

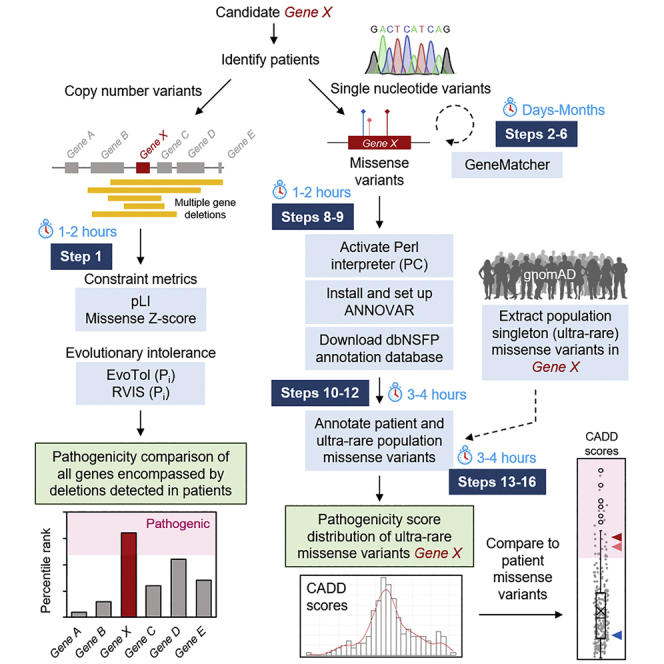
Summary
Identifying a disease gene and determining its causality in patients can be challenging. Here, we present an approach to predicting the pathogenicity of deletions and missense variants for an autosomal dominant gene. We provide online resources for identifying patients and determining constraint metrics to isolate the causal gene among several candidates encompassed in a shared region of deletion. We also provide instructions for optimizing functional annotation programs that may be otherwise inaccessible to a nonexpert or novice in computational approaches.
For complete details on the use and execution of this protocol, please refer to Gennarino et al. (2018).
Subject areas: Sequence analysis, Health Sciences, Clinical Protocol, Genetics, Sequencing
Graphical abstract
Highlights
-
•
Recruit affected patients harboring variation in a candidate gene of interest
-
•
Identify a single causal gene within a large genomic deletion spanning multiple loci
-
•
Annotate genetic variants with multiple pathogenicity prediction scores
-
•
Assess pathogenicity range of singleton missense variants from the general population
Identifying a disease gene and determining its causality in patients can be challenging. Here, we present an approach to predicting the pathogenicity of deletions and missense variants for an autosomal dominant gene. We provide online resources for identifying patients and determining constraint metrics to isolate the causal gene among several candidates encompassed in a shared region of deletion. We also provide instructions for optimizing functional annotation programs that may be otherwise inaccessible to a nonexpert or novice in computational approaches.
Before you begin
We begin with the assumption that the gene of interest has been found to produce a phenotype in the model organism, which in our case was a mouse. To then identify human patients and establish the pathogenicity of genetic variants will take some time, with particular attention to two critical points.
Assembling a cohort of patients begins with databases that contain the clinical features of interest in de-identified individuals. In our case, a haploinsufficient mouse model of Pum1 displayed ataxia reminiscent of spinocerebellar ataxia (SCA) type 1, and the gene of interest modifies ATAXIN1 (ATXN1) activity (Gennarino et al., 2015), so we searched databases of patients with ataxia but without identified genetic causes (see below, in key resources table). Because the Pum1−/− knockout mice had a more severe phenotype of neurodevelopmental delay (NDD), we also searched databases for cases of developmental or intellectual delays without known genetic causes. Lastly, you will need approval from your Institutional Review Board (IRB) and that of the clinicians with whom you share information about patients.
Key resources table
Step-by-step method details
Note: The process of patient recruitment will vary significantly from project to project. Readers should look for ways to adapt the process we describe below to their specific needs.
