

Abstract
Novel 5-pyridinyl-1,2,4-triazoles were designed as dual inhibitors of histone deacetylase 2 (HDAC2) and focal adhesion kinase (FAK). Compounds 5d, 6a, 7c, and 11c were determined as potential inhibitors of both HDAC2 (IC50 = 0.09–1.40 μM) and FAK (IC50 = 12.59–36.11 nM); 6a revealed the highest activity with IC50 values of 0.09 μM and 12.59 nM for HDAC2 and FAK, respectively. Compound 6a was superior to reference drugs vorinostat and valproic acid in its ability to inhibit growth/proliferation of A-498 and Caki-1 renal cancer cells. Further investigation proved that 6a strongly arrests the cell cycle at the G2/M phase and triggers apoptosis in both A-498 and Caki-1 cells. Moreover, the enhanced Akt activity that is observed upon chronic application of HDAC inhibitors was effectively suppressed by the dual HDAC2/FAK inhibitor. Finally, the high potency and selectivity of 6a towards HDAC2 and FAK proteins were rationalized by molecular docking. Taken together, these findings highlight the potential of 6a as a promising dual-acting HDAC2/FAK inhibitor that could benefit from further optimization.
Keywords: HDAC2; FAK; 1,2,4-triazoles; Anticancer; Molecular docking
1. Introduction
The approach “one agent multiple targets” or “multi-target agents” gained significant support in the field of drug discovery, and has come to be known as polypharmacology [1]. It is generally agreed upon that combination therapy has a considerable therapeutic relevance; however, such an approach is not without significant disadvantages. For example, patients taking combination therapy suffer from unpredictable pharmacokinetic profile, drug-drug interactions, and physical incompatibilities, leading to unpredictable and additive side effects and adverse reactions, unforeseen drug inactivation or precipitation of the formulation [2]. On the contrary, a single compound acting on dual or multiple targets can concurrently modulate various tumor pathways to get desirable therapeutic efficiency without such additive side effects [3]. Consequently, designing and developing multi-target drugs implemented an adequate strategy for cancer therapeutics.
Histone deacetylases (HDACs) are important therapeutic targets because their inhibition can reverse the aberrant epigenetic states linked to cancer. The several FDA approved inhibitors of HDAC for cancer treatment are vorinostat (SAHA), romidepsin, belinostat, panobinostat, and chidamide (Figure 1) [4]. The HDAC binding pocket consists of a hydrophobic cap, linker, and zinc binding group (ZBG). Fortunately, each part of HDAC pharmacophore can be modified to allow for the hybridization approach. The cap group can be modified with a variety of hydrophobic moieties, the linker could be aliphatic or aromatic, and the ZBG could be a carboxylic acid, hydroxamic acid, 2-aminobenzamide, or thiol [5]. Among HDAC inhibitions, those that are selective for the HDAC2 isoform are particularly important because HDAC2 is overexpressed in several cancers such as prostate, gastric, non-small lung, colon, hepatocellular, and renal [5–11]. In fact, selective inhibition of HDAC2 isoform inhibits tumor growth, promotes apoptosis, and results in G2/M cell cycle arrest and G1 delay [12, 13].
Figure 1:

Chemical structures of the FDA approved HDAC inhibitors
Despite their demonstrated anti-tumor efficacy, most of the studied HDAC inhibitors are associated with the emergence of resistance upon prolonged administration, such as Akt kinase activation and STAT3 phosphorylation [14–19]. Several researchers embarked upon developing dual or multi-target HDAC inhibitors as a promising research area. Currently, there are several dual/multi acting HDAC inhibitors in clinical trials such as CUDC-907 (PI3K and HDAC), 4SC-202 (LSD1 and HDAC), tinostamustine (alkylating agent and HDAC), and CUDC-101 (EGFR, HER2, HDAC), each combining a diverse array of targets with HDACs [4, 20, 21].
Focal adhesion kinase (FAK) is an essential cell signaling regulator inside the microenvironment of the tumor. It plays an indispensable role in regulating tumor cell survival, migration, metastasis, proliferation, and angiogenesis. FAK is recognized as a promising therapeutic target as it is overexpressed in several tumors [22, 23]. The human FAK gene is comprised of three domains: an amino-terminal FERM domain, central protein kinase, and a carboxy-terminal focal adhesion targeting (FAT) domain. Of these domains, the maximum effort has gone into targeting the kinase domain. The main pharmacophore of the FAK inhibitors is comprised of diphenyl five/six-membered nitrogenous heterocycles. The six-membered are the early discovered inhibitors and got the most attention, whereas the five-membered are growing as quite promising FAK kinase inhibitors. Compounds PF-573228 and TAE226 are potent FAK kinase inhibitors; however, they did not enter clinical trials because the former did not show remarkable antiproliferative activity, and the latter affected the glucose blood levels and glucose metabolism (Figure 2) [24]. Four FAK inhibitors completed phase І clinical trials CEP-37440, PF-562271, PND-1186, and GSK-2256098, while defactinib (VS-6063) completed phase ІІ clinical trials [25]. Meanwhile, the five-membered FAK kinase inhibitors revealed a substantial ability to inhibit FAK and tumor cells (Figure 3) [26]. Several discovered five/six-membered FAK kinase inhibitors revealed dual/multi-kinase inhibitory activity due to the various kinases’ structural similarities. However, till now, there are no reported dual acting FAK inhibitors that target another non-kinase protein.
Figure 2:
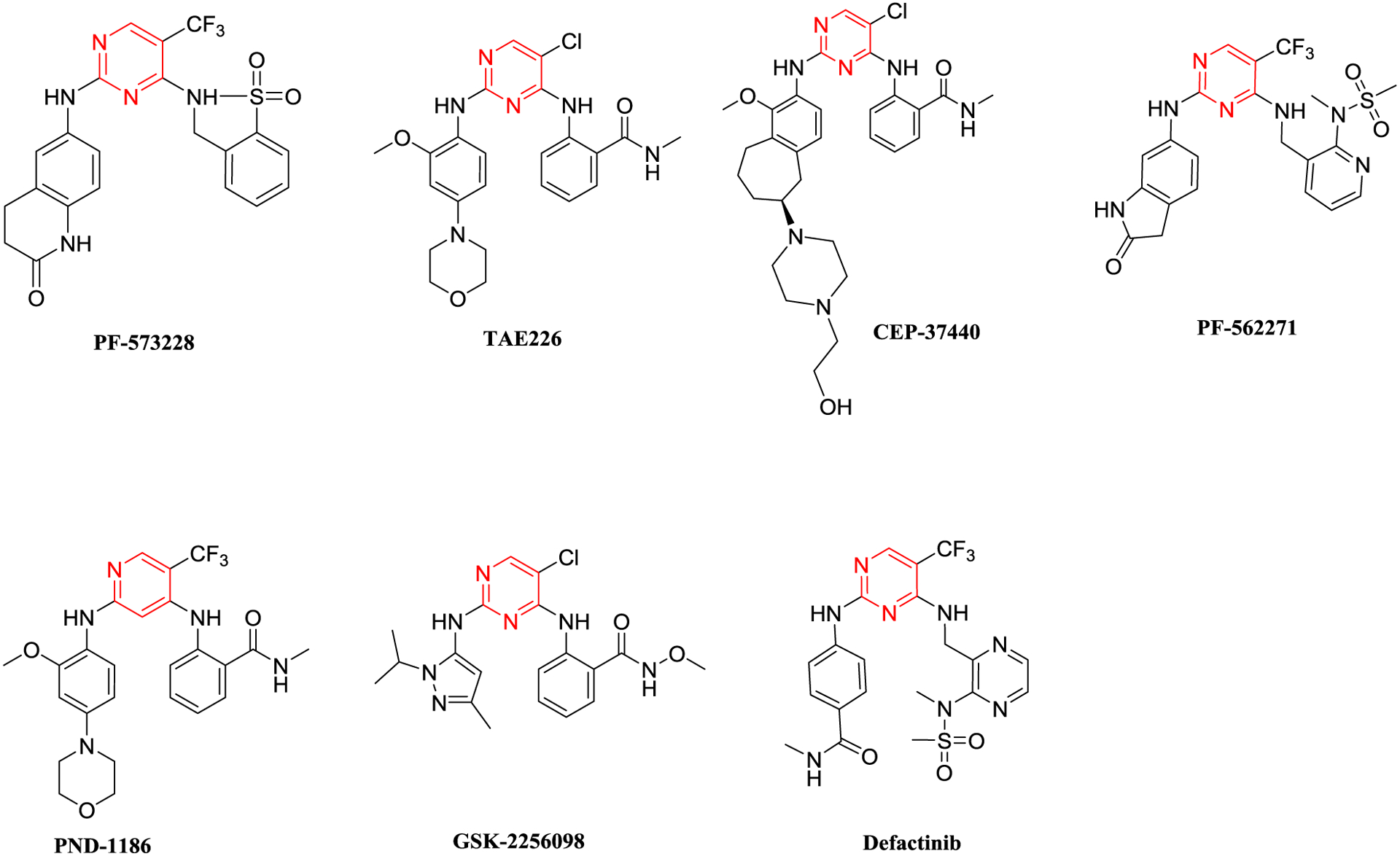
Chemical structure of the six-membered FAK kinase inhibitors
Figure 3:
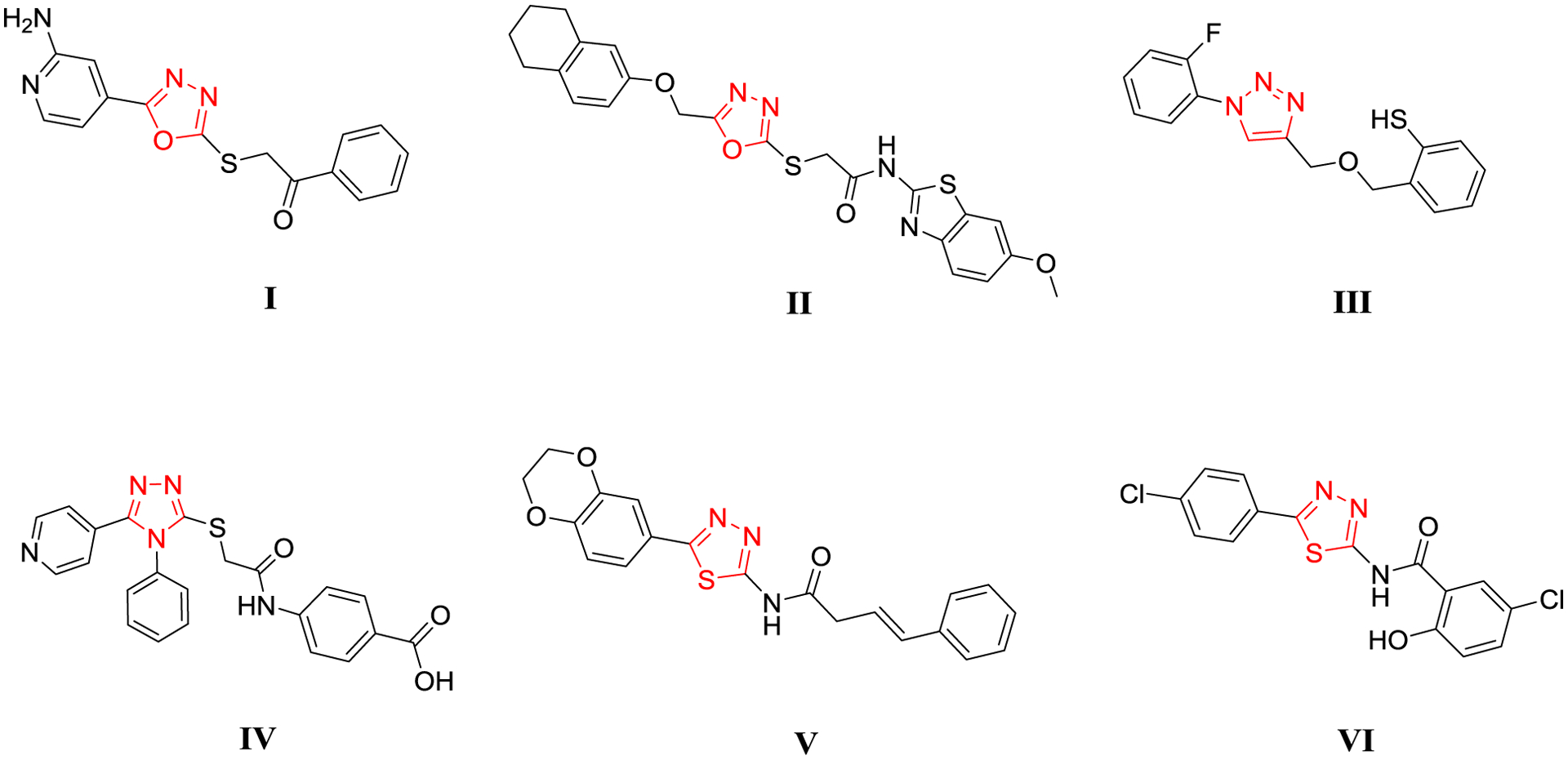
Chemical structure of the five-membered FAK kinase inhibitors
1,2,4-triazole and pyridine rings constitute useful heterocyclic moieties in developing several anticancer agents [27, 28]. Besides their known antiproliferative activity, 1,2,4-triazole and pyridine bearing compounds showed potent FAK and HDAC inhibitory activities. Moreover, pyridine moiety was widely used as a surface recognition group in developing potent HDAC and FAK inhibitors as well [5, 26, 29–33].
The administration of HDAC inhibitors is associated with Akt kinase activation, leading to pro-survival cell elicitation effects, whereas FAK kinase inhibition leads to Akt suppression [14, 15, 34]. All the reported dual/multi-acting FAK inhibitors targeted FAK and another different kinase; these multi-kinase targeting compounds are prone to have side effects. Because of this, the best is to merge non-kinase targets such as histone deacetylase with FAK kinase; unlike the non-selective multi-kinase inhibitors, derivatives that inhibit non-kinase proteins are expected to show high levels of specificity.
Given this underwhelming performance of HDAC and FAK inhibitors created to date, we set out to design and synthesis of 20 novel dual-acting HDAC2/FAK inhibitors that leverage the promising properties of 1,2,4-triazole and pyridine heterocyclic rings. The design of the dual inhibitors involved using 5-pyridinyl-1,2,4-triazole moiety as the surface recognition group and five carbon atom linker (aliphatic or aromatic) was implemented to the designed structures, while the ZBG was carboxylic acid, hydroxamic acid, or 2-aminobenzamide (Figure 4). Moreover, the variation of substituents (methyl, ethyl, allyl, or phenyl) was made on N4 of the 1,2,4-triazole ring Table 1.
