

Abstract
Background
Muscle weakness is a frequently occurring complication of sepsis, associated with increased morbidity and mortality. Interestingly, obesity attenuates sepsis‐induced muscle wasting and weakness. As the adipokine leptin is strongly elevated in obesity and has been shown to affect muscle homeostasis in non‐septic conditions, we aimed to investigate whether leptin mediates the protective effect of obesity on sepsis‐induced muscle weakness.
Methods
In a mouse model of sepsis, we investigated the effects of genetic leptin inactivation in obese mice (leptin‐deficient obese mice vs. diet‐induced obese mice) and of leptin supplementation in lean mice (n = 110). We assessed impact on survival, body weight and composition, markers of muscle wasting and weakness, inflammation, and lipid metabolism. In human lean and overweight/obese intensive care unit (ICU) patients, we assessed markers of protein catabolism (n = 1388) and serum leptin (n = 150).
Results
Sepsis mortality was highest in leptin‐deficient obese mice (53% vs. 23% in diet‐induced obese mice and 37% in lean mice, P = 0.03). Irrespective of leptin, after 5 days of sepsis, lean mice lost double the amount of lean body mass than obese mice (P < 0.0005). Also, irrespective of leptin, obese mice maintained specific muscle force up to healthy levels (P = 0.3) whereas lean mice suffered from reduced specific muscle force (72% of healthy controls, P < 0.0002). As compared with lean septic mice, both obese septic groups had less muscle atrophy, liver amino acid catabolism, and inflammation with a 50% lower plasma TNFα increase (P < 0.005). Conversely, again mainly irrespective of leptin, obese mice lost double amount of fat mass than lean mice after 5 days of sepsis (P < 0.0001), showed signs of increased lipolysis and ketogenesis, and had higher plasma HDL and LDL lipoprotein concentrations (P ≤ 0.01 for all). Muscle fibre type composition was not altered during sepsis, but a higher atrophy sensitivity of type IIb fibres compared with IIa and IIx fibres was observed, independent of obesity or leptin. After 5 days of critical illness, serum leptin was higher (P < 0.0001) and the net waste of nitrogen (P = 0.006) and plasma urea‐to‐creatinine ratio (P < 0.0001) was lower in overweight/obese compared with lean ICU human patients.
Conclusions
Leptin did not mediate the protective effect of obesity against sepsis‐induced muscle wasting and weakness in mice. Instead, obesity—independent of leptin—attenuated inflammation, protein catabolism, and dyslipidaemia, pathways that may play a role in the observed muscle protection.
Keywords: Leptin, Critical illness, Sepsis, Muscle weakness, Intensive care unit acquired weakness, Atrophy, Protein catabolism
Introduction
Intensive care unit acquired weakness (ICUAW) is a debilitating complication of critical illness, with no other plausible aetiology besides the illness itself. It affects both the respiratory and peripheral muscles, hampers recovery, and increases the risk of late death. 1 The prevalence of ICUAW is around 40% but varies depending on the population studied. Sepsis, multiple organ failure, high severity of illness, prolonged illness, and mechanical ventilation are the most important risk factors for the development of ICUAW. 1 , 2 Currently, clinical management is limited to preventive measures avoiding modifiable risk factors because effective treatments for ICUAW are still lacking.
Remarkably, obese critically ill patients and obese septic mice are protected against muscle wasting and weakness as compared with their lean counterparts, irrespective of whether they received nutrition or were fasted. 3 Large observational studies also have shown that premorbid obesity, although associated with the development of chronic comorbidities and premature death in the general population, is associated with a lower mortality rate in critically ill patients. 4 , 5 This phenomenon is called the obesity paradox of critical illness and is also observed in chronic illnesses like end‐stage renal disease, heart failure, and coronary artery disease. The mechanisms by which obesity might protect against muscle wasting and weakness are not clear, but an increased mobilization of fat from adipose tissue, and subsequent increased hepatic fatty acid oxidation and ketogenesis have shown to play an important role. 6 , 7
Besides its role in fat storage, adipose tissue also produces a variety of signalling molecules, adipokines, with important functions in the regulation of appetite, energy expenditure, glucose and lipid metabolism, and inflammation. Altered production of these adipokines in obese patients might be an additional mechanism by which excess adipose tissue protects against ICUAW. Leptin, an adipokine whose expression and secretion correlates strongly with the total amount of adipose tissue, has a pleiotropic role in the transition from the fed to fasted state and protects the organism against excessive energy deprivation. 8 In animal models of infection and sepsis, leptin‐deficient mice displayed an increased mortality, 9 , 10 , 11 whereas leptin supplementation in septic mice has shown conflicting results, both improving and worsening survival. 11 , 12 , 13 Importantly, leptin can promote muscle proliferation and growth while decreasing muscle atrophy, both in an insulin‐dependent and insulin‐independent fashion. 14 , 15 , 16 The interaction between obesity, leptin, and sepsis‐induced muscle weakness however is currently not well studied. Therefore, we here investigated whether leptin plays a role in the protective effect of obesity on muscle wasting and weakness in a validated mouse model of sepsis‐induced muscle weakness. We assessed both the impact of leptin deficiency in obese mice and leptin supplementation in lean mice in a mouse model of sepsis. In addition, we assessed serum leptin levels and net protein breakdown in lean and overweight/obese critically ill patients.
Methods
Animal study design
Male B6.V‐Lepob/ob/JRj (Ob‐KO) and male C57BL/6J mice were purchased at the age of 6 weeks (Janvier). Upon arrival, C57BL/6J mice were randomized to a high‐fat diet (60 KJ% fat, E15742‐34, ssniff) or regular chow (9 KJ% fat, V1535‐000) ad libitum, to generate diet‐induced obese mice (Ob‐DIO) and lean mice (Ln), respectively. Leptin‐deficient mice (Ob‐KO) received a control diet (10 KJ% fat, E157453) ad libitum but were calorie‐restricted from Week 11 to Week 16, to be weight matched with the Ob‐DIO mice (Figure 1A). At Weeks 17–18 (Figure 1B), Ob‐DIO and Ob‐KO obese mice were randomized to either healthy control or sepsis and lean mice were randomized to healthy control, septic placebo‐treated mice (Ln), or septic leptin‐treated mice (Ln‐lep). Ln‐lep mice received leptin treatment (1 mg/kg/day), divided in two doses per day. 14 Mice randomized to the sepsis group were anaesthetized, and a catheter was placed in the left central jugular vein, followed by caecal ligation and puncture (CLP) to induce sepsis. After surgery, mice received intravenous fluid resuscitation and after 24 h, standard mixed parenteral nutrition. Throughout the study, mice received antibiotics and analgesics. Pain/discomfort was assessed twice daily, and cumulative illness scores were calculated to assess illness severity. Mice randomized to healthy controls were individually caged receiving standard chow ad libitum. Mice were sacrificed 125 h (5 days) after CLP. The Institutional Ethics Committee for Animal Experimentation of the KU Leuven approved the protocol (Internal Project Number P181‐2016). More details on the set‐up can be found in the Supporting Information.
Figure 1.
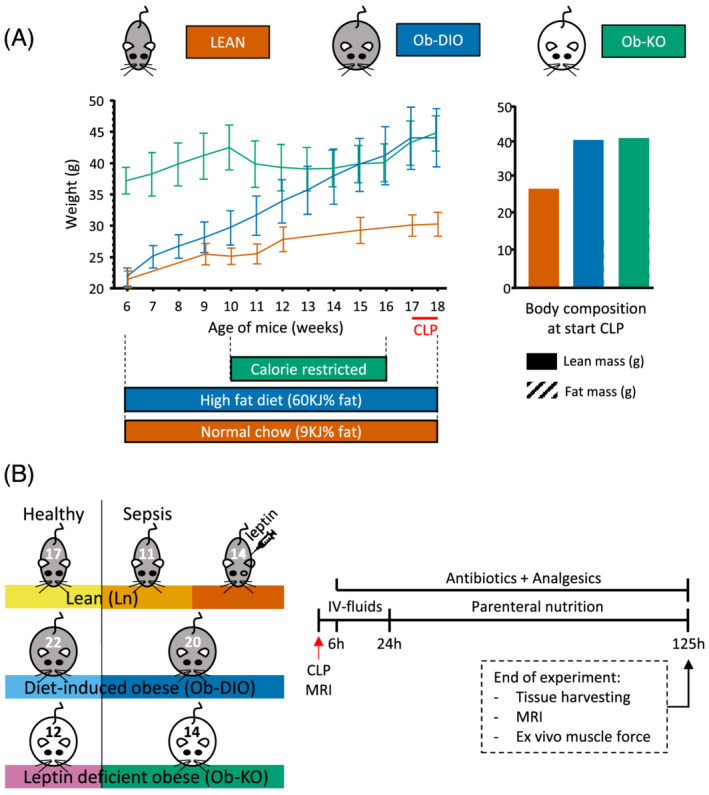
Experimental set‐up of the study. (A) Diet and weight set‐up prior to CLP. Prior to sepsis, diet‐induced obese mice (Ob‐DIO) and leptin‐deficient obese mice (Ob‐KO) were similar in body weight (43.9 ± 4.7 and 44.4 ± 2.8 g, respectively, P = 0.5) but 46% heavier than lean mice (30.2 ± 1.9 g). Ob‐KO mice started with on average 7.2 g more fat mass but less lean mass than Ob‐DIO mice (P < 0.0001). (B) Experimental set‐up of the sepsis model.