Identifying case-control association between PUM1-spanning copy number variations (CNV) and neurodevelopmental disorders (NDD)
Timing: Months
To validate the connection between loss-of-function (LOF) in PUM1 and neurodevelopmental disease (NDD), we screened several public and private consortia (Decipher, nstd102, see key resources table) (Firth et al., 2009; Kaminsky et al., 2011), in-house databases (Baylor Genetics), and the published literature (Gennarino et al., 2018; Riggs et al., 2012) for individuals for putatively pathogenic variation in PUM1 (see key resources table). Our search criteria were limited to individuals with deletions <20 Mb in size and sufficient clinical characterization to establish a diagnosis of NDD. We identified 9 individuals with de novo heterozygous deletions involving PUM1 that ranged in size from 0.6 to 5.60 Mb (Figure 1A). All deletions overlapped the entire PUM1 locus on chromosome 1.
Figure 1.

Comparison of constraint metrics and mutation intolerance scores among genes in the deleted regions
(A) Alignment of deletions from nine patients on chromosome 1p35.2; Mb, megabases. Dashed lines indicate the minimal region spanning PUM1, figure adapted from Gennarino et al. (2018).
(B–F) Bar graphs of constraint metrics and variant intolerance algorithms: (B) EvoTol (C) ExAC pLI, (D) RVIS (E) ExAC missense Z-score and (F) pcGERP. The dotted red arrow delineates the recommended threshold beyond which (pink shaded region) variants are predicted to be deleterious. Genes for which scores were not predictable or not available are denoted as not applicable (n/a).
Although all 9 deletions encompassed PUM1, each deletion also spanned adjacent genes (15 in total) which could create additive effects beyond the activities of PUM1. We evaluated the disease association for each gene using the three databases, the Online Mendelian Inheritance in Man (OMIM) (Amberger et al., 2015), Mouse Genome Informatics (MGI, The Jackson Laboratory), and DisGeNET (Pinero et al., 2017) (see key resources table), and, with the exception of FABP3, found no association with neurodevelopmental abnormalities. We then further showed that PUM1 was the gene most likely to cause the disease features by comparing population-based constraint and tolerance metrics:
Assess population constraint metrics and mutation tolerance sensitivity scores for all genes encompassed in the overlapping region of deletions
In this step, users will use a series of web-based tools to obtain constraint metrics and dosage sensitivity scores for a group of genes within a shared region encompassed by large deletion variants identified in patients. Comparing pathogenicity metrics will assist users in distinguishing which among the deleted candidates is the most likely shared causal gene in all patients.
-
1.Obtain pre-calculated scores using each of the URL provided below:
-
a.pLI and Missense Z-score
-
i.Go to URL: https://gnomad.broadinstitute.org/
-
ii.Choose ExAC in the dropdown menu
-
iii.Enter single Entrez gene ID
-
iv.Scores provided under “Constraint”
-
v.Repeat for each respective gene
-
i.
-
b.EvoTol (evolutionary intolerance) (Rackham et al., 2015)
-
i.Go to URL: http://www.evotol.co.uk/
-
ii.Enter comma delimited list of Entrez gene ID for all genes of interest
-
iii.“EvoTol percentile” scores will be tabulated and graphed.Note: Score may not be available for genes that are not expressed above
-
i.
-
c.RVIS (residual variation intolerance score) (Petrovski et al., 2013)
-
i.Go to URL: http://genic-intolerance.org/
-
ii.Enter comma delimited list of Entrez gene ID for all genes of interest
-
iii.RVIS scores provided with percentiles in parenthesesEach gene can be assessed as to whether its score falls within the predicted range for pathogenicity. For instance, genes with pLI scores ≥ 0.9 are interpreted as extremely intolerant to loss-of-function (Lek et al., 2016; Samocha et al., 2014). A description of each score and recommended pathogenicity thresholds are provided in Table 1. In addition, the scores for all genes can be directly compared to one another and ranked relative to each other, especially in situations where multiple genes are predicted to be pathogenic.Returning to the example in Gennarino et al. (2018), PUM1 was determined to be the most likely causal gene within the deleted regions (Figure 1A) because (1) its loss of function measures consistently surpassed the pathogenicity threshold, and, more importantly, (2) its effect was greater than that of any of the other 15 deleted genes (Figures 1B–1F).