Figure 4:
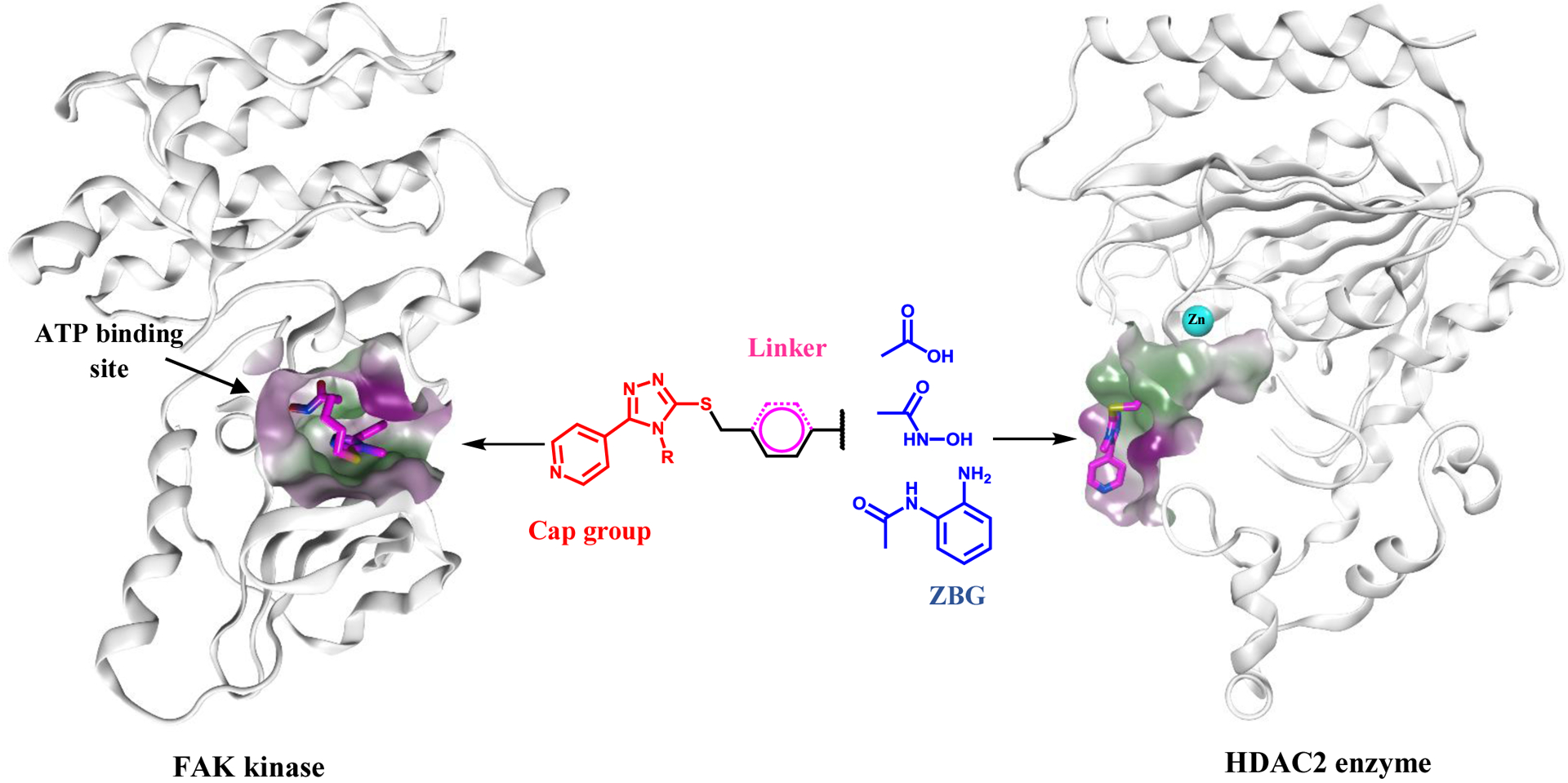
Design of the dual acting 5-pyridinyl-1,2,4-triazole derivatives
Table 1:
Growth inhibition (%) of 5-pyridinyl-1,2,4-triazole derivatives 5a-d, 6a-d, 7a-d, 10a-d, and 11a-d
| Cpd | R | HCT116 | HT-29 | K562 | KG-1 | A-498 | Caki-1 |
|---|---|---|---|---|---|---|---|
| 5a | Methyl | 20.12 | 34.19 | 18.25 | 55.94 | 45.28 | 34.69 |
| 5b | Ethyl | 2.15 | 9.34 | 1.58 | 7.34 | 4.13 | 1.06 |
| 5c | Allyl | 16.25 | 21.25 | 2.95 | 15.36 | 11.36 | 3.25 |
| 5d | Phenyl | 78.51 | 80.48 | 72.73 | 67.19 | 89.34 | 94.8 |
| 6a | Methyl | 60.14 | 68.34 | 40.12 | 84.17 | 99.81 | 98.14 |
| 6b | Ethyl | 21.35 | 9.15 | 4.09 | 17.28 | 5.26 | 2.06 |
| 6c | Allyl | 2.07 | 41.28 | 16.12 | 5.91 | 11.06 | 8.62 |
| 6d | Phenyl | 3.56 | 1.09 | 8.34 | 11.25 | 4.36 | 7.68 |
| 7a | Methyl | 54.36 | 71.36 | 44.94 | 63.15 | 45.34 | 50.82 |
| 7b | Ethyl | 6.35 | 18.64 | 10.36 | 3.64 | 22.34 | 8.91 |
| 7c | Allyl | 90.49 | 95.48 | 86.37 | 94.18 | 97.67 | 95.38 |
| 7d | Phenyl | 24.51 | 9.64 | 15.14 | 34.91 | 4.67 | 1.65 |
| 10a | Methyl | 32.36 | 54 | 16 | 72.08 | 31.84 | 48.19 |
| 10b | Ethyl | 1.03 | 8.34 | 14.29 | 7.12 | 1.67 | 9.34 |
| 10c | Allyl | 44.13 | 25.34 | 36.08 | 61.95 | 44.85 | 50.21 |
| 10d | Phenyl | 29.34 | 42.67 | 39.13 | 50.94 | 56.91 | 61.85 |
| 11a | Methyl | 18.03 | 47.91 | 54.09 | 24.11 | 55.61 | 75.16 |
| 11b | Ethyl | 1.85 | 1.22 | 8.67 | 5.91 | 1.18 | 12.09 |
| 11c | Allyl | 80.46 | 70.67 | 90.14 | 87.46 | 97.08 | 94.81 |
| 11d | Phenyl | 45.36 | 37.92 | 19.6 | 47.34 | 72.54 | 66.94 |
2. Results and Discussion
2.1. Chemistry
The synthesis of the 5-pyridinyl-1,2,4-triazole derivatives 5a-d, 6a-d, 7a-d, 10a-d, and 11a-d (Table 1) is outlined in Scheme 1. Isonicotinic acid hydrazide 1 was treated with the appropriate isothiocyanate derivative to give thiosemicarbazide 2. Cyclization of 2 was accomplished through the addition of 2 N NaOH followed by acidification with conc. HCl to afford the corresponding 1,2,4-triazole-3-thiol derivatives 3a-d [35]. Alkylation of 3a-d with ethyl-6-bromohexanoate or ethyl-4-bromomethyl benzoate in the presence of triethylamine afforded the ester intermediates 4a-d and 8a-d, respectively. Hydrolysis of the ester derivatives using LiOH afforded the corresponding carboxylic acid derivatives 5a-d and 9a-d, respectively. The acid intermediates 5a-d and 9a-d were treated with N,N’-carbonyldiimidazole (CDI) in acetonitrile at room temperature. The resultant imidazolide intermediate was either treated with hydroxylamine hydroxide to yield the target hydroxamic acid derivatives 6a-d and 10a-d or with 2-phenylenediamine in the presence of trifluoroacetic acid to afford the corresponding 2-aminobenzamide derivatives 7a-d and 11a-d, respectively [30, 36]. Chemical structures of the synthesized 5-pyridinyl-1,2,4-triazole derivatives were confirmed by 1HNMR, 13CNMR, and mass spectroscopy and their purity were checked elemental analysis.
Scheme 1:
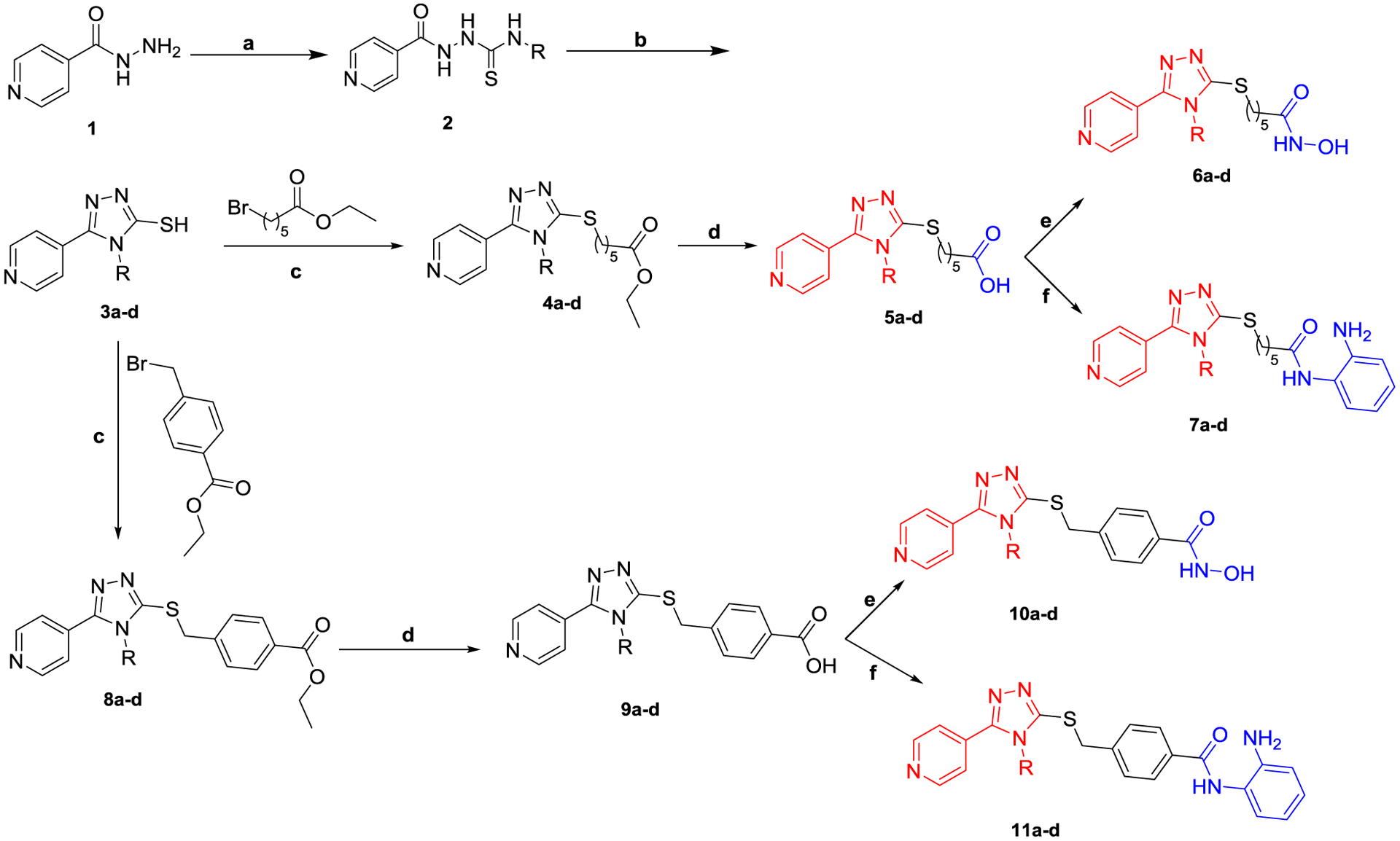
Synthetic approach of the target HDAC2/FAK inhibitors
(a) RNCS (R = methyl, ethyl, allyl, or phenyl), EtOH, 70 °C, 4h; (b) (i) 2N NaOH, (ii) HCl, 3h; (c) TEA, CH3CN, 70 °C, 8h; (d) (i) LiOH, EtOH, (ii) HCl; (e) CDI, DMF, NH2OH.HCl, rt, 10h; (f) CDI, DMF, 2-phenylenediamine, TFA, rt, 10h.
2.2. Biological investigation
2.2.1. In vitro antiproliferative activity
The synthesized compounds were evaluated for their anticancer activity as a single dose (10 μM) against six different cell lines derived from three tumor subpanels, including colon (HCT116 and HT-29), leukemia (K562, and KG-1), and renal (A-498 and Caki-1) cell lines, in which both HDAC2 and FAK are overexpressed, using the MTT assay (Table 1) [37–39]. Most compounds showed promising anticancer activity with growth inhibition percentages. Among them, compounds 5d, 6a, 7c, and 11c were the most potent against all the tested cells (GI% = 40.12–99.81%). The renal cancer cell lines A-498 and Caki-1 were the most sensitive toward 5d, 6a, 7c, and 11c (GI% = 89.34–99.81%). Therefore, we tested the effect of different concentrations (1, 5, 10, 25, 50, 100, 1000, 2000, and 4000 μM) of the synthesized compounds on renal cancer cells (A-498 and Caki-1), using DMSO as a negative control, whereas VPA, SAHA, and TAE226 were used as positive controls. The half maximal inhibitory concentration (IC50) was calculated for each compound (Table 2). Compounds 5d, 6a, 7c, and 11c displayed significant potency (IC50 = 0.95–2.35 μM) against the tested renal cancer cell lines. Interestingly, 6a showed a substantial ability to inhibit renal cancer growth; its IC50 values were comparable to TAE226 and superior to VPA and SAHA. On the other hand, compounds 7a, 5d, 10a, 10c, 10d, 11a, and 11d showed moderate potency (IC50 = 6.19–20.85 μM), whereas the derivatives 5b, 5c, 10b, 6b, 6c, 6d, 7d, and 11b showed low potency (IC50 = 76.36->100 μM). These results showed that the synthesized compounds possess promising anticancer activity, especially against renal cancer, among them 5d, 6a, 7c, and 11c were the most potent.
Table 2:
Concentration producing 50% growth inhibition (IC50, μM±SEM) of the 5-pyridinyl-1,2,4-triazole derivatives on A-498 and Caki-1 renal cancer cells
| Cpd | R | A-498 | Caki-1 | Cpd | R | A-498 | Caki-1 |
|---|---|---|---|---|---|---|---|
| 5a | Methyl | 12.3±1.2 | 16.54±2.49 | 10a | Methyl | 20.85±1.19 | 11.04±2.15 |
| 5b | Ethyl | 125.02±5.23 | 183.34±5.28 | 10b | Ethyl | 203.12±8.37 | 322.07±7.61 |
| 5c | Allyl | 85.34±3.97 | 164.3±3.16 | 10c | Allyl | 13.25±2.90 | 9.56±2.03 |
| 5d | Phenyl | 2.35±0.36 | 1.89±0.54 | 10d | Phenyl | 8.76±1.25 | 7.13±1.53 |
| 6a | Methyl | 0.95±0.17 | 1.23±0.35 | 11a | Methyl | 9.34±1.04 | 6.97±1.58 |
| 6b | Ethyl | 149.35±6.49 | 112.03±2.05 | 11b | Ethyl | 124.36±3.46 | 80.67±4.93 |
| 6c | Allyl | 76.36±2.37 | 89.36±2.56 | 11c | Allyl | 1.42±0.34 | 1.60±0.51 |
| 6d | Phenyl | 174.19±5.18 | 92.36±3.15 | 11d | Phenyl | 6.19±1.05 | 8.35±1.73 |
| 7a | Methyl | 13.40±2.31 | 9.87±2.49 | VPA | - | 2141.25±40.36 | 2825.37±22.46 |
| 7b | Ethyl | 77.63±3.08 | 98.3±5.30 | SAHA | - | 25.08±3.70 | 32.46±2.07 |
| 7c | Allyl | 1.19±0.28 | 1.34±0.34 | TAE226 | - | 0.81±0.14 | 1.05±0.34 |
| 7d | Phenyl | 341.85±2.79 | 214.97±6.01 |
The structure-activity relationship revealed that the N4 methyl derivatives showed promising anticancer activity with the carboxylic acid, the hydroxamic acid, and the 2-aminobenzamide ZBGs regardless of the type of the linker (aliphatic or aromatic). Except for the aliphatic derivatives bearing hydroxamic acid or the 2-aminobenzamide motifs, the N4 phenyl substituted derivatives showed considerable antiproliferative activity, while compounds containing N4 ethyl did not reveal notable activity. Moreover, the presence of 2-aminobenzamide ZBG is essential for the N4 allyl substituted derivatives to display potent antiproliferative activity.
