

Abstract
Background
Cancer cachexia, characterized by muscle and fat tissue wasting, is a major determinant of cancer‐related mortality without established treatment. Recent animal data revealed that cancer cells induce muscle wasting by releasing Hsp70 and Hsp90 as surface proteins on extracellular vesicles (EVs). Here, we test a therapeutic strategy for ameliorating cancer cachexia by inhibiting the release of Hsp70 and Hsp90 using proton pump inhibitor omeprazole.
Methods
Omeprazole effect on Hsp70/90 release through EVs by Lewis lung carcinoma (LLC) cells in vitro, serum levels of Hsp70/90 and Hsp70/90‐carrying EVs in LLC tumour‐bearing mice, and LLC‐induced muscle protein degradation pathways in C2C12 myotubes and mice were determined. Omeprazole effect on endolysosomal pH and Rab27b expression in LLC cells were analysed.
Results
Omeprazole treatment of LLC cells inhibited Hsp70/90 and Hsp70/90‐carrying EV release in a dose‐dependent manner (1 to 10 μM) and attenuated the catabolic activity of LLC cell‐conditioned medium on C2C12 myotubes. Systemic omeprazole administration to LLC tumour‐bearing mice (5 mg/kg/day subcutaneously) for 2 weeks blocked elevation of serum Hsp70, Hsp90, and Hsp70/90‐carrying EVs, abrogated skeletal muscle catabolism, and prevented loss of muscle function as well as muscle and epididymal fat mass without altering tumour growth. Consequently, median survival increased by 23.3%. Mechanistically, omeprazole increased cancer cell endolysosomal pH level dose‐dependently (0.1 to 1 μM) by inhibiting vacuolar H+‐ATPase. Further, omeprazole suppressed the highly elevated expression of Rab27b, a key regulator of EV release, in LLC cells.
Conclusions
Omeprazole ameliorates cancer cachexia by inhibiting cancer cell release of Hsp70 and Hsp90.
Keywords: Lewis lung carcinoma, Muscle wasting, Fat tissue wasting, Hsp70, Hsp90, Rab27b, Vacuolar H+‐ATPase
Introduction
Cachexia is a frequent complication seen in at least 50% of cancer patients. It is a complex metabolic syndrome characterized by loss of skeletal muscle mass and function known as muscle wasting. In addition, adipose tissue loss is frequently associated with cachexia. 1 Patients of many solid tumours including lung, pancreatic, colon, and gastric cancer are particularly prone to cachexia, resulting in progressive loss of muscle and fat mass leading to multiple organ failure and eventual death. Effective intervention of cancer cachexia could improve patient's physical condition, response to cancer therapies, and survival. However, because of the poor understanding of its aetiology, there has been no established treatment for cancer cachexia. Consequently, cachexia is the immediate cause of ∼30% cancer‐related death. 2 Previous attempts to intervene cancer cachexia with various strategies have not yielded a treatment that protects the loss of muscle mass and function. 3 , 4 The current study was designed to test a novel pharmacological strategy for ameliorating cancer cachexia by targeting cancer cell release of Hsp70 and Hsp90. 5
Cancer cachexia features a severe systemic inflammation, which causes muscle wasting through the activation of inflammatory signalling pathways including p38β MAPK and NF‐κB resulting in accelerated muscle protein degradation by the ubiquitin‐proteasome pathway (UPP) and the autophagy‐lysosomal pathway (ALP) in animal models of cancer cachexia. 6 Particularly, p38β MAPK is essential for the up‐regulation or activation of key components of UPP (atrogin1/MAFbx and UBR2) and ALP (LC3b, Gabarapl1, and ULK1), which mediate cancer‐induced muscle wasting. 7 , 8 , 9 However, the cancer milieu is highly complex, and there has been no effective way to intervene the systemic inflammation and cachexia. We recently discovered that cachectic tumour cells of mouse and human origin constitutively release high levels of extracellular vesicles (EVs) that carry Hsp70 and Hsp90 as surface proteins, resulting in elevated circulating Hsp70 and Hsp90 levels in mice bearing cachectic tumours. 5 , 10 These Hsp70/90‐carrying EVs are ∼110 nm in diameter and contain common exosome marker proteins CD9/CD81/TSG101 as well as acetylcholinesterase (AchE) 5 , 10 that is associated with a subpopulation of exosomes. 11 Similar increases of serum Hsp70/90 and EVs have been reported in patients with cachectic cancer including lung, 12 , 13 , 14 , 15 , 16 , 17 , 18 colorectal, 19 , 20 and pancreatic cancer. 21 , 22 In addition, serum Hsp70/90 levels in cancer patients increase with the development of pathological grade and clinical stage, 13 , 14 , 16 and the increase correlates with mortality. 12 , 19 In mice, elevated circulating Hsp70 and Hsp90 cause muscle wasting primarily by activating TLR4 on skeletal muscle cells. 5 , 23 TLR4 in turn activates UPP and ALP primarily through p38β MAPK‐mediated signalling. 23 , 24 In addition, TLR4 mediates endotoxin 25 or cancer‐induced 26 adipose tissue lipolysis and remodelling. Further, systemic activation of TLR4 increases circulating inflammatory cytokines that promote cachexia. 5 , 23 To translate the above findings to potential clinical interventions of cancer cachexia, we tested a pharmacological solution for cancer cachexia by inhibiting tumour cell release of Hsp70 and Hsp90.
Extracellular vesicles are released from a type of intracellular vacuolar compartment, multivesicular bodies (MVBs). 27 Microenvironmental pH is a key factor for EV traffic in tumour cells. 28 Proton pump inhibitors (PPIs) inhibit EV release by melanoma cells 29 and gastric cancer cells. 30 PPIs inhibit not only the H+/K+‐ATPase in gastric parietal cells but also the vacuolar H+‐ATPase (V‐H+‐ATPase) in other cell types, and they were thought to inhibit EV release by inhibiting V‐H+‐ATPase resulting in increased pH in intracellular vacuolar compartments including the MVB. 29 , 30 In the current study, we tested the hypothesis that cancer cachexia can be ameliorated by inhibiting cancer cell release of Hsp70 and Hsp90 through EVs using a PPI. We demonstrate here that omeprazole (OMP), an over‐the‐counter PPI, inhibits the release of Hsp70 and Hsp90 through EVs by Lewis lung carcinoma (LLC) cells and attenuates skeletal muscle as well as adipose tissue wasting in LLC tumour‐bearing mice. Mechanistically, in addition to increasing endolysosomal pH level, OMP abolishes the elevation of Rab27b expression in LLC cells. Thus, OMP has the potential to be repurposed for cancer cachexia due to its inhibition of cancer cell release of Hsp70 and Hsp90.