-
i.
-
a.
Table 1.
Description and recommended pathogenicity thresholds for gene-based constraint metrics and intolerance scores for the assessment of multiple genes encompassed with large copy-number variants in patients.
| Algorithm | Description | Score | Threshold interpretation | Reference |
|---|---|---|---|---|
| pLI | Dichotomous metric that reflects the probability of loss of function intolerance based on predicted protein truncating variation: nonsense, splice acceptor and splice donor variation | 0 to 1 | pLI ≥ 0.9 = Extremely intolerant | (Samocha et al., 2014) (Lek et al., 2016) |
| Missense Z-score | Standard deviation of the # of observed rare (<1% MAF) missense SNP's from the mean of the predicted number of expected rare (<1% MAF) missense SNP's for a given gene. (Synonymous variants typically have Z-score close to 0 and o/e close to 1) | -5 to 5 | Z-score > 0 = More Intolerant Z-score < 0 = Less intolerant |
(Samocha et al., 2014) (Lek et al., 2016) |
| EvoTol percentile | Percentile ranking of a gene's evolutionary intolerance relative to other genes based on the number of damaging versus non-damaging variants in dbSNP. | Percentile (priority ranking) | Top 25th percentile = Intolerant Top 1 percentile = Most intolerant |
(Rackham et al., 2015) |
| RVIS percentile | Percentile ranking of a gene's intolerance to functional variation relative to other genes based on observed versus expected frequency of loss-of-function variants from the NHLBI-ESP6500 data set in ExAC (release 0.3). | Percentile (priority ranking) | Bottom 25th percentile = Intolerant (Genes are ranked from most to least intolerant) |
(Petrovski et al., 2013) |
| pcGERP percentile | Percentile estimate reflecting how conserved the protein-coding sequence of a gene relative to other genes | Percentile estimate | Lower percentile = More intolerance (increased conservation) (Genes are ranked from most to least intolerant) |
(Davydov et al., 2010) (Cooper et al., 2005) (Petrovski et al., 2015) |
Abbreviation: pLI, probability of being Loss-of-function Intolerant; EvoTol, Evolutionary Intolerance; RVIS, Residual Variation Intolerance Score; pcGERP, protein-coding Genomic Evolutionary Rate Profiling; MAF, minor allele frequency.
Identifying NDD patients with pathogenic missense variants in PUM1
Here we used the publicly accessible GeneMatcher website (https://genematcher.org/) (Sobreira et al., 2015). The site allows users (clinicians, patients/families and researchers) to post a gene of interest and upload corresponding data such as clinical and demographic features. A match occurs when any of the following criteria are shared between two or more entries: (1) OMIM number, (2) gene symbol, (3) genomic location, or (4) phenotypic features. This search yielded three additional missense variants: two simplex cases, and a large family with multiple members segregating with a late-onset cerebellar ataxia. Below is a step-by-step guide on how to submit to GeneMatcher:
-
2.Go to URL https://genematcher.org/account/ and create a new user account
-
a.There are no email domain restrictions but it is recommended to provide academic institution-based credentials.
-
a.
-
3.
Login to the system portal with account credentials
-
4.
In the user account homepage, click the “Create a new submission” link under the “Submissions” tab
-
5.Under this “New submission” page, the match criteria for your gene can be specified
-
a.Under section “Results (human genes only, maximum of 10 genes only)”, we recommend providing a gene symbol without specifying the variant
-
a.
Note: Setting the minimum amount of match criteria at this stage is recommended. For novel genes, limiting criteria such as the type of variation, mode of inheritance, etc. are likely unknown, and overspecification will restrict the identification of cases.
-
6.
Gene “Match” notifications with information to contact submitters will be sent to the email address listed on the user account after which correspondence to discuss collaboration can begin.