2.2.2. In vitro HDAC inhibition assay
The HDAC (1, 2, 3, 6, and 8) inhibitory activities of the synthesized 5-pyridinyl-1,2,4-triazole derivatives were evaluated by fluorogenic enzymatic assays compared to valproic acid (VPA), TSA, and SAHA as positive controls. The results are summarized in Table 3. The dual-acting compounds showed strong to weak inhibitory activities against HDAC (1, 2, 3, 6, and 8). The IC50 values revealed that the synthesized compounds were more potent toward HDAC2 (IC50 range: 0.09–40.07 μM) than HDAC1 (IC50 range: 8.93->300 μM), HDAC3 (IC50 range: 3.41->300 μM), HDAC6 (IC50 range: 6.93->300 μM), and HDAC8 (IC50 range: 5.32->300 μM). Compounds 5d, 6a, 7c, and 11c were the most potent derivatives in this study toward HDAC2 with IC50 values ranging from 0.09 to 1.4 μM. The other compounds showed moderate to low activities. Moreover, compound 6a exhibited comparable HDAC2 inhibitory activity (IC50 = 0.090 μM), compared to the reference HDAC inhibitors TSA (IC50 = 0.035 μM) and SAHA (IC50 = 0.096 μM), and more potent than VPA (IC50 = 102.74 μM). Compound 6a was found to be more selective toward HDAC2 than HDAC1 (~137 fold), HDAC3 (~38 fold), HDAC6 (~154 fold), and HDAC8 (~79 fold). These data revealed that using the 5-pyridinyl-1,2,4-triazole scaffold, the five aliphatic/aromatic carbon linker, and various ZBGs improved HDAC2 selectivity and retained the antiproliferative activity. The hydroxamic acid derivative 6a, bearing N4 methyl moiety with the aliphatic chain, showed the most potent HDAC2 selectivity.
Table 3:
HDAC inhibitory activities of 5-pyridinyl-1,2,4-triazole derivatives (IC50, μM)
| Cpd | R | HDAC1 | HDAC2 | HDAC3 | HDAC6 | HDAC8 | HDAC1/ HDAC2 | HDAC3/ HDAC2 | HDAC6/ HDAC2 | HDAC8/ HDAC2 |
|---|---|---|---|---|---|---|---|---|---|---|
| 5a | Methyl | 8.93±1.25 | 7.36±0.36 | 84.60±4.32 | 78.39±2.15 | 11.95±2.34 | 1.21 | 11.49 | 10.65 | 1.62 |
| 5b | Ethyl | 184.36±8.36 | 26.39±2.78 | 156.40±5.94 | >300 | >300 | 6.99 | 5.93 | >11.37 | >11.37 |
| 5c | Allyl | 98.19±4.37 | 10.12±2.10 | 245.79±4.36 | >300 | >300 | 9.70 | 24.29 | >29.64 | >29.64 |
| 5d | Phenyl | 8.95±2.04 | 0.64±0.15 | 21.10±3.17 | 10.89±3.09 | 11.05±4.37 | 13.98 | 32.97 | 17.02 | 17.27 |
| 6a | Methyl | 12.31±1.64 | 0.09±0.02 | 3.41±0.36 | 13.87±1.54 | 7.10±0.86 | 136.78 | 37.89 | 154.11 | 78.89 |
| 6b | Ethyl | 76.34±2.15 | 28.93±6.35 | >300 | 269.72±11.23 | >300 | 2.64 | 10.37 | >9.32 | 10.37 |
| 6c | Allyl | >300 | 11.72±2.30 | 222.41±9.37 | 287.96±2.81 | 11.06±0.37 | >25.60 | 18.98 | 24.57 | 0.94 |
| 6d | Phenyl | >300 | 32.36±2.64 | >300 | 246.13±5.19 | >300 | >9.27 | >9.27 | 7.61 | >9.27 |
| 7a | Methyl | 40.18±5.02 | 14.36±0.82 | 84.13±1.39 | 14.20±2.70 | 7.28±0.49 | 2.80 | 5.86 | 0.99 | 0.51 |
| 7b | Ethyl | >300 | 27.13±1.72 | >300 | >300 | 198.07±5.32 | >11.06 | >11.06 | >11.06 | 7.30 |
| 7c | Allyl | 31.05±3.40 | 1.40±0.33 | 10.98±1.03 | 9.79±2.16 | 5.86±0.81 | 22.18 | 7.84 | 6.99 | 4.19 |
| 7d | Phenyl | 256.36±11.34 | 38.73±3.71 | >300 | 159.46±2.38 | >300 | 6.62 | >7.75 | 4.12 | >7.75 |
| 10a | Methyl | 14.2±1.22 | 4.35±0.76 | 56.39±4.39 | 14.36±2.32 | 29.76±1.87 | 3.26 | 12.96 | 3.30 | 6.84 |
| 10b | Ethyl | >300 | 30.39±2.49 | 174.13±8.69 | >300 | >300 | >9.87 | 5.73 | >9.87 | >9.87 |
| 10c | Allyl | 19.60±2.72 | 11.19±2.18 | 20.70±0.79 | 9.86±1.28 | 28.37±6.87 | 1.75 | 1.85 | 0.88 | 2.54 |
| 10d | Phenyl | 15.03±2.58 | 9.76±3.90 | 17.96±1.72 | 6.93±0.57 | 12.76±2.51 | 1.54 | 1.84 | 0.71 | 1.31 |
| 11a | Methyl | 7.64±2.79 | 12.97±0.63 | 71.08±2.79 | 11.03±3.76 | 7.85±3.19 | 0.59 | 5.48 | 0.85 | 0.61 |
| 11b | Ethyl | >300 | 40.07±5.37 | >300 | >300 | >300 | >7.49 | 7.49 | >7.49 | >7.49 |
| 11c | Allyl | 11.84±1.63 | 1.28±0.46 | 42.09±4.72 | 9.96±1.29 | 22.49±3.10 | 9.25 | 32.88 | 7.78 | 17.57 |
| 11d | Phenyl | 22.13±3.47 | 8.39±1.08 | 112.75±8.91 | 9.63±1.48 | 5.32±0.79 | 2.64 | 13.44 | 1.15 | 0.63 |
| VPA | - | 84.21±4.18 | 102.74±3.94 | 134.67±2.87 | 215.01±8.56 | 184.30±5.16 | 0.81 | 1.31 | 2.09 | 1.79 |
| TSA | - | 0.041±0.012 | 0.035±0.010 | 0.46±0.18 | 0.25±0.094 | 0.98 | 1.17 | 13.14 | 7.14 | 28.00 |
| SAHA | - | 0.062±0.019 | 0.096±0.015 | 0.03±0.012 | 0.015±0.008 | 0.42±0.10 | 0.65 | 0.31 | 0.16 | 4.38 |
2.2.3. In vitro activity against FAK kinase
As our rationale is to target HDAC2/FAK proteins, we evaluated the synthesized 5-pyridinyl-1,2,4-triazole derivatives for their activity against the FAK enzyme using Z’-LYTE® technology, which is based on FRET (Invitrogen/Life Technologies). TAE226 was used as a positive control. The results in Table 4 showed that all the synthesized compounds have FAK inhibitory activity in the nanomolar range (IC50 range 12.59–425.31 nM). Interestingly, with agree with the HDAC2 inhibition results, 5d, 6a, 7c, and 11c were found to be the most potent anti-FAK derivatives with IC50 values ranging from 12.6 to 36.11 nM. Compound 6a was also the most potent derivative in this study against FAK kinase (IC50 = 12.59 nM), and its activity was found to be comparable to the positive anti-FAK reference, TAE226 (IC50 = 5.20 nM). Besides, the prepared compounds showed a superior selectivity on FAK when evaluated against five different tyrosine kinases (Table S1). These results revealed that the synthesized compounds have potent anti-FAK activity; among them, 6a is the most potent.
Table 4:
The FAK enzymatic activities of the 5-pyridinyl-1,2,4-triazole derivatives (IC50, nM)
| Cpd | R | IC50 (nM) | Cpd | R | IC50 (nM) |
|---|---|---|---|---|---|
| 5a | Methyl | 50.30±2.14 | 7d | Phenyl | 312.11±3.05 |
| 5b | Ethyl | 76.21±5.36 | 10a | Methyl | 64.35±3.75 |
| 5c | Allyl | 124.30±8.71 | 10b | Ethyl | 254.36±5.28 |
| 5d | Phenyl | 36.11±1.09 | 10c | Allyl | 46.12±2.01 |
| 6a | Methyl | 12.59±1.41 | 10d | Phenyl | 22.40±1.39 |
| 6b | Ethyl | 124.03±5.79 | 11a | Methyl | 53.12±2.91 |
| 6c | Allyl | 189.36±6.42 | 11b | Ethyl | 112.15±5.30 |
| 6d | Phenyl | 425.31±9.54 | 11c | Allyl | 15.60±3.10 |
| 7a | Methyl | 57.08±3.04 | 11d | Phenyl | 96.12±5.84 |
| 7b | Ethyl | 140.64±6.18 | TAE226 | - | 5.20±0.94 |
| 7c | Allyl | 14.30±2.73 |
2.2.4. Inhibition of HDAC2 expression and FAK phosphorylation using Western blot analysis
To further investigate the biochemical effects of the most active compounds 5d, 6a, 7c, and 11c on HDAC2 and FAK, Western blot analysis was performed on A-498 and Caki-1 cells. The obtained data were attuned to β-actin expression to exclude the variations. Among the tested compounds, 6a maintained a promising HDAC2/FAK inhibitory activity; Treating cells with 6a decreased the expression level of HDAC2 and FAK phosphorylation (Figure 5). Moreover, 6a effectively reduced their expression in A498 than Caki-1 cells. Interestingly, FAK autophosphorylation was entirely blocked by treating A-498 and Caki-1 cells with 7c. Taken together, the mentioned results demonstrate that 6a represses the expression of HDAC2 and the phosphorylation FAK.
Figure 5:
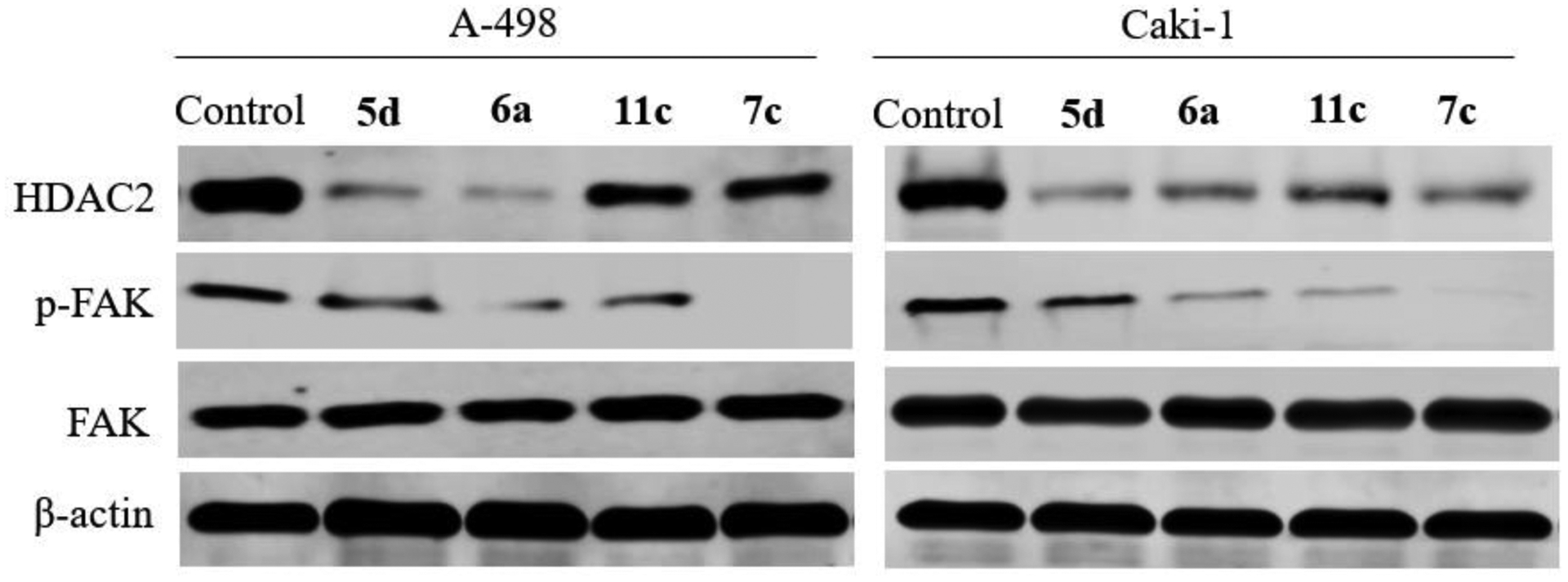
Western blot analysis of HDAC2, FAK, and total FAK in A-498 and Caki-1 cells treated with 5d, 6a, 7c, and 11c (β-actin was used as loading control)
2.2.5. Cell cycle and apoptosis analysis
The effect of compound 6a on cell cycle progression in A-498 and Caki-1 cells was analyzed using flow cytometry (Figure 6A and 6B). Compared to the negative control group, the percentages of cells in G2/M phase increased substantially from 18% to 65%, in A-498 cells and from 5.5% to 35%, in Caki-1 cells. Also, the percentage of cells in G1 phase decreased after treatment with 1.5 μM of compound 6a for 24 h, whereas the percentage of cells in S phase showed minor changes. Evidently, the derivative 6a induced the arrest of a significant percentage of cells in G2/M phase of the cell cycle compared with the untreated cells. To explore whether the antiproliferative activity of the synthesized compounds toward renal cancer cells (A-498 and Caki-1) was accompanied by an increase in cancer cell apoptosis, the apoptosis of A-498 and Caki-1 cells treated with different concentrations of the most potent derivative 6a (0.75 and 1.5 μM) was analyzed using Annexin V-FITC/PI assay. As illustrated in Figure 6C and 6D, it shows that 50% of A-498 cells experienced apoptosis with 6a treatment. Moreover, 6a induced apoptosis in Caki-1 cells to reach 37% at 1.5 μM concentration.