Methods
Muscle and tumour cell cultures
Murine C2C12 myoblasts were purchased from the American Type Culture Collection (ATCC, Rockville, MD), and were grown in growth medium (DMEM supplemented with 10% foetal bovine serum) at 37°C under 5% CO2. At 85–90% confluence, differentiation was induced by incubation for 96 h in differentiation medium (DMEM supplemented with 4% heat‐inactivated horse serum) to form myotubes. Conditioned medium of 48 h cultures of LLC cells (National Cancer Institute) cultured in DMEM supplemented with 10% foetal bovine serum or non‐tumorigenic NL20 cells (ATCC) cultured in Ham's F12 medium supplemented with 1.5 g/L sodium bicarbonate, 2.7 g/L glucose, 2.0 mM L glutamine, 0.1 mM non‐essential amino acids, 0.005 mg/mL insulin, 10 ng/mL epidermal growth factor, 0.001 mg/mL transferrin, 500 ng/mL hydrocortisone, and 4% foetal bovine serum were collected and centrifuged (1000g, 5 min). The supernatant was used to treat myotubes (25% final volume in fresh medium) when indicated, and replaced every 24 h. H1299 cells (ATCC) were cultured in RPMI‐1640 medium supplemented with 10% foetal bovine serum. OMP (Tci Americas) dissolved in DMSO (0.1% final concentration) was used to treat cells.
Animal use
All studies were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experimental protocols were approved in advance by the institutional Animal Welfare Committee at the University of Texas Health Science Center at Houston. Experimental mice were group‐housed, kept on 12:12 h light–dark cycle with access to standard rodent chow and water ad libitum. LLC cells (1 × 106 in 100 μL) or an equal volume of vehicle [phosphate‐buffered saline (PBS)] was injected subcutaneously into the flanks of 8‐week‐old male mice of C57BL/6 background. When indicated, OMP dissolved in 0.1% DMSO in PBS were administered from Day 7 of tumour implant, when the tumour became palpable, through a subcutaneously implanted Alzet® Osmotic Pump (Alzet, Cupertino, CA) at a rate of 5 mg/kg/day. Control animals were administered with the vehicle accordingly. Development of cachexia was monitored by body weight change and forelimb grip strength test. Tissue samples were collected after mice had been euthanized on Day 21. Tibialis anterior (TA) and extensor digitorum longus (EDL) were excised for various analyses. Subcutaneous and epididymal fat pad were collected according to a published protocol. 31
Western blotting
Tibialis anterior homogenate was prepared by homogenizing minced muscle in Bullet Blender (Next Advance, Troy, NY) with 0.2 mm beads at maximal speed for 3 min in 4°C RIPA buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1% Nonidet P‐40, 0.1% SDS, 2 mM phenylmethylsulfonylfluoride, 0.1% sodium deoxycholate, 1 mM NaF; 1:100 protease inhibitor cocktail (Sigma‐Aldrich) containing AEBSF, pepstatin A, E‐64, bestatin, leupeptin and aprotinin; and 1:100 phosphatase inhibitor cocktail (Sigma‐Aldrich) containing sodium vanadate, sodium molybdate, sodium tartrate, and imidazole. Myotube lysate was prepared in the same buffer with sonication for 5 × 5 s. Insoluble muscle or cellular debris was removed by centrifugation at 16 000g (4°C). The supernatant was collected, and protein concentration was determined using the Bio‐Rad protein assay (Bio‐Rad, Richmond, CA, USA) with BSA as a standard. Western blot analysis was carried out as previously described. 12 Antibodies to total (9212 L) and phosphorylated p38 MAPK (4511S) were from Cell Signaling Technology (Danvers, MA). Antibodies for myosin heavy chain (MHC) (MF20) and α‐tubulin (12G10) were from Developmental Studies Hybridoma Bank at University of Iowa, Iowa City, IA. Antibody for Atrogin1/MAFbx (AP2041) was from ECM Biosciences (Versailles, KY). Antibodies for UBR2 (NBP1‐45243) LC3 (NB100‐2220), and Rab27b (NBP1‐79631) were from Novus Biologicals (Littleton, CO). GAPDH antibody (MAB374) was from Millipore.
Fluorescence microscopy and histology study
C2C12 myotubes were stained with anti‐MHC antibody (MF‐20, Development Studies Hybridoma Bank at University of Iowa, Iowa City, IA) and FITC‐conjugated secondary antibody, and examined using a Zeiss Axioskop 40 microscope at 20x and photographed with a Zeiss Axiocam MRM camera system controlled by Axiovision Release 4.6 imaging software. Myotube diameter was measured in MHC‐stained myotubes as previously described. 24 The diameters were measured in a total of 200 myotubes from ≥10 random fields using the ImageJ software (National Institutes of Health). Myotubes were measured at 3 points along their length. Excised TA was fixed with 3.7% formaldehyde and embedded in paraffin. Haematoxylin‐and‐eosin‐stained TA sections (5 μm) were prepared by the Breast Center Pathology Core (Baylor College of Medicine, Houston, TX, USA). Cross‐sectional area of stained TA sections was quantified by using the ImageJ software (National Institutes of Health). Five view‐fields with ∼100 myofibres per field in each section were measured. Data are expressed as frequency histogram.
Quantification of extracellular Hsp70 and Hsp90
Hsp70 and Hsp90α levels in cell‐conditioned medium (concentrated 20 folds by centrifugation with 10 K filters from Millipore) or mouse sera were analysed by enzyme‐linked immunosorbent assay according to the manufacturer's instruction (Enzo Life Sciences, PA). Hsp70 and Hsp90α in the culture medium from foetal bovine serum supplement were also measured and subtracted from total Hsp70 and Hsp90α levels measured in cell‐conditioned medium.
Extracellular vesicle isolation and quantitation
Hsp70/90‐carrying EVs in concentrated cell‐conditioned media or serum were isolated using the ExoQuickTM kit (System Biosciences, Mountain View, CA) 32 and quantified by measuring the activity of AchE as described previously. 5 Hsp70/90‐carrying EVs from foetal bovine serum supplement in the culture medium were also measured and subtracted from total Hsp70/90‐carrying EV levels measured in cell‐conditioned media.