Using GeneMatcher, the c.3439C>T (p.Arg1147Trp) and c.3415C>T (p.Arg1139Trp) missense variants were identified in two patients. Whole exome sequencing (WES) and pre-/post-processing and genomic alignment (hg19) had already been performed. These two PUM1 variants were identified as the most likely cause of disease in each patient by systematically eliminating variants that are likely benign. Variants found in the probands with a minor allele frequency (MAF) >1% in gnomAD (Karczewski et al., 2020) or ExAC (Karczewski et al., 2017) were filtered out, and remaining variants were prioritized based on pathogenicity prediction algorithms.
The c.3103A>T (p.Thr1035Ser) variant required additional familial segregation analyses because it fell below established pathogenicity criteria and was associated with a milder, late-onset phenotype of ataxia rather than NDD (Figure 2). Direct Sanger sequencing of Exon 19 in 7 family members across two generations showed segregation between the disease phenotype and the variant in all except one member, suggesting incomplete penetrance (Gennarino et al., 2018). (We have since recruited additional patients with late-onset ataxia who bear the p.(Thr1035Ser) variant, which confirms its pathogenicity.) In addition, mildly hypomorphic variants such as p.(Thr1035Ser) may also sporadically appear in population databases (gnomAD) of “healthy” individuals in cases where disease-onset occurs late in life. For some genes, this is not an uncommon occurrence (Kryukov et al., 2007), although such variants are still typically “ultra-rare” in the general population. Nevertheless, such situations should be carefully considered when assessing the pathogenicity of novel disease variants.
Figure 2.

Pathogenicity scores for disease-associated PUM1 variants
CADDv1.6, REVEL, M-CAP and Eigen pathogenicity scores of p.(Arg1147Trp) (red arrowhead), p.(Arg1139Trp) (pink arrowhead) and p.(Thr1035Ser) (blue arrowhead), relative to the distribution (boxplot) of PUM1 singleton (ultra-rare) variant scores found in gnomAD (gray circles). The dotted red arrow delineates the recommended threshold beyond which (pink shaded region) variants are predicted to be deleterious.
Pathogenicity prediction scores were helpful in distinguishing p.(Arg1147Trp) and p.(Arg1139Trp), but p.(Thr1035Ser) was inconsistent (Figure 2). Using singleton missense variants (allele count = 1) extracted from the gnomAD database, we determined the pathogenicity score distribution of ultra-rare missense variants in PUM1 within which the p.(Thr1035Ser) variant falls. Prediction scores for all PUM1 missense variants were obtained using the genetic annotation program ANNOVAR (Wang et al., 2010). The follow is a step-by-step protocol for running ANNOVAR using either a Windows or Apple iOS operating system. We encourage users to adapt the provided script to their own needs and refer to https://annovar.openbioinformatics.org/ for additional resources.
Use ANNOVAR and dbnsfp (version 4.2a) dataset to annotate genetic variants with pathogenicity scores
In this step, PC/Apple iOS users will use the Perl programming language based genetic annotation program, ANNOVAR (Wang et al., 2010) (through an installed or intrinsic Perl interpreter), to obtain functional and pathogenic predictions for a list of variants found in patients. Rather than downloading the entire dataset of numerous functional prediction scores for annotation, which would require local disk space beyond the average capacity of individual work stations (>100 TB), annotations will come from a “lightweight” but comprehensive dataset, dbnsfa (Li et al., 2021; Liu et al., 2011) (∼50–60 Gb), containing all potential nonsynonymous and splice-site SNVs in the human genome.
Note: the scripts provided for annotating the p.(Arg1147Trp), p.(Arg1139Trp), p.(Thr1035Ser) and gnomAD singleton variants are for demonstration purposes. Users should modify and adapt this script according to their own needs.
-
7.Install (or activate) Perl language interpreter
-
a.Windows/PC users:
-
i.Visit the URL: https://strawberryperl.com/ and install program according to operating system (64 bit or 32 bit)
-
ii.Verify installation:perl -v
-
i.
-
b.Apple iOS users: a Perl interpreter comes pre-installed. Perl scripts can run by directly invoking the interpreter:perl myprogram.pl
-
a.
-
8.Install ANNOVAR annotation program
- a.
