and analyzed using FlowJo Software v.10.5.3 (Tree Star, Inc.). Dead cells were stained using LIVE/DEAD Fixable Yellow Dead Cell Stain dye (catalog no. L-34968, Life Technologies) and excluded from the analysis. Experiments and gating related to the presented results were conducted once. The associated gating strategies are shown in Extended Data Fig. 10. The results were graphed using Microsoft Excel v.2016 and GraphPad Prism v.8.00.
MIF staining and analysis.
The mIF staining was performed using similar methods and reagents to those previously described and validated38. Briefly, 4-μm-thick formalin-fixed, paraffin-embedded (FFPE) tumor sections were stained using an automated staining system (BOND-RX; Leica Microsystems) and two panels contained antibodies against: panel 1: cytokeratin (clone AE1/AE3, dilution 1:300, Dako), PD-L1 (clone E1L3N, dilution 1:3,000, Cell Signaling Technology), CD68 (clone PG-M1, dilution 1:450, Dako), CD3 (catalog no. IS503, dilution 1:100, Dako), CD8 (clone C8/144B, dilution 1:300, Thermo Fisher Scientific), and PD-1 (clone EPR4877-2, dilution 1:250, Abcam); panel 2: cytokeratin (clone AE1/AE3, dilution 1:300, Dako), CD3 (catalog no. IS503, dilution 1:100, Dako), CD8 (clone C8/144B, dilution 1:300, Thermo Fisher Scientific), CD45RO (clone UCHL1, Cell Signaling Technology), Granzyme B (clone 11F1, Cell Signaling Technology), and FOXP3 (clone D2W8E, Cell Signaling Technology). All the markers were stained in sequence using their respective fluorophore contained in the Opal 7 kit (catalog no. NEL797001KT; Akoya Biosciences/PerkinElmer)39. The stained slides were scanned using the multispectral microscope, Vectra v.3.0.3 imaging system (Akoya Biosciences/PerkinElmer), under fluorescence conditions in low magnification at x1039. After being scanned in low magnification, a pathology selected around five regions of interest (ROIs; each ROI: 0.3345 mm2) per sample to cover around 1.65 mm2 of tumor tissue using the phenochart v.1.0.9 viewer (Akoya Biosciences/PerkinElmer). The ROIs were analyzed by a pathologist using InForm v.2.8.2 image analysis software (Akoya Biosciences/PerkinElmer). Marker colocalization was used to identify malignant cells (AE1/AE3+), malignant cells expressing PD-L1 (AE1/AE3+PD-L1+), and populations of T cells (CD3+), cytotoxic T cells (CD3+CD8+), antigen-experienced T cells (CD3+PD-1+), cytotoxic antigen-experienced T cells (CD3+CD8+PD-1+), macrophages (CD68+), and macrophages expressing PD-L1 (CD68+PD-L1+) in panel 1, and T cells (CD3+), cytotoxic T cells (CD3+CD8+), cytotoxic activated T cells (CD3+CD8+Granzyme B+), effector/memory cytotoxic T cells (CD3+CD8+CD45RO+), and Treg cells (CD3+CD8−FOXP3+) in panel 2. Densities of each colocalized cell population were quantified as the average and the final data were expressed as number of cells per mm2 in two compartments: tumor nests and tumor stroma40. Malignant cells and macrophages expressing PD-L1 were also expressed as percentages. All the data were consolidated using the R studio v.3.5.3 (Phenopter v.0.2.2 packet, Akoya Biosciences/PerkinElmer) and SAS v.7.1 Enterprise. Experiments and scorings related to the presented micrographs were conducted once. The results were plotted using Microsoft Excel v.2016 and GraphPad Prism v.8.00.
IHC of PD-L1 and analysis.
FFPE tumor tissue was used for single chromogenic IHC analysis of PD-L1 (clone 28-8, dilution 1:100; Abcam) in malignant cells. The antibody’s conditions were previously optimized and validated41. The IHC staining was performed in a Leica Bond Max autostainer system (Leica Biosystems) according to standard automated protocols. Briefly, tissue sections were deparaffinized and rehydrated following the Leica Bond protocol; antigen retrieval was performed with Bond Solution no. 2 (Leica Biosystems, equivalent to ethylenediaminetetraacetic acid, pH 9.0) for 20 min; the primary antibody was incubated for 15 min at room temperature and detected using the Bond Polymer Refine Detection kit (Leica Biosystems) with 3,3′-diaminobenzidine as the chromogen. The slides were counterstained with hematoxylin, dehydrated and cover-slipped. Two pathologists evaluated PD-L1 expression in the membrane of viable malignant cells by standard microscopy according to the International Association for the Study of Lung Cancer guidelines42. The results were reported as percentage of malignant cells with any positive membrane staining. Experiments and scorings related to the presented micrographs were conducted once. The results were plotted using Microsoft Excel v.2016 and GraphPad Prism v.8.00.
TCR-sequencing analysis.
Genomic DNA was extracted from peripheral blood mononuclear cells (PBMCs, n = 7 patients) and tumor tissues (n = 7 patients) at pretherapy, as well as PBMCs (n = 7 patients) at post-therapy, and tumor-adjacent, uninvolved, resected lungs (n = 12 patients) and tumors (n = 20 patients) at surgery using QIAamp DNA Blood Mini Kit (blood DNA extractions), Qiagen QIAamp DNA Mini kit (frozen samples) and Qiagen AllPrep DNA/RNA FFPE kit (FFPE samples). DNA samples then underwent sequencing of the variable CDR3 region of the β-chain of the TCR involved in antigen binding, using the hsTCRβ immunoSEQ assay (Adaptive Biotechnologies Inc.). PCR products were then sequenced on a MiSeq (Illumina) before the data upload to Adaptive Biotechnologies using the immunoSEQ Data Assistant for data deconvolution (immunoSEQ Analyzer v.3). From CDR3β data, we extracted T-cell richness (a measure of T-cell diversity) and T-cell clonality (a measure of T-cell reactivity). To identify TCRs that were statistically enriched or contracted in one group of samples compared with another, we applied a differential abundance framework using the Adaptive Biotechnologies Analyzer platform’s default settings. Statistical significance was set up at two-sided P < 0.01, with Benjamini-Hochberg adjustment for false discovery rate. The top 100 most frequent clones in each tumor at surgery were compared with paired samples to determine whether they were shared or unique. Details on how all metrics were calculated have been previously reported22,43. The results were plotted using Microsoft Excel v.2016 and GraphPad Prism v.8.00.
Fecal microbiome specimen processing and analysis.
For fecal microbiome analysis, total DNA was extracted from fecal samples using the QIAamp DNA Stool Kit (QIAGEN), followed by a bead-beating lysis step. The V4 region of the bacterial 16S rRNA was amplified and sequenced on the Illumina MiSeq platform with 2 x 250 bp reads (Illumina, Inc.). We used VSEARCH to merge and de-replicate paired-end reads, and sorted them by length and size. Sequences were then error corrected and chimera filtered using the UNOISE algorithm44 v.3 and generated operational taxonomic units (OTUs) and presumed chimeras. Later, we added the chimeras sequences identified by the UNOISE algorithm44 v.3 but matched an entry in Silva database v.12845 with a perfect score back to the OTU list and generated a total of 1,849 OTUs. The sequencing depths ranged from 1,339 to 175,238, with a median of 11,656 reads per sample. A rarefaction cutoff of 1,339 reads was applied to the dataset for the calculation of α-diversity metrics reported herein as an inverse Simpson index. The results were plotted in R (R Core Team 2020; https://www.R-project.org) using ggplot2 package (https://ggplot2.tidyverse.org) and GraphPad Prism v.8.00.