Body composition analysis and ex vivo muscle force measurements
Body composition was measured immediately before CLP and immediately before sacrifice, at 125 h after CLP, with magnetic resonance imaging (echoMRI‐100H, Whole Body Magnetic Resonance Analyser, Zinsser Analytic GmbH). Directly after euthanasia, the hindlimb m. extensor digitorum longus (EDL) was carefully dissected to measure muscle force (300C‐LR Dual‐Mode muscle lever, Aurora Scientific). 3 More details on the methods can be found in the Supporting Information.
Blood and tissue analyses
Blood glucose and ketone concentrations were measured on whole blood drawn from the tail vein with Accu‐Check (Roche) and StatStrip Xpress 2 (Nova Biomedical), respectively. Plasma urea nitrogen (EIABUN, Invitrogen), free fatty acids (7010310, Cayman Chemical Company), glycerol (MAK117, Sigma‐Aldrich), LDL and HDL cholesterol (DZ128A‐K and DZ129A‐K, Diazyme Laboratories), TNFα (MHSTA50, R&D Systems), IL‐6 (M6000B, R&D Systems), corticosterone (EIA5186, DRG), and leptin (EZML82K, Millipore) were measured using commercial assays. From liver and muscle tissue samples, isolated RNA was reverse‐transcribed and relative gene expression was determined with the 2−ΔΔCt method with 18S ribosomal RNA (Rn18s) as housekeeping gene (Applied Biosystems). More details on the methods and an overview of the gene expression assays used for markers of muscle wasting and weakness, inflammation, and lipid metabolism are provided in the Supporting Information. For protein analysis, western blots were performed on tissue homogenates with primary antibodies for markers of autophagy LC3B (L7543, Sigma‐Aldrich) and p62 (H00008878‐M01, Novus Biologicals), and protein synthesis p70 S6 kinase (#9202 and #9205, Cell Signaling). For histology purposes, paraformaldehyde‐fixed paraffin embedded muscle (right tibialis anterior) and liver sections were stained with haematoxylin and eosin and scored semi‐quantitatively for structural changes. For muscle fibre type staining, cryosections of the left tibialis anterior muscle were stained with fibre type‐specific primary antibodies. Whole slides were scanned with TissueFAXS i PLUS microscope (TissueGnostics, Vienna, Austria) and analysed with MuscleJ software. More details on the methods can be found in the Supporting Information.
Assessment of protein breakdown and plasma leptin in human patients
We performed a post hoc analysis of the EPaNIC study, a large randomized trial that demonstrated that early parenteral nutrition to supplement insufficient enteral nutrition increased morbidity in the intensive care unit (ICU). 17 In these patients (n = 4640), protein breakdown during ICU stay was assessed by the plasma urea‐to‐creatinine ratio and by calculating the net nitrogen loss as the difference in nutritional nitrogen intake and the calculated nitrogen loss using individual plasma and urinary urea concentrations. 18 The study followed the Declaration of Helsinki and had been approved by the Institutional Review Board of the KU Leuven. Written informed consent was obtained from the patient or legal guardian. For the current study, we compared the urea‐to‐creatinine ratio and net nitrogen loss in lean and obese prolonged critically ill patients on ICU Day 5. To account for selection bias due to baseline differences between lean and overweight/obese patients, propensity score matching was performed. From critically ill patients with a minimum ICU stay of 5 days, and for whom nitrogen balance data were available (n = 1477), 694 of the 697 patients with a body mass index (BMI) < 25 (lean patients) were matched to 694 of the 780 patients with a BMI ≥ 25 (overweight/obese patients) (Supporting Information, Table S2). In addition, in 150 patients of this matched set (81 lean and 69 overweight/obese patients), serum leptin concentrations on admission and on Day 5 of ICU stay were available from an earlier post hoc analysis. 19
Statistical analysis
Data were compared with Kruskal–Wallis and Kaplan–Meier survival curves were compared using log‐rank test. Two‐sided P‐values below 0.05 were considered statistically significant. In mice, additional post hoc each pair Wilcoxon rank‐sum tests were performed to compare each septic group with its own healthy control group, to compare high leptin conditions with low leptin conditions, and to compare lean with obese conditions. No corrections for multiple comparisons were performed. Association of investigated markers with muscle wasting and muscle weakness was performed with Spearman's rank analysis. For the human data, propensity scores were estimated using logistic regression with age and APACHE‐II score upon admission as continuous covariates and randomization to early or late parenteral nutrition, gender, nutritional risk score, history of malignancy, and diagnostic category upon admission as categorical covariates (IBM SPSS Statistics Version 28.0.0.0). After estimation of the propensity scores, 1:1 nearest neighbour matching was performed, using a caliper of 0.2, which is the maximum allowable difference between two cases on their estimated propensity scores, defined in units of standard deviations of the logit of the estimated propensity score. Data of the matched patients were compared with Kruskal–Wallis. Data are presented as median (interquartile range, IQR), as mean (±standard deviation), or as numbers and percentages. When presented as box plots, the horizontal line within the box represents the median, the box the 25th and 75th quantiles, and the whiskers being drawn to the furthest point within 1.5 times the IQR from the box. Bar graphs and point curves represent the mean and the error bars represent the standard deviation. Statistical analyses were performed with JMP Pro 15 (SAS Institute Inc, Cary, NC, USA).
Results
Body composition prior to sepsis and plasma leptin values
Prior to sepsis, diet‐induced obese mice (Ob‐DIO) and leptin‐deficient obese mice (Ob‐KO) were matched for body weight (mean ± standard deviation 43.9 ± 4.7 and 44.4 ± 2.8 g, respectively, P = 0.5) but were 46% heavier than lean mice (30.2 ± 1.9 g). This difference in weight was mainly explained by a higher fat mass in obese compared with lean mice (P < 0.0001 for all, Figure 1A). Although similar in total body weight, Ob‐KO mice started with more fat mass but less lean body mass than Ob‐DIO mice (P < 0.0001 for both, Figure 1A).
In pre‐sepsis healthy mice, plasma leptin was undetectable in Ob‐KO mice and higher in Ob‐DIO mice than in lean mice (P < 0.0001, Figure 2A). After 5 days of sepsis, Ob‐DIO mice were not significantly different from their pre‐sepsis plasma values (P = 0.1), whereas plasma leptin values decreased in lean placebo‐treated mice (Ln, P = 0.0007). Septic lean mice who received exogenous leptin (Ln‐Lep) showed increased plasma leptin values up to those of Ob‐DIO, as expected (Figure 2A).
Figure 2.