-
b.Submit registration form and a download link will be emailed to you
-
c.Unzip file into your base directory
-
9.Download dbnsfp 4.2a dataset into the humandb folder
-
a.Open the command (CMD) prompt
-
b.Set working directory to annovar program folder
-
c.Running the following command will download two zipped folders:
-
i.hg19_dbnsfp42a.txt.gz
-
ii.hg19_dbnsfp42a.txt.idx.gzperl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar dbnsfp42a humandb/
 CRITICAL: The hg19 assembly was used here in accordance with the procedures in Gennarino et al., the -buildver argument in annotate_variation.pl command can be changed to hg18 and hg38 accordingly.
CRITICAL: The hg19 assembly was used here in accordance with the procedures in Gennarino et al., the -buildver argument in annotate_variation.pl command can be changed to hg18 and hg38 accordingly.
-
i.
-
d.Manually unzip folder using any generic decompression program (see key resources table)
-
e.Move the respective files in each folder (hg19_dbnsfp42a.txt and hg19_dbnsfp42a.txt.idx) into the humandb folder
-
a.
-
10.
Create .txt file listing the genomic location and nucleotide change of each patient variant. Required (tab-separated) column values for each variant are: chromosome number, start position, end position, reference nucleotide, alternate nucleotide. Headers should not be included in the input file. Depending on the number of variants, this may be done manually using any text editing software (see key resources table).
Note: ensure that the coordinates are consistent with the reference genome build of the annotation data—which in this case is hg19.
-
11.
Place the .txt input file into the annovar folder. (For this example, we will name the file “PUM1_subject_variants.txt” (see Data S1)
-
12.Run the table_annovar.pl script to annotate this file:perl table_annovar.pl PUM1_subject_variants.txt humandb/ -buildver hg19 -out PUM1_subject_variants_output -remove -protocol refGene,dbnsfp42a -operation gx,f -nastring . -csvout -polish -xref example/gene_xref.txtCRITICAL: The hg19 assembly was used here in accordance with the procedures in Gennarino et al., the -buildver” argument in table_annovar.pl command can be changed to hg18 and hg38 accordingly.
-
a.The file “PUM1_subject_variants_output.hg19_multianno.csv” (Data S2) will be generated in the user’s working directory
-
a.
Extract and annotate singleton missense variants in PUM1 from the healthy population for comparison to patient variant scores
-
13.Extract a list of all singleton variants (allele count = 1) from population database
- a.
-
b.Download a .csv file of all PUM1 variants by clicking the “Export variants to CSV” button on this URL page.Note: The downloaded file name is date- and time-stamped and therefore will vary for users. For this protocol, a sample file named: “gnomAD_v2.1.1_ENSG00000134644_2021_10_17_00_08_13.csv” will be used and is provided as Data S3
-
c.Singleton missense variants can be manually extracted from this file using any spreadsheet application (Excel) or text editor by setting the following column filters:
-
i.VEP Annotation = “missense_variant”
-
ii.Allele count = "1"
-
i.
-
14.
After filtering, the values from the Chromosome, Position, Reference and Alternate columns in this file can then be extracted to create a (table_annovar.pl) input file in the format described above.
-
15.Alternatively, this entire process can be performed in a single step using the statistical computing program R and the scripts provided:
-
a.Install the latest version of R and RStudio (version 4.1.1 recommended, see key resources table) and open RStudio application.
-
b.Import the .csv file into RStudio in one of two ways:
-
i.File>Import Database>From Text(base)…
-
ii.Run the following read.csv commandNote: adjust the relative path for “read.csv” function to the directory containing the “gnomAD_v2.1.1_ENSG00000134644_2021_10_17_00_08_13.csv file”,Data S3:gnomAD_v2.1.1_ENSG00000134644_2021_10_17_00_08_13 <- read.csv("∼/gnomAD_v2.1.1_ENSG00000134644_2021_10_17_00_08_13.csv")
-
i.