Extended Data
Extended Data Fig. 1. Trial schema.
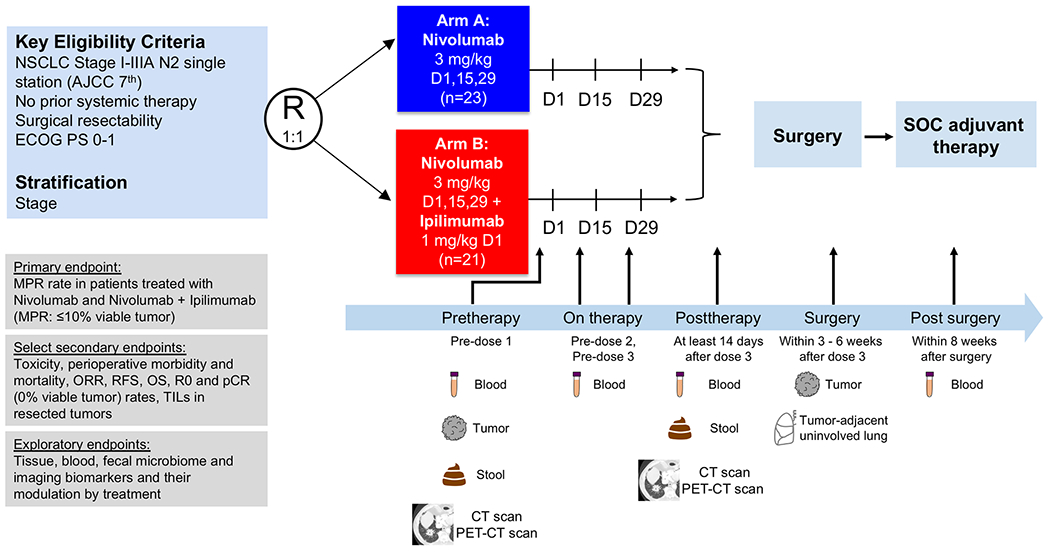
Patients with resectable, pathologically confirmed, clinical stage I-IIIA (N2 single station) NSCLC were stratified by stage and randomized in 1:1 ratio to neoadjuvant nivolumab 3 mg/kg IV every 14 days for up to three doses (arm A; D1, D15 and D29) or ipilimumab 1 mg/kg IV every 6 weeks plus nivolumab 3 mg/kg IV every 14 days for up to three doses (arm B; ipilimumab on D1 only, nivolumab on D1, D15 and D29), followed by surgical resection (at least 3 weeks and within 6 weeks after the last dose of nivolumab). Standard of care adjuvant chemotherapy and/or postoperative radiation therapy were allowed at the discretion of the treating physician. The primary endpoint of the trial was MPR, defined as ≤10% viable tumor in resected tumor specimens. Select secondary endpoints included toxicity, perioperative morbidity and mortality, objective response rates (ORR) by RECIST v.1.1, survival outcomes, radical resection (R0) rate, pathologic complete response (pCR) rate, defined as 0% viable tumor in resected tumor specimens, and quantification of TILs in resected tumor tissues. Select exploratory endpoints included analysis of biomarkers and their modulation by treatment. Imaging studies were performed with CT and PET-CT scans pretherapy (prior to first dose) and at least 14 days after the last dose of neoadjuvant therapy before surgical resection (posttherapy). Tumor samples were collected pretherapy and at surgery together with tumor-adjacent uninvolved lung tissue. Stool samples were collected pretherapy and posttherapy (prior to surgery). Longitudinal blood samples were collected pretherapy, prior to dose 2 and 3, posttherapy (prior to surgery) and within 8 weeks after surgery. NSCLC, non–small cell lung cancer; ECOG PS, Eastern Cooperative Oncology Group performance status; MPR, major pathologic response; ORR, objective response rate; RFS, recurrence-free survival; OS, overall survival; R0, complete surgical resection; pCR, pathologic complete response; TILs: tumor-infiltrating lymphocytes. D: day of therapy. CT: computed tomography, PET-CT: positron emission tomography-computer tomography scan.
Extended Data Fig. 2. Consolidated Standards of Reporting Trials (CONSORT) flow diagram.
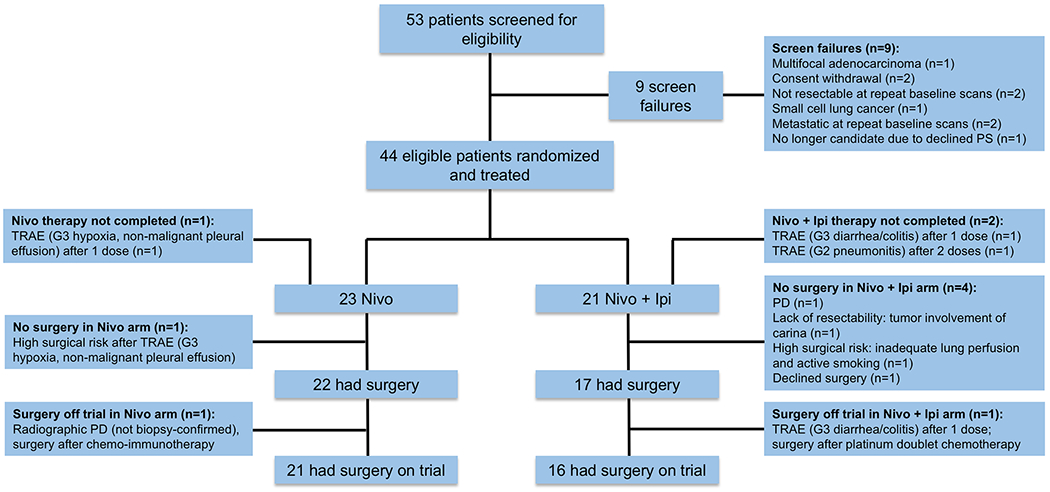
Flow diagram depicts the disposition of patients throughout the phases of the study, including screening, randomization to neoadjuvant treatment and surgery. Reasons for screen failures, no completion of planned neoadjuvant therapy and surgery not performed, or surgery performed off trial are shown. SAE, serious adverse event, TRAE, treatment-related adverse event; PD, progressive disease; PS, performance status.
Extended Data Fig. 3. Tumor size change from baseline after neoadjuvant ICIs by treatment arm and by MPR status.