Tyrosine release assay
Tyrosine release was measured as described previously. 24 Briefly, excised mouse EDL was preincubated for 30 min at 37°C in Krebs Henseleit buffer (120 mM NaCl, 4.8 mM KCl, 25 mM NaHCO3, 2.5 mM CaCl2, 1.2 mM KH2PO4, and 1.2 mM MgSO4, pH 7.4, supplemented with 5 mM glucose, 5 mM HEPES, 0.1% BSA, 0.17 mM leucine, 0.20 mM valine, 0.10 mM isoleucine, 0.1 U/mL insulin, and 0.5 mM cycloheximide; chemicals from Sigma‐Aldrich). The buffer was saturated with 95% O2 to 5% CO2. EDL was then transferred into fresh buffer and incubated for 2 h. Tyrosine released into the buffer was determined by a fluorometric method established by Waalkes and Udenfriend. 33
Ratiometric endolysosome pH measurement
Endolysosome pH was measured using a previously published protocol. 34 Cells were seeded on MatTek glass‐bottom dishes 24 h before imaging at density of 30–50%, then changed to fresh medium containing 0.2 mg/mL Oregon Green 488 conjugated dextran (Thermo Fisher Scientific # D‐7170). After overnight incubation, cells were washed three times with PBS and incubated with fresh medium for 2 h chase. The medium was then replaced with imaging buffer (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 10 mM HEPES, 10 mM glucose, pH 7.4). Images were acquired with an inverted microscope (Nikon) equipped with 40× oil immersion lens. Excitation wavelengths were switched between 440 ± 10 nm and 490 ± 10 nm by Sutter lambda‐10B Smartshutter system controlled by Incytim2 software (Intracellular Imaging). Emissions were filtered at 545 ± 50 nm and acquired with an CCD camera. Each individual endolysosomal cluster or cell was selected as one regions of interest (ROI). The mean intensity ratio (490/440) was calculated for each individual ROI. At the end of imaging, in situ pH calibration was performed in isotonic K+ solution [140 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM glucose, supplemented with 10 μM Nigericin (Cayman # 11437)], with pH of 4.0, 4.5, 4.75, 5.0, 5.25, 5.5, 6.0, 6.5, and 7.0, respectively. For each pH point, images were taken after 10 min incubation with the calibration buffer and the mean values of 490/440 of individual ROI were fitted as a function of pH to a Boltzmann sigmoid curve. The endolysosome pH values were then calculated from the equation obtained from the calibration curve. The frequency distribution of endolysosomal pH values was analysed using Prism 6 (GraphPad) and fitted into Gaussian distribution.
Real‐time PCR
Real‐time PCR was performed as described previously. 7 Sequences of specific primers are: mouse Rab27b (sense: 5′‐CTTACCACTGCCTTCTTCAGAG‐3′, antisense: 5′‐TGCAGTTGACTCATCCAGTTT‐3′) and mouse GAPDH (sense: 5′‐CATGGCCTTCCGTGTTCCTA‐3′, antisense: 5′‐GCGGCACGTCAGATCCA‐3′). Data were normalized to GAPDH.
Statistical analyses
Data were analysed with one‐way analysis of variance combined with Tukey's test, Student's t‐test or χ 2 test as appropriate using the SigmaStat software (Systat Software, San Jose, CA) as indicated. Survival data were analysed by the Kaplan–Meier curve and the log‐rank test. When applicable, control samples from independent experiments were normalized to a value of 1. All data were expressed as means ± standard deviation or standard error as indicated. Statistical significance was accepted at P < 0.05.
Results
Omeprazole inhibits LLC cell release of Hsp70 and Hsp90 through EVs and abrogates LLC cell induction of myotube atrophy
To determine whether OMP inhibits LLC cell release of Hsp70 and Hsp90 though EVs, OMP was incubated with LLC cell‐conditioned medium (LCM) for 48 h (1–10 μM). By measuring Hsp70 and Hsp90α content in the conditioned medium using enzyme‐linked immunosorbent assay, a dose‐dependent inhibition of Hsp70 and Hsp90α release by OMP was observed, indicating that OMP inhibits Hsp70/90 release by LLC cells (Figure 1A). To investigate whether OMP inhibits Hsp70/90 release by LLC cells through inhibiting the release of Hsp70/90‐carrying EVs, Hsp70/90‐carrying EVs were isolated from LCM and quantified using a previously validated protocol based on their characteristic association with AchE‐positive EVs. 5 We observed that OMP inhibited the release of Hsp70/90‐carrying EVs into the conditioned medium in a dose‐dependent manner similar to its inhibition of Hsp70/90 release (Figure 1B). These results suggest that OMP inhibits the release of Hsp70/90‐carrying EVs resulting in a blockade of Hsp70 and Hsp90 release by LLC cells.
Figure 1.
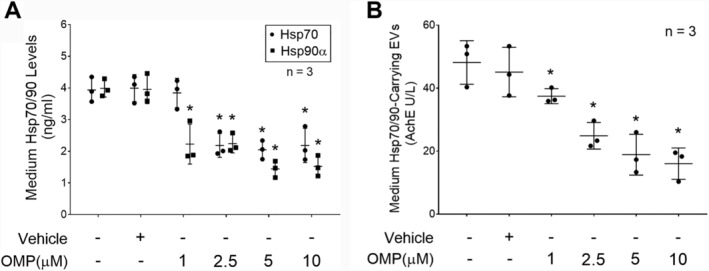
Omeprazole inhibits the release of Hsp70 and Hsp90 as well as Hsp70/90‐carrying EVs by Lewis lung carcinoma (LLC) cells in a dose‐dependent manner. LLC cells were treated with indicated concentrations of OMP for 48 h. (A) Contents of Hsp70 and Hsp90α in the culture medium were analysed by enzyme‐linked immunosorbent assay. (B) Contents of Hsp70/90‐carring EVs in the medium were quantified by measuring AchE activity in isolated EVs. Means ± SD of three independent experiments (n) were analysed by one‐way analysis of variance combined with Tukey's test. Three independent experiments were conducted. * denotes a difference from untreated LLC cell‐conditioned medium (P < 0.05). AchE, acetylcholinesterase; EVs, extracellular vesicles; OMP, omeprazole.
To determine whether OMP inhibition of Hsp70 and Hsp90 release from LLC cells resulted in impairment of their capacity to induce myotube atrophy, C2C12 myotubes were incubated with conditioned medium of LLC cells that had been treated with OMP (5 μM) or vehicle for 48 h. We observed that OMP treatment abrogated LCM‐induced catabolic response including activation of p38 MAPK (Figure 2A), up‐regulation of ubiquitin ligases atrogin1/MAFbx and UBR2, and loss of myofibrillar protein MHC (Figure 2B), as well as reduction of myotube diameters (Figure 2C). These data demonstrate that OMP abrogates LCM‐induced myotube atrophy by inhibiting LLC cell release of Hsp70 and Hsp90 through EVs.
Figure 2.