-
c.The following script will extract all singleton missense variants and generate a .txt input file (in this case, named “PUM1_gnomad_singleton_missense_annovar_input.txt”) (Data S4) in working directory of RStudio ready for annotation with ANNOVAR:PUM1_gnomad_singleton_missense_annovar_input <-subset(gnomAD_v2.1.1_ENSG00000134644_2021_10_17_00_08_13,Allele.Count == 1 & VEP.Annotation == "missense_variant",select = c(Chromosome,Position,Position,Reference,Alternate))write.table(PUM1_gnomad_singleton_missense_annovar_input,file = "PUM1_gnomad_singleton_missense_annovar_input.txt",sep = "∖t",row.names = FALSE,col.names = FALSE,quote = FALSE)
-
d.The file “PUM1_gnomad_singleton_missense_annovar_input.txt” (Data S4) will be generated in the working directory
-
e.Place this file in the “annovar” folder
-
a.
-
16.Annotate this file with the following table_annovar.pl command:perl table_annovar.pl PUM1_gnomad_singleton_missense_annovar_input.txt humandb/ -buildver hg19 -out PUM1_gnomad_singleton_missense_annovar_output -remove -protocol refGene,dbnsfp42a -operation gx,f -nastring . -csvout -polish -xref example/gene_xref.txt
-
a.The file “PUM1_gnomad_singleton_missense_annovar_output.hg19_multianno.csv” (Data S5) will be generated in the user’s working directory
-
a.
Expected outcomes
Step 1 should provide scores and percentile values for each gene of interest that can be recorded and analyzed. The results of this analysis in Gennarino et al. (2018) show that of the 16 genes encompassed by the deletion-spanning region (Figure 1A), PUM1 had the highest probability of being the primary causal gene in each case because the algorithms consistently predicted it to be the most intolerant to pathogenic variation of all the deleted genes (Table 1). These results are provided in Figures 1B–1F. Steps 7 and 8 download and install the Perl interpreter Strawberry and ANNOVAR annotation program. Step 9 downloads the dbnsfp (version 4.2a) annotation dataset within the humandb subfolder within the annovar main folder. Step 12 generates a .csv file named “PUM1_subject_variants_output.hg19_multianno.csv” (Data S2) containing 113 annotation columns for each variant listed in the manually created input file (step 5). Step 13 downloads a .csv file of all gnomAD listed variants for particular gene from the provided URL link (PUM1 example used: “gnomAD_v2.1.1_ENSG00000134644_2021_10_17_00_08_13.csv”, Data S3). Step 15 generates a .txt containing the data for all singleton missense variants across the 5 required tab-separated columns (without header) to be used as the raw input file for annotation using ANNOVAR. Step 16 generates a .csv file named “PUM1_gnomad_singleton_missense_annovar_output.hg19_multianno.csv” (Data S5) of 113 annotation columns for all gnomAD singleton missense variants listed in the input file (step 10). Users can compare the pathogenicity of scores of patient variants to the score distribution of these singleton variants found in the general population. Comparison of CADDv1.6, REVEL, M-CAP and Eigen scores for the PADDAS-associated variants p.(Arg1147Trp) and p.(Arg1139Trp) and the PRCA-associated variant p.(Thr1035Ser) variant scores to the distribution/interquartile range of PUM1 singleton (ultra-rare) missense variants from gnomAD are shown in Figure 2. Certain pathogenicity algorithms may not be available for annotation programs like ANNOVAR (or others) but have web-based interfaces for public use, such as Evolutionary Action (Katsonis and Lichtarge, 2014) (see key resources table).
Quantification and statistical analysis
Functional predictions, based simply on sequencing results from an individual, can help distinguish likely causal variants from non-causal (benign) variants. The current protocol provides users access to these resources and bioinformatic tools to gauge the pathogenicity of variants among a cohort of patients with shared clinical features.
All functional prediction scores have recommended pathogenicity thresholds (i.e., numerical cut-offs beyond which variants are predicted to be likely pathogenic or benign). The manner in which these thresholds are defined varies according to each algorithm. Methods such as REVEL (Ioannidis et al., 2016), are random forest classification algorithms trained to distinguish variants as pathogenic or benign based on data deposited in known clinical databases (ClinVar) (Landrum et al., 2020). Many algorithms base their pathogenicity scores on loss-of-function variants, which of course is not the only mutation type. Statistically comparing the score of one or more variants of an autosomal dominant gene to the confidence interval or interquartile range of the distribution of scores of singleton variants from the (presumably unaffected) general population provides a conservative measure for users to assess pathogenicity.