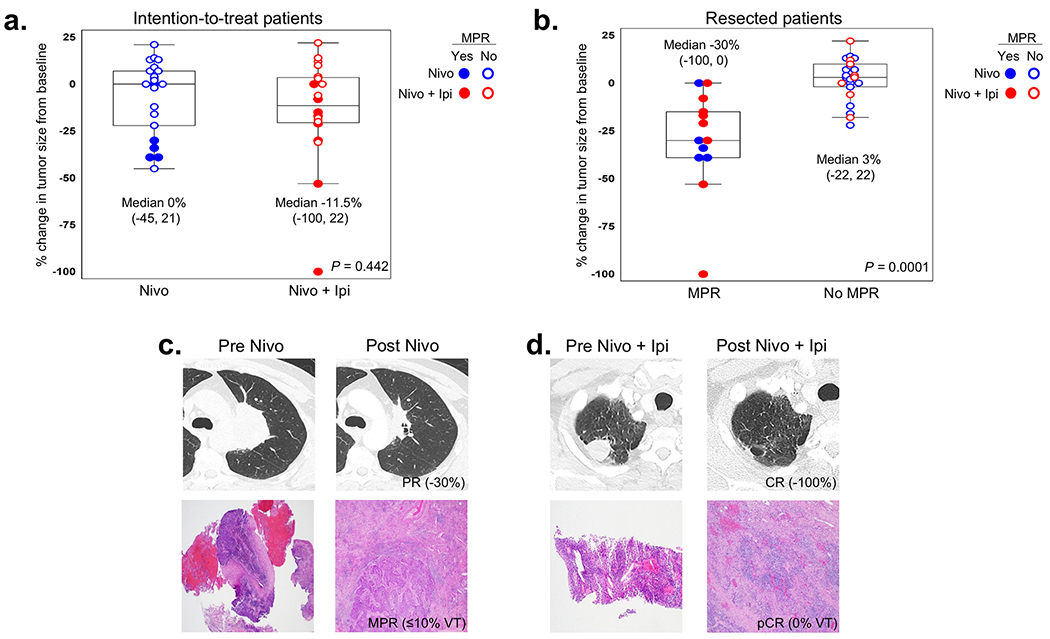
a,b, Boxplots depict the association between percent change in tumor measurement from pretherapy (baseline) in patients treated with neoadjuvant therapy by treatment arm in ITT (a) and in resected patients by MPR status (b). In one patient, the solid lesion was <1 cm following three doses of nivolumab monotherapy and did not change compared to baseline; response was considered SD. One patient developed TRAE (SAE) after one dose of nivolumab plus ipilimumab and RECIST response and percent change in tumor size from baseline were not evaluable. ITT patients: Nivo, n = 22; Nivo plus Ipi, n = 20. Resected patients: MPR, n = 13; No MPR, n = 23. Data are presented as median with minima, lower and upper quartiles, and maxima. The ends of the box are the upper and lower quartiles (75th and 25th percentiles), the median is the horizontal line inside the box. The whiskers are the two lines outside the box that extend to the maxima and minima. Two-sided P value is from Wilcoxon rank-sum test. c,d, Examples of radiographic (CT scan) and pathologic (H&E) images of NSCLC pre- and post-nivolumab (c) and pre- and post-nivolumab plus ipilimumab (d). CR, complete response; PR, partial response; MPR, major pathologic response; pCR, pathologic complete response; VT, viable tumor; CT, computed tomography; H&E, hematoxylin and eosin.
Extended Data Fig. 4. Impact of histology, stage, smoking status, responses and postoperative treatment on lung cancer-related RFS after neoadjuvant nivolumab and nivolumab plus ipilimumab.
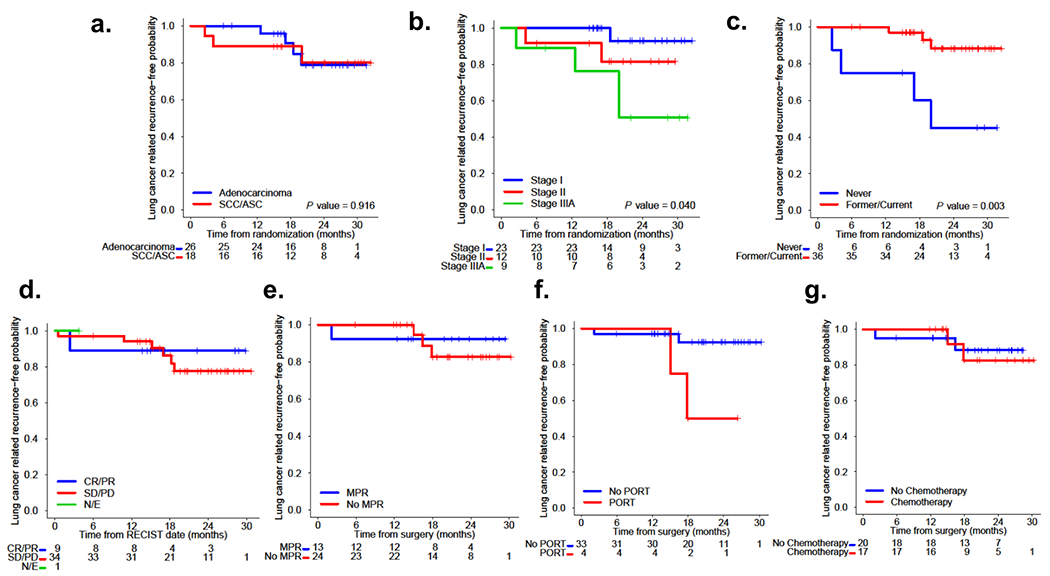
a, Kaplan-Meier curves of probability of lung cancer-related RFS after neoadjuvant nivolumab and nivolumab plus ipilimumab by tumor histology. Among 26 patients with adenocarcinoma (26/44, 59%), four patients (4/26, 15%) progressed, and among 18 patients with SCC/ASC (18/44, 41%), three patients (3/18, 17%) progressed/died. b, Kaplan-Meier curves of probability of lung cancer-related RFS after neoadjuvant nivolumab and nivolumab plus ipilimumab by stage. Among 23 patients with stage I disease (23/44, 52%), one patient (1/23, 4%) progressed, among 12 patients with stage II disease (12/44, 27%), two patients (2/12, 17%) progressed/died, and among nine patients with stage IIIA disease (9/44, 20%), four patients (4/9, 44%) progressed. c, Kaplan-Meier curves of probability of lung cancer-related RFS after neoadjuvant nivolumab and nivolumab plus ipilimumab by smoking status. Among 36 former/current smokers (36/44, 82%), three patients (3/36, 8%) progressed, and among eight never smokers (8/44, 18%), four patients (4/8, 50%) progressed/died. d, Kaplan-Meier curves from landmark analysis performed to explore the effects of radiographic (RECIST) responses to neoadjuvant nivolumab and nivolumab plus ipilimumab on lung cancer-related RFS. Among nine patients with CR/PR (9/44, 20%), one patient (1/9, 11%) died following steroid-treated pneumonitis complicated with BPF and empyema and respiratory failure, and among 34 patients with SD/PD (34/44, 77%), six patients (6/34, 18%) experienced disease recurrence, and, among those, one later died from the disease. One patient was not evaluable due to development of grade 3 TRAE after one dose of nivolumab plus ipilimumab. e, Kaplan-Meier curves from landmark analysis performed to explore the effects of pathologic response (MPR vs. No MPR) to neoadjuvant nivolumab and nivolumab plus ipilimumab on lung cancer-related RFS. Among 13 resected patients with MPR (13/37, 35%), one patient (1/13, 8%) died 2.2 months after surgery, and among 24 resected patients with no MPR (24/37, 65%), three (3/24, 13%) patients progressed 15.0, 16.4, and 17.9 months after surgery. f, Kaplan-Meier curves from landmark analysis performed to explore the effects of PORT on lung cancer-related RFS after neoadjuvant nivolumab and nivolumab plus ipilimumab. Among four resected patients who received PORT (4/37, 11%), two patients (2/4, 50%) progressed, and among 33 resected patients who did not receive PORT (33/37, 89%), two patients (2/33, 6%) progressed/died. g, Kaplan-Meier curves from landmark analysis performed to explore the effects of adjuvant chemotherapy on lung cancer-related RFS after neoadjuvant nivolumab and nivolumab plus ipilimumab. Among 17 resected patients who received adjuvant chemotherapy (17/37, 46%), two patients (2/17, 12%) progressed, and among 20 resected patients who did not receive adjuvant chemotherapy (20/37, 54%), two patients (2/20, 10%) progressed/died. SCC, squamous cell carcinoma; ASC, adenosquamous carcinoma; Never, never smokers; Former/Current, Former smokers/Current smokers; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. MPR, major pathologic response; N/E, not evaluable. PORT, postoperative radiation therapy. Two-sided P value is from logrank test.