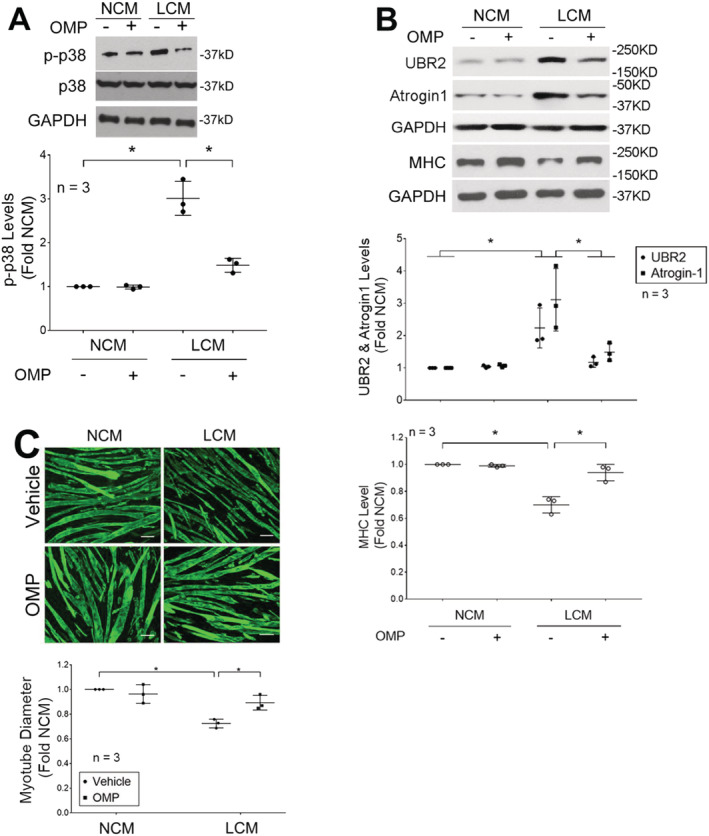
Omeprazole inhibits LCM‐induced myotube atrophy. Lewis lung carcinoma (LLC) cells were treated with omeprazole (5 μM) or vehicle for 48 h. NL20 cells were treated in parallel as control. Conditioned media of LLC cells (LCM) and NL20 cells (NCM) were then used to treat C2C12 myotubes for indicated period. (A) OMP treatment of LLC cells inhibits LCM induction of p38 MAPK activation at 1 h. (B) OMP treatment of LLC cells inhibits LCM induction of up‐regulation of atrogin1/MAFbx and UBR2 at 8 h, and loss of MHC a 72 h. (C) OMP treatment of LLC cells inhibits LCM‐induced reduction of myotube diameter at 72 h. scale bar = 50 μm. Means ± SD of three independent experiments (n) were analysed by one‐way analysis of variance combined with Tukey's test. * denotes a difference (P < 0.05). OMP, omeprazole.
Omeprazole blocks elevation of serum Hsp70 and Hsp90 and prevents cachexia in LLC tumour‐bearing mice
To investigate the in vivo effect of OMP on Hsp70 and Hsp90 release by LLC, OMP (5 mg/kg/day) or vehicle was administered subcutaneously through an osmotic pump to mice from Days 7 to 20 of LLC cell implant. On Day 21, mice were euthanized and analysed. A blockade of elevation of serum Hsp70 and Hsp90α were observed in LLC tumour‐bearing mice administered with OMP (Figure 3A). In addition, a similar blockade of elevation of serum Hsp70/90‐carrying EVs was observed (Figure 3B). Consistent to the in vitro data, these in vivo data support that OMP blocks the release of Hsp70 and Hsp90 through EVs. In addition, OMP treatment prevented the catabolic response in TA as indicated by attenuated p38 MAPK activation, atrogin1/mAFbx and UBR2 up‐regulation, LC3‐II increase, and MHC loss (Figure 3C). On the other hand, OMP did not alter tumour volume (Figure 3D). Consequently, OMP treatment ameliorated cachexia in LLC tumour‐bearing mice as measured by body weight (Figure 4A), muscle (TA and EDL) weight (Figure 4B), muscle protein degradation measured by tyrosine release from EDL (Figure 4C), muscle function measured by forelimb grip strength (Figure 4D), and myofibre size measured by myofibre cross‐sectional area of TA (Figure 4E). Notably, OMP treatment also attenuated the loss of adipose tissue mass (subcutaneous and epididymal fat pad) (Figure 4F). These data indicate that OMP ameliorates cancer cachexia by attenuating both skeletal muscle and adipose tissue wasting.
Figure 3.
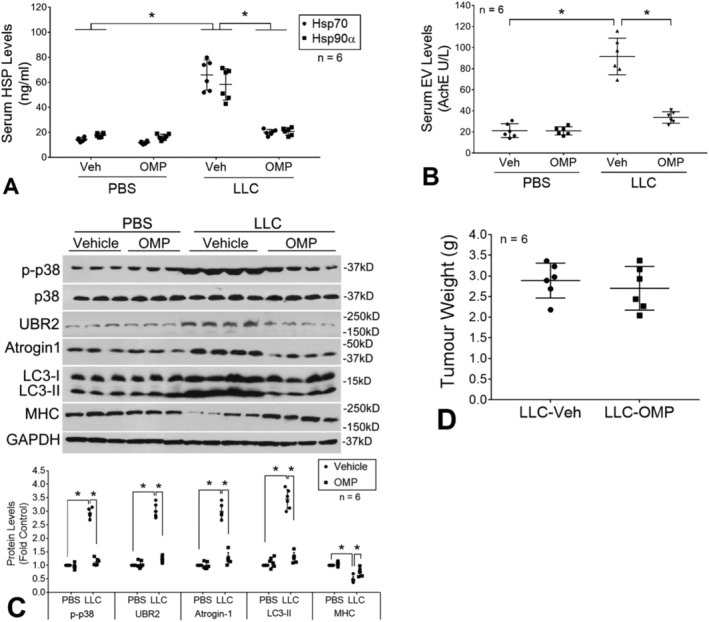
Omeprazole abrogates elevation of serum Hsp70 and Hsp90 as well as Hsp70/90‐carrying EVs resulting in abrogated muscle catabolism in LLC tumour‐bearing mice. OMP (5 mg/kg/day, SC) or vehicle was administered to mice bearing LLC tumor starting from Days 7 to 20 of LLC cell implant. On Day 21, mice were euthanized for analyses. OMP treatment blocked elevation of serum Hsp70 and Hsp90α (A) and Hsp70/90‐carrying EVs (B), and muscle catabolism in TA (C), but did not alter tumor growth (D). Means ± SD of six mice per group (n) were analysed by one‐way analysis of variance combined with Tukey's test. * denotes a difference (P < 0.05). LLC, Lewis lung carcinoma; OMP, omeprazole; PBS, phosphate‐buffered saline.
Figure 4.