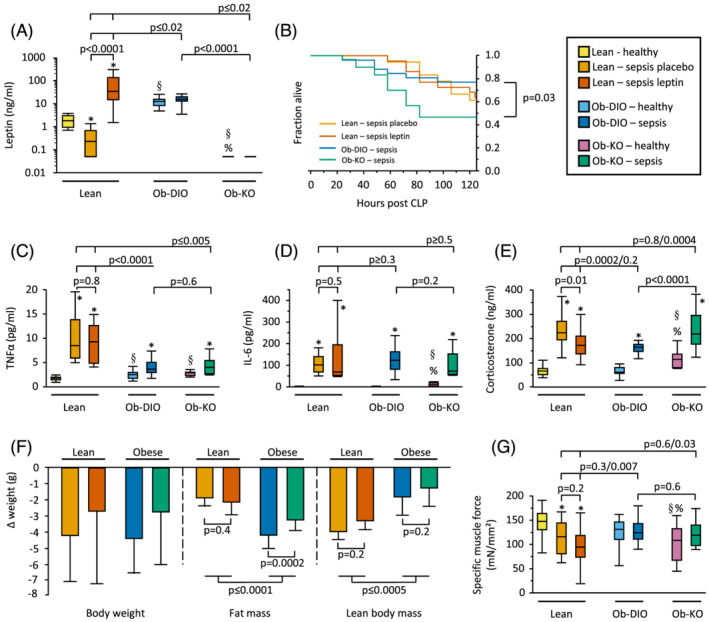
Plasma leptin, mortality, inflammation, body composition, and muscle force. (A) Plasma leptin levels (Kruskal–Wallis P < 0.0001). (B) Survival curves of septic mice, Ob‐DIO vs. Ob‐KO: P = 0.03, other groups not statistically different. Lean sepsis placebo n = 11/18, lean sepsis leptin n = 14/22, Ob‐DIO sepsis n = 20/26, Ob‐KO sepsis n = 14/30. (C) Plasma TNFα (Kruskal–Wallis P < 0.0001). (D) Plasma Il‐6 (Kruskal–Wallis P < 0.0001). (E) Plasma corticosterone (Kruskal–Wallis P < 0.0001). (F) Loss of total body weight (Kruskal–Wallis P = 0.2), fat mass (Kruskal–Wallis P < 0.0001), and lean body mass (Kruskal–Wallis P < 0.0001). (G) Specific muscle force of the right extensor digitorum longus (Kruskal–Wallis P = 0.004). (A, C–G) Lean healthy n = 17, lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. *P < 0.05 between septic mice and healthy controls; § P < 0.05 compared with lean healthy mice; % P < 0.05 compared with healthy Ob‐DIO mice.
Effect of obesity vs. leptin on severity of illness, glucose levels, and inflammation
After 5 days of sepsis, mortality was highest in Ob‐KO mice (P = 0.03 vs. Ob‐DIO), but not significantly different in Ln, Ln‐lep, and Ob‐DIO mice (Figure 2B). Similarly, severity of illness scores in surviving animals were highest in Ob‐KO mice (P = 0.03 vs. Ob‐DIO), but not significantly different in Ln, Ln‐lep, and Ob‐DIO mice (P > 0.5). Blood glucose was elevated prior to sepsis in Ob‐DIO mice only (P < 0.05, Figure S1). Blood glucose levels fell during the acute phase of sepsis and rose thereafter in all septic groups, but rose to higher levels in obese mice, irrespective of leptin (P < 0.05, Figure S1).
Plasma TNFα, which was elevated in pre‐sepsis in healthy Ob‐DIO and Ob‐KO mice, increased to a much higher extent during sepsis in lean mice compared with obese mice, irrespective of leptin (P ≤ 0.005, Figure 2C). Plasma IL‐6, which was elevated in pre‐sepsis healthy Ob‐KO mice, increased in all septic groups, irrespective of leptin (Figure 2D). Plasma corticosterone, which was elevated in pre‐sepsis healthy Ob‐KO mice, increased in all septic groups after 5 days of illness, but to a higher extent in the ‘low leptin’ Ln and Ob‐KO mice as compared with the ‘high leptin’ Ln‐lep and Ob‐DIO mice (P ≤ 0.01, Figure 2E).
Effect of obesity vs. leptin on changes in body composition and muscle force
Over 5 days of illness, all septic groups lost a comparable amount of body weight (Figure 2F). However, obese mice lost double the amount of fat mass as compared with lean mice (P < 0.0001, Figure 2F). This loss in fat mass was more pronounced in Ob‐DIO mice as compared with Ob‐KO mice (P = 0.0002) but was not affected by leptin supplementation in lean mice (P = 0.4). In contrast, lean mice lost double the amount of lean body mass as compared with obese mice (P < 0.0005), but this loss was not affected by leptin in lean or obese mice (P = 0.2, Figure 2F).
Functionally, the specific muscle force of Ob‐DIO and Ob‐KO mice was not significantly different from their healthy controls (98.8% and 116.5%, respectively), whereas Ln and Ln‐Lep mice suffered from reduced specific muscle force (77.9% and 65.4% of lean healthy controls, respectively, P ≤ 0.01, Figure 2G).
Effect of obesity vs. leptin on muscle protein breakdown and inflammation
Muscle mass is determined by a balance between protein synthesis (anabolism) and protein breakdown (catabolism). We first investigated whether the more severe loss of lean body mass in lean mice was due to an increase in protein breakdown. Plasma urea, as a marker for protein breakdown, was higher in septic lean mice compared with septic obese mice, irrespective of leptin (P ≤ 0.002, Figure 3A). Hepatic gene expression of amino acid catabolic enzymes was higher in septic lean mice compared with septic Ob‐DIO mice (Aass, Arg1, Got1, Sds) and septic Ob‐KO mice (Aass, Oat) (Figure 3B). Additionally, muscle gene expression of atrophy markers Fbxo32, Trim63, Ubb, and Ubc increased in all septic groups, but more so in septic lean mice compared with septic Ob‐DIO mice (Trim63, Ubb) and septic Ob‐KO mice (Ubc) (Figure 3C). In contrast, muscle proteasome activity was lower in septic lean mice compared with healthy control and septic obese mice (Figure 3D). The autophagosome marker LC3II increased in all septic groups with a higher expression in Ln‐lep compared with Ob‐DIO mice (Figure 3E). The protein level of the autophagy substrate P62 increased in both septic lean groups and in Ob‐DIO mice, but not in the OB‐KO mice. Muscle cathepsin activity was elevated in pre‐sepsis healthy Ob‐KO mice. Sepsis increased muscle cathepsin activity in lean and obese mice, irrespective of leptin (Figure 3E).
Figure 3.
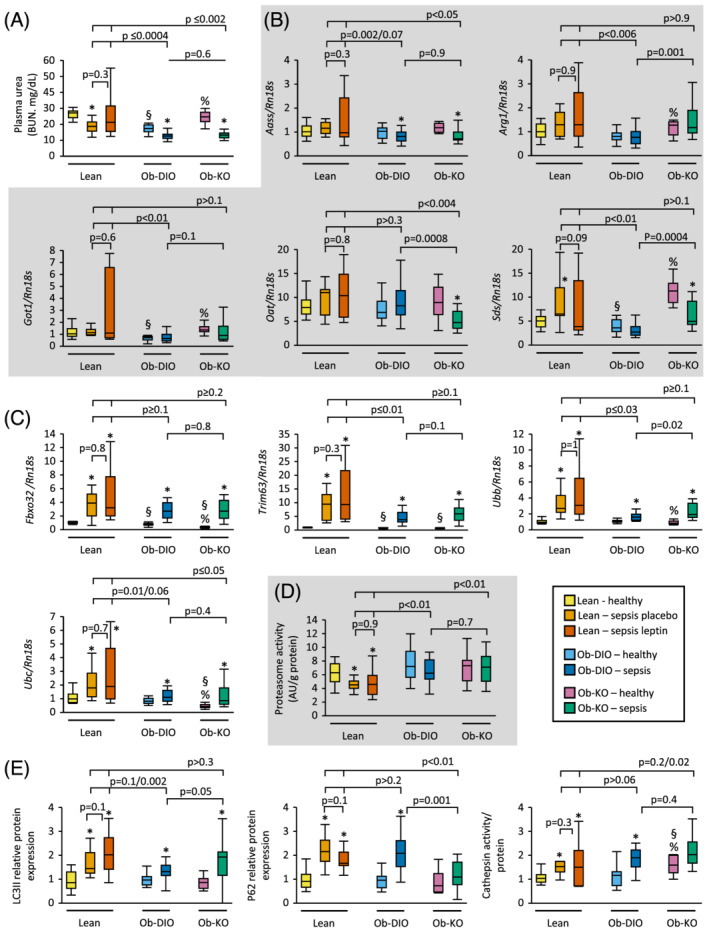
Amino acid catabolism and muscle protein breakdown. (A) Blood urea nitrogen (Kruskal–Wallis P < 0.0001). (B) Relative mRNA expression of enzymes involved in amino acid catabolism in the liver (Kruskal–Wallis P ≤ 0.005). (C) Relative mRNA expression of markers of atrophy in gastrocnemius muscle (GNM) (Kruskal–Wallis P < 0.0001). (D) GNM proteasome activity (Kruskal–Wallis P = 0.002). (E) Relative GNM protein expression of LC3II, P62, and muscle cathepsin activity (Kruskal–Wallis P < 0.0001). Gene and protein expression data are shown relative the median of lean healthy mice. Lean healthy n = 17, lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. *P < 0.05 between septic mice and healthy controls; § P < 0.05 compared with lean healthy mice; % P < 0.05 compared with healthy Ob‐DIO mice.