Extended Data Fig. 5. Association between tumor PD-L1 expression in malignant cells and response to neoadjuvant nivolumab and nivolumab plus ipilimumab.
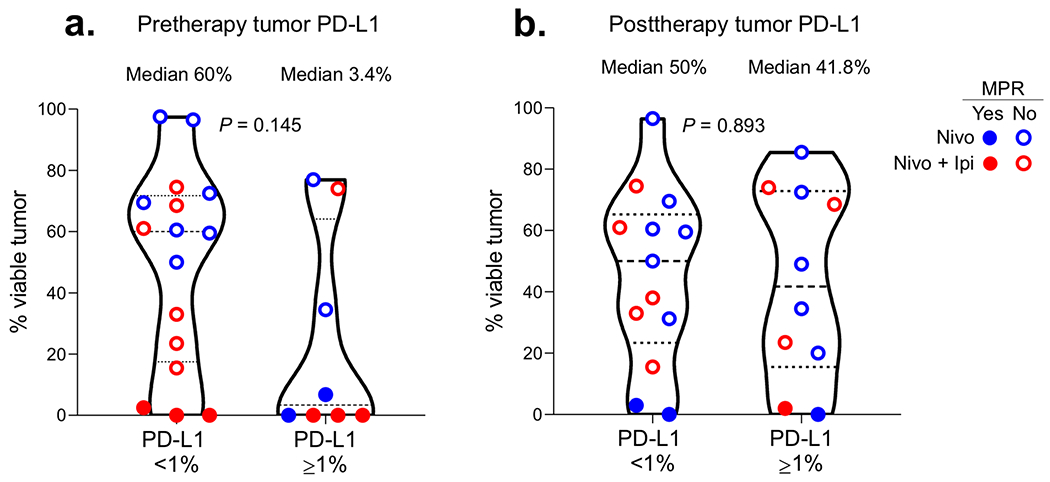
a,b, Percent viable tumor in tumor specimens resected after nivolumab and nivolumab plus ipilimumab according to pretherapy (a) and posttherapy (b) tumor PD-L1 IHC expression (< 1% vs. ≥ 1%) in malignant cells. Pretherapy tumor PD-L1: < 1%, n = 16; ≥ 1%, n = 8. Posttherapy tumor PD-L1: < 1%, n = 13; ≥ 1%, n = 10. Data are presented as median with minima, lower and upper quartiles, and maxima. All violin plots show single data points, dashed line shows the median value, dotted lines show lower quartile and upper quartile values of the range; top and bottom of the violin plots indicate the minima and maxima. Experiments and scorings related to the presented results were conducted once. Two-sided P value is from Wilcoxon rank-sum test.
Extended Data Fig. 6. Multiplex immunofluorescence (mIF) VECTRA staining of immune infiltrates in pre- and posttherapy tumors.
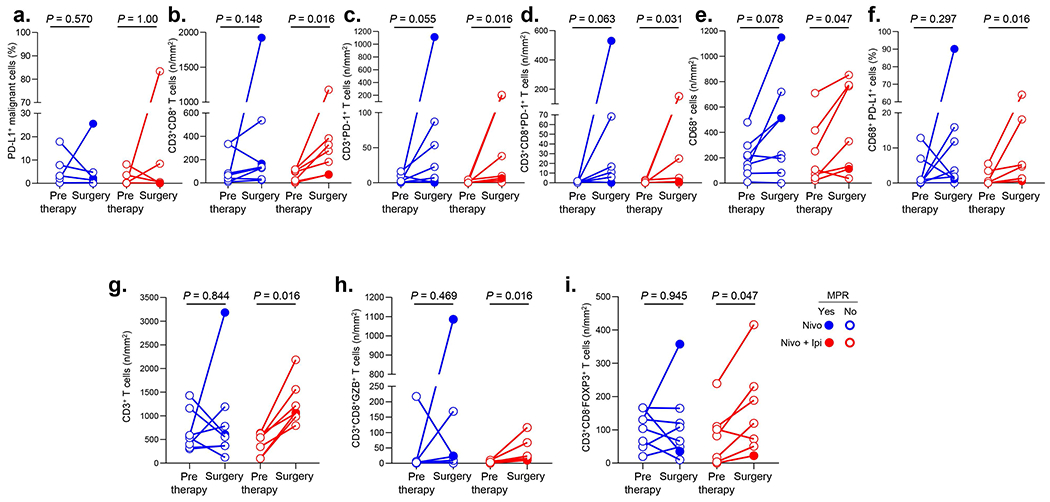
a-f, Staining of cell populations identified with co-expression markers in mIF VECTRA panel 1 as (a) PD-L1+ malignant cells (%), (b) CD3+CD8+ T cells (n/mm2), (c) CD3+PD-1+ T cells (n/mm2), (d) CD3+CD8+PD-1+ T cells (n/mm2), (e) CD68+ cells (n/mm2), (f) CD68+PD-L1+ cells (%) in resected (surgery) vs. pretherapy tumors treated with nivolumab (n = 8) or nivolumab plus ipilimumab (n = 7). g-i, Staining of cell populations identified with co-expression markers in mIF VECTRA panel 2 as (g) CD3+ T cells (n/mm2), (h) CD3+CD8+GZB+ T cells (n/mm2), (i) CD3+CD8−FOXP3+ T cells (n/mm2) in resected (surgery) vs. pretherapy tumors treated with nivolumab (n = 8) or nivolumab plus ipilimumab (n = 7). Experiments and scorings related to the presented results were conducted once. Two-sided P values are from Wilcoxon signed-rank test.
Extended Data Fig. 7. Changes in T cell clones after neoadjuvant treatment and correlation with tumor pathologic regression.
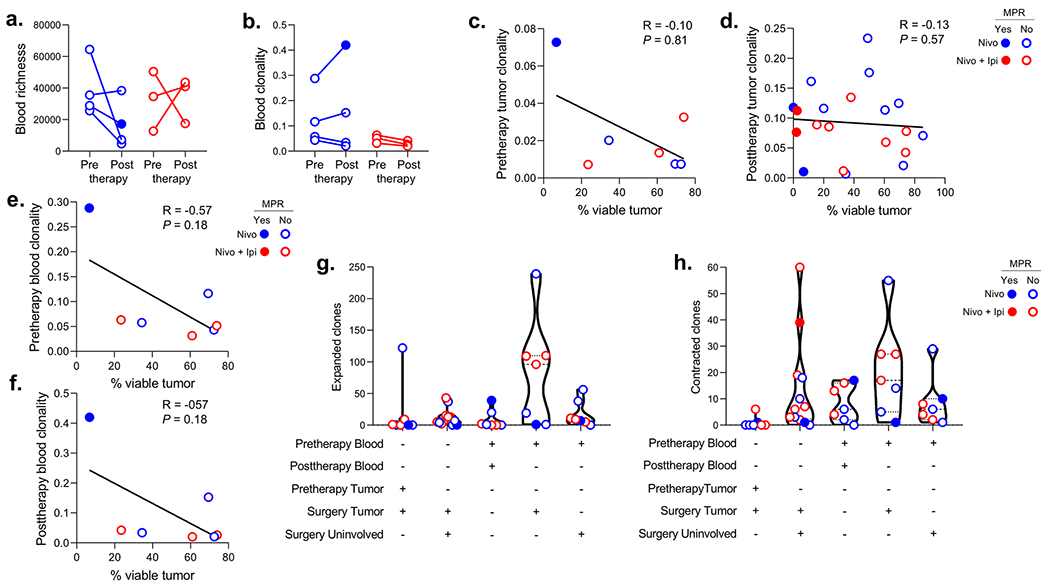
a,b, Changes in TCR repertoire richness (a) and clonality (b) in matched blood samples from pre- to posttherapy (prior to surgery) after neoadjuvant nivolumab (n = 4, blue) or nivolumab plus ipilimumab (n = 3, red). c-f, Correlation between percent viable tumor at surgery and T cell clonality in tumor (c,d) or blood (e,f) pretherapy (c,e) and posttherapy (d,f) with neoadjuvant nivolumab (blue) and nivolumab plus ipilimumab (red). Two-sided P value is from Spearman rank-order correlation. g,h, Number of significantly (two-sided P < 0.01 with Benjamini-Hochberg adjustment for false-discovery rate) expanded (g) and contracted (h) T cell clones in matched resected (surgery tumor) vs. pretherapy tumors (n = 7), matched resected tumors (surgery tumor) vs. tumor-adjacent uninvolved lungs (surgery uninvolved) (n = 12), matched posttherapy (prior to surgery) vs. pretherapy blood samples (n = 7), matched resected tumors (surgery tumor) vs. pretherapy blood samples (n = 7) and tumor-adjacent uninvolved lungs (surgery uninvolved) vs. pretherapy blood samples (n = 7) after neoadjuvant nivolumab (blue) and nivolumab plus ipilimumab (red). Data are presented as median with minima, lower and upper quartiles, and maxima. All violin plots show single data points, dashed line shows the median value, dotted lines show lower quartile and upper quartile values of the range; top and bottom of the violin plots indicate the minima and maxima. Closed dots: MPR; Open dots: No MPR.