Figure 6:
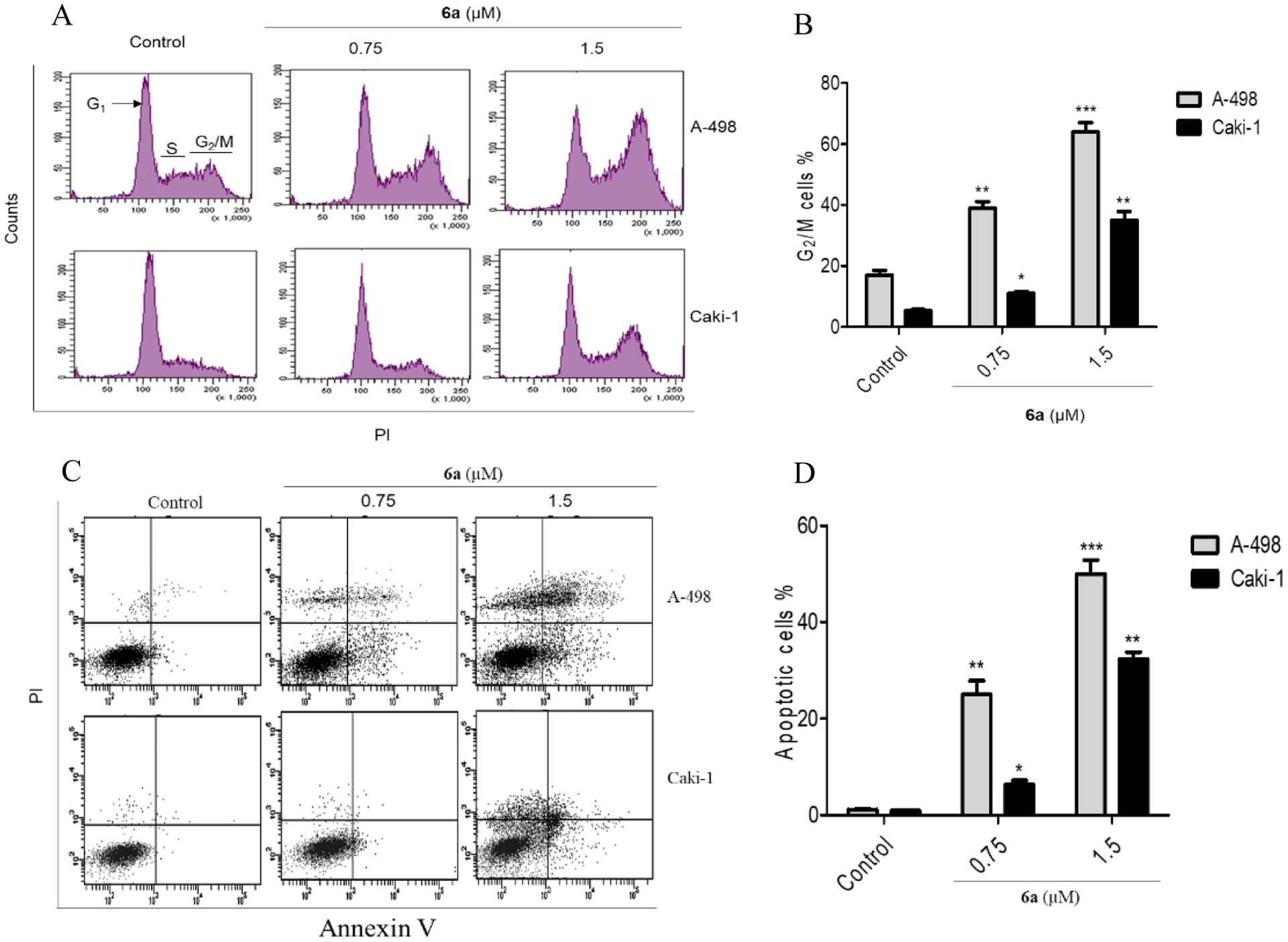
Cell cycle analysis and Flow cytometry of A-498 and Caki-1 cells at 0.75 and 1.5 μM concentrations. A) 6a induces G2/M arrest and decreases G1 phase concomitantly; B) Increasing the percentage of cells in G2/M phase parallel to increasing 6a concentration; C) 6a induces apoptosis in A-498 and Caki-1 cells; D) Percentage of apoptotic cells triggered by 6a administration
2.2.6. Relevance of Akt activation in HDAC inhibitor, VPA, resistance and relevance of Akt suppression in dual HDAC and FAK inhibitors sensitivity
It was reported that the chronic the HDAC inhibitor, VPA, application causes resistance via activation of Akt, and it is well known that FAK plays a vital role in Akt activation. Therefore, FAK inhibition will overcome HDAC inhibitors’ resistance via Akt activation suppression. We hypothesized that the synthesized dual HDAC2/FAK inhibitors might not allow resistance development upon prolonged use due to their ability to inhibit FAK and eventually suppress Akt activation. To test our hypothesis, A-498 cells were exposed to a fixed dose of either the synthesized dual HDAC2/FAK inhibitor 6a or the HDAC inhibitor VPA for 12 weeks, and then the antiproliferative activities of 6a and VPA were reassessed. There was no marked change in the IC50 on 6a-pretreated A-498 cells (IC50 = 1 μM) in comparison with its IC50 in cells that were not pretreated with 6a (0.95 μM, Figure 7A). In contrast, a significant change in the IC50 (from 2.1 mM to 3.09 mM) was observed in cells that were not pretreated with VPA compared to pretreated cells (Figure 7B). These data suggest that there is no observed resistance to 6a in A-498 cells, but the cells got resistant to VPA. Previously, researchers showed that the reason for the VPA resistance is due to Akt activation after chronic use. To elucidate the molecular mechanism by which VPA acquires resistance and 6a maintains its sensitivity, we examined HDAC2, p-FAK, FAK, p-Akt, and Akt proteins expression in A-498 cells pretreated or not pretreated with either 6a or VPA for 12 weeks for 24 h. The obtained results revealed that 6a showed a steady ability to inhibit HDAC2 expression and FAK phosphorylation for 12 weeks in both pretreated and not pretreated A-498 cells (Figure 7C). Also, it showed a substantial ability to inhibit Akt phosphorylation in both pretreated and not pretreated experiments. On the contrary, the VPA-pretreated cells showed a drastic overexpression of Akt phosphorylation, accompanied by a lower ability to inhibit HDAC2 expression (Figure 7D).
Figure 7:
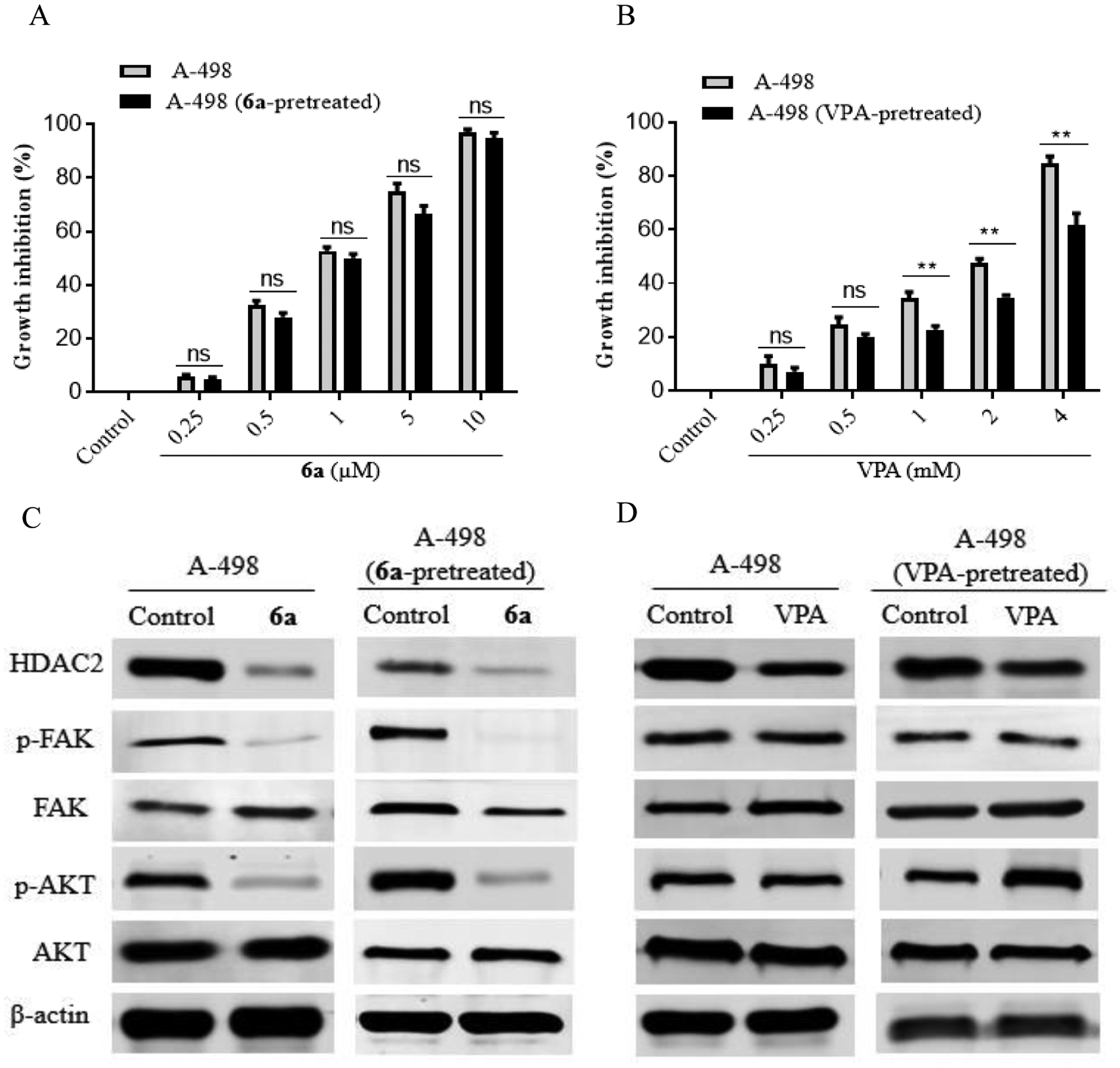
Effect of the hydroxamic acid derivative 6a on the pretreated A-498 cells. A) GI% of pretreated and not pretreated A-498 cells with 6a; B) GI% of pretreated and not pretreated A-498 cells with VPA; C) pretreatment of A-498 cells with 6a causes Akt activation suppression; D) pretreatment of A-498 cells with VPA causes significant Akt activation
2.2.7. Compound 6a inhibits STAT3 activation
The transcription factor, STAT3, plays a fundamental role in carcinogenesis, and is promising target for anticancer drug development. Moreover, the activation of STAT3 is well correlated with the overexpression of HDAC and FAK [17, 18]. To elucidate whether the anticancer effects of 6a are associated with STAT3 inhibition, this study examined the expression of STAT3. Western blotting results in Figure 8 showed the ability of 6a to inhibit the STAT3 activation.
Figure 8:
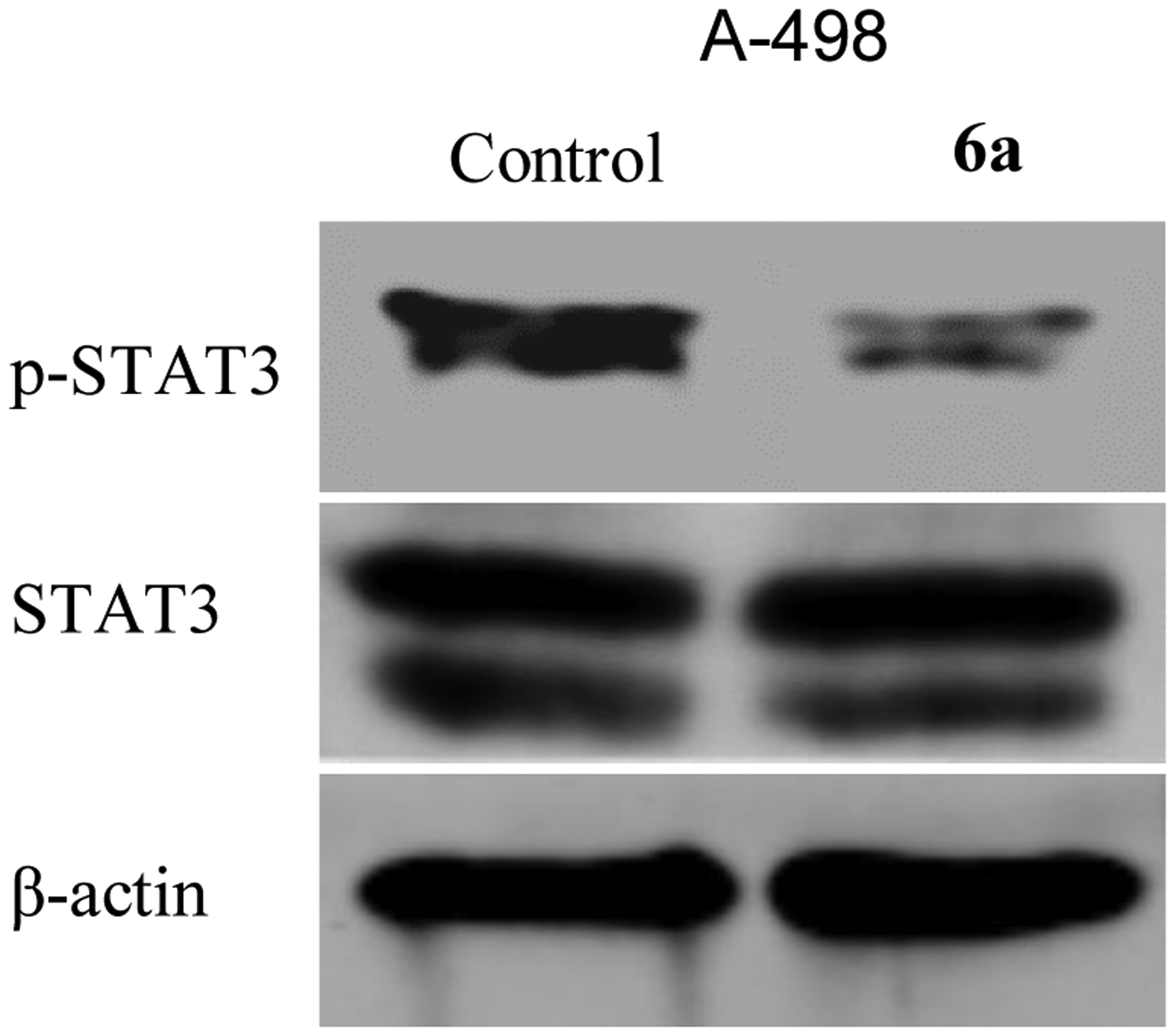
Effect of the hydroxamic acid derivative 6a on STAT3 activation. Treatment of A-498 cells with 6a causes STAT3 activation suppression.
2.2.8. Activation of different caspases as apoptotic executioners
Activation of caspases (2, 3, 6, 7, 8, 9, and 10) is a prominent biochemical feature representing a central executioner of apoptosis mediated by various inducers. HDAC/FAK inhibition is associated with caspase-8, caspase-9, and caspase-3 activation [40, 41]. Consequently, Compound 6a was evaluated for its potential to induce caspase-8, caspase-9, and caspase-3 activation in A-498 cell line (Table 5). The exposure of A-498 cells to 6a revealed a substantial activation of caspase-8 and caspase-9 by 3 and 6-folds, respectively, compared to the negative control. Interestingly, 6a activated caspase-3 dramatically by 11-folds in A-498 cells compared to the negative control. The caspases activation could explain the ability of the synthesized compounds to induce apoptosis via caspases pathways.