A major regulator of muscle atrophy is inflammation; for this reason, we examined changes in a panel of inflammatory markers. Sepsis induced gene expression of inflammatory markers Il‐1β and Nlrp3 in all groups (Figure 4). However, gene expression of Tnfα and Il‐6 increased in septic lean mice but did not increase in obese mice or showed an attenuated rise in Ob‐KO mice, respectively.
Figure 4.
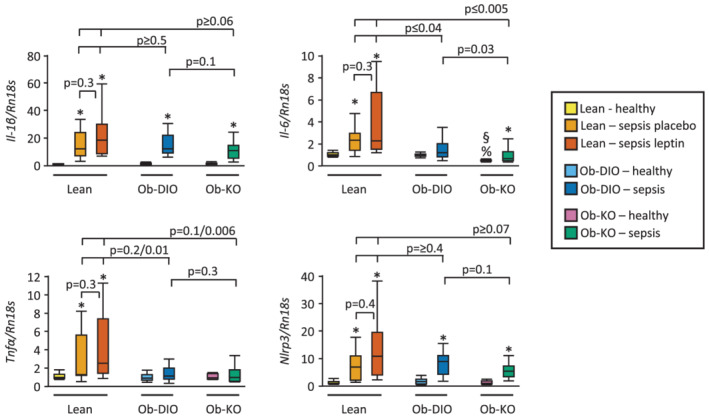
Muscle inflammation. Relative mRNA expression of inflammatory markers IL‐1β, Il‐6, TNFα, and the inflammasome Nlrp3 in gastrocnemius muscle (Kruskal–Wallis P ≤ 0.004). Data are shown relative the median of lean healthy mice. Lean healthy n = 17, lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. *P < 0.05 between septic mice and healthy controls; § P < 0.05 compared with lean healthy mice; % P < 0.05 compared with healthy Ob‐DIO mice.
Effect of obesity vs. leptin on muscle protein synthesis and proliferation
To evaluate muscle protein synthesis, we quantified muscle gene expression of the contractile proteins myosin and actin. Gene expression of Mhc type I was not affected by sepsis, obesity, or leptin, while gene expression of Mhc type IIa and IIx increased in lean placebo‐treated septic mice only (Figure 5A). In contrast, gene expression of Mhc type IIb decreased after 5 days of sepsis in all groups. Gene expression of Acta1 was not affected by sepsis, obesity, or leptin (Figure 5B). Additionally, we quantified the ratio of phosphorylation of p70S6K1 as a marker for mTOR activation. Phosphorylation of p70s6K1 increased in all septic groups, but to a greater extent in septic Ob‐KO mice and Ln‐lep mice (Figure 5C).
Figure 5.
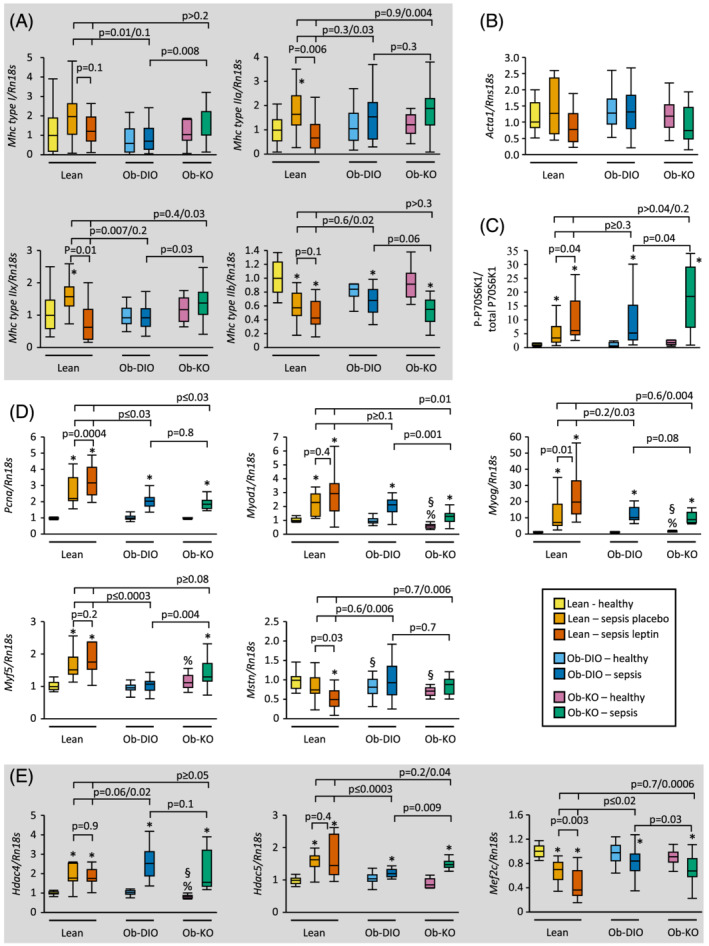
Muscle protein synthesis, growth, and regeneration. Relative mRNA expression of myosin isoform (Kruskal–Wallis P ≤ 0.02) (A) and actin (Kruskal–Wallis P = 0.07) (B) in gastrocnemius muscle (GNM). (C) Phosphorylated P70S6K1 over total P70S6K1 protein expression in GNM (Kruskal–Wallis P < 0.0001). (D, E) Relative mRNA expression of markers of muscle growth and regeneration in GNM (Kruskal–Wallis P < 0.0001). Gene expression data are shown relative the median of lean healthy mice. Lean healthy n = 17, lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. *P < 0.05 between septic mice and healthy controls; § P < 0.05 compared with lean healthy mice; % P < 0.05 compared with healthy Ob‐DIO mice.
Muscle mass accretion secondary to leptin treatment is associated with increased expression of PCNA and specific myogenic markers and decreased myostatin expression. 14 , 15 , 16 Sepsis increased gene expression of proliferation marker Pcna, most pronounced in lean mice compared with obese mice and with a further rise in Ln‐Lep vs. Ln mice (Figure 5D). Sepsis increased gene expression of regeneration markers Myod1, Myog, and Myf5 in all groups except for Myf5 in Ob‐DIO. Leptin administration in lean mice further increased Myog gene expression, while leptin deficiency in obese mice decreased Myod1 and increased Myf5 gene expression. Furthermore, leptin administration decreased Mstn, an inhibitor of muscle growth, without an effect of sepsis and obesity. Ketone body supplementation in septic mice was previously shown to inhibit class IIa HDAC expression, which are known suppressors of the myogenic MEF2 transcription factor. 6 , 20 Sepsis increased gene expression of Hdac4 and Hdac5 and concomitantly decreased gene expression of muscle regeneration inhibitor Mef2c in all groups, most pronounced in lean mice, irrespective of leptin (Figure 5E).
Effect of obesity vs. leptin on muscle fibre size and typing
To further characterize the atrophy and loss of lean body mass during sepsis, we determined muscle fibre size and type in the tibialis anterior muscle (Figure 6A). The tibialis anterior has a similar fibre type distribution as the EDL, which was used for muscle force analyses. Prior to sepsis, Ob‐KO mice had a lower average muscle fibre size compared with Ob‐DIO mice (Figure 6B and 6C). After 5 days of sepsis, average muscle fibre size decreased most pronounced in Ln‐lep and Ob‐DIO mice (Figure 6B and 6C).
Figure 6.