Extended Data Fig. 8. Association between fecal microbiome diversity and tumor pathologic responses and TRAEs.
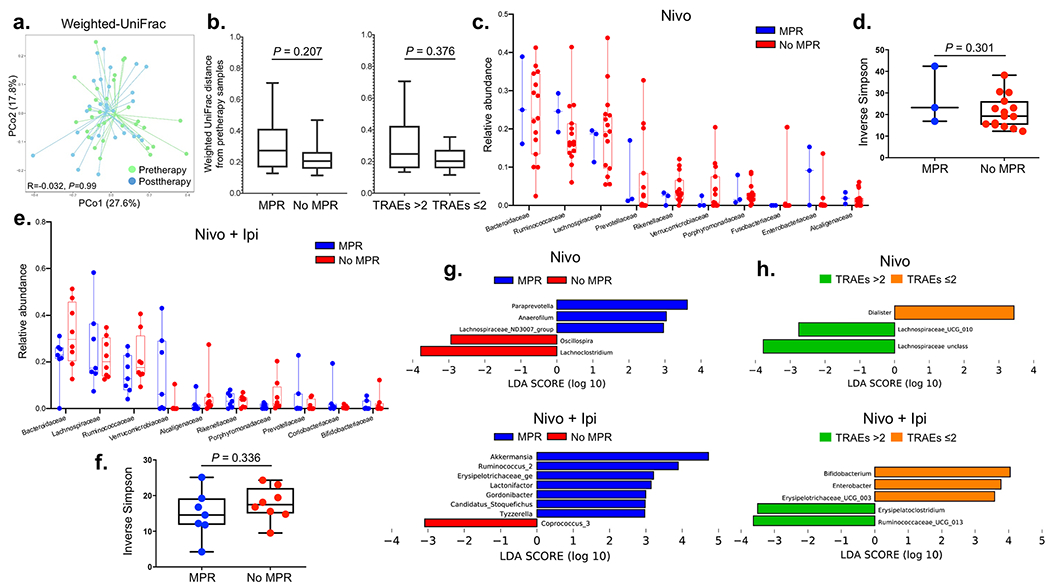
a, Ordination plots from principal coordinate analysis (PCoA) demonstrating clustering patterns of fecal microbiomes of patients at pre- (n = 30) and posttherapy (n = 28) using Weighted UniFrac distance. Two-sided P value is from analyses of similarities (ANOSIM) test performed with 999 permutations to calculate whether taxonomic composition between these two categories were significantly different. b, Box-and-whisker plots of pairwise distances between pre- and posttherapy samples within response and toxicity groups of patients having microbiome data (n = 25, MPR = 9, No MPR = 16; treatment-related adverse events (TRAEs) >2 = 12,TRAEs ≤2 = 13). The box portion of the plot is drawn from the first quartile to the third quartile with inside line indicating the median value. The whiskers extend from the ends of the box to the minimum and maximum data values. Two-sided P value is from Mann-Whitney U rank-sum test. c, Box-and-whisker plots of the relative distributions of the top ten most abundant bacteria at family level observed in MPR (n = 3) and No MPR (n = 15) in nivolumab-treated patients (top panel). The box portion of the plot is drawn from the first quartile to the third quartile with inside line indicating the median value. The whiskers extend from the ends of the box to the minimum and maximum data values. d, Inverse Simpson index estimating fecal bacterial diversity between MPR (n = 3) and No MPR (n = 15) in nivolumab-treated patients (bottom panel). The box portion of the plot is generated from the first quartile to the third quartile with inside line indicating the median value. The whiskers extend from the ends of the box to the minimum and maximum data values. Two-sided P value is from Mann-Whitney U rank-sum test. e, Box-and-whisker plots of the relative distributions of the top ten most abundant bacteria at family level observed in MPR (n = 7) and No MPR (n = 8) in nivolumab plus ipilimumab-treated patients. The box portion of the plot is generated from the first quartile to the third quartile with inside line indicating the median value. The whiskers extend from the ends of the box to the minimum and maximum data values. f, Inverse Simpson index estimating fecal bacterial diversities between MPR (n = 7) and No MPR (n = 8) in nivolumab plus ipilimumab-treated patients. The box portion of the plot is generated from the first quartile to the third quartile with inside line indicating the median value. The whiskers extend from the ends of the box to the minimum and maximum data values. Two-sided P value is from Mann-Whitney U rank-sum test. g, Linear Discriminant Analysis (LDA) Effective Size (LEfSe) used to estimate discriminative features in fecal microbiomes at genus level between MPR (n = 3) and No MPR (n = 15) in nivolumab (top) and nivolumab plus ipilimumab-treated patients (bottom; MPR, n = 7 and No MPR, n = 8) pretherapy. The length of the bar indicates the effect size associated with a genus. Alpha value of 0.05 for the factorial Kruskal-Wallis test and logarithmic LDA score of 2 were used to calculate the discriminative features. h, LDA Effect Size (LEfSe) plot of pairwise comparisons of bacterial taxa at genus level dichotomized by TRAE categories in nivolumab-treated patients (top panel) (TRAEs ≤2 (n = 10) and TRAEs >2 (n = 10)) and in nivolumab plus ipilimumab-treated patients (bottom panel) (TRAEs ≤2 (n = 9) and TRAEs >2 (n = 10)). Alpha value of 0.05 for the factorial Kruskal-Wallis test and logarithmic LDA score of 2 were used to calculate the discriminative features.
Extended Data Fig. 9. Association between fecal microbiome diversity and tumor TCR clonality and richness.