Table 5:
Effect of compound 6a on caspase-8, caspase-9, and caspase-3 activation
| Compound | Caspase-8 | Caspase-9 | Caspase-3 concentration, pg/ml |
|---|---|---|---|
| Concentration, ng/ml | |||
| 6a | 0.888 | 28.47 | 515.40 |
| Control | 0.274 | 4.53 | 45.57 |
2.3. Docking study
To further investigate the broad antiproliferative activity of the hydroxamic acid derivative 6a, molecular docking was performed on the crystallographic structures of HDAC2 (PDB code: 4LXZ) and FAK (PDB code: 2JKK), obtained from the Protein Data Bank (PDB) [42, 43]. The 4LXZ entry was selected for HDAC2 because the protein is bound with the potent HDAC inhibitor SAHA in this structure. Likewise, the entry 2JKK was chosen for FAK because, in this case, the protein is complexed with the potent FAK inhibitor TAE226. Compound 6a showed several significant interactions within HDAC2 active site, which may account for its potent inhibitory activity (Table 6). The hydroxamic acid moiety (ZBG) exhibited a coordinate bond with Zn atom and formed three hydrogen bonds with the key amino acids His145, Gly154, and Tyr308 (Figure 9A). Besides, Leu276 showed a hydrogen bonding interaction with the N2 of the 1,2,4-triazole ring. Moreover, the hydrophobic amino acids Phe155, Phe210, and His183 showed π…H interactions with the aliphatic linker (methylene groups). The 1,2,4-triazole and pyridine rings formed two π…H contacts with Phe155 and His33 amino acids, respectively. Additionally, 6a occupied the ATP binding site of FAK kinase perfectly and formed approximately the same interactions as TAE226 (Table 7). Favorably, the nitrogen in the pyridinyl moiety formed a hydrogen bond with Cys502 of the kinase hinge (Figure 9B). The pyridine ring formed four π…H contacts with Ile428, Cys502, Leu501, and Leu553 amino acids. The sulfur atom made a hydrogen bond with Gly429 amino acid, and the 5-pyridinyl-1,2,4-triazole moiety pointed toward the gatekeeper residue Met499. Moreover, the carbonyl oxygen formed two hydrogen bonds with Gly563 and Asn551 amino acids. The hydroxamic acid moiety was also in higher proximity to Asp564 of the DFG motif of the activation loop.
Table 6:
Energy scores (kcal/mol) and binding features for the HDAC2/FAK inhibitor 6a compared to SAHA, within HDAC2 binding site
| Compound | Energy score (S) (kcal/mol) | Ligand-receptor interactions | ||
|---|---|---|---|---|
| Residue | Type | Length (Ǻ) | ||
| Zn His145 |
Coordinate bond Hydrogen bond |
2.56 2.65 |
||
| Phe155 | π…H | 3.82 | ||
| SAHA | −9.43 | His183 | π…H | 4.06 |
| Phe210 | π…H | 4.44 | ||
| Zn | Coordinate bond | 2.69 | ||
| His145 | Hydrogen bond | 2.70 | ||
| Gly154 | Hydrogen bond | 1.97 | ||
| 6a | −8.82 | Tyr308 | Hydrogen bond | 1.78 |
| Leu276 | Hydrogen bond | 3.86 | ||
| His33 | π…H | 4.91 | ||
| His183 | π…H | 4.20 | ||
| Phe155 | π…H | 4.0 | ||
Figure 9:
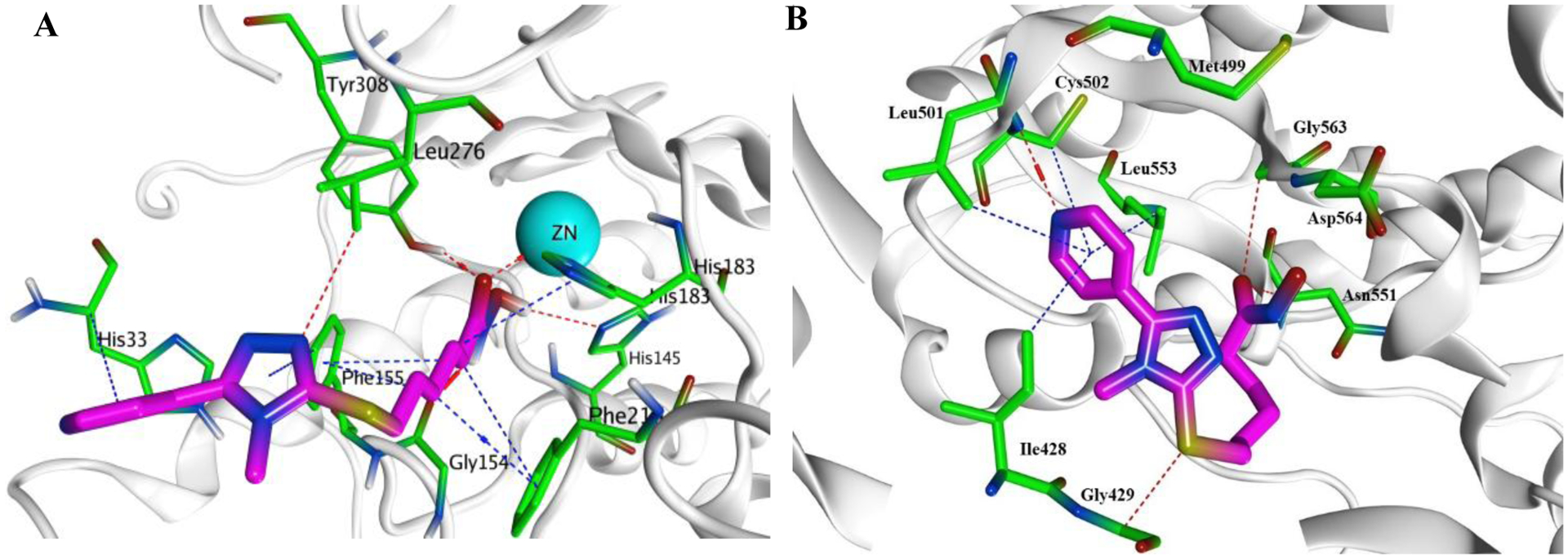
Presumptive binding modes of the compound 6a (magenta). A) Docking pose of 6a within HDAC2 active site (PDB code: 4LXZ); B) Docking pose of 6a within the ATP binding site of FAK (PDB code: 2JKK)
Table 7:
Energy scores (kcal/mol) and binding features for the HDAC2/FAK inhibitor 6a compared to TAE226, within the ATP binding site of FAK
| Compound | Energy score (S) (kcal/mol) | Ligand-receptor interactions | ||
|---|---|---|---|---|
| Residue | Type | Length (Ǻ) | ||
| Cys502 Glu500 |
Hydrogen bond Hydrogen bond | 2.99 3.24 |
||
| TAE226 | −9.51 | Ile428 | Hydrogen bond | 3.83 |
| Asp564 | Hydrogen bond | 3.00 | ||
| Leu501 | π…H | 4.45 | ||
| Leu553 | π…H | 3.33 | ||
| Cys502 | Hydrogen bond | 3.27 | ||
| Gly429 | Hydrogen bond | 3.69 | ||
| Ile428 | Hydrogen bond | 4.08 | ||
| 6a | −8.72 | Asn551 | Hydrogen bond | 3.24 |
| Gly563 | Hydrogen bond | 3.74 | ||
| Cys502 | π…H | 4.80 | ||
| Ile428 | π…H | 4.10 | ||
| Leu501 | π…H | 4.48 | ||
| Leu553 | π…H | 3.46 | ||
3. Conclusion
HDAC inhibitors comprise a diverse library of scaffolds that is amenable to several modifications, allowing the hybridization approach. FAK inhibition is emerging as a promising approach to treat cancer and prevent metastasis. The major discovery we report here is the rationale design and validation of a series of novel first-in-class dual HDAC2/FAK inhibitors containing 5-pyridinyl-1,2,4-triazole. The structural features of the HDAC scaffold included a surface recognition group (pyridine moiety), a linker (aliphatic or aromatic five-carbon linker), and a ZBG (carboxylic, hydroxamic acids, and 2-aminobenzamide). The prepared compounds showed remarkable HDAC2 selectivity among five tested HDAC isoforms. Compounds 5d, 6a, 7c, and 11c showed significant dual inhibition of HDAC2 and FAK enzymes. The same compounds revealed a promising ability to inhibit six different cancer cells (HCT116, HT-29, K562, KG-1, A-498, and Caki-1). The hydroxamic acid derivative 6a displayed the most potent HDAC2/FAK inhibitory activity and, subsequently, the highest antiproliferative activity. Unlike VPA, Western blot analysis revealed that 6a pretreated A-498 cells did not result in Akt activation, thus decreased the HDAC inhibition related side effects. Besides, 6a inhibited STAT3 phosphorylation, induced cellular apoptosis, and arrested the cell cycle. It activated caspase-8, caspase-9, and caspase-3. Compound 6a occupied HDAC2 and FAK active sites and formed all the essential interactions. These results suggest that developing a compound that could target HDAC/FAK proteins is considered a promising approach that warrants further investigation in animal models.
4. Experimental Methods
4.1. Chemistry
All chemicals were purchased from Aldrich, Merck, Alpha Aesar, and El-Nasr pharmaceutical chemicals companies, and were used without further purification. Reactions were monitored by thin layer chromatography (TLC), using Merck9385 pre-coated aluminum plate silica gel (Kieselgel 60) 5 × 20 cm plates with a layer thickness of 0.2 mm. The spots were detected by exposure to UV-lamp at 254 nm. Melting points were determined on Stuart electro thermal melting point apparatus and were uncorrected. NMR spectra were carried out using a Bruker Avance 400 MHz 1HNMR spectrometer and 100 MHz 13CNMR spectrometer (Beni Sweif, Egypt), using TMS as internal reference. Chemical shifts (δ values are given in parts per million (ppm) relative to TMS DMSO-d6 (2.5 and 3.5 ppm for 1HNMR and 76.9 ppm for 13CNMR) and coupling constants (J) in Hertz. Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Elemental analyses were recorded on Shimadzu GC/MS-QP5050A, Regional center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt. Mass spectra were recorded on Advion compact mass spectrometer (CMS) and reported as mass/charge (m/z), [ESI-H] samples were dissolved in methanol+water+TEA and [ESI+Na] samples were dissolved in methanol+water+sodium acetate, Nawah scientific center for research, Almokattam, Cairo, Egypt.
4.1.1. General procedure for the synthesis of the compounds 3a-d
The 1,2,4-triazole derivatives were prepared according to the general procedure in literatures [35]. Equimolar quantities of the isonicotinic acid hydrazide 1 (0.01 mol) and the appropriate isothiocyanate 2 (0.01 mol) were heated under reflux in absolute ethanol for 4 h. The solvent was evaporated under vacuum and 100 mL of 2N NaOH solution was added. The solution was refluxed for 3 h, then, the reaction mixture was cooled and acidified with conc. HCl. The formed precipitate was filtered off and recrystallized from ethanol to afford white solid with 92% yield.
4.1.2. General procedure for the synthesis of the compounds 5a-d and 9a-d
The prepared 1,2,4-triazoles 3a-d were reacted in equimolar quantities (1.4 mmol) with the substituted esters (1.4 mmol), in presence of triethylamine (1.7 mmol) as a base and CH3CN as a solvent to yield the ester derivatives 4a-d and 8a-d. The corresponding carboxylic acid derivatives 5a-d and 9a-d (1.3 mmol), were obtained through the hydrolysis of 4a-d and 8a-d with 20 equivalents of LiOH solution, followed by acidification with dil. HCl. The produced precipitate was recrystallized with aqueous ethanol with 90–95% yield.
4.1.2.1. 6-((4-methyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanoic acid 5a
White powder (0.38 g, 92% yield); mp 222–224°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.35–1.39(m, 2H, CH2 aliphatic), 1.45–1.51(m, 2H, CH2 aliphatic), 1.63–1.69(m, 2H, CH2 aliphatic), 2.17(t, 2H, J=7.21 Hz, CH2 aliphatic), 3.14(t, 2H, J=7.21 Hz, S-CH2), 3.63(s, 3H, N-CH3), 7.71(d, 2H, J=4.81 Hz, Ar-H), 8.72(d, 2H, J=5.61 Hz, Ar-H), 11.98(s, 1H, COOH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 24.49, 27.98, 29.40, 32.44, 32.99, 34.09, 122.76, 134.99, 150.86, 152.74, 153.71, 174.98 (C=O); Anal. Calcd for C14H18N4O2S: C, 54.88; H, 5.92; N, 18.29; S, 10.46. Found: C, 55.12; H, 6.16; N, 18.40; S, 10.53; ESI-MS: [M-H]−, 305.3, found: 305.2.
4.1.2.2. 6-((4-ethyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanoic acid 5b
White powder (0.39 g, 93% yield); mp 205–207°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.19(t, 3H, J=7.21 Hz, N-CH2-CH3), 1.34–1.39(m, 2H, CH2 aliphatic), 1.45–1.51(m, 2H, CH2 aliphatic), 1.65–1.71(m, 2H, CH2 aliphatic), 2.17(t, 2H, J=7.21 Hz, CH2 aliphatic), 3.19(t, 2H, J=7.21 Hz, S-CH2), 4.02(q, 2H, J=7.21 Hz, N-CH2-CH3), 7.66(d, 2H, J=5.61 Hz, Ar-H), 8.73(d, 2H, J=5.61 Hz, Ar-H), 12.03(s, 1H, COOH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 15.50, 24.49, 28.01, 29.40, 32.94, 34.02, 122.83, 135.14, 151.01, 152.18, 153.12, 174.97 (C=O); Anal. Calcd for C15H20N4O2S: C, 56.23; H, 6.29; N, 17.49; S, 10.01. Found: C, 56.45; H, 6.38; N, 17.71; S, 9.87; ESI-MS: [M-H]−, 319.4, found: 319.0.
4.1.2.3. 6-((4-allyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanoic acid 5c
Off white powder (0.41 g, 95% yield); mp 171–173°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.30–1.35(m, 2H, CH2 aliphatic), 1.43–1.48(m, 2H, CH2 aliphatic), 1.61–1.66(m, 2H, CH2 aliphatic), 2.14(t, 2H, J=6.41 Hz, CH2 aliphatic), 3.15(t, 2H, J=7.21 Hz, S-CH2), 4.81(d, 2H, Jcis=4.81 Hz, N-CH2-CH=CH2), 4.73(d, 1H, Jcis=16.83 Hz, -CH=CH2), 5.17(d, 1H, Jtrans=10.42 Hz, -CH=CH2), 5.88–5.97(m, 1H, CH2-CH=CH2), 7.61(d, 2H, J=5.61 Hz, Ar-H), 8.69(d, 2H, J=5.61 Hz, Ar-H); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 24.49, 27.98, 29.36, 33.10, 34.03, 47.11, 117.61, 122.52, 132.80, 134.87, 150.96, 152.97, 153.49, 174.94 (C=O); Anal. Calcd for C16H20N4O2S: C, 57.81; H, 6.06; N, 16.85; S, 9.64. Found: C, 58.04; H, 6.21; N, 16.97; S, 9.75; ESI-MS: [M-H]−, 331.4, found: 331.0.