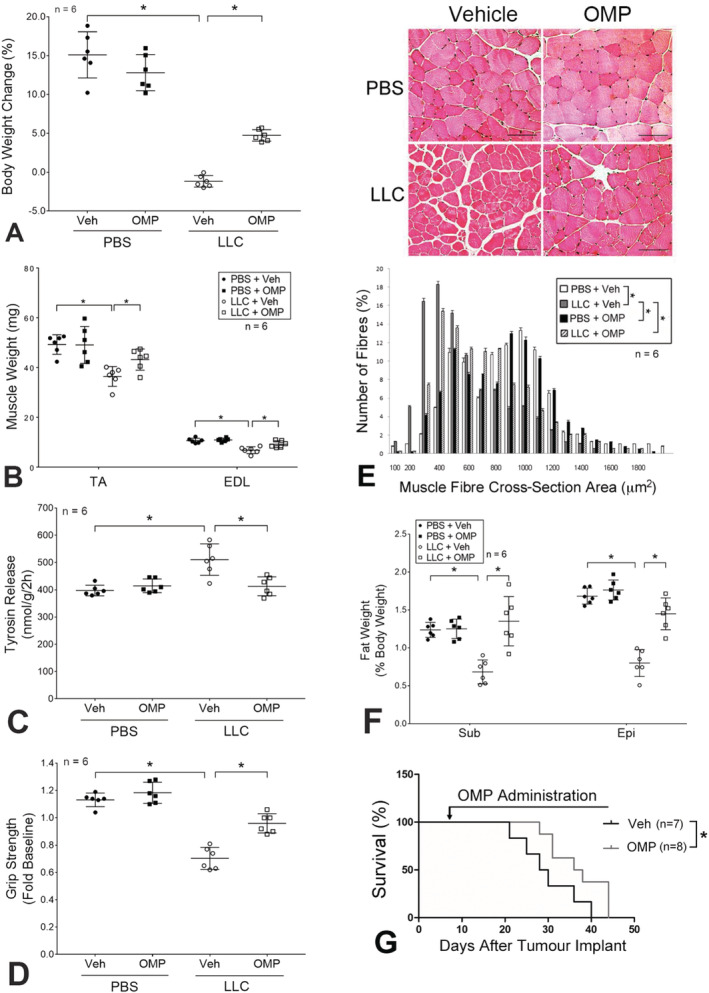
Omeprazole ameliorates cachexia and prolongs survival of LLC tumour‐bearing mice. Mice derived from Figure 3 (six mice per group) were analysed for cachexia by measuring body weight (A), muscle weight (B), protein degradation (tyrosine release) (C), muscle function (grip strength) (D), myofibre cross‐sectional area of TA (E), and subcutaneous (Sub) and epididymal (Epi) fat pad mass (F). (G) Omeprazole prolongs survival of LLC tumour‐bearing mice. OMP (5 mg/kg/day, SC, eight mice) or vehicle (seven mice) was administered to mice bearing LLC tumor from Day 7 of LLC cell implant and continued till pre‐determined humane endpoint was reached. Data were plotted using the Kaplan–Meier survival curve and analysed by the log‐rank test (P < 0.05). Data in (E) were means ± SD analysed by the χ 2 test. Scale bar = 100 μm. The rest of data were presented as means ± SD and analysed by one‐way analysis of variance combined with Tukey's test. * denotes a difference (P < 0.05). LLC, Lewis lung carcinoma; OMP, omeprazole; PBS, phosphate‐buffered saline.
To determine whether preservation of skeletal muscle and fat mass by OMP translates into longer survival of cachectic mice, OMP treatment was extended till the mice reached pre‐determined humane endpoint. As shown in Figure 4G, LLC tumour‐bearing mice treated with vehicle DMSO had a median survival of 30 days. However, OMP‐treated LLC tumour‐bearing mice survived longer to a median survival of 37 days. Thus, OMP treatment increased the survival of LLC tumour‐bearing mice by 23.3%.
Omeprazole alkalinizes endolysosomal pH and abolishes elevated expression of Rab27b in LLC cells
It was proposed that PPIs inhibit EV release by inhibiting V‐H+‐ATPase, although detailed mechanism remains undefined. 29 , 30 To verify whether OMP inhibited V‐H+‐ATPase at the concentrations we used in LLC cells, we investigated the effect of OMP (0.01–10 μM) on the pH levels in endolysosomes of two lung cancer cell lines. Endolysosomes are intracellular vacuolar compartments similar to MVB. Using the dextran loading method 34 to label endolysosomes with a ratiometric pH indicator, Oregon Green 488, we observed that OMP treatment of LLC cells as well as human lung carcinoma cell line H1299 increased endolysosomal pH in a dose and time‐dependent manner (Figure 5). Thus, OMP inhibition of EV release is associated with an inhibition of V‐H+‐ATPase activity, which alkalinized pH in intracellular vacuolar compartments. However, the dose‐dependent increase of endolysosomal pH by OMP treatment was largely in the 0.01–1 μM range, whereas dose‐dependent inhibition of Hsp70/90‐carrying EV release by OMP was largely from 1 to 10 μM (Figure 1A), suggesting that additional mechanisms might be involved in OMP inhibition of EV release.
Figure 5.
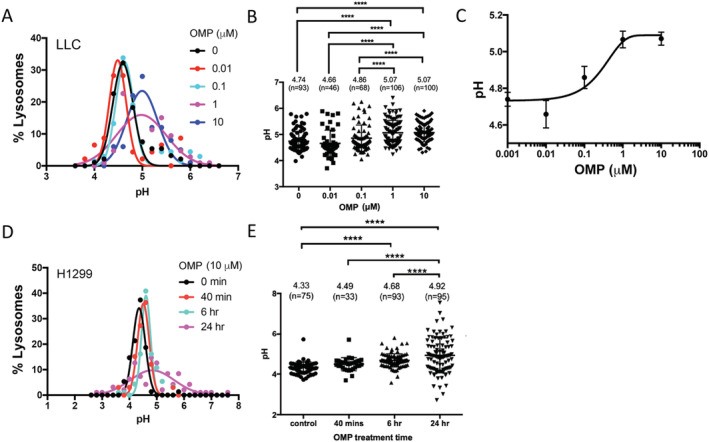
Omeprazole increases endolysosomal pH in LLC and H1299 cells. Cancer cells loaded with Oregon Green 488 conjugated dextran were subjected to OMP treatment for 24 h or indicated time period. The pH values in individual lysosomes ware determined by Oregon Green fluorescence ratios. (A–C) OMP treatment of LLC cells (0–10 μM) for 24 h increases endolysosomal pH in a concentration‐dependent manner. Data are presented as Gaussian distribution plots of pH values of individual lysosomes (A), individual data points and means ± SE (B), as well as a dose–response curve fitted by the Hill equation (C). Number of lysosomes measured at each time points ranges from 46 to 100 as indicated in panel (B). (D–E) OMP treatment of H1299 cells (10 μM) increases endolysosomal pH in a time‐dependent manner. Shown are pH distribution plots of individual lysosomes (D) and individual data points and means ± SE (E). Number of lysosomes measured at each time points ranges from 33 to 96 as indicated in panel (B). Data were analysed by one‐way analysis of variance combined with Tukey's test. ****P < 0.0001. LLC, Lewis lung carcinoma; OMP, omeprazole.