Limitations
Computational predictions, of course, have their limits, as they represent statistical simulations of biological effects. Different algorithms can produce contradictory predictions for the same variant. Predictions therefore need to be considered in conjunction with other factors such as functional studies and the nature of the phenotype. Please refer to de Prisco et al. STAR Protocols (submitted, STAR-PROTOCOLS-D-21-00615R1) for an approach to functional studies.
Troubleshooting
Problem 1
(Step 1) Missing or unsearchable values for web-based pathogenicity predictions. Unlike the annotation programs that identify genes based on genomic coordinates and human genome reference assemblies, these programs search for gene “symbols” of “ID’s,” which can be problematic for genes with multiple identifiers.
Potential solution
Verify the specific identifier for each program (e.g., EvoTol requires Entrez Gene ID’s) or input alternative ID’s or symbols for a particular gene of interest.
Problem 2
(Step 12 or 16) command prompt error:
'perl' is not recognized as an internal or external command, operable program or batch file.
Potential solution
For PC users, ensure that the Perl interpreter (Strawberry Perl) is correctly installed and, most importantly, within the path of the ANNOVAR command files. After installation, unzip both ANNOVAR and Strawberry Perl into the PC’s main hard drive folder (typically C:∖).
Problem 3
Errors in either of the five required data columns for an ANNOVAR input file (chromosome#, start/end position, and reference/alternate nucleotide) will result in an annotation error for that variant. The most common, particularly when input files are manually assembled, is the inclusion of variants from a different reference genome assembly.
Potential solution
Ensure that all variant coordinates in the input files represent the correct genome reference build (hg18, hg19 or hg 38) when using the ANNOVAR commands (-buildver argument) as they are presented in this protocol. Annotation datasets for each reference assembly should be downloaded separately with annotate_variation.pl:
-buildver hg18
-buildver hg19
-buildver hg38
Accordingly, the -buildver argument in table_annovar.pl should also be indicated in each case.
Various web-based tools to verify the genome reference build of a variants are available (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Users are encouraged to modify the scripts presented in this protocol for their own needs. Further information on using ANNOVAR can be found at https://annovar.openbioinformatics.org/en/latest/misc/faq/.
Problem 4
(Step 15) The subset() and write.table() functions in the Rscript provided at step 15 are included in R versions 3.6.2 and above. Errors may most likely be associated with outdated versions of R.
Potential solution
Verify the version of R by simply typing R.version.string in the R or Rstudio console to print/display the current R version. Visit https://www.r-project.org/ for further details on installing the most updated version of R.
Problem 5
Users who prefer to work with output files (.csv) generated in step 12 (Data S2) and step 16 (Data S5) using proprietary spreadsheet applications like Excel may encounter issues with numerical analyses due to encoding issues with the data. For instance, when importing these files to Excel, the values will likely be stored in each cell as “text” rather than numerical values.
Potential solution
For Excel users, convert the encoding of all values to “Number” by copying and pasting values as follows: Copy>Paste Special (Paste: Values; Operation: Add)
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Vincenzo A. Gennarino (vag2138@cumc.columbia.edu).
Materials availability
No biological reagents were used as part of this protocol.
Acknowledgements
We thank members of the Gennarino lab and Dr. Takayuki Nagasaki for helpful discussions. We also thank V. Brandt for essential input on the manuscript. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS; R01NS109858 to V.A.G.) and the Paul A. Marks Scholar Program, Columbia University Vagelos College of Physicians and Surgeons.
Author contributions
W.L. designed the protocol and wrote the manuscript. N.d.P. edited the protocol. V.A.G. supervised, wrote, and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101150.
Supplemental information
Sample ANNOVAR input file of PUM1 patient variants.
Output file of annotated PUM1 patient.