4.1.2.4. 6-((4-phenyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanoic acid 5d
White crystals (0.43 g, 90% yield); mp 180–182°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.26–1.32(m, 2H, CH2 aliphatic), 1.42–1.48(m, 2H, CH2 aliphatic), 1.61–1.67(m, 2H, CH2 aliphatic), 2.15(t, 2H, J=7.21 Hz, CH2 aliphatic), 3.12(t, 2H, J=7.21 Hz, S-CH2), 7.28(d, 2H, J=4.81 Hz, Ar-H), 7.42–7.55(m, 2H, Ar-H), 7.52–7.56(m, 3H, Ar-H), 8.5(d, 2H, J=5.61 Hz, Ar-H), 12.01(s, 1H, COOH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 24.47, 28.01, 29.23, 32.38, 34.00, 122.05, 128.20, 130.68, 130.97, 134.05, 134.47, 150.61, 152.70, 153.91, 174.97 (C=O); Anal. Calcd for C19H20N4O2S: C, 61.94; H, 5.47; N, 15.21; S, 8.70. Found: C, 61.72; H, 5.70; N, 15.44; S, 8.92; ESI-MS: [M-H]−, 367.4, found: 367.2.
4.1.3. General procedure for synthesis of 5-pyridinyl-4H-1,2,4-triazol-3-thiol derivatives 6a-d, 7a-d, 10a-d, and 11a-d.
The corresponding carboxylic acid derivatives 5a-d and 9a-d (1.24 mmol) were treated with CDI (1.6 mmol) in dimethylformamide or acetonitrile at 40°C. The resulting imidazolide derivative was then reacted with hydroxylamine hydrochloride (1.6 mmol) to yield the corresponding hydroxamic acid derivatives 6a-d and 10a-d, while treating the imidazolide derivatives with 2-phenylenediamine (1.6 mmol) in presence of TFA (0.13 g, 1.09 mmol) yielded the corresponding 2-aminobenzamide derivatives. After reaction completion (4–10 h), the resulting mixture was poured on dist. H2O. The formed precipitate was dried and recrystallized with aq. Methanol (Compounds 7a-c were purified with flash column using CH2Cl2:MeOH (9:1) as an eluent.
4.1.3.1. N-hydroxy-6-((4-methyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanamide 6a
White powder (0.35 g, 88% yield); mp 141–143°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.34–1.41(m, 2H, CH2 aliphatic), 1.48–1.56(m, 2H, CH2 aliphatic), 1.66–1.73(m, 2H, CH2 aliphatic), 1.96(t, 2H, J=7.27 Hz, CH2 aliphatic), 3.19(t, 2H, J=7.21 Hz, S-CH2), 3.68(s, 3H, N-CH3), 7.76(d, 2H, J=6.11 Hz, Ar-H), 8.77(d, 2H, J=6.11 Hz, Ar-H), 10.38(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 25.08, 27.96, 29.31, 32.38, 32.58, 32.97, 122.68, 134.92, 150.79, 152.67, 153.65, 169.45 (C=O); Anal. Calcd for C14H19N5O2S: C, 52.32; H, 5.96; N, 21.79; S, 9.98. Found: C, 52.49; H, 6.13; N, 22.06; S, 10.04; ESI-MS: [M+Na]+, 344.4, found: 344.2.
4.1.3.2. 6-((4-ethyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyhexanamide 6b
White powder (0.36 g, 86% yield); mp 233–235°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.24(t, 3H, J=7.21 Hz, N-CH2-CH3), 1.36–1.42(m, 2H, CH2 aliphatic), 1.49–1.57(m, 2H, CH2 aliphatic), 1.68–1.76(m, 2H, CH2 aliphatic), 1.96(t, 2H, J=7.27 Hz, CH2 aliphatic), 3.23(t, 2H, J=7.27 Hz, S-CH2), 4.07(q, 2H, J=7.21 Hz, N-CH2-CH3), 7.71(d, 2H, J=6.24 Hz, Ar-H), 8.78(d, 2H, J=6.24 Hz, Ar-H), 8.72(s, 1H, NH), 10.38(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 15.44, 25.09, 28.00, 29.32, 32.58, 32.91, 122.75, 135.08, 150.94, 152.12, 153.06, 169.45 (C=O); Anal. Calcd for C15H21N5O2S: C, 53.71; H, 6.31; N, 20.88; S, 9.56. Found: C, 53.98; H, 6.52; N, 20.74; S, 9.67; ESI-MS: [M+Na]+, 358.4, found: 357.9.
4.1.3.3. 6-((4-allyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)-N-hydroxyhexanamide 6c
Off white powder (0.35 g, 82% yield); mp 143–145°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.33–1.40(m, 2H, CH2 aliphatic), 1.48–1.55(m, 2H, CH2 aliphatic), 1.65–1.73(m, 2H, CH2 aliphatic), 1.96(t, 2H, J=7.23 Hz, CH2 aliphatic), 3.20(t, 2H, J=7.28 Hz, S-CH2), 4.72(d, 2H, Jcis=4.40 Hz, N-CH2-CH=CH2), 4.79(d, 1H, Jcis=17.45 Hz, -CH=CH2), 5.23(d, 1H, Jtrans=10.46 Hz, -CH=CH2), 5.97–6.02(m, 1H, CH2-CH=CH2), 7.68(d, 2H, J=5.99 Hz, Ar-H), 8.75(d, 2H, J=5.99 Hz, Ar-H), 8.72(s, 1H, NH), 10.38(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 25.08, 27.97, 29.28, 32.57, 33.05, 47.05, 117.55, 121.67, 122.46, 132.72, 134.79, 150.77, 150.89, 152.91, 153.43, 169.45 (C=O); Anal. Calcd for C16H21N5O2S: C, 55.31; H, 6.09; N, 20.16; S9.23. Found: C, 55.60; H, 6.23; N, 20.45; S, 9.26; ESI-MS: [M+Na]+, 370.1, found: 370.1.
4.1.3.4. N-hydroxy-6-((4-phenyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanamide 6d
White crystals (0.38 g, 85% yield); mp 163–165°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.29–1.35(m, 2H, CH2 aliphatic), 1.46–1.53(m, 2H, CH2 aliphatic), 1.64–1.72(m, 2H, CH2 aliphatic), 1.94(t, 2H, J=7.29 Hz, CH2 aliphatic), 3.17(t, 2H, J=7.23 Hz, S-CH2), 7.30(d, 2H, J=6.24 Hz, Ar-H), 7.47–7.50(m, 2H, Ar-H), 7.57–7.61(m, 3H, Ar-H), 8.56(d, 2H, J=6.24 Hz, Ar-H), 8.73(s, 1H, NH); 10.38(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 25.06, 28.00, 29.14, 32.35, 32.56, 121.99, 128.11, 130.62, 130.91, 133.97, 134.40, 150.53, 152.63, 153.85, 169.46 (C=O); Anal. Calcd for C19H21N5O2S: C, 59.51; H, 5.52; N, 18.26; S, 8.36. Found: C, 59.74; H, 5.59; N, 18.42; S, 8.45; ESI-MS: [M+Na]+, 406.4, found: 405.9.
4.1.3.5. N-(2-aminophenyl)-6-((4-methyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanamide 7a
Brown oil (0.40 g, 81% yield); 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.49–1.55(m, 2H, CH2 aliphatic), 1.64–1.72(m, 2H, CH2 aliphatic), 1.76–1.83(m, 2H, CH2 aliphatic), 2.38(t, 2H, J=7.34 Hz, CH2 aliphatic), 3.26(t, 2H, J=7.21 Hz, S-CH2), 3.72(s, 3H, N-CH3), 4.83(s, 2H, NH2), 6.58(t, 1H, J=7.52 Hz, Ar-H), 6.77(d, 1H, J=9.41 Hz, Ar-H), 6.94(t, 1H, J=7.27 Hz, Ar-H), 7.20(d, 1H, J=9.41 Hz, Ar-H), 7.81(d, 2H, J=6.11 Hz, Ar-H), 8.81(d, 2H, J=6.11 Hz, Ar-H), 9.20(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 25.24, 28.03, 29.36, 32.37, 33.02, 36.05, 116.35, 116.64, 122.17, 122.68, 123.98, 125.79, 135.64, 142.38, 150.78, 152.69, 153.65, 171.55 (C=O); Anal. Calcd for C20H24N6OS: C, 60.58; H, 6.10; N, 21.20; S, 8.09. Found: C, 60.41; H, 6.28; N, 20.98; S, 8.27; ESI-MS: [M+Na]+, 419.5, found: 419.2.
4.1.3.6. N-(2-aminophenyl)-6-((4-ethyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanamide 7b
Brown oil (0.43 g, 86% yield); 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.24(t, 3H, J=7.27 Hz, CH2-CH3), 1.46–1.50(m, 2H, CH2 aliphatic), 1.60–1.68(m, 2H, CH2 aliphatic), 1.74–1.81(m, 2H, CH2 aliphatic), 2.34(t, 2H, J=7.34 Hz, CH2 aliphatic), 3.27(t, 2H, J=7.21 Hz, S-CH2), 4.07(q, 2H, J=7.27 Hz, N-CH2-CH3), 4.84(s, 2H, NH2), 6.53(t, 1H, J=7.52 Hz, Ar-H), 6.72(d, 1H, J=9.54 Hz, Ar-H), 6.87–6.92(m, 1H, Ar-H), 7.16(d, 1H, J=9.54 Hz, Ar-H), 7.71(d, 2H, J=6.11 Hz, Ar-H), 8.77(d, 2H, J=6.11 Hz, Ar-H), 9.13(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 15.45, 25.24, 28.06, 29.37, 32.98, 30.05, 116.33, 116.61, 117.72, 122.75, 123.99, 125.76, 126.18, 135.08, 142.36, 150.94, 152.12, 153.06, 171.51 (C=O); Anal. Calcd for C21H26N6OS: C, 61.44; H, 6.38; N, 20.47; S, 7.81. Found: C, 61.68; H, 6.62; N, 20.71; S, 7.94; ESI-MS: [M+Na]+, 433.5, found: 433.3.
4.1.3.7. 6-((4-allyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)-N-(2-aminophenyl)hexanamide 7c
Brown oil (0.42 g, 92% yield); 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.44–1.50(m, 2H, CH2 aliphatic), 1.60–1.67(m, 2H, CH2 aliphatic), 1.72–1.79(m, 2H, CH2 aliphatic), 2.34(t, 2H, J=7.34 Hz, CH2 aliphatic), 3.24(t, 2H, J=7.21 Hz, S-CH2), 4.72(d, 2H, Jcis=4.52 Hz, N-CH2-CH=CH2), 4.78–4.88(m, 3H, Jcis=17.24 Hz, -CH=CH2 and NH2), 5.24(d, 1H, Jtrans=10.39 Hz, -CH=CH2), 5.94–6.03(m, 1H, CH2-CH=CH2), 6.54(t, 1H, J=7.46 Hz, Ar-H), 6.72(d, 1H, J=9.54 Hz, Ar-H), 6.90(t, 1H J=7.40 Hz, Ar-H), 7.16(d, 1H, J=7.95 Hz, Ar-H), 7.69(d, 2H, J=6.24 Hz, Ar-H), 8.76(d, 2H, J=6.24 Hz, Ar-H), 9.12(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 24.78, 27.58, 28.88, 32.67, 35.60, 46.59, 115.89, 116.18, 117.11, 121.76, 122.02, 123.44, 125.73, 132.28, 133.91, 134.12, 135.20, 141.88, 150.45, 170.79 (C=O); Anal. Calcd for C22H26N6OS: C, 62.53; H, 6.20; N, 19.89; S, 7.59. Found: C, 62.77; H, 6.39; N, 20.06; S, 7.73; ESI-MS: [M+Na]+, 445.5, found: 445.3.
4.1.3.8. N-(2-aminophenyl)-6-((4-phenyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)hexanamide 7d
yellowish brown powder (0.48 g, 92% yield); mp 179–181°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.37–1.44(m, 2H, CH2 aliphatic), 1.57–1.65(m, 2H, CH2 aliphatic), 1.70–1.78(m, 2H, CH2 aliphatic), 2.32(t, 2H, J=7.40 Hz, CH2 aliphatic), 3.20(t, 2H, J=7.21 Hz, S-CH2), 4.83(s, 2H, NH2), 6.53(t, 1H, J=7.52 Hz, Ar-H), 6.72(d, 1H, J=9.54 Hz, Ar-H), 6.89(t, 1H J=7.58 Hz, Ar-H), 7.15(d, 1H, J=9.54 Hz, Ar-H), 7.30(d, 2H, J=6.24 Hz, Ar-H), 7.47–7.50(m, 2H, Ar-H), 7.56–7.61(m, 3H, Ar-H), 8.57(d, 2H, J=6.24 Hz, Ar-H), 9.11(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 25.23, 28.07, 29.21, 32.38, 36.05, 116.34, 116.63, 121.98, 123.99, 125.75, 126.17, 128.12, 130.61, 130.89, 133.99, 134.41, 142.35, 150.54, 152.63, 153.85, 171.50 (C=O); Anal. Calcd for C25H26N6OS: C, 65.48; H, 5.71; N, 18.33; S, 6.99. Found: C, 65.74; H, 5.98; N, 18.50; S, 7.02; ESI-MS: [M+Na]+, 481.5, found: 481.3.
4.1.3.9. N-hydroxy-4-(((4-methyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)benzamide 10a
White powder (0.36 g, 81% yield); mp 190–192°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 3.64(s, 3H, N-CH3), 4.53(s, 2H, S-CH2), 7.51(d, 2H, J=6.24 Hz, Ar-H), 7.75(d, 2H, J=6.24 Hz, Ar-H), 7.79(d, 2H, J=4.28 Hz, Ar-H), 8.83(d, 2H, J=4.28 Hz, Ar-H), 9.1(s, 1H, NH), 11.27(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 32.38, 36.84, 122.67, 127.52, 129.94, 132.39, 135.47, 140.49, 150.83, 152.34, 155.07, 164.35 (C=O); Anal. Calcd for C16H15N5O2S: C, 56.29; H, 4.43; N, 20.51; S, 9.39. Found: C, 56.61; H, 4.56; N, 20.74; S, 9.51; ESI-MS: [M-H]−, 340.3, found: 340.0.
4.1.3.10. 4-(((4-ethyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-N-hydroxybenzamide 10b
White powder (0.35 g, 76% yield); mp 198–200°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.17(t, 3H, J=7.27 Hz, N-CH2-CH3), 4.03(q, 2H, J=7.27 Hz, N-CH2-CH3), 4.58(s, 2H, S-CH2), 7.52(d, 2H, J=8.31Hz, Ar-H), 7.73–7.76(m, 4H, Ar-H), 8.82(d, 2H, J=6.11 Hz, Ar-H), 9.10(s, 1H, NH-NH), 11.26(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 15.38, 36.89, 39.35, 122.73, 127.54, 129.47, 132.39, 135.00, 141.01, 150.97, 151.39, 153.22, 164.33 (C=O); Anal. Calcd for C17H17N5O2S: C, 57.45; H, 4.82; N, 19.71; S, 9.02. Found: C, 57.72; H, 5.06; N, 19.97; S, 9.14; ESI-MS: [M-H]−, 354.4, found: 353.8.