Rab27b is a GTPase that controls EV release. 35 Increased expression of Rab27b has been observed in pancreatic, breast, bladder, and liver cancers. 36 , 37 , 38 , 39 We previously showed that the high‐level expression of Rab27b in pancreatic cancer cells is responsible for their high‐level release of Hsp70/90‐carrying EVs. 10 Given the high‐level release of Hsp70/90‐carrying EVs by LLC cells, we postulated that LLC cells may also highly express Rab27b, which is suppressed by OMP. By examining both the mRNA and protein levels of Rab27b, we found that in LLC cells expression of the Rab27b mRNA was four times higher than the control cells, non‐tumorigenic human lung epithelial cell line NL20 that releases basal level of Hsp70/90 5 (Figure 6A), resulting in a 3.5 times higher level of Rab27b protein (Figure 6B). Remarkably, OMP treatment of LLC cells suppressed Rab27b mRNA and protein expression to almost the basal level in NL20 cells (Figure 6A and 6B). Given that Rab27b is required for EV release, the suppression of Rab27b expression by OMP is likely to contribute to its blockade of Hsp70/90‐carrying EV release in addition to its inhibition of V‐H+‐ATPase.
Figure 6.
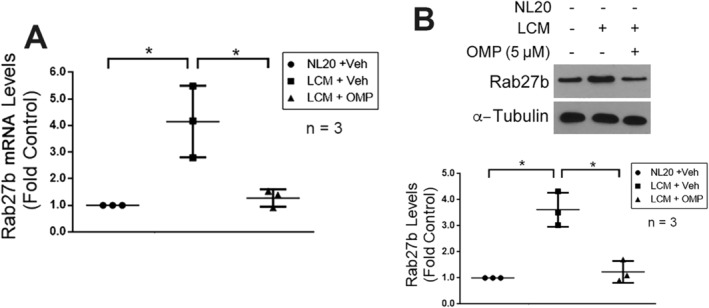
OMP suppresses elevated expression of Rab27b in LLC cells. (A) OMP blocks high‐level expression of the mRNA of Rab27b in LLC cells. Lewis lung carcinoma (LLC) cells were treated with OMP (5 μM) or vehicle for 4 h. The mRNA levels of RAB27b were determined by real‐time PCR. NL20 cells were used as control. (B) OMP blocks high level expression of Rab27b protein in LLC cells. LLC cells were treated with OMP (5 μM) or vehicle for 8 h. The protein levels of Rab27b were determined by western blot analysis. Means ± SD from three independent experiments (n) were analysed by one‐way analysis of variance combined with Tukey's test. * denotes a difference (P < 0.05). LCM, LLC cell‐conditioned medium; OMP, omeprazole.
Discussion
The current study suggests a therapeutic strategy for cancer cachexia by targeting cancer cell release of Hsp70 and Hsp90 pharmacologically utilizing an over‐the‐counter PPI, OMP. It also reveals that the underlying mechanism of the OMP action involves inhibition of cancer cell release of Hsp70/90‐carrying EVs by increasing intracellular vacuolar pH and suppressing Rab27b expression. Given the lack of an approved medication for cancer cachexia, OMP could be considered for off‐label use to improve cancer patient care and survival by alleviating cancer cachexia.
Cancer cachexia is defined as a multifactorial syndrome characterized by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and leads to progressive functional impairment. 1 The current study reveals that OMP ameliorates cancer cachexia by attenuating the loss of both skeletal muscle and adipose tissue mass. Consequently, OMP not only preserved muscle function but also prolonged survival of LLC tumour‐bearing mice.
Due to the lack of the signal peptide sequence that mediates protein secretion via the conventional route, the release of Hsp70 and Hsp90 by cancer cells is normally through EVs for intracellular communications, except that damaged cells can release free Hsp70/90. 40 , 41 , 42 , 43 The composition of EVs is highly heterogeneous and cell type‐dependent, therefore, there is not an ‘optimal’ method that is uniformly suitable for isolating and quantifying EVs. 11 , 44 Although AchE is not considered a generic marker of EVs, 11 it is associated with Hsp70/90‐carrying EVs as confirmed previously. 5 Therefore, to quantify Hsp70/90‐carrying EVs in an EV preparation that inevitably contains heterogeneous populations of EVs, measuring AchE activity could be more accurate than measuring the protein content or particle number of the EV preparation. 5 The high degree of fidelity between OMP inhibition of Hsp70/90 release and AchE‐positive EV release by LLC cells observed in vitro (Figure 1) and in vivo (Figure 3A and 3B) further verified that the detected Hsp70 and Hsp90 were associated with AchE‐positive EVs.
The effectiveness of OMP in blocking Hsp70 and Hsp90 release and ameliorating skeletal muscle wasting verifies the previous findings that cancer‐released Hsp70 and Hsp90 are key cachexins that cause muscle wasting and cachexia. 5 TLR4 has been identified as the Hsp70/90 receptor that mediates cancer‐induced muscle wasting. 5 , 23 In addition, our data suggest that Hsp70 and Hsp90 also mediate cancer‐induced adipose tissue wasting, which was also shown to be dependent on TLR4. 26 TLR4 stimulates skeletal muscle mass loss primarily through the activation of p38β MAPK‐regulated protein degradation pathways. 6 However, the intracellular signalling through which TLR4 stimulates adipose tissue mass loss remains to be defined.
The choice of using young adult mice in this study is based on the considerations that cancer cachexia has a different pathogenesis from sarcopenia (age‐related muscle wasting) 45 and that cancer occurs in a wide range of ages including young adults. Our data revealed that tumour blunted body weight gain in young adult mice, which was attenuated by OMP (Figure 4A). This protective effect of OMP can be attributed to its anti‐catabolic actions in skeletal muscle and visceral fat tissue. OMP abrogated the ∼30% increase in muscle protein degradation (Figure 4C), resulting in a robust protection against tumour‐induced loss of muscle myofibre cross‐sectional area (Figure 4E). In addition, LLC caused ∼50% loss of epididymal fat tissue mass, which was partially attenuated by OMP (Figure 4F).