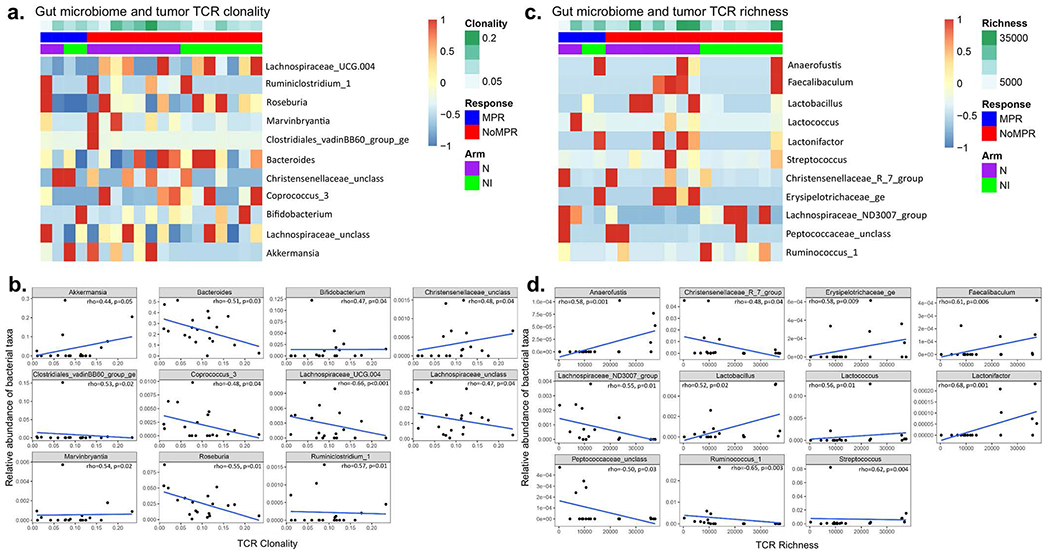
a,c, Heatmaps showing pretherapy taxonomic abundances at various levels in nivolumab (n = 10) and nivolumab plus ipilimumab (n = 9) arms and posttherapy tumor TCR clonality (a) and richness (c). b,d, The relationships between the microbiome and TCR clonality (b) and richness (d) were conducted using the linear regression model. Spearman correlation test (two-sided) was used to calculate the rho and P values. Here, the unadjusted P value cutoff of 0.05 was used.
Extended Data Fig. 10. Flow cytometry gating strategy for CD103 T cell and T cell memory panels.
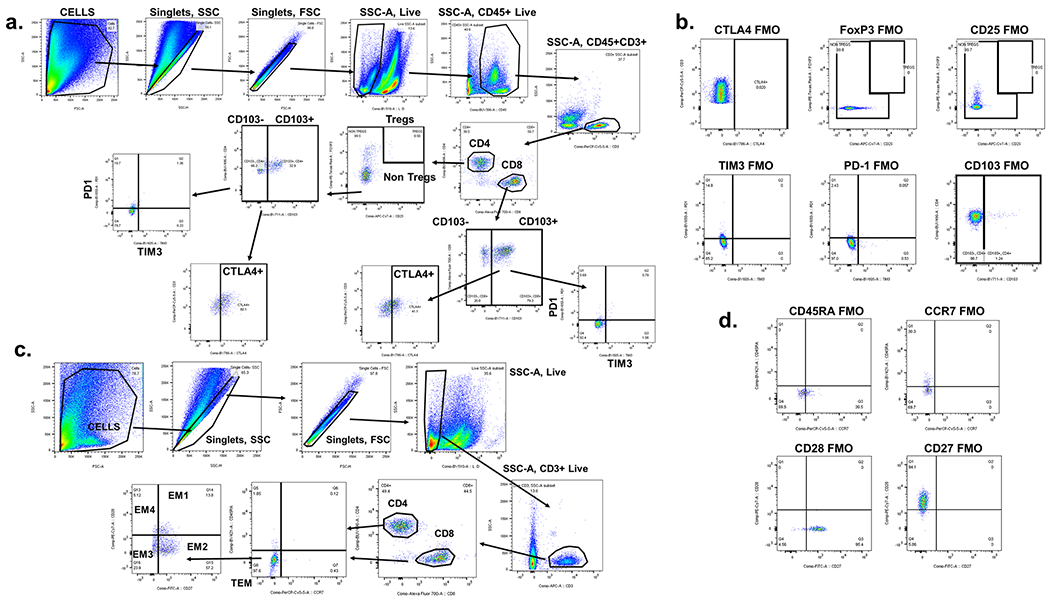
The gating strategy is shown including initial QC gates (SSC singlets, FSC singlets, live cells) followed by immune cell subsets included in the panel. The frequencies referenced for each subgated cell population shown are from the parental gate. a, Subgating is performed on CD4+ Tregs, CD4+ non-Tregs and CD8+ T cell subsets as shown. Subgating of checkpoint receptors was also assessed on the tissue-resident memory (TRM) (CD103+) and non-TRM (CD103−) T cell subsets. Arrows indicate the transition through the individual gates. b, Fluorescence minus one (FMO) gating for CTLA-4, FoxP3, CD25, TIM3, PD-1 and CD103 controls are shown. c, Subgating for T cell memory panel is performed on CD4+ and CD8+ T cell subsets as shown. Arrows indicate the transition through the individual gates. d, Fluorescence minus one (FMO) gating for CD45RA, CCR7, CD28 and CD27 controls are shown. Experiments and gating related to presented results were conducted once. Subgating was only performed when more than 100 events were present in parental gate.
Supplementary Material
Acknowledgements
We thank the patients and their families for participating in this study. We thank all the members of our regulatory, clinical, data coordination and translational research teams in the Departments of Thoracic/Head and Neck Medical Oncology and Thoracic Surgery at MD Anderson Cancer Center for their support on this trial. We thank the members of the strategic alliance teams at Bristol Myers Squibb and MD Anderson Cancer Center (E.B. Roarty and A. Spelman) for their support. We thank the members of the Translational Molecular Pathology Immune-Profiling Laboratory (TMP-IL) B. Sanchez Espiridon, S. Wijeratne, for their assistance with sample procurement and inventory, C-W. B. Chow, W. Lu, L. Kakarala, M. Jiang, A. Tamegnon, and J. Zhou for their technical assistance, and D. Lorenzini for pathology assistance in imaging analysis. We thank L. Little and C. Gumbs from the Department of Genomic Medicine for assistance with TCR sequencing. The authors would like to acknowledge MD Anderson’s Program for Innovative Microbiome and Translational Research (PRIME-TR) for supporting the analysis and interpretation of the microbiome results presented herein (Drs. J.A. Wargo and N.J. Ajami are the program director and executive scientific director for PRIME-TR, respectively). We thank Mr. D. Aten, Sr. Medical Illustrator in Creative Communications at MD Anderson Cancer Center, for his assistance with figure formatting. Funding support for the clinical trial was provided by Bristol Myers Squibb. Support for the study was also partially provided by the National Institutes of Health (NIH)/National Cancer Institute (NCI) through The University of Texas Lung Specialized Program of Research Excellence (SPORE; grant 5P50CA070907 to T.C., L.A.B., F.S., J.M.K., J.A.R., I.I.W., D.L.G. and J.V.H.), the NIH/NCI P30 CA016672 Cancer Center Support Grant, the Conquer Cancer Foundation of the American Society of Clinical Oncology Career Development Award 2018 Project ID 12895 (to T.C.), the Connie Rasor Endowment for Cancer Research (to D.L.G.), the Bruton Endowed Chair in Tumor Biology (to J.V.H.), and the TMP-IL at the Department of Translational Molecular Pathology, the University of Texas MD Anderson Cancer Center. The study was also partially supported by the generous philanthropic contributions to the University of Texas MD Anderson Cancer Center Lung Cancer Moon Shot Program, the University of Texas MD Anderson Cancer Center Physician Scientist Program (from the T.J. Martell Foundation, to T.C). the Khalifa Bin Zayed Al Nahyan Foundation (to T.C.), the Ford Petrin Donation (to J.V.H.), the Rexanna’s Foundation for Fighting Lung Cancer (to T.C., D.L.G., J.V.H. B.S.) and the Bob Mayberry Foundation (to T.C.).