4.1.3.11. 4-(((4-allyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-N-hydroxybenzamide 10c
White powder (0.41 g, 86% yield); mp 194–196°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 4.52(s, 2H, S-CH2), 4.64(d, 2H, J=4.65 Hz, N-CH2-CH=CH2), 4.73(d, 1H, Jcis=17.24 Hz, -CH=CH2), 5.17(d, 1H, Jtrans=10.39 Hz, -CH=CH2), 5.86–5.94(m, 1H, CH2-CH=CH2), 7.48(d, 2H, J=8.29 Hz, Ar-H), 7.66(d, 2H, J=6.11 Hz, Ar-H), 7.70(d, 2H, J=8.31 Hz, Ar-H), 8.76(d, 2H, J=6.11 Hz, Ar-H), 9.05(s, 1H, NH), 11.21(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 36.83, 47.04, 117.67, 122.49, 127.52, 129.48, 132.40, 132.59, 134.69, 140.89, 150.92, 152.20, 153.59, 164.34 (C=O); Anal. Calcd for C18H17N5O2S: C, 58.84; H, 4.66; N, 19.06; S, 8.73. Found: C, 59.11; H, 4.83; N, 19.28; S, 8.59; ESI-MS: [M+Na]+, 390.4, found: 390.2.
4.1.3.12. N-hydroxy-4-(((4-phenyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)benzamide 10d
White powder (0.48 g, 92% yield); mp 145–147°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 4.54(s, 2H, S-CH2), 7.32–7.35(m, 2H, Ar-H), 7.43–7.51(m, 4H, Ar-H), 7.61–7.65(m, 3H, Ar-H), 7.74(d, 2H, J=5.14 Hz, Ar-H), 8.61(d, 2H, J=6.11 Hz, Ar-H), 9.11(s, 1H, NH), 11.27(s, 1H, OH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 36.01, 121.98, 127.50, 128.04, 129.49, 130.61, 130.95, 132.37, 133.80, 134.31, 140.86, 150.59, 152.81, 153.22, 164.37 (C=O); Anal. Calcd for C21H17N5O2S: C, 62.52; H, 4.25; N, 17.36; S, 7.95. Found: C, 62.70; H, 4.42; N, 17.59; S, 8.06; ESI-MS: [M-H]−, 402.4, found: 402.2.
4.1.3.13. N-(2-aminophenyl)-4-(((4-methyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3- yl)thio)methyl)benzamide 11a
Light yellow crystals (0.47 g, 91% yield); mp 244–246°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 3.65(s, 3H, N-CH3), 4.55(s, 2H, S-CH2), 4.94(s, 2H, NH2), 6.64(t, 1H, J=7.52 Hz, Ar-H), 6.83(d, 1H, J=8.07 Hz, Ar-H), 7.02(t, 1H, J=7.85 Hz, Ar-H), 7.20(d, 1H, J=7.83 Hz, Ar-H), 7.56(d, 2H, J=6.24 Hz, Ar-H), 7.78(d, 2H, J=4.28 Hz, Ar-H), 7.97(d, 2H, J=8.31 Hz, Ar-H), 8.81(d, 2H, J=4.28 Hz, Ar-H), 9.70(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 32.43, 36.90, 116.57, 116.71, 122.66, 123.68, 127.02, 127.22, 128.44, 129.38, 134.20, 134.82, 141.24, 143.66, 150.83, 151.87, 153.85, 165.42 (C=O); Anal. Calcd for C22H20N6OS: C, 63.44; H, 4.84; N, 20.18; S, 7.70. Found: C, 63.71; H, 4.95; N, 20.37; S, 7.65; ESI-MS: [M+Na]+, 439.5, found: 439.2.
4.1.3.14. N-(2-aminophenyl)-4-(((4-ethyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)benzamide 11b
Brown oil (0.39 g, 73% yield); 1HNMR (400 MHz, DMSO-d6) δ (ppm): 1.16(t, 3H, J=7.34 Hz, CH2-CH3), 4.01(q, 2H, J=7.34 Hz, CH2-CH3), 4.58(s, 2H, S-CH2), 4.93(s, 2H, NH2), 6.61(t, 1H, J=7.70 Hz, Ar-H), 6.80(d, 1H, J=8.19 Hz, Ar-H), 6.98(t, 1H, J=7.70 Hz, Ar-H), 7.18(d, 1H, J=8.19 Hz, Ar-H), 7.55(d, 2H, J=8.07 Hz, Ar-H), 7.70(d, 2H, J=5.50 Hz, Ar-H), 7.96(d, 2H, J=8.31 Hz, Ar-H), 8.78(d, 2H, J=5.50 Hz, Ar-H), 9.71(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 15.44, 36.86, 39.34, 116.59, 116.72, 122.73, 123.70, 127.01, 127.21, 128.46, 129.37, 134.21, 134.99, 141.25, 143.65, 150.98, 151.40, 153.23, 165.42 (C=O); Anal. Calcd for C23H22N6OS: C, 64.17; H, 5.15; N, 19.52; S, 7.45. Found: C, 64.40; H, 5.39; N, 19.78; S, 7.62; ESI-MS: [M+Na]+, 453.5, found: 453.2.
4.1.3.15. 4-(((4-allyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-N-(2-aminophenyl)benzamide 11c
Yellowish brown powder (0.51 g, 93% yield); mp 182–184°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 4.61(s, 2H, S-CH2), 4.71(d, 2H, J=4.52 Hz, N-CH2-CH=CH2), 4.79(d, 1H, Jcis=17.24 Hz, -CH=CH2), 4.96(s, 2H, NH2), 5.24(d, 1H, Jtrans=10.64 Hz, -CH=CH2), 5.92–6.01(m, 1H, CH2-CH=CH2), 6.65(t, 1H, J=7.58 Hz, Ar-H), 6.83(d, 1H, J=8.07 Hz, Ar-H), 7.03(t, 1H, J=7.58 Hz, Ar-H), 7.21(d, 1H, J=7.95 Hz, Ar-H), 7.59(d, 2H, J=8.19 Hz, Ar-H), 7.71(d, 2H, J=4.65 Hz, Ar-H), 7.99(d, 2H, J=8.19 Hz, Ar-H), 8.80(d, 2H, J=4.65 Hz, Ar-H), 9.71(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 36.84, 47.07, 116.57, 116.71, 117.71, 122.49, 123.69, 127.01, 127.20, 128.43, 129.38, 132.62, 134.22, 134.69, 141.15, 143.65, 150.93, 152.20, 153.60, 165.42 (C=O); Anal. Calcd for C24H22N6OS: C, 65.14; H, 5.01; N, 18.99; S, 7.24. Found: C, 65.38; H, 5.17; N, 19.23; S, 7.45; ESI-MS: [M+Na]+, 465.5, found: 465.2.
4.1.3.16. N-(2-aminophenyl)-4-(((4-phenyl-5-(pyridin-4-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)benzamide 11d
Off white powder (0.54 g, 91% yield); mp 230–232°C; 1HNMR (400 MHz, DMSO-d6) δ (ppm): 4.58(s, 2H, S-CH2), 4.96(s, 2H, NH2), 6.65(t, 1H, J=7.58 Hz, Ar-H), 6.83(d, 1H, J=7.95Hz, Ar-H), 7.02(t, 1H, J=7.58 Hz, Ar-H), 7.21(d, 1H, J=7.82 Hz, Ar-H), 7.35(d, 2H, J=6.24 Hz, Ar-H), 7.47(d, 2H, J=8.19 Hz, Ar-H), 7.56–7.64(m, 5H, Ar-H), 7.97(d, 2H, J=8.19 Hz, Ar-H), 8.62(d, 2H, J=4.52 Hz, Ar-H), 9.70(s, 1H, C=O-NH); 13CNMR (100 MHz, DMSO-d6) δ (ppm): 34.07, 114.63, 114.77, 120.03, 121.77, 125.06, 125.26, 126.10, 126.46, 127.44, 128.70, 129.02, 131.88, 132.35, 139.18, 141.70, 148.64, 148.73, 150.87, 151.26, 163.50 (C=O); Anal. Calcd for C27H22N6OS: C, 67.76; H, 4.63; N, 17.56; S, 6.70. Found: C, 68.02; H, 4.80; N, 17.82; S, 6.83; ESI-MS: [M+Na]+, 501.5, found: 501.2.
4.2. Biological investigations
Cell culture and reagents
Six different cell lines derived from three tumor subpanels, including colon (HCT116 and HT-29), leukemia (K562, and KG-1), and renal (A-498 and Caki-1) cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and were cultured in their suitable media containing 10% fetal bovine serum (FBS; Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life technologies) in a humidified atmosphere with 5% CO2 at 37°C. 5-pyridinyl-1,2,4-triazole derivatives were chemically synthesized (5a-d, 6a-d, 7a-d, 10a-d, and 11a-d). All chemicals were purchased from Sigma-Aldrich Co., LLC except TAE226 was purchased from Selleck Chemicals (Houston, TX, USA). All chemicals used in this study were of the analytical or cell-culture grade.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
The proliferation assay for six different cell lines derived from three tumor subpanels, including colon (HCT116 and HT-29), leukemia (K562, and KG-1), and renal (A-498 and Caki-1) cell lines was quantified using MTT assay. Cells were seeded as 1 × 104 cells/well and cultured overnight in a 96-well plate. The cells were treated with either 10 μM of 5-pyridinyl-1,2,4-triazole derivatives (5a-d, 6a-d, 7a-d, 10a-d, and 11a-d), or DMSO as a negative control. After 24 h, the cells washed with phosphate buffered saline (PBS, Invitrogen Gibco) and incubated with 20 μl of MTT solution (2 mg/ml) for 4 hr at 37°C. Then, 150 μl DMSO was used to solubilize MTT formazan crystals. Finally, the plates were shaken, and the optical density was determined at 570 nm using ELISA plate reader (Model 550, Bio-Rad, USA). At least, three independent experiments were performed. Percentage of growth inhibition was determined as (1− [OD of treated cells/OD of control cells]). On the other hand, using the MTT assay, we tested the effect of different concentrations (1, 5, 10, 25, 50, 100, 1000, 2000, and 4000 μM) of the synthesized compounds on renal cancer cells (A-498 and Caki-1), using DMSO as a negative control, whereas VPA, SAHA, and TAE226 were used as positive controls. The IC50 values were calculated using Prism v.8 software (GraphPad Software Inc., La Jolla, CA).
Fluorogenic enzymatic assays for HDAC isoforms:
To determine the ability of the synthesized compounds to inhibit the activity of HDACs, we did invitro optimize fluorogenic enzymatic assays for HDAC isoforms in 96-well opaque half-area microplate (Corning). Recombinant, full-length human HDAC1, 2, 3, and 8 (BPS Bioscience, CA, USA) were diluted in assay buffer A (25 mM Tris–HCl, 137 mM NaCl, 2.7 mM KCl, 1mM MgCl2, and 1 mg/mL BSA, pH 8.0) to give 4, 5, 1, 8.5 ng/μL stock solutions, respectively. Serial dilutions of inhibitors were made in assay buffer B (25 mM Tris–HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0). Then, we mixed 10 μL of each enzyme stock to 30 μL of inhibitor and incubated for 5 min at room temperature. After incubation, 10 μL of HDAC substrate was added and the mixture incubated for 45 min at room temperature. For HDAC1, 2, 3 and 8, the reaction was quenched with trypsin and trichostatin A dissolved in assay buffer B and incubated at room temperature for 45 min. Fluorescence measurements were obtained with Synergy 4 Hybrid microplate reader (Bio-Tek) at excitation wavelength 360 nm and emission wavelength 460 nm. On the other hand, to determine the ability of the synthesized compounds to inhibit the activity of HDAC6, we used fluorogenic Assay kit (BPS Biosciences) per the manufacturer’s instructions. Valproic acid, trichostatin A and SAHA were utilized as inhibitor positive controls. The experiments were repeated at least three times with each experiment containing three replicates. The IC50 values were determined using Prism v.8 software.
kinase selectivity assays
IC50 values of the synthesized compounds and TAE226 on five different kinases (FAK, FLT, FRK, IGF-1R, and BTK) were determined using Z’-LYTE® technology, which is based on FRET (Invitrogen/Life Technologies).
Western blotting
A-498 and Caki-1 cells were harvested, washed twice with chilled PBS, and lysed with ice-cold lysis buffer containing 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2 μg/ml aprotinin, 5 μg/ml pefabloc SC (4-(2-Aminoethyl) benzenesulfonyl fluoride hidrochloride), a protease inhibitor cocktail, 1 % phosphatase inhibitor cocktail and 50 mM Tris–HCl (pH 7.4) after 24 h incubation with the IC50 value of the potent compounds (5d, 6a, 11c, and 7c) or DMSO (control). The cell lysates were kept on ice for 30 min after gently vortex, and then centrifuged at 14,000g for 15 min at 4°C. The supernatants were loaded onto SDS-PAGE gel immediately after protein extraction, or otherwise the supernatant was stored at −80°C until use. The protein concentration was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. For western blotting analysis, 40 μg protein-weight of cell lysate were loaded onto a SDS-PAGE gel. Proteins separated on a SDS-PAGE gel were transferred onto a PVDF membrane. The PVDF membrane was incubated in blocking buffer containing either 5% non-fat milk powder or 5% bovine serum albumin (Sigma-Aldrich) for 1 h. Subsequently, the PVDF membrane was incubated with primary antibodies for overnight and followed by HRP-conjugated anti-rabbit IgG (Cell Signaling Technologies) or HRP- conjugated anti-mouse IgG (Becton Dickinson Co, Durham, NC, USA) for 1 h with agitation at room temperature. Finally, the specific bindings to each primary antibody were detected on an X-ray film (Konica Minolta Medical Imaging, Wayne, NJ, USA) with ECL prime immunodetection reagent (GE Healthcare, Little Chalfont, UK).
Cell Cycle Assay
The fiow cytometer was used to support the anti-proliferative activity of the synthesized compounds towards A-498 and Caki-1 cells. Briefiy, cells at a density of 1 × 105 cells/mL were cultured with different concentrations of 6a (0.75 and 1.5 μM) or DMSO as a negative control and incubated for 24 hrs. The cells were harvested and washed with PBS. Subsequently, 80% ice cold ethanol was added to the cell pellets drop by drop with continuous vortexing to prevent clumping and aggregation of cells and then kept at 20∘C for overnight. Then, the cell pellets washed twice, stained with PI, and incubated in the dark for 20 minutes. Flow cytometric analysis was conducted using FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Apoptosis assay
Induction of apoptosis of A-498 and Caki-1 cells was analyzed using the annexin V/propidium iodide (PI) staining kit (BioLegend, San Diego, CA, USA) according to the manufacturer’s instructions. A-498 and Caki-1 cells were treated with different concentrations of 6a (0.75 and 1.5 μM) or DMSO as a negative control and incubated for 24 hrs. Flow cytometric analysis was conducted using FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Quantitative analysis of the FACS data was performed with FlowJo software (FlowJo, Ashland, OR, USA).