Microenvironmental pH is a key factor for EV traffic in tumour cells. 28 We demonstrate that OMP blocks cancer cell release of Hsp70/90‐carrying EVs while increasing endolysosomal pH, a result of its inhibition of V‐H+‐ATPase. V‐H+‐ATPase pumps protons across the plasma membrane and across the membranes of intracellular vacuolar compartments including the MVB 46 and is responsible for the establishment and maintenance of the acidic pH of endocytic and secretory organelles. 47 V‐H+‐ATPase is also present in Rab27b‐positive EVs and regulates the size and peripheral distribution of Rab27b‐positive EVs that mediates Hsp90 release by breast cancer cells, 48 although the detailed mechanism is unknown. Our data suggest that OMP inhibition of V‐H+‐ATPase contributes to its inhibition of Hsp70/90‐carrying EV release.
We previously showed that Rab27b is required for Hsp70/90‐carrying EV release from cachexia‐prone cancer cells. 5 , 10 However, the regulation of Rab27b is still poorly understood. We found in the current study that LLC cells expressed very high levels of Rab27b, which is consistent to the high‐level release of Hsp70/90‐carrying EVs. Remarkably, OMP abolished the heightened Rab27b expression. Given the dependence of Hsp70/90‐carrying EV release on Rab27b, 5 , 10 this OMP action is likely to contribute to its capacity of blocking Hsp70/90‐carrying EV release. Considering the time course of OMP inhibition of Rab27b mRNA expression (<4 h, Figure 6A) this action of OMP is unlikely a result of its increase of endolysosomal pH level, because that OMP treatment increased endolysosomal pH gradually and at 6 h of treatment endolysosomal pH value increased only moderately (Figure 5E).
Proton pump inhibitors are extensively used in clinical settings for gastric‐acid‐related diseases, and considered relatively safe for use over extended periods. 49 Approximately 20% of cancer patients are treated with PPIs to alleviate the symptoms of gastroesophageal reflux. 50 In addition, by inhibiting V‐H+‐ATPase, PPIs sensitize cancer cells to chemotherapy 51 , 52 and have been proposed for repositioning to increase the efficacy and safety of chemotherapy. 53 The current study demonstrates that OMP could be effective for the very‐difficult‐to‐treat cancer cachexia. Among the existing PPIs, OMP is considered particularly safe as an OTC drug and of very low cost. Thus, OMP could be an inexpensive addition to cancer therapy for patients with cachexia.
Funding
This study is supported by a grant from National Institute of Arthritis and Musculoskeletal and Skin Diseases to Y‐P.L. (R01 AR063786), and a grant from National Cancer Institute to Y‐P.L. and M.L. (R01 CA203108).
Conflict of interests
The authors declare that they have no competing interests.
Acknowledgements
We thank James Z. Zhu for technical support.
Liu Z., Xiong J., Gao S., Zhu M. X., Sun K., Li M., Zhang G., and Li Y.‐P. (2022) Ameliorating cancer cachexia by inhibiting cancer cell release of Hsp70 and Hsp90 with omeprazole, Journal of Cachexia, Sarcopenia and Muscle, 13, 636–647, 10.1002/jcsm.12851
Contributor Information
Guohua Zhang, Email: zhgh71@163.com.
Yi‐Ping Li, Email: yi-ping.li@uth.tmc.edu.
References
- 1. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 2. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 3. Advani SM, Advani PG, VonVille HM, Jafri SH. Pharmacological management of cachexia in adult cancer patients: a systematic review of clinical trials. BMC Cancer 2018;18:1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dingemans AM, de Vos‐Geelen J, Langen R, Schols AM. Phase II drugs that are currently in development for the treatment of cachexia. Expert Opin Investig Drugs 2014;23:1655–1669. [DOI] [PubMed] [Google Scholar]
- 5. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KW, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 2017;8:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sin TK, Zhang G, Zhang Z, Gao S, Li M, Li YP. Cancer takes a toll on skeletal muscle by releasing heat shock proteins—an emerging mechanism of cancer‐induced cachexia. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang G, Jin B, Li YP. C/EBPbeta mediates tumour‐induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J 2011;30:4323–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang G, Lin RK, Kwon YT, Li YP. Signaling mechanism of tumor cell‐induced up‐regulation of E3 ubiquitin ligase UBR2. FASEB J 2013;27:2893–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Z, Sin KWT, Ding H, Doan HA, Gao S, Miao H, et al. p38beta MAPK mediates ULK1‐dependent induction of autophagy in skeletal muscle of tumor‐bearing mice. Cell Stress 2018;2:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J, Zhang Z, Zhang Y, Ni X, Zhang G, Cui X, et al. ZIP4 promotes muscle wasting and cachexia in mice with orthotopic pancreatic tumors by stimulating RAB27B‐regulated release of extracellular vesicles from cancer cells. Gastroenterology 2019;156:722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lotvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014;3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzuki K, Ito Y, Wakai K, Kawado M, Hashimoto S, Seki N, et al. Serum heat shock protein 70 levels and lung cancer risk: a case‐control study nested in a large cohort study. Cancer Epidemiol Biomarkers Prev 2006;15:1733–1737. [DOI] [PubMed] [Google Scholar]
- 13. Shi Y, Liu X, Lou J, Han X, Zhang L, Wang Q, et al. Plasma levels of heat shock protein 90 alpha associated with lung cancer development and treatment responses. Clin Cancer Res 2014;20:6016–6022. [DOI] [PubMed] [Google Scholar]
- 14. Rong B, Zhao C, Liu H, Ming Z, Cai X, Gao W, et al. Identification and verification of Hsp90‐beta as a potential serum biomarker for lung cancer. Am J Cancer Res 2014;4:874–885. [PMC free article] [PubMed] [Google Scholar]
- 15. Gunther S, Ostheimer C, Stangl S, Specht HM, Mozes P, Jesinghaus M, et al. Correlation of Hsp70 serum levels with gross tumor volume and composition of lymphocyte subpopulations in patients with squamous cell and adeno non‐small cell lung cancer. Front Immunol 2015;6:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Balazs M, Zsolt H, László G, Gabriella G, Lilla T, Gyula O, et al. Serum heat shock protein 70, as a potential biomarker for small cell lung cancer. Pathol Oncol Res 2016;23:377–383. [DOI] [PubMed] [Google Scholar]
- 17. Ren B, Luo S, Xu F, Zou G, Xu G, He J, et al. The expression of DAMP proteins HSP70 and cancer‐testis antigen SPAG9 in peripheral blood of patients with HCC and lung cancer. Cell Stress Chaperones 2017;22:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rabinowits G, Gercel‐Taylor C, Day JM, Taylor DD, Kloecker GH. Exosomal microRNA: a diagnostic marker for lung cancer. Clin Lung Cancer 2009;10:42–46. [DOI] [PubMed] [Google Scholar]