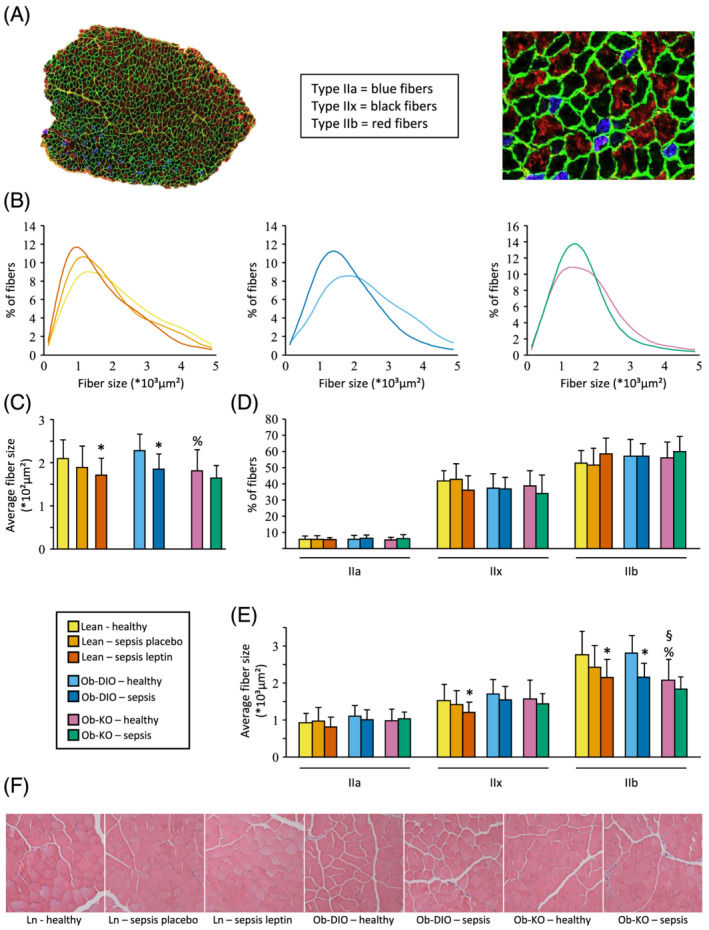
Tibialis anterior muscle histology analysis. (A) Tibialis anterior section stained for muscle fibre types and laminin; type I fibres were only sporadically present and were omitted in the analyses. (B) Frequency histograms showing the distribution of cross‐sectional areas (mm2). (C) Average fibre size area for all fibres (Kruskal–Wallis P = 0.0006). (D) Percentage of fibres expressing myosin heavy chain types IIA, IIB, and IIX proteins (Kruskal–Wallis P ≥ 0.15). (E) Average fibre size area splitted by fibre type. Type IIa Kruskal–Wallis P = 0.09; type IIx and IIb Kruskal–Wallis P ≤ 0.02. (F) Representative H&E staining showing no morphological differences between groups. Lean healthy n = 17, lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. *P < 0.05 between septic mice and healthy controls; § P < 0.05 compared with lean healthy mice; % P < 0.05 compared with healthy Ob‐DIO mice.
Fibre type composition was similar in lean and obese groups, with a predominance of fast glycolytic type IIb fibres and type IIx fibres, only a small fraction of slower oxidative IIa fibres, and virtually no slow oxidative type I fibres (Figure 6D). The average fibre size of type IIb fibres was significantly smaller in healthy Ob‐KO mice compared with healthy Ob‐DIO and lean mice (Figure 6E). With sepsis, the decrease in fibre size was mainly due to a decrease in type IIb fibres. Semi‐quantitative scoring of muscle histology revealed no overt morphological differences (Figures 6F and S2).
Effect of obesity vs. leptin on plasma and hepatic markers of lipid metabolism
The doubling in loss of fat mass observed in obese septic mice (Figure 2F) was accompanied by a different lipid profile in obese vs. lean septic mice, largely irrespective of leptin (Figure 7A and 7B). Plasma free fatty acids and glycerol were reduced in lean septic mice, irrespective of leptin, whereas this reduction was blunted or even prevented in obese septic mice (Figure 7A). Also, the pronounced reduction in plasma lipoproteins HDL and LDL observed in lean septic mice, again irrespective of leptin, was blunted in obese septic mice, with even a small increase in LDL in Ob‐DIO mice (Figure 7B).
Figure 7.
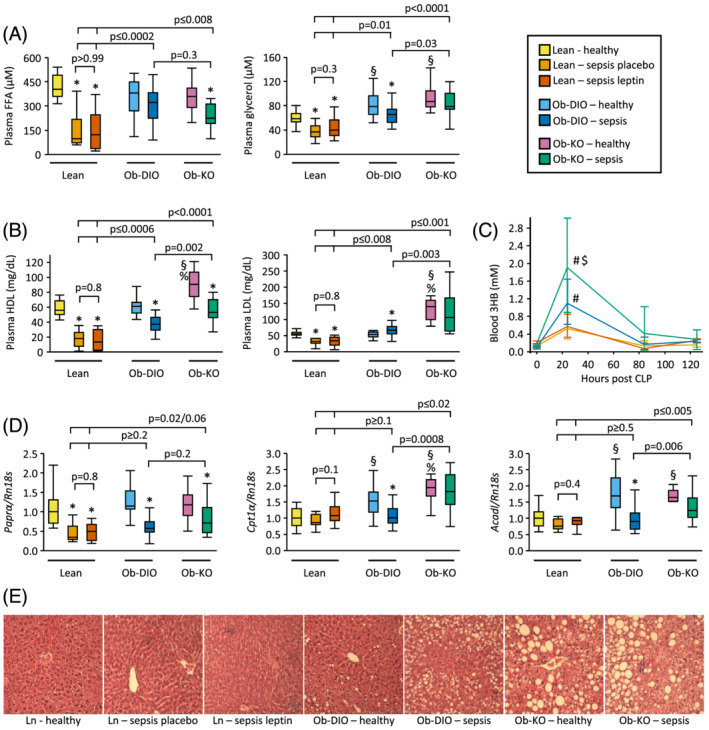
Plasma lipid profile and liver histological analysis. (A) Plasma free fatty acids and plasma glycerol (Kruskal–Wallis P < 0.0001). (B) Plasma HDL and LDL cholesterol (Kruskal–Wallis P < 0.0001). (C) Plasma 3‐hydroxybutyrate (3HB). Kruskal–Wallis P ≤ 0.01 at 24 and 84 h; P > 0.05 at other time points. # P < 0.05 compared with lean septic mice; $ P < 0.05 compared with septic Ob‐DIO mice. (D) mRNA expression of genes involved in fatty acid oxidation (Kruskal–Wallis P < 0.0001). (E) Representative H&E stained liver sections. Lean healthy n = 17, lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. *P < 0.05 between septic mice and healthy controls; § P < 0.05 compared with lean healthy mice; % P < 0.05 compared with healthy Ob‐DIO mice.
Plasma ketone levels peaked 24 h after induction of sepsis but to a higher extent in obese compared with lean septic mice (P ≤ 0.0002, Figure 7C). Ob‐KO peaked even higher than Ob‐DIO mice (P = 0.007). Sepsis decreased hepatic gene expression of Pparα in all groups. Healthy obese mice displayed a higher hepatic expression of ketogenic markers Cpt1α and Acadl compared with lean healthy mice, which remained elevated after 5 days of sepsis in the Ob‐KO group only (Figure 7D).
Histologically, all septic groups displayed signs of liver inflammation, but the Ob‐KO and Ob‐DIO mice also showed signs of liver inflammation in the healthy condition (Figures 7E and S3). The degree of vacuolization, a marker of glycogen content, decreased in all septic groups. Sinusoidal dilation, a marker of oedema, increased only in septic lean mice. Lean mice never showed signs of intrahepatic fat accumulation, whereas obese mice always did, even more during sepsis than during health. The highest fat accumulation was observed in septic Ob‐KO mice.
Rank correlation analysis of potential regulatory pathways of muscle wasting and muscle weakness
As leptin did not explain the protection against muscle wasting and muscle weakness observed in obese mice, we assessed the impact of the tested regulators and pathways of muscle wasting and weakness for their explanatory role by univariate rank correlation analysis.