Competing Interests
T.C. reports speaker’s fees from Society for Immunotherapy of Cancer and Bristol Myers Squibb, consulting fees from MedImmune/AstraZeneca and Bristol Myers Squibb, advisory role fees from EMD Serono and Bristol Myers Squibb, and research funding to MD Anderson Cancer Center from Boehringer Ingelheim, MedImmune/AstraZeneca, EMD Serono and Bristol Myers Squibb. W.N.W.Jr reports consulting or advisory role fees from Clovis Oncology and AstraZeneca, speaker’s fees from Boehringer Ingelheim, honoraria from Roche/Genentech, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Merck, Bayer, Pfizer and Eli Lilly, and research funding from OSI Pharmaceuticals, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, and Merck. M.C.B.G. has received research funding from Siemens Healthcare. H.T.T. reports research funding from Bayer-AS, Bristol Myers Squibb, Ziopharm and Guardant Health. N.K. reports consulting or advisory role fees from Merck, Bristol Myers Squibb, Abbvie and Roche. A.S.T. reports advisory board/consultant fees from Bristol Myers Squibb, Eli Lilly, Genentech, Roche, Novartis, Ariad, EMD Serono, Merck, Seattle Genetics, AstraZeneca, Boehringer Ingelheim, Sellas Life Science, Takeda, Epizyme and Huron, and receives research grants from Eli Lilly, Millennium, Polaris, Genentech, Merck, Boehringer-Ingelheim, Bristol Myers Squibb, Ariad, Epizyme, Seattle Genetics, Takeda, and EMD Serono. G.B.Jr receives personal fees and research funding from Amgen, Bayer, Bristol Myers Squibb, Celgene, Daiichi Sankyo, Genentech, MedImmune, Merck, Roche, Xcovery; research funding from Adaptimmune, Exelixis, GlaxoSmithKline, Immatics, Immunocore, Incyte, Kite pharma, Macrogenics, Torque, AstraZeneca, Tmunity, Regeneron, Beigene, Novartis, Repertoire Immune Medicines, and personal fees from Abbvie, Adicet, Amgen, Araid, Clovis Oncology, AstraZeneca, Bristol Myer Squibb, Celgene, Genentech, Gilead, Merck, Novartis, Roche, Virogin Biotech, John & Johnson/Janssen, and Maverick Therapeutics. X.L. receives consultant and advisory fees from Eli Lilly, AstraZeneca and EMD Serono, and research funding from Eli Lilly, Boehringer Ingelheim and Spectrum Pharmaceuticals. J.Z. reports grants from Merck, Johnson and Johnson, and consultant fees, advisory fees or honoraria from Bristol Myers Squibb, AstraZeneca, GenePlus, Innovent, OrigMed and Roche outside the submitted work. M.A. reports research funding to MD Anderson Cancer Center from Genentech, Nektar therapeutics, Merck, GlaxoSmithKline, Novartis, Jounce therapeutics, Bristol Myers Squibb, Eli Lilly, and Adaptimmune, and receives advisory fees from GlaxoSmithKline and Shattuck labs. B.G. reports research funding to MD Anderson Cancer Center from Pfizer Inc., ISA Pharmaceuticals, MedImmune/AstraZeneca and Cue Bio. L.A.B. receives advisory/consultant fees and research funding from AstraZeneca, AbbVie, GenMab, PharmaMar and Sierra Oncology, advisor/consultant fees from Genentech, Bristol Myers Squibb, Alethia, Merck and Pfizer, as well as research funding from ToleroPharmaceuticals. W.L.H. receives research funding from Johnson & Johnson. J.A.R. reports fees as consultant, scientific advisor, ownership interest, inventor on intellectual property licensed by Genprex and PI on Genprex-sponsored research. H.K. receives funding to MD Anderson Cancer Center from Johnson and Johnson. C.H. serves as an advisory board member for Briacell. R.R.J. receives consultant role fees from Merck, Karius and Microbiome DX, advisory member role fees from Seres, and Kaleido and patent licensing fees from Seres. P.S. reports consulting, advisory roles and/or stocks/ownership for Achelois, Apricity Health, BioAlta, Codiak BioSciences, Constellation, Dragonfly Therapeutics, Forty-Seven Inc., Hummingbird, ImaginAb, Jounce Therapeutics, Lava Therapeutics, Lytix Biopharma, Marker Therapeutics, Oncolytics, Infinity Pharma, BioNTech, Adaptive Biotechnologies and Polaris, and owns a patent licensed to Jounce Therapeutics. J.P.A. reports consulting, advisory roles and/or stocks/ownership for Achelois, Apricity Health, BioAtla, Codiak BioSciences, Dragonfly Therapeutics, Forty-Seven Inc., Hummingbird, ImaginAb, Jounce Therapeutics, Lava Therapeutics, Lytix Biopharma, Marker Therapeutics, Polaris, BioNTech and Adaptive Biotechnologies, and owns a patent licensed to Jounce Therapeutics. J.A.W. is an inventor on a US patent application (PCT/US17/53.717) submitted by the University of Texas MD Anderson Cancer Center that covers methods to enhance immune checkpoint blockade responses by modulating the microbiome, reports compensation for speaker’s bureau and honoraria from Imedex, Dava Oncology, Omniprex, Illumina, Gilead, PeerView, Physician Education Resource, MedImmune, Exelixis, and Bristol Myers Squibb, serves as a consultant/advisory board member for Roche/Genentech, Novartis, AstraZeneca, GlaxoSmithKline, Bristol Myers Squibb, Merck, Biothera Pharmaceuticals and Microbiome DX, and receives research support from GlaxoSmithKline, Roche/Genentech, Bristol Myers Squibb and Novartis. I.I.W. reports honoraria from Genentech/Roche, Bayer, Bristol Myers Squibb, AstraZeneca/Medimmune, Pfizer, HTG Molecular, Asuragen, Merck, GlaxoSmithKline, Guardant Health, Platform Health, Daiichi, Merck, Flame, Oncocyte, and MSD, and research support from Genentech, Oncoplex, HTG Molecular, DepArray, Merck, Bristol Myers Squibb, Medimmune, Adaptive, Adaptimmune, EMD Serono, Pfizer, Takeda, Amgen, Karus, Johnson & Johnson, Bayer, Iovance, 4D, Novartis and Akoya. S.G.S. reports speaker, travel and lodging expenses – Egyptian Society of Surgical Oncology / Best of SSO Cairo; West Hawaii Cancer Symposium; review panel participant, travel and lodging expenses - Peter MacCallum Cancer Centre; unpaid advisory board participant – Ethicon. D.L.G. reports honoraria for scientific advisory boards from AstraZeneca, Sanofi, Alethia Biotherapeutics and Janssen, and research support from Janssen, Takeda, Ribon Therapeutics and AstraZeneca. J.V.H. reports fees for advisory committees from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Catalyst, EMD Serono, Foundation Medicine, Hengrui Therapeutics, Genentech, GSK, Guardant Health, Eli Lilly, Merck, Novartis, Pfizer, Roche, Sanofi, Seattle Genetics, Spectrum and Takeda, research support from AstraZeneca, GlaxoSmithKline, Spectrum, and royalties and licensing fees from Spectrum. B.S. reports consulting fees from Bristol Myers Squibb. The remaining authors have no competing interest to report.