- 19. Kocsis J, Madaras B, Toth EK, Fust G, Prohaszka Z. Serum level of soluble 70‐kD heat shock protein is associated with high mortality in patients with colorectal cancer without distant metastasis. Cell Stress Chaperones 2010;15:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Silva J, Garcia V, Rodriguez M, Compte M, Cisneros E, Veguillas P, et al. Analysis of exosome release and its prognostic value in human colorectal cancer. Genes Chromosomes Cancer 2012;51:409–418. [DOI] [PubMed] [Google Scholar]
- 21. Dutta SK, Girotra M, Singla M, Dutta A, Stephen FO, Nair PP, et al. Serum HSP70: a novel biomarker for early detection of pancreatic cancer. Pancreas 2012;41:530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Q, Ni Q, Wang X, Zhu H, Wang Z, Huang J. High expression of RAB27A and TP53 in pancreatic cancer predicts poor survival. Med Oncol 2015;32:372. [DOI] [PubMed] [Google Scholar]
- 23. Zhang G, Liu Z, Ding H, Miao H, Garcia JM, Li YP. Toll‐like receptor 4 mediates Lewis lung carcinoma‐induced muscle wasting via coordinate activation of protein degradation pathways. Sci Rep 2017;7:2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll‐like receptor 4 mediates lipopolysaccharide‐induced muscle catabolism via coordinate activation of ubiquitin‐proteasome and autophagy‐lysosome pathways. FASEB J 2011;25:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zu L, He J, Jiang H, Xu C, Pu S, Xu G. Bacterial endotoxin stimulates adipose lipolysis via toll‐like receptor 4 and extracellular signal‐regulated kinase pathway. J Biol Chem 2009;284:5915–5926. [DOI] [PubMed] [Google Scholar]
- 26. Henriques F, Lopes MA, Franco FO, Knobl P, Santos KB, Bueno LL, et al. Toll‐like receptor‐4 disruption suppresses adipose tissue remodeling and increases survival in cancer cachexia syndrome. Sci Rep 2018;8:18024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol 2014;29:116–125. [DOI] [PubMed] [Google Scholar]
- 28. Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem 2009;284:34211–34222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS ONE 2014;9:e88193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guan XW, Zhao F, Wang JY, Wang HY, Ge SH, Wang X, et al. Tumor microenvironment interruption: a novel anti‐cancer mechanism of proton‐pump inhibitor in gastric cancer by suppressing the release of microRNA‐carrying exosomes. Am J Cancer Res 2017;7:1913–1925. [PMC free article] [PubMed] [Google Scholar]
- 31. Tan P, Pepin E, Lavoie JL. Mouse adipose tissue collection and processing for RNA analysis. J Vis Exp 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor DD, Zacharias W, Gercel‐Taylor C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol Biol 2011;728:235–246. [DOI] [PubMed] [Google Scholar]
- 33. Waalkes TP, Udenfriend S. A fluorometric method for the estimation of tyrosine in plasma and tissues. J Lab Clin Med 1957;50:733–736. [PubMed] [Google Scholar]
- 34. Cai M, He J, Xiong J, Tay LW, Wang Z, Rog C, et al. Phospholipase D1‐regulated autophagy supplies free fatty acids to counter nutrient stress in cancer cells. Cell Death Dis 2016;7:e2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 2010;12:19–30. [DOI] [PubMed] [Google Scholar]
- 36. Zhao H, Wang Q, Wang X, Zhu H, Zhang S, Wang W, et al. Correlation between RAB27B and p53 expression and overall survival in pancreatic cancer. Pancreas 2016;45:204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hendrix A, Maynard D, Pauwels P, Braems G, Denys H, van den Broecke R, et al. Effect of the secretory small GTPase Rab27B on breast cancer growth, invasion, and metastasis. J Natl Cancer Inst 2010;102:866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ho JR, Chapeaublanc E, Kirkwood L, Nicolle R, Benhamou S, Lebret T, et al. Deregulation of Rab and Rab effector genes in bladder cancer. PLoS ONE 2012;7:e39469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong WW, Mou Q, Chen J, Cui JT, Li WM, Xiao WH. Differential expression of Rab27A/B correlates with clinical outcome in hepatocellular carcinoma. World J Gastroenterol 2012;18:1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL, et al. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Biol Chem 2012;287:15874–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCready J, Sims JD, Chan D, Jay DG. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 2010;10:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li X, Wang S, Zhu R, Li H, Han Q, Zhao RC. Lung tumor exosomes induce a pro‐inflammatory phenotype in mesenchymal stem cells via NFkappaB‐TLR signaling pathway. J Hematol Oncol 2016;9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy‐Martin JP, et al. Membrane‐associated Hsp72 from tumor‐derived exosomes mediates STAT3‐dependent immunosuppressive function of mouse and human myeloid‐derived suppressor cells. J Clin Invest 2010;120:457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larsson L, Degens H, Li M, Salviati L, Lee YI, Thompson W, et al. Sarcopenia: aging‐related loss of muscle mass and function. Physiol Rev 2019;99:427–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nishi T, Forgac M. The vacuolar (H+)‐ATPases—nature's most versatile proton pumps. Nat Rev Mol Cell Biol 2002;3:94–103. [DOI] [PubMed] [Google Scholar]
- 47. Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 2007;8:917–929. [DOI] [PubMed] [Google Scholar]
- 48. Hendrix A, Sormunen R, Westbroek W, Lambein K, Denys H, Sys G, et al. Vacuolar H+ ATPase expression and activity is required for Rab27B‐dependent invasive growth and metastasis of breast cancer. Int J Cancer 2013;133:843–854. [DOI] [PubMed] [Google Scholar]
- 49. Malfertheiner P, Kandulski A, Venerito M. Proton‐pump inhibitors: understanding the complications and risks. Nat Rev Gastroenterol Hepatol 2017;14:697–710. [DOI] [PubMed] [Google Scholar]
- 50. Smelick GS, Heffron TP, Chu L, Dean B, West DA, DuVall SL, et al. Prevalence of acid‐reducing agents (ARA) in cancer populations and ARA drug‐drug interaction potential for molecular targeted agents in clinical development. Mol Pharm 2013;10:4055–4062. [DOI] [PubMed] [Google Scholar]
- 51. Lindner K, Borchardt C, Schöpp M, Bürgers A, Stock C, Hussey DJ, et al. Proton pump inhibitors (PPIs) impact on tumour cell survival, metastatic potential and chemotherapy resistance, and affect expression of resistance‐relevant miRNAs in esophageal cancer. J Exp Clin Cancer Res 2014;33:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst 2004;96:1702–1713. [DOI] [PubMed] [Google Scholar]
- 53. Ikemura K, Hiramatsu S, Okuda M. Drug repositioning of proton pump inhibitors for enhanced efficacy and safety of cancer chemotherapy. Front Pharmacol 2017;8:911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the journal of cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]