For each of the regulatory processes of muscle wasting under investigation, protein breakdown, protein synthesis, and muscle proliferation, we first assessed association with loss in lean body mass of all investigated markers. For protein breakdown, plasma urea reached highest association (ρ = −0.60, P < 0.0001), for protein synthesis, none of the parameters reached a significant association, whereas for muscle proliferation, the association with Hdac5 gene expression (ρ = −0.43, P = 0.0007) was most pronounced. The investigated regulators of muscle wasting plasma TNFα (ρ = 0.31, P = 0.01), plasma IL‐6 (ρ = 0.39, P = 0.003), and plasma corticosterone (ρ = 0.35, P = 0.007) associated with lean body mass loss.
To identify potential regulators of muscle weakness explaining the observed protective effect of obesity, we assessed markers of inflammation, lipolysis, and ketogenesis for their explanatory role in a univariate analysis. To correct for the difference in muscle force in healthy lean and obese mice, and to correct for the role of muscle mass in muscle weakness, the specific muscle force for each mouse was expressed as a ratio of its specific muscle force and the mean value of its own healthy control group. Univariate analysis indicated a significant association of this corrected muscle force with decrease in fat mass (indicative of endogenous lipolysis, ρ = −0.40, P = 0.003), peak plasma 3‐hydroxybutyrate (ρ = 0.37, P = 0.005), plasma LDL cholesterol (ρ = 0.48, P = 0.0003) and HDL cholesterol (ρ = 0.55, P < 0.0001), and plasma TNFα (ρ = −0.51, P = 0.0001).
Association of overweight/obesity in human critically ill patients with markers of protein catabolism and on leptin concentrations
Lean patients [median BMI (IQR) 22.5 (20.7–23.8) kg/m2] were matched with overweight/obese patients [median BMI (IQR) 28.6 (26.7–31.2) kg/m2] for demographics and severity and type of critical illness (Table S2). Compared with lean patients, patients who were overweight/obese showed higher leptin serum concentrations on admission and on Day 5 in ICU (Figure 8A). On Day 5 in ICU, overweight/obese critically ill patients showed lower net protein catabolism than lean critically ill patients as indicated by a reduced net nitrogen loss (Figure 8B) and by a reduced urea‐to‐creatinine ratio (Figure 8C).
Figure 8.
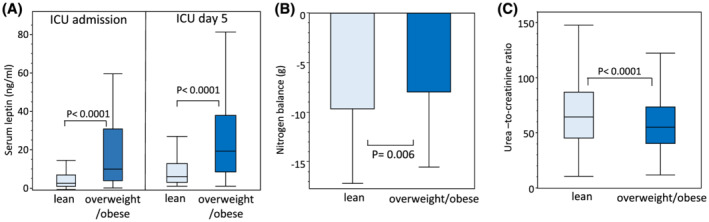
Serum leptin and net protein loss in human lean and overweight/obese patients. (A) Serum leptin on admission and Day 5 in ICU in lean (n = 81) and overweight/obese (n = 69) patients. (B) Nitrogen balance on ICU Day 5 is the calculated net nitrogen loss, and (C) urea‐to‐creatinine ratio is the plasma concentration of urea (mg/dL) divided by the plasma concentration of creatinine (mg/dL) on Day 5 in lean (n = 694) and overweight/obese (n = 694) patients.
Discussion
In this study, we showed that obesity, but not leptin levels, protected against sepsis‐induced muscle wasting and weakness, despite higher mortality of leptin‐deficient obese mice. As compared with lean septic mice, obese mice had a decreased inflammatory response and showed less signs of protein breakdown, together with an increased mobilization of adipose tissue fat stores, increased ketogenesis, and higher plasma lipoprotein levels. Muscle fibre type composition was not altered during sepsis, but a higher atrophy sensitivity of large fast type IIb fibres compared with IIa and IIx fibres was observed, independent of obesity or leptin levels. In matched human overweight/obese patients, we confirmed decreased protein breakdown compared with lean patients.
Similar to previous studies in septic mice and human critically ill patients, 3 , 6 we observed a protective effect of obesity on both the loss of lean body mass and muscle force. Here, we extend this finding and showed that plasma leptin levels do not explain this protective effect of obesity. While leptin‐deficient obese mice have a higher mortality than diet‐induced obese mice when confronted with sepsis, the reduction in lean body mass loss and maintenance of muscle force was comparable between the two groups. These data indicate that leptin is necessary for sepsis survival and might also contribute to the increased survival observed in premorbid obese critically ill patients. 4 , 5 While investigation of the survival disadvantage of ob/ob mice was beyond the scope of this paper, it is interesting to point out that pre‐septic healthy ob/ob mice had elevated plasma levels of IL‐6. Interestingly, mice who are stressed prior to an inflammatory challenge also suffer from a survival disadvantage and this has recently been linked to IL‐6. 21 Furthermore, supplementation of ob/ob mice with leptin normalized IL‐6 plasma levels and increased survival while leptin supplementation increased mortality in IL‐6−/− mice. 11 , 12
During the course of sepsis, irrespective of leptin levels, lean mice lost more lean body mass than obese mice. Loss of muscle mass occurs when the rate of proteolysis exceeds the rate of muscle protein synthesis. The ubiquitin‐proteasome system (UPS) is a major mechanism of muscle proteolysis. Several markers of the UPS were more up‐regulated in lean septic mice compared with obese septic mice, irrespective of leptin. In contrast with the other proteolysis markers, proteasome activity was lower in septic lean mice compared with lean healthy control and septic obese mice. While muscle proteasome activity is typically reported as being increased in experimental sepsis, these studies investigated earlier time points. 22 Potentially, in the prolonged phase of sepsis which we investigated, proteasome activity decreases after an initial acute rise, as is observed in animal models of cancer cachexia and alloxan‐induced diabetes mellitus. 23 , 24 Autophagy, another major mechanism of proteolysis, has a controversial role in muscle wasting disorders as both excessive as impaired autophagy may lead to profound muscle wasting. 25 , 26 Additionally, impaired activation of autophagy, as observed in animal models and human biopsies during critical illness, leads to the accumulation of cellular damage and reduced muscle quality. 26 , 27 The concomitant increase in LC3II (marker of autophagosome formation) and P62 protein (a cargo protein normally cleared by autophagy) confirms this phenotype of insufficient activated autophagy in muscle of lean and diet‐induced obese septic mice. Leptin‐deficient obese septic mice did not show this concomitant increase in P62, which might suggest increased autophagic flux, as described earlier in this genotype. 28 Such increase in autophagy would improve the muscle quality during sepsis and could have contributed to the more pronounced protection of muscle weakness we observed in these mice.
Concerning muscle protein synthesis, changes in phosphorylation of P70S6K1, as downstream target of mTORC1, or muscle gene expression of contractile proteins could not explain the difference in muscle mass loss we observed between lean and obese septic mice. Similarly, changes in markers of muscle cell proliferation could not explain the differences we observed in muscle mass loss. Our findings are in contrast to the known stimulatory effect of leptin on muscle mass in healthy mice. 14 , 15 , 16 While leptin supplementation did lead to a higher phosphorylation of P70S6K1 and hence mTORC1 activity, higher muscle gene expression of contractile proteins and lower myostatin gene expression, and increased the expression of PCNA and MyoG, this did not translate to an increase in muscle mass. The shorter duration of supplementation, difference in feeding regimen, concomitant increase in muscle atrophy, and the illness itself all may have contributed to the lack of leptin effect in our study.
Based on our findings, we conclude that decreased muscle breakdown rather than a difference in protein synthesis may be responsible for the reduced muscle wasting observed in obese mice during sepsis. A likely strong contributor to the reduced wasting in obese mice is the lower plasma TNFα increase, a known stimulator of muscle atrophy. 29 Also, human obese patients displayed a blunted inflammatory response in acute respiratory distress syndrome and septic shock with lower plasma levels of inflammatory cytokines when compared with lean patients. 30 , 31 In addition to inflammatory cytokines, also glucocorticoids are powerful catabolic stimulators. 32 Plasma corticosterone levels increased in all septic mice, but were, as expected, 8 regulated by leptin levels, and as such did not explain the observed protection in obese compared with lean septic mice. The observed changes in plasma urea and hepatic expression of genes involved in liver amino acid catabolism are a further indication of reduced urea cycle activity and hence amino acid catabolism in the obese mice. Our observation that overweight/obese human critically ill patients display reduced net protein catabolism compared with lean critically ill patients further strengthens our findings in obese mice. Indeed, in addition to the earlier observed reduced incidence of ICUAW in obese patients, 3 obese patients also displayed reduced net nitrogen loss and reduced urea‐to‐creatinine ratio, both markers of reduced protein catabolism. 18 , 33 Increased loss of muscle protein of lean mice and patients and hence a greater release of amino acids into the circulation could be the primary driver of such increased amino acid catabolism. Indeed, a rise in circulating amino acids stimulates glucagon, by itself the main regulator of amino acid catabolism. 34 In addition, glucagon resistance as observed with obesity 35 could have strengthened this effect.