Sample file containing all PUM1 variants downloaded from https://gnomad.broadinstitute.org/ (accessed October 17th, 2021).
Sample ANNOVAR input file of PUM1 singleton missense variants generated from step 15.
Output file of annotated PUM1 singleton missense variants generated by step 16.
Data and code availability
We provide R and Perl scripts in this protocol. Links to publicly available software are provided in key resources table.
References
- Amberger J.S., Bocchini C.A., Schiettecatte F., Scott A.F., Hamosh A. OMIM.org: online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G.M., Stone E.A., Asimenos G., Program N.C.S., Green E.D., Batzoglou S., Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino V.A., Palmer E.E., McDonell L.M., Wang L., Adamski C.J., Koire A., See L., Chen C.A., Schaaf C.P., Rosenfeld J.A., et al. A mild PUM1 mutation is associated with adult-onset ataxia, whereas haploinsufficiency causes developmental delay and seizures. Cell. 2018;172:924–936.e911. doi: 10.1016/j.cell.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino V.A., Singh R.K., White J.J., De Maio A., Han K., Kim J.Y., Jafar-Nejad P., di Ronza A., Kang H., Sayegh L.S., et al. Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell. 2015;160:1087–1098. doi: 10.1016/j.cell.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D., et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- Kaminsky E.B., Kaul V., Paschall J., Church D.M., Bunke B., Kunig D., Moreno-De-Luca D., Moreno-De-Luca A., Mulle J.G., Warren S.T., et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet. Med. 2011;13:777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alfoldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski K.J., Weisburd B., Thomas B., Solomonson M., Ruderfer D.M., Kavanagh D., Hamamsy T., Lek M., Samocha K.E., Cummings B.B., et al. The ExAC browser: displaying reference data information from over 60000 exomes. Nucleic Acids Res. 2017;45:D840–D845. doi: 10.1093/nar/gkw971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsonis P., Lichtarge O. A formal perturbation equation between genotype and phenotype determines the evolutionary action of protein-coding variations on fitness. Genome Res. 2014;24:2050–2058. doi: 10.1101/gr.176214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov G.V., Pennacchio L.A., Sunyaev S.R. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C., et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835–D844. doi: 10.1093/nar/gkz972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Zhi D., Wang K., Liu X. MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. bioRxiv. 2021 doi: 10.1101/2021.04.09.438706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Jian X., Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Li C., Mou C., Dong Y., Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Medicine. 2020;12:103. doi: 10.1186/s13073-020-00803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S., Gussow A.B., Wang Q., Halvorsen M., Han Y., Weir W.H., Allen A.S., Goldstein D.B. The intolerance of regulatory sequence to genetic variation predicts gene dosage sensitivity. Plos Genet. 2015;11:e1005492. doi: 10.1371/journal.pgen.1005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S., Wang Q., Heinzen E.L., Goldstein D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinero J., Bravo A., Queralt-Rosinach N., Gutierrez-Sacristan A., Deu-Pons J., Centeno E., Garcia-Garcia J., Sanz F., Furlong L.I. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45:D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham O.J., Shihab H.A., Johnson M.R., Petretto E. EvoTol: a protein-sequence based evolutionary intolerance framework for disease-gene prioritization. Nucleic Acids Res. 2015;43:e33. doi: 10.1093/nar/gku1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs E.R., Jackson L., Miller D.T., Van Vooren S. Phenotypic information in genomic variant databases enhances clinical care and research: the International Standards for Cytogenomic Arrays Consortium experience. Hum. Mutat. 2012;33:787–796. doi: 10.1002/humu.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnstrom K., Mallick S., Kirby A., et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sample ANNOVAR input file of PUM1 patient variants.
Output file of annotated PUM1 patient.
Sample file containing all PUM1 variants downloaded from https://gnomad.broadinstitute.org/ (accessed October 17th, 2021).
Sample ANNOVAR input file of PUM1 singleton missense variants generated from step 15.
Output file of annotated PUM1 singleton missense variants generated by step 16.
Data Availability Statement
We provide R and Perl scripts in this protocol. Links to publicly available software are provided in key resources table.