Data availability
The data supporting the findings of the present study are available within the paper and its supplementary information files. Taxonomy was assigned using the Silva database (https://www.arb-silva.de/) for 16S rRNA sequences. TCR-sequencing data (supporting the findings in Fig. 5 and Extended Data Figs. 7 and 9) have been deposited and are publicly available at the immuneACCESS platform (DOI: 10.21417/TC2020NM; http://clients.adaptivebiotech.com/pub/cascone-2020-nm). The 16S fecal microbiome sequencing data (supporting the findings in Extended Data Figs. 8 and 9) have been deposited and are publicly available at the National Center for Biotechnology Information Sequence Read Archive (SRA BioProject ID PRJNA665109; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA665109). All other relevant deidentified data related to the present study are available from the corresponding author (T.C.) upon reasonable academic request and will require the researcher to sign a data access agreement with the University of Texas MD Anderson Cancer Center after approval. Source data are provided with this paper.
References
- 1.Martin J, et al. Long-term results of combined-modality therapy in resectable non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20, 1989–1995 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Pignon JP, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 26, 3552–3559 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Group, N.M.-a.C. Preoperative chemotherapy for non-small-cell lung cancer: a systematic review and meta-analysis of individual participant data. Lancet 383, 1561–1571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pataer A, et al. Histopathologic response criteria predict survival of patients with resected lung cancer after neoadjuvant chemotherapy. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 7, 825–832 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hellmann MD, et al. Pathological response after neoadjuvant chemotherapy in resectable non-small-cell lung cancers: proposal for the use of major pathological response as a surrogate endpoint. Lancet Oncol 15, e42–50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaft JE, et al. Phase II trial of neoadjuvant bevacizumab plus chemotherapy and adjuvant bevacizumab in patients with resectable nonsquamous non-small-cell lung cancers. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 8, 1084–1090 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cascone T, et al. Induction Cisplatin Docetaxel Followed by Surgery and Erlotinib in Non-Small Cell Lung Cancer. Ann Thorac Surg 105, 418–424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei SC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 170, 1120–1133 e1117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hellmann MD, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 18, 31–41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellmann MD, et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine 381, 2020–2031 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Sepesi B, et al. Nodal Immune Flare (NIF) Following Neoadjuvant Anti-PD-1 and Anti-CTLA-4 Therapy in Non-Small Cell Lung Cancer. Journal of Thoracic Oncology 14, S745–S745 (2019). [Google Scholar]
- 12.Weissferdt A, et al. Agreement on Major Pathological Response in NSCLC Patients Receiving Neoadjuvant Chemotherapy. Clin Lung Cancer (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cascone T, et al. A Phase I/II Study of Neoadjuvant Cisplatin, Docetaxel and Nintedanib for Resectable Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forde PM, et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. The New England journal of medicine 378, 1976–1986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwiatkowski DJ, et al. Neoadjuvant atezolizumab in resectable non-small cell lung cancer (NSCLC): Interim analysis and biomarker data from a multicenter study (LCMC3). Journal of Clinical Oncology 37, 8503–8503 (2019). [Google Scholar]
- 16.Gao S, et al. Neoadjuvant PD-1 inhibitor (Sintilimab) in NSCLC. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 15, 816–826 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Amaria RN, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nature medicine 24, 1649–1654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djenidi F, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194, 3475–3486 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Duhen T, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nature communications 9, 2724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei SC, et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proceedings of the National Academy of Sciences of the United States of America 116, 22699–22709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu CA, et al. Neoadjuvant atezolizumab and chemotherapy in patients with resectable non-small-cell lung cancer: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol (2020). [DOI] [PubMed] [Google Scholar]
- 22.Reuben A, et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nature communications 11, 603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu TD, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Blank CU, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nature medicine 24, 1655–1661 (2018). [DOI] [PubMed] [Google Scholar]
- 25.William WN Jr., et al. Computed tomography RECIST assessment of histopathologic response and prediction of survival in patients with resectable non-small-cell lung cancer after neoadjuvant chemotherapy. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 8, 222–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Ramalingam SS, et al. Nivolumab + ipilimumab versus platinum-doublet chemotherapy as first-line treatment for advanced non-small cell lung cancer: Three-year update from CheckMate 227 Part 1. Journal of Clinical Oncology 38, 9500–9500 (2020). [Google Scholar]
- 29.Provencio M, et al. Neoadjuvant chemotherapy and nivolumab in resectable non-small-cell lung cancer (NADIM): an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol 21, 1413–1422 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Reck M, et al. Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): CheckMate 9LA. Journal of Clinical Oncology 38, 9501–9501 (2020). [Google Scholar]
Methods-only References
- 31.Simon R Optimal two-stage designs for phase II clinical trials. Control Clin Trials 10, 1–10 (1989). [DOI] [PubMed] [Google Scholar]
- 32.Thall PF, Simon RM & Estey EH New statistical strategy for monitoring safety and efficacy in single-arm clinical trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 14, 296–303 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Eisenhauer EA, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45, 228–247 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Woolson R a.C WR. Statistical Methods for the Analysis of Biomedical Data, (New York, 2002). [Google Scholar]
- 35.Kaplan EL & Meier P Nonparametric-Estimation from Incomplete Observations. J Am Stat Assoc 53, 457–481 (1958). [Google Scholar]
- 36.Mantel N Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 50, 163–170 (1966). [PubMed] [Google Scholar]
- 37.Bentebibel SE, et al. A First-in-Human Study and Biomarker Analysis of NKTR-214, a Novel IL2Rbetagamma-Biased Cytokine, in Patients with Advanced or Metastatic Solid Tumors. Cancer discovery 9, 711–721 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Parra ER, et al. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci Rep 7, 13380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parra ER, Francisco-Cruz A & Wistuba II. State-of-the-Art of Profiling Immune Contexture in the Era of Multiplexed Staining and Digital Analysis to Study Paraffin Tumor Tissues. Cancers (Basel) 11(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parra ER, et al. Procedural Requirements and Recommendations for Multiplex Immunofluorescence Tyramide Signal Amplification Assays to Support Translational Oncology Studies. Cancers (Basel) 12(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parra ER, Villalobos P, Mino B & Rodriguez-Canales J Comparison of Different Antibody Clones for Immunohistochemistry Detection of Programmed Cell Death Ligand 1 (PD-L1) on Non-Small Cell Lung Carcinoma. Appl Immunohistochem Mol Morphol 26, 83–93 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsao MS, et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 13, 1302–1311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reuben A, et al. TCR Repertoire Intratumor Heterogeneity in Localized Lung Adenocarcinomas: an Association with Predicted Neoantigen Heterogeneity and Postsurgical Recurrence. Cancer discovery (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv, 081257 (2016). [Google Scholar]
- 45.Quast C, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of the present study are available within the paper and its supplementary information files. Taxonomy was assigned using the Silva database (https://www.arb-silva.de/) for 16S rRNA sequences. TCR-sequencing data (supporting the findings in Fig. 5 and Extended Data Figs. 7 and 9) have been deposited and are publicly available at the immuneACCESS platform (DOI: 10.21417/TC2020NM; http://clients.adaptivebiotech.com/pub/cascone-2020-nm). The 16S fecal microbiome sequencing data (supporting the findings in Extended Data Figs. 8 and 9) have been deposited and are publicly available at the National Center for Biotechnology Information Sequence Read Archive (SRA BioProject ID PRJNA665109; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA665109). All other relevant deidentified data related to the present study are available from the corresponding author (T.C.) upon reasonable academic request and will require the researcher to sign a data access agreement with the University of Texas MD Anderson Cancer Center after approval. Source data are provided with this paper.