Morphologically, muscle of lean and obese mice with high or low leptin levels appeared very similar, although fibre size was smaller in ob/ob mice, as predicted. 14 Interestingly, while we did not observe a fibre type switch with sepsis, fast glycolytic type IIb fibres displayed a greater sensitivity for the atrophic effects of sepsis, compared with the slower type IIx and IIa fibre types. Specific type II fibre atrophy was also observed in patients with non‐excitable muscle membranes. 36 This could be due to fibre type differences in muscle breakdown or muscle protein synthesis. Indeed, basal protein synthesis rate is smallest in fast type IIb fibres, and during food deprivation, fast type IIx/IIb fibres show the strongest decrease in muscle protein synthesis compared with type I and type IIa. 37
Obese mice lost a considerable higher amount of fat mass than lean mice after 5 days of sepsis, which was accompanied by increased ketogenesis. As described previously, 6 the increased mobilization of fat stores and subsequent ketogenesis in obese contributed to the maintenance of muscle force during sepsis. As the skeletal muscle appears to be bio‐energetically inert for delivered lipids during critical illness, ketones rather than the released fatty acids are the likely effectors. 38 Obese septic mice also displayed higher plasma cholesterol levels, whereas these were strongly suppressed in lean mice. A link between low cholesterol and the development of ICUAW was recently demonstrated. 39 In addition, higher plasma HDL and LDL cholesterol could also have protected against excessive inflammation and so indirectly reduced muscle atrophy. Indeed, lipoproteins bind and neutralize LPS and lipoteichoic acid, and HDL has direct anti‐inflammatory properties. 40 Although we cannot exclude that obese mice had a milder disease course than lean mice and as such displayed lower inflammatory markers and higher plasma cholesterol levels, the higher mortality and illness severity scores observed in the ob/ob mice argue against a milder disease course in these mice. Nonetheless, the association between plasma lipoproteins, sepsis, and obesity remains intriguing and warrants further research.
Our study has some limitations. While our prolonged mouse model mimics several aspects of human prolonged critical illness and ICUAW, extrapolation to the human setting has to be done with caution. Our mice are not mechanically ventilated nor completely immobilized. Furthermore, while critical illness‐induced muscle wasting in humans is hallmarked by a preferential loss of myosin, 1 myosin and actin is lost to similar extents in our mouse model of sepsis. In addition, not all metabolic syndrome‐related aspects of obesity are similarly present in humans and rodents. 41 Second, we did not include sham‐operated controls, nor did we administer fluid resuscitation, parenteral nutrition, antibiotics, or pain medication to healthy mice as it was our aim to study the complete phenotype of critical illness, including the complex interplay of disease and treatment received in the ICU. Third, we used congenital leptin‐deficient obese mice, and pre‐existing differences developed before the initiation of sepsis, such as the difference in body mass composition could have influenced the results. Fourth, the study was powered to detect an improved muscle function; all other secondary parameters that were investigated have to be considered as hypothesis‐generating and require further investigation.
In conclusion, leptin did not mediate the protective effect of obesity against sepsis‐induced muscle wasting and weakness in mice. Instead, obesity—independent of leptin—attenuated inflammation, protein catabolism, and dyslipidaemia, pathways that may play a role in the observed muscle protection.
Conflict of interest
The authors have no conflicts of interest to disclose.
Funding
G.V.d.B. and L.L., via the KU Leuven, receive long‐term structural research support from the Methusalem Program funded by the Flemish Government (METH14/06) and from the Research Foundation Flanders (G.0C78.17N). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (AdvG 2017‐785809). J.G. holds a postdoctoral research fellowship supported by the University Hospitals Leuven. Nova Biomedical and Menarini Diagnostics kindly provided the point‐of‐care ketone meters and accompanying analytical disposables.
Supporting information
Table S1. list of commercially used gene expression assays (Applied Biosystems)
Table S2. baseline characteristics of lean and overweight/obese ICU patients
Figure S1. Blood glucose. Blood glucose concentration. # p < 0.05 compared to lean septic placebo treated mice; $ p < 0.05 compared to lean septic leptin treated mice; & p < 0.05 compared to septic Ob‐DIO mice. The colour of the mark refers to the comparator.
Figure S2. semi‐quantitative scoring of tibialis anterior H&E sections. Data are shown as cumulative percentages of the respective group. Number (n) of animals: Lean healthy n = 17; lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. * p < 0.05 between septic mice and healthy controls.
Figure S3. semi‐quantitative scoring of liver H&E sections. Data are shown as cumulative percentages of the respective group. Number (n) of animals: Lean healthy n = 17; lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. * p < 0.05 between septic mice and healthy controls; § p < 0.05 compared to lean healthy mice; % p < 0.05 compared to healthy Ob‐DIO mice; brackets between groups indicated p < 0.05.
Acknowledgements
We thank Marissa Boone, Ruben Weckx, and Arno Téblick for their technical assistance with the animal experiments. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle. 42
Vankrunkelsven W., Derde S., Gunst J., Vander Perre S., Declerck E., Pauwels L., Derese I., Van den Berghe G., and Langouche L. (2022) Obesity attenuates inflammation, protein catabolism, dyslipidaemia, and muscle weakness during sepsis, independent of leptin, Journal of Cachexia, Sarcopenia and Muscle, 13, 418–433, 10.1002/jcsm.12904
Greet Van den Berghe and Lies Langouche have equal contribution.