Resistance development assays
To examine whether A-498 cells can develop resistance to 6a or valproic acid, A-498 cells were exposed to 30% of IC50 of 6a or valproic acid for 12 weeks, followed by treatment with different concentrations of 6a (0.25, 0.5, 1, 5, and 10 μM) or valproic acid (VPA; 0.25, 0.5, 1, 2, and 4 mM) or DMSO as a negative control for 24 h. Then, the cells were subjected to the MTT assay to detect cell growth inhibition or to the Western blot assay to detect HDAC2, p-FAK, FAK, p-Akt, and Akt proteins expression. Control cell cultures were not pretreated with 6a or VPA.
Determination of caspase-8 & caspase-9
The determination of caspase-8 and caspase-9 was performed using human caspase-8 ELISA kit (EIA-4863) and human caspase-9 ELISA kit DRG® (EIA-4860) (DRG International Inc., USA). Cells were lysed with cell extraction buffer. The lysate was diluted in standard diluent buffer over the range of the assay and measured for human caspase-8 and caspase-9 content, separately. For each assay, the cells were plated in a density of 1.2–1.8 × 10,000 cells/well in a volume of 100 μl complete growth medium +100 μL of the tested compound per well in a 96-well plate for 24 h before the enzyme assay for caspase-8 and caspase-9 according to the manufacturer’s protocol. Finally, the absorbance of each microwell was read using a microplate reader set at 450 nm.
Determination of caspase-3
The level of human active caspase-3 protein was evaluated using Invitrogen (Catalog KHO1091) ELISA kit. The manufacturer’s instructions were followed in the following procedures. Add 100 μL of the standard diluent buffer to the zero standard wells. Add 100 μL of standards and controls or diluted samples to the appropriate microtitre wells. Cover wells and incubate for 2 h at room temperature. Thoroughly aspirate or decant solution from wells and discard the liquid. Pipette 100 μL of caspase-3 (active) detection antibody solution into each well. Cover plate and incubate for 1 h at room temperature. Add 100 μL Anti-Rabbit IgG HRP working solution to each well. Cover wells with the plate cover and incubate for 30 min at room temperature. Add 100 μL of stabilised chromogen to each well. The liquid in the wells will begin to turn blue. Incubate for 30 min at room temperature. Stop solution has been added to each well. The solution in the wells should change from blue to yellow. Read the plate within 2 h after adding the stop solution. Use a curve fitting software to generate the standard curve.
Docking study
Molecular modeling and visualization processes were performed within the colchicine binding site of β-subunit of tubulin using Molecular Operating Environment (MOE) 2019.0102 software (Chemical Computing Group, Montreal, QC, Canada). The co-crystal structure was retrieved from the RCSB Protein Data Bank (PDB code 5LYJ). First, 6a was prepared with the standard protocol designated in MOE 2019. However, the 6a structure’s energy was minimized using Amber10:EHT forcefield with a gradient RMSD of 0.0001kcal/mol. Then the protein structures were prepared by using the MOE QickPrep protocol. To validate the docking study at the binding sites, the native ligands were re-docked into the binding site using the same set of parameters as described above. The RMSD values of the best-docked poses were 0.8172 Å (SAHA) and 0.8762 Å (TAE226) in HDAC2 and FAK active sites, respectively, thus validating the docking using MOE. Compound 6a was then docked into the binding sites using the Alpha triangle placement method. Refinement was carried out using Forcefield and scored using the Affinity dG scoring system. The resulting docking poses were visually inspected, and the pose of the lowest binding free energy value was considered.
Statistical analysis
Data are represented as means ± SEM. Statistical significance was assessed using one-way analysis of variance (ANOVA) including a Tukey’s test for multiple comparisons. *p < 0.05, **p < 0.01, and ***p < 0.001. The data shown in the figures are representative data for three independent experimental results.
Supplementary Material
Acknowledgements
A.A.A was supported by an NIH-funded Cancer Therapeutics Training Program (CT2, T32 CA121938) and P.G by the NIH (CA238042, CA100768 and CA160911).
References
- [1].Anighoro A, Bajorath J, Rastelli G, Polypharmacology: challenges and opportunities in drug discovery: miniperspective, Journal of medicinal chemistry, 57 (2014) 7874–7887. [DOI] [PubMed] [Google Scholar]
- [2].Hesham HM, Lasheen DS, Abouzid KA, Chimeric HDAC inhibitors: Comprehensive review on the HDAC‐based strategies developed to combat cancer, Medicinal Research Reviews, 38 (2018) 2058–2109. [DOI] [PubMed] [Google Scholar]
- [3].Yun F, Cheng C, Ullah S, Yuan Q, Design, synthesis and biological evaluation of novel histone Deacetylase1/2 (HDAC1/2) and cyclin-dependent Kinase2 (CDK2) dual inhibitors against malignant cancer, European Journal of Medicinal Chemistry, (2020) 112322. [DOI] [PubMed] [Google Scholar]
- [4].kA Bass A, El-Zoghbi MS, Nageeb E-SM, Mohamed MF, Bader M, Abuo-Rahma GE-DA, Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors, European Journal of Medicinal Chemistry, (2020) 112904. [DOI] [PubMed] [Google Scholar]
- [5].Sangwan R, Rajan R, Mandal PK, HDAC as onco target: reviewing the synthetic approaches with SAR study of their inhibitors, European journal of medicinal chemistry, 158 (2018) 620–706. [DOI] [PubMed] [Google Scholar]
- [6].Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche F, Niesporek S, Denkert C, Dietel M, Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy, British journal of cancer, 98 (2008) 604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, Increased expression of histone deacetylase 2 is found in human gastric cancer, Apmis, 113 (2005) 264–268. [DOI] [PubMed] [Google Scholar]
- [8].Mutze K, Langer R, Becker K, Ott K, Novotny A, Luber B, Hapfelmeier A, Göttlicher M, Höfler H, Keller G, Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in gastric cancer, Annals of surgical oncology, 17 (2010) 3336–3343. [DOI] [PubMed] [Google Scholar]
- [9].Li L, Mei T, Zeng Y, HDAC2 promotes the migration and invasion of non-small cell lung cancer cells via upregulation of fibronectin, Biomedicine & Pharmacotherapy, 84 (2016) 284–290. [DOI] [PubMed] [Google Scholar]
- [10].Ler SY, LEuNG CHW, Khin LW, Lu G-D, Salto-Tellez M, Hartman M, IAu PTC, Yap CT, Hooi SC, HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a Southeast Asian population, Oncology reports, 34 (2015) 2238–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kiweler N, Brill B, Wirth M, Breuksch I, Laguna T, Dietrich C, Strand S, Schneider G, Groner B, Butter F, Heinzel T, Brenner W, Krämer OH, The histone deacetylases HDAC1 and HDAC2 are required for the growth and survival of renal carcinoma cells, Archives of Toxicology, 92 (2018) 2227–2243. [DOI] [PubMed] [Google Scholar]
- [12].Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ, Xie HJ, Chang YG, Kim MG, Park WS, Lee JY, Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcinoma cells by deregulating expression of G1/S cell cycle proteins, PloS one, 6 (2011) e28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang B, Laban M, Leung CH, Lee L, Lee C, Salto-Tellez M, Raju G, Hooi S, Inhibition of histone deacetylase 2 increases apoptosis and p21 Cip1/WAF1 expression, independent of histone deacetylase 1, Cell Death & Differentiation, 12 (2005) 395–404. [DOI] [PubMed] [Google Scholar]
- [14].Kong D, Chen F, Sima N, Inhibition of focal adhesion kinase induces apoptosis in bladder cancer cells via Src and the phosphatidylinositol 3-kinase/Akt pathway, Experimental and therapeutic medicine, 10 (2015) 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lv J, Bai R, Wang L, Gao J, Zhang H, Artesunate may inhibit liver fibrosis via the FAK/Akt/β-catenin pathway in LX-2 cells, BMC Pharmacology and Toxicology, 19 (2018) 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gonneaud A, Turgeon N, Jones C, Couture C, Lévesque D, Boisvert F-M, Boudreau F, Asselin C, HDAC1 and HDAC2 independently regulate common and specific intrinsic responses in murine enteroids, Scientific reports, 9 (2019) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Visavadiya NP, Keasey MP, Razskazovskiy V, Banerjee K, Jia C, Lovins C, Wright GL, Hagg T, Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3, Cell Communication and Signaling, 14 (2016) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Keasey MP, Kang SS, Lovins C, Hagg T, Inhibition of a novel specific neuroglial integrin signaling pathway increases STAT3-mediated CNTF expression, Cell Communication and Signaling, 11 (2013) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jin KL, Park J-Y, Noh EJ, Hoe KL, Lee JH, Kim J-H, Nam J-H, The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells, Journal of gynecologic oncology, 21 (2010) 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bhatia S, Krieger V, Groll M, Osko JD, Reßing N, Ahlert H, Borkhardt A, Kurz T, Christianson DW, Hauer J, Discovery of the first-in-class dual histone deacetylase–proteasome Inhibitor, Journal of medicinal chemistry, 61 (2018) 10299–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Peng X, Sun Z, Kuang P, Chen J, Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment, Eur J Med Chem, 208 (2020) 112831. [DOI] [PubMed] [Google Scholar]
- [22].Sulzmaier FJ, Jean C, Schlaepfer DD, FAK in cancer: mechanistic findings and clinical applications, Nature reviews cancer, 14 (2014) 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Béraud C, Dormoy V, Danilin S, Lindner V, Béthry A, Hochane M, Coquard C, Barthelmebs M, Jacqmin D, Lang H, Targeting FAK scaffold functions inhibits human renal cell carcinoma growth, International journal of cancer, 137 (2015) 1549–1559. [DOI] [PubMed] [Google Scholar]
- [24].Lu Y, Sun H, Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK), Journal of Medicinal Chemistry, (2020). [DOI] [PubMed] [Google Scholar]
- [25].Ott GR, Cheng M, Learn KS, Wagner J, Gingrich DE, Lisko JG, Curry M, Mesaros EF, Ghose AK, Quail MR, Discovery of clinical candidate CEP-37440, a selective inhibitor of focal adhesion kinase (FAK) and anaplastic lymphoma kinase (ALK), Journal of medicinal chemistry, 59 (2016) 7478–7496. [DOI] [PubMed] [Google Scholar]
- [26].Mustafa M, Abuo-Rahma GEA, Abd El-Hafeez AA, Ahmed ER, Abdelhamid D, Ghosh P, Hayallah AM, Discovery of antiproliferative and anti-FAK inhibitory activity of 1,2,4-triazole derivatives containing acetamido carboxylic acid skeleton, Bioorg Med Chem Lett, 40 (2021) 127965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mustafa M, Abdelhamid D, Abdelhafez EM, Ibrahim MA, Gamal-Eldeen AM, Aly OM, Synthesis, antiproliferative, anti-tubulin activity, and docking study of new 1, 2, 4-triazoles as potential combretastatin analogues, European journal of medicinal chemistry, 141 (2017) 293–305. [DOI] [PubMed] [Google Scholar]
- [28].Mustafa M, Anwar S, Elgamal F, Ahmed ER, Aly OM, Potent combretastatin A-4 analogs containing 1, 2, 4-triazole: Synthesis, antiproliferative, anti-tubulin activity, and docking study, European journal of medicinal chemistry, 183 (2019) 111697. [DOI] [PubMed] [Google Scholar]
- [29].Zhang Y-B, Liu W, Yang Y-S, Wang X-L, Zhu H-L, Bai L-F, Qiu X-Y, Synthesis, molecular modeling, and biological evaluation of 1, 2, 4-triazole derivatives containing pyridine as potential anti-tumor agents, Medicinal Chemistry Research, 22 (2013) 3193–3203. [Google Scholar]
- [30].Aboeldahab AM, Beshr EA, Shoman ME, Rabea SM, Aly OM, Spirohydantoins and 1, 2, 4-triazole-3-carboxamide derivatives as inhibitors of histone deacetylase: Design, synthesis, and biological evaluation, European journal of medicinal chemistry, 146 (2018) 79–92. [DOI] [PubMed] [Google Scholar]
- [31].Wen J, Bao Y, Niu Q, Yang J, Fan Y, Li J, Jing Y, Zhao L, Liu D, Identification of N-(6-mercaptohexyl)-3-(4-pyridyl)-1H-pyrazole-5-carboxamide and its disulfide prodrug as potent histone deacetylase inhibitors with in vitro and in vivo anti-tumor efficacy, European journal of medicinal chemistry, 109 (2016) 350–359. [DOI] [PubMed] [Google Scholar]
- [32].Tang G, Wong JC, Zhang W, Wang Z, Zhang N, Peng Z, Zhang Z, Rong Y, Li S, Zhang M, Identification of a novel aminotetralin class of HDAC6 and HDAC8 selective inhibitors, Journal of medicinal chemistry, 57 (2014) 8026–8034. [DOI] [PubMed] [Google Scholar]
- [33].Ghosh B, Zhao W-N, Reis SA, Patnaik D, Fass DM, Tsai L-H, Mazitschek R, Haggarty SJ, Dissecting structure–activity-relationships of crebinostat: Brain penetrant HDAC inhibitors for neuroepigenetic regulation, Bioorganic & medicinal chemistry letters, 26 (2016) 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Neel VA, Todorova K, Wang J, Kwon E, Kang M, Liu Q, Gray N, Lee SW, Mandinova A, Sustained Akt activity is required to maintain cell viability in seborrheic keratosis, a benign epithelial tumor, Journal of Investigative Dermatology, 136 (2016) 696–705. [DOI] [PubMed] [Google Scholar]
- [35].Abuo-Rahma GE-DA, Abdel-Aziz M, Beshr EA, Ali TF, 1, 2, 4-Triazole/oxime hybrids as new strategy for nitric oxide donors: Synthesis, anti-inflammatory, ulceroginicity and antiproliferative activities, European journal of medicinal chemistry, 71 (2014) 185–198. [DOI] [PubMed] [Google Scholar]
- [36].Mohamed MF, Shaykoon MSA, Abdelrahman MH, Elsadek BE, Aboraia AS, Abuo-Rahma GE-DA, Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors, Bioorganic Chemistry, 72 (2017) 32–41. [DOI] [PubMed] [Google Scholar]
- [37].Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer, BMC cancer, 8 (2008) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ahmadzadeh A, Khodadi E, Shahjahani M, Bertacchini J, Vosoughi T, Saki N, The role of HDACs as leukemia therapy targets using HDI, International journal of hematology-oncology and stem cell research, 9 (2015) 203. [PMC free article] [PubMed] [Google Scholar]
- [39].Stypula-Cyrus Y, Damania D, Kunte DP, Cruz MD, Subramanian H, Roy HK, Backman V, HDAC up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure, PloS one, 8 (2013) e64600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barbero S, Mielgo A, Torres V, Teitz T, Shields DJ, Mikolon D, Bogyo M, Barilà D, Lahti JM, Schlaepfer D, Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis, Cancer research, 69 (2009) 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shankar S, Singh TR, Fandy TE, Luetrakul T, Ross DD, Srivastava RK, Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: involvement of both death receptor and mitochondrial pathways, International journal of molecular medicine, 16 (2005) 1125–1138. [PubMed] [Google Scholar]
- [42].Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability, Journal of Biological Chemistry, 288 (2013) 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lietha D, Eck MJ, Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation, PloS one, 3 (2008) e3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.