References
- 1. Schefold JC, Wollersheim T, Grunow JJ, Luedi MM, Z'Graggen WJ, Weber‐Carstens S. Muscular weakness and muscle wasting in the critically ill. J Cachexia Sarcopenia Muscle 2020;11:1399–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, et al. Acute skeletal muscle wasting in critical illness. JAMA 2013;310:1591–1600. [DOI] [PubMed] [Google Scholar]
- 3. Goossens C, Marques MB, Derde S, Vander Perre S, Dufour T, Thiessen SE, et al. Premorbid obesity, but not nutrition, prevents critical illness‐induced muscle wasting and weakness. J Cachexia Sarcopenia Muscle 2017;8:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sakr Y, Alhussami I, Nanchal R, Wunderink RG, Pellis T, Wittebole X, et al. Being overweight is associated with greater survival in ICU patients: results from the intensive care over nations audit. Crit Care Med 2015;43:2623–2632. [DOI] [PubMed] [Google Scholar]
- 5. Pickkers P, de Keizer N, Dusseljee J, Weerheijm D, van der Hoeven JG, Peek N. Body mass index is associated with hospital mortality in critically ill patients: an observational cohort study. Crit Care Med 2013;41:1878–1883. [DOI] [PubMed] [Google Scholar]
- 6. Goossens C, Weckx R, Derde S, Dufour T, Vander Perre S, Pauwels L, et al. Adipose tissue protects against sepsis‐induced muscle weakness in mice: from lipolysis to ketones. Crit Care 2019;23:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lipina C, Hundal HS. Lipid modulation of skeletal muscle mass and function. J Cachexia Sarcopenia Muscle 2017;8:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos‐Flier E, et al. Role of leptin in the neuroendocrine response to fasting. Nature 1996;382:250–252. [DOI] [PubMed] [Google Scholar]
- 9. Ikejima S, Sasaki S, Sashinami H, Mori F, Ogawa Y, Nakamura T, et al. Impairment of host resistance to Listeria monocytogenes infection in liver of db/db and ob/ob mice. Diabetes 2005;54:182–189. [DOI] [PubMed] [Google Scholar]
- 10. Faggioni R, Fantuzzi G, Gabay C, Moser A, Dinarello CA, Feingold KR, et al. Leptin deficiency enhances sensitivity to endotoxin‐induced lethality. Am J Physiol 1999;276:R136–R142. [DOI] [PubMed] [Google Scholar]
- 11. Tschop J, Nogueiras R, Haas‐Lockie S, Kasten KR, Castaneda TR, Huber N, et al. CNS leptin action modulates immune response and survival in sepsis. J Neurosci 2010;30:6036–6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Negrin LL, Jahn A, van Griensven M. Leptin protects against mortality and organ dysfunction in a two‐hit trauma/sepsis model and is IL‐6‐dependent. Shock 2017;48:130–137. [DOI] [PubMed] [Google Scholar]
- 13. Shapiro NI, Khankin EV, Van Meurs M, Shih SC, Lu S, Yano M, et al. Leptin exacerbates sepsis‐mediated morbidity and mortality. J Immunol 2010;185:517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sainz N, Rodriguez A, Catalan V, Becerril S, Ramirez B, Gomez‐Ambrosi J, et al. Leptin administration favors muscle mass accretion by decreasing FoxO3a and increasing PGC‐1α in ob/ob mice. PLoS ONE 2009;4(9):e6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burgos‐Ramos E, Canelles S, Rodriguez A, Frago LM, Gomez‐Ambrosi J, Chowen JA, et al. The increase in fiber size in male rat gastrocnemius after chronic central leptin infusion is related to activation of insulin signaling. Mol Cell Endocrinol 2018;470:48–59. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez A, Becerril S, Mendez‐Gimenez L, Ramirez B, Sainz N, Catalan V, et al. Leptin administration activates irisin‐induced myogenesis via nitric oxide‐dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int J Obes (Lond) 2015;39:397–407. [DOI] [PubMed] [Google Scholar]
- 17. Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med 2011;365:506–517. [DOI] [PubMed] [Google Scholar]
- 18. Gunst J, Vanhorebeek I, Casaer MP, Hermans G, Wouters PJ, Dubois J, et al. Impact of early parenteral nutrition on metabolism and kidney injury. J Am Soc Nephrol 2013;24:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Langouche L, Vander Perre S, Marques M, Boelen A, Wouters PJ, Casaer MP, et al. Impact of early nutrient restriction during critical illness on the nonthyroidal illness syndrome and its relation with outcome: a randomized, controlled clinical study. J Clin Endocrinol Metab 2013;98:1006–1013. [DOI] [PubMed] [Google Scholar]
- 20. Miska EA, Langley E, Wolf D, Karlsson C, Pines J, Kouzarides T. Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res 2001;29:3439–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qing H, Desrouleaux R, Israni‐Winger K, Mineur YS, Fogelman N, Zhang C, et al. Origin and function of stress‐induced IL‐6 in murine models. Cell 2020;182:372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stana F, Vujovic M, Mayaki D, Leduc‐Gaudet JP, Leblanc P, Huck L, et al. Differential regulation of the autophagy and proteasome pathways in skeletal muscles in sepsis. Crit Care Med 2017;45:e971–e979. [DOI] [PubMed] [Google Scholar]
- 23. Khal J, Wyke SM, Russell ST, Hine AV, Tisdale MJ. Expression of the ubiquitin‐proteasome pathway and muscle loss in experimental cancer cachexia. Br J Cancer 2005;93:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Galban VD, Evangelista EA, Migliorini RH, de Carmo Kettelhut I. Role of ubiquitin‐proteasome‐dependent proteolytic process in degradation of muscle protein from diabetic rabbits. Mol Cell Biochem 2001;225:35–41. [DOI] [PubMed] [Google Scholar]
- 25. Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll‐like receptor 4 mediates lipopolysaccharide‐induced muscle catabolism via coordinate activation of ubiquitin‐proteasome and autophagy‐lysosome pathways. FASEB J 2011;25:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab 2009;10:507–515. [DOI] [PubMed] [Google Scholar]
- 27. Vanhorebeek I, Gunst J, Derde S, Derese I, Boussemaere M, Guiza F, et al. Insufficient activation of autophagy allows cellular damage to accumulate in critically ill patients. J Clin Endocrinol Metab 2011;96:E633–E645. [DOI] [PubMed] [Google Scholar]
- 28. Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun 2014;5:4934. [DOI] [PubMed] [Google Scholar]
- 29. Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, et al. TNF‐α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J 2005;19(3):362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stapleton RD, Dixon AE, Parsons PE, Ware LB, Suratt BT. Network NARDS: the association between BMI and plasma cytokine levels in patients with acute lung injury. Chest 2010;138(3):568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wacharasint P, Boyd JH, Russell JA, Walley KR. One size does not fit all in severe infection: obesity alters outcome, susceptibility, treatment, and inflammatory response. Crit Care 2013;17:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tiao G, Fagan J, Roegner V, Lieberman M, Wang JJ, Fischer JE, et al. Energy‐ubiquitin‐dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest 1996;97:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haines RW, Zolfaghari P, Wan Y, Pearse RM, Puthucheary Z, Prowle JR. Elevated urea‐to‐creatinine ratio provides a biochemical signature of muscle catabolism and persistent critical illness after major trauma. Intensive Care Med 2019;45:1718–1731. [DOI] [PubMed] [Google Scholar]
- 34. Thiessen SE, Derde S, Derese I, Dufour T, Vega CA, Langouche L, et al. Role of glucagon in catabolism and muscle wasting of critical illness and modulation by nutrition. Am J Respir Crit Care Med 2017;196:1131–1143. [DOI] [PubMed] [Google Scholar]
- 35. Suppli MP, Bagger JI, Lund A, Demant M, van Hall G, Strandberg C, et al. Glucagon resistance at the level of amino acid turnover in obese subjects with hepatic steatosis. Diabetes 2020;69:1090–1099. [DOI] [PubMed] [Google Scholar]
- 36. Bierbrauer J, Koch S, Olbricht C, Hamati J, Lodka D, Schneider J, et al. Early type II fiber atrophy in intensive care unit patients with nonexcitable muscle membrane. Crit Care Med 2012;40:647–650. [DOI] [PubMed] [Google Scholar]
- 37. Goodman CA, Kotecki JA, Jacobs BL, Hornberger TA. Muscle fiber type‐dependent differences in the regulation of protein synthesis. PLoS ONE 2012;7:e37890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Puthucheary ZA, Astin R, McPhail MJW, Saeed S, Pasha Y, Bear DE, et al. Metabolic phenotype of skeletal muscle in early critical illness. Thorax 2018;73:926–935. [DOI] [PubMed] [Google Scholar]
- 39. Goossens C, Weckx R, Derde S, Vander Perre S, Derese I, Van Veldhoven PP, et al. Altered cholesterol homeostasis in critical illness‐induced muscle weakness: effect of exogenous 3‐hydroxybutyrate. Crit Care 2021;25:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka S, Couret D, Tran‐Dinh A, Duranteau J, Montravers P, Schwendeman A, et al. High‐density lipoproteins during sepsis: from bench to bedside. Crit Care 2020;24:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Preguica I, Alves A, Nunes S, Fernandes R, Gomes P, Viana SD, et al. Diet‐induced rodent models of obesity‐related metabolic disorders—a guide to a translational perspective. Obes Rev 2020;21:e13081. [DOI] [PubMed] [Google Scholar]
- 42. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. list of commercially used gene expression assays (Applied Biosystems)
Table S2. baseline characteristics of lean and overweight/obese ICU patients
Figure S1. Blood glucose. Blood glucose concentration. # p < 0.05 compared to lean septic placebo treated mice; $ p < 0.05 compared to lean septic leptin treated mice; & p < 0.05 compared to septic Ob‐DIO mice. The colour of the mark refers to the comparator.
Figure S2. semi‐quantitative scoring of tibialis anterior H&E sections. Data are shown as cumulative percentages of the respective group. Number (n) of animals: Lean healthy n = 17; lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. * p < 0.05 between septic mice and healthy controls.
Figure S3. semi‐quantitative scoring of liver H&E sections. Data are shown as cumulative percentages of the respective group. Number (n) of animals: Lean healthy n = 17; lean sepsis placebo n = 11, lean sepsis leptin n = 14, Ob‐DIO healthy n = 22, Ob‐DIO sepsis n = 20, Ob‐KO healthy n = 12, Ob‐KO sepsis n = 14. * p < 0.05 between septic mice and healthy controls; § p < 0.05 compared to lean healthy mice; % p < 0.05 compared to healthy Ob‐DIO mice; brackets between groups indicated p < 0.05.